 CHAPTER 181 Seizures (Paroxysmal Disorders)
CHAPTER 181 Seizures (Paroxysmal Disorders)
Paroxysmal disorders of the nervous system produce sudden, reversible changes in mental status or somatosensory function that tend to be stereotyped and repetitive in nature. They have a variable duration from seconds to minutes (rarely hours), end abruptly, and are followed by a gradual return to baseline. There may be a warning before (aura) or an altered state of awareness afterward (postictal state). The differential diagnosis of paroxysmal disorders includes epileptiform disorders, specifically seizures, and nonepileptiform disorders such as migraine, syncope, transient ischemic attack, paroxysmal dyskinesia, vertigo, hypoglycemia, breath-holding spells, tics, and conversion reactions (Table 181-1). A thorough medical history from the patient and primary witnesses is the most reliable tool for establishing the correct diagnosis.
The electroencephalogram (EEG) is the most useful neurodiagnostic test in distinguishing seizures from nonepileptic paroxysmal disorders. The EEG must be interpreted in the context of the clinical history because many normal children have epileptiform EEG patterns. Children with seizures may have normal EEGs between attacks. When the diagnosis is still unclear, more sophisticated EEGs with prolonged recordings with simultaneous video or ambulatory EEG monitoring of the patient in an attempt to capture a spell, may be necessary.
ETIOLOGY AND EPIDEMIOLOGY
Seizures represent the abnormal and excessive discharging of the neuroglial network. A diverse group of disturbances of cerebral function or homeostasis can lead to seizures (Table 181-2). Epilepsy is defined as recurrent, unprovoked seizures. Epileptic seizures are generally classified into types on the basis of the electrical mechanism that arises from one region of the cortex (focal, partial, or localization-related) and seizures that arise from both hemispheres, simultaneously (generalized). The epileptic syndromes represent clinical entities wherein age, pattern of clinical event, EEG abnormality, natural history, and prognosis are distinctive (Table 181-3).
TABLE 181-3 Classification of Seizures and Epileptic Syndromes
PARTIAL SEIZURES
GENERALIZED SEIZURES
EPILEPTIC SYNDROMES
Partial or focal seizures constitute 40% to 60% of the classifiable epilepsies of childhood. Focal brain lesions (tumors, infarct, dysgenesis) may cause partial seizures, but most partial seizures in children are due to genetic influences. Generalized tonic, clonic, and biphasic tonic-clonic seizures are common during childhood. Although it is often difficult to distinguish primary generalized tonic and clonic seizures from secondary generalized partial seizures on purely clinical grounds, this distinction is important. While the majority of children with primary generalized or partial onset epilepsy have a genetic basis for their seizures, brain lesions must be considered, especially when focal clinical or electrographic features are present. The presence of an aura indicates a focal origin of the attack. Young children often are unaware of an aura or focal onset of their seizure and caregivers frequently witness only the generalized aspects of the event.
Approximately 6% to 20% of epileptic children have typical generalized absence seizures (petit mal epilepsy). There is a 75% concordance rate in monozygotic twins, suggesting a genetic etiology. Approximately 40% to 50% of children with absence seizures have associated generalized convulsive seizures; 60% occur before and 40% occur after the onset of absence seizures.
Myoclonic, tonic, atonic, and atypical absence seizures compose 10% to 15% of childhood epilepsies. These seizures frequently are associated with underlying structural brain disease and are difficult to treat and classify. They often occur in combination with each other and with generalized tonic-clonic seizures. The peak age of occurrence of myoclonic absence seizures is within the first year. Status epilepticus can be the first ictal manifestation in patients with myoclonic features.
CLINICAL MANIFESTATIONS
Partial Seizures
Simple partial seizures arise from a specific anatomic focus. Clinical symptoms include motor, sensory, psychic, or autonomic abnormalities, but consciousness is preserved. Location and direction of spread of the seizure focus determine the clinical symptoms. Complex partial seizures are similar, but in addition, consciousness is impaired. When partial seizures spread to involve the whole brain and produce a generalized tonic-clonic seizure, they show secondary generalization (jacksonian seizures). Partial seizures that manifest only with psychic or autonomic symptoms can be difficult to recognize. Uncinate seizures arising from the medial temporal lobe manifest with an olfactory hallucination of an extremely unpleasant odor (burning rubber). Gelastic seizures originating from hypothalamic tumors are spells of uncontrolled laughter. Lip-smacking seizures arise from the anterior temporal lobe and episodes of macropsia, micropsia, altered depth perception, and vertigo from the posterior temporal lobe. Limbic temporal lobe discharges result in dreamlike states (déjà vu and bizarre psychic abnormalities). Episodic autonomic phenomena, such as fever, tachycardia, shivering, and increased gastrointestinal motility, may rarely be seizures of temporal lobe origin.
Generalized Seizures
Generalized Tonic, Clonic, and Tonic-Clonic Seizures (Generalized Major Motor Seizures)
Tonic, clonic, and biphasic tonic-clonic seizures may occur alone or be associated with other seizure types. Typically the attack begins abruptly, but occasionally is preceded by a series of myoclonic jerks. During a tonic-clonic seizure, consciousness and control of posture are lost, followed by tonic stiffening and upward deviation of the eyes. Pooling of secretions, pupillary dilation, diaphoresis, hypertension, and piloerection are common. Clonic jerks follow the tonic phase, then the child is briefly tonic again. Thereafter, the child remains flaccid and urinary incontinence may occur. As the child awakens, irritability and headache are common. During an attack, the EEG shows repetitive synchronous bursts of spike activity followed by periodic paroxysmal discharges. Generalized tonic-clonic activity lasting longer than 20 minutes, or repeated seizures without restoration of consciousness for more than 30 minutes, is defined as status epilepticus and may lead to irreversible brain injury.
Absence Seizures
Staring spells can be either primary generalized absence (petit mal) or complex partial seizures (temporal lobe epilepsy). The clinical hallmark of generalized absence seizures is a brief loss of environmental awareness accompanied by eye fluttering or simple automatisms, such as head bobbing and lip smacking. Seizures usually begin between 4 and 6 years of age. Neurologic examination and brain imaging are normal. The characteristic EEG patterns consist of synchronous 3-Hz spike-and-wave activity. A clinical seizure can be provoked by hyperventilation or strobe light stimulation. Differentiating absence epilepsy from partial complex staring seizures can be difficult. Both seizure types are characterized by cessation of activity, staring, and alteration of consciousness and may include automatisms. The automatisms of partial complex seizures are usually more complicated and may involve repetitive swallowing, picking of the hands, or walking in nonpurposeful circles. Partial complex seizures are often followed by postictal confusion; absence seizures are not. Absence seizures are provoked by hyperventilation and usually last a few seconds; partial complex seizures occur spontaneously and usually last several minutes. Children may have dozens of absence seizures per day; children rarely have more than one or two partial complex seizures in a day. The distinction is important because the choice of anticonvulsant treatment is different.
Myoclonic, Tonic, Atonic, and Atypical Absence Seizures
Atypical absence seizures manifest as episodes of impaired consciousness with automatisms, autonomic phenomena, and motor manifestations, such as eye opening, eye deviation, and body stiffening. The EEG shows slow spike-and-wave activity at 2 to 3 Hz. Myoclonus is a sudden jerk of all or part of the body; not all myoclonus is epileptic in nature. Nonepileptic myoclonus may originate in the basal ganglia, brainstem, or spinal cord. It may be benign, as in sleep myoclonus, or indicate serious disease. Myoclonic epilepsy usually is associated with multiple seizure types. The underlying illness producing myoclonic epilepsy may be developmental and static or progressive and associated with neurologic deterioration (neuronal ceroid lipofuscinosis). Myoclonic absence refers to the body jerks that commonly accompany absence seizures and atypical absence seizures.
EPILEPTIC SYNDROMES
Benign focal epilepsy, also known as rolandic epilepsy, usually begins between ages 5 and 10 years. The incidence is 21 per 100,000, accounting for 16% of all afebrile seizures in children under 15 years of age. They are usually focal motor seizures involving the face and arm and tend to occur only during sleep or on awakening in more than half of patients. Symptoms commonly include abnormal movement or sensation around the face and mouth with drooling and a rhythmic guttural sound. Speech and swallowing are impaired. A family history of similar seizures is found in 13% of patients. The disorder is called benign because seizures usually respond promptly to anticonvulsant therapy; intellectual outcome and brain imaging are normal, and epilepsy resolves after puberty. Daily antiepileptic drug therapy may not be needed.
Benign neonatal convulsions are an autosomal dominant genetic disorder localized to chromosome 20. Generalized clonic seizures occur toward the end of the first week of life (3-day fits or familial 5-day fits). Response to treatment varies, but the outlook generally is favorable.
Juvenile myoclonic epilepsy (of Janz) occurs in adolescence and is an autosomal dominant disorder localized on chromosome 6 with variable penetrance. The patient may have absence, generalized tonic or clonic, and myoclonic seizures. The hallmark is morning myoclonus occurring predominantly within 90 minutes of awakening. Seizures usually resolve promptly with therapy with valproic acid, but therapy must be maintained for life.
Infantile spasms (West syndrome) are brief contractions of the neck, trunk, and arm muscles, followed by a phase of sustained muscle contraction lasting 2 to 10 seconds. The initial phase consists of flexion and extension in various combinations such that the head may be thrown either backward or forward. The arms and legs may be either flexed or extended. Spasms occur most frequently when the child is awakening from or going to sleep. Each jerk is followed by a brief period of relaxation, repeated multiple times in clusters of unpredictable and variable duration. Many clusters occur each day. The EEG during the waking state, hypsarrhythmia, is dramatically abnormal, consisting of high-voltage slow waves, spikes, and polyspikes accompanied by background disorganization. The peak age at onset is 3 to 8 months; 86% of infants experience the onset of seizures before age 1 year. When flexion of the thighs and crying are prominent, the syndrome may be mistaken for colic. Infantile spasms have a poor prognosis. The etiology is not determined in 40% of children. This idiopathic or “cryptogenic” group has a better response to therapy than the group with a clear etiology; 40% have a good intellectual outcome. Etiology is determined in 60% (Table 181-4). This symptomatic group responds poorly to anticonvulsant therapy and has a poor intellectual prognosis. Tuberous sclerosis is the most common recognized cause. Treatment options for infantile spasms include adrenocorticotropic hormone (ACTH), oral corticosteroids, benzodiazepines, valproic acid, and vigabatrin.
Lennox-Gastaut syndrome is an epileptic syndrome with variable age of onset. Most children present before age 5 years. Multiple seizure types, including atonic-astatic, partial, atypical absence, and generalized tonic, clonic, or tonic-clonic varieties characterize the disorder. Many children have underlying brain injury or malformations. These seizures usually respond poorly to treatment, but some patients have a good response to valproic acid.
Astatic-akinetic or atonic seizures have their onset between 1 and 3 years of age. The seizures last 1 to 4 seconds and are characterized by a loss of body tone, with falling to the ground, dropping of the head, or pitching forward or backward. A tonic component usually is associated. The seizures frequently result in repetitive head injury if the child is not protected with a hockey or football helmet. They are most frequent on awakening and on falling asleep; 50 or more daily seizures are usual. Children with astatic-akinetic seizures usually have developmental delay and underlying brain abnormalities. Tuberous sclerosis is a frequent cause.
Acquired epileptic aphasia (Landau-Kleffner syndrome) is characterized by the abrupt loss of previously acquired language in young children. The language disability is an acquired cortical auditory deficit (auditory agnosia). Some patients develop partial and generalized epilepsy. The EEG is highly epileptiform in sleep, the peak area of abnormality often being in the dominant perisylvian region (language areas). It is unclear whether frequent temporal lobe seizures cause the language disability or whether unknown, perhaps inflammatory temporal lobe disease is responsible for the seizures and language loss.
Rasmussen encephalitis is a chronic, progressive focal inflammation of the brain and is of unknown origin. An autoimmune origin and focal viral encephalitis have been postulated. The usual age at onset is 6 to 10 years. The disease begins with focal, persistent motor seizure activity, including epilepsia partialis continua. Over months, the child develops hemiplegia and cognitive deterioration. EEG shows focal spikes and slow wave activity. Brain imaging studies are initially normal, then show atrophy in the involved area. Hemispherectomy has been the only successful therapy as measured by seizure eradication and prevention of cognitive deterioration, but permanent hemiparesis is an inevitable consequence.
SPECIAL CONDITIONS
Seizures in the setting of fever may be caused by infections of the nervous system (meningitis, encephalitis, or brain abscess), unrecognized epilepsy triggered by fever, or simple febrile convulsions. The latter represents a common genetic predisposition to seizures in children between 6 months and 6 years that is precipitated by fever. They occur in 2% to 4% of children; most occur between ages 1 and 2 years (mean age 22 months). Simple febrile convulsions are generalized major motor seizures lasting less than 15 minutes that occur only once in a 24-hour period in a neurologically and developmentally normal child. If there are focal features, the seizures last longer than 15 minutes, the child has preexisting neurologic challenges, or the seizures occur multiple times within one febrile event, the seizure is referred to as a complex or atypical febrile seizure.
The prognosis of children with simple febrile convulsions is excellent. Intellectual achievements are normal. Many children have further febrile seizures, but the risk of subsequent epilepsy is no greater than that for the general population (approximately 1%). Febrile seizures recur in 30% to 50% of children who have their first febrile seizure before 1 year of age and in 28% of children who have their first seizure after 1 year of age. About 10% of children with febrile seizures have three or more recurrences. Children with complex febrile seizures have only a 7% risk of having further complicated febrile seizures. Factors that increase the risk for the development of epilepsy include abnormal neurologic examination or development, family history of epilepsy, and complex febrile seizures. The probability of developing epilepsy is 2% if one risk factor is present and 10% if two or three risk factors are present.
Because febrile seizures are brief and the outcome is benign, most children require no treatment. Rectal diazepam can be administered during a seizure to abort a prolonged event. Daily administration of phenobarbital or valproic acid prevents febrile seizures, but the potential for serious side effects limits the use of these agents. Intermittent treatment with oral diazepam three to four times per day during a febrile illness has shown variable effectiveness.
Status epilepticus is a neurologic emergency and is defined as ongoing seizure activity for greater than 20 minutes or repetitive seizures without return of consciousness for greater than 30 minutes. Status epilepticus can cause hypoxemia and decreased cortical perfusion resulting in irreversible brain injury. In 50% of children presenting with status epilepticus, there is no definable etiology, but in 50% of this group, status is triggered by fever. About 25% have an acute brain injury, such as purulent or aseptic meningitis, encephalitis, electrolyte disorder, or acute anoxia. Twenty percent have a history of brain injury or congenital malformation. Sudden cessation of anticonvulsant medication is another frequent cause. Overall the mortality rate of status epilepticus is less than 10% and is related to the etiology of the seizure pattern.
IMMEDIATE TREATMENT
The first priority of treatment is to ensure an adequate airway and to assess the cardiovascular status (see Chapter 38). Oxygen is administered. If there is any doubt concerning the adequacy of the airway, the child should be intubated. An intravenous (IV) infusion should be started, and laboratory evaluation should be undertaken. Hypoglycemia and electrolyte abnormalities must be addressed. Several pharmacologic options exist for management of status epilepticus (Table 181-5). Initial management is usually with a benzodiazepine. Lorazepam (0.05–0.1 mg/kg), diazepam (0.1–0.3 mg/kg), and midazolam (0.2 mg/kg) all are effective agents. Diazepam distributes rapidly to the brain, but has a short duration of action. Alternatively, or even simultaneously, administration of either phenytoin (10–15 mg/kg) or fosphenytoin (10–20 mg/kg) at a rate of 1 mg/kg/min is effective. Phenytoin and fosphenytoin distribute more slowly but have a much longer duration of action. If the seizures persist with these measures, a continuous IV infusion of diazepam, loading dose of 10 to 20 mg/kg of phenobarbital, or IV valproic acid at a dose of 20 mg/kg is appropriate (see Table 181-5). If this approach is ineffective, preparations for general anesthesia are undertaken. While awaiting anesthesia, continuously infused diazepam or pentobarbital is recommended. When status epilepticus stops, maintenance therapy is initiated with the appropriate anticonvulsant.
TABLE 181-5 Management of Status Epilepticus
STABILIZATION
PHARMACOLOGIC MANAGEMENT
ECG, electrocardiogram.
Pseudoseizures may occur in children with hysteria or malingering. Children with genuine seizure disorders may, consciously or subconsciously, exhibit activity that simulates their own seizures. Although the clinical differentiation can be difficult, pseudoseizures differ from epileptic seizures in that movement is tremulousness or thrashing rather than true tonic-clonic activity. Verbalization and pelvic thrusting are seen more commonly in pseudoseizures; urinary and fecal continence is preserved, and injury does not occur. Pseudoseizures are more likely to be initiated or terminated by suggestion. An EEG performed during pseudoseizure activity does not show typical epileptiform patterns.
LABORATORY AND DIAGNOSTIC EVALUATION OF SEIZURES
A complete laboratory evaluation of a child with the new onset of seizures includes a complete blood count; measurement of blood chemistries, including glucose, calcium, sodium, potassium, chloride, bicarbonate, urea nitrogen, creatinine, magnesium, and phosphorus; blood or urine toxicology screening; analysis of cerebrospinal fluid (CSF); and EEG and brain imaging (magnetic resonance imaging [MRI]). Neonates also may require testing for inborn errors of metabolism, blood ammonia, CSF glycine, lactate, and herpes simplex polymerase chain reaction; urine and stool culture (cytomegalovirus and enterovirus), and a clinical trial of pyridoxine. Analysis of CSF is not necessary if the patient is afebrile and has no other neurologic signs or if the history does not suggest a meningeal infection or subarachnoid hemorrhage. Children with simple febrile seizures who have recovered completely may require little or no laboratory evaluation other than studies necessary to evaluate the source of the fever.
MRI is superior to computed tomography (CT) in showing brain pathology, but in the emergency department setting, CT can be performed rapidly and shows acute intracranial hemorrhage more clearly than MRI. MRI is likely to be normal in patients with the primary generalized epilepsies, such as typical absence and juvenile myoclonic epilepsy. Lesions (tumors, arteriovenous malformations, cysts, strokes, gliosis, or focal atrophy) in 25% of other patients may be identified even when the clinical examination and EEG do not suggest focal features. Identification of some static lesions, such as cortical dysplasia, hamartoma, and mesial temporal sclerosis, may allow consideration of surgical correction of medically refractory epilepsy.
LONG-TERM THERAPY
The decision to institute daily seizure medications for a first unprovoked seizure must be based on the likelihood of recurrence balanced against the risk of long-term drug therapy. Determination of the recurrence risk is based on the clinical history and results of neurodiagnostic testing. Absence seizures, infantile spasms, atypical absence seizures, and astatic-akinetic seizures are universally recurrent at the time of diagnosis, indicating the need for therapy. The overall risk of recurrence for children whose first seizure is generalized tonic-clonic is approximately 50%. It seems reasonable to wait for recurrence before therapy is instituted. An otherwise healthy child with a single unprovoked partial seizure with a normal neurodiagnostic evaluation, including a normal EEG, may have a recurrence risk of 25%. In such a case, it is reasonable to educate family members regarding first aid techniques for seizures and to withhold daily antiepileptic agents. Conversely a child with preexisting neurologic abnormality and an abnormal EEG may have a recurrence risk of 75%, in which case institution of a daily antiepileptic agent may be justified after the first seizure.
When treatment is initiated, the goal is to maintain an optimal functional state. Medication toxicity should be weighed against the risk of seizure itself. Initial drug selection is based on the mechanism of the seizure (Fig. 181-1). A single agent limits toxicity, contains cost, and improves compliance. Approximately 50% of children obtain satisfactory seizure control with the initial drug. If seizure control is not achieved with confirmed therapeutic anticonvulsant levels, addition of a second drug must be considered. When available, measuring anticonvulsant blood levels is helpful in adjusting medication and monitoring compliance, but the levels should be interpreted in light of the patient’s clinical state. Antiepileptic drug levels should be drawn at trough, usually early in the morning before the morning doses. When hepatic or renal disease is present, drug binding is likely to be altered. In this instance, free and bound anticonvulsant levels can be helpful. Treatment is not necessary for benign febrile seizures.
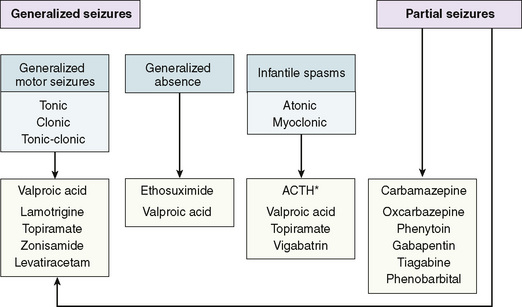
The duration of anticonvulsant treatment varies with seizure type. Children with generalized tonic, clonic, and tonic-clonic seizures; absence seizures; and certain partial seizures may not require therapy for more than 2 to 4 years. The risk of recurrence is higher with partial complex seizures. Children with juvenile myoclonic epilepsy, progressive myoclonic epilepsy, atypical absence seizures, and Lennox-Gastaut syndrome usually require treatment for life. As a rule, children who are neurologically abnormal, have seizures that were initially difficult to control, and have persistently epileptiform EEGs are at highest risk for recurrence when therapy is discontinued.
General guidance for families with children with epilepsy is to be careful with heights, head injury, and swimming. Children with epilepsy have a greater risk of submersion accidents. This risk can be minimized by maintaining therapeutic anticonvulsant drug levels and by appropriate adult supervision.