 CHAPTER 55 Fat Metabolic Disorders
CHAPTER 55 Fat Metabolic Disorders
DISORDERS OF FATTY ACID OXIDATION
Fatty acids are derived from hydrolysis of triglycerides and catabolism of fat. The catabolism of fatty acids (Fig. 55-1) proceeds through the serial, oxidative removal of two carbons at a time as acetyl groups (each as acetyl-CoA). The reactions are catalyzed by a group of enzymes that exhibit specificities related to the chain length and other properties of the fatty acids: very long chain acyl-CoA dehydrogenase (VLCAD), long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) or trifunctional protein, medium-chain acyl-CoA dehydrogenase (MCAD), and short-chain acyl-CoA dehydrogenase (SCAD). MCAD deficiency is the most common inborn error of β-oxidation. Hypoketotic hypoglycemia is a common manifestation, as is Reye syndrome–like illness with hypoglycemia and elevated liver enzymes. Fatty infiltration of the liver also occurs. True hepatic failure is rare. Episodes may be recurrent in the patient or the family. Sudden infant death syndrome is reported in infants with MCAD deficiency, perhaps related to hypoglycemia. Treatment requires avoidance of fasting and provision of calories with fever or other metabolic stress. Medium-chain triglycerides must be avoided.
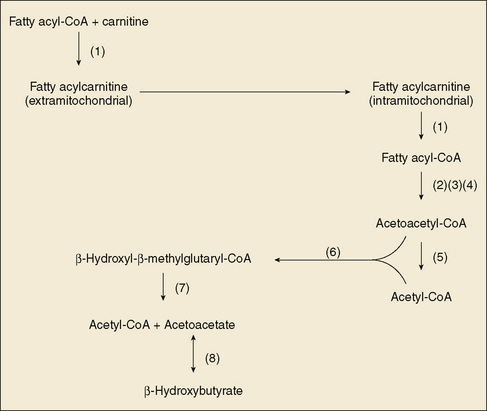
FIGURE 55-1 Scheme of fatty acid and catabolism and ketone body formation. (1) Carnitine acyl-CoA dehydrogenases, (2) long-chain fatty acyl-CoA dehydrogenase (trifunctional protein), (3) medium-chain fatty acyl-CoA dehydrogenase, (4) short-chain fatty acyl-CoA dehydrogenase, (5) β-ketothiolase, (6) β-hydroxy-β-methylglutaryl-CoA synthase, (7) β-hydroxy-β-methylglutaryl-CoA lyase, (8) β-hydroxybutyrate dehydrogenase. CoA, coenzyme A.
VLCAD deficiency and LCHAD (trifunctional protein) deficiency result in significant myopathy and cardiomyopathy, and LCHAD deficiency is accompanied by a retinopathy in later childhood. In all of the disorders of β-oxidation, carnitine depletion can occur through excessive urinary excretion of carnitine esters of the incompletely oxidized fatty acids. Measurement of plasma carnitine is helpful in monitoring for this deficiency, which results in weakness and muscle pain, along with myoglobinuria in some people.
Hydroxymethylglutaryl-CoA lyase deficiency, although not a disorder of β-oxidation, interferes profoundly with hepatic adaptation to fasting by impairing ketogenesis (see Fig. 55-1). The clinical manifestations are those of MCAD deficiency, except that carnitine depletion is less prominent.
The diagnosis of disorders involving a deficiency of β-oxidation is suggested by the clinical picture and by hypoketotic hypoglycemia. The diagnosis is confirmed by analysis of urinary organic acid and acylglycine profiles, along with plasma acylcarnitine and free fatty acid profiles. Enzyme measurements and DNA testing complete the confirmatory testing. The profile of acylcarnitines in cultured skin fibroblasts may be helpful if other testing is not conclusive. In MCAD deficiency, a single mutation, 985A-G, accounts for a significant percentage of cases. Treatment includes avoidance of fasting, and fluid and calorie supplementation during periods of metabolic stress, such as fever. In MCAD deficiency, medium-chain triglycerides must be avoided. In the long-chain fatty acid metabolic disorders, provision of medium-chain fatty acids improves muscle energy metabolism. SCAD deficiency has a variable and poorly understood phenotype that can include little in the way of symptoms or, in other patients, muscle weakness and hypoglycemia when fasting. Avoidance of fasting is recommended. Carnitine supplementation may be helpful. Careful monitoring is recommended.
GLUTARIC ACIDURIA TYPE II
Glutaric aciduria type II (multiple acyl-CoA dehydrogenase deficiency) is a clinical disease produced by a defect in the transfer of electrons from flavine adenine nucleotides to the electron transport chain (electron transfer flavoprotein [ETF], or ETF dehydrogenase); this defect results in a deficiency of multiple fatty acyl CoA dehydrogenases (see Fig. 55-1). It should not be confused with glutaric acidemia type I (see Chapter 54). When the enzyme essentially is nonfunctional, congenital anomalies are common, including renal cysts, facial abnormalities, rocker-bottom feet, and hypospadias. Severely affected infants have nonketotic hypoglycemia, metabolic acidosis, and the odor of sweaty feet soon after birth; these infants may die within the neonatal period. Less severely affected infants may have a more episodic, Reye syndrome–like illness. Skeletal and cardiac myopathy can be prominent in this complex, multisystemic disease. Onset in later childhood may be marked by recurrent hypoglycemia and myopathy. Treatment has not been effective in infants with complete deficiency. Milder forms respond to avoidance of fasting and caloric support during metabolic stress. Some patients respond to administration of riboflavin. Glutaric aciduria type II exhibits autosomal recessive inheritance. Confirmatory testing is similar to that for the other fatty acid oxidation disorders.
CARNITINE DEFICIENCY
Carnitine is a crucial cofactor in the transport of long-chain fatty acids across the mitochondrial inner membrane (see Fig. 55-1). It is synthesized from lysine by humans and is present in dietary red meat and dairy products. Carnitine deficiency is either primary (caused by failure of intake, synthesis, or transport of carnitine) or secondary (caused by the excretion of excessive amounts of carnitine as carnityl esters in patients with other inborn errors of metabolism or treatment with drugs that complex carnitine, such as valproic acid). Primary systemic carnitine deficiency results from inadequate renal reabsorption of carnitine secondary to a mutation in the sodium-dependent carnitine transporter. It responds well to carnitine supplementation. There are numerous examples of secondary carnitine deficiency among the organic acidurias, most prominently in disorders of the propionate pathway and in disorders of the β-oxidation of long-chain and medium-chain fatty acids. Clinical manifestations of carnitine deficiency include hypoketotic hypoglycemia, lethargy, lassitude, muscle weakness, and cardiomyopathy.