Chapter 27 Biotechnologically produced products
Proteins as drugs
Introduction
Proteins have been used successfully as drugs since the early twentieth century when insulin was first used to treat diabetes. The isolation of insulin was followed by the development of other protein drugs that were mainly extracted from animal tissues until the 1970s when smaller peptides, which had been discovered by extraction from animal tissues, became available via chemical synthesis. Table 27.1 lists some of these older protein drugs with their therapeutic actions.
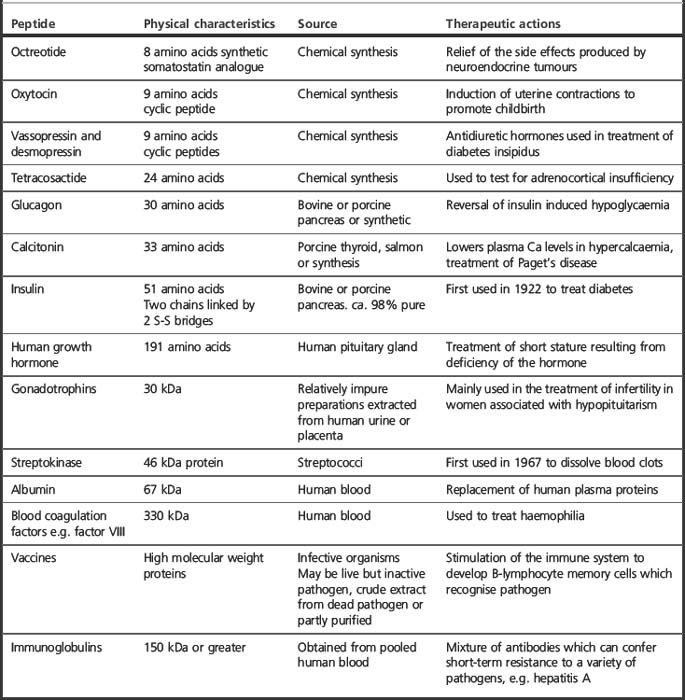
In the case of small peptides with fewer than 20 amino acid residues, it is usually more economical to carry out chemical synthesis rather than to rely on biotechnology for production. Oxytocin with 9 amino acid residues is made synthetically. Calcitonin with 32 amino acid residues can be made synthetically but the rDNA product salcitonin (salmon calcitonin) is the main product in use. For peptides with more than 50 amino acid residues, it is unlikely that synthesis will ever compete with rDNA/biotechnological methods.
Disadvantages and advantages of protein drugs
After the difficulties of producing a drug using biotechnology have been overcome, there are other major difficulties in both registration and administration of the drug:
Production of peptide drugs by chemical synthesis
Automated peptide synthesis is quite routine now, although the expense of the reagents and difficulty of purification when more than 20 amino acids are incorporated into a peptide means that it is only viable for smaller peptides.
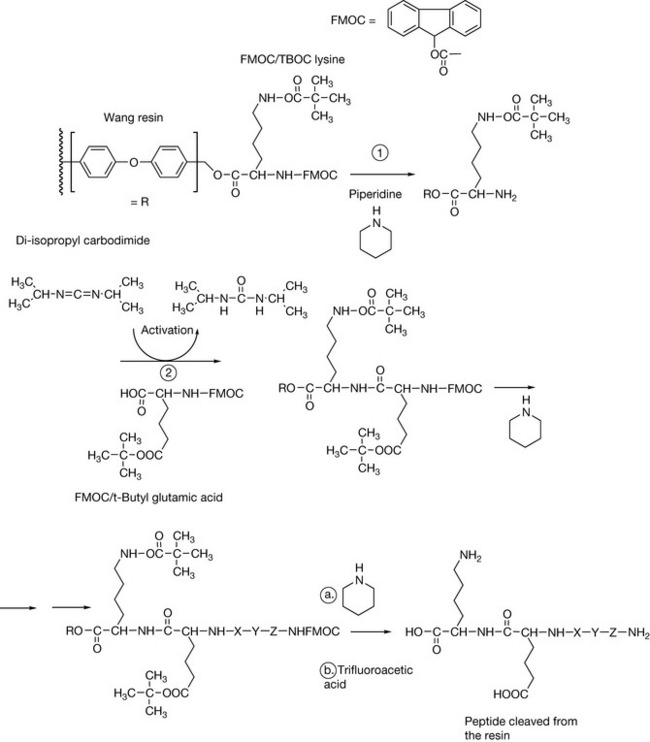
In order to synthesise a peptide, one end has to be protected so that the amino acid does not polymerise with itself. Also, if the side chain of the amino acid has an acidic or a basic group, it also has to be protected so that it does not become modified by the amino acid which is being added to extend the chain. The peptide is synthesised by protecting the carboxylic acid group of the first amino acid in the sequence by linking via esterification onto a resin (Fig. 27.1). The most commonly used resins these days are the Wang resins. The carboxylic acid ester is formed with a benzyl alcohol group which makes it a bit more labile than a link to an aliphatic alcohol. The amine group on each amino acid used in extending the chain is protected by a fluorenyloxy carbonyl (FMOC) group. This group can be removed in order to extend the chain using the base piperazine. If the amino acids contain side chains with either an acid or an amine group in them, e.g. lysine or glutamic acid, these groups are protected with either tertiary butyloxy carbonyl (tBOC) or by esterification with tertiary butanol, respectively. Importantly, the groups protecting the side chain are not removed by piperazine treatment and remain in place until the finished peptide is cleaved from the resin with trifluoroacetic acid which also removes the side chain protecting groups. As indicated above, this process is generally only viable for peptides with less than 20 amino acids. However, technology advances, and Roche have recently licensed Fuzeon™ which is a 36 amino acid synthetic peptide used in the treatment of AIDS. Figure 27.2 shows two examples of synthetic peptides used as drugs. Although these drugs are made by peptide synthesis methods, some of the amino acids have been modified so they are not entirely made up of naturally occurring amino acids. Octreotide is used to treat the symptoms produced by neuroendocrine tumours and also in reducing vomiting during palliative care. It is an analogue of somastatin, a tetradecapeptide which acts on the hypothalamus, inhibiting release of various hormones. It is used particularly to treat carcinoid tumour, which is a gastrointestinal tumour which secretes large amounts of serotonin. The tumour expresses somastatin receptors, and octreotide as a potent agonist for these receptors is effective in suppressing serotonin secretion. Lanreotide is another synthetic peptide which is used in treating carcinoid tumour and also thyroid tumours.
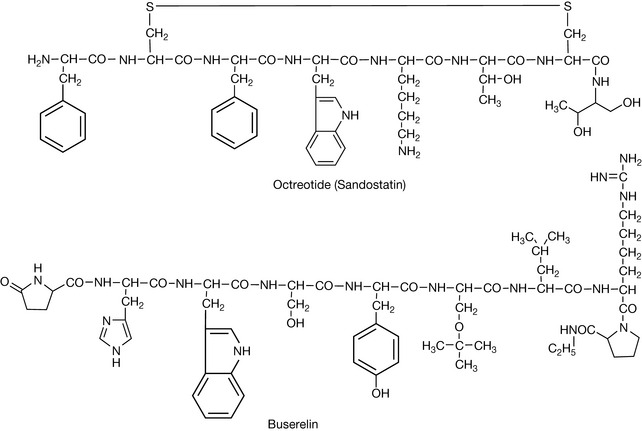
Buserelin is, again, a peptide-like structure with some natural and some unnatural amino acids in its structure. It is an agonist of gonadotropin releasing hormone and is used to treat prostate cancer (see Ch. 21) by suppressing the release of testosterone by the tumour. There are a number of other peptide analogues in the same category: goserelin, leuprorelin and triptorelin.
Production of protein and peptide drugs by genetically modified bacteria
Introduction
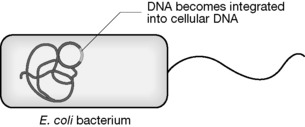
The technology for large-scale culture of bacteria is over 50 years old and bacteria can be cultured in stirred tank ‘reactors’ which may have a capacity of up to 10 000 litres. Escherichia coli is by far the most extensively used bacterium for the expression of rDNA. Human insulin (Humulin, Eli Lilly), the first biotechnologically produced peptide, was produced using transformed E. coli and this was quickly followed by the production of human growth hormone by this method.
Disadvantages of the production of rDNA drugs in bacteria
Strategies for improving the production of rDNA drugs by bacteria
An example of the production of a rDNA drug by E. coli
Insulin is secreted by the pancreatic β-cells and is required for the synthesis of the glucose storage polymer glycogen and is also required for the entry of glucose into tissues. Reduced glucose entry into tissues is the major effect of insulin deficiency and this produces an increased level of glucose into the circulation due to inhibition of glycogen biosynthesis. The effects of glucose deficiency in tissues are: accelerated protein catabolism, accelerated lipid metabolism and decreased lipid synthesis. Increased glucose levels in plasma lead to hyperosmolarity and dehydration and some complications may arise from the reactivity of glucose itself. The effects of insulin deficiency can be eliminated by insulin injection.
Diabetes is the third leading cause of death in the USA. Treatment of diabetes with insulin began in 1921 following its discovery at the University of Toronto and, as a consequence, rather than facing certain death diabetics were able to have a normal lifespan. The early insulin preparations were crude extracts from animal pancreas. They tended to promote allergic reaction due to the presence of additional proteins and required frequent injection because of their low purity. Purification steps were later introduced involving alcohol/acid precipitation of the protein and later crystallisation as the zinc salt (the basic crystal form involves six insulin molecules and two zinc atoms). Modern extracted insulins are subjected to a chromatographic step, gel-filtration, in order to remove allergenic contaminating proteins. Human insulin produced by transformed E. coli became available in 1982.
The structure of insulin
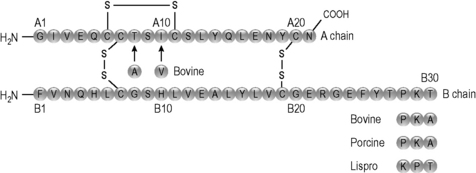
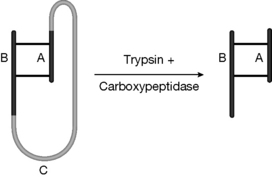
Insulin is a 50 amino acid protein which, unlike many peptides, is not glycosylated. It is produced naturally as three chains, the A, B and C chains. The C chain links the A and B chains and is removed by the body when the insulin is activated, leaving the A and B chains linked by two S–S bridges (Fig. 27.3). The metal ion zinc is also involved in stabilising the peptide, and insulin is stored in pancreatic β-cells as a hexamer complexed with two zinc atoms.
The need for biotechnologically produced insulins
There was a strong requirement for a good production process for insulin because:
The biotechnological process leading to the production of human insulin
In the process used by Lilly, insulin is produced in the form of proinsulin as a fusion protein where it is joined to the protein tryptophan synthase via a terminal methionine. The tryptophan synthase is then removed by cleavage with cyanogen bromide. The proinsulin is then converted to insulin via treatment with a mixture of the protease enzymes trypsin and carboxypeptidase.
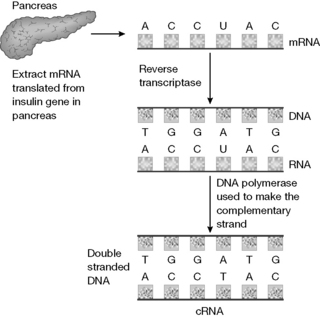
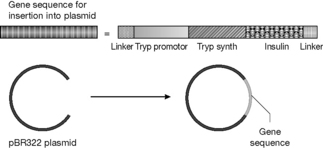
Quality control of recombinant insulin
Particularly stringent checks are needed to control the quality of the insulin produced because of the complexity of the molecule and the fact that it is derived from a biological system that could potentially be variable. High-performance liquid chromatography (HPLC) systems have to be capable of separating human insulin from the closely related animal insulins which differ from it by only one or two amino acids.
HPLC provides a highly specific technique which can distinguish between human insulin and animal insulins differing from it by one or two amino acids. In addition to making sure that the primary structure of the peptide is correct, the secondary structure of the peptide also has to be checked to ensure that it is correctly folded. This is carried out using a technique called circular dichroism which is related to the optical rotation technique used to determine the relative configuration (i.e. [+] or [−]) of small drug molecules. When the circular dichroism ‘fingerprint’ of a standard for insulin and a test sample of insulin are identical, they can be said to be arranged in three-dimensional space in the same way. The product is also quality controlled for biological activity by injection into a rabbit followed by the monitoring of blood glucose levels.
A major advance in simplifying quality control of biotechnologically produced drugs has been the development of electrospray mass spectrometry, which can measure exactly the molecular weight of a particular protein and also detect and characterise small amounts of contaminating proteins. Figure 27.8 shows the electrospray mass spectrum of human insulin.
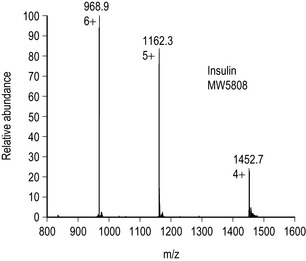
Figure 27.8 Electrospray mass spectrum of human insulin showing multiply charged ions. Insulin contains 6 basic centres: 2 × histidine, 1 × arginine, 1 × lysine and terminal amine groups on the A and B chains hence the 6+ ion. 6 × 968.9 −6 (for the 6 added protons) = 5808 which is the MW of human insulin.
Modern insulins
Formulation types
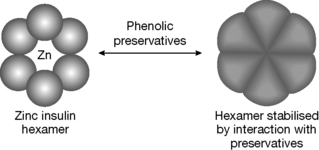
Recent developments in insulin presentation have been reviewed.1 Initially, insulins were formulated in solution under acidic conditions in order to improve their stability. However, there was a tendency for the amide bond in the C-terminal asparagine residue to hydrolyse, with consequent loss of potency. As a result, the use of zinc to stabilise soluble formulations became common. The stability of such formulations is further enhanced by the presence of phenolic preservatives in the formulation which cause a change in the conformation of the zinc hexamers (Fig. 27.9) to render them even more stable.
Sources of insulin
Bovine insulin
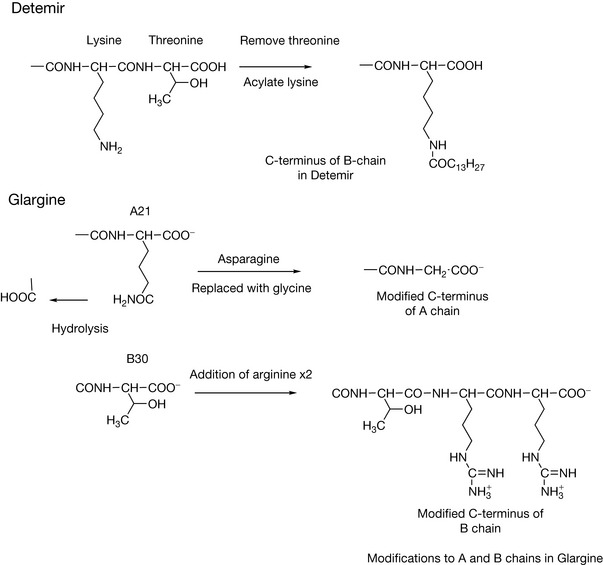
Bovine insulin differs from human insulin by three amino acids (alanine for threonine A-chain 8, isoleucine for valine A-chain 10, alanine for threonine B-chain 30; see Fig. 27.3). Bovine insulin does promote antibody production to some extent and doses may need to be adjusted to compensate for this.
Porcine insulin
Porcine insulin differs from human insulin by one amino acid having alanine instead of threonine at B-chain 30 (see Fig. 27.3). The difference in structure from human insulin is not sufficient for antibody production to occur.
Human insulin types
There has been some concern that human insulin is more likely to produce hypoglycaemic shock but, on the other hand, it is less likely to produce wasting away of fatty tissue at the injection site in comparison with animal insulins. Concern over the tendency of human insulin to produce hypoglycaemic shock has led to the production of analogues of human insulin that have fewer tendencies to cause this.
Human insulin analogues
Since insulin binds to its receptor as a monomer, any structural alteration which reduces insulin self-association results in more rapid action. Rapid-acting analogues were developed so that insulin could be injected immediately before a meal so that carbohydrate absorption and insulin action correlated more closely.
Rapid-acting analogues (reduced self-association)
Long-acting insulins
In recent years, analogues of insulin with modified structures have been produced which have prolonged duration of action, thus reducing the need for frequent injection.
Insulin formulations
Long/lntermediate acting
 Self Test 27.2
Self Test 27.2
Human growth hormone
Human growth hormone (HGH) is secreted by the pituitary gland and plays a key role in somatic growth through its effects on the metabolism of proteins, carbohydrates and lipids; it stimulates cell proliferation at growth plates in bones by direct binding to receptors in these tissues. It is present at high levels in children and also becomes elevated in response to exercise. Low levels of HGH are associated with obesity. HGH also increases calcium absorption by the gut, increases glomerular filtration and stimulates erythrocyte production by bone marrow. HGH is well established for the treatment of short stature in children. It is being tested in a number of other applications including: osteoporosis, renal failure, treatment of severe burns and wound healing. Intranasal delivery is being evaluated. Side effects are infrequent, although the safety of the product is not absolutely established.
HGH was originally extracted from the pituitary gland of human cadavers and the extracted product was found in some cases to produce the Creutzfeld-Jacob disease, which is caused by a prion protein. HGH is a 191 amino acid non-glycosylated protein. The amino acid sequences vary greatly between species and non-primate growth hormones have little activity in man. The commercial products are produced by transformed E. coli: Genotropin™ (Pharmacia), Humatrope™ (Lilly), Norditropin™ (Novo Nordisk), NutropinAq™ (Ipsen) and Zomacton™ (Ferring) or mammalian cultures Saizen™ (Serono). All of the commercial products are chemically equivalent.
Human growth hormone antagonists
Pegvisomant is a mutated form of HGH.2,3 The protein is altered so that it functions as an antagonist of HGH by binding strongly at one binding site of the HGH receptor but only weakly at the other binding site (Figs 27.11, 27.12).
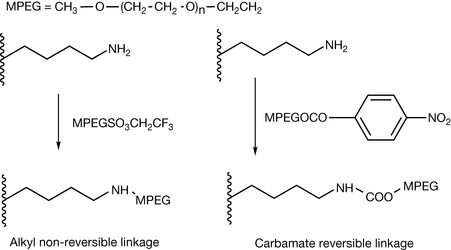
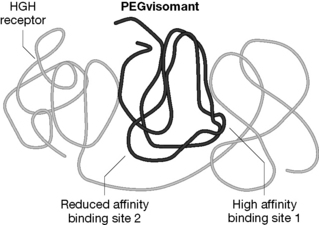
In order to achieve the reduced binding to the HGH receptor, 8 amino acids at the binding site were altered. In addition, the protein is PEGylated at 4-6 lysine residues (Box 27.1). One of the amino acids replaced at binding site 1 was a lysine residue. This was carried out in order to ensure that the presence of PEG did not reduce the binding of the protein to the receptor to a great extent. Additionally, a lysine amino acid was introduced at binding site 2, thus increasing the likelihood of PEGylation at this site with consequent reduced binding affinity. The presence of PEG at the other positions reduces the overall binding affinity of the protein but greatly increases in circulating half-life compared with HGH. Pegvisomant thus functions as an antagonist of HGH and is used to treat acromegaly.
Reaction of proteins with polyethylene glycol (PEGylation) is of increasing importance for manipulating the pharmacokinetics of therapeutic proteins.2 The polyethylene glycol chains are largely linear polymers around 12 kDa; however, the use of branched chains is becoming more common. PEG chains are very bulky because of the water associating with them and thus shield sites of proteins which might cause immunogenic reaction or be susceptible to proteolytic degradation. PEGylation generally results in increased circulating half-life for a therapeutic protein. The disadvantage of PEGylation is that it can reduce the activity of the therapeutic protein by preventing it binding efficiently to its site of action. The PEGs used are usually methylated at one end to avoid producing reagents reacting at both ends which would crosslink the protein. The PEGs can either be linked by a strong covalent bond which is not easily reversible, e.g. C–N, or via a linkage such as carbamate which is reversible (Fig. 27.11). Reversible linkages are used where the presence of the PEG chain reduces the activity of the protein. These chains are removed by enzymatic hydrolysis in the body. Lysine residues are a popular target for modification because they are both reactive and common in proteins.
Colony stimulating factors
Granulocyte colony stimulating factor (G-CSF) is a haemopoietic growth factor which is involved in promoting the differentiation of stem cells formed in the bone marrow into granulocytes (neutrophils, basophils, eosinophils) which are responsible for killing bacteria in the blood. It is used to rectify myelosuppression in patients undergoing cancer chemotherapy where white cells in the blood are depleted by the chemotherapeutic agent. The protein occurs either as a 174 or a 180 amino acid protein. The commercial product filgrastim (Neupogen™) is produced in non-glycosylated form by transformed E. coli. Pegfilgrastim is a PEGylated version of the protein which has an increased circulating half-life, reducing the necessity for daily injections. Lenograstim is another form of G-CSF produced by CHO cells; unlike filgrastim it is glycosylated and is slightly more potent.
Interferons and other cytokines
Interferons (IFNs) are a group of inducible cytokines which have antiviral properties. IFN-α and -β are inducible in several cells while IFN-γ is produced by T lymphocytes.
α-Interferon
Recombinant α-interferon2a (Roferon-A, Roche) is produced in the form of a 165 amino acid protein that is unglycosylated. The drug is used to treat Kaposi’s sarcoma and also hepatitis B and hepatitis C. Interferon α2a (IntronA, Schering-Plough) has similar indications. It is produced by genetically transformed E. coli. It is also produced in a PEGylated form in order to increase its circulating half-life.
β-Interferon
Interferon-β1b is an 18.5 kDa protein that is produced commercially using genetically transformed E. coli. The gene inserted into the E. coli was originally isolated from human fibroblasts and was engineered so that a cysteine residue present at position 17 was replaced by a serine residue which improves its stability during synthesis in E. coli. The commercial product, Betaferon® (Schering), is predominantly used for the treatment of relapsing-remitting multiple sclerosis. The discovery of its use in MS therapy was based on the unproven theory that MS might have a viral aetiology. Interferon β1a is produced by CHO cells (Avonex®, Biogen). It is identical to human interferon β and has similar indications to betaferon. Rebif™ is a differently formulated version of interferon β1a.
γ-Interferon
γ-Interferon is produced in the form a of a 140 amino acid single-chain polypeptide γ-Interferon1b. It occurs naturally in a glycosylated form but the commercial product is unglycosylated and is expressed in E. coli. It is used in the treatment of chronic granulomatous disease and severe malignant osteoporosis where it is believed to increase the activity of phagocytic cells.
Interleukin-2
Interleukin-2 is a 15.5 kDa protein which stimulates the production of T lymphocytes. The protein occurs naturally in glycosylated form but the unglycosylated form is also active. The commercial product is produced in the unglycosylated form by genetically transformed E. coli. The drug is used to treat metastatic renal cell carcinoma.
Production of peptide drugs by animal cell cultures
Introduction
Animal cells are capable of producing peptides which are closer to naturally occurring human peptides, i.e. they are capable of carrying out post-translational modifications of proteins such as glycosylation, phosphorylation and alterations in the tertiary structure of the protein. It is not always necessary to insert rDNA into animal cells to get them to express a required protein. A line of cells in culture may naturally produce the protein or the cells may be stimulated to produce the protein, e.g. using a virus. This means that the techniques to stimulate peptide production by animal cells are more diverse than those used for production using E. coli.
Post-translational modification
In animals, once the protein has been synthesised within the endoplasmic reticulum, it must be folded correctly, sorted and finally transported. Proteins that are secreted by a cell or are incorporated into the cell membrane undergo five principal types of modification: the formation of disulphide bonds, proper folding of the protein, addition and modification of carbohydrates, specific proteolytic cleavages, and formation of multiple chain proteins. Proteins that have not been assembled properly are likely to be enzymatically degraded. In bacterial cells, the formation of disulphide bonds may not occur correctly. In a protein containing several cysteine residues a number of permutations of S–S bond formation are possible and a mammalian cell production system is more likely to produce the correct combination of S–S bonds. It is possible to refold the protein post-production but there is no guarantee that the correctly folded protein can be easily produced. Often, the most critical modification, which mammalian cells are required for, in order to obtain full biological activity for a biotechnologically produced protein, is the post-translational glycosylation of a protein. This is not always critical for the biological activity of the protein but, where it is required, the protein must be expressed in mammalian cells. The sugar chains on proteins are fairly small, up to ca. 15 monosaccharide units in the most complex cases. Such short chains of sugar units are known as oligosaccharides. The glycoside chains are either O-linked, linked to serine of threonine side chains, or N-linked to the amide nitrogen of asparagine. There are principally eight sugars involved in building the oligosaccharide chains. These are: glucose, galactose, mannose, fucose, xylose, N-acetylglucosamine, N-acetylgalactosamine and neuramic (sialic) acid (Fig. 27.13). Small variations in such chains can produce large differences in the way that the body responds to a protein. For instance, variation in a single residue in a glycoside chain within the glycoproteins in the membrane of erythrocytes determines blood group. Thus the immune system of people who cannot synthesise the A or B antigens will destroy erythrocytes which contain the antigen which is not present in their cells. People with the O blood group can safely donate to people with A and B antigens since the O antigen lacks the extra sugar present in the A and B antigens and thus does not trigger an immune response.
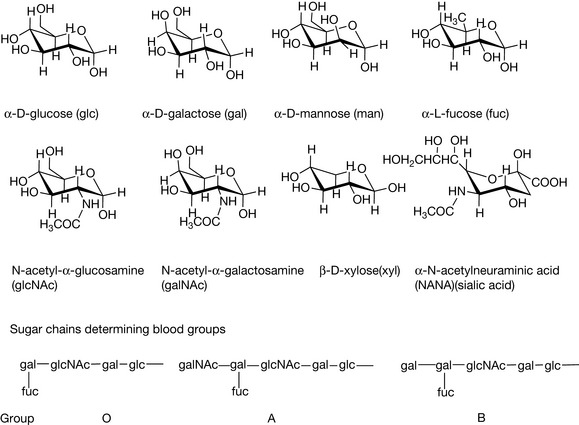
Figure 27.13 Monosaccharides commonly found in oligosaccharide chains in glycoproteins and glycosidic determinants of blood groups.
For biotechnologically produced mammalian proteins such as erythropoietin and tissue plasminogen activating factor the correct glycosylation pattern is essential for activity.
Advantages of producing peptides using animal cell cultures
Disadvantages of producing peptides using animal cell cultures
Strategies for improving the production of peptides by animal cell cultures
An example of the production of a peptide drug by animal cell cultures in detail
Introduction
Erythropoietin (EPO) is produced mainly by the kidneys and acts on stem cells in the bone marrow, stimulating mitosis of erythroid progenitor cells (Fig. 27.14). It increases red cell production in response to reduced oxygen delivery to the kidney. Thus administration of the drug can correct anaemia caused by EPO deficiency such as occurs in chronic renal failure (CRF). There are many causes of CRF and to date the only means of correcting it are either transplantation or dialysis. A major side effect of the disease is anaemia that is due to EPO deficiency. This is caused in part by dialysis. Prior to the advent of biotechnologically produced EPO the only means of treating the anaemia associated with CRF were frequent blood transfusions or the administration of anabolic steroids; both methods of treatment have attendant risks and side effects. EPO is highly effective in correcting anaemia and eliminates the need for transfusions. The therapeutic goal is a haematocrit (packed red cell volume) of >0.3. Side effects are generally not serious but may include: exacerbation of hypertension; extracorporeal blood clotting; iron deficiency; seizures; flulike syndromes and headache. The most commonly reported side effect is exacerbation of hypertension and this may be treated with antihypertensive drugs. EPO is an ideal drug of abuse in sport since it raises red blood cell levels yet is very difficult to distinguish from the EPO that is naturally present. However, variations exist in the glycosylation pattern between the natural and the biotechnologically produced drug and these can be used for detection of this form of drug abuse.
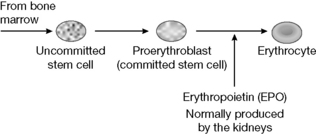
The structure of EPO
EPO is a 166 amino acid glycoprotein that is heavily glycosylated (almost 50% of the molecular weight [MW] is due to glycosylation) and it has a molecular weight of 34 kDa. Glycosylation is essential for biological activity, and removal of terminal sialic residues from the glycan chains in the protein greatly reduces its half-life in the body. Animal cells have to be used in the production process in order to make a product with the correct glycosylation pattern.
The biotechnological process leading to EPO production

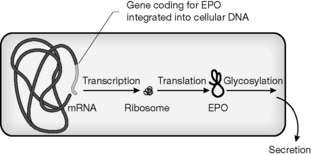
A commercial supply of EPO was first produced by Amgen. The two products sold in the UK are Eprex® (Jannsen-Cilag) and NeoRecormon (Roche). These products are almost identical in structure, having the same amino acid sequence but very slight differences in glycosylation pattern. A version of EPO which is more extensively glycosylated called Darbepoetin is also sold by Amgen. The increased level of glycosylation gives this version of EPO a longer circulating half-life which means that it does not have to be administered as frequently.
Other rDNA drugs produced using animal cell culture
Tissue plasminogen activator (TPA)
Tissue plasminogen activator (TPA) is a 527 amino acid serine protease glycoprotein with a molecular weight of 64 kDa. About 8% of the MW is due to carbohydrate. The gene has been expressed in both E. coli and CHO cells. The commercial products Alteplase® (Genentech) and Tenecteplase® (Boehringer Ingleheim) are produced by CHO cells. Related products include Reteplase® (Roche) which is expressed in E. coli and only contains part of the TPA structure but is equally effective. Different products may have different glycosylation patterns but such variations do not greatly affect biological activity.
TPA is the most important physiological activator of plasminogen, the clot-dissolving enzyme. It is locally released from blood vessels and binds to the fibrin clot with simultaneous binding of plasminogen, and this results in the digestion of the clot.
There is a danger of haemorrhage although contraindications are well established as being: a history of cerebrovascular accident, internal bleeding, and severe uncontrolled hypertension. TPAs with varying glycosylation patterns are being produced in order to try to reduce some of the side effects of the drug and in order to increase the plasma half-life of the drug so that it can be administered as a bolus injection rather than by continuous infusion.
Factor VIII, factor VIIa and factor IX
Factor VIII is a complex of proteins involved in the blood-clotting cascade that results in the production of fibrin. A genetic abnormality resulting in a lack of factor VIII production results in haemophilia. The protein prepared by fractionation from human plasma is still extensively used but recombinant forms are also now available. The most important elements in the structure of factor VIII are two protein chains of 80 kDa and 90 kDa which are glycosylated to various extents. The two commercially available products are: Recombinate® (Baxter Health Care) which is prepared using genetically transformed CHO cells, and Kogenate® (Bayer/Miles) which is prepared using transformed baby hamster kidney cells. Recombinant versions of the blood-clotting proteins factor VIIa and factor IX are now also available.
Etanercept
Etanercept (Enbrel™) is used in the treatment of rheumatoid and psoriatic arthritis. It is produced using CHO cells. The protein acts as a soluble version of the tumour necrosis factor (TNF-α) receptor, TNF is involved in the pathology of rheumatoid arthritis (RA) and psoriasis. The soluble receptor binds to TNF before it can bind to TNF receptors on the inflammatory cells within the body, thus interrupting the inflammatory cascade.
Anakinra
Anakinra (Kineret™) provides another strategy for the treatment of RA. It is a recombinant version of the native interleukin-1 (IL) receptor antagonist IL-1Ra. IL-1 may have an even greater role than TNF-α in promoting RA through inhibiting proteoglycan synthesis and stimulating bone resorption. There is evidence to suggest that an imbalance between IL-1 and IL-1Ra exists in the rheumatic joint. Recombinant IL-1Ra differs from native IL-1Ra in that it has a methionine residue at its N-terminus.
Growth factors
Growth factors are involved in tissue repair (Box 27.2) and are a potentially exciting class of drugs. They include: epidermal growth factor (EGF); fibroblast growth factor (FGF); platelet derived growth factor (PDGF); transforming growth factor (TGF) and insulin like growth factor (IGF).
Box 27.2
The following events occur in response to injury of tissue:
The only agent that has made it to the market is PDGF, which produces a chemotactic response in fibroblasts and smooth muscle, is an attractant for inflammatory cells and induces collagen synthesis. PDGF is available as a gel Regranex® for use in the treatment of diabetic foot ulcers where it promotes wound healing.
Antibodies and antibody therapy
What are antibodies?
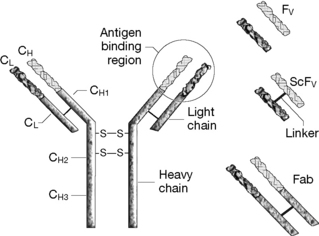
Antibodies are immunoglobulin proteins, composed of four protein chains, two heavy chains (MW >53 000) and two light chains (MW ca. 23 000) linked together by S–S bridges. They constitute 20% of the proteins circulating in the blood. Antibodies are Y-shaped molecules having a constant region, which classifies the antibody as being of the IgA, IgD, IgE, IgG or IgM type (the effector domain) and a variable or idiotype region which is responsible for recognition and binding to a specific antigen (Fig. 27.18). The idiotype region is within the arms of the Y and an antigen binds in a lock-and-key fashion to the sites at the end of each arm. An antigen tagged in this way is recognised as foreign by the immune system and destroyed. The constant region determines the function carried out by the antibody as detailed below. Antibodies are produced by B lymphocytes and there are five types:
Production of antibodies
Antibodies are produced by B lymphocytes which circulate in the blood stream. The B cells are responsible for recognising antigens and tagging them so that they can be destroyed by white blood cells and by T lymphocytes.
Antibodies contain a number of discrete regions (Fig. 27.18), each of which is composed of about 110 amino acids, and the biotechnology of antibody production has developed through expression of parts of the antibody in various different cell systems. The various regions of importance are summarised in Table 27.2.
| Region | Function | Comments |
|---|---|---|
| VL | Variable region responsible for antigen recognition at the N-terminus of the light chain | Composed of four β-pleated sheets joined by 3 loops referred to as hypervariable loops or complementarity determining regions Two types of light chain are coded for by κ or λ genes About 60% of human antibodies contain κ chains |
| VH | Variable region responsible for antigen recognition at the N-terminus of the heavy chain | As for the light chain composed of four β-pleated shoots joined by 3 loops VH displays even greater diversity VL |
| CL | Single constant region of the light chain | Invariant part of the κ or λ gene sequence |
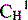 |
First of three constant regions within the heavy chain above the hinge region | |
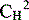 |
Second of the constant regions of the heavy chain Contains glycosylation sites with sugar chains N-linked to asparagine residues |
Glycosylation pattern may be important for the structural integrity of the antibody and for its effector functions such as recruitment of immune cells and complement activation |
 |
Third constant region of the heavy chain containing the C-terminus region | Contains the effector binding sites |
| VL VH CL  |
The Fab′ fragment is half of the antigen recognition site plus part of the constant chain | Has one antigen binding site |
| VL VH | The ScFV fragment consisting of the variable regions of the light and heavy chains | Retains the full binding capacity of the antibody but lacks the effector functions Joined together with a linker sequence |
Therapeutic antibodies in the BNF
Antisera
Antibodies can be seen immediately to represent a method for therapeutic intervention since in cases of infection they can cause the body to recognise the infective agent and destroy it. Perhaps their use is most familiar in the dramatic circumstances of the use of antisera against snakebite. In this case, a fairly crude preparation containing antibodies prepared from the serum of animals exposed to the toxin is used to trigger the body into rapidly recognising and destroying the toxin. Such antisera are available for treatment of serious infectious diseases such as botulism and diphtheria. The use of antibodies is distinct from vaccination, which is discussed later in this chapter, in that the treatment is not dependent on stimulation of B cells to produce antibodies but immediately provides the body with a means of recognising the infection. They are only used in serious conditions since the antisera are produced in animals and have a high potential for triggering an allergic reaction to the animal protein.
Immunoglobulins
The use of partially fractionated human immunoglobulins (IgGs) represents a more refined approach to passive vaccination than the use of antisera. Normal immunoglobulin is harvested from pooled serum and confers immunity to a number of common infectious diseases, most prominently hepatitis A, measles and rubella. For many years it was the only source of protection against hepatitis A before the advent of a vaccine against the virus. More specific IgGs are prepared from pooled serum from selected donors with a high level of an antibody against a particular condition. More specific IgGs are available against hepatitis B, tetanus, rabies, tetanus, cytomegalovirus and varicella-zoster. Prophylaxis using IgGs is particularly effective because IgGs have a circulating half-life of several weeks. Of course, vaccination offers much longer-term protection.
Monoclonal antibodies
Introduction
Since antibodies are able to recognise and modulate the activity of other proteins within the body, they have great therapeutic potential. In 1975, Kohler and Milstein in a remarkable experiment showed that antibody-producing B lymphocytes could be fused with malignant rapidly proliferating myeloma cells (i.e. which contain oncogenes conferring immortality) and the hybrid myeloma cells or hybridomas could both express lymphocyte-specific antibodies and continue to proliferate. This provided the biotechnological basis for producing antibodies on a large scale.
Processes used for monoclonal antibody production (e.g. production of an antibody to tumour necrosis factor)
In the original experiment leading to monoclonal antibody (MAb) production the antibody-producing B lymphocytes were obtained by injecting a rabbit or mouse with the appropriate antigen (Fig. 27.19). For example, an antibody to tumour necrosis factor, a peptide involved in the autoimmune response, might be required. The spleen of the animal was removed and lymphocytes isolated from the spleen were fused with mouse myeloma cells by treatment with polyethylene glycol or Sendai virus.
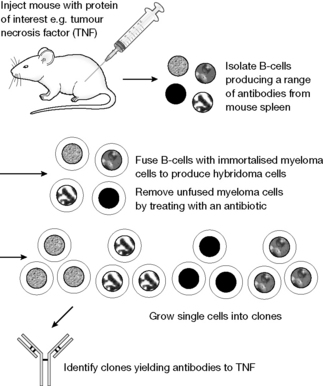
The hybridoma cells were selected by poisoning any remaining unfused myeloma cells with aminopterin, which selectively blocked their DNA synthesis. The hybridoma cells survived the antibiotic treatment and single cells were then grown into clones and these clones were screened for production of the required antibody (a range of antibodies will be expressed by the mixture of lymphocytes originally isolated). In the example shown, clone 1 is selected as the one producing TNF antibodies. MAbs could then be produced on a large scale in air-lift fermentors of up to 10 000 litres. This volume of culture can produce up to 1 kg of MAb. Now the methodology for producing MAbs is more variable and in part depends on the application for which the antibody is required. In recent years there has been a move away from this type of production process towards the genetic engineering processes, described below, where the monoclonal antibodies are produced in mammalian cell lines such as CHO cells.4
Chimaeric antibodies
Pure murine antibodies produced as described above are allergenic in humans and thus are either toxic or have reduced half-lives in the body. In order to reduce these effects, chimaeric antibodies are produced where the constant regions of the heavy and light chains are replaced by human protein. The genes for all the human immunoglobulin subtypes have been cloned and this allows the replacement of all the genetic material coding for a murine antibody, apart from that coding for the variable region, with DNA coding for human protein. These hybrid genes can be expressed in a number of different cell cultures including mammalian and insect cell cultures. The most complete replacement of murine protein with human protein is in the humanised antibodies, where all the genetic material coding for the antibody is replaced with human gene sequence apart from the hypervariable regions. The disadvantage of replacing all but the hypervariable regions with human protein is that some binding specificity may be lost since the conformation of the β-pleated sheets present in the variable region of the original murine antibody may be optimal for binding of the hypervariable regions to the antigen. One way around this is to introduce the gene coding for human IgG into mouse embryos. When the mice developed from these embryos are challenged with an antigen, they produce B cells secreting fully human antibodies. These B cells may be isolated and fused with myeloma cells, thus producing human monoclonal antibodies.
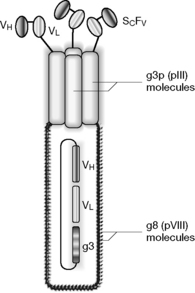
Phage display technology
Phage display technology and related techniques such as ribosomal and yeast display systems form the basis of modern monoclonal antibody production.5-9 Bacteriophages are viruses that infect a range of bacteria. They only have 11 genes. Figure 27.20 shows a schematic diagram of a phage. The genetic material which is to be expressed as a protein is incorporated into the phage genetic material. A typical sequence involved in genetic engineering of a monoclonal antibody is as follows:
Fab′ fragments
Fab′ fragments lack the Fc chain and thus do not stimulate the immune response in the same way as antibodies which have the Fc chain to act as an effector. In some cases binding affinity with a lack of immune response is an advantage, e.g. where it is preferable to block a receptor protein on cell surface but not stimulate the immune system to destroy the cell. Expression of the smaller Fab′ fragments is much simpler than the expression of a full antibody. These fragments can be produced using phage display technology.
Types of MAb
There are currently 18 monoclonal antibodies which have been licensed for therapeutic use and this therapeutic category is expanding rapidly. The majority of these antibodies have been produced by recombinant DNA technology such as the phage display system but there are three which are produced by the more old-fashioned murine antibody technology produced in hybridomas. There is currently quite a number of Fab′ fragments in clinical trials.
Monoclonal antibodies targeted against receptor proteins
The most successful application of MAbs is in the targeting of receptor proteins on particular cell types. The result of the binding of the MAb may be blocking of the cell’s function or destruction of the cell.
Anticancer antibodies
Immunosuppressive antibodies
Graft rejection occurs when organs are transplanted between humans, e.g. kidney transplant, or between animals and humans, and the immune system of the recipient of the transplant recognises the transplanted organ as being antigenic. The immune response can be suppressed by conventional drugs and a combination of ciclosporin and corticosteroids is used to prevent graft rejection. However, ciclosporin is quite toxic to the kidney. Another strategy is to target the immune response more directly. However, in summary, a major cause of rejection is that the body directs cytotoxic T lymphocytes against the transplanted tissue in order to destroy it. The cytotoxic T-lymphocyte response is triggered by another type of T cell called a T-helper lymphocyte. The T-helper cells are responsible for immune surveillance and they have several receptors on their surfaces which are sensitive to antigens. These receptors are known as cell differentiation clusters (CDs). One particular receptor on the surface of the T-helper cells, the CD3 receptor, is a useful target for reducing the T-helper cell response (which in turn triggers the cytotoxic T cells). The specificity of monoclonal antibodies means that they can be directed to very specifically bind to a particular antigen, and in order to modulate the immune response in transplant rejection the monoclonal antibody OKT3 was produced which binds the CD3 receptor. OKT3 is a first-generation MAb and was produced using hybridoma cells which were screened for OKT3 antibody production by measuring the strength of binding of the antibodies they were producing to mature T-helper cells. High-yielding hybridoma clones were cultured in the peritoneal cavities of mice, which resulted in a very high-yielding system where the OKT3 is produced at a level of 12–15 mg/mL. OKT3 is purely based on murine protein and as a consequence there is the risk of human anti-mouse antibody (HAMA) formation in patients. The consequence of the administration of OKT3 is that the levels of T-helper cells in the body are reduced through the immune system removing them from the circulation. There are, of course, side effects since the agent compromises the natural immune response, but in the specific case of kidney transplant the benefits can outweigh the side effects because the effects of ciclosporin on the kidney cause some confusion since it is difficult to tell whether a poorly functioning transplanted kidney results from rejection or is due to the toxicity of ciclosporin. The main indications of the drug are in the early stages after transplant or as rescue therapy after standard immunosuppressant therapy has failed to prevent rejection. The risk of HAMA has meant that the use of OKT3 has declined. Two antibodies with similar indications have recently been licensed. Darlizumab (Zenopax™) is used to reduce the likelihood of rejection following kidney transplantation. It binds to part of the IL-2 receptor produced on the surface of activated T cells, thus interrupting their role in tissue rejection. It has a very good side effect profile and a circulating half-life of 20 days. Basiliximab (Simulect™) has a similar effect to Darlizumab, acting on T cells to reduce their activity in tissue rejection.
Anticlotting
Abciximab (ReoPro™) is used to prevent platelet aggregation and consequent clot formation in unstable angina and during angioplasty. It binds to the GPIIb/IIIa receptors on platelets, blocking the binding of fibrinogen, which acts to bind the platelets together into a plug that results in clot formation. The drug was developed from antiplatelet antibodies which were generated in mice. It consists of the Fab fragment of a human/murine MAb which is produced using a hybridoma cell line into which the gene for a chimaeric antibody has been inserted. The Fab fragment is released by treatment with papain, followed by chromatographic purification. It consists of ca. 50% murine and 50% human amino acid sequences.
Monoclonal antibodies targeted against protein mediators
Problems can occur in the use of antiprotein antibodies due to the following factors:
Monoclonal antibodies used as passive vaccines
The most widely used monoclonal antibody in this category is palivizumab (Synagis™) which is used in the prevention of lower respiratory tract infection caused by respiratory syncytial virus in premature infants, which can prove fatal.13,14 As yet, there is no successful vaccine against infection. Palivizumab is a humanised mouse MAb which binds to the RSVF protein which is present on the surface of the virus. Binding to the virus stimulates the immune response to remove the virus.
Monoclonal antibodies used in targeted therapy
Work has been carried out on the conjugation of toxic agents to antibodies for directed targeting of particular cell types. The only licensed product so far is a conjugate of the complex oligosaccharide calicheamicin and a humanised monoclonal antibody (gemtuzumab-ozogamicin or Mylotarg™) to the CD33 receptor which is present on myeloid leukaemia cells but is not present on normal stem cells, which are needed to provide a supply of blood cells. Thus the drug acts as a selective toxin against the cancerous cells.
A conjugate between Rituximab and iodine-131 (Bexxar™) is in the final stages of clinical trials for treatment, as is the indication for Rituximab itself, advanced follicular lymphoma. A similar conjugate between Rituximab and yttrium-90 is also undergoing clinical trials.
Self Test 27.3
Explain in more detail the nature of the following MAbs which have entered clinical trials. For example, in the light of the modes of action discussed in the previous pages, what is the likely mode of action for these antibodies?
Vaccines
Introduction
Vaccines provide the most cost-effective method of disease prevention. Even during the mid 1960s fifteen million cases of smallpox occurred. The disease was finally eradicated, apart from the threat of bioterrorism, in the 1970s. The goal of vaccination is to generate memory cells from B lymphocytes that enable a heightened immune response to occur upon exposure to the pathogen. There are still many infectious diseases in the world that are not adequately controlled. These diseases include: malaria, 270 million cases worldwide, 2 million deaths per annum with 2 billion at risk from the disease; tuberculosis, 20 million of those infected have symptoms of the disease and it is projected that 30 million will die this decade from TB; trypanosomiasis, 20 million infected, mainly in South America; and schistosomiasis, 200 million infected worldwide. Biotechnology has brought with it the prospect of the improvement of existing vaccines and the development of new ones against the remaining serious infectious diseases.
Box 27.3 gives a brief summary of the immune system.
Box 27.3 The immune system in brief
Aspects of the immune system have been covered in relation to the action of monoclonal antibodies. There is a range of non-specific immune defences based on the action of immune defence cells such as macrophages. The specific immune response that recognises a particular antigen is based on the activities of T and B lymphocytes. A brief summary of the immune response required for the understanding of vaccine action is given below.
B cells
B cells circulate in the blood. Their surfaces have a number of receptors (CD35, CD21, etc.) and also immunoglobulins are present on the surface of the cell. Each ‘naive’ B cell expresses an immunoglobulin (Ig) that is fairly specific for a particular antigen. If the B cell encounters the antigen that binds to its Ig it will form a clone of plasma cells, which are rapidly dividing and which secrete large amounts of the specific Ig; the ability of the Ig to recognise its antigen will be refined as the plasma cells continue to divide. As part of the immune response, a subpopulation of B cells forms memory cells, and these circulate for a very long time after the antigen has disappeared and retain the ability to recognise antigens more rapidly than the ‘naive’ B cells.
T cells
The function of the immune system is complex and thus it is difficult to determine where recognition of an antigen begins. T cells have a surveillance role and it is easiest to cast the T-helper cells as the front line of the defence. T-helper cells have a receptor called the CD4 receptor that binds to the complexes formed between the major histocompatibility complex II (MHCII) and foreign peptides that have been partially digested by antigen-presenting cells. Complexes formed with the T cells and recognised as non-self persist and cause the T-helper cells to multiply, secrete cytokines and thus trigger the division of B cells and cytotoxic T cells.
Types of vaccine
Attenuated live vaccines
These were the first type of vaccine. The attenuated organism has much in common with the infective form but due to genetic alterations it is no longer pathogenic. Genetic alterations are produced by selection of non-virulent strains upon repeated culturing of the organism or by chemical treatment. Examples include the bacille Calmette-Guérin (BCG) tuberculosis vaccine, mumps, rubella, polio and measles vaccines. The live organism gives a full immune response, penetrating into cells to produce tissue immunity as well as humoral immunity in the blood stream. Live organisms persist in the body long enough to produce both T- and B-memory cells. The organism could revert to its virulent form and those with compromised immune systems may be challenged even by the weakened pathogen. Nowadays, genetic engineering techniques can be used to specifically delete specific virulence genes.
Inactivated (dead) pathogen vaccines
The pathogen is inactivated by heat or chemicals such as formaldehyde or glutaraldehyde while ensuring that the surface antigens remain intact. Examples include cholera, whooping cough, influenza, rabies and polio vaccines. With these vaccines there is no risk of pathogenicity developing. Complex immune response can be induced by multiple antigens. Such vaccines are not necessarily completely safe; e.g. the whooping cough vaccine has occasionally produced hypersensitive responses. These vaccines do not enter cells and thus do not produce cellular immunity; they only confer humoral immunity.
Purified subunit vaccines
The major determinants of the immune response are extracted from the organism and purified. For example, some organisms such as those causing tetanus and diphtheria produce toxins and the main defence of the body against these organisms is the production of antibodies against these toxins. Treatment of these toxins with formaldehyde (Box 27.4) to produce toxoids renders them safe for use as vaccines while preserving their ability to induce an immune response. A similar approach has been used in attempting to reduce the risks of the whole cell pertussis vaccine but without a wholly successful vaccine being produced. Another approach is to utilise the surface polysaccharides from bacteria. On their own, such polysaccharides are poorly immunogenic, and thus their immunogenicity is increased by conjugating them to tetanus or diphtheria toxoids. Some of the vaccines against meningitis are based on such polysaccharide–toxoid conjugates. The advantage of these types of vaccine is that there is no risk of pathogenicity and hypersensitivity, but the vaccine may only stimulate humoral immunity and is expensive to produce.
Box 27.4 Formaldehyde inactivation
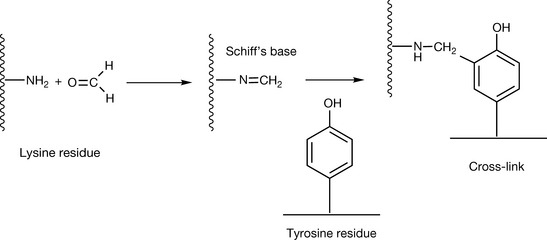
The process of toxin inactivation by formaldehyde treatment is poorly understood. However, it does involve the formation of a Schiff’s base, either with lysine or arginine residues in the protein, followed by cross-linking to residues such as tyrosine, lysine and tryptophan. The quality control for this reaction is bioassay of the resultant toxoid using an animal model but there is the possibility of using mass spectrometry to follow the modification (Fig. 27.21).15
Advances in oral vaccination
The development of oral vaccines for stimulation of mucosal immunity is an important goal because of the simplicity of administration. The mucosal system is the first barrier a pathogen has to penetrate, and mucosal immunity is based on the production of IgA antibodies. The oral polio vaccine was an early example. The types of vaccines used for oral dosage are the same as those available for administration by injection. Formulation of oral vaccines presents a particular challenge for the pharmacist and is likely to see major progress in the future.
Licensed vaccines
Live attenuated
Poliomyelitis (oral vaccine), measles, mumps, rubella (the attenuated virus strains are now combined in a single vaccine), tuberculosis (BCG vaccine), varicella-zoster, yellow fever.
Inactivated
Influenza (inactivated by formaldehyde or propiolactone treatment), hepatitis A (inactivated by formaldehyde treatment), pertussis (inactivated by formaldehyde treatment, usually combined with diphtheria and tetanus toxins into a single vaccine), rabies, tick-borne encephalitis (formaldehyde inactivated).
Applications of biotechnology to vaccine production
A logical extension of the use of purified subunit vaccines is to try to produce antigenic components which are capable of producing an immune response using biotechnology. The first successful example of this was the hepatitis B subunit vaccine which has been very successful. The production of this is described in detail below.
The hepatitis B vaccine and example in detail
Hepatitis B virus infects the liver and causes progressive liver disease and ultimately liver cancer. There are about 250 million sufferers worldwide. Unlike hepatitis A, which can be contracted from contaminated food, hepatitis B is predominantly maternally or sexually transmitted or is transmitted by intravenous drug use. Infection can also arise from contact with physiological fluids and people working with these materials, e.g. in biochemistry labs, would be advised to receive vaccination against it. The first hepatitis B vaccines based on Dane particles were derived from plasma from human carriers of the disease, but supply was limited by the availability of such plasma. In addition, extensive processing of the material extracted from plasma was necessary to ensure its non-infectivity. From the point of view of patients, there was a reluctance to accept a vaccine derived from human plasma. The biotechnologically produced hepatitis B coat protein is now the most commonly used vaccine.
The vaccine is based on a surface glycoprotein of the virus coat and is thus an antigenic component vaccine as opposed to a live vaccine. Since the vaccine is not live it does not invade tissues and thus tissue immunity is not stimulated. Despite some limitations, the antigenic component hepatitis B vaccine does appear to be a highly effective vaccine.
The biotechnological process leading to the production of a hepatitis B vaccine
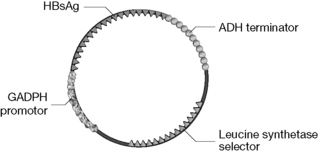
Figure 27.22 pBR322 based expression vector used to transform yeast cells so that they produce a hepatitis B vaccine.
Hepatitis B vaccines have also been produced by genetically transformed Chinese hamster ovary (CHO) cells. These vaccines contain both glycosylated and unglycosylated forms of the coat protein and are thus indistinguishable from the natural antigen. However, it has not been established that glycosylation is important with regard to antigenicity. A CHO-derived HBV (GenHevac B) is licensed for use in France.
Other recombinant vaccines
There have been no major developments in recombinant vaccine production since the hepatitis B vaccine. Recombinant vaccines were produced against Lyme disease and rotavirus but have been subsequently withdrawn from the market. Recently, an oral cholera vaccine (Dukoral) based on mixture on inactive cholera bacteria and a cholera toxin B subunit produced by recombinant technology has been launched.
Recent developments in vaccine production by biotechnology
There still remain many challenges in vaccine development and the examples discussed below indicate strategies provided by biotechnology which are being used to develop new vaccines.
Approaches to the development of an AIDS vaccine
No safe attenuated form of the virus has been recognised and thus a live attenuated vaccine is unlikely to be developed. The most promising approach is based on subunit vaccines based on the viral glycoproteins gp 160 and gp 120. These recombinant proteins have been expressed in a number of systems including yeast and mammalian cells. In order to stimulate T as well as B cell response, the genes coding these subunit vaccines have been coupled to the vaccinia virus, which has a track record of use in humans since it was used as a smallpox vaccine. A number of AIDS vaccines have undergone small-scale clinical trials.
Approaches to the development of a malaria vaccine
Vaccination against malaria is difficult because of the three stages in the development of the parasite. An effective vaccine might need to contain antigens from each stage. Examples of vaccines which have been tested have been based on: three merozite stage surface proteins, a seven-antigen vaccine containing proteins from each stage of the life cycle and a recombinant vaccine consisting of an antigen found in the sporozite stage fused with a hepatitis B antigen producing overall a stronger immune response than the antigen on its own.
Anticancer vaccines
The difficulty in developing this type of vaccine is in finding immunogens since cancers are often not recognised by the immune system as being foreign. There are three categories of tumour associated antigen: tumour-specific antigens, tumour-associated differentiation antigens which are found in normal tissues but are overexpressed in cancer cells, and antigenic peptides which are involved in the development of the cancer. A number of vaccines based on these approaches have been tested.
 Self Test 27.2
Self Test 27.2
2. T. The single amino acid threonine is released from the C-terminal of the B-chain.
3. Six charges in total. Two on histidine, one on arginine, one on lysine and two at the N-terminus of the A and B chains.
4. Insulin has four aspartate residues and two terminal carboxyl groups making a total of six acidic residues. This is balanced by six basic residues. Thus the p/ value = (3+8)/2 = 5.5, i.e. that insulin will be charge neutral at that pH with the acids and bases carrying equal charges.
Self Test 27.3
LymphoCide: Promotes lysis of malignant B lymphocytes by binding to a specific receptor on their surface.
Vitaxin: Binds to vascular integrin protein thus promoting its removal by the immune system and reducing angiogenesis in tumours.
Anti-hepatitis B monoclonal antibody: Binds to hepatitis B thus promoting removal of the virus, acting as a passive vaccine.
Anti-CD5: Toxic antibody targeted therapy specifically aimed at the T-cells overproduced in T-cell lymphoma.
1 Bhatnagar S., Srivastava D., Jayadev M.S.K., Dubey A.K. Molecular variants and derivatives of insulin for improved glycemic control in diabetes. Progress Biophys Mol Biol. 2006;91:199-228.
2 Roberts M.J., Bently M.D., Harris J.M. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54:459-476.
3 Pradhananga S., Wilkinson I., Ross R.J.M. Pegvisomant: structure and function. J Mol Endocrinol. 2002;29:11-14.
4 Birch J.R., Rachner A.J. Antibody production. Adv Drug Del Rev. 2006;58:671-685.
5 Popov S., Hubbard J.G., Ward E.S. Novel and efficient method for the isolation of antibodies that recognise T cell receptor Vσs. Mol Immunol. 1996;33:493-502.
6 Azzazy H.M.E., Highsmith W.E. Phage display technology: clinical applications and recent innovations. Clin Biochem. 2002;35:425-455.
7 Ladner R.C., Sato A.K., Gorzelany J., de Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9:525-529.
8 Kehoe J.W., Kay B.K. Filamentous phage display in the new millennium. Chem Rev. 2005;105:4056-4072.
9 Nishbori N., Hiroyuki H., Furusawa S., Matsuda H. Humanisation of chicken monoclonal antibody using phage-display system. Mol Immunol. 2006;43:634-642.
10 Vilcek J., Feldmann M. Historical review: cytokines as therapeutics and targets of therapeutics. Trends Pharmacol Sci. 2004;25:201-209.
11 Taylor P.C. Antibody therapy for rheumatoid arthritis. Current Opinion Pharmacol. 2003;3:323-328.
12 Bayry J., Siberil S., Triebel F., et al. Rescuing CD4+CD25+ regulatory T-cell functions in rheumatoid arthritis by cytokine-targeted monoclonal antibody therapy. Drug Discov Today. 2007;12:548-552.
13 Wu H., Pfarr D.S., Johnson S., et al. Development of Motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368:652-665.
14 Bussel J.B., Giulino L., Lee S., et al. Update on therapeutic monoclonal antibodies. Curr Probl Pediatr Adolesc Health Care. 2007;37:118-135.
15 Thaysen-Andersen M., Jørgensen S.B., Wilhelmsen E.S., et al. Investigation of the detoxification mechanism of formaldehyde-treated tetanus toxin. Vaccine. 2007;25:2213-2227.