 CHAPTER 61 Respiratory Diseases of the Newborn
CHAPTER 61 Respiratory Diseases of the Newborn
Respiratory distress that becomes manifested by tachypnea, intercostal retractions, reduced air exchange, cyanosis, expiratory grunting, and nasal flaring is a nonspecific response to serious illness. Not all of the disorders producing neonatal respiratory distress are primary diseases of the lungs. The differential diagnosis of respiratory distress includes pulmonary, cardiac, hematologic, infectious, anatomic, and metabolic disorders that may involve the lungs directly or indirectly. Surfactant deficiency causes respiratory distress syndrome (RDS), resulting in cyanosis and tachypnea; infection produces pneumonia, shown by interstitial or lobar infiltrates; meconium aspiration results in a chemical pneumonitis with hypoxia and pulmonary hypertension; hydrops fetalis causes anemia and hypoalbuminemia with high-output heart failure and pulmonary edema; and congenital or acquired pulmonary hypoplasia causes pulmonary hypertension and pulmonary insufficiency. It also is clinically useful to differentiate the common causes of respiratory distress according to gestational age (Table 61-1).
TABLE 61-1 Etiology of Respiratory Distress
PRETERM INFANT
Respiratory distress syndrome (RDS)*
Erythroblastosis fetalis
Nonimmune hydrops
Pulmonary hemorrhage
FULL-TERM INFANT
Primary pulmonary hypertension of the neonate*
Meconium aspiration pneumonia*
Polycythemia
Amniotic fluid aspiration
PRETERM AND FULL-TERM INFANT
Bacterial sepsis (GBS)*
Transient tachypnea*
Spontaneous pneumothorax
Congenital anomalies (e.g., congenital lobar emphysema, cystic adenomatoid malformation, diaphragmatic hernia)
Congenital heart disease
Pulmonary hypoplasia
Viral infection (e.g., herpes simplex, CMV)
Inborn metabolic errors
CMV, cytomegalovirus; GBS, group B streptococcus.
In addition to the specific therapy for the individual disorder, the supportive care and evaluation of the infant with respiratory distress can be applied to all the problems mentioned earlier (Table 61-2). Blood gas monitoring and interpretation are key components of general respiratory care.
TABLE 61-2 Initial Laboratory Evaluation of Respiratory Distress
| Test | Rationale |
|---|---|
| Chest radiograph | To determine reticular granular pattern of RDS; to determine presence of pneumothorax, cardiomegaly, life-threatening congenital anomalies |
| Arterial blood gas | To determine severity of respiratory compromise, hypoxemia, and hypercapnia and type of acidosis; the severity determines treatment strategy |
| Complete blood count | Hemoglobin/hematocrit to determine anemia and polycythemia; white blood cell count to determine neutropenia/sepsis; platelet count and smear to determine DIC |
| Blood culture | To recover potential pathogen |
| Blood glucose | To determine presence of hypoglycemia, which may produce or occur simultaneously with respiratory distress; to determine stress hyperglycemia |
| Echocardiogram, ECG | In the presence of a murmur, cardiomegaly, or refractory hypoxia; to determine structural heart disease or PPHN |
DIC, disseminated intravascular coagulation; ECG, electrocardiogram; PPHN, primary pulmonary hypertension of the newborn; RDS, respiratory distress syndrome.
Treatment of hypoxemia requires knowledge of normal values. In term infants, the arterial PaO2 level is 55 to 60 mm Hg at 30 minutes of life, 75 mm Hg at 4 hours, and 90 mm Hg at 24 hours. Preterm infants have lower values. PaCO2 levels should be 35 to 40 mm Hg, and the pH should be 7.35 to 7.40. It is imperative that arterial blood gas analysis be performed in all infants with significant respiratory distress, whether or not cyanosis is perceived. Cyanosis becomes evident when there is 5 g of unsaturated hemoglobin; anemia may interfere with the perception of cyanosis. Jaundice also may interfere with the appearance of cyanosis. Capillary blood gas determinations are useful in determining blood pH and the PaCO2 level. Because of the nature of the heel-stick capillary blood gas technique, venous blood may mix with arterial blood, resulting in falsely low blood PaO2 readings. Serial blood gas levels may be monitored by an indwelling arterial catheter placed in a peripheral artery or through the umbilical artery to the aorta at the level of the T6–10 or the L4–5 vertebrae. This placement avoids catheter occlusion of the celiac (T12), superior mesenteric (T12–L1), renal (L1–2), and inferior mesenteric (L2–3) arteries. Another method for monitoring blood gas levels is to combine capillary blood gas techniques with noninvasive methods used to monitor oxygen (pulse oximetry or transcutaneous oxygen diffusion).
Metabolic acidosis, defined as a reduced pH (<7.25) and bicarbonate concentration (<18 mEq/L) accompanied by a normal or low PCO2 level, may be caused by hypoxia or by insufficient tissue perfusion. The origin of the disorder may be pulmonary, cardiac, infectious, renal, hematologic, nutritional, metabolic, or iatrogenic. The initial approach to metabolic acidosis is to determine the cause and treat the pathophysiologic problem. This approach may include, as in the sequence of therapy for hypoxia, increasing the inspired oxygen concentration; applying continuous positive airway pressure nasally, using oxygen as the gas; or initiating mechanical ventilation using positive end-expiratory pressure and oxygen. Patients with hypotension produced by hypovolemia require fluids and may need inotropic or vasoactive drug support. If metabolic acidosis persists despite specific therapy, sodium bicarbonate (1 mEq/kg/dose) may be given by slow intravenous infusion. Near-normal or low PCO2 levels should be documented before sodium bicarbonate infusion. The buffering effect of sodium bicarbonate results in increased PCO2 levels, unless adequate ventilation is maintained.
Respiratory acidosis, defined as an elevated PCO2 level and reduced pH without a reduction in the bicarbonate concentration, may be caused by pulmonary insufficiency or central hypoventilation. Most disorders producing respiratory distress can lead to hypercapnia. Treatment involves assisted ventilation but not sodium bicarbonate. If central nervous system depression of respirations is caused by placental passage of narcotic analgesics, assisted ventilation is instituted first, then the central nervous system depression is reversed by naloxone.
RESPIRATORY DISTRESS SYNDROME (HYALINE MEMBRANE DISEASE)
RDS occurs after the onset of breathing and is associated with an insufficiency of pulmonary surfactant.
Lung Development
The lining of the alveolus consists of 90% type I cells and 10% type II cells. After 20 weeks of gestation, the type II cells contain vacuolated, osmophilic, lamellar inclusion bodies, which are packages of surface-active material (Fig. 61-1). This lipoprotein surfactant is 90% lipid and is composed predominantly of saturated phosphatidylcholine (lecithin), but also contains phosphatidylglycerol, other phospholipids, and neutral lipids. The surfactant proteins SP-A, SP-B, SP-C, and SP-D are packaged into the lamellar body and contribute to surface-active properties and recycling of surfactant. Surfactant prevents atelectasis by reducing surface tension at low lung volumes when it is concentrated at end expiration as the alveolar radius decreases; surfactant contributes to lung recoil by increasing surface tension at larger lung volumes when it is diluted during inspiration as the alveolar radius increases. Without surfactant, surface tension forces are not reduced, and atelectasis develops during end expiration as the alveolus collapses.
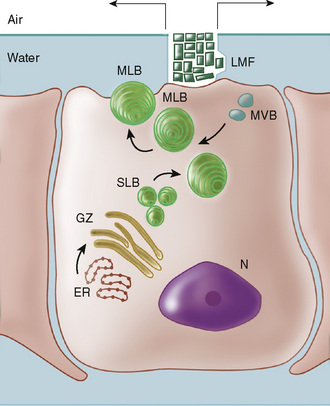
FIGURE 61-1 Proposed pathway of synthesis, transport, secretion, and reuptake of surfactant in the type II alveolar cell. Phospholipids are synthesized in the smooth endoplasmic reticulum (ER). The glucose/glycerol precursor may be derived from lung glycogen or circulating glucose. Phospholipids and surfactant proteins are packaged in the Golgi apparatus (GZ), emerge as small lamellar bodies (SLB), coalesce to mature lamellar bodies (MLB), migrate to the apical membrane, and are released by exocytosis into the liquid hypophase below the air-liquid interface. The tightly coiled lamellar body unravels to form the lattice (tubular) myelin figure (LMF), the immediate precursor to the phospholipid monolayer at the alveolar surface. Reuptake by endocytosis forms multivesicular bodies (MVB) that recycle surfactant. The enzymes, receptors, transporters, and surfactant proteins are controlled by regulatory processes at the transcriptional level in the nucleus (N). Corticosteroid and thyroid hormones are regulatory ligands that may accelerate surfactant synthesis.
(From Hansen T, Corbet A: Lung development and function. In Taeusch HW, Ballard R, Avery ME, editors: Diseases of the Newborn, 6th ed, Philadelphia, 1991, Saunders, p 465.)
The timing of surfactant production in quantities sufficient to prevent atelectasis depends on an increase in fetal cortisol levels that begins between 32 and 34 weeks of gestation. By 34 to 36 weeks, sufficient surface-active material is produced by the type II cells in the lung, is secreted into the alveolar lumen, and is excreted into the amniotic fluid. The concentration of lecithin in amniotic fluid indicates fetal pulmonary maturity. Because the amount of lecithin is difficult to quantify, the ratio of lecithin (which increases with maturity) to sphingomyelin (which remains constant during gestation) (L/S ratio) is determined. An L/S ratio of 2:1 usually indicates pulmonary maturity. The presence of minor phospholipids, such as phosphatidylglycerol, also is indicative of fetal lung maturity and may be useful in situations in which the L/S ratio is borderline or possibly affected by maternal diabetes, which reduces lung maturity. The absence of phosphatidylglycerol suggests that surfactant might not be mature.
Clinical Manifestations
A deficiency of pulmonary surfactant results in atelectasis, decreased functional residual capacity, arterial hypoxemia, and respiratory distress. Surfactant synthesis may be reduced as a result of hypovolemia, hypothermia, acidosis, hypoxemia, and rare genetic disorders of surfactant synthesis. These factors also produce pulmonary artery vasospasm, which may contribute to RDS in larger premature infants who have developed sufficient pulmonary arteriole smooth muscle to produce vasoconstriction. Surfactant deficiency–induced atelectasis causes alveoli to be perfused but not ventilated, which results in a pulmonary shunt and hypoxemia. As atelectasis increases, the lungs become increasingly difficult to expand, and lung compliance decreases. Because the chest wall of the premature infant is very compliant, the infant attempts to overcome decreased lung compliance with increasing inspiratory pressures, resulting in retractions of the chest wall. The sequence of decreased lung compliance and chest wall retractions leads to poor air exchange, an increased physiologic dead space, alveolar hypoventilation, and hypercapnia. A cycle of hypoxia, hypercapnia, and acidosis acts on type II cells to reduce surfactant synthesis and, in some infants, on the pulmonary arterioles to produce pulmonary hypertension.
Infants at greatest risk for RDS are premature and have an immature L/S ratio. The incidence of RDS increases with decreasing gestational age. RDS develops in 30% to 60% of infants between 28 and 32 weeks of gestation. Other risk factors include delivery of a previous preterm infant with RDS, maternal diabetes, hypothermia, fetal distress, asphyxia, male sex, white race, being the second born of twins, and delivery by cesarean section without labor.
RDS may develop immediately in the delivery room in extremely immature infants at 26 to 30 weeks of gestation. Some more mature infants (34 weeks’ gestation) may not show signs of RDS until 3 to 4 hours after birth, correlating with the initial release of stored surfactant at the onset of breathing accompanied by the ongoing inability to replace the surfactant owing to inadequate stores. Manifestations of RDS include cyanosis, tachypnea, nasal flaring, intercostal and sternal retractions, and grunting. Grunting is caused by closure of the glottis during expiration, the effect of which is to maintain lung volume (decreasing atelectasis) and gas exchange during exhalation. Atelectasis is well documented by radiographic examination of the chest, which shows a ground-glass haze in the lung surrounding air-filled bronchi (the air bronchogram) (Fig. 61-2). Severe RDS may show an airless lung field or a whiteout on a radiograph, even obliterating the distinction between the atelectatic lungs and the heart.
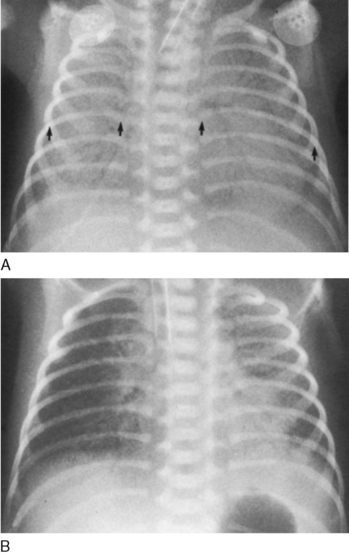
FIGURE 61-2 Respiratory distress syndrome. The infant is intubated, and the lungs show a dense reticulonodular pattern with air bronchograms (A). To evaluate rotation on the frontal chest, the lengths of the posterior ribs are compared from left to right (arrows). Because the infant is supine, the side of the longer ribs indicates to which side the thorax is rotated. In this case, the left ribs are longer, and this radiograph is a left posterior oblique view. Surfactant was administered, resulting in significant improvement in the density of the lung (B). The right lung is slightly better aerated than the left. Uneven distribution of clearing is common.
(From Hilton S, Edwards D: Practical Pediatric Radiology, 2nd ed, Philadelphia, 1994, Saunders.)
During the first 72 hours, infants with RDS have increasing distress and hypoxemia. In infants with severe RDS, the development of edema, apnea, and respiratory failure necessitates assisted ventilation. Thereafter, uncomplicated cases show a spontaneous improvement that often is heralded by diuresis and a marked resolution of edema. Complications include the development of a pneumothorax, a patent ductus arteriosus (PDA), and bronchopulmonary dysplasia (BPD). The differential diagnosis of RDS includes diseases associated with cyanosis and respiratory distress (see Table 58-10).
Prevention and Treatment
Strategies to prevent preterm birth include maternal cervical cerclage, bed rest, treatment of infections, and administration of tocolytic medications. Additionally, prevention of neonatal cold stress, birth asphyxia, and hypovolemia reduces the risk of RDS. If premature delivery is unavoidable, the antenatal administration of corticosteroids (e.g., betamethasone) to the mother (and thus to the fetus) stimulates fetal lung production of surfactant; this approach requires multiple doses for at least 48 hours.
After birth, RDS may be prevented or its severity reduced by intratracheal administration of exogenous surfactant immediately after birth in the delivery room or within a few hours of birth. A mammalian-derived surfactant is currently preferred. Exogenous surfactant can be administered repeatedly during the course of RDS in patients receiving endotracheal intubation, mechanical ventilation, and oxygen therapy. Additional management includes the general supportive and ventilation care presented in Table 61-3.
TABLE 61-3 Potential Causes of Neonatal Apnea
| Central nervous system | IVH, drugs, seizures, hypoxic injury |
| Respiratory | Pneumonia, obstructive airway lesions, atelectasis, extreme prematurity (<1000 g), laryngeal reflex, phrenic nerve paralysis, severe RDS, pneumothorax |
| Infectious | Sepsis, necrotizing enterocolitis, meningitis (bacterial, fungal, viral) |
| Gastrointestinal | Oral feeding, bowel movement, gastroesophageal reflux, esophagitis, intestinal perforation |
| Metabolic | ↓ Glucose, ↓ calcium, ↓ PO2, ↓↑ sodium, ↑ ammonia, ↑ organic acids, ↑ ambient temperature, hypothermia |
| Cardiovascular | Hypotension, hypertension, heart failure, anemia, hypovolemia, change in vagal tone |
| Idiopathic | Immaturity of respiratory center, sleep state, upper airway collapse |
IVH, intraventricular hemorrhage; RDS, respiratory distress syndrome.
The PaO2 level should be maintained between 60 and 70 mm Hg (oxygen saturation 90%), and the pH should be maintained greater than 7.25. An increased concentration of warm and humidified inspired oxygen administered by an oxygen hood or nasal cannula may be all that is needed for larger premature infants. If hypoxemia (PaO2 <50 mm Hg) is present, and the needed inspired oxygen concentration is 70% to 100%, nasal continuous positive airway pressure should be added at a distending pressure of 8 to 10 cm H2O. If respiratory failure ensues (PCO2 >60 mm Hg, pH <7.20, and PaO2 <50 mm Hg with 100% oxygen), assisted ventilation using a respirator is indicated. Conventional rate (25 to 60 breaths/min), high-frequency jet (150 to 600 breaths/min), and oscillator (900 to 3000 breaths/min) ventilators all have been successful in managing respiratory failure caused by severe RDS. Suggested starting settings on a conventional ventilator are fraction of inspired oxygen, 60% to 100%; peak inspiratory pressure, 20 to 25 cm H2O; positive end-expiratory pressure, 5 cm H2O; and respiratory rate, 30 to 50 breaths/min.
In response to persistent hypercapnia, alveolar ventilation (tidal volume − dead space × rate) must be increased. Ventilation can be increased by an increase in the ventilator’s rate or an increase in the tidal volume, which is the gradient between peak inspiratory pressure and positive end-expiratory pressure. In response to hypoxia, the inspired oxygen content may be increased. Alternatively, the degree of oxygenation depends on the mean airway pressure. Mean airway pressure is directly related to peak inspiratory pressure, positive end-expiratory pressure, flow, and inspiratory-to-expiratory ratio. Increased mean airway pressure may improve oxygenation by improving lung volume, enhancing ventilation-perfusion matching. Because of the difficulty in distinguishing sepsis and pneumonia from RDS, broad-spectrum parenteral antibiotics (ampicillin and gentamicin) are administered for 48 to 72 hours, pending the recovery of an organism from a previously obtained blood culture.
COMPLICATIONS OF RESPIRATORY DISTRESS SYNDROME
Patent Ductus Arteriosus
PDA is a common complication that occurs in many low birth weight infants who have RDS. The incidence of PDA is inversely related to the maturity of the infant. In term newborns, the ductus closes within 24 to 48 hours after birth. However, in preterm newborns, the ductus frequently fails to close, requiring medical or surgical closure. The ductus arteriosus in a preterm infant is less responsive to vasoconstrictive stimuli, which when complicated with hypoxemia during RDS, may lead to a persistent PDA that creates a shunt between the pulmonary and systemic circulations.
During the acute phase of RDS, hypoxia, hypercapnia, and acidosis lead to pulmonary arterial vasoconstriction and increased pressure. The pulmonary and systemic pressures may be equal, and flow through the ductus may be small or bidirectional. When RDS improves and pulmonary vascular resistance declines, flow through the ductus arteriosus increases in a left-to-right direction. Significant systemic-to-pulmonary shunting may lead to heart failure and pulmonary edema. Excessive intravenous fluid administration may increase the incidence of symptomatic PDA. The infant’s respiratory status deteriorates because of increased lung fluid, hypercapnia, and hypoxemia. In response to poor blood gas levels, the infant is subjected to higher inspired oxygen concentrations and higher peak inspiratory ventilator pressures, both of which can damage the lung and cause chronic lung disease.
Clinical manifestations of a PDA usually become apparent on day 2 to 4 of life. Because the left-to-right shunt directs flow to a low-pressure circulation from one of high pressure, the pulse pressure widens; a previously inactive precordium shows an extremely active precordial impulse, and peripheral pulses become easily palpable and bounding. The murmur of a PDA may be continuous in systole and diastole, but usually only the systolic component is auscultated. Heart failure and pulmonary edema result in rales and hepatomegaly. A chest radiograph shows cardiomegaly and pulmonary edema; a two-dimensional echocardiogram shows patency; and Doppler studies show markedly increased left-to-right flow through the ductus.
Treatment of a PDA during RDS involves initial fluid restriction and diuretic administration. If there is no improvement after 24 to 48 hours, indomethacin or ibuprofen, prostaglandin synthetase inhibitors, is administered. Contraindications to using indomethacin include thrombocytopenia (platelets <50,000/mm3), bleeding, serum creatinine measuring more than 1.8 mg/dL, and oliguria. Because 20% to 30% of infants do not respond initially to indomethacin and because the PDA reopens in 10% to 20% of infants who do, a repeat course of indomethacin or surgical ligation is required in some patients.
Pulmonary Air Leaks
Assisted ventilation with high peak inspiratory pressures and positive end-expiratory pressures may cause overdistention of alveoli in localized areas of the lung. Rupture of the alveolar epithelial lining may produce pulmonary interstitial emphysema as gas dissects along the interstitial space and the peribronchial lymphatics. Extravasation of gas into the parenchyma reduces lung compliance and worsens respiratory failure. Gas dissection into the mediastinal space produces a pneumomediastinum, occasionally dissecting into the subcutaneous tissues around the neck, causing subcutaneous emphysema.
Alveolar rupture adjacent to the pleural space produces a pneumothorax (Fig. 61-3). If the gas is under tension, the pneumothorax shifts the mediastinum to the opposite side of the chest, producing hypotension, hypoxia, and hypercapnia. The diagnosis of a pneumothorax may be based on unequal transillumination of the chest and may be confirmed by chest radiograph. Treatment of a symptomatic pneumothorax requires insertion of a pleural chest tube connected to negative pressure or to an underwater drain. Prophylactic or therapeutic use of exogenous surfactant has reduced the incidence of pulmonary air leaks.
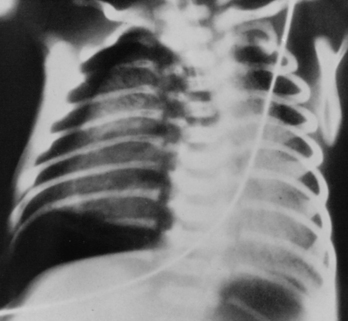
FIGURE 61-3 Pneumothorax. Right-sided hyperlucent pleural air is obvious. The findings of linear interstitial air and the resultant noncompliant but collapsed lung are noted.
(From Heller RM, Kirchner SG: Advanced exercises in diagnostic radiology: The Newborn, Philadelphia, WB Saunders, 1979).
Pneumothorax also is observed after vigorous resuscitation, meconium aspiration pneumonia, pulmonary hypoplasia, and diaphragmatic hernia. Spontaneous pneumothorax is seen in less than 1% of deliveries and may be associated with renal malformations.
Bronchopulmonary Dysplasia (Chronic Lung Disease)
BPD is a clinical diagnosis defined by oxygen dependence at 36 weeks’ postconceptual age and accompanied by characteristic clinical and radiographic findings that correspond to anatomic abnormalities. Oxygen concentrations greater than 40% are toxic to the neonatal lung. Oxygen-mediated lung injury results from the generation of superoxides, hydrogen peroxide, and oxygen free radicals, which disrupt membrane lipids. Assisted ventilation with high peak pressures produces barotrauma, compounding the damaging effects of highly inspired oxygen levels. In most patients, BPD develops after ventilation for RDS that may have been complicated by PDA or pulmonary interstitial emphysema. Inflammation from prolonged assisted ventilation and repeated systemic and pulmonary infections may play a major role. Failure of RDS to improve after 2 weeks, the need for prolonged mechanical ventilation, and oxygen therapy required at 36 weeks’ postconception age are characteristic of patients with RDS in whom BPD develops. BPD also may develop in infants weighing less than 1000 g who require mechanical ventilation for poor respiratory drive in the absence of RDS. Fifty percent of infants of 24 to 26 weeks’ gestational age require oxygen at 36 weeks’ corrected age.
The radiographic appearance of BPD is characterized initially by lung opacification and subsequently by development of cysts accompanied by areas of overdistention and atelectasis, giving the lung a spongelike appearance. The histopathology of BPD reveals interstitial edema, atelectasis, mucosal metaplasia, interstitial fibrosis, necrotizing obliterative bronchiolitis, and overdistended alveoli.
The clinical manifestations of BPD are oxygen dependence, hypercapnia, compensatory metabolic alkalosis, pulmonary hypertension, poor growth, and development of right-sided heart failure. Increased airway resistance with reactive airway bronchoconstriction also is noted and is treated with bronchodilating agents. Severe chest retractions produce negative interstitial pressure that draws fluid into the interstitial space. Together with cor pulmonale, these chest retractions cause fluid retention, necessitating fluid restriction and the administration of diuretics.
Patients with severe BPD may need treatment with mechanical ventilation for many months. To reduce the risk of subglottic stenosis, a tracheotomy may be indicated. To reduce oxygen toxicity and barotrauma, ventilator settings are reduced to maintain blood gases with slightly lower PaO2 (50 mm Hg) and higher PaCO2 (50 to 75 mm Hg) levels than for infants during the acute phase of RDS. Dexamethasone therapy may reduce inflammation, improve pulmonary function, and enhance weaning of patients from assisted ventilation. However, dexamethasone may increase the risk of cerebral palsy or abnormal neuromotor developmental outcome. Older survivors of BPD have hyperinflation, reactive airways, and developmental delay. They are at risk for severe respiratory syncytial virus pneumonia and as infants should receive prophylaxis against respiratory syncytial virus.
Retinopathy of Prematurity (Retrolental Fibroplasia)
Retinopathy of prematurity (ROP) is caused by the acute and chronic effects of oxygen toxicity on the developing blood vessels of the premature infant’s retina. The completely vascularized retina of the term infant is not susceptible to ROP. ROP is a leading cause of blindness in very low birth weight infants (<1500 g). Excessive arterial oxygen tensions produce vasoconstriction of immature retinal vasculature in the first stage of this disease. Vaso-obliteration follows if the duration and extent of hyperoxia are prolonged beyond the time when vasoconstriction is reversible. Hypercarbia and hypoxia may contribute to ROP. The subsequent proliferative stages are characterized by extraretinal fibrovascular proliferation, forming a ridge between the vascular and avascular portions of the retina, and by the development of neovascular tufts. In mild cases, vasoproliferation is noted at the periphery of the retina. Severe cases may have neovascularization involving the entire retina, retinal detachment resulting from traction on vessels as they leave the optic disc, fibrous proliferation behind the lens producing leukokoria, and synechiae displacing the lens forward, leading to glaucoma. Both eyes usually are involved, but severity may be asymmetrical.
The incidence of ROP may be reduced by careful monitoring of arterial blood gas levels in all patients receiving oxygen. Although there is no absolutely safe PaO2 level, it is wise to keep the arterial oxygen level between 50 and 70 mm Hg in premature infants. Infants who weigh less than 1500 g or who are born before 28 weeks’ gestational age (some authors say 32 weeks) should be screened when they are 4 weeks of age or more than 34 weeks’ corrected gestational age, whichever comes first. Laser therapy or (less often) cryotherapy may be used for vitreous hemorrhage or for severe, progressive vasoproliferation. Surgery is indicated for retinal detachment. Less severe stages of ROP resolve spontaneously and without visual impairment in most patients.
TRANSIENT TACHYPNEA OF THE NEWBORN
Transient tachypnea of the newborn is a self-limited condition characterized by tachypnea, mild retractions, hypoxia, and occasional grunting, usually without signs of severe respiratory distress. Cyanosis, when present, usually requires treatment with supplemental oxygen in the range of 30% to 40%. Transient tachypnea of the newborn usually is noted in larger premature infants and in term infants born by precipitous delivery or cesarean section without prior labor. Infants of diabetic mothers and infants with poor respiratory drive as a result of placental passage of analgesic drugs are at risk. Transient tachypnea of the newborn may be caused by retained lung fluid or slow resorption of lung fluid. Chest radiographs show prominent central vascular markings, fluid in the lung fissures, overaeration, and occasionally a small pleural effusion. Air bronchograms and a reticulogranular pattern are not seen; their presence suggests another pulmonary process, such as RDS or pneumonia.
MECONIUM ASPIRATION SYNDROME
Meconium-stained amniotic fluid is seen in 15% of predominantly term and post-term deliveries. Although the passage of meconium into amniotic fluid is common in infants born in the breech presentation, meconium-stained fluid should be considered clinically as a sign of fetal distress in all infants. The presence of meconium in the amniotic fluid suggests in utero distress with asphyxia, hypoxia, and acidosis.
Aspiration of amniotic fluid contaminated with particulate meconium may occur in utero in a distressed, gasping fetus; more often, meconium is aspirated into the lung immediately after delivery. Affected infants have abnormal chest radiographs, showing a high incidence of pneumonia and pneumothoraces.
Meconium aspiration pneumonia is characterized by tachypnea, hypoxia, hypercapnia, and small airway obstruction causing a ball-valve effect, leading to air trapping, overdistention, and extra-alveolar air leaks. Complete small airway obstruction produces atelectasis. Within 24 to 48 hours, a chemical pneumonia develops in addition to the mechanical effects of airway obstruction. Abnormal pulmonary function may be caused by the meconium in part through inactivation of surfactant. Primary pulmonary hypertension of the newborn (PPHN) frequently accompanies meconium aspiration, with right-to-left shunting caused by increased pulmonary vascular resistance. The chest radiograph reveals patchy infiltrates, overdistention, flattening of the diaphragm, increased anteroposterior diameter, and a high incidence of pneumomediastinum and pneumothoraces. Comorbid diseases include those associated with in utero asphyxia that initiated the passage of meconium.
Treatment of meconium aspiration includes general supportive care and assisted ventilation. Infants with a PPHN-like presentation should be treated for PPHN. If severe hypoxia does not subside with conventional or high-frequency ventilation, surfactant therapy, and inhaled nitric oxide, extracorporeal membrane oxygenation (ECMO) may be beneficial.
Prevention of meconium aspiration syndrome involves careful in utero monitoring to prevent asphyxia. When meconium-stained fluid is observed, the obstetrician should suction the infant’s oropharynx before delivering the rest of the infant’s body. If the infant is depressed with poor tone, minimal respiratory effort, and cyanosis, the infant’s oropharynx should be suctioned, the vocal cords visualized, and the area below the vocal cords suctioned to remove any meconium from the trachea. This procedure can be repeated two to three times as long as meconium is present, before either stimulating the infant to breathe or initiating assisted ventilation. Saline intrauterine amnioinfusion during labor may reduce the incidence of aspiration and pneumonia.
PRIMARY PULMONARY HYPERTENSION OF THE NEWBORN
PPHN occurs in post-term, term, or near-term infants. PPHN is characterized by severe hypoxemia, without evidence of parenchymal lung or structural heart disease that also may cause right-to-left shunting. PPHN is often seen with asphyxia or meconium-stained fluid. The chest radiograph usually reveals normal lung fields rather than the expected infiltrates and hyperinflation that may accompany meconium aspiration. Additional problems that may lead to PPHN are congenital pneumonia, hyperviscosity-polycythemia, congenital diaphragmatic hernia, pulmonary hypoplasia, congenital cyanotic heart disease, hypoglycemia, and hypothermia. Total anomalous venous return associated with obstruction of blood flow may produce a clinical picture that involves severe hypoxia and that is indistinguishable from PPHN; however, a chest radiograph reveals severe pulmonary venous engorgement and a small heart. Echocardiography or cardiac catheterization confirms the diagnosis.
Significant right-to-left shunting through a patent foramen ovale, through a PDA, and through intrapulmonary channels is characteristic of PPHN. The pulmonary vasculature often shows hypertrophied arterial wall smooth muscle, suggesting that the process of or predisposition to PPHN began in utero as a result of previous periods of fetal hypoxia. After birth, hypoxia, hypercapnia, and acidosis exacerbate pulmonary artery vasoconstriction, leading to further hypoxia and acidosis. Some infants with PPHN have extrapulmonary manifestations as a result of asphyxia. Myocardial injuries include heart failure, transient mitral insufficiency, and papillary muscle or myocardial infarction. Thrombocytopenia, right atrial thrombi, and pulmonary embolism also may be noted.
The diagnosis is confirmed by echocardiographic examination, which shows elevated pulmonary artery pressures and sites of right-to-left shunting. Echocardiography also rules out structural congenital heart disease and transient myocardial dysfunction.
Treatment involves general supportive care; correction of hypotension, anemia, and acidosis; and management of complications associated with asphyxia. If myocardial dysfunction is present, dopamine or dobutamine is needed. The most important therapy for PPHN is assisted ventilation. Reversible mild pulmonary hypertension may respond to conventional assisted ventilation. Patients with severe PPHN do not always respond to conventional therapy. Paralysis with pancuronium may be needed to assist such vigorous ventilation. Surfactant replacement seems to have no effect when PPHN is the primary diagnosis. If mechanical ventilation and supportive care are unsuccessful in improving oxygenation, inhaled nitric oxide, a selective pulmonary artery vasodilating agent, should be administered. If hypoxia persists, the patient may be a candidate for ECMO. Infants who require extremely high ventilator settings, marked by an alveolar-to-arterial oxygen gradient greater than 620 mm Hg, have a high mortality rate and benefit from ECMO if they do not respond to nitric oxide. In addition, the oxygenation index (OI) is used to assess the severity of hypoxemia and to guide the timing of interventions such as inhaled nitric oxide and ECMO. The OI is calculated using the equation: OI = [(mean airway pressure × fraction of inspired oxygen)/PaO2] × 100. A high OI indicates severe hypoxemic respiratory failure.
APNEA OF PREMATURITY
Although apnea typically is associated with immaturity of the respiratory control system, it also may be the presenting sign of other diseases or pathophysiologic states that affect preterm infants (see Table 61-3). A thorough consideration of possible causes is always warranted, especially with the onset or unexpected increase in the frequency of episodes of apnea (or bradycardia).
Apnea is defined as the cessation of pulmonary airflow for a specific time interval, usually longer than 10 to 20 seconds. Bradycardia often accompanies prolonged apnea. Central apnea refers to a complete cessation of airflow and respiratory efforts with no chest wall movement. Obstructive apnea refers to the absence of noticeable airflow but with the continuation of chest wall movements. Mixed apnea, a combination of these two events, is the most frequent type. It may begin as a brief episode of obstruction followed by a central apnea. Alternatively, central apnea may produce upper airway closure (passive pharyngeal hypotonia), resulting in mixed apnea.
A careful evaluation to determine the cause of apnea should be performed immediately in any infant with apnea. The incidence of apnea increases as gestational age decreases. Idiopathic apnea, a disease of premature infants, appears in the absence of any other identifiable disease states during the first week of life and usually resolves by 36 to 40 weeks of postconceptional age (gestational age at birth + postnatal age). The premature infant’s process of regulating respiration is especially vulnerable to apnea. Preterm infants respond paradoxically to hypoxia by developing apnea rather than by increasing respirations as do mature infants. Poor tone of the laryngeal muscles also may lead to collapse of the upper airway, causing obstruction. Isolated obstructive apnea also may occur as a result of flexion or extreme lateral positioning of the premature infant’s head, which obstructs the soft trachea.
Treatment of apnea of prematurity involves administration of oxygen to hypoxic infants, transfusion of anemic infants, and physical cutaneous stimulation for infants with mild apnea. Methylxanthines (caffeine or theophylline) are the mainstay of pharmacologic treatment of apnea. Xanthine therapy increases minute ventilation, improves the carbon dioxide sensitivity, decreases hypoxic depression of breathing, enhances diaphragmatic activity, and decreases periodic breathing. Treatment usually is initiated with a loading dose followed by maintenance therapy. High-flow nasal cannula therapy and nasal continuous positive airway pressure of 4 to 6 cm H2O also are effective and relatively safe methods of treating obstructive or mixed apneas; they may work by stimulating the infant and splinting the upper airway. Continuous positive airway pressure also probably increases functional residual capacity, improving oxygenation.
MISCELLANEOUS RESPIRATORY DISORDERS
Pulmonary hypoplasia becomes manifested in the delivery room as severe respiratory distress that progresses rapidly to pulmonary insufficiency. The lungs, which are very small, develop pneumothoraces with normal resuscitative efforts. A small chest size radiographically, joint contractures, and early onset of pneumothoraces (often bilateral) are early clues to the diagnosis. Pulmonary hypertension results from the decreased lung mass and hypertrophy of pulmonary arteriole muscle.
Pulmonary hypoplasia may be a result of asphyxiating thoracic dystrophies (a small chest wall) or of decreased amniotic fluid volume that causes uterine compression of the developing chest wall, inhibiting lung growth. The latter failure of lung development may be associated with renal agenesis (Potter syndrome) or with a chronic leak of amniotic fluid from ruptured membranes. Isolated agenesis of one lung (usually the left lung) often is asymptomatic and familial. In contrast, serious pulmonary hypoplasia is noted in patients with a diaphragmatic hernia. Herniated bowel interferes with lung growth and leads to hypertrophy of the muscle layers of small pulmonary arteries, producing pulmonary hypertension. Pneumothoraces are common.
Treatment of congenital diaphragmatic hernia involves surgical evacuation of the chest and repair of the diaphragmatic defect after the pulmonary hypertension is treated. Respiratory care is similar to that for patients with PPHN.
Neuromuscular diseases that interfere with fetal breathing movements also may produce pulmonary hypoplasia (e.g., congenital anterior horn cell disease [Werdnig-Hoffmann syndrome]). Another cause of bilateral pulmonary hypoplasia is hydrops fetalis (see Chapter 60). Hydrops from any cause is characterized by anasarca, ascites, and pleural and pericardial effusions. Bilateral pleural effusions act as space-occupying masses that interfere with lung growth, resulting in pulmonary hypoplasia.