Stroke
STROKE
Overview and Definition
Stroke, or cerebral vascular accident, often occurs in well-appearing adults as a sudden, devastating vascular event that results in destruction of surrounding brain tissue. Stroke is a leading cause of serious long-term disability, with estimated direct and indirect costs totaling $62.7 billion annually. Stroke is primarily a consequence of changes in both the function of the heart and in the integrity of the vessels providing blood to the brain.
Transient ischemic attack (TIA) has been the term used for focal neurologic symptoms that completely resolve within 24 hours. However, because the etiology is the same as stroke, and it is becoming more common for the symptoms to be regarded and named as stroke, resulting in hospitalization, closer observation and early use of imaging to determine level of brain damage are warranted. Because there is less microvascular bleeding in TIA than stroke, early management often includes blood thinners.
Cerebrovascular disease, the primary cause of stroke, is caused by one of several pathologic processes involving the blood vessels of the brain. The damage may be intrinsic to the vessel, or the stroke may originate remotely, such as when an embolus from the heart or extracranial circulation lodges in an intracranial vessel. The stroke may result from the rupture of a vessel in the subarachnoid space or intracerebral tissue. Fig. 32-1 shows the effects of different types of stroke on brain tissue.98
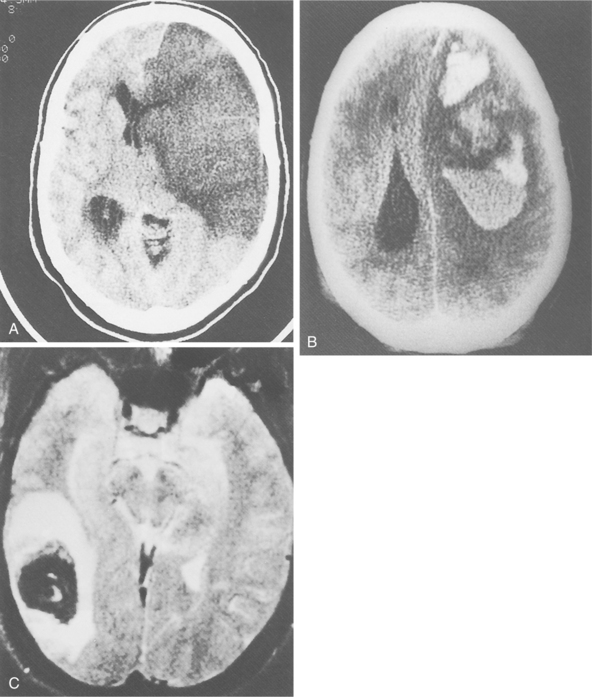
Figure 32-1 Radiographic images of the brain after stroke. A, An acute infarct with mass effect and compression of the ventricle. B, An acute intracerebral hemorrhage in the hemisphere. C, Amyloid angiopathy with acute hemorrhage; the edema surrounding the area results in a slight mass effect on the midbrain. (Reprinted from Ramsey R: Neuroradiology, Philadelphia, 1994, WB Saunders.)
Incidence
The average incidence rate of first strokes is 114 per 100,000 persons, which accounts for 750,000 first strokes each year in the United States. First strokes account for about 75% of acute events, and recurrent strokes account for about 25%. An estimated 700,000 persons in the United States will have a stroke, and approximately 160,000 will die from stroke. Currently close to 4 million stroke survivors are alive. Two thirds of strokes occur in low-income and middle-income countries, where the average age of stroke onset is 15 years below that in high-income countries.
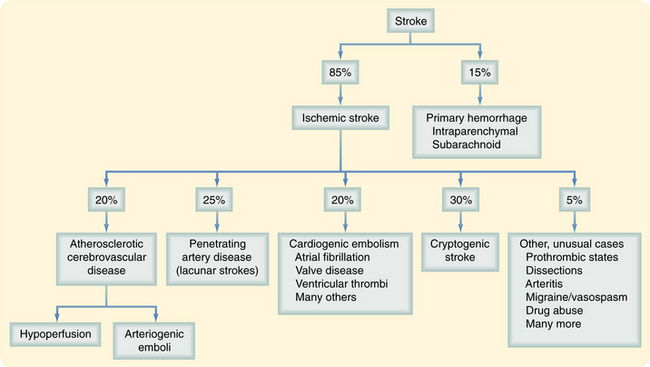
Mexican-American and African-American men have a 50% higher chance of having a stroke than do Caucasian men; this is associated with large artery occlusive disease, which is most pronounced in strokes at earlier ages.32 Sickle cell disease is increased in African-Americans and leads to a higher incidence of stroke.61 Stroke death rates are higher in the southeastern United States compared with other regions of the country; African-Americans, American Indians/Alaska Natives, Asians/Pacific Islanders, and Hispanics die from stroke at younger ages than Caucasians. The prevalence of stroke differs depending on the area of the country, ranging from 1.5% in Connecticut to 4.3% in Mississippi. Strokes occur more often in the spring.69 There are several stroke types with different etiology and risk factors; therefore management is driven by the stroke subtype. Fig. 32-2 shows the prevalence of stroke types.
Risk Factors
Risk factors for stroke can be divided into those that are potentially modifiable and those that are not. Among the nonmodifiable risk factors (age, race, and sex), age constitutes the greatest risk. The incidence of stroke doubles with every decade after age 55 years. Approximately 5% of men aged 65 to 69 years have had stroke compared with the 10% of men aged 80 to 84 years. Women have a 20% less chance of stroke than men, but age increases the risk just as with men. Incidence of stroke is increased with a family history of stroke, with both paternal and maternal influence.52 Cognitive function and incident cognitive decline appear to be associated with increased risk for stroke. Additional studies are needed to determine whether modification of stroke risk factors can reduce the cognitive decline that is often attributed to normal aging.
Stroke is a heterogeneous multifactorial disorder, but epidemiologic data provide substantial evidence for a genetic component to the disease and the extent of predisposition is unknown. The identification of NOTCH3 mutations has been found in individuals with cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Studies of sickle cell disease have drawn attention to the importance of modifier genes and of gene-gene interactions in determining stroke risk. There are probably many alleles with small effect sizes associated with multifactorial stroke. Genetic association studies on a wide range of candidate path- ways are currently underway. Fig. 32-3 shows the interaction of genetics, disease, and environment.16
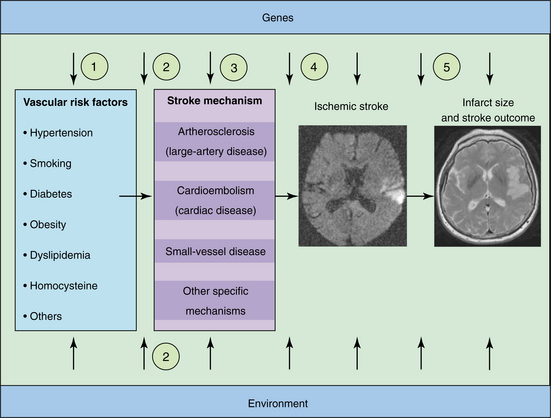
Figure 32-3 Genetic factors and ischemic stroke. Genetic factors may affect stroke risk at various levels. They could act through conventional risk factors (1), interact with conventional and environmental risk factors (2), or contribute directly to an established stroke mechanism such as atherosclerosis or small-vessel disease (3). Genetic factors could further affect the latency to stroke (4) or infarct size and stroke outcome (5). Similarly, environmental factors and interactions between genes and the environment could occur at various levels. (Reprinted from Dichgans M: Genetics of ischaemic stroke, Lancet Neurol 6(2):149-161, 2007.)
Hypertension (blood pressure above 160/95 mm Hg) is the most prevalent and modifiable risk factor for stroke. Decreasing diastolic blood pressure by 5 to 6 mm Hg decreases risk of stroke by up to 40%.20 Prehypertension describes blood pressure at the upper end of normal range (between 120/80 and 129/89 mm Hg), which in combination with other risk factors as described below may increase the risk of cardiovascular disease and stroke.
Various cardiac diseases have been shown to increase risk of stroke, including coronary heart disease, left ventricular hypertrophy, and cardiac failure. The stroke risk increases with the degree of stenosis. Death is more often from fatal coronary artery disease than stroke. The risk of stroke after myocardial infarction (MI) is 30% in the first month.98 If cerebral microembolism occurs after stroke, there is increased risk of an embolic event.65 Development of neurologic deficits preceded by brief symptoms of focal symptoms in the same vascular territory usually suggests atherothrombosis as the vascular mechanism. Cardiac valve abnormalities such as mitral stenosis and mitral annular calcification are moderate risk factors. Structural abnormalities of the heart, such as patent foramen ovale and atrial septal aneurysm increase risk. Atrial fibrillation is an important risk factor for stroke.83 It increases the stroke risk by a factor of six, and 8% of individuals over the age of 80 years have atrial fibrillation. See Chapter 12 for cardiac structure and function and related pathogenesis.111
Fibrinogen is a coagulation factor that has been demonstrated to be associated with increased stroke risk. Fibrinogen plays a crucial role in platelet aggregation. Platelets initiate thrombosis by attracting fibrin and other clot-forming substances. Conditions associated with increased fibrin deposition or increased blood viscosity include rheumatic heart disease, endocarditis, atherosclerosis, polycythemia, and thrombocytosis.
Leukemia is complicated occasionally by brain hemorrhages and microinfarcts. When the white blood cell count is very high (increased leukocrit), the white blood cells can pack capillaries, leading to microinfarcts and vascular rupture with small hemorrhages in the brain. Larger intraparenchymatous hemorrhages and subarachnoid hemorrhages (SAHs) are most often related to thrombocytopenia because of replacement of the bone marrow with leukocyte precursors.27
Diabetes mellitus, a common endocrine disorder, has long been established as a risk factor for stroke. Diabetes is known to cause large artery atherosclerosis, increased cholesterol levels, and plaque formation. The vasodilatory ability of the cerebral arterioles is reduced in long-standing type 2 diabetes.47
Cholesterol has long been considered a part of the stroke risk profile; however, the relation between raised lipid level and stroke remains unclear.94 High total cholesterol levels create increased risk of ischemic stroke. These include the large artery and lacunar subtypes that have atherosclerotic pathogeneses. Younger individuals and those with low high-density lipoprotein levels are at greater risk; higher levels of high-density lipoprotein cholesterol are associated with decreased risk of ischemic stroke. Low-density lipoprotein cholesterol levels currently are believed to be biochemical predictors of coronary artery disease associated with carotid athrerosclerosis.99
High plasma homocysteine levels and low levels of folate and vitamin B6 have been associated with increased risk of carotid disease and heart attack and therefore may have an effect on the risk of stroke. Homocysteine is a sulfur-containing acid formed during the metabolism of methionine. The use of folic acid, pyridoxine, and vitamin B12 has been reported to lower levels of homocysteine in the blood. Homocysteine-lowering treatment is cheap and well tolerated; it has been considered a rational approach in individuals at high risk for stroke. However, the strength of association of homocysteine with risk of cardiovascular disease may be weaker than had previously been believed. Clearly more studies are warranted.3
A number of nutrients have been suggested to be important to reduce the risk of stroke, including flavonoids, carotene, and folic acid.20 Beneficial effects of omega-3 fatty acid consumption on coronary artery disease, fatal and nonfatal MI, and stroke have been determined through many studies. Cardioprotective benefits have been found with consumption of even modest amounts of omega-3 fatty acids provided by an average intake of 1 to 2 oz of fish daily. Fish consumption five times per week decreased coronary artery disease mortality by 38%. Magnesium sulfate is believed to be neuroprotective, and extensive study has been done on its potential ability to preserve function if given within 8 hours of the onset of stroke. The results of these studies have not shown significant protection to this point.78
Significant obesity, particularly abdominal adiposity, worsens the prognosis of individuals with coronary disease. The protective effect of physical activity may be related to its role in controlling other risk factors such as hypertension, diabetes mellitus, and obesity. Reductions in plasma fibrinogens levels and platelet activity with corresponding increases in high-density platelet concentrations also are believed to add to the benefit. The beneficial effect of physical activity appears to be more apparent for men than women.
Cigarette smoking increases the risk of stroke by approximately 50%; the risk is directly related to the number of cigarettes smoked per day. Alcohol consumption has a direct dose-dependent effect on the risk of stroke. Three or more alcoholic drinks per day increase risk by 45%. Heavy consumption of alcohol—more than 14 drinks per week or more than four drinks per occasion in men and more than seven drinks per week or more than three drinks per occasion in women—causes increased risk through hypertension, hypercoagulable states, arrhythmia, and decreased cerebral blood flow. Evidence suggests that light to moderate drinking may have beneficial effects by increasing high-density lipoprotein cholesterol levels and decreasing platelet aggregation and fibrinogen levels, with a 32% decrease in risk.
Cocaine use is associated with hemorrhagic stroke by increased risk related to focal arterial vasoconstriction and occasionally to inflammatory vasculitis. Although the evidence remains inconclusive, other recreational drugs such as lysergic acid diethylamide (LSD) and marijuana are believed to increase the risk. Some concern exists regarding the use of over-the-counter cold medications, diet pills, ephedrine, and pseudoephedrine.
Young women have a low absolute risk of stroke, and use of oral contraceptives seems to be insignificant. However, the absolute risk of stroke is much greater in postmenopausal women simply because they are older. In older women, a modest relative increase in stroke risk when using oral contraceptives may produce a much larger absolute increase in risk of stroke. Thus differences in prescribing estrogen-containing compounds to premenopausal versus postmenopausal women appears be justified. Women with hypertension or a history of smoking have a higher absolute risk. Individuals who have had an ischemic stroke are at risk of a second stroke. Thus oral contraceptive use should be discouraged in women with prior stroke and should certainly be stopped in women who have had a stroke while taking oral contraceptives.
Recognition of the multiple risk factors that can interact to increase the probability of stroke is important. Use of risk profiles can assist in the ability to predict stroke in a single individual, such as the one established as a part of the Framingham study.110 The percentage of individuals with “no known risk factors” is declining, and the prevalence of high blood pressure, type 2 diabetes, and obesity among U.S. adults is increasing.
Clinical Manifestations
The first indication of the onset of stroke may be transient with focal symptoms, but it is the first warning that a stroke is about to occur. Early warning signs are listed in Box 32-1. Although the risk factors and early warning signs have been well publicized, individuals at highest risk do not appear to be aware of warning signs and may not consult a physician when the symptoms occur. Clinical manifestations are related to the type of stroke that occurs and are addressed later in the chapter.
Prognosis
The University of Oxford ABCD scale can be used to determine the chance of progression to a stroke with greater consequences (Box 32-2). Risk of second stroke varies between 3% and 5% in 5 years and is related to the concomitant cardiac and vascular disorders. The recurrence rate of stroke is highest in the first 30 days after the first stroke and remains higher for 1 year. The presence of atherosclerosis increases the chance of another thrombotic event. Men have 30% to 80% higher rates of recurrence than do women.84
Living with stroke can be a challenge, with the onset of impairments causing difficulty eating, urinary incontinence, and difficulty speaking or understanding language. Hemiparesis remains a long-term consequence in almost half of stroke survivors. Cognitive changes, anxiety, and confusion can be very disturbing for the individual and add to caregiver stress. Quality of life often is reported as poor.
Concomitant diseases of aging such as arthritis, diabetes, osteoporosis, and decreased plasticity of the nervous system associated with aging often make the recovery from stroke a challenge. Medical complications occur in up to 85% of stroke survivors and present potential barriers to optimal recovery. Infections occur in almost 25% of stroke survivors, primarily in the urinary tract and chest. Incidence of deep vein thrombosis and pulmonary emboli is increased. Falls resulting from impaired mobility are also prevalent, with serious injury reported in 5% of the reported falls. Approximately 75% of stroke survivors will have a fall within the first 6 months after stroke. Pain and depression are reported in more than 30% of stroke survivors.57 Urinary incontinence after stroke is a bad prognostic feature, both for survival and functional recovery.
Mortality associated with stroke has decreased in the past 20 years in all age groups. However, stroke still remains the number one cause of disability in the adult population. The mortality rate after stroke in African-Americans is higher than in whites and appears to be increasing.38
The Global Stroke Initiative is proposed as an international collaboration of the World Health Organization in partnership with the International Stroke Society and the World Federation of Neurology on behalf of other related professional civic groups. The primary focus will be to harness the necessary resources to implement existing knowledge and strategies for stroke prevention, especially in low-income and middle-income countries and in disadvantaged populations in high-income countries.6
Pathogenesis
Occlusion of Major Arteries.: Thrombosis and embolic occlusion of a major vessel are the most common causes of ischemic stroke. The heart is the most common source of embolic material as a result of damage to heart tissue from atherothrombotic disease. Atrial fibrillation is believed to cause thrombus formation in the fibrillating atrium. Left ventricular MI can be a source of emboli, especially in the first few weeks following the event when thrombus formation is most prevalent.88 Mitral valve prolapse or congenital septal defects are also sources of emboli. Formation of emboli during or after coronary artery surgery or intracardiac surgery is a well-recognized complication.
Artery-to-artery embolism, usually arising from an atherothrombotic lesion in the carotid or vertebrobasilar system, may lead to stroke. The emboli from this lesion may travel along the course of circulation and may cause occlusion in the smaller branches. The proximal internal carotid artery is the most common site of atherosclerosis and atherothrombosis leading to stroke. Other causes of emboli may be thrombus in the pulmonary vein, fat emboli in the blood, and tumor emboli from a neoplastic process.24 Sources of emboli are shown in Fig. 32-4.
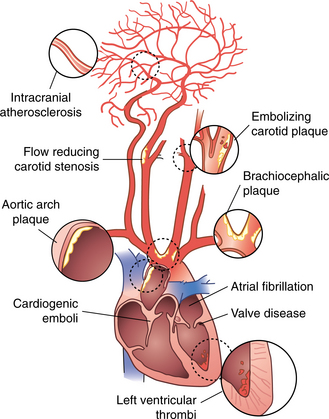
Figure 32-4 Cardiogenic and arterial atherosclerotic sources for stroke. (Reprinted from Townsend CM: Sabiston textbook of surgery, ed 17, Philadelphia, 2004, Saunders.)
Changes in the collateral pathways of the circle of Willis are apparent in response to internal carotid artery obstruction that may provide some protection against neurologic damage associated with occlusion. The anterior circle of Willis and the posterior communicating artery show increased diameter in some individuals when the internal carotid artery is blocked.40 Fig. 32-5 shows the distribution of the circle of Willis.
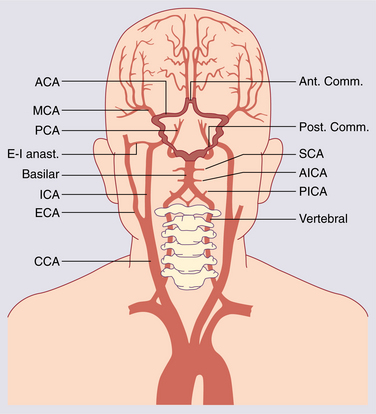
Figure 32-5 Extracranial and intracranial arterial supply to the brain. Vessels forming the circle of Willis are highlighted. ACA, Anterior cerebral artery; AICA, anterior inferior cerebellar artery; Ant. Comm., anterior communicating artery; CCA, common carotid artery; ECA, external carotid artery; E-I anast., extracranial-intracranial anastomosis; ICA, internal carotid artery; MCA, middle cerebral artery; PCA, posterior cerebral artery; PICA, posterior inferior cerebellar artery; Post. Comm., posterior communicating artery; SCA, superior cerebellar artery. (Modified from Lord R: Surgery of occlusive cerebrovascular disease, St Louis, 1986, C.V. Mosby.)
Secondary Vascular Responses.: When a cerebral artery is occluded, the formation of thromboemboli probably begins in the distal vessels of that artery. These presumed microvascular occlusions progressively increase in number and continue to impair blood flow in the brain. Cell death surrounding the area of blocked blood flow may be due to the squeezing effects of microvessels by the swelling of the astrocyte, one of the cellular support structures of the nervous system. Fig. 32-6 shows how the emboli can affect the brain tissue (see also the section on Cellular Dysfunction in Chapter 28). Astroglial swelling is one of the earliest cell changes induced by single-artery occlusion. The formation of fibrin in the grey matter surrounding the occluded vessel also may contribute to the lack of reperfusion of microvessels. Other factors include bleeding into the parenchyma, increased platelet aggregation, endothelial cells swelling in the walls of the vessels, and vasospasm.31,70
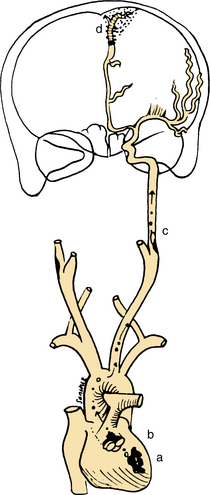
Figure 32-6 Examples of potential sources of embolism: cardiac mural thrombus (a); vegetation on heart valve (b); and emboli from carotid plaque (c). Also shown is an infarcted cortex from an embolism (d) in the area supplied by the terminal anterior cerebral artery. (Modified from Caplan LR: Caplan’s stroke: a clinical approach, ed 3, Boston, 2000, Butterworth-Heinemann.)
Secondary Neuronal Damage.: The tissue of the brain, or the parenchyma, is highly vulnerable to an interruption in its blood supply. When the cerebral blood flow falls below 20 mL/100 mg of tissue per minute, neuronal functioning is impaired. Neuronal death, or infarction, occurs when the brain receives less than 8 to 10 mL/100 mg/min. Frequently in an acute infarction a portion of the affected brain receives no blood, while a surrounding area receives sufficient blood from collateral circulation to maintain viability but not to sustain function. This territory has been termed the ischemic penumbra.100 Although the major injury to the neurons in the brain is the hypoxia-ischemia related to the occlusion of the artery causing cell death near the core, further damage to the brain tissue and neurons occurs as a secondary response. There is a characteristic uncoupling between cerebral blood flow and metabolism in the infarcted area. There is decreased perfusion relative to the necessary oxygen requirements. If blood flow to this ischemic area is restored before irreversible damage occurs, then the tissue will likely recover and resume normal function.
Distant from the ischemic and stroke sites are regions that also show alterations in metabolism despite being normal on anatomic imaging studies such as computed tomography or magnetic resonance imaging (see below). The most distinctive and characteristic example of such remote effects is crossed cerebellar diaschisis. There appears to be an uncoupling of oxygen consumption and glucose use that may reflect a change in brain metabolism caused by deafferentation. There are also other areas that are hypometabolic after a cortical infarct. These areas include the ipsilateral thalamus, the ipsilateral caudate nucleus, and the ipsilateral primary visual cortex (if the infarct is in the anterior visual pathways). There appears to be a decline in oxygen metabolism in the unaffected hemisphere from the acute to the subacute stage, which suggests a delayed effect from the corpus callosum fiber degeneration. There can be a delayed remote hypometabolism that develops during recovery that seems to be related to infarct size. For example, neurologic recovery does not appear to be a function of thalamic hypometabolism but appears to be influenced by prefrontal metabolism, possibly because this region is part of a network that has an important compensatory role in motor recovery.66
Changes in the neurotransmitter substances normally present in the brain can cause damage in the hypoxic-ischemic state believed to reflect mitochondrial dysfunction and energy failure of the damaged tissue. In keeping with its widespread role in central excitatory neurotransmission, glutamate is normally present throughout central nervous system grey matter. The glutamate is stored in synaptic terminals, and when it is released into the extracellular space, rapid uptake normally occurs, so the resting level of glutamate outside the synaptic terminal is minimal. After an ischemic event, the cells that normally clear the excess glutamate are compromised, and excess glutamate is found in the extracellular space. Depolarization of the postsynaptic cell occurs in response to this increase in glutamate. The ultimate effect of the excess glutamate is to facilitate the entry of calcium ions into the cells. Excessive numbers of calcium ions begin the process that causes cell death. Cell death is related to edema within the neuron associated with excess levels of calcium, water, and chloride. Catabolic enzymes are activated by the release of calcium ions and can cause damage of the proteins that support neurons, the glial cells. It appears that other mechanisms cause excess calcium during the ischemic process in addition to glutamate, including the dysfunction of the electrochemical gradient in the damaged membrane. Evidence exists of apoptosis, programmed cell death that occurs in response to the hypoxic damage.62
The changes in the perfusion pressure associated with hypoxia can also cause the endothelial cells to trigger the release of neurotoxic substances such as free radicals. Oxygen free radicals can initiate many destructive processes in the brain tissue. See Chapter 28 for further information on oxygen free radicals. The overall result of the hypoxic event is a chain of reactions, some that occur simultaneously, extending the damage and death of brain tissue beyond the area of vascular supply.
Clinical Manifestations
Syndromes.: Syndromes reflect the dysfunction associated with disruption of blood flow in specific areas of the brain.51,59 The syndromes are named according to the arteries that feed the specific areas. The syndrome can be partial or complete. When the blockage is in the more proximal component of the artery, the resulting area of hypoxia is greater than if the clot is lodged in a more distal part of the artery. Because of the collateral circulation provided by the circle of Willis, some areas of the brain are supplied by more than one artery. When one artery is blocked, circulation is provided to the tissues through the blood supply of other arteries. In this case, the clinical syndromes are not as extensive. The actual configuration of arteries is different in each individual, so the syndromes described here are not to be considered all encompassing. This is an overview of the types of symptoms that might be encountered when a particular artery is blocked.
Middle Cerebral Artery Syndrome.: If the entire middle cerebral artery is occluded at its stem, blocking both the penetrating and cortical branches, the clinical findings are contralateral hemiplegia and hemianesthesia, or the loss of movement and sensation on one half of the body. If the dominant hemisphere is affected, global aphasia, or the loss of fluency, ability to name objects, comprehend auditory information, and repeat language, is the result. (See the section on Parietal Lobe Syndromes in Chapter 28.)
Partial syndromes resulting from embolic occlusion of a single branch include brachial syndrome, or weakness of the upper extremity, and frontal opercular syndrome, or facial weakness with motor aphasia with or without arm weakness. A combination of sensory disturbance, motor weakness, and motor aphasia suggests that an embolus has occluded the proximal superior division branch and has infarcted large portions of the frontal and parietal cortices.
If Wernicke’s aphasia occurs without weakness, the inferior division of the middle cerebral artery supplying the temporal cortex of the dominant hemisphere has been occluded. Jargon speech and an inability to comprehend written and oral language are prominent features. Hemiplegia or spatial agnosia without weakness indicates that the inferior division of the middle cerebral artery in the nondominant hemisphere is involved. Fig. 32-7 represents the area of the middle cerebral artery.
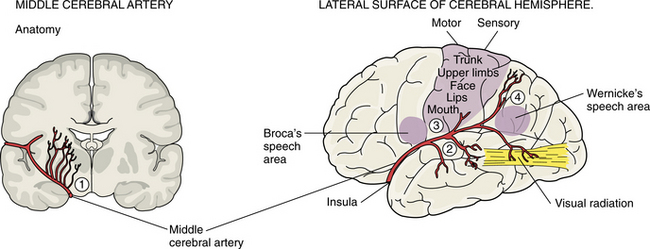
Figure 32-7 The middle cerebral artery is the largest branch of the internal carotid artery and the most common site of emboli. Its deep branches feed the internal capsule and basal ganglia. On the lateral surface the branches feed areas of the parietal, frontal, and temporal lobes. (Reprinted from Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.)
Anterior Cerebral Artery Syndrome.: Infarction in the territory of the anterior cerebral artery is uncommon and is more often the result of embolism than to atherothrombosis. Collateral flow is able to compensate for most occlusion of the artery so that dysfunction is minimal. If both segments of the artery arise from a single anterior cerebral stem, the occlusion affects both hemispheres. Contralateral hemiparesis and sensory loss are usually seen with the lower extremity more involved. Profound abulia, a delay in verbal and motor response, is common. Akinetic mutism also can result in significant disability. Fig. 32-8 represents the area of blood flow of the anterior cerebral artery.
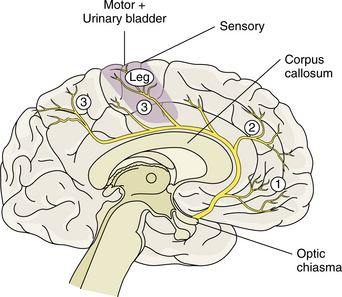
Figure 32-8 The anterior cerebral artery branches from the internal carotid. Deep branches supply the internal capsule and basal ganglia. Superficial branches supply the frontal and parietal lobes. (Reprinted from Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.)
Internal Carotid Artery Syndrome.: The clinical picture of internal carotid occlusion varies depending on whether the cause of ischemia is thrombus, embolus, or low flow. The cortex supplied by the middle cerebral territory is affected most often. Occasionally, the origins of both the anterior and middle cerebral arteries are occluded at the top of the carotid artery. Symptoms consistent with both syndromes result. With a competent circle of Willis producing adequate collateral circulation, the occlusion can be asymptomatic.
Posterior Cerebral Artery Syndrome.: If the proximal posterior cerebral artery is occluded, including penetrating branches, the areas of the brain that are affected are the subthalamus, medial thalamus, and ipsilateral (same side) cerebral peduncle and midbrain. Signs include thalamic syndrome, including abnormal sensation of pain, temperature, proprioception, and touch. Sensations may be exaggerated and light pressure may be interpreted as painful stimuli. This may develop into intractable, searing pain, which can be incapacitating. The anterior pattern consists mainly of perseverations and superimposition of unrelated information, apathy, and amnesia. After paramedian infarct, the most frequent features are disinhibition syndromes with personality changes, loss of self-activation, amnesia and, in the case of extensive lesions, thalamic dementia; this pattern may often be difficult to distinguish from primary psychiatric disorders, especially when neurologic dysfunction is lacking. After inferolateral lesion, executive dysfunction may develop but is often overlooked, although it may occasionally lead to severe long-term disability. After posterior lesion, cognitive dysfunction with neglect and aphasia are well known.12
If the posterior cerebral artery is completely occluded at its origin, hemiplegia results from infarction of the cerebral peduncle. Involvement of the red nucleus or dentatorubrothalamic tract can produce contralateral ataxia. When palsy of cranial nerve III occurs with contralateral ataxia, it is known as Claude syndrome. Third nerve palsy occurring with contralateral hemiplegia is known as Weber syndrome. Hemiballismus, or flailing of the extremity, usually results from a deep penetrating vessel causing infarct in the contralateral subthalamic nucleus. Paresis of upward gaze, drowsiness, and abulia (the lack of interest in movement) can be attributed to occlusion of the artery of Percheron. If the posterior cerebral stem is occluded, causing extensive infarction of the subthalamus, coma and decerebrate rigidity may result.
Peripheral supply of the posterior cerebral artery includes the temporal and occipital lobes. Occlusion of this component of the artery often affects the occipital lobe with homonymous hemianopsia, in which the visual field defect is on the side opposite to the lesion. Cortical blindness, the inability of the brain to record an image although the optic nerve is intact, is one of the visual disturbances that is seen with infarct in this region.
Medial temporal lobe involvement (including the hippocampus) can cause an acute disturbance in memory, particularly if it occurs in the dominant hemisphere. This resolves because memory has dual representation. Memory is represented on both sides of the brain; if one area is affected the intact side can compensate to a considerable extent. If the dominant hemisphere is affected and the infarct extends to involve the splenium of the corpus callosum, the individual may demonstrate alexia without agraphia, or impairment of reading without the impairment of writing. Agnosia, or difficulty in identification or recognition, affecting the ability to identify faces, objects, mathematical symbols, and colors, may occur. Anomia, impaired ability to identify objects by name, and visual hallucinations of brightly colored scenes and objects can occur with peripheral posterior cerebral infarction. Embolic occlusion of the top of the basilar artery can produce a clinical picture that includes any or all of the central or peripheral territory symptoms. Fig. 32-9 represents the area of blood flow of the posterior cerebral artery.
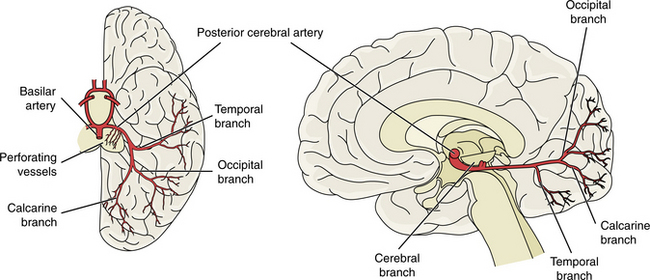
Figure 32-9 The posterior cerebral arteries branch from the basilar artery. The small perforating branches supply the midbrain structures and posterior thalamus. The temporal branch supplies the temporal lobe, and the occipital and calcarine supply the occipital lobe, including the visual cortex. (Reprinted from Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.)
Vertebral and Posterior Inferior Cerebellar Artery Syndrome.: Blood supply to the brainstem, medulla, and cerebellum is provided by the vertebral and posterior cerebellar arteries. Collateral circulation is provided by the bilateral component of the vertebral artery so that ischemia often is not manifested in the presence of atherothrombosis.
When infarction ensues, the lateral medulla and the posteroinferior cerebellum are affected, resulting in Wallenberg syndrome, which is characterized by vertigo, nausea, hoarseness, and dysphagia (difficulty swallowing). Other symptoms include ipsilateral ataxia (uncoordinated movement), ptosis (eyelid droop), and impairment of sensation in the ipsilateral portion of the face and contralateral portion of the torso and limbs. Individuals with lateral medullary infarction complain of numbness, burning, and cold in the face and limbs, with symptoms exaggerated by a cold environment, reflecting the spinal thalamic tract involvement.50 The onset of symptoms may be delayed for up to 6 months.43
A medial medullary infarction of the pyramid can result in contralateral hemiparesis of the arm and leg, sparing the face. If the medial lemniscus and the hypoglossal nerve fibers are involved, loss of joint position sense and ipsilateral tongue weakness can occur.
The edema associated with cerebellar infarction can cause sudden respiratory arrest from raised intracranial pressure (ICP) in the posterior fossa. Gait unsteadiness, dizziness, nausea, and vomiting may be the only early symptoms. Fig. 32-10 shows the area of distribution of the superior cerebellar, anterior inferior cerebellar, and posterior inferior cerebellar arteries.
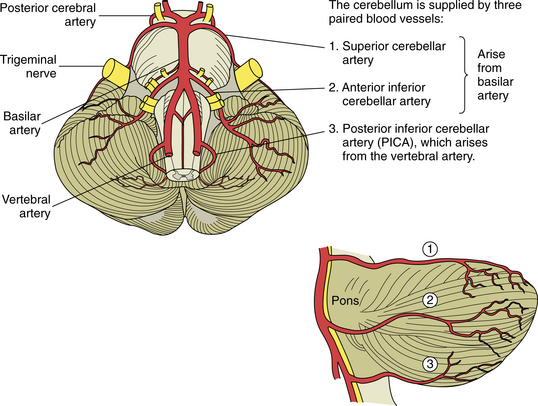
Basilar Artery Syndrome.: Atheromatous lesions can occur anywhere along the basilar trunk, but they occur most often in the proximal basilar and distal vertebral area. Ischemia as a result of occlusion of the basilar artery can affect the brainstem, including the corticospinal tracts, corticobulbar tracts, medial and superior cerebellar peduncles, spinothalamic tracts, and cranial nerve nuclei.
If the basilar artery is occluded, the brainstem symptoms are bilateral. When a branch of the basilar artery is occluded, the symptoms are unilateral, involving the sensory and motor aspects of the cranial nerves.
Superior Cerebellar Artery Syndrome.: Occlusion of the superior cerebellar artery results in severe ipsilateral cerebellar ataxia, nausea and vomiting, and dysarthria, or slurring of speech. Scanning speech, a drawn-out and monotone speech pattern, reflects damage to the cerebellum. Loss of pain and temperature in the contralateral extremities, torso, and face occurs. Dysmetria, characterized by the inability to place the extremity at a precise point in space, is common, affecting the ipsilateral upper extremity.
Anterior Inferior Cerebellar Artery Syndrome.: Principal symptoms include ipsilateral deafness, facial weakness, vertigo, nausea and vomiting, nystagmus (rhythmic oscillations of the eye), and ataxia. Horner syndrome ptosis, miosis (constriction of the pupil), and loss of sweating over the ipsilateral side of the face may occur. A paresis of lateral gaze may be seen. Pain and temperature sensation are lost on the contralateral side of the body.
Lacunar Syndrome.: Lacunar infarcts are small infarcts of the end arteries found in the basal ganglia, internal capsule and pons. The lacunar infarcts have the characteristics of ischemic necrosis and the cysts are surrounded by astrocytic gliosis, or scarring of the support structures of the brain.44 These small, cystic spaces resulting from healed ischemic infarcts are common in individuals with hypertension or diabetes. A large majority are asymptomatic, but in about 20% of cases a stroke syndrome occurs with a slowly progressive (over 24 to 36 hours) dysfunction of the cells in the area of the lacune.64 The lacunar syndrome is representative of the area of infarct in which the lacunae are predominant often in the deep structures of the brain and have their effect often on white matter. If the posterior limb of the internal capsule is affected, a pure motor deficit may result; in the anterior limb of the internal capsule, weakness of the face and dysarthria may occur. If the posterolateral thalamus is affected there is a pure sensory stroke. When the lacunae occur predominantly in the pons, ataxia, clumsiness, and weakness may be seen. Fig. 32-11 shows the areas of predilection for lacunae to develop.
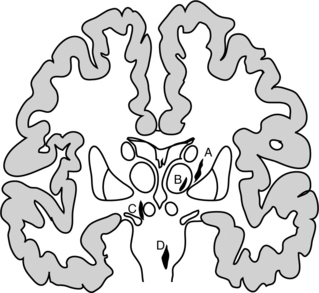
Figure 32-11 Usual sites of lacunar infarcts in the deep white matter. A, Internal capsule/putamen. B, Thalamus. C, Mesencephalon. D, Pons. (Reprinted from Pryse-Phillips W, Murray TJ: Essential neurology: a concise textbook, ed 4, New York, 1992, Medical Examination Publishing.) Medical Examination Publishing
MEDICAL MANAGEMENT
History of the neurologic event should be obtained, including timing, pattern of onset, and course. An embolic stroke occurs rapidly, with no warning. A more progressive and uneven onset is typical with thrombosis. The presenting symptoms will help to determine the location of the lesion.
Studies of the carotid artery and vertebral arteries are performed using ultrasound evaluation. Doppler ultrasound looks at the flow velocity of the blood through the artery. Plaque accumulation and ulceration can be identified by Doppler. Doppler studies are used to determine the need for carotid endarterectomy.37
Neuroimaging of the brain has become a standard procedure in the diagnosis of stroke. Computed tomographic (CT) scan is the fastest, most convenient and widely available test to use for the diagnosis and early treatment of acute stroke. It can confirm the diagnosis and rule out other pathologies and extent of the lesion. Fig. 32-12 shows how an acute stroke looks on CT. However, CT scans may be normal in the acute stage of an embolic stroke. Bleeding into the brain tissue is seen acutely in a hemorrhagic stroke. Displacement of brain structures, such as the ventricles, by edema sometimes can be seen early in a large infarct. In ischemic stroke, CT scans reveal the area of decreased density and loss of grey/white matter differentiation resulting from edema. Cortical lesions appear wedge shaped and deeper lesions appear to be round or oval. Potential for hemorrhagic transformation of the ischemic infarct can be seen on CT.75 Lacunar infarcts are sometimes visible on CT scans as small, punched-out, hypodense areas. Images of lacunae and be seen in Fig. 32-13. Identification of the penumbra and infarct core on hyperacute noncontrast and perfusion CT may lead to potentially more aggressive treatments related to reperfusion and to arrest progression of stroke damage in the early part of the stroke.74 Fig. 32-14 demonstrates how the use of new imaging techniques may assist in this goal.
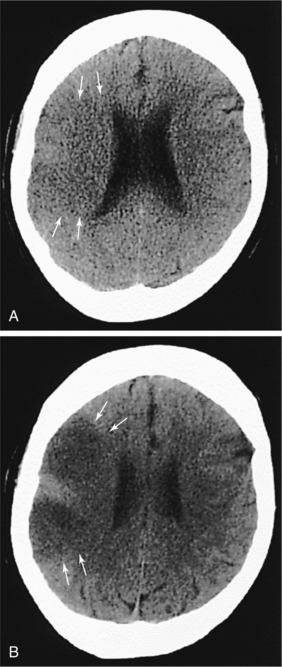
Figure 32-12 CT scan taken 2 hours, 50 minutes after large right middle cerebral artery occlusion. There are subtle, ultra-early ischemic changes, including loss of the grey-white interface (arrows) and subtle evidence of sulcal effacement. B, CT scan of same patient approximately 8 hours after symptom onset shows acute hypodensity (arrows) and more prominent sulcal effacement. (Reprinted from Marx JA: Rosen’s emergency medicine: concepts and clinical practice, ed 6, St Louis, 2006, Mosby.)
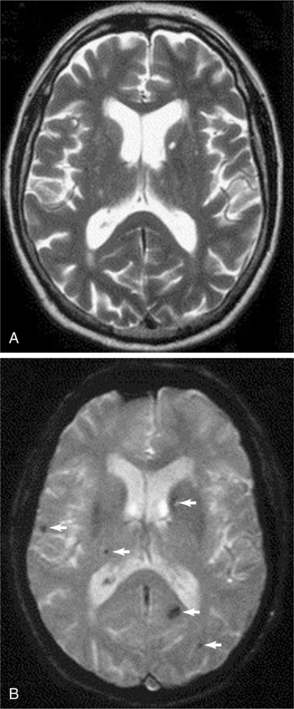
Figure 32-13 In these images, the left side of the brain is on the right of the panel. Axial T2-weighted fast spin-echo sequence (A) and corresponding axial T2*-weighted gradient echo sequence (B) from a 57-year-old man who presented with a left lacunar syndrome; his risk factors included hypertension and smoking. The T2-weighted fast spin-echo sequence shows several hyperintense foci in the cerebral white matter and basal ganglia but no microbleeds. The T2*-weighted gradient echo image shows several areas of focal signal loss consistent with microbleeds (arrows) in the right frontal lobe, right thalamus, left parietal lobe, and left caudate nucleus. (Reprinted from Werring DJ, et al: Cerebral microbleeds are common in ischemic stroke but rare in TIA, Neurology 65(12):1914-1918, 2005.)
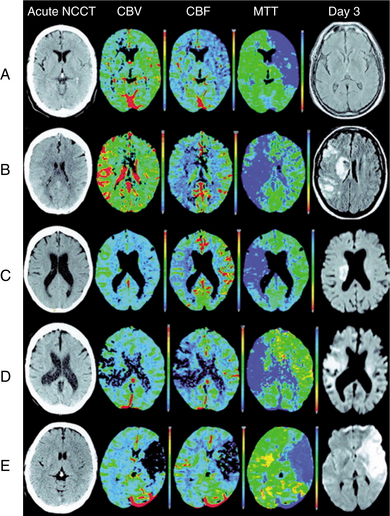
Figure 32-14 Patient A: Isolated focal swelling (IFS) on noncontrast CT (NCCT), hypoperfusion on mean transit time (MTT), and increased cerebral blood volume (CBV) on acute CT perfusion (CTP) maps, and no progression to infarction with subsequent major reperfusion. Patient B: IFS on NCCT, hypoperfusion on MTT, and increased CBV on acute CTP maps, but progression to infarction occurred without major reperfusion. Patient C: Hypoperfusion on MTT and increased CBV on acute CTP maps without any change apparent on acute NCCT. No infarction in cortical regions on follow-up with major reperfusion. Patient D: Hypoperfusion on MTT and decreased CBV on acute CTP maps without any apparent change on NCCT. Subsequent infarction present in reduced CBV regions on follow-up MRI. Patient E: Profound decrease in CBV and CBF on acute CTP maps with associated parenchymal hypoattenuation on NCCT. Extensive infarction on follow-up MRI. (Reprinted from Parsons M, Pepper EM, Bateman GA, et al: Identification of the penumbra and infarct core on hyperacute noncontrast and perfusion CT, Neurology 68(10):730-736, 2007.)
Magnetic resonance imaging (MRI) allows for the identification of an ischemic event within 2 to 6 hours of onset. The soft tissue contrast and multiplanar imaging capability offered by MRI have led to its wide acceptance as the method of choice for high-resolution brain imaging. Diffusion-weighted MRI (DWI) provides an indication of the brain tissue’s physiologic response to ischemia and can document the evolution of stroke. Because halting the evolution of the stroke is the therapeutic goal, DWI may be useful in the evaluation of therapeutic effectiveness. Perfusion imaging uses a tight bolus of paramagnetic contrast agent and a sequence of rapid MRI scans to detect the passage of the agent through the brain tissue.21 MRI stroke sequences can be used as a measure of ischemic penumbra and can help pinpoint potentially salvageable brain tissue, helping to identify who is going to be a good candidate for the later window of intervention using thrombolysis and who is not.86
Positron emission tomographic (PET) imaging has been of great benefit in advancing the understanding of the pathophysiology of cerebrovascular disorders. PET imaging allows for the detection of stroke earlier and with higher sensitivity than anatomic imaging with either MRI or CT. Furthermore, PET imaging has been useful in evaluating the extent of the functional damage because areas not immediately affected by the infarct may show hypometabolism or decreased blood flow. Initial stroke severity has been shown to correlate with the initially affected volume as determined by PET, whereas neurologic deterioration during the first week after stroke correlates with the proportion of the initially affected volume that infarcted, and functional outcome correlates with the final infarct volume. Crossed cerebellar diaschisis is seen as hypometabolism and hypoperfusion in the cerebellar cortex contralateral to the site of the infarct and usually occurs during the first 2 months after infarction (Fig. 32-15).66
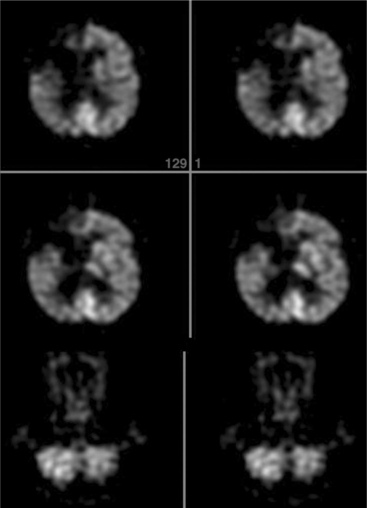
Figure 32-15 Fluorodeoxyglucose PET scan of a patient after embolic stroke in the distribution of the right anterior cerebral artery. There is severely decreased metabolism in the right frontal lobe extending to the midline. There is also crossed cerebellar diaschisis with decreased metabolism in the left cerebellum. (Reprinted from Newburg AB, Alavi A: The role of PET imaging in the management of patients with central nervous system disorders, Radiol Clin North Am 43(1):49-65, 2005.)
Cerebral angiography can be used in the absence of CT or MRI but is an invasive procedure and used only when other forms of imaging are not appropriate.
TREATMENT.
Treatment of individuals with ischemic stroke consists of managing the stroke and preventing further embolic strokes. Cerebral perfusion, or the blood flow around the area of the stroke, is the main concern when the cause is embolic. Blood pressure should not be lowered unless it is as high as 230/120 mm Hg. The goal is not to try to normalize the pressure, but to bring it down from dangerously high levels. If the blood pressure is low, raising it is appropriate in the first few hours after the stroke. An excessive rise in blood pressure may cause an increase in edema. When clinically stable, individuals with blood pressure over 140/90 mm Hg should be given medication to lower blood pressure. Unless contraindicated, use of diuretics and β-blockers should be the medication of choice. Angiotensin-converting enzyme inhibitors have been used with diabetes mellitus, heart failure, and MI.67
Emboli that lodge in the artery stem can cause edema. If not controlled the edema can spread and create pressure in the area of the cerebellum and brainstem. Even a small amount of edema in the cerebellum can cause respiratory arrest from compression of the brainstem and lead to coma and death. It is the most common fatal complication. Water restriction and agents that raise the serum osmolarity should be considered with the onset of significant edema.
Thrombolytic and antithrombotic agents form the cornerstone of ischemic stroke treatment and prevention.2 Recombinant tissue plasminogen activator is used for the emergent care of embolic stroke. Tissue plasminogen activator activates plasminogen to form plasmin, which actively digests fibrin strands, and is effective in dissolving the thrombosis or blood clot responsible for the blockage. By promoting early recanalization of occluded vessels and early reperfusion of ischemic fields, there is potential to salvage penumbral neuronal tissue. If it is received within 3 hours after the initial stroke, the person is 30% more likely to recover from the stroke. The greatest risk factor is the chance of hemorrhage and the inappropriate use in a stroke that is hemorrhagic. It must be determined on CT that the stroke is purely embolic; guidelines are established by the National Institute of Neurological Disorders and Stroke study.27 With the use of DWI imaging and further understanding of the status of the ischemic penumbra, the window of opportunity may become larger and therapies more effective.
Several prognostic factors must be considered for selecting candidates for intravenous thrombolysis. Younger age, absence of cardiac disease or diabetes, lower blood pressure on admission, lower neurologic score, absence of early ischemic parenchymal changes, large artery thrombus visible on baseline brain CT, and a developed collateral circulation are all factors associated with a more favorable outcome. Risk factors for developing brain hemorrhage include time to treatment, dose of thrombolytics, blood pressure level, severity of neurologic deficit, and severity of ischemia. Besides hemorrhage, potential complications of thrombolysis include reperfusion injury, arterial reocclusion, and secondary embolization due to thrombus fragmentation. Thus adequate hospital facilities and personnel are required for administration of thrombolytic therapy as well as for monitoring and managing potential complications. Following tissue plasminogen activator administration, blood pressure should be closely monitored and kept at less than 180/105 mm Hg and antithrombotic agents should be avoided for 24 hours.
Intracranial clot retrieval is now possible with the Mechanical Embolus Removal in Cerebral Ischemia (MERCI) retrieval system (Concentric Medical, Inc, Mountain View, CA). When outcomes using MERCI were compared with those in individuals who were treated and those who were not, good outcomes occurred in 49% versus 10%, respectively, and mortality rate was 25% versus 52%, respectively. The retriever device goes in as a straight wire that turns into a corkscrew when it comes out of the guide catheter that is screwed into the clot; a balloon is pumped proximal to the clot to prevent antegrade flow. The clot is then pulled out. Most stroke centers are now offering this system with trials done up to 8 hours after symptom onset when used with very large clots.91
PROPHYLAXIS
Anticoagulation therapy has played a prominent role in the prevention of acute infarction for several decades, and current research supports its use in high-risk individuals. Large, randomized trials have also highlighted the effectiveness and safety of early and continuous antiplatelet therapy in reducing atherothrombotic stroke recurrence. Large, randomized trials have shown the benefit of four different agents: (1) aspirin, (2) ticlopidine, (3) clopidogrel, and (4) dipyridamole.71
Aspirin has become the antiplatelet standard for individuals with acute ischemic stroke who are not receiving thrombolysis. Several other antiplatelet agents such as ticlopidine, clopidogrel, and aspirin-dipyridamole have been shown to be more effective. The prevention of cardioembolic stroke is best accomplished with oral anticoagulation, barring any contraindications. Antiplatelets such as aspirin are used to decrease risk of second MI and may reduce the chance of stroke after MI.
Clopidogrel, a platelet adenosine diphosphate receptor antagonist, appears to be an effective antiplatelet drug that has rare interactions with other medication, although it should be used with caution in conjunction with heparin or warfarin or nonsteroidal antiinflammatory drugs.19
Anticoagulation should not be used with high blood pressure or other risk factors of hemorrhagic stroke. Studies are underway to determine use in acute poststroke management.46 Heparin can be used prophylactically against deep venous thrombosis and pulmonary embolism.
Warfarin sodium (Coumadin, Panwarfin) appears to be about twice as effective as aspirin in the prevention of stroke in individuals with atrial fibrillation. Use of warfarin after MI has shown reduced stroke risk overall but increases the chance for hemorrhagic stroke. Persons older than 75 years have the highest risk of stroke and can benefit from anticoagulation. The risk of bleeding increases with age, and the ability to determine the risk/benefit ratio is complex. These medications are probably currently underutilized in the older individuals at risk for stroke.73 When there are contraindications to warfarin, aspirin is an alternative therapy. Fig. 32-16 shows an algorithm for treatment options in stroke.
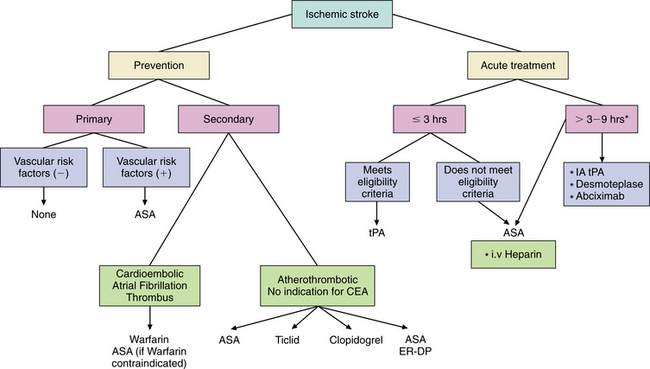
Figure 32-16 Prevention and treatment of ischemic stroke.*Experimental or ongoing trials. †Used in clinical practice for specific stroke syndromes. ASA, Aspirin; ER-DP, extended-release dipyridamole; IA tPA, intra-arterial tissue plasminogen activator; Ticlid, ticlopidine. (Reprinted from Ocava LC: Antithrombotic and thrombolytic therapy for ischemic stroke, Clin Geriatr Med 22(1):135-154, 2006.)
Lipid-Lowering Agents.
Cholesterol-lowering agents such as statins decrease the risk of stroke after MI. Studies show that the anti-stroke effects may be separate from the lipid-lowering properties through changes in the endothelium, inflammatory response, plaque stabilization, and thrombus formation. Several organizations have endorsed the use of statins for stroke prevention. The United States Food and Drug Administration has added ischemic stroke as an indication for statin therapy. The use of statins, in accordance with National Cholesterol Education Program Adult Treatment Panel III guidelines, is endorsed by the American Heart Association and American Academy of Neurology for both primary and secondary prevention of stroke.90
PET studies have also been used to monitor the success of various treatment regimens. PET has been used to evaluate the effects of thrombolytic therapy in acute stroke and has found that critically hypoperfused tissue can be preserved by early reperfusion and that large infarcts can be prevented by early reperfusion to viable tissue. In the future, functional imaging modalities that could eventually include tracers for neuronal integrity might be used to help in the selection of individuals for thrombolytic therapy, possibly permitting the extension of the critical time period for inclusion of individuals to aggressive stroke management strategies.66
Neuroprotection.
Medications aimed at creating neuroprotection to decrease the amount of cell death secondary to excitotoxicity are being developed, and clinical studies are underway.25 Approaches directed at presynaptic reduction of pathologic glutamate release may control the damage caused by excess extracellular glutamate. Reducing the amplification of excitotoxic calcium ion release also may help control cell death. In this category are endogenous growth factors, which may improve recovery from calcium overload and appear to improve outcome after focal brain ischemia. Enhancing gamma-aminobutyric acid activity may limit the neurotoxic effects of increased glutamate. Serotonin agonists appear to decrease infarction size in animal studies.25 Clinical trials have been disappointing for many of these medications.
A tetracycline derivative, minocycline, reduces inflammation and appears to protect against focal cerebral ischemia after stroke. It is most effective when started before ischemia develops but can be effective after the onset of ischemia. It has no therapeutic effect on astrogliosis or spreading depression but may provide some protection from glutamate toxicity.113 Timing is critical in the administration of these drugs. The window of opportunity may be 2 to 6 hours after infarction. These antiexcitotoxic therapies may be suitable for either hemorrhagic or ischemic stroke and someday may be given by paramedics before full neurologic evaluation.103
Nerve Growth.
Animal studies show positive results when stem cells have been implanted into the brain. Although the ability to repair damaged tissue and reform neural connections is limited when vast amounts of parenchyma are lost, seeded lesions show the potential for growing and differentiating into neurons. Enhancement of neural processes, reformation of cortical tissue, and promotion of connectivity all appear to be possible.
Surgical Intervention.
Carotid endarterectomy is the treatment of choice for low-flow or embolic TIA in individuals less than 80 years old.4 If stenosis is greater than 70% in a sclerotic lesion at the origin of the internal carotid artery, endarterectomy is indicated. Because women have arteries that are 10% smaller than men to begin with, the absolute size of the artery becomes significantly smaller with 70% occlusion. Therefore this equation may need to be looked at differently in women.
Careful selection of candidates for surgery is essential, as is an experienced team of surgeons and other health care providers to manage the postoperative course. Controversy surrounds the role of prophylactic endarterectomy in persons with asymptomatic extracranial carotic stenosis.22
Control of Symptoms.
Pharmacotherapy of spasticity in stroke is controversial. Weakness of the extremities can result, and if the spasticity is contributing to stability, that may be lost with use of medications. Baclofen and benzodiazepines work at the level of the spinal cord; dantrolene works on the muscle fibers.
Treatment to decrease the firing of specific muscles with botulinum toxin gives more discrete control of the choice of muscle to be injected. Choosing the appropriate muscle or group of muscles is critical in a successful outcome. The effects of botulinum toxin usually last for approximately 3 to 6 months, so it is a temporary solution.
Urinary incontinence can be a disabling sequela of stroke. Urge incontinence is treated with behavioral therapy and anticholinergics. An areflexic bladder can be managed with self-catheterization or use of a Foley catheter.
Depression after stroke is common and does not appear to be related to the area of lesion. Depression after stroke responds to treatment and should be guided by the other concomitant medical conditions and the side effects of the particular medication. Use of tricyclic antidepressants shows improvement within 3 to 6 weeks and should be continued for a minimum of 6 months.39
PROGNOSIS.
The prognosis for survival after cerebral infarction is better than after cerebral or SAH. Loss of consciousness after an ischemic stroke implies a poorer prognosis than remaining conscious. Individuals with ischemic stroke are at risk for other strokes or MIs. The risk factors and type of damage related to the stroke syndrome relate to degree of disability and mortality.
Neurobiochemical markers of brain damage are being studied to determine a relation with outcome after stroke. Two such markers, protein S-100B and neuron-specific enolase, show a relation between the blood level 2 to 4 days after stroke and functional impairment and discharge from acute level of care.112
Recovery from stroke is the fastest in the first few weeks after onset, with the most measurable neurologic recovery (approximately 90%) in the first 3 months.72 However, movement patterns can continue to be influenced by intervention with goal-directed activities, and repetition of movement appears to improve the speed and control of the movement in the individual up to 5 or more years after stroke. (See Special Implications for the Therapist, Stroke Rehabilitation, later in this chapter.)
Intracerebral Hemorrhage
Intracerebral hemorrhage (ICH) is bleeding from an arterial source into brain parenchyma (often referred to as an intraparenchymal hemorrhage) and is regarded as the most deadly of stroke subtypes. Primary ICH describes spontaneous bleeding in the absence of a readily identifiable precipitant and is usually attributable to microvascular disease associated with hypertension or aging. Secondary ICH occurs most often in association with trauma, impaired coagulation, toxin exposure, or an anatomic lesion. Chronic hypertension causes fibrinoid necrosis in the penetrating and subcortical arteries, weakening of the arterial walls, and formation of small aneurysmal outpouchings, or microaneurysms, that predispose to spontaneous ICH. Bleeding usually arises from the deep penetrating arteries of the circle of Willis, including the lenticulostriate, thalamogeniculate, and thalamo-perforating arteries and perforators of the basilar artery. Acute rises in blood pressure and blood flow can also precipitate ICH even in the absence of preexisting severe hypertension. A ruptured vascular malformation is the second most common cause of ICH.
Bleeding is limited by the resistance of tissue pressure in the surrounding brain structures. If a hematoma is large, distortion of structures and increased ICP cause headache, vomiting, and decreased alertness. Because the cranial cavity is a closed system, enlargement of a hematoma or development of severe edema may shift brain tissues into another compartment, or herniate, and cause deterioration in the clinical condition.
Supratentorial ICH, so named because it occurs above the cerebellar tentorium, is classified as being lobar (i.e., involving the hemispheres of the cerebrum) or deep (i.e., implying involvement of structures of the midbrain, such as the thalamus, putamen, or caudate nucleus). Infratentorial, below the tentorium, refers to involvement of either the brainstem, usually the pons, or cerebellum.85
Incidence
The incidence of ICH is low among persons younger than 45 years, and it increases dramatically after the age of 65 years. In one study the incidence of ICH doubled with each advancing decade until age 80 years, after which the incidence became 25 times higher.84 ICH tends to occur more frequently in men. In the United States, African-Americans are more likely to have an ICH than are whites. Worldwide rates are higher in Asian populations than in Western populations. ICH is a major cause of morbidity and death and accounts for 10% to 15% of all strokes in whites and about 30% of strokes in African-Americans and individuals of Asian origin. Locations of hypertensive ICHs are the putamen (40%), lobar (22%), thalamus (15%), pons (8%), cerebellum (8%), and caudate (7%). Figs. 32-17 and 32-18 represent the areas most likely to be involved in ICH and occurrences.
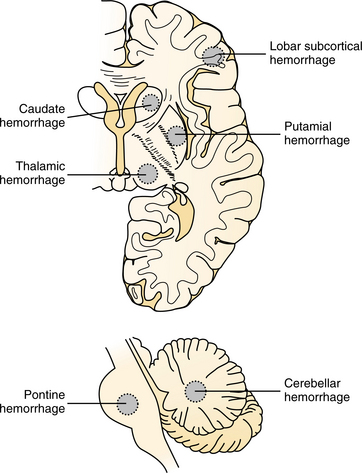
Figure 32-17 Horizontal cerebral section (top) and sagittal brainstem section (bottom) showing most common sites of ICH. (Modified from Caplan LR: Caplan’s stroke: a clinical approach, ed 3, Boston, 2000, ButterworthHeinemann.)
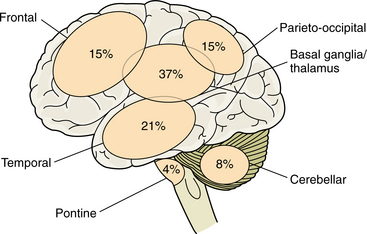
Figure 32-18 Sites of predilection of ICH. (Modified from Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.)
Spontaneous ICH can also occur in association with the prescription of anticoagulants, primary or metastatic brain tumors or granulomas, and use of sympathomimetic drugs. Aneurysms rarely bleed only into the brain, but when they do, they cause a local hematoma near the brain surface.
Etiologic and Risk Factors
Spontaneous ICH in the parenchyma of the brain usually is from an anomaly of the vessel structure or changes brought on by hypertension. Hypertension represents the single most important modifiable risk factor for ICH. Cerebral amyloid angiopathy (CAA) causing abnormal changes in the vessels of the brain accounts for approximately 10% of ICHs. CAA is recognized as an important cause of ICH in elderly persons.29
Excessive use of alcohol has been associated with massive spontaneous ICH. Alcohol has a number of acute and chronic effects that may contribute to hemorrhagic stroke, such as direct effects on cerebral vessels, hypertension, and impaired coagulation. Cocaine and amphetamine use is acknowledged as an important cause of ICH.
ICH is the most important adverse effect of thrombolytic therapy. Hemorrhage from fibrinolytic agents can occur within 12 to 24 hours and are typically lobar, occurring in the cortex and subcortical white matter.89
Long-term anticoagulant therapy is associated with an increased risk for ICH. Many individuals with anticoagulant-associated ICH are also hypertensive, so that the extent of increased risk is difficult to clearly identify independently.83
Use of other medications in conjunction with thrombolytic therapy may place individuals at risk for ICH. A number of drugs, such as nonsteroidal antiinflammatory agents, nitrates, and propranolol, may affect platelet function and contribute to bleeding.114
Other possible risk factors include liver disease, prior ischemic stroke, and cigarette smoking. Obesity, sickle cell anemia, mitral valve prolapse, patent foramen ovale, and polycythemia have been identified as possible risk factors. More research in this area is ongoing and will provide more insight into the relation of chronic disease and lifestyle to ICH.
Pregnancy may increase the risk of ICH. Eclampsia accounts for more than 40% of ICHs in pregnancy and is a common cause of death secondary to eclampsia.
Pathogenesis
Histopathologic changes in the cerebral microvasculature of hypertensive individuals include processes that affect both the contents and the walls of the blood vessels of the brain. These changes are seen in small cerebral arteries and arterioles where they branch, and they are more severe in the small penetrating vessels in the deep white matter than in cortical vessels of similar size. Changes are more severe distally than proximally. Smooth muscle cells are progressively replaced by collagen (hyalinization). Altered permeability of the vessel wall leads to fibrinoid changes in the vessel wall. This results in accumulation of proteinaceous material and fat deposits on the subintimal wall. The vessel wall becomes prone to leakage or rupture.108
Those with hypertension have a substantial reduction in the percentage of smooth muscle in the vessel wall. This decreased smooth muscle mass most likely represents structural weakening, resulting in rupture and hemorrhage. Necrosis of the endothelium may be a result of vessel ischemia. The changes in smooth muscle and the thickening of the intimal wall increase the metabolic requirements and impede the flow of oxygen to the outermost part of the vessel wall.
CAA is characterized by protein fibrils in the arterioles and small cerebral arteries and is formed by aberrant protein synthesis. Amyloid replaces smooth muscle in the media, separating the elastic membranes. Lymphocytic infiltrates, hyaline arteriole degeneration, and fibrinoid necrosis are characteristic changes in the vessel wall. The parenchymal changes seen in CAA reflect the consequences of the vascular pathology and direct deposition of amyloid in the brain tissue. Brains with evidence of CAA frequently demonstrate periventricular demyelination, believed to be caused by ischemia from amyloid deposition in the vessels supplying the deep white matter.30,39
In drug-related ICH, some underlying vascular pathologic lesion may be present, such as an arteriovenous malformation (AVM) or chronic vasculitis. The ICH occurs as a result of a sudden increase in blood pressure triggered by the drug. The proposed mechanism for the increased incidence of ICH in the individual with increased alcohol ingestion is decreased circulating levels of clotting factors produced by the liver. Thrombocytopenia, which is often associated with alcoholism, may underlie or potentiate hemorrhage.35
Hemorrhagic transformation, or conversion of an ischemic cerebral infarction, refers to secondary bleeding believed to occur either with early reperfusion into a damaged vascular bed with impaired autoregulation or as a result of development of collateral circulation into the same vascular bed.
When hemorrhage occurs, it spreads along a path of least resistance, primarily following the fiber tracts of the white matter. Grey matter, with its dense cell structure, is more resistant to the shearing forces of the growing hematoma and is more likely than white matter to be compressed rather than infiltrated by the spreading hematoma. Edema forms in the parenchyma surrounding the hematoma. Blood is reabsorbed by macrophages at the periphery of the hemorrhage, leaving a cavity surrounded by necrotic tissue. This process usually takes weeks to months.107
Clinical Manifestations
Neurologic symptoms occur gradually in most cases, representing the expansion of the hematoma. In some cases (approximately 30%) onset is sudden, which is also characteristic of an ischemic stroke. The earliest signs relate to blood issuing into parenchymatous structures. For example, a hematoma in the left putamen and internal capsule would first cause weakness of the right limbs; a cerebellar hematoma would cause gait ataxia. As the hematomas enlarge, the focal symptoms increase. If the hematoma becomes large enough to raise ICP, headache, vomiting, and decreased alertness develop. Some hematomas remain small and the only symptoms relate to the focal collection of blood. Once the condition is stabilized, the symptoms improve in parallel with the resorption of the hematoma.
Although headache is an important symptom of ICH, it is present in severe form in only 30% to 40% of cases. Headache is most common as a sign of superficial and large hemorrhages.
The incidence of seizure correlates with the location of the hemorrhage. Cerebral cortex hemorrhage causes the most prevalent seizure activity. Two thirds of the seizures are generalized and one third are focal. Focal seizures affect the body on the contralateral side (see Chapter 36). The level of consciousness at onset is unrelated to the occurrence of seizure.
Syndromes.: Syndromes associated with ICH are representative of the area of bleed and reflect brain activity of the particular site.8 Table 32-1 gives typical signs in individuals with intracerebral hemorrhages at various sites.
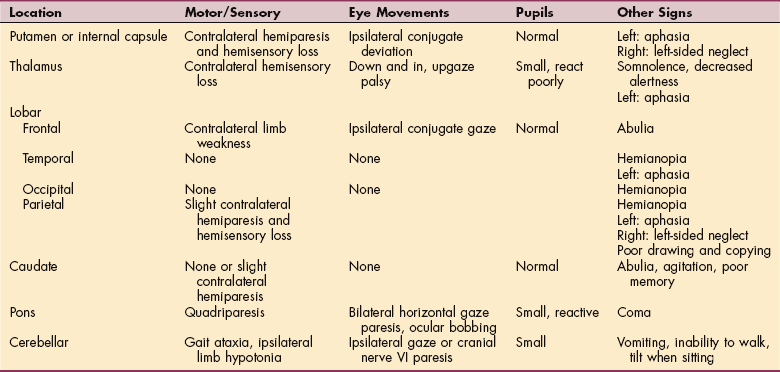
Putamen.: Approximately 50% to 80% of hemorrhages occur in the putamen. The result is contralateral sensorimotor deficit resulting from its proximity to the internal capsule. Small putamen hemorrhages may mimic lacunar syndromes, such as pure motor weakness. A type of aphasia may be present that mimics Broca aphasia when the lesion is in the mid-putamen. Wernicke’s aphasia is seen with a posterior putamen lesion. Pupillary abnormalities, visual field loss, and oculomotor deficits are common. Conjugate gaze deviation toward the side of the lesion may be present. Abulia and motor impersistence are seen when the anterior putamen is affected. Headache and vomiting also occur in about 25% of cases.
Thalamus.: Sensory losses, or dysesthesias, are common with thalamic hemorrhage, and some motor deficit occurs secondary to internal capsular involvement. The lateral thalamus abuts the posterior limb of the internal capsule. Oculomotor dysfunction also is seen, the most frequent abnormalities being vertical gaze palsies, often with downward eye deviation and convergence spasm. Constriction (miosis) of the pupil is seen in 50% of cases. In dominant hemisphere thalamic lesions, aphasia, disorientation, and memory disturbances may be seen. With nondominant lesions, apraxia (impairment of a learned motor activity) may exist. Midline hematomas are associated with alterations in the level of consciousness during the acute phase followed by prefrontal signs, such as change in character, speaking to oneself, memory disturbance, and impaired learning. Although not as common, the individual may experience symptoms such as headache, nausea, and vomiting to the same degree as with putamen hemorrhage.
Cerebellum.: A hallmark of cerebellar hemorrhages is ataxia. Additional symptoms may be nausea and vomiting, and dizziness with nystagmus and vertigo often is present. The individual may be dysarthric. Brainstem signs, such as facial paresis, can be present with a hemorrhage that extends to the brainstem. The signs of cerebellar hemorrhage should be carefully monitored, as the progression to compression of vital structures in the region of the fourth ventricle and medulla can be rapid and can produce life-threatening changes.
Pons.: Brainstem hemorrhages commonly arise in the midline of the pons, leading to coma, quadriparesis, and nonreactive pupils with absent horizontal eye movement. Lateral pontine hemorrhage can result in extraocular paresis with deviation away from the side of the lesion. Quick, downward jerks of the eye occur with a slow, upward drift. Pupils are small but reactive. Contralateral sensory and motor symptoms and ipsilateral cerebellar signs also are seen.
Caudate.: Caudate hemorrhages can rupture into the ventricles and therefore have a presentation like that of a SAH. Headache, vomiting, and loss of consciousness may occur. The internal capsule may be involved, causing sensorimotor involvement.
Internal Capsule.: Internal capsule hemorrhages often result in a pure motor, pure sensory, or sensorimotor stroke with ataxia. The internal capsule is a white matter tract that is situated lateral to the basal ganglia as it connects to the cerebral cortex.
Lobar.: Lobar hematomas are centered in the immediate subcortical white matter. Symptoms are lobe-specific (see the section on Clinical Manifestations of Ischemic Stroke in this chapter and Higher Brain Disorders in Chapter 28). Seizures are more common with lobar hemorrhages than with deeper bleeds.
MEDICAL MANAGEMENT
The availability of CT allows for prompt diagnosis of ICH. The specific area of damage can be imaged and the amount of blood identified. CT also has documented acute clot retraction progression of hyper tensive ICH and early hemorrhagic infarction. It accurately documents the size and location of the hematoma, the presence and extent of any mass effect, and the presence of hydrocephalus and intraventricular hemorrhage. CT scans should be performed immediately in individuals suspected of having an ICH. Follow-up CT scans are requested when there is a change in clinical signs or state of alertness in order to monitor changes in the size of the lesion and ventricular system and to detect important pressure shifts. If the clinical syndrome and CT findings are typical of hypertensive hemorrhage in the basal ganglia, caudate nucleus, thalamus, pons, or cerebellum, angiography is usually not necessary. If the hemorrhage is in an atypical location, the individual is young and not hypertensive, angiography is indicated to exclude an AVM, aneurysm, vasculitis, or tumor. Individuals who have ICH after cocaine use have a high likelihood of vascular malformations and aneurysms and need angiography. Particular attention should be directed to the presence of a coagulopathy. A drug screen should be obtained to evaluate use of sympathomimetics if substance abuse is suspected. Increased sympathetic outflow due to the hemorrhage may lead to an increase in dysrhythmias. Dysrhythmias also may signal impending brainstem compression from an expanding hemorrhage.
MRI can provide multiplanar views and can discriminate subtle tissue changes and rapidly flowing blood. However, it is of limited usefulness in the first 24 hours after ICH. MRI has the capability to detect previous hemorrhage, and it can image the posterior fossa more clearly than can CT.
Prothrombin time, partial thromboplastin time, and platelet count should be performed in all individuals to rule out a bleeding disorder. Coagulation factor deficiencies can be detected by evaluation of liver enzymes. Bleeding time, platelet aggregation studies, and fibrinogen assay also can be indicators of disorders related to possible repeat hemorrhage.
The differential diagnosis for ICH is similar to that of ischemic stroke and includes migraine, seizure, tumor, abscess, hypertensive encephalopathy, and trauma. Hypertensive encephalopathy and migraine also can present with headache, nausea, and vomiting. Although focal neurologic signs are uncommon, they may occur with these entities. With hypertensive encephalopathy, individuals usually have marked elevation in blood pressure and other evidence of end-organ injury, including proteinuria, cardiomegaly, papilledema, and malignant hypertensive retinopathy. These individuals usually improve significantly with treatment of hypertension. Migraines often are associated with an aura, and the individual often has a history of similar headaches.
The difference between ICH and labyrinthitis can be especially difficult to determine in the elderly. The abrupt onset of vertigo, vomiting, and nystagmus can represent a peripheral process, such as labyrinthitis, or a central process, such as cerebellar or brainstem infarct or hemorrhage. Age older than 40 years and a history of hypertension or other risk factors for ICH increase the possibility of a cerebellar hemorrhage. Findings specifically referable to the brainstem must be sought; these include hiccups, diplopia, facial numbness, dysphagia, and ataxia. Vertiginous individuals often have a strong desire to remain immobile with their eyes closed, but this must not preclude a thorough cranial nerve and cerebellar examination, including gait. Gross ataxia should be present with cerebellar stroke and absent with labyrinthine disease. A head CT scan should be strongly considered in individuals older than age 40 years to assist in differentiating labyrinthitis and cerebellar hemorrhage.
A screen for toxic substances in the blood should be performed, especially in the younger population. Acquired immunodeficiency syndrome should be considered as a possible cause of ICH. Biopsy of brain tissue can be diagnostic of CAA, cerebral vasculitis, and neoplasm. Evaluation of cerebrospinal fluid may indicate levels of toxicity; however, individuals with increased ICP are at risk during the procedure for possible herniation causing compression of the brainstem.
TREATMENT.
The acute reduction of elevated blood pressure is advisable and is most readily accomplished with rapid-acting, potent antihypertensive medication along with effective control of increased ICP, which exacerbates blood pressure elevation.49 A major issue in the management of ICH is control of edema (see the section on Treatment of Ischemic Stroke in this chapter). Anticonvulsant therapy should be considered with lobar hemorrhage.
The frequency of ICH may increase with the use of long-term anticoagulation therapy. Treatment with vitamin K is useful to correct an elevated prothrombin time; however, this takes 12 to 24 hours. Fresh-frozen plasma immediately restores diminished clotting factors. Protamine sulfate is the treatment of choice for reversal of the heparin effect. Thrombocytopenia responds to plasma infusion and plasma exchange.
An individual with a potential ICH requires rapid assessment and transport to a facility that has CT scanning capability and intensive care management. The prehospital management is similar to that for ischemic stroke. The circumstances surrounding the event and other concomitant medical conditions also should be ascertained. An evaluation of the initial level of consciousness, Glasgow Coma Scale, any gross focal deficits, difficulty with speech, clumsiness, gait disturbance, or facial asymmetry should be noted.
Supportive care involving attention to airway management and perfusion is of the highest priority. Individuals with hemorrhagic stroke are more likely to have an altered level of consciousness that may progress rapidly to unresponsiveness, requiring emergent endotracheal intubation. Intravenous access and cardiac monitoring should be initiated. Evaluation of blood glucose and appropriate dextrose and naloxone administration should be considered in any patient with altered mental status.
There is disagreement regarding optimal blood pressure management in a individual with ICH. Hypertension may cause deterioration by increasing ICP and potentiating further bleeding from small arteries or arterioles. Hypotension may decrease cerebral blood flow, worsening brain injury. In general, recommendations for treatment of hypertension in individuals with ICH are more aggressive than those for individuals with ischemic stroke. The current consensus for ICH is to recommend antihypertensive treatment with parenteral agents for systolic pressures greater than 160 to 180 mm Hg or diastolic pressures greater than 105 mm Hg. Treatment for lower blood pressures is controversial. Disadvantages include the need for careful monitoring (ideally with an indwelling arterial catheter) and the theoretical risk of worsening the hemorrhage secondary to the vasodilatory effects of nitroprusside on cerebral vessels. Labetalol is another therapeutic option.
Hyperventilation and diuretics such mannitol move fluid from the intracranial compartment, thereby reducing cerebral edema. These interventions should not be used prophylactically. Although this effect may be temporarily helpful in the acute setting, the brain tissue re-equilibrates, and rebound swelling can occur and worsen the individual’s clinical status. These agents also can cause dehydration and lead to hypotension. The use of steroids in cerebral hemorrhage, previously a common practice, may be harmful and is not recommended. Other experimental modalities include barbiturate coma and hypothermia. Seizure activity can cause neuronal injury, elevations in ICP, and destabilization of an already critically ill individual. In addition, nonconvulsive seizure may contribute to coma. Seizure prophylaxis (18 mg/kg phenytoin) should be considered for individuals with ICH, especially individuals with lobar hemorrhage.
Selected individuals with sizable lobar hemorrhage and progressive neurologic deterioration may benefit from surgical drainage. Surgery is more efficacious in individuals with cerebellar hemorrhage. The clinical course in cerebellar hemorrhage is notoriously unpredictable. Individuals with minimal findings may deteriorate suddenly to coma and death with little warning. For this reason, most neurosurgeons consider emergent surgery for individuals with cerebellar hemorrhage within 48 hours of onset.
PROGNOSIS.
Although the overall mortality from ICH is high, functional recovery among survivors is also high. The most important predictor of mortality is hemorrhage size. The individuals who are comatose at onset or who have a wide spectrum of neurologic deficits tend to do poorly compared with those who remain alert and have focal neurologic symptoms. Survival depends on the location, size, and rapidity of development of the hematoma. ICHs are at first soft and dissect along white matter fiber tracts. If the individual survives the initial changes in ICP, blood is absorbed and a cavity or slit forms that may disconnect brain pathways. Individuals with small hematomas located deep and near midline structures often develop secondary herniation and mass effect and have a high mortality rate. Survivors invariably have severe neurologic deficits. In individuals with medium-sized hematomas, the deficit varies with the location and size of the hematomas. Most individuals survive with some residual neurologic signs. The older the individual, the less complete the expected recovery.
Survival for individuals with hemorrhage in the posterior fossa is more dependent on location of hemorrhage than size. Midline pontine hemorrhage is often fatal, whereas lateral hemorrhages carry a better prognosis.
Subarachnoid Hemorrhage
SAH can begin with the sudden onset of a “thunderclap” with searing pain; sometimes the headache begins with exertion. SAH results in frank blood in the subarachnoid space between the arachnoid and the pia, which are contiguous membranes that surround the brain tissue. SAH can be spontaneous, is often seen in normotensive persons, and results in a sudden, severe headache. One percent to 4% of all individuals presenting to the emergency department with a headache have an SAH.
Etiologic and Risk Factors
Aneurysm and vascular malformations are responsible for most SAHs. SAH can be the result of trauma, developmental defects, neoplasm, or infections that cause rupture into the subarachnoid space. Hypertension may be seen in 32% and fever (higher than 37.5° C) in 5%. Vascular malformations are responsible for approximately 6% of hemorrhages into the subarachnoid space. Included in vascular malformations are venous malformation, AVM, and cavernous malformation. The highest incidence of SAH is in women over the age of 70 years. Risk factors for SAH include smoking, excessive alcohol consumption, and hypertension. Definite genetic transmission of the tendency to develop berry aneurysm has not been proven; however, a tendency for familial occurrence of aneurysms is apparent. First-degree relatives of persons who have experienced an SAH have a threefold to sevenfold increased risk of an SAH. If transmitted genetically the effect will most likely be on the connective tissue. Aneurysms are also more common among individuals with hereditary connective tissue disorders. Individuals with hemorrhage resulting from an aneurysm tend to be younger than those with hemorrhage secondary to hypertension.
Individuals with fibromuscular dysplasia, polycystic kidney disease, or connective tissue diseases have a higher incidence of aneurysms. Embolization, often from endocarditis, can cause mycotic aneurysms. Fungi or tumor tissue can travel to the brain. About one fifth of individuals with aneurysms have more than one vascular anomaly or other aneurysms.
Clinical Manifestations
Forty percent of individuals who have SAH present with a sentinel headache. A sentinel headache results from a minor aneurysm leak that precedes rupture by days or weeks. Individuals who experience a sentinel headache typically report headache as the only symptom and have a normal physical examination
Common associated symptoms include nausea and vomiting (75%), syncope (36%), neck pain (24%), coma (17%), confusion (16%), lethargy (12%), and seizures (7%). Physical examination in individuals who have SAH can have variable findings. For example, nuchal rigidity may take several hours to develop and is present in only 35% to 52% of individuals Thirty-six percent have a normal level of consciousness, whereas 28% are somnolent or confused. Focal motor weakness is detected in only 10%, and cranial nerve palsies are seen in 9%. Often noted initially is cessation of physical and intellectual activity, vomiting, and alteration of consciousness. Drowsiness, restlessness, and agitation are especially common. Severe focal neurologic signs such as hemiplegia and hemianopia are absent at onset unless the aneurysm also bleeds into the brain.
MEDICAL MANAGEMENT
Up to 38% of individuals who have an SAH are misdiagnosed initially. Misdiagnosis of SAH is associated with increased morbidity and mortality. The most common misdiagnoses for SAH are viral meningitis, migraine, and headache of uncertain etiology. Often individuals have subtle presentations and normal neurologic examinations. It is important to realize that the headache of SAH may occur in any location, may be mild, may resolve spontaneously, or may be relieved by analgesics. Prominent vomiting may lead to a misdiagnosis of viral syndrome, gastroenteritis, influenza, or viral meningitis. The presence of blood irritating the cervical or lumbar theca may lead to a misdiagnosis of cervical strain or sciatica.
A CT scan of the head without intravenous contrast is the diagnostic modality of choice in individuals suspected of having SAH. The sensitivity of CT for SAH is approximately 90% to 95%. The longer the duration is from onset of symptoms, the lower is the sensitivity of CT for SAH. Therefore a lumbar puncture should be performed in all individuals suspected of having an SAH when the CT scan is negative or inadequate. The hematoma caused by an aneurysm tends to be of a different character than that caused by hypertension. The location of hemorrhage is also a clue to the diagnosis of aneurysm. Primary sites for hematoma resulting from aneurysms are in the area of the corpus callosum and anterior horns, the frontal lobe, and the temporal lobes. Angiography is required to establish that an intracerebral hematoma has been caused by a ruptured aneurysm.
TREATMENT.
Once the diagnosis of SAH is made, the physician should obtain prompt neurosurgical consultation and arrange for transport to the closest emergency department. The treatment of individuals with SAH involves the prevention and management of the relatively common secondary complications of SAH: rebleeding, vasospasm, hydrocephalus, hyponatremia, and seizures. About one half of individuals with SAH have vasospasm, and this problem may resolve or progress to cerebral infarction. Fifteen percent to 20% of individuals with vasospasm die despite maximal therapy. Angiographic vasospasm has a typical temporal course: onset between 3 to 5 days after hemorrhage, maximal narrowing at 5 to 14 days, and gradual resolution over 2 to 4 weeks. Measures of proven value in decreasing the risk of delayed cerebral ischemia are a liberal supply of fluids, avoidance of antihypertensive drugs, and administration of the calcium antagonist nimodipine.
PROGNOSIS.
Mortality rate from SAH is high in elderly persons. Functional outcomes are poor, and few individuals 75 years or older are able to live independently at discharge. Early aggressive surgical treatment of elderly individuals admitted in good condition may lead to better outcomes. Seizure-like episodes occur in 25% of individuals after SAH.
If the resulting hematoma is less than 3 cm, the prognosis is good. Evacuation of hematomas that are larger should include resection of the causative aneurysm. Prompt removal may result in dramatic and early improvement of neurologic function. Repeat hemorrhage is more likely to occur if the hematoma is evacuated without treatment of the ruptured aneurysm.73,92
Types of Subarachnoid Hemorrhage
Berry aneurysm is a congenital abnormal distention of a local vessel that occurs at a bifurcation, where the medial layer of the vessel is the weakest. About 90% of SAHs are due to berry aneurysms. Aneurysms are probably caused by a combination of congenital defects in the vascular wall and degenerative changes. Aneurysms usually occur at branching sites on the large arteries of the circle of Willis at the base of the brain. When an aneurysm ruptures, blood is released under arterial pressure into the subarachnoid space and quickly spreads through the cerebrospinal fluid around the brain and spinal cord. Aneurysms are less often caused by arterial dissection through the adventitia of arterial walls, embolism of infected material of distal cerebral arteries (mycotic aneurysms), and degenerative elongation and tortuosity of arteries (dolichoectasia). Fig. 32-19 shows the typical formations of berry aneurysms.
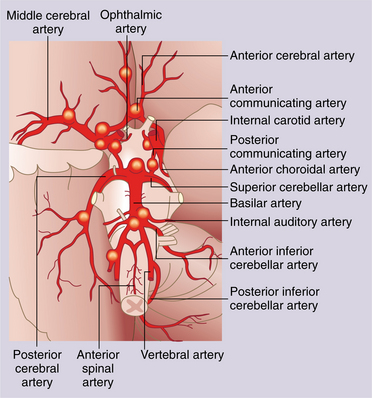
Figure 32-19 Berry aneurysms typically develop at the bifurcations of arteries on the undersurface of the brain. (Reprinted from Goldman L: Cecil textbook of medicine, ed 22, Philadelphia, 2004, WB Saunders.)
Venous malformations are composed entirely of veins, which are usually thickened and hyalinized, with minimal elastic tissue or smooth muscle. The veins converge on a draining vein. Normal brain parenchyma is interspersed among the vessels. Venous malformation is the most common form of vascular malformation, constituting approximately 50% of malformations. The risk of hemorrhage from venous malformation has been estimated at 20% per year. Individuals with a cerebellar malformation have the greatest risk of hemorrhage. Occasionally, seizures may be associated with venous malformations. Headaches and focal neurologic deficits are manifested according to the area of the brain that is disrupted.
Treatment.: Spontaneous rupture of a venous malformation is not common, and the resulting bleed is often not of great consequence. Surgical resection of the hematoma may be necessary if it is significantly extensive. With evacuation of the hematoma, the malformation is left intact in most cases.
Arteriovenous malformation (AVM) is characterized by direct artery-to-vein communication without an intervening capillary bed. The blood vessel contains elastin and smooth muscle cells. Brain parenchyma is found within the AVM, and it is usually gliotic and nonfunctional. AVMs are the result of abnormal fetal development at approximately 3 weeks’ gestation. More than 90% of AVMs occur in the cerebral hemispheres. Most AVMs are sporadic, but familial AVMs occur; these appear to have an autosomal-dominant inheritance with incomplete penetrance.
The annual risk of bleeding from an AVM is 1% to 4%. AVMs result in an SAH more than 50% of the time. Seizures, headache, or an audible bruit may precede a hemorrhage. Progressive focal neurologic deficits develop in some individuals, and cognitive decline may also precede a hemorrhage. AVM-associated hemorrhages occur predominantly in the third and fourth decades of life.
Malformations less than 3 cm in diameter are more likely to bleed than are larger AVMs because arterial pressure is higher in the smaller vessels. A single draining vein, obstructed drainage, or a periventricular or intraventricular location increases the risk of hemorrhage.
Angiography is the definitive diagnostic procedure for an AVM. CT scanning is diagnostic for a dense lesion in the brain. Suspicion of an AVM arises when an area of decreased density is seen around the hematoma and heterogeneous densities appear within the hematoma. MRI is not useful in the diagnosis; however, AVM can be suspected based on MRI with evidence of intravascular moving blood. An AVM should be suspected as a cause of hemorrhage in persons younger than 40 years, especially if they are normotensive. Figs. 32-20 and 32-21 represent the vascular disorder and its appearance on imaging.79
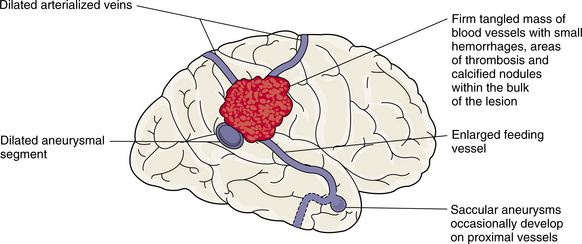
Figure 32-20 Typical deformation of blood vessels and brain tissue in relation to an AVM. (Reprinted from Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.)
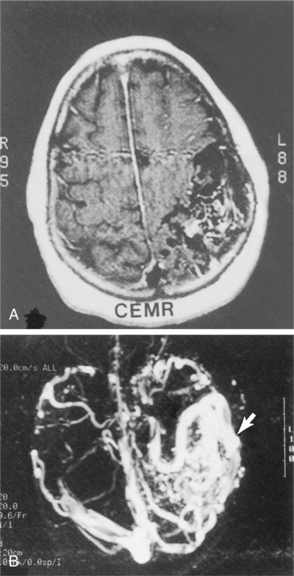
Figure 32-21 The AVM as seen with MRI (A) and MR angiography (B). Arrow points to enlarged vessel in periphery of AVM. (Reprinted from Ramsey R: Neuroradiology, Philadelphia, 1994, WB Saunders.)
Approximately 10% of individuals die from an AVM hemorrhage. In the first year following the hemorrhage, the chance of recurrence is 6%. A concomitant aneurysm increases the chance of recurrent bleeding to 7%.7
Neuroradiologic embolization, stereotactic radiotherapy, and surgery are the current treatments for AVM. These techniques are used alone or in combination depending on the size and site of the lesion. Vasospasm after surgery is a side effect, and it appears that surgeries in the posterior circulation are better tolerated than those in the anterior circulation.55
Cavernous malformations consist of dilated, endotheliumlined, fibrous channels. No smooth muscle or elastin is present in the vascular walls. Neural tissue is present only at the periphery of the lesion. Thrombosis and calcification within the malformation occur, and gliosis (scarring) often surrounds the malformation.
Cavernous malformations represent approximately 10% of vascular malformations. Multiple malformations can occur in the same person. It is believed that cavernous malformations are inherited through autosomal dominance, with close to 100% penetration. Hispanics are at particular risk for the familial disorder. No apparent relation exists between malformation size and age and the likelihood of hemorrhage. Women tend to be more susceptible to hemorrhage than men. Subclinical bleeding frequently occurs around the malformation.
MRI is the imaging procedure of choice for visualizing cavernous malformations. The malformation appears as a well-defined region with a central area of mixed signal intensity surrounded by a rim of hypointensity.
The majority of individuals recover from a cavernous malformation hemorrhage, and the risk of repeat bleeding is low. Spontaneous rupture of a venous malformation is not common, and the resulting hemorrhage is often not of great consequence. Surgery is the treatment of choice for malformations that have hemorrhaged and are in an accessible part of the brain. The majority of individuals recover from a cavernous malformation hemorrhage, and the risk of recurrence is low.
Syndromes are associated with the location of the hemorrhage, as they are in ICH.
Subdural Hemorrhage
A subdural hemorrhage, or hematoma, is most often the result of tearing of the bridging veins between the brain surface and dural sinus. It results in accumulation of blood in the dural space. If it is a small amount, the body can reabsorb the fluid; if the blood is of great enough volume, as can occur with trauma, it becomes a space-occupying lesion. The lesion is reflected in the area of the hemorrhage and the result can be herniation of the cortex into the adjoining spaces. Fig. 32-22 illustrates the actual spaces and potential spaces in the cranial meninges. Compression of the brain tissue can result in both localized lesions and general decrease in the level of consciousness. Fig. 32-23 represents the pressures on the brain that accompany a subdural hemorrhage. Chronic subdural hematoma (CSH) is defined as a subdural hemorrhage that is more than 20 days old. The peak incidence for CSH occurs in the sixth and seventh decades, with up to 80% occurring in elderly men. In elderly persons, CSH often is caused by minor trauma, especially falls. In the majority of cases there is no underlying brain injury. Fragility of the bridging veins and cerebral atrophy allow increased movement of the brain within the skull, thereby predisposing elderly individuals to CSH. Anticoagulant therapy is a recognized risk factor for CSH. Fig. 32-24 shows the change in brain pressures before and after removal of a CSH.104
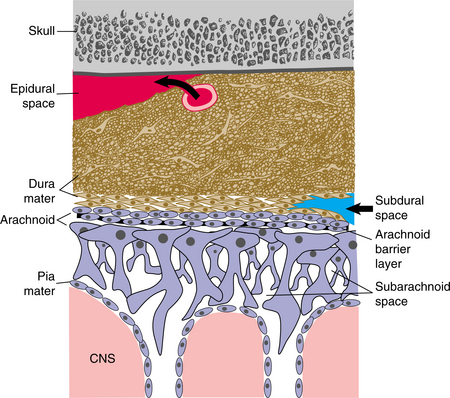
Figure 32-22 Actual spaces and potential spaces in the cranial meninges. Epidural space between dura and skull can be opened up by blood from a ruptured meningeal artery. Subdural space may be opened up by blood from a vein that tears as it cross the arachnoid to enter a dural sinus. (Reprinted from Nolte J: The human brain, St Louis, 2002, Mosby.)
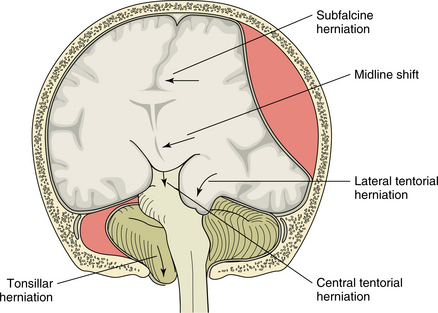
Figure 32-23 Compression of brain tissue with herniation into adjacent structures produced by the subdural hemorrhage. (Reprinted from Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.)
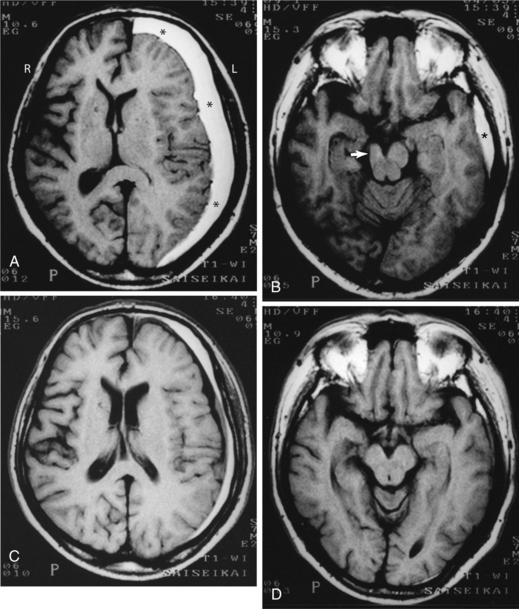
Figure 32-24 A, CSH (*) over the surface of the left cerebral hemisphere, compressing its subarachnoid spaces and lateral ventricle. B, Shifting midline structures to the right and deforming the cerebral peduncle (arrow) by pressing it against the tentorium cerebelli, causing left-sided weakness. C and D, Return to midline of structures and release of pressure after surgical evacuation. (Reprinted from Itoyama Y, Fujioka S, Ushio Y: Kernohan’s notch in chronic subdural hematoma: findings on magnetic resonance imaging: case report, J Neurosurg 82(4):645-646, 1995.)
Elderly individuals who have CSH typically present with a complaint of headache and/or changes in mental status. Mild generalized headache is present in up to 90% of individuals who have CSH. Any elderly individual who has headache, especially with a change in mental or functional status, should be evaluated for CSH. The diagnostic modality of choice for CSH is a non–contrast-enhanced CT scan of the head. When a hematoma is dense, delayed contrast-enhanced CT scan or MRI may be helpful. Once the diagnosis is confirmed, the physician should consult a neurosurgeon promptly, and the individual should be transported rapidly to the closest emergency department.
Epidural Hematoma
The meningeal arteries run in the periosteal layer of the dura. They can be torn during a traumatic skull injury, and bleeding occurs between the periosteum and the skull, resulting in an epidural hematoma. The damage comes from compression of the brain. Because there is potential for extensive pooling of blood, this is considered a medical emergency and should be evacuated immediately to prevent compression on the posterior structures, which may cause death. Fig. 32-25 presents an MRI showing an epidural hematoma.
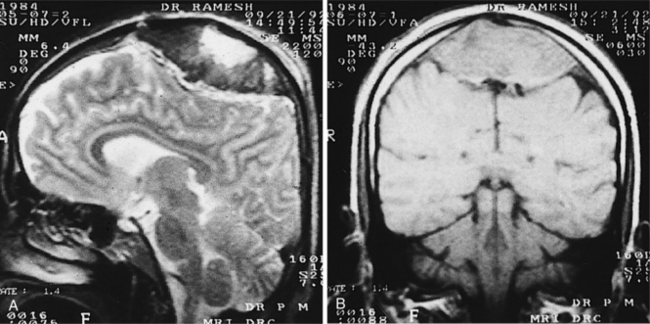
VASCULAR DISORDERS OF THE SPINAL CORD
Vascular disorders of the spinal cord are rare; however, vascular disorders of the brain and spinal cord have many common factors. Because of anatomic differences, some special issues must be considered in vascular disorders of the spinal cord. Infarctions in the spinal cord can be a result of any of the same causes as those in the brain; however, some of the symptoms appear to affect the lower motor neuron at the site of the anterior horn, and the result may be more flaccid extremities and muscle atrophy.
An AVM in the spine can cause pain and burning below the level of the lesion with progressive spastic paraparesis, with or without lower motor neuron lesion. Bowel and bladder dysfunction are seen when the AVM occurs in the lumbar region.
Transverse myelitis, the dysfunction of both halves of the spinal cord in a transverse section, can be related to vascular disorders. In addition to congenital vascular malformation, transverse myelitis can be caused by viral infection, multiple sclerosis, and degenerative disorders. Necrosis of the spinal cord often occurs at several levels, resulting in sensory and motor loss and pain. The lesion may involve nerve roots or just the central structures. The majority of individuals with transverse myelitis experience some degree of recovery (see also Chapter 34).
32-1 SPECIAL IMPLICATIONS FOR THE THERAPIST
Impaired Motor Function and Sensory Integrity Associated with Nonprogressive Disorders of the Central Nervous System—Acquired in Adolescence or Adulthood
Impaired Arousal, Range of Motion, and Motor Control Associated with Coma, Near Coma, or Vegetative State
Impaired Ventilation and Respiration/Gas Exchange Associated with Ventilatory Pump Dysfunction or Failure
The disability resulting from stroke is a major problem for the stroke survivor, the family, and health care providers. A team of professionals is necessary to address the multitude of problems present after stroke. Therapists play a key role in this team, and there is now stronger evidence relating therapeutic intervention to decreased levels of disability. Stroke is highly heterogeneous in its effects, and each individual must be managed according to the particular impairment and disability that remains after the onset of stroke.13
The organization of movement changes after stroke. Mobility can be limited by a number of impairments. These changes reflect damage in specific areas of the brain. Basic reflex patterns can change and limit the ability to rely on automatic postural control mechanism for balance. A lack of normal inhibition of the H-reflex in the soleus during activation of tibialis anterior during balance activity has been noted in individuals after stroke. The increased H-reflex activity disrupts the normal on-off cycle in the muscles of the distal extremity and can cause an abnormal balance strategy.54,58 Box 32-3 gives examples of typical motor impairments after stroke.
Identification of which impairments may be causing an inability to perform functional activities and cause disability is critical. There is a degree of spontaneous recovery after stroke, but some impairment persists over time. A good understanding of the typical characteristics of the different stroke syndromes is critical to establishing the appropriate strategies for intervention. Fig. 32-26 represents areas of brain damage and associated clinical signs.
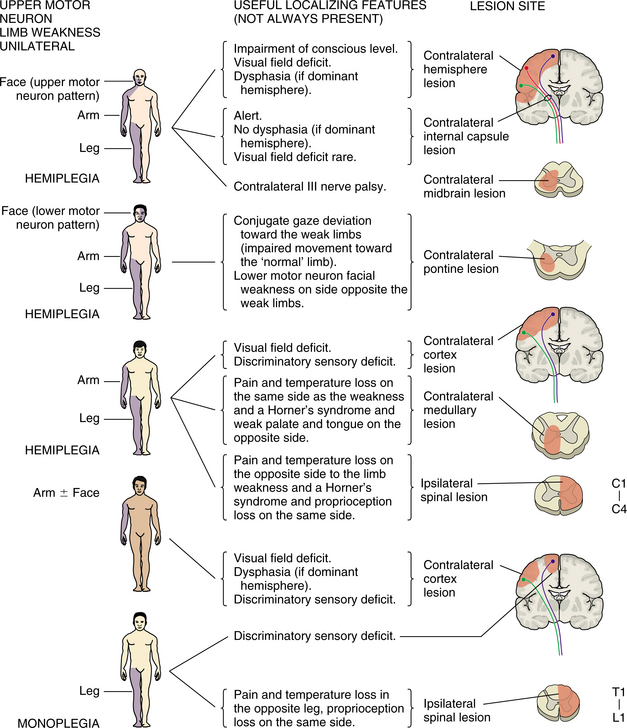
Figure 32-26 Localizing features of damage to specific areas of the brain and spinal cord. (Reprinted from Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.).
Qualitative as well as quantitative measures should show sensitivity to change over time and identify relevant issues for the stroke survivor, including quality of life.106 Many measures are currently used and there are efforts to benchmark levels of function that can help determine rehabilitation potential as early in the process as possible.18,45,93
Predicting upper limb recovery after stroke appears to be related to the early ability to shoulder shrug and perform at least synergistic hand movement.48 Measurement of movement with a computer-assisted reach can quantify constraint forces and range of motion of the upper extremity. This may lead to more precise monitoring of movement and ability to determine recovery.80
The focus of measurement tools has traditionally been directed toward the contralateral side; however, the degree of impairment and disability of the ipsilateral side always has been questioned.11 Unilateral tapping speed of the ipsilateral fingers may point toward ability to determine if the unaffected side has maintained functional integrity.76 MRI demonstrates ipsilateral activation in the primary sensory cortex during contralateral tasks performed in an individual after stroke.38
Discrete differences exist according to the side of hemispheric lesion, with a right hemisphere lesion causing impairment of pacing, transporting, and coordinating two body parts and left hemisphere lesions resulting in impairment in calibrating movements. Despite these differences, there does not appear to be an overall difference in functional level as measured by instrumental activities of daily living between individuals according to which hemisphere the stroke affected.10
Brain reorganization after neural injury is of great interest to therapists, as understanding the relation between alterations in neural structure and functional recovery are critical to the interventions chosen for each individual. It appears that the nature of the infarct may drive the plasticity related to recovery, and while a recovery after a small infarct may result in reorganization around the area of the infarct, a large lesion may require more extensive changes in remote metabolism, blood flow, neurotransmitter function, and axonal sprouting that may not follow the same pattern of plasticity. Inherent in the processes of rehabilitation-dependent motor recovery are restoration and reorganization of motor maps within the cortex surrounding the lesion.26,68,95
Skill acquisition for recovery of function is the main goal in movement retraining. Increased knowledge of neural plasticity will lead to more precision in determining the optimal task practice for motor learning. Problem solving is critical for improving skill, but impairments of motor control such as decreased force production, increased tone, and poor control of degrees of freedom in movement must be adequately addressed. Implicit motor learning also depends on a neural network that can be affected in the pathogenesis of stroke.7,53
Constraint-induced training with the unaffected limb is effective and is being used in a variety of formats. Increased dexterity and function are possible even after extended time after stroke.101 The specifics of training, including problem solving and manipulating the spatial and temporal aspects of the performance, appear to be inherent in the successful outcomes.
Movement of the upper extremity facilitated with electromyography-triggered stimulus and use of electrical stimulation orthoses continues to be studied with positive effects on movement and functional use of the upper extremity.28,105 Robotics are used to assist and retrain movement.1 There has been a rapid increase in available robotic devices that allow augmentation to hands-on therapy. Robots that assist threedimensional movement are being developed to mimic the trajectories of human movement.23
Virtual reality applications are being developed with a focus on attention, executive function, memory, and spatial ability. Functional training for activities such as crossing the street, driving, preparing meals, and navigating by wheelchair are possible with virtual reality.81
Fall prevention is always a primary goal for the individual sustaining a stroke. Falls are frequent and in more than 5% of cases result in significant injury. Falls are common during the transition from sit to stand, and the kinematics of this activity has been studied. Evidence shows that people who fall take more time to rise and to sit down and demonstrate increased center of pressure sway in mediolateral directions. Weight on the nonparetic leg is significantly increased during the activity.14,41
Obtaining functional and normal-looking gait has long been the goal of therapists and stroke survivors. Research continues in this area.64,97 Treadmill training with partial body weight support shows functional changes over typical terrain, with more normal movement of the affected limb during both stance and swing.43,102 Computer-assisted gait training results in increased stride length and speed of walking.96,97 Balance retraining with center of pressure feedback provides more even distribution and control of center of gravity.88
Although a therapeutic goal is often to move clients out of assistive devices, studies show that for some parameters of gait the orthosis or assistive device still provides control over some of the critical components of gait. Rigid ankle braces resulted in longer relative single-stance duration, improved swing symmetry, and ankle excursions. The decreased activity in the anterior tibialis continues to be a drawback.42 Use of a cane improves weight shift to prepare for the next step and results in decreased circumduction. Joint angles were more normal during the swing phase.56
Control of spasticity and contractures by antispastic pattern positioning, range-of-motion exercise, stretching, and splinting has long been a part of the rehabilitation process. Maintaining soft tissue mobility in the distal extremities is a critical link to the performance of a motor activity. Traditionally it was thought that the activity of the spastic muscle should be limited to prevent further increase in tone; however, it has been shown that increased workload in a spastic muscle can be performed without increasing the spasticity.9
Cardiovascular endurance training is indicated for the stroke survivor; programs incorporating such activities should be part of rehabilitation programs. Treadmill training has been shown to reduce the energy expenditure and cardiovascular demands of gait within the stroke population.63 Submaximal all-extremity exercise shows potential in the rehabilitation programs for stroke.60 Research is ongoing to establish the parameters of cardiovascular training within the limits created by neurologic deficits.
Based on the theory of motor learning, the ability to learn a new motor program or a different way of moving does not follow a specific time frame following brain damage.5 It appears that the stroke-related sensorimotor deficits affect the processes underlying the control execution of motor skills but not the learning of those skills. Potential for adaptation based on learning goes beyond the time frame of spontaneous recovery.109
Failure to maintain functional gains after the course of therapy is a concern for all individuals involved in the management of poststroke rehabilitation. Functional exercise done on a regular basis has been shown to have a positive impact on recovery.17 Early and consistent involvement of the family or primary caretakers is paramount, as is the follow-through of a home management program of activity and exercise. Compliance of stroke survivors and caregivers continues to be low despite efforts toward better education.82 The functional consequence of fatigue in physical, professional, and social activities should be considered.36
The clinician and family should watch for symptoms related to angina, peripheral vascular disease, and deep vein thrombosis that may arise after stroke. Osteoporotic bone loss has been demonstrated with immobility of the upper extremity, especially in women, and should be addressed as a part of a standard protocol.15
References
1. Aisen, ML, et al. The effect of robot-assisted therapy and rehabilitative training on motor recovery following stroke. Arch Neurol. 1997;54(4):443–446.
2. Albers, GW. Antithrombotic and thrombolytic therapy for ischemic stroke: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(3 Suppl):483S–512S.
3. B-Vitamin Treatment Trialists’ Collaboration. Homocysteine-lowering trials for prevention of cardiovascular events: a review of the design and power of the large randomized trials. Am Heart J. 2006;151(2):282–287.
4. Barnett, HJ, et al. Do the facts and figures warrant a 10-fold increase in the performance of carotic endarterectomy on asymptomatic patients? Neurology. 1996;46:603–608.
5. Barton, LA, Black, Sullivan K. Learning treatment strategies applied to stroke rehabilitation. In: Gordon WA, ed. Advances in stroke rehabilitation. Boston: Andover Medical Publishers; 1993:63–78.
6. Bonita, R, et al. The global stroke initiative. Lancet Neurol. 2004;3(7):391–393.
7. Boyd, LA, Winstein, CJ. Cerebellar stroke impair temporal but not spatial accuracy during implicit motor learning. Neurorehabil Neural Repair. 2004;18:134–143.
8. Brass, LM. Clinical syndromes of intracerebral hemorrhage. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:223–256.
9. Brown, DA, Kautz, SA. Increased workload enhances force output during pedaling exercise in persons with poststroke hemiplegia. Stroke. 1998;29(3):598–606.
10. Burnspang, B, Fisher, AG. Differences between persons with right or left cerebral vascular accident on the assessment of motor and process skills. Arch Phys Med Rehabil. 1995;76(12):1144–1151.
11. Cao, Y, et al. Pilot study of functional MRI to assess cerebral activation of motor function after poststroke hemiparesis. Stroke. 1998;29(1):112–122.
12. Carrera, E. The thalamus and behavior: effects of anatomically distinct strokes. Neurology. 2006;66(12):1817–1823.
13. Cavanaugh, JT, Schenkman, M. Physical therapy evaluation and treatment in stroke rehabilitation. Phys Ther Case Rep. 1998;1(4):200–209.
14. Cheng, PT, et al. The sit-to-stand movement in stroke patients and its correlation with falling. Arch Phys Med Rehabil. 1998;79:1043–1046.
15. Del Puente, A, et al. The determinants of bone mineral density in hemiplegic patients. Preliminary data. Annali Italiani di Medicina Interna. 1995;10(3):163–166.
16. Dichgans, M. Genetics of ischaemic stroke. Lancet Neurol. 2007;6(2):149–161.
17. Duncan, PW, Sudenski, SA, et al. Randomized clinical trial of therapeutic exercise in subacute stroke. Stroke. 2003;34:2173–2180.
18. Duncan, PW, Jorgensen, HS, Wade, DT. Outcomes measures in acute stroke trials: a systematic review and some recommendations to improve practice. Stroke. 2000;31:1429–1438.
19. Easton, JD. Clinical aspects of the use of clopidogrel, a new antiplatelet agent. Semin Thromb Haemost. 1999;25(suppl 2):77–82.
20. Ebrahim, S, Harwood, R. Stroke: epidemiology, evidence, and clinical practice, ed 2. Oxford, UK: Oxford University Press, 1999.
21. Elkins, JS. Stroke risk factors and loss of high cognitive function. Neurology. 2004;63(5):793–799.
22. Eugene, JR, et al. Carotid occlusive disease: primary care of patients with or without symptoms. Geriatrics. 1999;54(5):24–26.
23. Fasoli, SE, Krebs, HI, Hogan, N. Robotic technology and stroke rehabilitation: translating research into practice. Top Stroke Rehabil. 2004;11(4):11–19.
24. Feinberg, WM. Coagulation. In: Caplan LR, ed. Brain ischemia: basic concepts and clinical relevance. London: Springer-Verlag; 1995:85–96.
25. Finklestein, SP. Growth factors in stroke. In: Caplan LR, ed. Brain ischemia: basic concepts and clinical relevance. London: Springer-Verlag; 1995:37–42.
26. Fisher, BE, Sullivan, KJ. Activity-dependent factors affecting poststroke functional outcomes. Top Stroke Rehabil. 2001;8(3):31–44.
27. Fisher, M, Bogousslavsky, J. Further evolution toward effective therapy for acute ischemic stroke. JAMA. 1998;279(16):1298–1303.
28. Francisco, G, et al. Electromyogram triggered neuromuscular stimulation for improving the arm function of acute stroke survivors. Arch Phys Med Rehabil. 1998;79(5):570–575.
29. Frank, JI, Biller, J. Laboratory evaluation of intracerebral hemorrhage. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:257–290.
30. Furie, K, Feldmann, E. Cerebral amyloid angioplasty. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:49–64.
31. Garcia, JH. Mechanisms of cell death. In: Caplan LR, ed. Basic concepts and clinical relevance. London: Springer-Verlag; 1995:7–18.
32. Gillum, RF. Stroke mortality in African-Americans: disturbing trends. Stroke. 1999;30(8):1711–1715.
33. Goetz, CG. Textbook of clinical neurology, ed 2. Philadelphia: WB Saunders, 2003.
34. Torrecillas, Gonzales J.I, Mendlewicz, J, et al. Analysis of intensity of post-stroke depression and its relationship with the cerebral lesion location. Medicina Clinica. 1997;109(7):241–244.
35. Gorelick, PB, Kelly, MA. Ethanol. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:195–208.
36. Gramigna, S. Fatigue in neurological disease: different patterns in stroke and multiple sclerosis. Rev Neurol (Paris). 2007;163(3):341–348.
37. Grant, EG, Duerinck. Ultrasound imaging in cerebrovascular disease. In: Cohen SN, ed. Management of ischemic stroke. New York: McGraw-Hill, 2000.
38. Green, JB, et al. High resolution EEG in poststroke hemiparesis can identify ipsilateral generators during motor tasks. Stroke. 1999;30(12):2659–2665.
39. Harrison, MJ. Neurological complications of hypertension. In: Aminoff MJ, ed. Neurology and general medicine. ed 2. New York: Churchill Livingstone; 1995:119–136.
40. Hartcamp, MJ, van Der Grond, J, van Everdingen, AJ, et al. Circle of Willis collateral flow investigated by magnetic resonance angiography. Stroke. 1999;30(12):2671–2678.
41. Hesse, S, et al. Sit-to-stand manoeuvre in hemiparetic patients before and after a four week rehabilitation programme. Scand J Rehab Med. 1998;30(2):81–86.
42. Hesse, S, et al. Non-velocity-related effects of a rigid double-stopped ankle-foot orthosis on gait and lower limb muscle activity of hemiparetic subjects with an equinovarus deformity. Stroke. 1999;30(9):1855–1861.
43. Hesse, S, Konrad, M, Uhlenbrock, D. Treadmill walking with partial body support versus floor walking in hemiparetic subjects. Arch Phys Med Rehabil. 1999;80(4):421–427.
44. Hommel, M, Gray, F. Microvascular pathology. In: Caplan LR, ed. Brain ischemia: basic concepts and clinical relevance. London: Springer-Verlag; 1995:215–222.
45. Horgan, NF, Finn, AM. Motor recovery following stroke: a basis for evaluation. Disabil Rehabil. 1997;19(2):64–70.
46. Kalafut, MA, Saver, JL. The acute stroke patient: the first six hours. In: Cohen SN, ed. Management of ischemic stroke. New York: McGraw-Hill; 2000:17–52.
47. Karapanayiotides, T. Stroke patterns, etiology, and prognosis in patients with diabetes mellitus. Neurology. 2004;62(9):1558–1562.
48. Katrak, P, et al. Predicting upper limb recovery after stroke: the place of early hand movement. Arch Phys Med Rehabil. 1998;79(7):758–761.
49. Kelly, RE. Medical therapy. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:291–302.
50. Kim, JS, Choi-Kwon, S. Sensory sequelae of medullary infarction: differences between lateral and medical medullary syndrome. Stroke. 1999;30(12):2697–2703.
51. Kistler, JP, Ropper, AH, Martin, JB. Cerebrovascular diseases. In: Isselbacher KJ, Braunwald E, Wilson JD, et al, eds. Harrison’s principles of internal medicine. ed 13. New York: McGraw-Hill; 1994:2233–2252.
52. Kistler, JP. Brain attack: preventing and treating stroke. Princeton, NJ: Harvard Medical School, 2002.
53. Kleim, JA. Motor learning dependent synaptogenesis is localized to functionally reorganized motor cortex. Neurobiol Learn Mem. 2002;77(1):63–67.
54. Krebs, HI, et al. Quantization of continuous arm movements in humans with brain injury. Proc Natl Acad Sci U S A. 1999;96(8):4645–4649.
55. Kremer, C, et al. Outcome after endovascular treatment of Hunt and Hess grade IV or V aneurysms: comparison of anterior versus posterior circulation. Stroke. 30(12), 1999. [2617-1622].
56. Kuan, TS, Tsou, JY, Su, FC. Hemiplegic gait of stroke patients: the effect of using a cane. Arch Phys Med Rehabil. 1999;80(7):777–784.
57. Langhorne, P, Stott, DJ, Robertson, L, et al. Medical complications after stroke, a multicenter study. Stroke. 2000;32(2):1223–1229.
58. Leonard, CT, et al. H-reflex modulations during voluntary and automatic movements following upper motor neuron damage. Electroencephalogr Clin Neurophysiol. 1998;109(6):475–483.
59. Lindsay, KW, Bone, I, Callander, R. Neurology and neurosurgery illustrated. New York: Churchill Livingstone, 1986.
60. Loudon, JK, et al. A submaximal all-extremity exercise test to predict maximal oxygen consumption. Med Sci Sports Exerc. 1998;30(8):1299–1303.
61. Lulovits, TG, Gorelick, PB. Stroke in African Americans. In: Cohen SN, ed. Management of ischemic stroke. New York: McGraw-Hill; 2000:485–497.
62. Lyden, PD, Zivin, JA. Cytoprotective therapies in ischemic stroke. In: Cohen SN, ed. Management of ischemic stroke. New York: McGraw-Hill; 2000:225–241.
63. Macko, RF, et al. Treadmill aerobic exercise training reduces the energy expenditure and cardiovascular demands of hemiparetic gait in chronic stroke patients. Stroke. 1997;28(2):326–330.
64. Mercier, C, et al. Description of a new motor re-education programme for the paretic lower limb aimed at improving the mobility of stroke patients. Clin Rehabil. 1999;13(3):199–206.
65. Nadareishvili, ZG, et al. Cerebral microembolism in acute myocardial infarction. Stroke. 1999;30(12):2679–2682.
66. Newberg, AB. The role of PET imaging in the management of patients with central nervous system disorders. Radiol Clin North Am. 2005;43(1):49–65.
67. Norrving, B. Medical therapy to prevent stroke. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura, 1994.
68. Nudo, R, Wise, BM, et al. Neural substrates for the effects of rehabilitative training on motor recovery after ischemic infarct. Science. 1996;272(5269):1791–1794.
69. Oberg, AL, et al. Incidence of stroke season of the year: evidence of an association. Am J Epidemiol. 2000;152(6):558–564.
70. O’Brien, MD. Ischemic cerebral edema. In: Caplan LR, ed. Brain ischemia: basic concepts and clinical relevance. London: Springer-Verlag; 1995:43–50.
71. Ocava, LC. Antithrombotic and thrombolytic therapy for ischemic stroke. Clin Geriatr Med. 2006;22(1):135–154.
72. O’Sullivan, SB. Stroke. In: O’Sullivan SB, Schmitz TJ, eds. Physical rehabilitation: assessment and treatment. ed 3. Philadelphia, FA: Davis; 1994:327–360.
73. Pambianco, GP, Orchard, MB, Landau, P. Deep vein thrombosis: prevention in stroke patients during rehabilitation. Arch Phys Med Rehabil. 1995;76:324–330.
74. Parsons, MW. Identification of the penumbra and infarct core on hyperacute noncontrast and perfusion CT. Neurology. 2007;68(10):730–736.
75. Pelz, AD. Computed tomography scanning in ischemic stroke. In: Cohen SN, ed. Management of ischemic stroke. New York: McGraw-Hill, 2000.
76. Prigatano, GP, Wong, JL. Speed of finger tapping and goal attainment after unilateral cerebral vascular accident. Arch Phys Med Rehabil. 1997;78(8):847–852.
77. Pryse-Phillips, WE, Murray, TJ. Essential neurology. New York: Medical Examination Publishing Company, 1992.
78. Psota, TL. Dietary omega-3 fatty acid intake and cardiovascular risk. Am J Cardiol. 2006;98(4A):3i–18i.
79. Ramsey, R. Neuroradiology. Philadelphia: WB Saunders, 1994.
80. Reinkensmeyer, DJ, Dewald, JP, Rymer, WZ. Guidance-based quantifications of arm impairment following brain injury: a pilot study. IEEE Transactions Rehab Engin. 1999;7(1):1–11.
81. Rizzo, AA, Schultheis, MT, et al. Analysis of assets for virtual reality: applications in neuropsychology. Neropsychol Rehabil. 2004;14(1):207–239.
82. Rodgers, H, et al. Randomized controlled trial of a comprehensive stroke education program for patients and caregivers. Stroke. 1999;30(12):2585–2591.
83. Ryder, KM, Benjamin, EJ. Epidemiology and significance of atrial fibrillation. Am J Cardiol. 1999;84(9A):131R–138R.
84. Sacco, RL, Wolf, PA, Gorelick, PB. Risk factors and their management for stroke prevention: outlook for 1999 and beyond. Neurology. 1999;53(Suppl 4):S15–S24.
85. Sacco, RL, Mayer, SA. Epidemiology of intracerebral hemorrhage. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:3–26.
86. Schwab, M. Extending the potential of perfusion imaging with MRI to prevent major stroke. Lancet Neurol. 2007;6(2):102–104.
87. Schwammenthal, Y. Homocysteine, B-vitamin supplementation, and stroke prevention: from observational to interventional trials. Lancet Neurol. 2004;3(8):493–495.
88. Simmons, RW, et al. Balance retraining in a hemiparetic patient using center of gravity biofeedback: a single case study. Percept Mot Skills. 1998;87(2):603–609.
89. Sloan, MA. Thrombolysis and intracranial hemorrhage. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:99–150.
90. Smith, EE. Serum lipid profile on admission for ischemic stroke: failure to meet National Cholesterol Education Program Adult Treatment Panel (NCEP-ATPIII) guidelines. Neurology. 2007;68(9):660–665.
91. Smith, W. Safety and efficacy of mechanical embolectomy in acute ischemic stroke: results of MERCI trial. Stroke. 2005;36:1432–1438.
92. Stern, BJ, Kahn, A. Vascular malformations and aneurysms. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:169–194.
93. Stineman, MG, et al. Functional task benchmarks for stroke rehabilitation. Arch Phys Med Rehabil. 1998;79(5):497–504.
94. Streja, D. The subacute stroke patient: lipid management. In: Cohen SN, ed. Management of ischemic stroke. New York: McGraw-Hill; 2000:119–131.
95. Strens, LH, Asselman, P, et al. Corticocortical coupling in chronic stroke: its relevance to recovery. Neurology. 2004;63(3):475–484.
96. Sullivan, K, Knowlton, B, Dobkin, B. Step training with body weight support: effect of treadmill speed and practice paradigms on poststroke locomotor recovery. Arch Phys Med Rehabil. 2002;83(5):683–691.
97. Suzuki, K, et al. Relationship between stride length and walking rate in gait training for hemiparetic stroke patients. Am J Phys Med Rehabil. 1999;78(2):147–152.
98. Tanne, D, et al. Frequency and prognosis of stroke/TIA among 4804 survivors of acute myocardial infarction. Stroke. 1993;24:1490–1495.
99. Tirschwell, DL. Association of cholesterol with stroke risk varies in stroke subtypes and patient subgroups. Neurology. 2004;63(10):1868–1875.
100. Tuhrim, S. Medical therapy of ischemic stroke. In: Gordon WA, ed. Advances in stroke rehabilitation. New York: Andover; 1993:3–18.
101. Van der Lee, JH, et al. Forced use of the upper extremity in chronic stroke patients: results from a single-blind randomized clinical trial. Stroke. 1999;30(11):2369–2375.
102. Visitin, M, et al. A new approach to retrain gait in stroke patients through body weight supported and treadmill stimulation. Stroke. 1998;29(6):1122–1128.
103. Wahlgren, NG. Cytoprotective therapy for acute ischemic stroke. In: Fisher M, ed. Stroke therapy. Boston: Butterworth-Heinemann; 1995:315–350.
104. Walker, RA. Headache in the elderly. Clin Geriatr Med. 2007;23(2):291–305.
105. Weingarden, HP, et al. Hybrid functional electrical stimulation orthosis system for the upper limb: effects on spasticity in chronic stable hemiplegia. Am J Phys Med Rehabil. 1998;77(4):276–281.
106. Williams, LS, et al. Measuring the quality of life in a way that is meaningful to stroke patients. Neurology. 1999;53(8):1839–1843.
107. Wilterdink, JL. Hypertensive intracerebral hemorrhage. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:27–48.
108. Wilterdink, JL, Feldmann, E. Neuropathology and pathophysiology of intraparenchymal hemorrhage. In: Gorelick PB, ed. Atlas of cerebrovascular disease. Philadelphia: Current Medicine, 1996.
109. Winstein, CJ, et al. Motor learning after unilateral brain damage. Neuropsychologia. 1999;37:975–987.
110. Wolf, PA, et al. Probability of stroke: a risk profile from the Framingham Study. Stroke. 1991;22:312–318.
111. Wolf, PA, Abbott, RD, Kannel, WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke. 1991;22:983–988.
112. Wunderlich, MT, et al. Early neurobehavioral outcome after stroke is related to release of neurobiochemical markers of brain damage. Stroke. 1999;30:1190–1195.
113. Yrjanheikki, J, et al. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96(23):13496–13500.
114. Zoppo, GL, Hamann, RF, Fitrigde, A, et al. Thrombolytic therapy. In: Feldman E, ed. Intracerebral hemorrhage. Armonk, NY: Futura; 1994:291–314.