 CHAPTER 177 Disorders of Sexual Differentiation
CHAPTER 177 Disorders of Sexual Differentiation
NORMAL SEXUAL DEVELOPMENT
The successive sequence of chromosomal sex, gonadal sex, and phenotypic sex leads to gender identity of the individual. Genes usually determine the morphology of internal organs and of gonads (gonadal sex); this directs the appearance of the external genitalia which form the secondary sex characteristics (phenotypic sex); self-perception of the individual (gender identity) and the perception of the individual by others (gender role) follow last. In most children, these features blend and conform, but in some patients, one or more features may not follow this sequence, leading to an intersex condition (see Chapter 23).
Ambiguous genitalia in a newborn must be attended to with as little delay as possible and with informed sensitivity to the psychosocial context as required. The laboratory evaluations required might take days or weeks to complete, delaying a sex assignment and naming of the infant, such that choice often precedes diagnosis. Beyond infancy and childhood, and to offset any gender uncertainty in the patient and confusion in the parents, health care providers must help families come to an appropriate closure and gender choice.
Diagnosis and treatment of disorders of sex differentiation are best understood in terms of the embryology and hormonal control of normal sex differentiation. The internal and external genitalia are formed between 9 and 13 weeks of gestation. Fetal gonad and external genitalia are bipotential and have the capacity to support development of a normal male or female phenotype (Fig. 177-1). In the presence of a gene called SRY for sex-determining region on the Y gene on the Y chromosome, the primitive fetal gonad differentiates into a testis (Fig. 177-2). The testis secretes testosterone, which has direct effects (stimulating development of the wolffian ducts), but also is locally converted to dihydrotestosterone (DHT) by the 5α-reductase enzyme. DHT causes enlargement, rugation, and fusion of the labioscrotal folds into a scrotum; fusion of the ventral surface of the penis to enclose a penile urethra; and enlargement of the phallus with ultimate development of male external genitalia. Testicular production and secretion of müllerian-inhibitory substance cause the regression and disappearance of the müllerian ducts and their derivatives, such as the fallopian tubes and uterus. In the presence of testosterone, the wolffian ducts develop into the vas deferens, seminiferous tubules, and prostate.
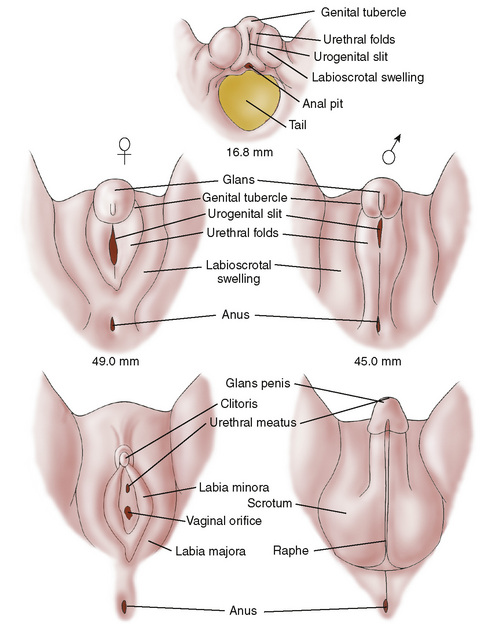
FIGURE 177-1 Differentiation of male and female external genitalia as proceeding from a common embryonic anlage. Testosterone acts at 9 to 13 weeks of gestation to virilize the bipotential anlage. In the absence of testosterone action, the female phenotype develops.
(From Grumbach MM, Conte FA: Disorders of sexual differentiation. In Wilson JD, Foster DW [eds]: Textbook of Endocrinology, 8th ed. Philadelphia, WB Saunders, 1990, p 873. Adapted from Spaulding MH: Contrib Embryol Instit 13:69–88, 1921.)
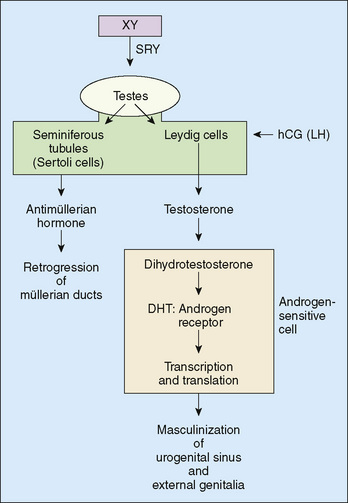
FIGURE 177-2 A diagrammatic scheme of male sex determination and differentiation. DHT, dihydrotestosterone; SRY, the gene for the testis-determining factor. SRY is the master gene controlling male sex differentiation, but there are many other genes and their products that control male and female sexual differentiation.
(From Wilson JD, Foster DW [eds]: Williams Textbook of Endocrinology, 8th ed. Philadelphia, WB Saunders, 1992, p 918.)
The female phenotype develops unless specific male influences alter development. In the absence of SRY, an ovary spontaneously develops from the bipotential, primitive gonad. In the absence of fetal testicular secretion of müllerian-inhibitory substance, a normal uterus, fallopian tubes, and posterior third of the vagina develop out of the müllerian ducts, and the wolffian ducts degenerate. In the total absence of androgens, the external genitalia appear female.
A child with ambiguous genitalia may have a male karyotype or a female karyotype. In the female pseudohermaphrodite, the genotype is 46,XX, and the gonads are ovaries, but the external genitalia are virilized. In the male pseudohermaphrodite, the genotype is 46,XY, and the external genitalia are undervirilized. Reasons can include abnormal development of the testes; defects of sex steroid biosynthesis, including testosterone or DHT; or androgen receptor defects. On physical examination, it is essential to note where the urethral opening lies and whether there is fusion of the anterior portion of the labioscrotal folds. Endogenous excessive production of androgen (as in congenital adrenal hyperplasia [CAH]) in a female fetus between 9 and 13 weeks of gestation leads to ambiguous genitalia. If the vaginal opening is normal, and there is no fusion, but the clitoris is enlarged without ventral fusion of the ventral urethra, the patient had later exposure to androgens. A patient with a fully formed scrotum, even if small, and a normally formed but small penis, termed a microphallus, must have had normal exposure to and action of androgen during 9 to 13 weeks of gestation.
ABNORMAL SEXUAL DEVELOPMENT
Virilization of the 46,XX Female (Female Pseudohermaphroditism)
Masculinization of the external genitalia of genotypic females (except for isolated enlargement of the clitoris, which can occur from later androgen exposure) is always caused by the presence of excessive androgens during the critical period of development (8–13 weeks of gestation) (Table 177-1). The magnitude of the changes reflects the quantity and duration of exposure to androgens. The degree of virilization can range from mild clitoral enlargement to the appearance of a male phallus with a penile urethra and fused scrotum with raphe. Congenital virilizing adrenal hyperplasia is the most common cause of ambiguous genitalia; it is most commonly the result of an enzyme deficiency that impairs synthesis of glucocorticoids, but does not affect androgen production. The impaired cortisol secretion leads to adrenocorticotropic hormone (ACTH) hypersecretion, which stimulates hyperplasia of the adrenal cortex and excessive adrenal production of androgens (see Chapter 178).
TABLE 177-1 Causes of Virilization in the Female
| Condition | Additional Feature(s) |
|---|---|
| P-450c21 deficiency | Salt loss in some |
| 3β-Hydroxysteroid dehydrogenase deficiency | Salt loss |
| P-450c11 deficiency | Salt retention/hypertension |
| Androgenic drug exposure (e.g., progestins) | Exposure between 9 and 12 weeks of gestation |
| Mixed gonadal dysgenesis or mosaic Turner syndrome | Karyotype = 46,XY/45,X |
| True hermaphrodite | Testicular and ovarian tissue present |
| Maternal virilizing adrenal or ovarian tumor | Rare, positive history |
Inadequate Masculinization of the 46,XY Male (Male Pseudohermaphroditism)
Underdevelopment of the male external genitalia occurs because of a relative deficiency of testosterone production or action (Table 177-2). The penis is small, with various degrees of hypospadias (penile or perineal) and associated chordee or ventral binding of the phallus; unilateral, but more often bilateral, cryptorchidism may be present. The testes should be sought carefully in the inguinal canal or labioscrotal folds by palpation or ultrasound. Rarely a palpable gonad in the inguinal canal or labioscrotal fold represents a herniated ovary, or an ovotestis in true hermaphroditism. The latter patients have ovarian and testicular tissue and usually ambiguous external genitalia. Production of testosterone by a gonad implies that testicular tissue is present and that at least some cells carry the SRY gene.
TABLE 177-2 Causes of Inadequate Masculinization in the Male
| Condition | Additional Feature(s) |
|---|---|
| P-450scc (StAR) deficiency | Salt loss |
| 3β-Hydroxysteroid dehydrogenase deficiency | Salt loss |
| P-450c17 deficiency | Salt retention/hypokalemia/hypertension |
| Isolated P-450c17 deficiency with 17,20-desmolase deficiency | Adrenal function normal |
| 17β-Hydroxysteroid oxidoreductase deficiency | Adrenal function normal |
| Dysgenetic testes | Possible abnormal karyotype |
| Leydig cell hypoplasia | Rare |
| Complete androgen insensitivity or testicular feminization | Female external genitalia, absence of müllerian structures |
| Partial androgen insensitivity | As above with ambiguous external genitalia |
| 5α-Reductase deficiency | Autosomal recessive, virilization at puberty |
StAR, steroid acute regulatory protein.
Testosterone production can be reduced by specific deficiencies of the enzymes needed for androgen biosynthesis or by dysplasia of the gonads. In the latter, if müllerian-inhibiting substance production also is reduced, a rudimentary uterus and fallopian tubes are present. Enzyme defects in testosterone biosynthesis, which also block cortisol production, produce adrenal hyperplasia. Congenital gonadotropin deficiency may produce a normally formed but small penis (microphallus without hypospadias). Hypopituitarism with luteinizing hormone (LH) deficiency does not result in ambiguous genitalia because placental human chorionic gonadotropin (hCG) in the fetal circulation stimulates fetal gonadal testosterone synthesis during the critical 9 to 13 weeks of gestation to allow development of a normal male phallus. Later in gestation, fetal LH is needed to stimulate the testes to produce adequate androgen to enlarge the fetal penis. Hypopituitarism is often combined with deficiencies of growth hormone (GH) and ACTH in neonatal hypopituitarism, causing neonatal hypoglycemia. Microphallus due to this cause responds to testosterone treatment, resulting in an enlargement of the penis.
The complete form of androgen resistance or androgen insensitivity syndrome is the most dramatic example of resistance to hormone action by defects in the androgen receptor. Affected patients have a 46,XY karyotype, normally formed testes (usually located in the inguinal canal or labia majora), and feminine-appearing external genitalia with a short vagina and no internal müllerian structures. At the time of puberty, testosterone concentrations increase to normal or above normal male range. Because a portion of the testosterone is normally converted to estradiol in peripheral tissues and the estrogen cannot be opposed by the androgen, breast development ensues at the normal age of puberty without growth of pubic, facial, or axillary hair or the occurrence of menstruation. Gender identity and gender role are unequivocally female.
5α-Reductase deficiency presents at birth with predominantly female phenotype or with ambiguous genitalia, including perineoscrotal hypospadias. The defect is in 5α reduction of testosterone to its metabolite DHT. At puberty, spontaneous secondary male sexual development occurs, and the individual, if raised as a girl until this age, in most cases converts to a male gender identity and male gender role.
APPROACH TO THE INFANT WITH GENITAL AMBIGUITY
The major goal is a rapid identification of any life-threatening disorders (salt loss and shock caused by the salt-losing form of CAH). The decision of female sex assignment can be rendered more complex by the realization that prenatal androgen exposure (in an individual without complete androgen resistance) causes a tendency toward a male gender identity and male gender role. Although the classic approach to sex assignment has been based on the feasibility of genital reconstruction and potential fertility rather than on karyotype or gonadal histology, the effects of prenatal androgen must be considered. And yet, it may be inappropriate to raise a female infant who is severely virilized from virilizing CAH as a male; in most reported cases, sex assignment and adult gender role remain female and fertility can be retained because the internal organs are female. A 46,XY male with ambiguous genitalia and an extremely small phallus that does not increase in size with androgen therapy (partial androgen resistance) traditionally has been raised as a female because surgical construction of a fully functional phallus is difficult. Some of these patients frequently revert spontaneously to a male gender identity. Present management of ambiguous genitalia involves extensive open discussion with parents involving the biology of the infant and the likely prognosis. Treatment should be individualized and managed by a team including an experienced pediatric endocrinologist, urologist or gynecologist, and the primary care physician.
Diagnosis
The first step toward diagnosis is to determine if the disorder represents virilization of a genetic female (androgen excess) or underdevelopment of a genetic male (androgen deficiency) (see Fig. 177-2). Inguinal gonads that are evident on palpation usually are testes and indicate that incomplete development of a male phenotype has occurred; this pattern is not consistent, and ovaries and ovotestes may feel similar. Similarly, absence of female internal genitalia (detected by ultrasound) implies that müllerian-inhibiting substance was present and secreted by fetal testes. Karyotype determination is only one of many factors in deciding the sexual identity for purposes of rearing; the SRY gene may be found on chromosomes other than the Y chromosome, and, conversely, a Y chromosome may lack an SRY gene (it may have been translocated to an X chromosome, leading to the development of a 46,XX male).
Statistically, most virilized females have CAH; 90% of these females have 21-hydroxylase deficiency. The diagnosis is established by measuring the plasma concentration of 17-hydroxyprogesterone and androstenedione (see Chapter 178), which typically is hundreds of times above the normal range. Other enzymatic defects also may be diagnosed by quantifying circulating levels of the adrenal steroid precursor proximal to the defective enzyme block.
Establishing an accurate diagnosis is more difficult in underdeveloped males. When certain types of adrenal hyperplasia coexist with defects in androgen production of the testes, excessive ACTH secretion elevates levels of specific adrenal steroid precursors substantially, allowing diagnosis. If the defect is restricted to testosterone biosynthesis, the measurement of testosterone and its precursors in the basal state and after stimulation by hCG may be required. Patients with normal levels of testosterone either have persistent androgen resistance or have had an interruption of normal morphogenesis of the genitalia. Abnormalities of the sex chromosomes may be associated with dysgenetic gonads, which may be associated with persistence of müllerian structures.
Treatment
Treatment consists of replacing deficient hormones (cortisol in adrenal hyperplasia or testosterone in a child with androgen biosynthetic defects who will be raised as male), surgical restoration to make the individual look more appropriate for the gender of rearing, and psychological support of the whole family. Gonads and internal organs discordant for the gender of rearing are removed. Dysgenetic gonads with Y-genetic material always should be removed because gonadoblastomas or dysgerminomas may develop in the organ. Reconstructive surgery is usually by 2 years of age so that genital structure reflects gender of rearing. This recommendation for reconstructive surgery is controversial; some advocate that surgery not be performed in infancy or early childhood so that the child or young adolescent can be involved in the decision. A decision for gender of rearing is recommended from birth, however, and the knowledge that the intersexed person may change gender later on is shared with parents from the outset.