 CHAPTER 178 Adrenal Gland Dysfunction
CHAPTER 178 Adrenal Gland Dysfunction
The adrenal gland consists of an outer cortex, responsible for the synthesis of steroids, and an inner medulla derived from neuroectodermal tissue, which synthesizes catecholamines. The adrenal cortex consists of three zones: an outer glomerulosa whose end product is the mineralocorticoid, aldosterone, which regulates sodium and potassium balance; a middle zone, the fasciculata, whose end product is cortisol; and an inner reticularis, which synthesizes sex steroids. The general scheme of these synthetic steps is shown in Figure 178-1.
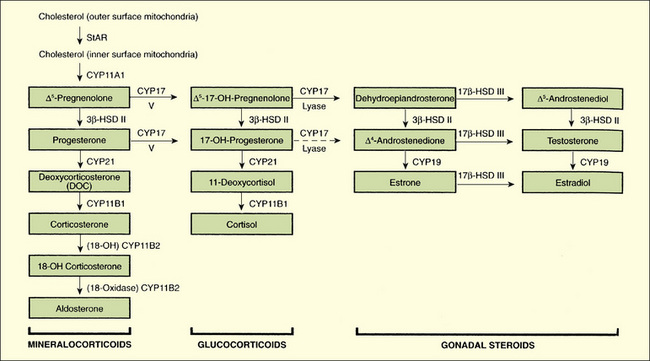
FIGURE 178-1 Diagram of the steroid biosynthetic pathways and the biosynthetic defects that result in congenital hyperplasia. The defect in patients with lipoid adrenal hyperplasia is not (except for one reported case) in the CYP11A1 (cholesterol side-chain cleavage) enzyme, but in StAR, the steroidogenic acute regulatory protein. This protein is involved in the transport of cholesterol from the outer mitochondrial membrane to the inner membrane, where the CYP11A1 enzyme is located. CYP11B1 (11β-hydroxylase) catalyzes 11β-hydroxylation of deoxcorticosterone and 11β-deoxycortisol primarily. CYP17 (17α-hydroxylase/17,20-lyase) catalyzes 17α-hydroxylation and splitting of the 17,20 bond, but for the latter it has preferential Δ5-17,20-lyase activity. CYP19 (aromatase) catalyzes the conversion of corticosterone to aldosterone. 3β-HSD I and 3β-HSD II, 3β-hydroxysteroid dehydrogenase/Δ4,5–isomerase types I and II; CYP21 (P450c21), 21-hydroxylase; 17β-HSD 3, 17β-hydroxysteroid dehydrogenase type 3. In the human, deletion of a homozygous null mutation of CYP11A (P450ccc) is probably lethal in utero, but a heterogeneous mutation caused congenital lipoid adrenal hyperplasia.
(From Melmed S, Polonsky K, Kronenberg H, Larsen R [eds]: Williams Textbook of Endocrinology, 10th ed. Philadelphia, Elsevier, 2003, p 917.)
Hypothalamic corticotropin-releasing hormone (CRH) stimulates the release of pituitary adrenocorticotropic hormone (ACTH, corticotropin), derived by selective processing from pro-opiomelanocortin. ACTH governs the synthesis and release of cortisol and adrenal androgens. Primary adrenal insufficiency or cortisol deficiency from any defect in the adrenal gland results in an oversecretion of ACTH; cortisol deficiency also may occur from ACTH (secondary) or CRH (tertiary) deficiency, causing low serum ACTH concentrations and low cortisol. Endogenous (or exogenous) glucocorticoids feed back to inhibit ACTH and CRH secretion. The renin-angiotensin system and potassium regulate aldosterone secretion; ACTH has little effect on aldosterone production except in excess, when it may increase aldosterone secretion.
Steroids that circulate in the free form (not bound to cortisol-binding protein [transcortin]) may cross the placenta from mother to fetus, but ACTH does not. The placenta plays an important role in steroid biosynthesis in utero, acting as a metabolic mediator between mother and child. Because the fetal CRH-ACTH-adrenal axis is operational in utero, deficiencies in cortisol synthesis lead to excessive ACTH secretion. If a virilizing adrenal enzyme defect is present, such as 21-hydroxylase deficiency, the fetal adrenal gland secretes excess androgens, virilizing the fetus.
Normal variation of serum cortisol and ACTH levels leads to values that are high early in the morning and lower at night. This normal diurnal variation may not be established until the child is 1 to 4 years of age.
ADRENAL INSUFFICIENCY
The clinical manifestations of inadequate adrenal function result from inadequate secretion or action of glucocorticoids, mineralocorticoids, or both (Table 178-1). In addition, in the case of enzyme defects that affect the gonad and the adrenal gland, overproduction or underproduction of potent androgens can occur, depending on the site of enzyme blockade (see Fig. 178-1). Progressive prenatal virilization of the external genitalia may occur in females; incomplete virilization may occur in males. Ambiguity of the external genitalia is a common manifestation of disordered fetal adrenal enzyme function. Precise diagnosis is essential for the prescription of appropriate therapy, long-term outlook, and genetic counseling. In patients with enzyme defects, an elevation in the precursor steroid is present proximal to the enzyme block and is metabolized through remaining normal alternate enzyme pathways, while a deficiency of steroids is present subsequent to the block.
The dominant clinical features of congenital adrenal mineralocorticoid deficiency are hyponatremia and hyperkalemia, usually developing by 5 to 7 days after birth but not usually immediately after birth. Vomiting, dehydration, and acidosis soon follow, as do hypotensive shock from glucocorticoid deficiency. Death may occur if the disorder remains undiagnosed and untreated. In females, the ambiguity of the external genitalia is an obvious clue that salt-losing congenital adrenal hyperplasia (CAH), or simple virilizing CAH, must be ruled out. Because these forms cannot be distinguished clinically, all presentations of ambiguous genitalia should involve evaluation for mineralocorticoid deficiency. In males, the most common form of CAH, 21-hydroxylase deficiency, does not cause abnormal genitalia. There may be hyperpigmentation of the scrotal skin, but this is a subtle sign. In all infants, the diagnosis of adrenal insufficiency may be overlooked or confused with pyloric stenosis. In pyloric stenosis, in contrast to salt-losing CAH, vomiting of stomach contents results in hypochloremia, serum potassium is normal or low, and alkalosis is present. This distinction may be lifesaving in preventing unnecessary investigations or inappropriate therapy.
Not all forms of adrenal hyperplasia present at birth; the spectrum of disorder ranges from severe, or classic, to mild, late-onset, or nonclassic. Milder forms may manifest in childhood, adolescence, or even young adulthood, not as glucocorticoid or mineralocorticoid deficiencies, but as androgen excess. In patients with congenital adrenal hypoplasia or adrenal hemorrhage, the secretion of all adrenal steroids is low. In contrast, CAH leads to a diagnostic steroid pattern in blood and urine (see Fig. 178-1). Deficiency of 21-hydroxylase is the most common form (95%) and serves as a paradigm for these disorders.
21-HYDROXYLASE DEFICIENCY
The incidence of classic 21-hydroxylase deficiency is about 1 in 12,000 among various white populations. A higher incidence occurs in Yupik Eskimos, Yugoslavians, and Ashkenazi Jews. Nonclassic CAH may occur with an incidence of 1 in 50 in certain populations. The gene for 21-hydroxylase lies on the short arm of chromosome 6; the genotype may be determined in a proband, permitting prenatal diagnosis in a subsequent pregnancy.
Deficient 21-hydroxylase activity (P-450c21 deficiency) impairs the conversion of 17-hydroxyprogesterone to 11-deoxycortisol and, in the salt-losing form, of progesterone to deoxycorticosterone, a mineralocorticoid proximal in the pathway to the production of aldosterone. The decreased production of cortisol causes hypersecretion of ACTH, which stimulates the synthesis of steroids immediately proximal to the block and shunting of these to overproduction of androgens. The primary clinical manifestation is the virilization of the external genitalia of the affected female fetus, in whom the development of the uterus, ovaries, and fallopian tubes remains unaffected by the androgens. The degree of virilization varies, ranging from mild clitoromegaly to complete fusion of labioscrotal folds, with severe clitoromegaly simulating a phallus (see Chapter 177). A male infant with this defect appears normal at birth, although penile enlargement may be apparent thereafter. The deficiency in aldosterone, found in about 75% of patients, causes salt wasting with shock and dehydration until the diagnosis is established and appropriate treatment is given.
The treatment of 21-hydroxylase deficiency requires hydrocortisone, and fludrocortisone in the case of the salt-losing form. Therapy must be adjusted throughout childhood at regular intervals. Overtreatment will cause growth stunting and weight gain (cushingoid features), whereas undertreatment will cause excessive height gain, skeletal advance, and early appearance puberty, ultimately jeopardizing adult height potential. Late-onset CAH is noted years after birth. Affected subjects have milder manifestations without ambiguous genitalia, but they do have acne, hirsutism, and, in girls, irregular menstrual cycles or amenorrhea. Late-onset CAH in girls may be confused with polycystic ovary disease.
Biochemical diagnostic studies show elevated levels of serum 17-OHP, the substrate for the defective 21-hydroxylase enzyme activity. In newborns with CAH, the values are elevated a hundredfold to a thousandfold. In late-onset CAH, an ACTH stimulation test is necessary to show an abnormally high response of 17-OHP. Serum cortisol and aldosterone levels (in salt losers) are low, whereas the testosterone level is elevated because it is derived from 17-OHP.
The goals of treatment are to achieve normal linear growth and bone age advancement. Long-term therapy consists of providing glucocorticoids at a dose of approximately 10 to 15 mg/m2/24 hours in three divided doses of oral hydrocortisone or its equivalent. Mineralocorticoid therapy for salt losers consists of fludrocortisone (Florinef) at a dose of 0.1 to 0.2 mg/24 hours often with sodium chloride supplementation in infancy and early childhood. Surgical correction of ambiguous external genitalia begins by 1 to 2 years of age. The adequacy of glucocorticoid replacement therapy is monitored by determining serum concentrations of adrenal precursors, including androstenedione and 17-OHP for 21-hydroxylase deficiency. In addition, the assessment of linear growth and skeletal age, by bone age determination, is required as a reflection of appropriate therapy. To avoid adrenal insufficiency, threefold higher doses of glucocorticoids are given during stressful states, such as febrile illnesses and surgery, and subcutaneous glucocorticoid (Solu-Cortef) is used in severe emergencies. Mineralocorticoid therapy is monitored with serum sodium and potassium and plasma renin activity levels. Prenatal treatment with dexamethasone to suppress fetal ACTH-induced androgen production can reduce or eliminate the ambiguity of the external genitalia in affected female fetuses, if begun at approximately 7 weeks of gestation; this remains experimental.
OTHER ENZYME DEFECTS
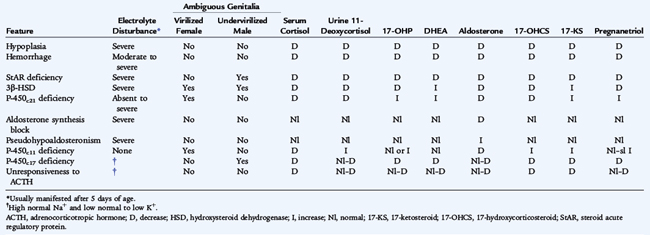
Other enzyme defects are rare in contrast to 21-hydroxylase deficiency. In 11-hydroxylase deficiency, the next most common cause of CAH, virilization occurs with salt retention and hypokalemia, as a result of the buildup of deoxycorticosterone (see Fig. 178-1), a potent mineralocorticoid. Hypertension develops as a result of excessive mineralocorticoid production. Table 178-2 summarizes the clinical and biochemical features of adrenal insufficiency in infancy.
ADDISON DISEASE
Addison disease is a rare acquired disorder of childhood, usually associated with autoimmune destruction of the adrenal cortex. It is a form of primary adrenal insufficiency with absence of glucocorticoid and mineralocorticoid.
Clinical manifestations are hyperpigmentation, salt craving, postural hypotension, fasting hypoglycemia, anorexia, weakness, and episodes of shock during severe illness. Baseline and ACTH-stimulated cortisol values are subnormal, confirming the diagnosis; hyponatremia, hyperkalemia, and elevated plasma renin activity indicate mineralocorticoid deficiency. Addison disease may occur within the context of autoimmune polyglandular syndromes APS I and APS II. APS I includes hypoparathyroidism, mucocutaneous candidiasis, occasionally type 1 diabetes, and, often, hypothyroidism. Associated autoimmune disorders include oophoritis, pernicious anemia and malabsorption, chronic hepatitis, vitiligo, and alopecia. In contrast, APS II includes type 1 diabetes, and autoimmune thyroid disease. It is a genetically distinct entity. Other rare causes of adrenal insufficiency include congenital adrenal hypoplasia, some of the rarer causes of congenital adrenal hyperplasia, and conditions that affect the hypothalamus-pituitary, whether acquired, such as in craniopharyngioma, or iatrogenic, such as in irradiation for treatment of malignancy.
Replacement treatment with 10 to 15 mg/m2/24 hours of hydrocortisone is indicated, with supplementation during stress at three times the maintenance dosage or the use of subcutaneous glucocorticoid. The dose is titrated to allow a normal growth rate. Mineralocorticoid replacement with fludrocortisone is monitored by plasma renin activity and serum sodium and potassium determinations.
CUSHING SYNDROME
Classic clinical manifestations of Cushing syndrome in children include progressive central or generalized obesity, marked failure of longitudinal growth, hirsutism, weakness, a nuchal fat pad (buffalo hump), acne, striae, hypertension, and, often, hyperpigmentation when ACTH is elevated. The most frequent cause is exogenous administration in the context of numerous conditions requiring long-term pharmacologic doses of glucocorticoids. Endogenous causes include adrenal adenoma, carcinoma, nodular adrenal hyperplasia, an ACTH-secreting pituitary microadenoma resulting in bilateral adrenal hyperplasia (Cushing disease), or an extremely rare ACTH-secreting tumor. A high-dose dexamethasone suppression test (20 μg/kg orally every 6 hours for 48 hours) suppresses glucocorticoid secretion in Cushing disease, but not in autonomous adrenal production of cortisol or in an ectopic ACTH-secreting tumor. Parenteral glucocorticoid therapy is necessary during and immediately after surgical treatment to avoid acute adrenal insufficiency.
Treatment of Cushing syndrome is directed to the etiology and may include excision of autonomous adrenal, pituitary, or ectopic ACTH-secreting tumors. Rarely, adrenalectomy or adrenal ablative agents (mitotane) are needed to control the symptoms.
Arlt W., Allolio B. Adrenal insufficiency. Lancet. 2003;361:1881-1892.
Carel J.-C., Léger J. Precocious puberty. N Engl J Med. 2008;358:2366-2377.
Chi C., Chong Lee H., Neely E.K. Ambiguous genitalia in the newborn. Neo Rev. 2008;9:e78-e84.
Cowell K.M. Focus on diagnosis: Type 2 diabetes mellitus. Pediatr Rev. 2008;29:289-292.
Cutler G.B.Jr. Treatment of hypopituitary children. J Pediatr. 2004;144:415-416.
Devendra D., Liu E., Eisenbarth G.S. Type 1 diabetes: Recent developments. BMJ. 2004;328:750-754.
Jospe N: Hyperthyroidism. In McInerny T, et al, editor: AAP Textbook of Pediatric Care
Kliegman R.M., Behrman R.E., Jenson H.B., et al. Nelson Textbook of Pediatrics, 18th ed. Philadelphia: WB Saunders, 2007.
Loscalzo M.L. Turner syndrome. Pediatr Rev. 2008;29:219-227.
Misra M., Pacaud D., Petryk A., et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008;122:398-417.
Roberts C.G.P., Landenson P.W. Hypothyroidism. Lancet. 2004;363:793-802.
Rose S.R., Vogiatzi M.G., Copeland K.C. A general pediatric approach to evaluating a short child. Pediatr Rev. 2005;26:410-420.