Chapter 21 Anticancer drugs
Introduction
Antineoplastic drugs have a major role in cancer treatment, sometimes alone and sometimes in conjunction with surgery or radiotherapy. The modern era of chemotherapy in cancer treatment began after the Second World War, following observations of the effects of nitrogen mustards, which had been developed as chemical warfare agents, on transplanted lymphomas in mice. The nitrogen mustards were the first drugs in the alkylating agent class of anticancer drugs and they were followed soon by antimetabolites, the first one of which was aminopterin, a folate antagonist. The antineoplastic drugs can be classified as follows: (1) alkylating agents, (2) antimetabolites, (3) natural products, (4) organometallic compounds, (5) hormones, (6) angiogenesis inhibitors, (7) signal transduction inhibitors, and (8) miscellaneous including biotechnologically produced drugs.
Alkylating agents
Nitrogen mustards
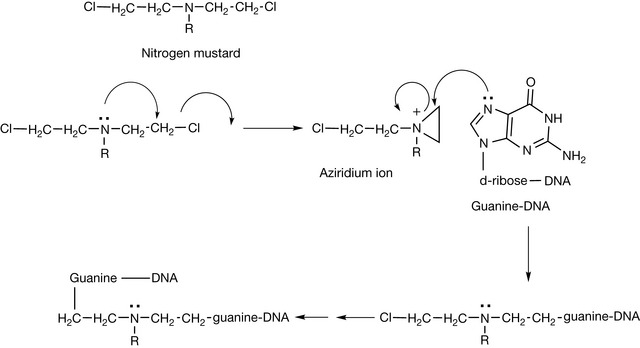
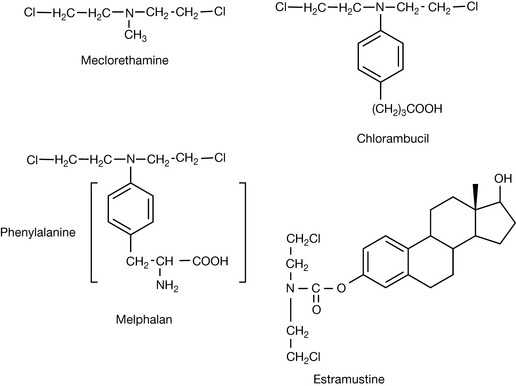
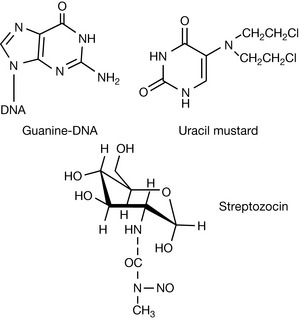
Nitrogen mustards have the general structure shown in Figure 21.1. They form an aziridinium ion which is subject to attack by the nucleophilic positions within DNA bases. The positions within DNA which can be subject to attack include: N-2, N-3, O-6 and N-7 of guanine; N-1, N-3 and N-7 of adenine; O-6 of thymine; N-3 of cytosine and phosphate oxygen atoms (see Ch. 7). The availability of the lone pair on the nitrogen is critical for formation of the aziridinium ion and the nitrogen must have a pKa of 6 or less so that the drug is largely un-ionised at pH 7.4. The low pKa is ensured by the R groups, being electron withdrawing; in addition, the chloroethyl groups alone reduce the pKa of the nitrogen. The damaged DNA formed by the cross-linking reaction shown in Figure 21.1 prevents cell division from occurring. This class of drugs relies on cancer cells being rapidly dividing in order to target them, but also targets other fast-growing cells such as white blood cells, resulting in myelosuppression. Nitrogen mustards include meclorethamine (less used because it is highly toxic and reactive) and chlorambucil and melphalan, where the reactivity of the nitrogen atom is reduced by the benzene ring withdrawing electrons, thus producing compounds which are slow alkylators (Fig. 21.2). In the case of melphalan there was a hope that it would be useful for targeting melanoma, since phenylalanine is a biosynthetic precursor of melanin; however, clinical data gave no indication of such targeting. This type of targeting is also used in the case of estramustine phosphate, which is used to treat prostate cancer. In this case, the nitrogen mustard is linked to estradiol which has some effect in targeting the oestrogen-dependent tumour.
Other types of reactive alkylating reagents have been developed. Some of these have the aziridine ring already built into them such as thiotepa (Fig. 21.3), which is a slow alkylator, since the ring has to be activated by protonation before it will alkylate.
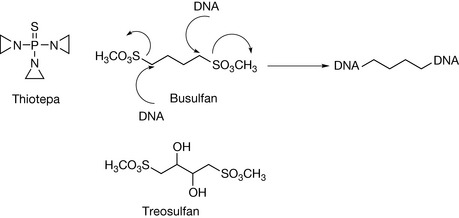
In busulfan (Fig. 21.3) it is the sulphonate group which is a good leaving group and which promotes alkylation of DNA. The four carbon atom spacing between the alkylating groups is optimal for cross-linking DNA strands. The mode of action of treosulfan is similar to that of busulfan.
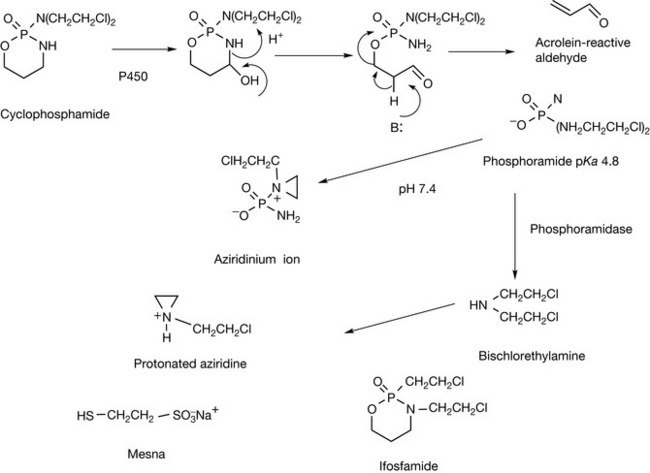
Cyclophosphamide is closely related in its mode of action to the nitrogen mustards. It is a prodrug which is activated via the action of a P450 enzyme as shown in Figure 21.4. The product of the enzyme action then breaks down into a phosphoramide mustard and acrolein which may be responsible for some of the side effects of the drug. The side effects of acrolein can be reduced by co-administration of a sulfhydryl reagent such as mesna, which converts the acrolein into an inactive product. The phosphoramide metabolite is strongly acidic which means that it is completely ionised within cells and becomes trapped there. The phosphoramide is a reactive nitrogen mustard and can form the aziridinium ion which reacts with DNA bases as was shown in Figure 21.1. The selectivity for neoplastic cells may be due to their lower pH, which results in slower formation of the aziridinium ion and hence longer persistence of the trapped drug within the cells. Alternatively, neoplastic cells have high levels of phosphoramidases, which results in the formation of chloroethylamine which alkylates DNA via protonated aziridine.
Isophosphamide is an isomer of cyclophosphamide which is activated in a similar manner via a P450 enzyme. It is metabolised more slowly than cyclophosphamide, which may offer some advantages.
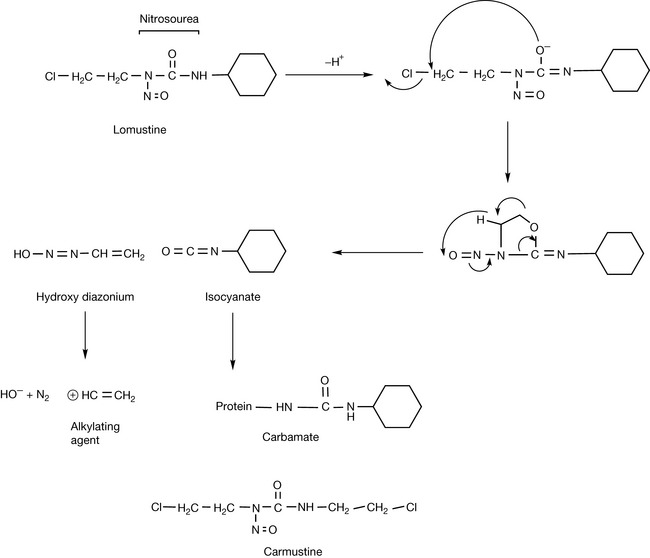
The final category of alkylating reagents is the nitrosoureas. The two commonly used drugs are carmustine and lomustine (Fig. 21.5). As shown in Figure 21.5 for lomustine, the nitrosoureas undergo spontaneous decomposition in an aqueous environment to produce two reactive species: a reactive diazohydroxide which can alkylate DNA and an isocyanate which is reactive towards amine groups within proteins.
Temozolomide is an alkylating agent but in this case the alkylating group is methyl. The methyl group derives from diazomethane which is produced by the spontaneous decomposition of the drug in an aqueous environment (Fig. 21.6). The diazomethane then reacts with the 7 position of guanine. However, resistant cells are able to repair DNA which has been damaged in this way, which limits the effectiveness of the drug. However, it is the standard treatment for brain tumours in combination with initial intensive radiotherapy.
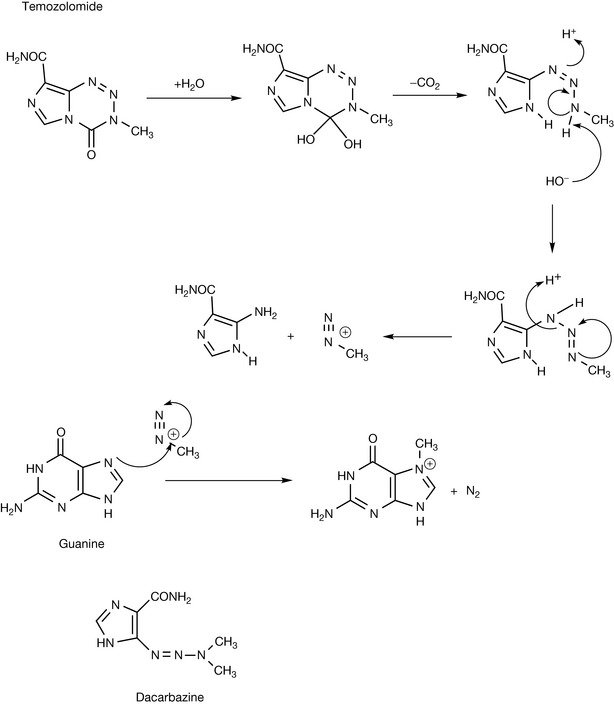
Dacarbazine acts in a similar manner to temozolomide, generating diazomethane, but it requires metabolic activation by a cytochrome P450 enzyme before it hydrolyses.
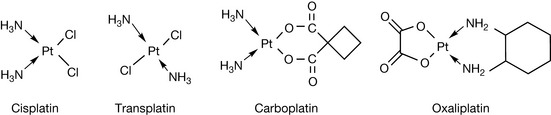
Cisplatin (Fig. 21.7) behaves in a manner analogous to an alkylating agent in that it cross-links guanine nucleotides. The N7 positions of adjacent guanines on a single strand of DNA react with cisplatin. Cisplatin only has a short half-life in plasma since it is hydrolysed in water quite rapidly. Transplatin is less effective as a cytotoxic agent. In part this may be because it is less effectively taken up by cells but also it can only cross-link between two DNA strands which is less effective as a cytotoxic mechanism since the lesion is more readily repaired. Carboplatin is less reactive than cisplatin and has reduced renal toxicity in comparison with it, while remaining an effective cytotoxic. Oxaliplatin similarly has reduced toxicity and altered selectivity.
Antimetabolites
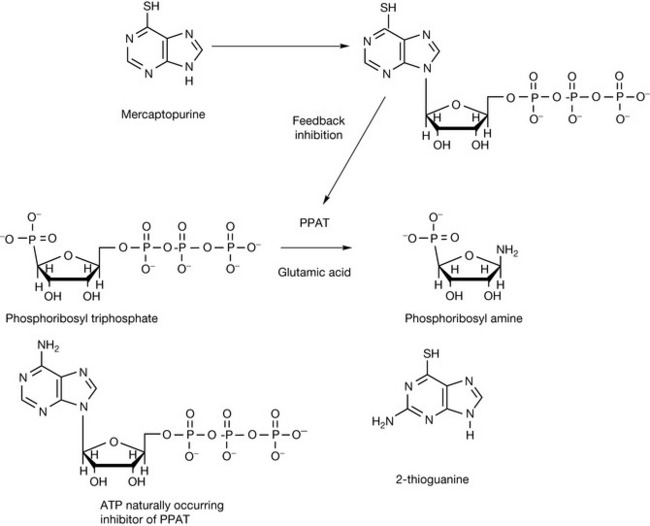
Most antimetabolites are enzyme inhibitors and inhibit key enzymes involved in nucleotide synthesis by acting as false substrates. Mercaptopurine (Fig. 21.8) is one of the most effective drugs for treating childhood leukaemia. It is a prodrug and requires activation to form its ribosyl pyrophosphate derivative in order to become effective. The activated form of the drug inhibits several enzymes involved in nucleotide biosynthesis but the most important of these is phosphoribosyltriphosphate amidotransferase (PPAT). The reaction promoted by PPAT is the rate-limiting step in purine biosynthesis. The enzyme is subject to feedback inhibition by ATP, which binds to a specific site on the enzyme, and mecaptopurine ribosyltriphosphate produces inhibition by a similar mechanism. 2-Thioguanine is similar to mercaptopurine and probably acts via a similar mechanism, mimicking the feedback inhibition produced by GTP.
Pyrimidine antimetabolites act via the inhibition of the synthesis of pyrimidine nucleotides, particularly the inhibition of the biosynthesis of deoxythyminemonophosphate (dTMP). The biosynthesis of dTMP is shown in Figure 21.9. A nucleophilic SH group in thymidylate synthetase attacks carbon-6 in deoxyuridinemonophosphate, precipitating attack on the methylene bridge in 5,10-methylenetetrahydrofolate. The ternary complex then undergoes oxidative breakdown, releasing deoxythymidine monophosphate. In Figure 21.10 the same reaction is shown for the antimetabolite 5-fluorouracil (5-FU), which is converted to 5-fluorouridinemonophosphate, which forms a complex with 5,10-methylenetetrahydrofolate which does not break down to release deoxythymidine monophosphate and dihydrofolate, and this results in inhibition of DNA formation.
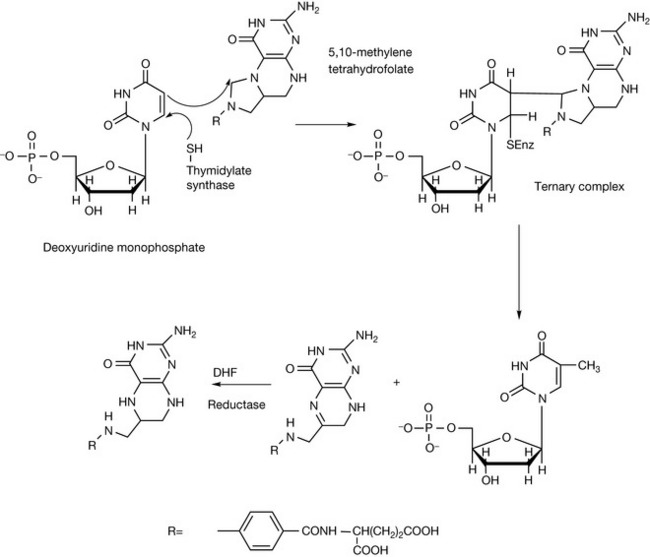
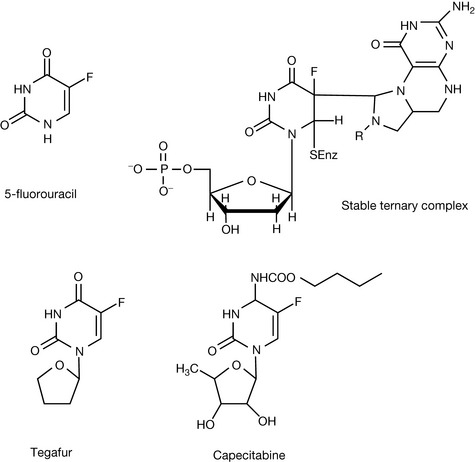
The oral bioavailability of 5-FU is variable due it being enzymatically reduced in the gut wall. Tegafur (Fig. 21.10) was developed as a prodrug of 5-FU with better oral bioavailability resulting from its resistance to reduction during absorption. It is slowly metabolised to 5-FU in the liver. Capecitabine is another prodrug of 5-FU which has improved oral bioavailability and is converted to 5-FU by three enzymes in the liver: carboxyesterase, cytidine deaminase and uridine phosphorylase. Apart from improved oral bioavailability, the drug has some selectivity for tumours expressing a high level of uridine phosphorylase activity.
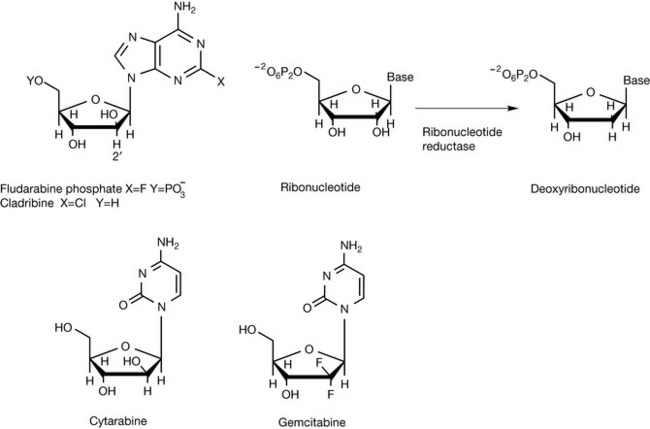
A number of drugs have been targeted at DNA polymerase. The adenine analogue fludarabine (Fig. 21.11) binds to DNA polymerase and ribonucleotide reductase, thus interfering with DNA synthesis. Cladribine is similar in structure to fludarabine but is much more toxic. It acts by inhibiting enzymes involved in DNA repair. The potential for interfering with ribonucleotide reductase arises from the replacement of ribose with its epimer arabinose. Thus the 2′ position can not be reduced as shown in Figure 21.11 for ribose. Cytarabine is a pyrimidine nucleoside analogue. It is phosphorylated in cells and acts via inhibition of DNA polymerase and via incorporation into DNA and RNA, producing miscoding. It also incorporates arabinose in place of ribose and thus can interfere with ribonucleotide reductase. Gemcitabine is an analogue of deoxycytidine in which fluorine has been introduced into the sugar ring. Since fluorine is very similar in size to hydrogen, it is a substrate for DNA polymerase and becomes incorporated into DNA, thus inhibiting DNA synthesis and, again, is an inhibitor of ribonucleotide reductase.
Pentostatin also belongs in the antimetabolite category although its mechanism of action is totally different from that of other antimetabolites. Pentostatin is a natural product isolated from the fermentation broth of Streptomyces. It is an example of a drug which is a transition-state analogue which is believed to mimic the transition state of 2-deoxyadenosine when it is being deamidated (Fig. 21.12). The conversion of 2-deoxyadenosine into 2-deoxyinosine is an important metabolic step in controlling DNA synthesis. If the conversion does not occur, 2-deoxyadenosine accumulates and is converted into its triphosphate (dATP), which is a potent inhibitor of ribonucleotide reductase. Inhibition of the formation of deoxyribonucleotides inhibits DNA synthesis. It is believed the transition state involved in the deamination of adenosine is as shown in Figure 21.12. Pentostatin resembles the transition of 2-deoxyadenosine deamination and thus binds to the deamidase enzyme, causing inhibition.
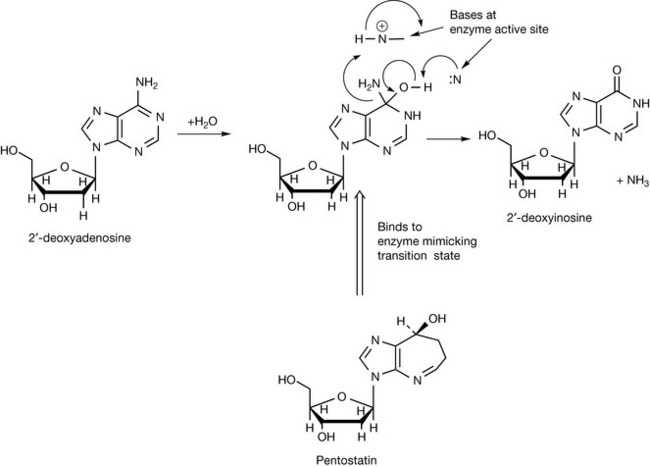
Antimetabolites interfering with the function of folic acid
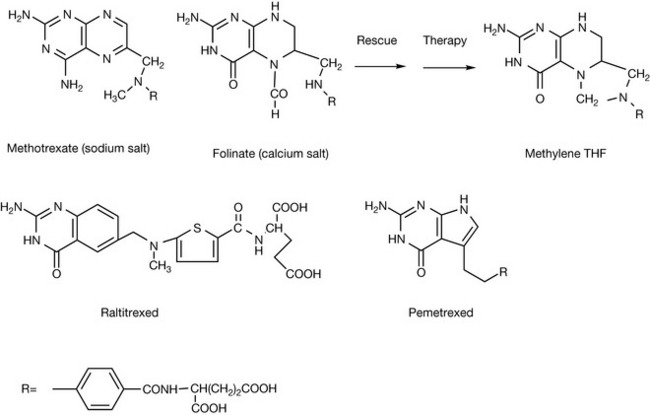
Folic acid is the vitamin responsible for providing single carbon units in the biosynthesis of DNA and RNA bases (see Ch. 26). Interfering with the action of folic acid results in the inhibition of DNA biosynthesis and thus with cell proliferation. An important group of antimetabolites used in cancer chemotherapy interferes with the action of folic acid. Figure 21.13 shows the biosynthetic steps involved in the conversion of folic acid to methylene tetrahydrofolate, which is responsible for transferring a methylene unit to precursors of nucleic acid bases. DHF is generated during methylene transfer. Methotrexate is close in structure to folic acid. The presence of an additional amino group in the structure increases its basicity so that it is protonated at physiological pH, and this allows it to inhibit binding of DHF to the enzyme dihydrofolate reductase.
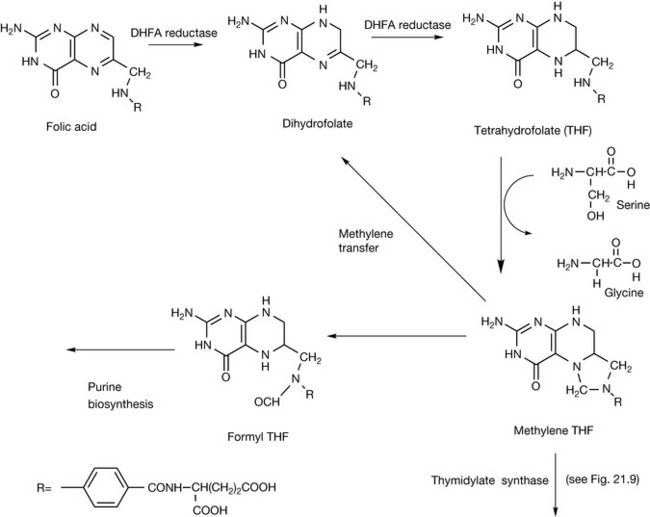
Methotrexate (Fig. 21.14) is often used in conjunction with calcium folinate which is injected 24 hours after administration of methotrexate in order to reduce its cytotoxic action to rescue normal cells while not reducing the cytotoxic effect on cancer cells. The selective toxicity of this combination for cancer cells rests on the fact that the normal cells are able to reduce the formyl group in folinate, yielding methylene THF, whereas the cancer cells are unable to complete this step. Raltitrexed inhibits the synthesis of nucleotides through a specific inhibition of thymidylate synthetase where it prevents the binding of the normal co-factor, methylene THF. Pemetrexed is a less specific inhibitor of folate action and acts on thymidylate synthetase, dihydrofolate reductase and formyl transferase.
Cytotoxic antibiotics
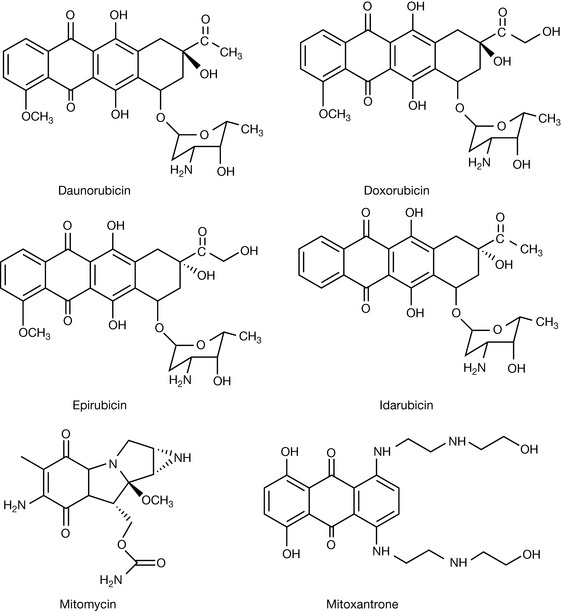
Daunorubicin (Fig. 21.15) was isolated from Streptomyces peucetius. It was found to be highly cytotoxic but its usefulness was limited by severe cardiotoxicity. The basis of the cytotoxic action of daunorubicin was found to be its ability to interacalate with DNA. Intercalation is a feature of flat aromatic and heteroaromatic molecules which bind perpendicular to the DNA axis. They are held in place by non-covalent charge transfer interactions (see Ch. 1) in combination with electrostatic and hydrogen bonding interactions. Intercalation causes distortion of the DNA structure by causing the base pairs to be pushed apart. It is more energetically favourable for pyrimidine-3′,5′purine sequences than purine-3′,5′pyrimidine sequences.
The structure of the complex formed between daunorubicin and DNA determined by X-ray diffraction indicates that the daunorubicin forms a complex with GC sequences in DNA. The daunorubicin molecule is orientated at ca. 90° to the long axis of the GC base pair. It interacts with GC pairs both above and below it in the helix (Fig. 21.16). The sugar ring attached to the molecule sits in the minor groove of the DNA.
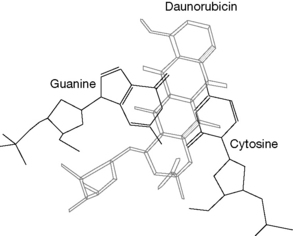
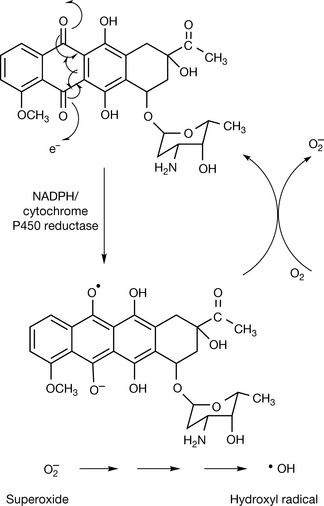
Daunorubicin may prevent DNA replication via two mechanisms. It may inhibit the action of topoisomerase II which causes double-stranded breaks in the sugar phosphate backbone of the DNA allowing conformational change within coiled DNA (see Ch. 7). It is not clear exactly how the intercalated drug inhibits replication but part of the process involves a ternary complex formed between DNA, the daunorubicin and topoisomerase II. However, another component in the mechanism of action is oxygen-dependent DNA damage. This might be due to the enzymatic reduction of the daunorubicin which can occur followed by the production of superoxide which can generate reactive hydroxyl radicals. The process can be promoted by the presence of the ferrous ion (Fig. 21.17). The hydroxyl radicals can cause strand breaks in DNA. The generation of reactive hydroxyl radicals is probably the mechanism that causes the severe cardiotoxicity of daunorubicin and related drugs.
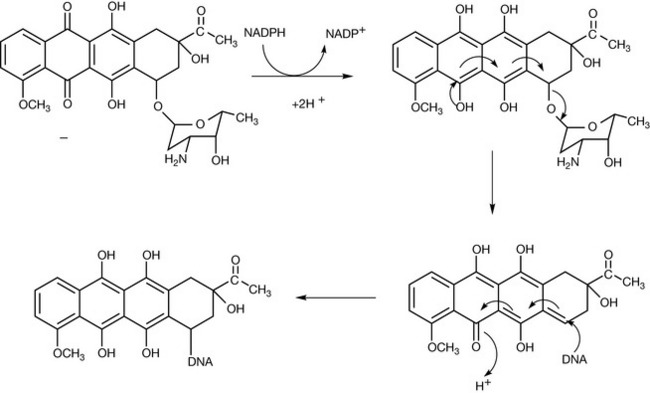
Another possible mechanism of DNA damage is by alkylation with daunorubicin which is promoted via a two electron oxidation (Fig. 21.18) where an adduct is formed between DNA and the drug. Apart from daunorubicin, there are three other similar antibiotics commonly used in cancer chemotherapy: doxorubicin, idarubicin and epirubicin (see Fig. 21.16). Their mode of action is the same as that of daunorubicin. Mitomycin-C is another antitumour antibiotic and it acts in a similar manner to the anthracylines. It functions as a prodrug which is activated via reduction to a semi-quinone which rearranges to form an alkylating agent. An advantage of bioreductive activation is that this means that the drug is targeted at solid tumours which are hypoxic at their centre.
Mitoxantrone (see Fig. 21.16) is a synthetic compound which is related in structure to the anthracycline antibiotics. It acts via a similar mechanism to the anthracyclines but it has a slightly higher reduction potential than these drugs and thus does not generate reactive oxygen species to the same extent. Its primary mode of action is via intercalation with DNA and consequent interference with topoisomerase II. The amine side chains may bind with the phosphate backbone of the DNA. The cardiotoxic side effects of mitoxantrone are lower than those of the anthracylines.
Plant derivatives
The search for anticancer compounds from natural sources can be dated back at least as far as the Ebers papyrus in 1550 bc. The use of plants in folk remedies can provide a lead for discovery, although in many cases the therapeutic indication which led to an interest in the plant was not anticancer activity.1
Vinca alkaloids
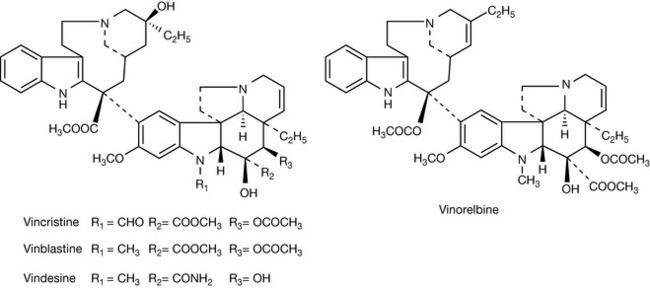
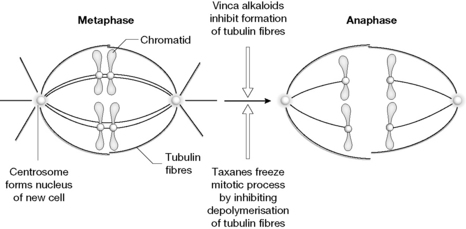
The Madagascar periwinkle (Catharantheus roseus) has a reputation in folk remedy in the treatment of diabetes. When the antidiabetic activity of extracts was tested in rats it was found that the rats succumbed to infection due to the death of white blood cells. The alkaloids vincristine and vinblastine (Fig. 21.19) were found to be responsible for the cytotoxicity of the plant extracts.2 The compounds work by inhibiting the formation of microtubules which must be formed as part of the cell division process. Figure 21.20 illustrates the process of mitosis where tubulin fibres attach themselves to chromatids formed from DNA replication within the cell and separate the genetic material into two halves which will form the genetic material in the nucleus of the two daughter cells produced. In order for the cell division process to occur, the microtubules undergo both elongation and depolymerisation so that the cell division process can move from the metaphase to the anaphase. Since the vinca alkaloids exhibit general toxicity via inhibition of cell division, they cause myelosuppression which affects the rapidly proliferating white blood cells the most. Even though the structures of the alkaloids are very similar, they exhibit a different spectrum of activities. Vincristine is more effective against Hodgkin’s disease and paediatric solid tumours than adult solid tumours. Vinorelbine is a semi-synthetic analogue of the Vinca alkaloids and is most effective against non-small cell lung, breast and ovarian cancer.
Taxanes
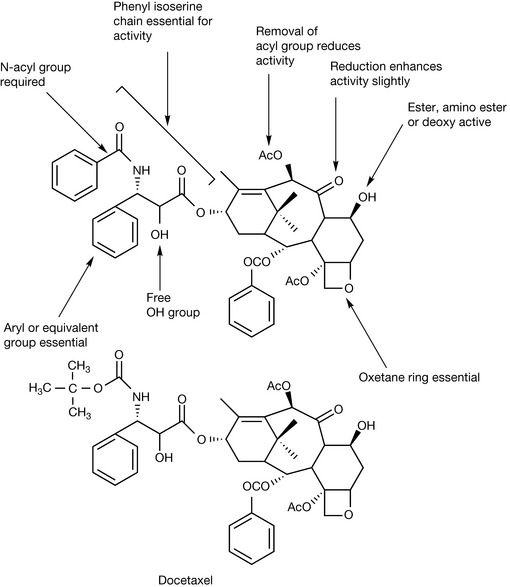
The discovery of taxol originated from a screening programme instituted in 1960 by the US national cancer institute. One of plants screened was the Pacific yew tree Taxus brevifolia where extracts were found to have in vitro cytotoxic activity against tumour cells. Initially, there was a lack of interest in the compound since it was only found in very small amounts in T. brevifolia but eventually better sources were found in other Taxus species. Figure 21.21 shows the important structural features for taxol which contribute to its activity. As with the Vinca alkaloids there is little rationale for its particular activity against tubulin. As indicated in Figure 21.20, it acts in a different manner to the Vinca alkaloids in that it binds to tubulin and promotes its polymerisation rather than inhibiting it. However, in the course of promoting polymerisation, it reduces the flexibility in the tubulin structure which is required for elongation of the tubulin fibres. Taxol promotes lateral contacts within the tubulin dimer at the expense of longitudinal contacts which normally promote the elongation of the microtubule and which are essential for completion of the cell division process.3 Taxol (paclitaxel) is poorly water soluble and therefore it has to be formulated in polysorbate or polyoxyl castor oil which can cause hypersensitivity. Docetaxel (Fig. 21.21) is a semi-synthetic analogue of taxol which has better water solubility and is also more potent than taxol. Research is ongoing into finding a potent water soluble derivative of taxol but so far none of the candidate compounds has a product license.
Topoisomerase inhibitors
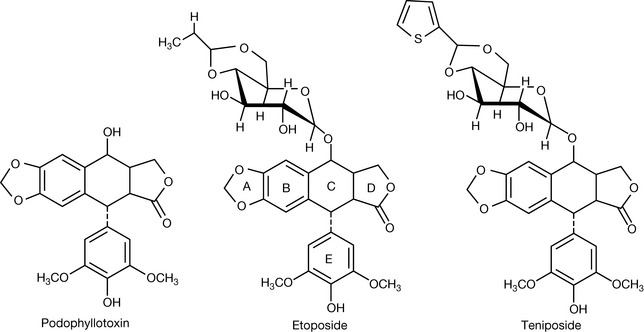
The podophyllotoxin (Fig. 20.22) was isolated from the American mandrake (Podophyllum pelatum), which had been traditionally used by American Indians as a laxative and anthelmintic, in 1880. It was subsequently shown to be a potent cytotoxic agent but was too toxic for use in cancer chemotherapy. Chemists at Sandoz in the 1950s further investigated Podophyllum species for analogues of podophyllotoxin. This eventually led to the development of the semi-synthetic compounds tenipinoside and etoposide (Fig. 21.22). These agents are topoisomerase II inhibitors. Teniposide is more cytotoxic than etoposide although it is not orally bioavailable.4
If the DNA in chromosomes were stretched out to its full length it would be far too long to fit into a cell. Thus the DNA helix coils up rather like the twisted cord of a telephone receiver where the torque due to the twist of the flex makes it twist round itself. This supercoiling can be either clockwise or anticlockwise (positive or negative). Replication of the DNA can only occur when it is negatively supercoiled, and topoisomerases such as topoisomerase II can break the double-stranded DNA and reform the strands to convert a positive strand into a negative strand (Fig. 21.23). In addition, DNA cutting is required to resolve tangles in the DNA during chromosome replication.
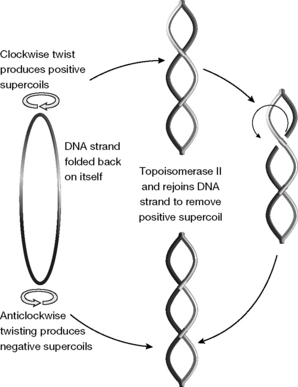
Mutations in topoisomerase genes are fatal to cells and likewise drugs which inhibit topisomerase activity. Etoposide and teniposide act by binding to the covalent topoisomerase/DNA complex, formed by reaction of the phosphate groups in DNA with a tyrosine residue in the enzyme, and stabilising it so that the DNA break is not repaired. This inhibits DNA replication, and thus cell division, and leads to cell death. Etoposide and teniposide are used to treat lymphomas, acute leukaemia, testicular cancer, small cell lung cancer, ovarian, bladder and brain cancers.
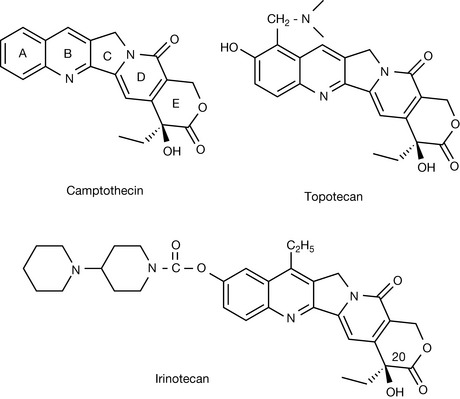
Camptothecin (Fig. 21.24) was isolated from the Chinese tree Camptotheca acuminate during screening for sterols which could be employed in the synthesis of cortisone. It was found to be cytotoxic but was unfortunately too toxic to be clinically useful. Rings A–D are essential for anticancer activity. The lactone ring E is essential for activity and the hydroxyl substituent in this ring at position 20 is also essential for activity and cannot be substituted by another element. For highest activity, the configuration at the 20 position must be S, the R isomer is up to 100 times less active. Modifications of the A and B rings can improve activity. Camptothecin inhibits topoisomerase I which has a similar type of activity to topoisomerase II except that it promotes single-strand breaks in DNA. Through its action in inhibiting topoisomerase I, camptothecin promotes fragmentation of chromosomal DNA. The toxicity of camptothecin was reduced by modification of rings A and B. Irinotecan and topotecan (Fig. 21.24) are clinically licensed analogues of camptothecin and have better water solubility than camptothecin. Irinotecan is a prodrug in which the ester linkage to the A ring has to be hydrolysed before it displays cytotoxicity. Irinotecan is used to treat metastatic colorectal cancer and topotecan is used to treat ovarian cancer. As a class of agents, camptothecin analogues show good promise for further development.
New targets
Imatinib
Protein kinase inhibitors (PKIs) provide a promising therapeutic strategy for cancer chemotherapy.5,6 Phosphorylation of proteins is a universal mechanism for the control of many cellular processes. The phosphorylation of proteins occurs when the γ-phosphate group of ATP is transferred to either tyrosine or serine/threonine within proteins. One family of enzymes catalyses tyrosine phosphorylation and another serine/threonine phosphorylation. Tyrosine phosphorylation is up-regulated in tumour cells and thus provides a selective target for therapy. Although there are numerous protein kinase structures, they have some features in common and the active site of the enzyme consists of two domains which hinge together in order to bind ATP. The majority of PKIs inhibit the binding of ATP to its binding site within the enzyme. However, the only licensed PKI, imatinib (Fig. 21.25) was discovered by development of lead compound discovered in a random screen. Imatinib inhibits protein kinase activity by binding to the enzyme so it becomes conformationally immobilised and is not itself able to undergo the phosphorylation required to activate it. The particular protein kinase targeted by imatinib is BCR-ABL protein kinase, which is an oncogene protein which enhances cell growth rates and resistance to apoptosis. This oncogene is found in 95% of patients with chronic myeloid leukaemia (CML). Imatinib has minimal effects on normal cells and is very effective in the selective treatment of CML. There are some indications of resistance occurring via mutations in BCR-ABL, and new analogues of imatinib are being developed in order to counteract these. In addition to inhibiting BCR-ABL, imatinib inhibits the c-KIT and PDGFR protein kinases which are dysregulated in gastrointestinal stromal tumours, and the drug is also being used to treat this tumour.
Hormone analogues
Cancers such as breast cancer and prostate cancer are associated with hormonal imbalances. Various hormone antagonists and inhibitors of hormone biosynthesis are used in cancers in which hormone activity is a component in the disease.
Tamoxifen
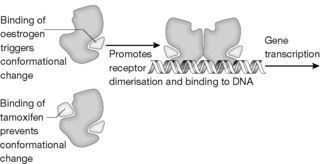
The tamoxifen structure (Fig. 21.26) was derived from the observation made in 1937 that triphenylethylene had weak oestrogenic activity. Tamoxifen was synthesised in 1962 and consisted of two geometrical isomers. It was found that while the Z-isomer functioned as an agonist at the oestrogen receptor, the E-isomer was an antagonist, and during the 1970s was adopted for the treatment of oestrogen-dependent breast cancer. In fact, subsequently it has been discovered that the action of tamoxifen is tissue dependent and, although it functions as an antagonist (modulator) in uterine tissue where it acts at the oestrogen-α receptor, in bone it functions as an agonist where it is binding to the oestrogen-β receptor.7,8
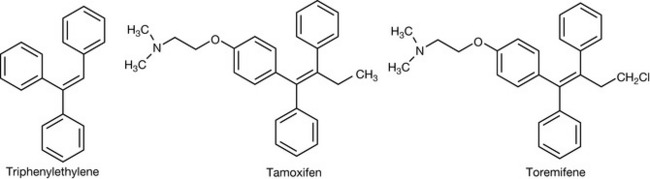
Like all steroids (apart from aldosterone), oestrogen binds to a hormone receptor which controls gene expression by binding to DNA. This type of receptor interacts with a variety of lipophilic molecules including vitamin D, retinoic acid, thyroxine and some prostaglandins. Figure 21.27 shows the primary sequence of part of the oestrogen receptor. A highly conserved domain of ca. 70 amino acids is present in all steroid receptors and forms two helices. Each helix contains four cysteine residues and four of these residues at the N-terminus of each helix which contains two zinc-binding Cys2–Cys2 sequence motifs. The structure consists of two helices perpendicular to each other. A zinc ion, coordinated by four conserved cysteines, holds the base of a loop at the N-terminus of each helix. This structural domain seems to be a general structure for protein–DNA recognition. The ligand binding region is composed of a long sequence of amino acids over 250 residues in length and oestrogen binds in this region, causing conformational change in the receptor and subsequent receptor dimerisation (Fig. 21.28). Following this, binding of the receptor to DNA occurs, which triggers off the gene transcription which, in the context of breast cancer, triggers of cell proliferation. Binding of an antagonist (or, more strictly, modulator) such as tamoxifen to the receptor blocks the conformational change which would normally be induced by oestrogen and thus blocks dimerisation and binding to DNA (Fig. 21.28). Toremifene is structurally close to tamoxifen and has the same mode of action.

Figure 21.27 Part of the amino acid sequence of the oestrogen receptor showing the cysteine-rich helices (in bold) that bind zinc and the oestrogen binding region.
 Self Test 21.3
Self Test 21.3
List the basic amino acids (see Ch. 6) in the basic region of the oestrogen receptor (bold in Fig. 21.27) which is an important component in the receptor binding region.
Aromatase inhibitors
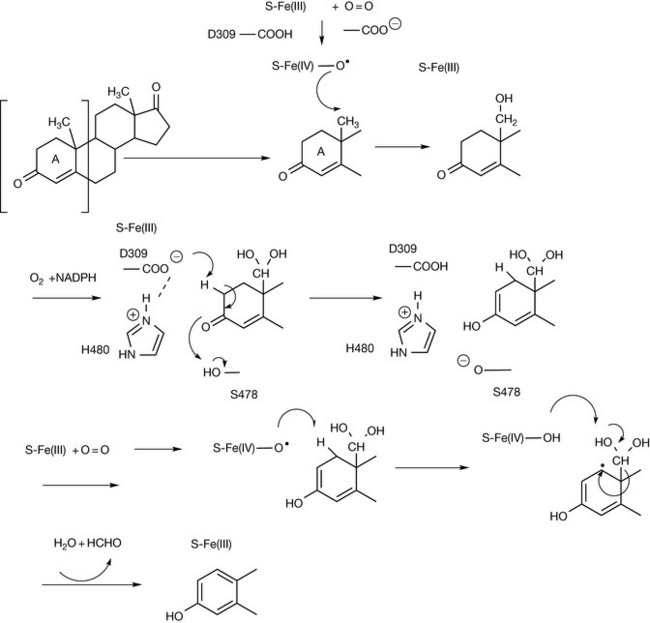
Oestrogens have a pivotal role in the development of breast cancer and 70% of breast cancers have been found to produce oestrogens in vivo. Drugs such as tamoxifen, discussed earlier, are active against breast cancer via blocking oestrogen receptors. The action of aromatase converts androstene dione to oestrogen (Fig. 21.29). The enzyme carrying out this conversion is a cyp450 which has the enzyme cyp450 reductase as a co-factor and oxidises the 19 methyl group through to formaldehyde. An aspartate (D309), histidine (H480) and a serine residue are involved in removing the first proton from ring A, and the second proton is removed by the cyp450 haem group, resulting in the formation of the estrone aromatic ring (Fig. 21.29).
The earliest aromatase inhibitor was aminoglutethimide (Fig. 21.30) which was originally synthesised as an anti-epileptic drug. However, the observation was made subsequently that it produced side effects similar to the symptoms of Addison’s disease. Addison’s disease results from a deficiency in steroid hormones, in most cases due to the deficiency of various enzymes in the biosynthetic pathway producing steroid hormones. Aromatase inhibitors can be divided into two classes: those binding to active site of the aromatase enzyme by competing with androstene dione (type I inhibitors), and those binding to the haem group (type II inhibitors). Aminoglutethimide belongs to the latter category and is rather non-specific in which haem-containing enzymes it targets. Thus it produces a general deficiency in steroid hormones, which has to be compensated for by co-administration with a corticosteroid such as dexamethasone. Various other drugs have been produced including later-generation type II inhibitors and type I inhibitors. The aromatase inhibitors on the market are shown in Figure 21.30. It is easier to model the interaction of the type I inhibitors with the enzyme active site rather than interaction with the haem group.
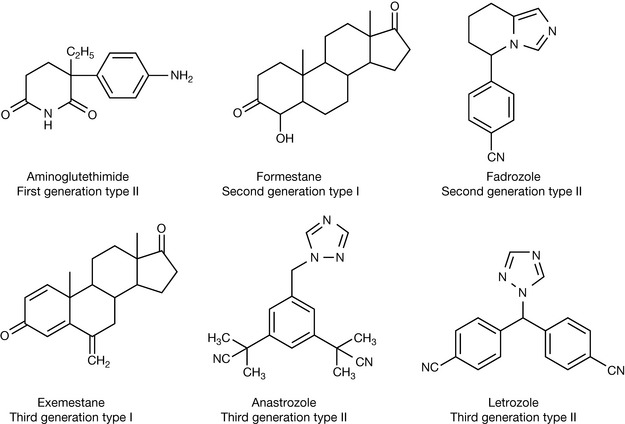
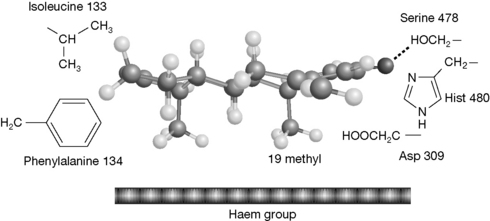
Figure 21.31 shows the proposed interaction between exemestane and the aromatase enzyme.9 The structure of exemestane is close to that of androstenedione and it fits the active site of the enzyme very closely, interacting with the lipophilic phenylalanine and isoleucine side chains and hydrogen bonding to serine 478. However, because of the extra double bond, it cannot complete the aromatisation reaction. It has been suggested that exemestane is a suicide inhibitor which reacts with the aromatase enzyme and this may occur via a reactive species generated at the C-19 position which would normally be eliminated upon formation of an aromatic ring. Release of the androstenedione substrate from the enzyme depends on the removal of the C-19 methyl group.
The third-generation type II aromatase inhibitors interact with the iron in the haem ring of the Cyp450. This prevents the haem group from reacting with oxygen and thus blocks hydroxylation of the C-19 methyl group. In the case of anastrozole, apart from the primary interaction of the molecule with the haem group, the binding to the active site is reinforced by hydrogen bonding between a threonine at position 310 and the triazole ring and between one of the CN groups in the molecule and asp 309.
Hormone analogues used to treat prostate cancer
Like breast cancer, prostate cancer is driven by a steroid hormone, in this case testosterone, and treatments block the effects of testosterone. The disease can be treated surgically by orchidectomy but the alternative front-line treatment is to use analogues of the peptide hormone gonadorelin (gonadotrophin releasing hormone, GnRH). This decapeptide has the amino acid sequence shown in Figure 21.32 with the unusual pyroglutamate residue at its N-terminus. It is responsible for releasing gonadotrophins, which have a range of actions including promoting testosterone production. The normal mode of action is for GnRH to be released by the body in pulses so that the receptor is not continuously exposed to the hormone. Testosterone production is suppressed by the use of GnRH agonists which, somewhat counterintuitively, due to the sustained circulating levels used, eventually cause down-regulation of receptor activity by overstimulation. In the initial phase of the treatment there is an increased production of testosterone, which may cause a flare-up in the cancer which is managed by using testosterone antagonists (see below). There are four commonly used GnRH agonists: buserelin, goserelin, leuprorelin and triptorelin. They are all modifications of the basic GnRH structure where the sixth amino acid from the N-terminus has been substituted with a D-amino acid, either tBu-serine, leucine or tryptophan, and the C-terminus has either an ethylamide or a diazane group.

Thus the structures of the agonists are very similar to that of GnRH where the N-terminus and C-terminus residues are conserved across many species. The GnRH receptor is a transmembrane G-coupled receptor and, as is the case with all of these receptors, is activated by several points of contact with its natural ligand. The gonadorelin analogues all bind more strongly to the receptor than GnRH and cause its activation. The GnRH analogues all have to be given by injection but there is the option of formulations designed to provide depot administration or an implant which release the hormone over several weeks.
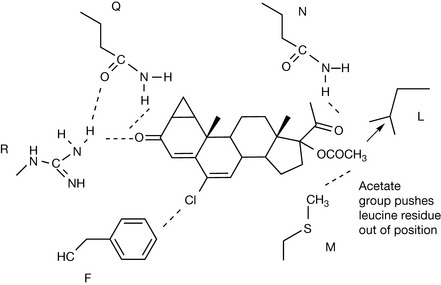
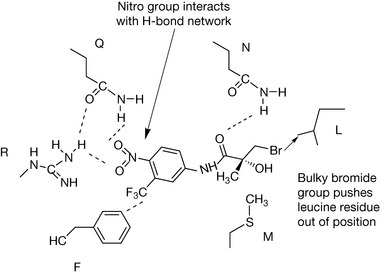
The flare effect of the GnRH analogues is managed by using antagonists of testosterone. In addition they are used as treatments on their own to directly block the action of testosterone. The three commonly used analogues are shown in Figure 21.33. Cyproterone acetate (CA) is a steroid analogue based on the structure of progesterone and, in addition to binding to the testosterone receptor, it also has some activity at the corticosteroid receptor and at the progesterone receptor. Despite being a much bulkier molecule than testosterone it binds strongly to its receptor and a co-crystallisation of CA with the cloned receptor has elucidated its binding mode.
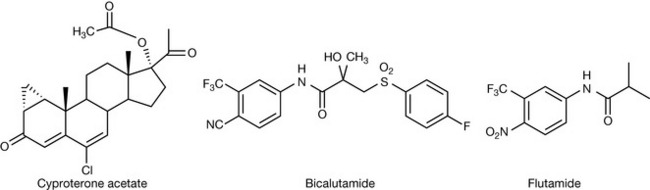
Figure 21.34 shows the interactions of CA with a number of amino acids in the receptor forming a hydrogen bond network with arginine and glutamine residues and having a van der Waals interaction between the chlorine in the structure and a phenylalanine residue.10 Although CA binds strongly to the receptor it does not activate it. The bulky acetate group displaces a leucine residue in the protein causing a conformational change without activating the receptor.11 In bicalutamide the CN group interacts with the hydrogen bonding network in the receptor in the same way as the carbonyl group of CA. The rest of the structure destabilises with normal structure of the receptor, thus binding without activation. In flutamide, the CN group is replaced by a nitro group.
1 Srivastana V., Negi A.S., Kumar J.K., Gupta M.M., Khanuja S.P.S. Plant based anticancer molecules: Chemical and biological profile of some important leads. Bioorg Med Chem. 2005;13:5892-5908.
2. Hait WH, Rubin E, Alli E, Goodin S. Tubulin targeting agents. Update on Cancer Therapeutics
3 Fiser S.B.H., Orr G.A. Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci. 2006;103:10166-10173.
4 Montecucco A., Biamonti G. Cellular response to etoposide treatment. Cancer Lett. 2006.
5 Scapin G. Structural biology in drug design: selective protein kinase inhibitors. Drug Discov Today. 2002;7:601-611.
6 Arora A., Scholar E.M. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Therap. 2005;315:971-979.
7 Schwabe J.W.R., Neuhaus D., Rhodes D. Solution structure of the DMA-binding domain of the oestrogen receptor. Nature. 1990;348:458-461.
8 McDonell D.P. The molecular determinants of estrogen receptor pharmacology. Maturitas. 2004;48:S7-S12.
9 Wijayaratne A.I.L., Nagel S.C., Paige L.A., et al. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology. 1999;140:5828-5840.
10 Bohl C.E., Wu Z., Miller D.D., Bell C.E., Dalton J.T. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J Biol Chem. 2007;282:3648-13655.
11 Bisson W.H., Abagyan R., Cavasotto C.N. Molecular basis of agonicity and antagonicity in the androgen receptor studied by molecular dynamics simulations. J Mol Graph Model. 2008;27:452-458.