MEDICAL MANAGEMENT
No clinical sign or diagnostic test is unique to MS, but a typical clinical syndrome with typical MRI of the brain or spinal cord and exclusion of other similar illnesses can result in a correct diagnosis very rapidly. MRI of the brain and spinal cord is critical to the diagnosis of MS. It is, however, important to first exclude other potentially treatable causes of the presenting symptoms before making a diagnosis of MS. Box 31-8 show some examples of differential diseases for which screening should be done. The corpus callosum is usually involved in MS, whereas this is not as common in hypoxic-ischemic diseases. This is because this structure receives a unique double blood supply, and with short arterioles, perfusion deficits may be less likely to result in injury.
The whole spinal cord can be imaged with high resolution and phased-array coils, showing abnormalities in 80% to 90% of individuals with MS, usually without accompanying neurologic symptoms or signs. Hypoxic-ischemic disease does not present with spinal cord abnormalities. Incidental spinal cord lesions do not occur with aging and are rarely reported in other immune-mediated disorders. Most individuals with early MS have lesions within the spinal cord. But imaging may not be performed, so they may not be isolated. Spinal cord lesions tend to increase as the number of brain lesions rises; this is associated with higher risk for a second attack and a diagnosis of clinical MS. Ultimately, abnormal brain MRI scans are present in more than 90% of individuals with clinically definite MS. Normal brain MRI scans may represent disease that is relatively restricted to the spinal cord. Reductions in nerve fiber density are seen in the spinal cord, including in otherwise normal-appearing tissue, and are likely related to permanent disability. Axonal loss can be profound in later stages of disease.
Fig. 31-8 shows the plaques seen on MRI. The lesions do not always correlate with the clinical signs, and there can be evidence of focal lesions in the absence of disease. In fact, the vast majority of enhancing lesions are considered to be asymptomatic when they first appear on the brain scan. However, there is a correlation between periods of clinical worsening of the disease and increases in the total number of lesions, the number of new lesions, and the total area of enhancement on MRI.23 Thus, a single brain MRI scan after a first event is highly prognostic of development of clinically definite MS.137 Fig. 31-9 shows an example of aggressive MS over 2 years revealed in MRI imaging.
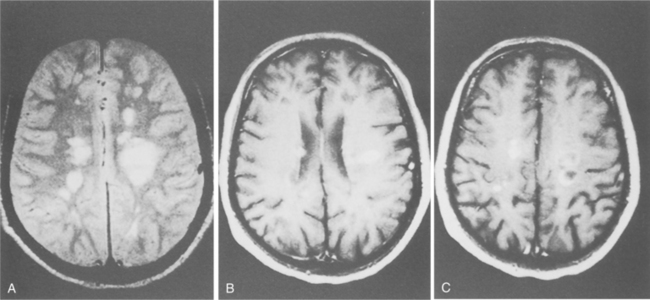
Figure 31-8 A, Typical scattered, variably sized plaques in the brain associated with the diagnosis of multiple sclerosis (MS). B, Contrast-enhanced magnetic resonance imaging reveals scattered area of solid and ring-shaped enhancement. C, Note the atrophy, greater than would be expected for the person’s age, a common finding in MS. (From Ramsey R: Neuroradiology, Philadelphia, 1994, Saunders.)
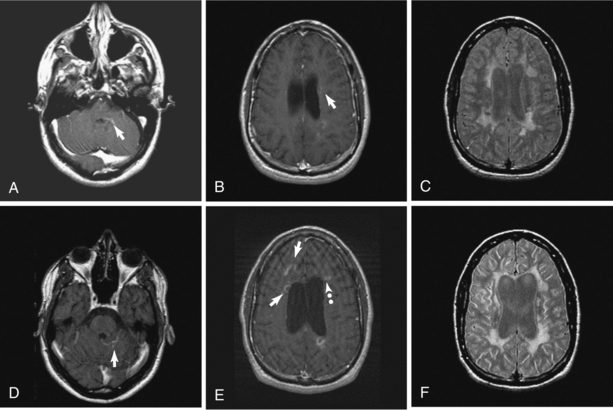
Figure 31-9 Aggressive multiple sclerosis over 2 years. Disease was initially relapsing-remitting but converted relatively quickly to secondary progressive MS. Top row: Contrast-enhanced left pons (A) and left frontal-parietal white matter (B), both showing a relatively rare edge enhancement pattern (arrows). Typical confluent T2 hyperintensities and mild-moderate volume loss based on lateral ventricle size (C). Bottom row: Two years later magnetic resonance image shows different edge-enhancing lesions (arrows) in posterior fossa (D) and both edge enhancement (arrows) and ring enhancement (dotted arrow) in deep white matter along the lateral ventricles (E). Progressive volume loss based on moderately large lateral ventricles and more extensive confluent T2 hyperintensity is seen in (F). (From Radiologic Clinics of North America, Volume 44, Number [1], January 2006. Copyright © 2006 WB Saunders Company.)
Contrast enhancement in CT and MRI suggests inflammation but is more accurately a measure of leakage of moderate-size molecules across the damaged tight junctions of the CNS endothelium. The enhancement pattern (size, shape, solid versus ring) may be variable within and more so between individuals, which reflects a heterogeneous pathology. Ring enhancement, for example, may suggest a more severe pathology. Fig. 31-10 demonstrates the development of a T2-weighted hyperintense lesion by serial MRI. The correlation between the pattern of enhancement, the underlying pathology, and the clinical course in given individuals may not be straightforward. Monitoring serial MRI studies with enhancement helps to identify agents that may be active against the early inflammatory stage of MS.170
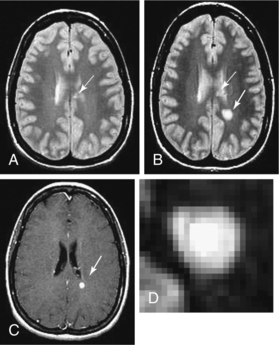
Figure 31-10 Development of a T2 hyperintense lesion by serial magnetic resonance imaging. A, Case of relapsing multiple sclerosis with low T2 hyperintense lesion burden, including chronic lesions in the corpus callosum (arrow). B, One month later, a new T2 hyperintense lesion develops in the left parietal-occipital white matter (solid arrow), whereas the corpus callosum lesions remain stable (dotted arrow). C, Corresponding enhancement in acute lesion (arrow) from blood-brain barrier breakdown and concurrent inflammation. D, Exploded view of the new lesion showing the complex structure, centrally hyperintense, most likely from mixed pathology including demyelination, matrix including glial change, and, importantly, axonal degeneration. The intermediate black ring may be a zone of macrophage infiltration, and the outer ring is likely from edema. (From Radiologic Clinics of North America Volume 44, Number 1, January 2006 Copyright © 2006 WB Saunders Company.)
Individuals with MS have significant atrophy of both white matter and grey matter. Atrophy is considered an important measure in MS because it likely reflects in most cases irreversible injury, much of which is from axon loss, but with additional contributions from myelin loss and structural changes from astrogliosis. The demyelinated cortex contains apoptotic neurons. Cortical demyelination could affect neurons, dendrites, and axons, which may lead to disease progression. The cerebral cortex may be affected by tissue loss and atrophy, particularly in areas adjacent to severe white matter pathology. Neurons in such lesions may show signs of retrograde damage as described earlier. Fig. 31-11 shows changes related to progressive forms of MS.
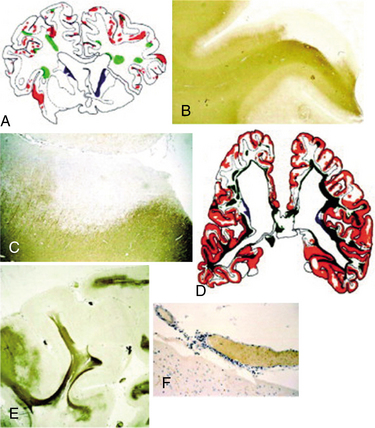
Figure 31-11 Cortical plaques in progressive forms of multiple sclerosis (MS). Cortical demyelination and diffuse white matter inflammation are hallmarks of primary progressive MS (PPMS) and secondary progressive MS (SPMS). A and D, Schematic lesion maps based on whole hemispheric sections from two archival cases of progressive MS. Case A (PPMS): A 37-year-old man with a history of gradually progressive hemiparesis (left greater than right), sphincter dysfunction, and dysarthria, requiring use of a wheelchair within 6 years of disease onset. Patient died at age 72 of aspiration pneumonia and acute myocardial infarction. Case D (SPMS): A 33-year-old woman initially presenting with diplopia and hemiataxia that partially resolved following short course of corticosteroids. Subsequent course characterized by gradually progressive dysarthria, dysphagia, ophthalmoplegia, and limb and gait ataxia, requiring use of a wheelchair within 7 years of disease onset. She also developed a focal seizure disorder 4 years prior to death and died at age 46 of aspiration pneumonia. B and C, Subpial cortical demyelination is demonstrated in case A at low (B) and high (C) magnification. E, Extensive subpial demyelination involving multiple gyri is illustrated in case D at low magnification. F, Meningeal inflammation may be prominent, often in close proximity to areas with subpial cortical demyelination. Proteolipid protein immunocytochemistry; green = focal demyelinated plaques in the white matter; orange = cortical demyelination; blue = demyelinated lesions in the deep grey matter. (From Pirko I: Gray matter involvement in multiple sclerosis, Neurology 68[9]:634-642, 2007.)
Functional MRI maps the brain areas activated using a task paradigm. Functional disturbances have been the basis for hypotheses suggesting that compensatory mechanisms develop in early MS, which initially may mask injury and delay the appearance of dysfunction. Functional disturbance may only become apparent after exhaustion of these adaptive mechanisms. Fatigue severity is correlated with the reduction in thalamic and cerebellar activation. Although abnormal functional MRI patterns may be observed in given individuals with MS, their interpretation may not be straightforward. Functional MRI findings reflect functional adaptation, they do not necessarily serve as direct evidence for grey matter pathology.
Functional MRI appears to identify sensorimotor and cognitive disturbances in MS. The most consistent finding by functional MRI studies in populations of individuals with MS is impairment in sensorimotor activation indicated by abnormally increased contralateral blood oxygenation level and ipsilateral supplementary motor activation. Sensorimotor functional MRI is sensitive even in the early stages of disease.
Measures of regional brain atrophy are useful to determine neuropsychologic dysfunction. Imaging data suggest that neuropsychologic impairment in MS is related, in part, to atrophy of grey matter regions and in the juxtacortical areas. Grey matter atrophy has been associated with impairments in verbal memory, euphoria, and disinhibition.
Whole-brain atrophy reflects the destructive aspects of the disease. The data linking brain atrophy to clinical impairments suggest that irreversible tissue destruction is a major determinant of disease progression, whereas white matter lesion activity has less correlation. The strongest correlations between MRI measures and disability may be those provided by atrophy measures. Confounding factors must be considered when assessing whether loss of brain volume directly indicates tissue atrophy. Secondary progressive disease causes significantly more atrophy of both white matter and grey matter and a significantly higher lesion load than relapsing-remitting disease.174 Primary progressive disorders show decreased numbers and volume of enhancing lesions, related to the less intense inflammation. Spinal cord pathology has been hypothesized to be an important factor in disease progression in primary progressive MS but is not always predictive (see Fig. 31-11).
Cellular imaging based on superparamagnetic iron oxide–tagged cells is a more specific probe of the migration occurring at the level of the blood-brain barrier basic to the inflammatory process. The superparamagnetic iron oxide particles are used to follow macrophages as they pass into the CNS. The particles exert a strong influence on the local magnetic field, which is detected as signal loss on T2-weighted and T2-weighted pulse sequences. The location of lesions and their time course based on superparamagnetic iron oxide imaging does not correlate strongly with that based on conventional contrast-enhanced MRI, suggesting that it provides different quantitative and qualitative information. Studies of fiber degeneration and connections between lesions and fiber pathways are becoming feasible in the clinical imaging environment through diffusion tensor MRI.
Although the mechanisms that contribute to brain atrophy are unclear, degenerative processes that underlie brain atrophy will hopefully provide novel therapeutic targets to help individuals. Several initiatives are under way to define criteria for successful or acceptable treatment versus treatment failure, based on both clinical and MRI activity. MRI monitoring may increase confidence in a clinical impression of stable disease or help discount borderline symptoms or signs, supporting maintenance of the current therapy. MRI activity may support a clinical impression or uncover a need to initiate aggressive and more risky therapy with immunosuppressive agents.
CSF analysis often shows increased mononuclear cell pleocytosis, an elevation in total immunoglobulins, and the presence of oligoclonal bands. These responses suggest an inflammatory response in the CNS. In 85% to 95% of individuals with clinically definite MS, these values are abnormal. In people with suspected MS, the number is much lower. Abnormal values, including oligoclonal bands, also may be seen in a smaller percentage of those with a variety of disorders, especially other inflammatory or infectious disorders that may affect the CNS. Metabolites from the breakdown of myelin may also be detected in CSF but are very nonspecific. Measurement of these myelin basic proteins in CSF is more useful as a predictor of response to steroids used during acute exacerbations than as a diagnostic tool.
Evoked potential response testing may detect slowed or abnormal conduction in visual, auditory, somatosensory, or motor pathways. These tests employ computer averaging techniques to record the electrical response evoked in the nervous system following repetitive sensory or motor stimuli. Evoked potentials are abnormal in up to 90% of individuals who have clinically definite MS. Testing can provide evidence of a second lesion in an individual with a single clinically apparent lesion, or it can demonstrate an objective abnormality in a individual with subjective complaints and a normal examination.
Myelography and CT are both insensitive to the pathologic changes in MS and should not be used where the diagnosis of MS is a possibility. Both may still be used to rule out other disorders that mimic MS symptoms.
TREATMENT.
Keeping an activated immune system from getting to the central myelinated fibers slows the process of demyelination in MS. Therefore, the principle of treatment in MS today revolves around immune modulation. Different steps of the pathologic cascade in MS may eventually be targeted therapeutically for optimal treatment of MS. Understanding the inflammatory process in MS is important, because it is becoming clear that inflammation includes destructive components that should be targeted for inactivation and potentially beneficial mechanisms that should be enhanced or not disturbed by treatment. Despite earnest research efforts, there is no cure for or immunization against MS.
Current drug therapy can diminish approximately one third of the attacks in the actively affected MS population 2 years beyond onset. The “ABC” drugs are used. A is for interferon beta-1a (Avonex, Biogen Idec), and B is for interferon beta-1b (Betaseron, Berlex Laboratories). The higher the dose of interferon, the more potent is the response. Rebif, another interferon beta-1a drug, is used in higher doses. Rebif is given subcutaneously at a 46% higher dose three times weekly, for a total of 4.4 times as much drug as Avonex. The potent antiinflammatory effects of interferons have a dramatic effect on the MRI scans, with a decrease in contrast-enhancing lesions.101,106 Interferon beta-1a also has been shown in relapsing individuals to slow progression of disability, brain atrophy, and cognitive dysfunction.88 The treatment effect can be delayed by at least several months. This delay might indicate the fact that the atrophy occurring in the first months is the culmination of a cascade of events that began before the onset of therapy. Alternatively, the ongoing loss of brain volume might be the result of “pseudoatrophy,” such as that due to treatment-related resolution of brain edema and inflammation. Proposed mechanisms by which interferon might limit brain atrophy include increasing nerve growth factors, limiting immune-mediated destructive inflammation, and limiting toxic mechanisms such as pathologic iron deposition.122 Thus, the question remains as to whether controlling acute relapses and inflammation will ultimately be adequate. Other factors may have an influence on demyelination and axon injury.134
The C drug, glatiramer acetate (Copaxone, Teva Pharmaceutical Industries) is a polypeptide that appears to fool the immune system. It seems to decrease the attack by blocking immune cells headed toward myelin and thereby preventing damage. It also has a partial and delayed but significant effect of limiting the rate of brain atrophy in relapsing-remitting MS. Glatiramer acetate has gained wide acceptance as one option for the treatment of relapsing-remitting MS as there is less occurrence of the flulike symptoms that are associated with the interferons. Although its precise immunologic mechanisms continue to be investigated, current views suggest that its principal effect may be mediated by a shift from a proinflammatory cell bias to an antiinflammatory cell bias. Ongoing studies are exploring the possibility that glatiramer acetate confers neuroprotection and are seeking novel ways to use the agent alone or in combination with other medications.120 Localized panniculitis or inflammation of lipid stores at the sites of subcutaneous injections seems to be a rare but characteristic side effect.
Mitoxantrone (Novantrone, Immunex) is used to modify relapsing and secondary progressive MS. Mitoxantrone is the only drug approved for treatment of secondary progressive MS. It presumably works by depression of T-cell counts and removal of activated T cells from the immune repertoire. Because of potential side effects, its use is being limited at present to individuals whose MS is clearly advancing in spite of aggressive ABC therapy or who already are in a secondary progressive phase. The ABC drugs have not yet been found conclusively to be helpful in secondary progressive disease, and no drugs have been found effective in primary progressive disease.169
Natalizumab (Tysabri) is a monoclonal antibody that prevents immune cells from moving from the blood to the CNS. It was originally approved by the FDA based on a dramatic lowering of relapse rate and a 50% reduction in the development of a sustained increase in disability. This treatment is a once-monthly intravenous infusion. Unfortunately, shortly after approval of the treatment by the FDA, there were two fatal cases of progressive multifocal leukoencephalopathy (PML) when it was used in conjunction with interferon beta-1a. As a result, the distribution of natalizumab was halted, pending further evaluation. Studies continue, and it may emerge again for use in treating MS, perhaps as a monotherapy.187
Fingolimod (FTY720) is a new oral immunomodulating agent under evaluation for the treatment of relapsing MS. Its final effect is also to reduce the normal circulation and trafficking of leukocytes. It also nears the 50% mark for decreasing the mean cumulative number of lesions. A phase III trial is under way to definitively assess clinical efficacy. Encephalopathy is a risk, and fin golimod is also associated with an initial reduction in the heart rate.92
Despite the time and effort given to slow the disease, the bulk of current intervention is devoted to symptomatic management. Improvement in the ability to control symptoms through therapy and medications can enhance quality of life in the individual with MS. Amantadine, pemoline, modafinil, and other medicines can reduce fatigue. Depression and sleep disorders may contribute to fatigue and must be recognized and treated appropriately. Centrally acting and peripherally acting muscle relaxants, such as baclofen (Lioresal), tizanidine, and dantrolene, decrease hypertonicity and leg spasms. Anticonvulsants and antidepressant medicines are used to treat pain. Intrathecal baclofen pumps reduce severe spasticity. Oxybutynin and tolterodine diminish bladder hyperactivity. Focal injections of BTX can be helpful in decreasing muscle spasticity.1 Repetitive transcranial magnetic stimulation may improve spasticity in MS. The antiepileptic agents and antidepressant treatments often are effective in modulating the painful symptoms. Gabapentin in relatively high doses often is necessary for the desired effect. Spasms occurring during the day usually are handled best by the addition of the antiepileptic medications, including gabapentin and topiramate.
Amitriptyline is helpful, especially at night as it can sedate and provide pain relief. Clonazepam, given at bedtime, can aid in sleep initiation and decrease spasms with minimum side effects. Diazepam has a similar effect. Dose escalation should be avoided. Dopamine agonists and dopamine itself also decrease nocturnal spasms reasonably effectively at low dosages.
All of these treatments require adjustment from time to time to maintain some relief. All medications currently available to control symptoms of MS have potential side effects and therefore must be used judiciously. Careful monitoring of systems affected by MS is essential to medical management.157
Both corticosteroids (prednisone, cortisone, methylprednisolone) and adrenocorticotrophic hormone (ACTH) are known to shorten the recovery period after an acute MS attack. There appears to be no consensus about the optimal form, dosage, route, or duration of corticosteroid therapy, but there is now a consensus statement from the American Academy of Neurology regarding treatment of acute optic neuritis with methylprednisolone. Oral prednisone should not be used. Corticosteroids can alter almost every aspect of the immune system. Corticosteroid-induced restoration of the blood-brain barrier, which becomes less effective during active demyelination or plaque formation, has an antiedema benefit and may prevent circulating toxins, viruses, or immunoactive cells from entering the CNS. Decreased activity of the macrophages and lymphocytes results in less damage to the myelin in response to steroid therapy.61 Individuals with severe demyelination who do not respond to corticosteroids may improve with plasma exchange.95,194
Based on the rapid advances in our understanding of the immunopathogenesis of MS, a variety of experimental approaches presently are under study. These include the hormonal agent estriol, matrix metalloproteinase inhibitors, statin drugs, adhesion molecule antibodies, T-cell peptides, combination therapies (especially ABC drugs with other types of agents), intravenous immunoglobulin, and stem cell transplantation. All these are preventive, not restorative of previously damaged CNS function. Growth factors that enhance CNS remyelination are being studied as a method to restore the loss of oligodendrocytes in MS.139 Findings of neural stem cells in the adult CNS and the potential of blood-derived stem cells to become neural cells offer the possibility of transplanting cells into the CNS that will restore function. Studies on intense immunosuppression with bone marrow transplantation (autologous and stem cell) continue, but results are not positive enough to recommend its use.
PROGNOSIS.
The average frequency of attacks of MS is approximately 1 per year. The attacks vary in severity; therefore close observation is required to reliably track the attack frequency. Attacks tend to be most common in the early years of MS and become less frequent in later years, regardless of the disability. The risk for rapid development of moderate disability may be greater in persons in whom the frequency of attacks is higher than average.
Multiple factors may predict a severe course, such as motor and cerebellar symptoms, disability after the first attack, and short time interval between attacks. Numerous relapses within the first year negatively influence the clinical course. Conversely, sensory symptoms, infrequent attacks, full neurologic recovery after a relapse, and a low level of disability after 5 to 7 years may be associated with an improved prognosis.
Burden of disease on MRI scans may be the strongest predictor of clinical outcomes. Over 14 years, there was no significant disability accrued in individuals with normal MRI findings at the time of diagnosis, whereas MRI scans with greater than 10 lesions predicted that individuals would require a cane for walking within that same time frame. A change in lesion load within the first year also is a negative predictor of outcome. Late-onset MS is not necessarily associated with a worse outcome. Progression of primary progressive and relapsing MS differed little between late-onset and early-adult-onset disease. The individuals with late-onset disease were older when reaching an Expanded Disability Status Scale score of 6.183
Because disability is often significant in individuals with MS, lifestyle changes are frequently necessary. Movement impairment is frequently associated with MS, and difficulty in walking is a major disability. If MS is untreated, 15 years after diagnosis 50% of individuals with MS will require the use of an assistive device to walk, and at 20 years 50% will be wheelchair bound. About one fourth of persons with MS will require human assistance with ADLs.94
It is the coexistence of physical and cognitive impairments together with emotional and social issues in a disease with an uncertain course that makes MS rehabilitation unique and challenging. Individual rehabilitation improves functional independence but has only limited success in improving the level of neurologic impairment. Severely disabled people derive as much as or more benefit than those who are less disabled, but cognitive problems and ataxia tend to be refractory. Cost and utility are significantly correlated with functional capacity.97 There is now good evidence that exercise can improve fitness and function for those with mild MS and helps to maintain function for those with moderate to severe disability. Several different forms of exercise have been investigated. For most individuals, aerobic exercise that incorporates a degree of balance training and socialization is most effective. Time constraints, access, impairment level, personal preferences, motivations, and funding sources influence the prescription for exercise and other components of rehabilitation. Just as immunomodulatory drugs must be taken on a continual basis and be adjusted as the disease progresses, so should rehabilitation be viewed as an ongoing process to maintain and restore maximum function and quality of life.27
Life expectancy is reduced by a modest amount in MS; the risk of dying of MS is strongly associated with severe disability. The death rate in persons who are unable to stand or walk is more than four times that in persons the same age without MS. In mildly disabled individuals, the death rate is approximately 1.5 times that of the age-matched population. Persons with more frequent initial episodes with rapidly developing disability have a poorer long-term outcome. Individuals with primary progressive disease also have decreased life expectancy. Suicide is more than seven times more common than in age-matched controls, and depression must be treated aggressively.
PARKINSONISM AND PARKINSON’S DISEASE
Overview and Definition
Atrophy of the brain leading to degeneration of neurons in the basal ganglia can be caused by a variety of disorders that are not well understood. These include striatonigral degeneration, Shy-Drager syndrome, progressive supranuclear palsy, olivopontocerebellar atrophy, corticobasal ganglionic degeneration, and diffuse Lewy body disease. Parkinsonian features can be manifested as a part of other diseases affecting the CNS, such as atherosclerosis, ALS, and HD.192
Parkinson’s disease (PD), or idiopathic parkinsonism, is a chronic progressive disease of the motor component of the CNS, characterized by rigidity, tremor, and bradykinesia and postural instability. The disease is thought to result from a complex interaction between multiple predisposing genes and environmental effects, although these interactions are still poorly understood. PD is still regarded as a sporadic neurodegenerative disorder, characterized by the loss of midbrain dopamine neurons and presence of Lewy body inclusions.
Incidence
Parkinsonism, including PD, affects more than 800,000 adults in the United States, with prevalence rates of 350 per 100,000. Approximately 42% of parkinsonism is related to PD. The lifetime risk of developing parkinsonism is 7.5% according to a Mayo study. There appears to be a higher rate among white Americans and Europeans compared with black Africans. Black persons in America and Chinese in Taiwan have higher rates of disease than their counterparts in West Africa or China.13 PD becomes increasingly common with advancing age, affecting more than 1 person in every 100 over the age of 75 years. A possible explanation of the correlation between age and prevalence may be the age-related neuronal vulnerability. Because of the increase in life expectancy, the aging of the baby boomers, and the precision of diagnosis, the incidence of PD is expected to rise. It is estimated that there will be more than 1.5 million persons living with PD in the United States and close to 40 million worldwide by the year 2020. The majority of cases begin between the ages of 50 and 79 years. Approximately 10% will develop initial symptoms before the age of 40 years.104
Etiologic and Risk Factors
An increasing number of chromosomal features linked to familial parkinsonism have been found, notably PARK1 to PARK11. Among these, seven genes have been identified, four causing autosomal dominant parkinsonism (synuclein, UCHL1, NURR1, LRRK2) and three causing autosomal recessive disease (DJ1, PINK1, parkin). These provide insights into the molecular pathogenesis of the disease, but genetic testing for these mutations is of little clinical relevance. The chance of identifying parkin mutations is less than 5% in sporadic cases with onset at younger than 45 years. The probability is much greater in those with onset at younger than 30 years and in those with an affected sibling. Confirmation of this recessive form of disease might be helpful in genetic counseling, because it renders transmission to the subsequent generation very unlikely.49 Given the late onset of typical PD, it is likely that by the time individuals become symptomatic, many first-degree relatives are deceased from other causes.
Many potential exposures have been cited as possible risk factors for PD. Three major groups include toxic exposures, infection exposures, and a heterogenic group of miscellaneous exposures. Some toxic agents such as carbon monoxide, manganese, cyanide, and methanol can damage the basal ganglia and produce parkinsonian symptoms. A rapidly developing Parkinson-like disease has been linked to the use of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a synthetic narcotic related to heroin. Some neuroleptics can produce a parkinsonian syndrome. In drug-induced parkinsonism, the symptoms can usually be reversed by withdrawal of the drug.
The link to infection exposure remains unresolved, despite years of study. There may still be a possibility that infection plays a role, based on observations of serum antibody titers for measles virus, rubella virus, herpes simplex virus types 1 and 2, and cytomegalovirus in persons with PD. Pesticides and herbicides may be environmental causes and are likely to produce between 2% and 25% increased risk. If some individuals are determined to be particularly susceptible to low environmental exposure, then pesticides may pose a more serious risk. In every age group, men are more likely than women to develop parkinsonism. This finding is not completely understood, but perhaps hormones may protect women, whereas men may be exposed to more environmental toxins according to their occupational choices. Long-term exposure to either manganese or copper has been linked to an increased incidence of parkinsonism.
More years of formal education appear to increase the risk of PD. Physicians are at significantly increased risk of PD when occupational data are used. By contrast, four occupational groups show a significantly decreased risk of PD according to one source: construction and extractive workers (miners, oil well drillers), production workers (machine operators, fabricators), metalworkers, and engineers.65
There is a relatively well established relationship between PD and a history of smoking. Individuals with a history of smoking seem to have a lower risk of developing PD.
High levels of physical exercise may lower PD risk. The risk of PD appears to be lower among women who report strenuous exercise during early adulthood. Physical exercise can promote secretion of growth factors in the CNS that in turn may contribute to the survival and neuroplasticity of dopaminergic neurons. Moreover, exercise decreases the ratio between dopamine transporter and vesicular monoamine transporter; a decrease in this ratio may lower the susceptibility of dopaminergic neurons to neurotoxins and reduce dopamine oxidation. Finally, physical exercise may activate the dopaminergic system and increase dopamine availability in the striatum. Any of these or other mechanisms may be responsible for the beneficial effects of forced exercise in animal experiments; however, the relevance of these short-term animal findings to possible neuroprotective effects of leisure physical exercise in human PD pathogenesis remains to be established. In the rat model of PD, forced exercise prior to chemically induced parkinsonism caused a significant increase of glial-derived neurotrophic factor that has neuroprotective effects for dopaminergic neurons.41
Pathogenesis
Parkinsonian symptoms come primarily from dysfunction within the subcortical grey matter in the basal ganglia. Physiologic studies have shown the basal ganglia to be actively involved in almost all types of movement, including postural responses, alternating movements, and spontaneously occurring movements. The basal ganglia are active prior to recorded EMG activity in the muscles involved in a movement. Lesions do not produce paralysis or weakness but rather change the character of movement, leading to loss of adaptive control, slowing of movement, and poor coordination. The motor loop that determines the initiation and scaling of motor activity derives its input from the premotor, motor, and somatosensory cortices. This is the process of preparation for forthcoming movement, and when disrupted it can cause a reduction in size and speed of the movement.
Basal ganglia–cerebral cortex interactions are disrupted by the abnormal function of the basal ganglia. This reflects a delay in motor programming related to the unconscious initiation of motor preparation, or lack of “response set” or readiness to move. The complex loop that includes the basal ganglia is involved in motivation and in planning global aspects of behavior.101 The basal ganglia interact with the frontal cortex and with the limbic system, including the hippocampus and amygdala, and therefore have a role in cognitive and emotional function. The basal ganglia, in association with the frontal lobe, appear to play an important role in the integration of sensory information. It is now recognized that diffuse neuronal loss in the cerebral cortex may also contribute to changes observed in PD.
The basal ganglia are large subcortical structures that are interconnected and functionally interposed between the cortex and the thalamus. They also have direct connections to the limbic lobe, frontal cortex, and brainstem. It is likely that the fundamental principles that will be described here in relationship to the cortex–basal ganglia–thalamus–cortex system can also explain the disorders related to the connections between the basal ganglia and the frontal cortex, limbic lobe, and brainstem. The failure to facilitate desired behaviors and simultaneously inhibit unwanted behaviors may be responsible for the cognitive, emotional, and memory problems that coexist with movement disorders.
The signs and symptoms of parkinsonism are neurochemical in origin. The pathologic hallmark is the degeneration of a nucleus that is part of the basal ganglia, the substantia nigra. It loses its ability to produce dopamine, a neurotransmitter necessary to normal function of basal ganglia neurons. A depletion of 70% to 80% of the dopamine is estimated to occur before clinical signs of the disease are noted.39,85 Initially, the system adapts, and there is increased efficiency in the pathways that depend on dopamine, but over time, as the dopamine depletion continues, function declines.
Abnormal protein breakdown, which may occur spontaneously or in relationship to a gene mutation, contributes to the neurodegeneration of PD. It appears that there are many possible triggers for the programmed cell death that results in mitochondrial dysfunction and oxidative stress. Despite significant research in this area, many observations are correlative in nature, and a precise process has not been identified. The inherited, early-onset forms of the disease may have a mutation which causes degradation of protein that may mimic the changes found in those individuals with sporadic, later-onset disorder. The changes in neurochemistry and protein are both consistent with aging. There is some overlap of degeneration that is similar to the processes seen in the dementias, including AD.
Free radical or oxidative stress appears to also have a role in the dysfunction of the basal ganglia.16 Compared to the rest of the brain, the substantia nigra is exposed to higher levels of oxidative stress. This is not a direct response and may be the result of dying cells.
Evidence of inflammation is typically found in the area of the substantia nigra pars compacta in conjunction with programmed cell death. It is proposed that neuroinflammation does not initiate PD but can promote progression and add to the worsening of symptoms.
The motor pattern generators, thoughts and behaviors, and processes for memory are all initiated through the cerebral cortex. The parietal-occipital-temporal lobes, prefrontal areas, thalamic nuclei, limbic lobe, amygdala, and hippocampus use glutamate projections into the striatum (caudate and putamen). The input into the striatum comes in a topographic organization that is maintained to some degree throughout the basal ganglia.
Dopamine is produced in the substantia nigra pars compacta. Dopamine has more than one configuration, and the D1 configuration either increases the efficiency or decreases the effect of cortical input to the striatum depending on the context of the desired movement. The D2 family primarily decreases the effect of cortical input to the striatum. The striatum (caudate and putamen) has medium spiny neurons that project outside of the striatum. Dopamine input to the striatum terminates largely on the shafts of these dendritic spines and is able to modulate transmission from the cerebral cortex to the striatum. Cholinergic interneurons and GABA interneurons also synapse on these dendrites. Although there are fewer GABA interneurons, they have a powerful inhibitory effect. Through long-term potentiation and long-term depression, dopamine may be involved in the neural mechanism of habit learning. Depletion of dopamine in the striatum impairs the learning of new movement sequences.
D1 dopamine goes from the striatum primarily to the internal globus pallidus and substantia nigra pars reticulata. (These two groups of neurons are functionally related and often grouped together.) The second population contains GABA and enkephalin and expresses D2 dopamine receptors. These neurons project to the external globus pallidus. The external globus pallidus sends inhibitory input via GABA receptors to the internal globus pallidus.
The primary basal ganglia output comes from the internal globus pallidus, representing the body below the neck, and the substantia nigra pars reticulata, representing the head and eyes. The output is to the thalamus and, via several additional pathways, eventually to the brainstem and spinal cord. The output to the brainstem and spinal cord is through GABA neurotransmitters and is inhibitory.
There is another parallel circuit via the subthalamic nucleus. Excitatory input using glutamate via the hyperdirect pathway comes from the frontal lobe and motor areas and goes directly to the subthalamic nucleus, forming a somatotopic organization. There also appears to be a topographic separation of motor and cognitive inputs to the subthalamic nucleus. The output of the subthalamic nucleus is excitatory, using glutamate, and facilitates the external globus pallidus and the substantia nigra pars reticulata, producing a GABA inhibition to the thalamus.
The subthalamic nucleus also participates in a third or indirect pathway. This pathway involves signaling from the striatum (caudate and putamen) to the external globus pallidus to the subthalamic nucleus to the basal ganglia outputs.
The basal ganglia intrinsic circuitry is complex, with the direct pathway through the striatum (caudate and putamen) and a hyperdirect pathway through the subthalamic nucleus to the basal ganglia outputs. The hyperdirect pathway through the subthalamic nucleus is excitatory and fast, and the direct pathway through the striatum (caudate and putamen) is slower and inhibitory but more powerful.124
The cumulative inhibitory output of the basal ganglia acts as a brake on the motor pattern generators in the cerebral cortex and brainstem. The interaction among these pathways allows for a planned movement to be executed while competing movements are prevented, thereby increasing the precision of the movement without losing the power necessary to perform an activity. This is thought also to allow one part of a movement sequence to be activated in order for another sequence to begin. Through these mechanisms, movement and thought can be adapted quickly when the environment requires a different response.
When dopamine stimulation is decreased or withdrawn from the cycle, the overall effect is increased inhibition of input into the thalamus. The final effect of increased inhibition of the thalamus decreases the ability of the thalamus to send excitatory input back to the frontal cortex. So the ultimate outcome is less activity in the cortex, resulting in slowed movement and less power generated through the musculoskeletal system (Fig. 31-12).
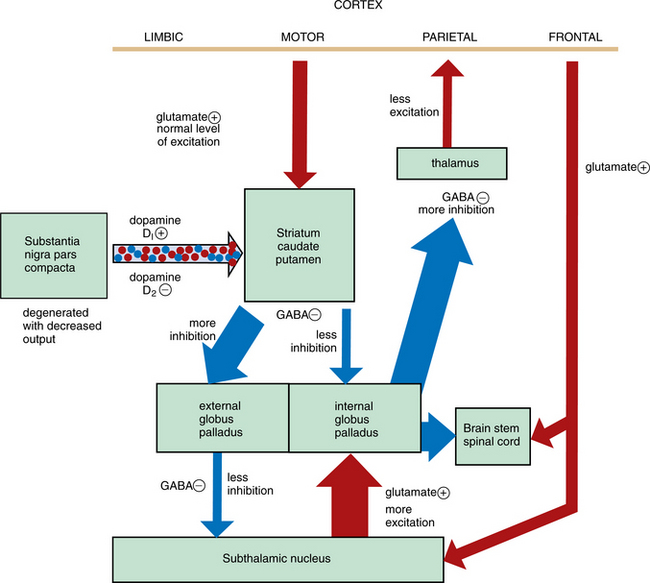
Figure 31-12 How the loss of dopamine production in the substantia nigra causes the eventual decrease in excitation of the cortex in the motor loop that involves the basal ganglia. Glutamate (positive transmission) is red and GABA (negative transmission) is blue. The decreased dopamine causes less than normal inhibition of the striatum (caudate/putamen), so there is more output to the internal globus pallidus. This output is inhibitory, so the cycle of inhibition is already started. Because the internal globus pallidus has been inhibited, there is less output, resulting in less inhibition of the subthalamic nucleus. This results in increased output of the subthalamic nucleus, this time causing more facilitation of the external globus pallidus. The increased output of the external globus pallidus, because it is inhibitory, causes greater inhibition of the thalamus, and the final end result is decreased excitation of the frontal or motor cortex. So the movement coming out of the cortex is less forceful than intended.
Note the other available pathway from the frontal cortex directly to the subthalamic nucleus. The subthalamic nucleus then facilitates the external globus pallidus, and the output of the external globus pallidus is inhibitory to the internal globus pallidus. The output of the internal globus pallidus is inhibitory to the thalamus as well as the brainstem and spinal cord. In this case, because of the double inhibition (sometimes referred to as disinhibition) the final output is excitatory. This pathway is not dependent on dopamine, but because it sends signals to the same nuclei that have already been affected by the decreases in dopamine, the system gets out of balance and the result is loss of normal modulation. Note also the pathway out of the basal ganglia to the brainstem and spinal cord.
These abnormal responses also influence the pathways to the brainstem that travel through the superior colliculus to the nucleus raphe magnus. Abnormalities in facial expression, blinking, and eye and eyelid movements result. Although less clearly understood, abnormal firing or sequencing to the pedunculopontine tegmental nucleus can influence locomotion, sleep cycle regulation, attention, arousal, and startle.
Although the relationship of the substantia nigra and the striatum (caudate and putamen) are foremost in the disorder of parkinsonism, neuropathologic findings can be found in many other dopaminergic and nondopaminergic cell groups. The pathway responsible for postural stability may be affected outside of the basal ganglia.
Clinical Manifestations
Movement disorder is the hallmark of parkinsonism, although other symptoms are evident and may actually precede the impairment of movement. The ability to move is not lost, but there is a problem with movement activation and loss of reflexive or automatic movement. Movement becomes reliant on cortical control. The ability to perform known tasks, such as walking, changing direction, writing, and basic ADLs, is diminished. The considerable variation among individuals in the clinical manifestations and the level of deterioration in movement over time can be explained by the complex mechanism of dysfunction defined in the Pathogenesis section.52
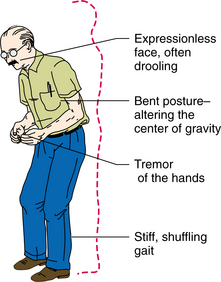
The tremor of PD, the most common initial manifestation, often appears unilaterally and may be confined to one upper limb for months or even years. It is first seen as a rhythmic, back-and-forth motion of the thumb and finger, referred to as the pill-rolling tremor. It is most obvious when the arm is at rest or during stressful periods. The tremor starts unilaterally but can eventually spread to all four limbs as well as neck and facial muscles. Tension or exertion will cause the tremor to increase, and it will disappear during sleep. Tremor does not usually impact the functioning of the individual.
Rigidity is an increased response to muscle stretch that appears in both antagonist and agonist muscle groups. Rigidity, like tremor, usually appears unilaterally and proximally in an upper limb and then spreads to the other extremities and trunk. One of the earliest signs of rigidity is the loss of associated movements of the arms when walking. Rigidity is identified when another person is trying to passively move the extremity and there is a jerky response, known as cogwheel rigidity, or a slow and sustained resistance, known as lead-pipe rigidity. Rigidity does not appear to have a direct effect on volitional movement. Axial rigidity usually limits rotation and extension of the trunk and spine. Reduced variability and less adaptation of movement between thoracic rotation and pelvic motion appears early in the onset of PD. This rigidity can decrease the ability to make adjustments of the extremities during functional tasks, such as transfers, reaching, and bed mobility as well as gait.
Bradykinesia is the slowness of movement seen in parkinsonism. Impairment of the normal mechanisms that scale the output of agonist muscles causes the inability to produce, modulate, and terminate quick movements. Persons diagnosed with PD show relatively small EMG bursts in agonist muscles and move the legs in a series of small steps rather than in a single movement. Bradykinesia results from disruption of the neurotransmitter from the internal globus pallidus to the motor cortical regions known as the supplementary motor area and the primary motor cortex.
The slowing of lip and tongue movements during talking causes a garbled speech pattern. There is loss of fine motor skills with the gradual development of small, cramped writing, or micrographia. Parkinsonism is accompanied often by diminished efficiency of pursuit eye movements, so that small accelerations of eye movement (saccades) are required to catch up with a moving target, which causes smooth pursuit eye movements to be jerky instead of smooth. Eye movement in the vertical plane may be reduced.
Akinesia is a disorder of movement initiation and is seen in parkinsonism as a paucity of natural and automatic movements, such as crossing the legs or folding the arms. Small gestures associated with expression are reduced. The face is masklike, with infrequent blinking and lack of expression. Freezing, or gait akinesia, is the sudden cessation of movement in the middle of an action sequence, as if the foot is stuck to the floor. Sometimes it is the environment that seems to trigger freezing, such as when the individual walks through a doorway or over a change in surface like stepping from a carpet to a hard floor. Freezing most often affects walking, but it can affect speech, arm movements, and blinking. Freezing is uncommon in the early phase but increases over time.58,128
The gait pattern in parkinsonism is highly stereotyped and characterized by impoverished movement. ROM in the joints of the lower extremity is often limited. Trunk and pelvic movement is diminished, resulting in a decreased step length and reciprocal arm swing. The gait is narrow based and shuffling. The speed is decreased. Persons with plantar flexion contractures will toe-walk, and this further narrows the available base of support. In gait there is a loss of heel strike, reduced toe elevation, reduced movement at the knee joints, loss of dynamic vertical force, reversal of ankle flexion-extension movement, and loss of backward-directed shear force.148 Festination is common when attempting to stop or change direction; the stride becomes smaller but more rapid, and instead of stopping, the individual actually increases speed and is usually stopped by running into something or by falling. Preparatory postural responses to move from a bipedal to single-limb stance are frequently absent for induced steps, which may increase instability during first step.152 There is reduced ability to adapt to changes of environments or to perform new tasks.
The posture in PD is characterized by flexion of the neck, trunk, hips, and knees (Fig. 31-13). Kyphosis, or extensive flexion of the spine, is the most common postural deformity. Scoliosis, an abnormal lateral curvature of the spine, can result from the unequal distribution of rigidity in posture. Persistent posturing of a forward head and trunk tends to pull the center of gravity forward. This may result in a tendency to keep weight shifted posteriorly in order to compensate. Postural instability is also associated with abnormal patterns of postural responses, including excessive antagonist activity that results in coactivation of distal and proximal muscles. Adapting to changing support conditions is less efficient in individuals with PD.127 The ability to sequence motor activity appears to have an impact on postural correction. Abnormal control of center of mass results in decreased limits of stability.79 Lateral postural stability is compromised by lack of trunk flexibility. During posterior perturbations, lack of stability appears to be the result of lack of appropriate knee flexion. Box 31-10 outlines some of the contributions to imbalance seen in individuals with parkinsonism.
Performing dual tasks causes more slowing in individuals who have parkinsonism. Smooth performance of sequential motor tasks is broken down into distinct components. Changing the “set” for an activity is more difficult for the individual with parkinsonism when the context or environment requires a sudden change in activity.
Olfactory function is diminished along with impaired color vision and visual perception.185 Spatial organization is often disturbed, resulting in difficulty with orientation to the environment. The inability to distinguish self-movement from movement in the environment can contribute to abnormal balance reactions.24 There is an increased dependence on visual information for motor control. There is strong visual dependency for balance, resulting in the inability to choose a balance strategy based on vestibular information even when the visual surround is unavailable for visual stability.19
Most people with PD experience weakness and fatigue once the disease becomes generalized. The person has difficulty sustaining activity and experiences increasing weakness and lethargy as the day progresses. Repetitive motor acts may start out strong but decrease in strength as the activity progresses. This compounds bradykinesia and increases immobility.
Nonmotor symptoms, such as those related to autonomic dysfunction, are common and potentially disabling manifestations of the disease. Loss of neurons in the sympathetic ganglia may cause autonomic dysfunction. This results in excessive sweating, excessive salivation, incontinence, and disabling orthostatic hypotension.51 There is a greasy appearance to the skin of the face and occasional drooling because of loss of the swallowing movements that normally dispose of saliva. Olfactory dysfunction is an early sign of PD in most individuals, and overlaps with multiple system atrophy and progressive supranuclear palsy.
Rapid eye movement sleep behavior disorders result in lack of the normal muscle atonia and jerking of body and limbs causing disrupted sleep. Restless leg syndrome appears to be associated, mostly because of the similarities in treatment response. Abnormal sleep patterns may also contribute to the daily fatigue.
Fatigue is related to other nonmotor features such as depression and excessive daytime sleepiness. In more than half of the individuals mental fatigue is persistent and seems to be an independent symptom that develops parallel to the progressive neurodegenerative disorder of PD.4
Many persons with PD experience pain that is poorly localized but is generally described as cramping in the axial muscles or the limbs. Paresthesias are reported by many persons, including tingling, numbness, and abnormal temperature sensation.
Dementia and intellectual changes occur in almost 50% of persons with PD. Development of dementia is associated with more rapid progression of disability and potential for need for assisted living. Bradyphrenia, a slowing of thought processes, with lack of concentration and attention may also occur. Coexisting AD, organic brain disease, and vascular compromise may also contribute to the dementia.143
Depression is common and is probably related to the dopamine depletion. Loss of serotonin in the brainstem and limbic lobes has been found using PET studies. Behavioral changes, such as apathy, lack of ambition, indecisiveness, and anhedonia, are common and may be related to depression. Depressive episodes or panic attacks can precede onset of motor symptoms.
Although reduced motor activity by itself would not seem to be a functional disorder, many of the small automatic muscular adjustments are important for successfully carrying out functional activities. For example, in attempting to rise from a chair a person may fail to make the small initial adjustments of legs that are crucial to standing up and fail to be able to get from sitting to standing without assist.
The person with PD typically becomes deconditioned. Rapid heart rate and difficulty breathing are common. Vital capacity is reduced as the kyphosis increases and the intercostal muscles develop rigidity.145 Respiratory complications are the leading cause of death. The Hoehn and Yahr classification (Table 31-3) is a common scale used to define the level of disability associated with PD.
Table 31-3
Hoehn and Yahr Classification of Disability
| Stage | Character of Disability |
| I | Minimal or absent; unilateral if present |
| II | Minimal bilateral or midline involvement; balance not impaired |
| III | Impaired righting reflexes; unsteadiness when turning or rising from chair; some activities restricted, but patient can live independently and continue some forms of employment |
| IV | All symptoms present and severe; standing and walking possible only with assistance |
| V | Confined to bed or wheelchair |
Modified from Hoehn MM, Yahr MD: Parkinsonism: onset, progression and mortality, Neurology 17:427, 1967.
MEDICAL MANAGEMENT
The classic triad of major signs of PD is made up of tremor, rigidity, and akinesia.179 The combination of asymmetry of symptoms and signs, the presence of a resting tremor, and a good response to levodopa best differentiates PD from parkinsonism due to other causes. Diagnostic problems may occur in mild cases. Other movement disorders that do not fall under the category of parkinsonism need to be recognized by clinicians to establish a differential diagnosis. See Box 31-11 for features of parkinsonism due to causes other than PD.
Depression, with its associated expressionless face, poorly modulated voice, and reduction in voluntary activity, may be difficult to distinguish from mild parkinsonism. Olfaction is frequently impaired in PD, suggesting that deficiencies in smell may be a potentially useful test to distinguish PD from related disorders.
CT or MRI is not helpful in diagnosis of PD but can identify other causes of symptoms, such as Wilson’s disease, or mass effects causing disruption of the basal ganglia function, such as stroke or hydrocephalus.
Functional imaging through PET is highly sensitive to regional changes in brain metabolism and receptor binding associated with movement disorders. SPECT shows differences in the posterior putamen, contralateral to the predominantly affected limb. Asymmetric scan findings have been observed in individuals with mild, newly recognized symptoms. Unilateral disease produces a significant difference in striatal uptake between the ipsilateral and contralateral sides in both the caudate and putamen nuclei. One explanation is that there is a preceding unequal functional reactivity of the basal ganglia, which results in an asymmetrical clinical response.
Altropane (a close cousin of cocaine), a component of radioactive technetium 99m, is a compound that can measure the concentration of dopamine transporters imaged by SPECT. This may lead to diagnosis of PD based on identifying decreasing levels in the brains of persons when only mild symptoms appear.
Assessing progression of PD using clinical rating scales such as the United Parkinson’s Disease Rating Scale (UPDRS) is a common way to track progression. However, the progression may be masked by medication, and because of the multitude of symptoms that may change at different rates, it is hard to determine a change in the course of the disease.
TREATMENT.
The current therapeutic approach to PD is symptomatic; major studies to determine possible thera- peutic neuroprotection are under way, but no single intervention has proven to be disease modifying. Drug therapy is adapted to the person’s needs, which may vary with the stage of the disease and the predominant manifestations.5 When mobility becomes affected to the degree that walking and self-care activities become difficult, medications improve the control of movement. As the disease progresses over time, the effectiveness of medication changes, leaving the individual with a shorter “on” time during which symptoms are reduced and more rigidity during “off” times when symptoms are active. Long-term use of medication can also increase the dyskinesia or chorea-like movement resulting from the change in activity in the basal ganglia. Side effects can become more problematic as the dosages needed to control symptoms are increased. The management of these medications becomes the focus of intervention.59
Levodopa (l-dopa), which is taken up by remaining dopaminergic neurons in the basal ganglia and converted to dopamine, improves most of the major features of parkinsonism, including bradykinesia. Initially it leads to nearly complete reversal of symptoms, with effects lasting up to 2 weeks, known as long-duration levodopa response. As the disease progresses, the length of the effect becomes shorter, it takes longer for the effect to be noticed after dosing, and symptoms increase during the end of the dose period. Eventually there is dose failure or lack of any effect at all. Levodopa can cause dyskinesias that produce chorea, athetosis, dystonia, tics, and myoclonus. Predictable fluctuations include a wearing-off effect and early-morning akinesia. The duration of effect of each dose becomes shorter and will often match the drug’s half-life of less than 2 hours. Although levodopa is the most effective drug for PD, the time to start taking it is controversial. Early use may contribute to greater activity levels and employability but cause more disability at later stages when the effect fluctuates and ultimately decreases. Protein in food uses the same mechanism as levodopa for crossing the blood-brain barrier. When levodopa is given with protein, the protein blocks the ability of the levodopa to cross the blood-brain barrier. This is usually managed by having the individual eat most of the daily protein in the evening, when immobility will cause the least inconvenience. Caffeine administered before levodopa may improve its pharmacokinetics in some individuals with parkinsonian symptoms.48 Infusion of levodopa directly into the intestines gives a more stable response but is expensive and invasive. Levodopa should be avoided in persons with malignant melanoma and in persons with active peptic ulcers, which may bleed.
Carbidopa inhibits the breakdown of levodopa and is often used in combination with levodopa. Carbidopa reduces the amount of levodopa required daily for beneficial effects and is often combined with levodopa in a single preparation (Sinemet).
Catechol-O-methyl transferase (COMT) inhibitors are reported to provide smoother and more sustained levels of levodopa to the brain. Of these, entacapone and tolcapone reduce the off time, and allow for decreased dosing of levodopa.141 Liver function must be monitored regularly with the use of tolcapone. Tolcapone must be used with levodopa but can decrease the amount of levodopa needed.
Dopamine agonists act directly on dopamine receptors. Bromocriptine seems to be the best tolerated, and its use in parkinsonism is associated with a lower incidence of response fluctuations. It is often given in combination with levodopa and carbidopa. Pramipexole or ropinirole can be used either to delay starting levodopa or to decrease the amount needed. Transdermal application of dopamine agonists can be provided with rotigotine and lisuride.
Selegiline (Eldepryl) and rasagiline have been used to inhibit monoamine oxydase type B (MAO-B) enzyme in the basal ganglia (MAO-B inactivates dopamine). It was thought that selegiline also may be able to delay the neuronal degeneration, but studies do not support that claim.42
Persons with mild symptoms but no disability may be helped by amantadine. Amantadine is also effective for the dyskinesia that develops later in the course of the disease. Coadministration of levodopa and amantadine controls dyskinesia without disrupting the effect of levodopa.
In the striatum the low level of dopamine is accompanied by increased cholinergic transmission. Accordingly, motor functions in individuals are improved by anticholinergic drugs. The side effects of the anticholinergic medications, including sedation, confusion, and psychosis, limit their usefulness, especially with advancing age. MAO-B inhibitors have replaced the use of anticholinergics in treatment of PD.36
Antioxidants have been studied for neuroprotection, such as coenzyme Q10, which helps stabilize mitochondria and appears to decrease the worsening of symptoms. Trophic factors, antiinflammatories, antiapoptotics, and antioxidants have been identified by the National Institutes of Health for further study for control of neuronal death.
Deep brain stimulation uses a pacemaker-like device surgically implanted with electrodes in the nuclei of choice and a pulse generator implanted in the chest. The generator is controlled externally through a magnetic field. When a tremor begins, the client activates the low-voltage, high-frequency generator by passing a magnet over it. Stimulation through the electrodes can be applied to the internal globus pallidus and the subthalamic nucleus or thalamus. Thalamic stimulation is most effective for tremor, with less effect on dyskinesia and rigidity. Electrode implantation in the globus pallidus appears to have good initial effect; however, there is a chance of psychosis and punding activity over time. Most centers are now stimulating the subthalamic nucleus bilaterally, but the individual’s profile leads the decision. Preoperative response to levodopa predicts better outcome after deep brain stimulation of the subthalamic nucleus. The ability to perform ADLs is improved, and there is typically improved sleep time. Apathy and abulia can occur over time; this may be related to withdrawal of levodopa. There can be an increase in sadness or the opposite response with excessive hilarity that may be related to stimulation of the surrounding area or change in subthalamic limbic activity. Edema around the electrode may contribute to the psychotropic effects. The implant is believed to last approximately 5 years and can be removed if another more effective type of treatment is found.8 Bilateral subthalamic stimulation, alone or in combination with levodopa, causes improvement in axial signs for posture and postural stability.
While there is little evidence for drug effect on postural stability and gait disorders, researchers in motor control are making progress in identifying the nature of the abnormal responses both inside and outside of the basal ganglia.161 (See Special Implications for the Therapist: Parkinson’s Disease later in the chapter.) Based on the strong evidence that relates prior exercise and activity status to risk of PD, and the recent knowledge gained about neuroplasticity in the brain, it is likely that changes in postural control may come through interventions that drive neuroplastic changes.
Although orthostatic hypotension affects less than 20% of individuals with parkinsonism, it can limit activity. Use of midodrine, fludrocortisone, and etilefrine can be helpful in maintaining normal blood pressure. Supine hypertension may result and must be monitored. Urinary dysfunction is treated via antimuscarinic agents or αagonists. Anticholinergics or scopolamine patches may be helpful for drooling, and use of intraparotid injection of BTX can help. Constipation is common and may precede the motor symptoms in PD; it is usually managed by fluids, fiber, stool softeners and exercise.
Depression is found in over 40% of individuals with PD. Medication interactions must be looked at carefully. Use of serotonin uptake inhibitors may interact with selegiline. Tricyclics can be useful, but the central effects must be monitored more carefully than in the healthy younger population.
Respiratory complications, which are the leading cause of death, can be prevented to some extent with an early aggressive aerobic exercise program, followed by regular moderate activity as the disease progresses. Control of breathing can be facilitated using verbal and tactile stimuli and should be integrated into any intervention.
Behavioral abnormalities can be associated with high doses of dopaminergic replacement therapy, including the phenomenon of punding, characterized by fascination with technical equipment and excessive sorting of objects, grooming, hoarding, or use of a computer. This may be related to the impaired frontal lobe function and a result of psychomotor stimulation. Other abnormalities in reward-seeking behavior related to dopamine are hypersexuality and excessive gambling. Reducing the level of medication is helpful, and some neuroleptics such as clozapine will lessen symptoms of psychosis.
Experimental therapeutics targeted at improving dopaminergic drugs to increase selectivity for various receptor subtypes and at controlling the uptake of dopamine are currently under study. Improved plasma stability is achieved through transdermal application, which bypasses the fluctuations in gastric release. Studies are aimed at potential substances that evoke antiparkinsonism through neurotransmitter systems outside of dopamine. Pharmacologic manipulation of glutamate and GABA neurotransmission includes the drug istradefylline, which is currently undergoing phase III study.13 Opiate, serotonin, and histamine receptors are possible sites for intervention. A more careful look at the cholinergic system may help to manage the issues of dementia and find possibilities for reducing the apparent dysfunction at the level of the brainstem that affects sleep wake cycles and orthostasis.
Cell transplantation of grafted dopaminergic neurons in PD continues to hold promise. The striatum (caudate and putamen) are primary targets for the implants. Individual selection, cell-handling variation in surgical techniques, and immunosuppression currently make it difficult to determine success. Graft-induced dyskinesia much like that associated with long-term levodopa use appears in some recipients. It is not clear which individual will develop the dyskinesia, and at this time there is not any medication to control the abnormal movements. Fetal stem cells remain the most successful; however, alternative types are being studied. There is hope for cells derived from the individual’s own body, but the response has been less than hoped for and the current graft survival is low. Rejection of the graft continues to limit effects, and immunosuppression has to be considered as a possible long-term adjunctive therapy with grafts.184
PROGNOSIS.
In general, all the clinical manifestations in PD worsen progressively, although not to the same extent. Tremor as a presenting symptom may be used to predict a more benign course and longer therapeutic benefit to levodopa. In individuals with newly diagnosed PD, older age at onset and rigidity/hypokinesia as an initial symptom can be used to predict more rapid rate of motor progression.171
The presence of associated comorbidities, stroke, auditory deficits, and visual impairments as well as male sex may be used to predict faster rate of motor progression. Older age at onset and initial hypokinesia/rigidity may be used to predict earlier development of cognitive decline and dementia. Older age at onset, dementia, and decreased dopamine responsiveness may be used to predict earlier nursing home placement as well as decreased survival. Lack of mobility, loss of balance reactions, and weakness result in more falls than in an age-matched normal population. Osteoporosis can result from prolonged inactivity and may be present secondary to advanced age at onset. Falls more often lead to fractures owing to the prevalence of osteoporosis. Fracture healing may be delayed. Posture and gait abnormalities are the most difficult to control in advanced cases.
PD does not significantly reduce life span in most persons who develop the generalized form between 50 and 60 years of age. However, since there is progressive neuronal loss despite the response to treatment, deterioration continues until death occurs, often from infection or other conditions associated with debilitation.
Since the onset of disease is typically in the fifth or sixth decade of life and is progressive despite medication, the economic cost of the disease can be quite high because of loss of income, cost of drugs, assistive devices, and assisted living. Pain, fatigue, and depression also adversely affect the quality of life compared with that of age-matched normal subjects.159
Secondary Parkinson’s Syndrome
Parkinsonian syndromes, also called atypical parkinsonism or Parkinson’s plus syndromes, are a family of neurodegenerative disorders that result from neuronal loss in different components of the basal ganglia, the brain system of which the dopaminergic midbrain neurons affected in PD are a part. All of these disorders can be difficult to differentiate from PD early in the course of the illness. These disorders have distinctive clinical features, which may emerge only after the onset of parkinsonism. Important clinical clues that one of these disorders is present are symmetric onset of parkinsonism, absence of typical resting tremor, early autonomic dysfunction, prominent dystonia, significant early cognitive impairment, and prominent early falls.2
Iatrogenic parkinsonism or drug-induced parkinsonism results from the use of pharmacologic agents that block dopamine effects or interfere with dopamine metabolism. The most common causes of drug-induced parkinsonism are dopamine antagonist antipsychotic medications. The risk of drug-induced parkinsonism is reduced significantly with newer atypical antipsychotic agents. Another group of drugs that can cause drug-induced parkinsonism is older (non–serotonin antagonist) dopamine antagonist antiemetics. Agents interfering with dopamine production or synaptic vesicular storage can cause drug-induced parkinsonism. These include methyl-para-tyrosine, methyldopa, and reserpine. Flunarizine and cinnarizine, when they are used as vestibular suppressants or cerebral vasodilators, can cause parkinsonism. Sodium valproate may cause tremor that can progress to parkinsonism. Features of iatrogenic parkinsonism are bilateral onset and predominant bradykinesia with increased involvement in the arm compared to the legs in the younger population, but more consistent with PD in older individuals. If drug-induced parkinsonism is suspected, the suspected offending agent is withdrawn, and the individual should improve. With dopamine antagonists or reserpine, improvements can occur within days to weeks after medication withdrawal, but there is sometimes a prolonged latency of months before marked improvement occurs.
Vascular parkinsonism involves primarily the lower extremities. It is associated with lacunar infarcts (see Chapter 32) and probably represents small infarcts in the basal ganglia or corticobasilar pathways. A stroke in the region of the striatum (caudate and putamen) and hemiparesis of the arm is common. Systemic lupus erythematosus may also cause cerebral vasculitis. Vascular parkinsonism presents typically with start hesitation, a broad-based shuffling gait (rather than the narrow-based gait associated with PD), and frequent falls. Depending on the level of damage and the cause, the response to levodopa will vary.
Infectious causes of parkinsonism are suspected when the symptoms develop during the acute or recovery phase of an illness with fever. Cases of parkinsonism have been reported as a result of West Nile virus infection and have historically been associated with encephalitis. Human immunodeficiency virus (HIV) infection can cause parkinsonism via the viral damage in encephalopathy or opportunistic infections.
Toxicity, often related to manganese accumulation in the substantia nigra, can cause parkinsonism and dystonia, seen in miners, factory workers making dry cell batteries, and those exposed to some fungicides.
Disorders with Parkinsonian Characteristics
Benign essential tremor is not associated with any underlying cause, is common after the age of 50 years, and is usually hereditary. This tremor is of a different character, and there is a lack of other neurologic signs.
Progressive Supranuclear Palsy
Progressive supranuclear palsy has symptoms of bradykinesia, rigidity, and postural instability similar to those of PD and is frequently misdiagnosed as PD. Neurofibrillary tangles are the main pathology in progressive supranuclear palsy; oligodendrocytes are also affected. Postural instability is the most pronounced symptom, with falls that are not associated with obstacles or change in surface. Gait freezing and apraxia are common. Dysarthria and dysphagia are on a continuum, with dysphagia typically occurring later than 2 years after onset. Loss of upward gaze, saccades, and smooth pursuit eye tracking progresses over time. Inhibition of eyelid opening and closure, or blepharospasm, can cause functional blindness. Inability to perform vestibulo-ocular reflex cancellation is lost. Apathy, intellectual slowing, and impairment of executive function progress, and there can be pseudobulbar laughter or crying. The autonomic nervous system maintains near-normal function.
Levodopa and deep brain stimulation are effective for the movement disorder, and BTX can help to improve blepharospasm.
Multiple System Atrophy
There is extreme clinical variability within the multiple system atrophy group of disorders that is primarily familial but can be sporadic. Neuronal atrophy is seen to a variable degree in the brainstem, cerebellum, spinal cord, and peripheral nerves. The differential pathology is associated with gliosis and cytoplasmic inclusion in the glia. Multiple system atrophy typically has its onset in the fifth to seventh decade, and parkinsonism is the primary condition; however, there is more evidence of cerebellar involvement, and autonomic dysfunction is greater and more disabling that that found in PD. Levodopa is used in the treatment, but with less success than when it is used in PD. Cerebellar and autonomic nervous system dysfunction respond poorly to anticholinergics. Large European studies are under way to examine pathogenesis and intervention strategies.
Olivopontocerebellar Atrophy
Olivopontocerebellar atrophy is one of the most common and variable of the non-PD parkinsonian conditions. Neuronal loss with gross atrophy is concentrated in the pons, medullary olives, and cerebellum. Ataxia, rigidity, spasticity, and oculomotor movement disturbances are present in variable degrees and combinations. The intracytoplasmic inclusions are predominantly oligodendrocytic, and there is modest tau and synuclein immunoreactivity.
Wilson’s Disease
Wilson’s disease, or progressive hepatolenticular degeneration, is rare but also represents degeneration of the basal ganglia and is related to excess deposition of copper. Cysts or cavities form in the basal ganglia with necrosis. The lateral ventricles can be enlarged with associated brain atrophy. This can be imaged using MRI, PET, or SPECT studies. Cerebellar and brainstem damage is common, and there can be spheroid bodies in the cerebral cortex. The symptoms of Wilson’s disease go far beyond movement disorder mimicking PD and include profound affective disorders. Ophthalmologic signs of brownish or greenish rings in the periphery of the cornea are a hallmark sign. The disorder is treated via copper chelating.
Restless Leg Syndrome
Restless leg syndrome is reported as the desire to move the extremities associated with paresthesia, motor restlessness with worsening of symptoms at rest (typically at night), and relief with activity or sensory stimulation. It is familial in 60% of cases, and the effect is related to reduced iron stores in the substantia nigra. There is no loss of dopaminergic neurons as is seen in PD, but the dysfunction lies within the presynaptic and postsynaptic junction of dopaminergic neurons. Levodopa is the traditional treatment, and it is effective when movement is the most problematic symptom. Opioids such as methadone can help when dopamine agents are not effective. For the individual with pain or dysesthesia, neuroleptics can be of benefit.
References
1. Adler, CH, Zimmerman, RS, Lyons, MK, et al. Perioperative use of botulinum toxin for movement disorder-induced cervical spine disease. Mov Disord. 1996;11:79–81.
2. Albin, RL. Parkinson’s disease: background, diagnosis, and initial management. Clin Geriatr Med. 2006;22(4):735–751.
3. Agency for Health Care Policy and Research. Guideline no. 19: early Alzheimer’s disease: recognition and assessment. Rockville, MD: U.S. Department of Health and Human Services, 1996.
4. Alves, G. Is fatigue an independent and persistent symptom in patients with Parkinson disease? Neurology. 2004;63(10):1908–1911.
5. Aminoff, M. Nervous system. In: Tierney LM, McPhee SJ, Papadakis MA, eds. Current medical diagnosis and treatment. ed 33. Norwalk, CT: Appleton & Lange; 1994:799–854.
6. Appel, SH, Appel, LV. Treatment of amyotrophic lateral sclerosis. In: Calne DB, ed. Neurodegenerative diseases. Philadelphia: Saunders; 1994:523–542.
7. Bagley, S, Kelly, B, Tunnicliffe, N, et al. The effect of visual cues on the gait of independently mobile Parkinson’s disease patients. Physiotherapy. 1991;77(6):415–420.
8. Barry, M. Reliability and responsiveness of the Barry-Albright Dystonia Scale. Devel Med Child Neurol. 1991;41:404–411.
9. Beal, MF, Richardson, EP, Martin, JB. Alzheimer’s disease and other dementias. In: Isselbacher KJ, Braunwald E, Wilson JD, et al, eds. Harrison’s principles of internal medicine. ed 13. New York: McGraw-Hill; 1994:2269–2295.
10. Behrman, AL, Teitelbaum, P, Cauraugh, J. Verbal instructional sets to normalize the temporal and spatial variables in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1998;65:580–582.
11. Bejjani, BP, Gervais, D, Arnulf, I, et al. Axial parkinsonian symptoms can be improved: the role of levodopa and bilateral subthalamic stimulation. J Neurol Neurosurg Psychiatry. 2000;68:595–600.
12. Belsh, JM. Diagnostic challenges in ALS. Neurology. 1999;53(8 suppl 5):S26–S30.
13. Ben-Shlomo, Y. How far are we in understanding the cause of Parkinson’s disease? J Neurol Neurosurg Psychiatry. 1996;61:4–16.
14. Betts, CD, D’Mellow, MT, Fowler, CJ. Urinary symptoms and the neurological features of bladder dysfunction in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1993;56:245–250.
15. Bickel, U. Diagnosis of CAA during life. In: Verbeek MM, deWaal RM, Vinters AJ, eds. Cerebral amyloid angiopathy in Alzheimer’s disease and related disorders. Dordrecht, Netherlands: Kluwer Academic Publishers; 2000:21–41.
16. Blandini, F, Nappi, G, Tassorelli, B, et al. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol. 2000;62:63–88.
17. Bohannon, RW. Results of resistance exercise on a patient with amyotrophic lateral sclerosis: a case report. Phys Ther. 1983;63:965–968.
18. Bond, JM, Morris, M. Goal-directed secondary motor tasks: their effects on gait in subjects with Parkinson’s disease. Arch Phys Med Rehabil. 2000;81:110–116.
19. Bowlby, C. Therapeutic activities with persons disabled by Alzheimer’s disease and related disorders. Gaithersburg, MD: Aspen, 1993.
20. Braak, H, Braak, E. Pathology of Alzheimer’s disease. In: Calne DB, ed. Neurodegenerative disease. Philadelphia: Saunders, 1994.
21. Brazier, J, Harper, R, Jones, N, et al. Validating the SF-36 health survey questionnaire: new outcomes measure for primary care. BMJ. 1992;305:160–164.
22. Bressman, S, Sabatti, C, Raymond, D, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. 2001;54:1746–1752.
23. Brex, PA, Ciccarelli, O, O’Riordan, JI, et al. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med. 2002;346(3):158.
24. Bronstein, AM, Hood, JD, Gresty, MA, et al. Visual control of balance in cerebellar and parkinsonian syndromes. Brain. 1990;113:767–779.
25. Brooks, BR. Introduction: defining optimal management in ALS: from first symptoms to announcement. Neurology. 1999;53(8 suppl 5):S1–S3.
26. Brown, RH. Motor neuron disease and the progressive ataxias. In: Isselbacher KJ, Braunwald E, Wilson JD, et al, eds. Harrison’s principles of internal medicine. ed 13. New York: McGraw-Hill; 1994:2280–2283.
27. Brown, TR. Exercise and rehabilitation for individuals with multiple sclerosis. Phys Med Rehabil Clin North Am. 2005;16(2):513–555.
28. Bruumlatock, W, Schmied, M, Suchanek, G, et al. Oligodendrocytes in the early course of multiple sclerosis. Ann Neurol. 1994;35:65–73.
29. Burt, A. Textbook of microanatomy. Philadelphia: Saunders, 1993.
30. Burton, A. Are men more likely to pass on multiple Sclerosis? Lancet Neurol. 2006;5(9):728.
31. Butcher, J. Implicit memory is retained by people with Alzheimer’s disease. Lancet Neurol. 2004;3(8):451.
32. Butler, K. Focal hand dystonia affecting musicians: Part II: an overview of current rehabilitative treatment techniques. Br J Hand Ther. 2006;11:79–87.
33. Byl, N, Merzenich, M, Cheung, S, et al. A primate model for studying focal dystonia and repetitive strain injury: effects on the primary somatosensory cortex. Phys Ther. 1997;77(3):269–283.
34. Byl, N, Wilson, F, Scott, P, et al. Sensory dysfunction associated with repetitive strain injuries of tendonitis and focal hand dystonia: a comparative study. J Orthop Sports Phys Ther. 1996;7:27–39.
35. Byl, N, Nagajaran, S, McKenzie, A. Effect of sensory discrimination training on structure and function in patients with focal hand dystonia: a case series. Arch Phys Med Rehabil. 2003;84:1505–1514.
36. Calabresi, P. A convergent model for cognitive dysfunctions in Parkinson’s disease: the critical dopamine-acetylcholine synaptic balance. Lancet Neurol. 2006;5(11):974–983.
37. Caselli, RJ, Boeve, BF. The degenerative dementias. In: Goetz CG, ed. Textbook of clinical neurology. ed 2. Philadelphia: Saunders; 2003:682–692.
38. Cashman, NR. Do the benefits of currently available treatments justify early diagnosis and announcement? Neurology. 1999;53(8 suppl 5):S50–S52.
39. Centonze, D, et al. Repetitive transcranial magnetic stimulation of the motor cortex ameliorates spasticity in multiple sclerosis. Neurology. 2007;68(13):1045–1050.
40. Chase, TN. A gene for Parkinson’s disease. Arch Neurol. 1997;54:1156–1157.
41. Chen, H. Physical activity and the risk of Parkinson disease. Neurology. 2005;64(4):664–669.
42. Ciccone, CD. Free-radical toxicity and antioxidant medications in Parkinson’s disease. Phys Ther. 1998;78(3):313–319.
43. Comella, CL, Stebbins, GT, Brown-Toms, MA, et al. Physical therapy and Parkinson’s disease: a controlled clinical trial. Neurology. 1994;44:376–378.
44. Consky, E, Basinski, A, Belle, L, et al. The Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS): assessment of validity and inter-rater reliability. Neurology. 1990;40(suppl):445.
45. Dai, Q, Borenstein, AR, Wu, Y, et al. Fruit and vegetable juices and Alzheimer’s disease: the Kame Project. Am J Med. 2006;119(9):751–759.
46. Dal Bello-Haas, VD, Kloos, AD, Mitsumoto, H. Physical therapy for a patient through six stages of amyotrophic lateral sclerosis. Phys Ther. 1998;78:1312–1324.
47. Deepak, K, Behari, M. Specific muscle EMG biofeedback for hand dystonia. Appl Psychophysiol Biofeedback. 1999;24(4):267–280.
48. Deleu, D. Effects of caffeine on levodopa pharmacokinetics and pharmacodynamics in Parkinson disease. Neurology. 2006;67(5):897–899.
49. Delval, A. Movement-related cortical activation in familial Parkinson disease. Neurology. 2006;67(6):1086–1087.
50. Desai, AK. Diagnosis and treatment of Alzheimer’s disease. Neurology. 2005;64(12 suppl 3):S34–S39.
51. Dewey, RB. Autonomic dysfunction in Parkinson’s disease. Neurol Clin. 2004;22(3 suppl):S127–S139.
52. Djaldetti, R. The mystery of motor asymmetry in Parkinson’s disease. Lancet Neurol. 2006;5(9):796–802.
53. Drake, D. Spasmodic torticollis: clinical and biological features and their implications for focal dystonia. Adv Neurol. 1988;50:473–492.
54. Dubinsky, R. Trends in hospital utilization and outcome for patients with ALS: analysis of a large U.S. cohort. Neurology. 2006;67(5):777–780.
55. Edison, P. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68(7):501–508.
56. Edwards, LL, Normand, MM, Wszolek, ZK. Cervical dystonia: a review of the role of botulinum toxin. Nebr Med J. 1995;80:109–115.
57. Eimeren, T, Siebner, H. An update on functional neuroimaging of parkinsonism and dystonia. Curr Opin Neurol. 2006;19:412–419.
58. Fahn, S. The freezing phenomenon in parkinsonism. Adv Neurol. 1995;67:53–63. [In Fahn S, Hallett M, Luders O, et al, eds: Negative motor phenomenon].
59. Fearnley, J, Lees, A. Pathology of Parkinson’s disease. In: Calne DB, ed. Neurodegenerative diseases. Philadelphia: Saunders; 1994:545–554.
60. Flaherty-Craig, C. A rapid screening battery to identify frontal dysfunction in patients with ALS. Neurology. 2006;67(11):2070–2072.
61. Folstein, SE. Huntington’s disease: a disorder of families. Baltimore: Johns Hopkins University Press, 1989;37–80.
62. Fox, EJ. Immunopathology of multiple sclerosis. Neurology. 2004;63(12 suppl 6):S3–S7.
63. Francis, DA, Compston, D.A.S, Batchcor, JR, et al. A reassessment of the risk of multiple sclerosis developing in patients with optic neuritis after extended follow up. J Neurol Neurosurg Psychiatry. 1987;50:758–765.
64. Freal, JE, Kraft, GH, Coryell, JK. Symptomatic fatigue in multiple sclerosis. Arch Phys Med Rehabil. 1984;65:135–138.
65. Frigerio, R. Education and occupations preceding Parkinson disease: a population-based case-control study. Neurology. 2005;65(10):1575–1583.
66. Gordon, PH, Cheung, YK. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS [Comment on. Neurology. 2006;66(2):265–267. Neurology. 2006;67(7):1314–1315. [author reply, 1314-1315. 2006].
67. Gilroy, J, Holliday, P. Basic neurology. New York: Macmillan, 1982;177–178.
68. Ghilardi, M, Carbon, M, Sivestri, G, et al. Impaired sequence learning in carriers of the DYT1 dystonia mutation. Ann Neurol. 2003;54:102–109.
69. Hallett, M. Is dystonia a sensory disorder? Ann Neurol. 1995;38(2):139–140.
70. Halliday, GM, McRitchie, DA, Macdonald, V, et al. Regional specificity of brain atrophy in Huntington’s disease. Exp Neurol. 1998;154:663–672.
71. Harper, PS, Morris, M. The epidemiology of Huntington’s disease. In: Harper PS, ed. Huntington’s disease. Philadelphia: Saunders; 1991:1–36.
72. Hawker, K, Frohman, E. Multiple sclerosis. Prim Care. 2004;31(1):201–226.
73. Hauser, SI. Multiple sclerosis and other demyelinating diseases. In: Isselbacher KJ, Braunwald E, Wilson JD, et al, eds. Harrison’s principles of internal medicine. ed 13. New York: McGraw-Hill; 1994:2287–2294.
74. Herndon, RM. Multiple sclerosis: basic science issues. MS Q Rep. 1994;12:3–5.
75. Heiman-Patterson, TD. NIPPV: a treatment for ALS whose time has come. Neurology. 2006;67(5):736–737.
76. Hof, PR, Morrison, JH. Hippocampal and neocortical involvement in normal brain aging and dementia: morphological and neurochemical profile of the vulnerable circuits. J Am Geriatr Soc. 1996;44:857–864.
77. Homberg, V. Motor training in the therapy of Parkinson’s disease. Neurology. 1993;43(suppl 6):45–46.
78. Horner, RD. Prospective study of military service and mortality from ALS [Comment on. Neurology. 2005;64(1):32–37. Neurology. 2005;65(1):180–181. [author reply 180-181].
79. Horak, FB, Nutt, JG, Nashner, LM. Postural inflexibility in parkinsonian subjects. J Neurol Sci. 1992;111:46–58.
80. Horak, F, Dimitrova, D, Nutt, JG. Direction-specific postural instability in subjects with Parkinson’s disease. Exp Neurol. 2005;193:504–521.
81. Hurley, AC, et al. Alzheimer’s discomfort rating scale. Res Nurs Health. 1992;15:369–377.
82. Hurwits, A. The benefit of a home exercise regimen for ambulatory Parkinson’s disease patients. J Neurosci Nurs. 1989;21(3):180–184.
83. Inada, T, Yagi, G. Current topics in neuroleptic-induced extrapyramidal symptoms in Japan. Keio J Med. 1996;45:95–99.
84. Jankovic, J, Orman, J. Botulinum A toxin for cranial-cervical dystonia: a double-blind, placebo-controlled study. Neurology. 1987;37:616–623.
85. Jankovic, J. Dystonia: medical therapy and botulinum toxin. Adv Neurol. 2004;94:275–286.
86. Jinnah, H, Hess Ellen, J. A new twist on the anatomy of dystonia: the basal ganglia and the cerebellum? Neurology. 2006;67:1740–1741.
87. Johns Hopkins Medical Letter: Health after. 1999;50(8):7.
88. Johnson, K. MRI studies confirm advantages of interferon beta-1b. Peer. 1994;1:3–9.
89. Johnson, KA, Cunninton, R, Bradshaw, JL, et al. Bimanual co-ordination in Parkinson’s disease. Brain. 1998;121:743–753.
90. Kaji, R, Ikeda, A, Ikeda, T, et al. A physiological study of cervical dystonia: task-specific abnormality in contingent negative variation. Brain. 1995;118:511–522.
91. Kaji, R, Shibasaki, H, Kimure, J. Writer’s cramp: a disorder of motor subroutine? Ann Neurol. 1995;38(6):837–838.
92. Kappos, L. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–1140.
93. Kartha, N, Dystonia. Clin Geriatr Med. 2006;22:899–914.
94. Kaufman, DI, Fratkin, J. Multiple sclerosis and the eye. Ophthalmol Clin North Am. 1992;5:513–530.
95. Keegan, M, Pineda, AA, McClelland, RL, et al. Plasma exchange for severe attacks of CNS demyelination: predictors of response. Neurology. 2002;58(1):143–146.
96. Kirsch, NR, Myslinski, MJ. The effect of a personally designed fitness program on the aerobic capacity and function for two individuals with multiple sclerosis, Phys Ther. Case Rep. 1999;2(1):21–27.
97. Kobelt, G. Costs and quality of life in multiple sclerosis: a cross-sectional study in the United States. Neurology. 2006;66(11):1696–1702.
98. Korein, J, Brudny, J, Grynbaum, B, et al. Sensory feedback therapy of spasmodic torticollis and dystonia: results in treatment of 55 patients. Adv Neurol. 1976;14:375–402.
99. Krivickas, LS. Pulmonary and respiratory failure. In: Mitsumoto H, Chad DA, Pioro P, eds. Amyotrophic lateral sclerosis. Philadelphia: FA Davis; 1998:382–404. [Contemporary neurology series].
100. ALS Kumar. Robbins and Cotran pathologic basis of disease, ed 7. Philadelphia: Saunders, 2005.
101. Kupersmith, MJ, Kaufman, D, Paty, DW. Megadose corticosteroids in multiple sclerosis. Neurology. 1994;44:1–4.
102. Kurland, L, Kurupath, R, Williams, DB. Amyotrophic lateral sclerosis-parkinsonism-dementia complex on Guam: epidemiologic and etiological perspectives. In: Williams D, ed. Motor neuron disease. London: Chapman & Hall; 1994:109–130.
103. Lagueny, A, Burbaud, P, Le Masson, G, et al. Involvement of respiratory muscles in adult-onset dystonia: a clinical and electrophysiological study. Mov Disord. 1995;10:708–713.
104. Lang, AE, Lozano, AM. Parkinson’s disease. N Engl J Med. 1998;339(15):1044–1053.
105. Lagueny, A, Burbaud, P, Le Masson, G, et al. Involvement of respiratory muscles in adult-onset dystonia: a clinical and electrophysiological study. Mov Disord. 1995;10:708–713.
106. Lassmann, H, Bruck, W, Lucchinetti, C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. 2001;7(3):115–121.
107. Lavretsky, EP, Jarvik, LF. Etiology and pathogenesis of Alzheimer’s disease: current concepts. In: Hamdy RC, Turnbull JM, Clark W, et al, eds. Alzheimer’s disease: a handbook for care givers. St Louis: Mosby-Year Book; 1994:80–92.
108. Lee, MS, Rinne, JO, Ceballos-Baumann, A, et al. Dystonia after head trauma. Neurology. 1994;44:1374–1378.
109. Lee, H-G, Casadesus, G, Zhu, X, et al. Challenging the amyloid cascade hypothesis: senile plaques and amyloid-? as protective adaptations to Alzheimer’s disease. Ann N Y Acad Sci. 2004;1019:1–4.
110. Leigh, PN. Pathogenic mechanisms in amyotrophic lateral sclerosis and other motor neuron disorders. In: Calne DB, ed. Neurodegenerative diseases. Philadelphia: Saunders; 1994:473–488.
111. Lenz, F, Jaeger, C, Seike, M, et al. Thalamic single neuron activity in patients with dystonia: dystonia-related activity and somatic sensory reorganization. J Neurophysiol. 1999;82:2372–2392.
112. Lucchinetti, C, Bruck, W, Parisi, J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):691–693.
113. Ludolph, AC, Reipe, MW. Do the benefits of currently available treatments justify early diagnosis and treatment of amyotrophic lateral sclerosis? Arguments against. Neurology. 1999;53(8 suppl 5):S46–S49.
114. Majsak, MJ, Kaminski, T, Gentile, AM, et al. The reaching movements of patients with Parkinson’s disease under self-determined maximal speed and visually cued conditions. Brain. 1998;121:755–766.
115. Mahendra, N. Exercise and behavioural management training improves physical health and reduces depression in people with Alzheimer’s disease. Evid Based Healthc. 2004;8(2):77.
116. Marchese, R, Diveria, M, Zucchi, F, et al. The role of sensory cues in the rehabilitation of parkinsonian patients: a comparison of two physical therapy protocols. Mov Disord. 2000;15(2):879–883.
117. McCance, KL, Heuther, SE. Alterations of neurologic function. In: McCance KL, Heuther SE, eds. Pathophysiology: the biological basis for diseases in adults and children. ed 2. St Louis: Mosby-Year Book; 1994:571–575.
118. Mertin, J, Paeth, B. Physiotherapy and multiple sclerosis-application of the Bobath concept. MS Q Rep. 1994;1:1.
119. Miller, AE. Clinical features. In: Cook SD, ed. Handbook of multiple sclerosis. New York: Marcel Dekker; 1990:169–175.
120. Miller, AE. Glatiramer acetate in the treatment of multiple sclerosis. Neurol Clin. 2005;23(1):215–231. [viii].
121. Miller, BL, Chang, L, Oropilla, G, et al. Alzheimer’s disease and frontal lobe dementias. In: Coffey CE, Cummings JL, eds. Textbook of geriatric neuropsychiatry. Washington, DC: American Psychiatric Press; 1994:390–400.
122. Miller, D. Multiple sclerosis: new insights and therapeutic progress. Lancet Neurol. 2007;6(1):5–6.
123. Miller, RG, Green, AT, Moussavi, RS, et al. Excessive muscular fatigue in patients with spastic paraparesis. Neurology. 1990;40:1271–1274.
124. Mink, JW. Functional organization of the basal ganglia. In: Janovic J, Tolosa E, eds. Parkinson’s disease and movement disorders. ed 5. Philadelphia: Lippincott Williams & Wilkins; 2007:1–6.
125. Mitsumoto, H, Chad, DA, Pioro, P. Amyotrophic lateral sclerosis. Philadelphia: FA Davis, 1998. [Contemporary neurology series].
126. Mitsumoto, H, Hanson, M, Chad, D. Amyotrophic lateral sclerosis: recent advances in pathogenesis and therapeutic trials. Arch Neurol. 1988;45:189–202.
127. Morris, M. Movement disorders in people with Parkinson disease: a model for physical therapy. Phys Ther. 2000;80(6):579–597.
128. Morris, M. Locomotor training in people with Parkinson’s disease. Phys Ther. 2006;86(10):1426–1435.
129. Morris, M. Psychiatric aspects of Huntington’s disease. In: Harper P, ed. Huntington’s disease. Philadelphia: Saunders; 1991:81–126.
130. Morris, M, Huxham, F, McGinley, J. Strategies to prevent falls in people with Parkinson’s disease. Physiother Singapore. 1999;2(4):135–141.
131. Morris, M, Tyler, A. Management and therapy. In: Harper P, ed. Huntington’s disease. Philadelphia: Saunders; 1991:205–250.
132. Murray, TJ, Pryse-Phillips, W. Amyotrophic lateral sclerosis. In: Noble J, ed. Textbook of primary care medicine. ed 3. St Louis: Mosby; 2001:1765–1768.
133. Nutt, J, Muenter, MD, Aronson, A, et al. Epidemiology of focal and generalized dystonia in Rochester, MN. Mov Disord. 1988;3:188–194.
134. Noseworthy, JH, Lucchinetti, C, Rodriguez, M, et al. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952.
135. Olafsson, I, Thorsteinsson, L. Genetics and neuropathology of hereditary cystatin C amyloid angiopathy (HCCAA). In: Verbeek MM, deWaal RM, Vinters AJ, eds. Cerebral amyloid angiopathy in Alzheimer’s disease and related disorders. Dordrecht, Netherlands: Kluwer Academic Publishers; 2000:121–136.
136. Olazarán, J. Benefits of cognitive-motor intervention in MCI and mild to moderate Alzheimer disease. Neurology. 2004;63(12):2348–2353.
137. Optic Neuritis Study Group. The 5-year risk of MS after optic neuritis: experience of the optic neuritis treatment trial. Neurology. 1997;49(5):1404–1413.
138. O’Sullivan, SB. Multiple sclerosis. In: O’Sullivan SB, Schmitz TJ, eds. Physical rehabilitation: assessment and treatment. ed 3. Philadelphia: FA Davis; 1994:451–467.
139. Pleasure, D. New direction in MS therapy: protein growth factors may enhance CNS remediation. Peer. 1994;1:12–13.
140. Ponchitera-Mulcare, JA, Bglaser, RM. Evaluation of muscle performance and cardiopulmonary fitness in patients with multiple sclerosis. Neurorehabilitation. 1993;3(17):17–29.
141. Pahwa, R. Practice parameter: treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66(7):983–995.
142. Pirko, I. Gray matter involvement in multiple sclerosis. Neurology. 2007;68(9):634–642.
143. Pontone, G. Clinical features associated with impulse control disorders in Parkinson disease. Neurology. 2006;67(7):1258–1261.
144. Prineas, JW, Barnard, RO, Kwon, EE. Multiple sclerosis: remyelination of nascent lesions. Ann Neurol. 1993;33:137–151.
145. Protas, EJ, Stanley, RK, Jankovic, PT, et al. Cardiovascular and metabolic responses to upper and lower extremity exercise in men with idiopathic Parkinson’s disease. Phys Ther. 1996;76(1):34–40.
146. Quarrell, O. The neurobiology of Huntington’s disease. In: Harper P, ed. Huntington’s disease. Philadelphia: Saunders; 1991:81–109.
147. Quarrell, O, Harper, P. The clinical neurology of Huntington’s disease. In: Harper P, ed. Huntington’s disease. Philadelphia: Saunders; 1991:37–80.
148. Rajput, AH. Clinical features and natural history of Parkinson’s disease (special considerations of aging). In: Calne DB, ed. Neurodegenerative diseases. Philadelphia: Saunders; 1994:555–572.
149. Reisberg, B, Ferris, SH, de Leon, MJ, et al. The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry. 1982;139:1136–1139.
150. Reuter, I, Engelhardt, M, Stecker, K, et al. Therapeutic value of exercise training in Parkinson’s disease. Med Sci Sports Exerc. 1997;31:1544–1549.
151. Rodriguez, M, Siva, A, Stolp-Smith, K. Impairment, disability and handicap in multiple sclerosis: a population-based study in Olmsted County, Minnesota. Neurology. 1994;44:28–33.
152. Rogers, MW. Disorders of posture balance and gait in Parkinson’s disease. Clin Geriatr Med. 1996;14(4):825–833.
153. Rothstein, JD, Jin, L, Dykes-Hoberg, M. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci U S A. 1993;90:6591–6595.
154. Rothwell, JC. Physiological study of cervical dystonia: task-specific abnormality in contingent negative variation. Brain. 1995;118(pt 2):511–522.
155. Rovio, S. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705–711.
156. Rowland, LP. Natural history and clinical features of amyotrophic lateral sclerosis and related motor neuron diseases. In: Calne DB, ed. Neurodegenerative diseases. Philadelphia: Saunders; 1994:507–522.
157. Rudick, RA, Cohen, JA, Weinstock-Guttman, et al. Management of multiple sclerosis. N Engl J Med. 1997;337(22):1604–1611.
158. Sadovnick, AD, Yee, IM, Ebers, GC, et al. Effect of age at onset and parental disease status on sibling risks for MS. Neurology. 1998;50:719–723.
159. Schenkman, M, Morey, M, Kuchibhatla, M. Spinal flexibility and balance control among community-dwelling adults with and without Parkinson’s disease. J Gerontol Med Sci. 2000;55A(8):441–445.
160. Schenkman, M, Zhu, CW, Cutson, TM, et al. Longitudinal evaluation of economic and physical impact of Parkinson’s disease. Parkinsonism Relat Disord. 2001;8:41–50.
161. Seigel, KL, Metman, LV. Effects of bilateral posteroventral pallidotomy on gait of subjects with Parkinson’s disease. Arch Neurol. 2000;57:198–204.
162. Shapiro, RT, Laven, L. Multiple sclerosis. In: Good DC, Couch JR, eds. Handbook of neurorehabilitation. New York: Marcel Dekker; 1994:551–559.
163. Schapiro, RT. Managing symptoms of multiple sclerosis. Neurol Clin. 2005;23(1):177–187.
164. Simmons, Z, Felgoise, SH, Bremer, BA, et al. The ALSSQOL: balancing physical and nonphysical factors in assessing quality of life in ALS. Neurology. 2006;67(9):1659–1664.
165. Simon, JH. Update on multiple sclerosis. Radiol Clin North Am. 2006;44(1):79–100.
166. Simpson, EP. Antioxidant treatment for amyotrophic lateral sclerosis. Lancet Neurol. 2005;4(5):266.
167. Siva, N. Can ketogenic diet slow progression of ALS? Lancet Neurol. 2006;5(6):476.
168. Smith, RA. Treatment of pseudobulbar affect in ALS. Lancet Neurol. 2005;4(5):270.
169. Sipe, JC, Romine, JS, Koziol, JA. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet. 1994;344:9–13.
170. Smith, ME, Stone, LA, Albert, PS. Clinical worsening in MS is associated with increased frequency of area of gado-pentetate dimeglumine enhancing MRI lesions. Ann Neurol. 1994;33:480–489.
171. Suchowersky, O. Practice parameter: diagnosis and prognosis of new onset Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66(7):968–975.
172. Svensson, B, Gerdle, B, Elert, J. Endurance training in patients with multiple sclerosis: five case studies. Phys Ther. 1994;74:1017–1026.
173. Tassorelli, C, Mancini, F, Balloni, L, et al. Botulinum toxin and neuromotor rehabilitation: an integrated approach to idiopathic cervical dystonia. Mov Disord. 2006;21:2240–2243.
174. Tedeschi, G, et al. Brain atrophy and lesion load in a large population of patients with multiple sclerosis. Neurology. 2005;65(2):280–285.
175. Thaut, MH, McIntosh, GC, Rice, RR, et al. Rhythmic stimulation in gait training for Parkinson’s disease patients. Mov Disord. 1996;11(2):193–200.
176. Third International Dystonia Symposium proceedings Miami, October 9-11, 1996.
177. Tierney, M, Yao, C, Kiss, A, et al. Neuropsychological tests accurately predict incident Alzheimer disease after 5 and 10 years. Neurology. 2005;64(11):1853.
178. Tinazzi, M, Frasson, E, Polo, A, et al. Evidence for an abnormal cortical sensory processing dystonia: selective enhancement of lower limb P37-N50 somatosensory evoked potential. Mov Disord. 1999;14(3):473–480.
179. Tolosa, E. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006;5(1):75–86.
180. Traynor, BJ. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology. 2006;67(1):20–27.
181. Tranchant, C, Bhatia, KP, Marsden, CD. Movement disorders in multiple sclerosis. Mov Disord. 1995;10:418–423.
182. Trapp, BD, Peterson, BS, Ransohoff, RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338(5):278–285.
183. Tremlett, H. Is late-onset multiple sclerosis associated with a worse outcome? Neurology. 2006;67(6):954–959.
184. Tsui, JC. Treatment of Parkinson’s disease. In: Calne DB, ed. Neurodegenerative diseases. Philadelphia: Saunders; 1994:573–582.
185. Uc, EY. Visual dysfunction in Parkinson disease without dementia. Neurology. 2005;65(12):1907–1913.
186. Uncini, A, DiMuzio, A, Thomas, A, et al. Hand dystonia secondary to cervical demyelinating lesion. Acta Neurol Scand. 1994;90:51–55.
187. Ursell, MR, O’Connor, PW. Natalizumab and other monoclonal antibodies. Neurol Clin. 2005;23(1):233–246. [viii].
188. Utz, U, McFarland, HF. The role of T cells in multiple sclerosis: implications for therapies targeting the T cell receptor. J Neuropathol Exp Neurol. 1994;53:351–358.
189. van de Pol, LA. Hippocampal atrophy in Alzheimer disease: age matters. Neurology. 2006;66(2):236–238.
190. Van Zandijcke, M. Cervical dystonia: some aspects of the natural history. Acta Neurol Belg. 1995;95:210–215.
191. Vinters, HV, Vonsattel, JG. Neuropathological features and grading of Alzheimer-related and sporadic CAA. In: Verbeek MM, deWaal RM, Vinters F, eds. Cerebral amyloid angiopathy in Alzheimer’s disease and related disorders. Dordrecht, Netherlands: Kluwer Academic Publishers; 2000:137–147.
192. Waxman, SG, deGroot, J. Correlative microanatomy. Norwalk, CT: Appleton & Lange, 1995.
193. Weinshenker, BG, Bass, B, Rice, GP. The natural history of multiple sclerosis: a geographically based study: II. Predictive value of the early clinical course. Brain. 1988;112:1419–1428.
194. Weinshenker, BG, O’Brien, PC, Petterson, TM, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46(6):878–886.
195. Whyte, S, Beyreuther, K, Masters, C. Rational therapeutic strategies for Alzheimer’s disease. In: Calne DB, ed. Neurodegenerative diseases. Philadelphia: Saunders; 1994:647–664.
196. Winkler, P., Bowker, B. & Brady, M. et al Mild idiopathic spasmodic torticollis treatment. Unpublished manuscript, Denver, 1997, Regis University.
197. Winkler, P., Olsen, L. & Maass, D. Surface EMG for treatment of mild idiopathic cervical dystonia: eliciting normal muscle activity by converting “sensory trick” to visual feedback. Unpublished manuscript, Denver, 1996, Regis University.
198. Wojcieszek, JM, Lang, AE. Hyperkinetic movement disorders. In: Coffey CE, Cummings JL, eds. Textbook of geriatric neuropsychiatry. Washington, DC: American Psychiatric Press; 1994:406–415.