Vascular Disorders and Thrombosis
Free-living unicellular organisms obtain nutrients and eliminate metabolic waste products directly into the external environment. Multicellular organisms require a circulatory system to deliver nutrients to and remove waste products from cells. The movement of fluid and cells through the circulatory system maintains homeostasis and integrates functions of cells and tissues in complex, multicellular organisms. In this chapter, the basic abnormalities that affect fluid circulation and balance within an animal are described.
Circulatory System
The circulatory system consists of blood, a central pump (heart), blood distribution (arterial) and collection (venous) networks, and a system for exchange of nutrients and waste products between blood and extravascular tissue (microcirculation) (Fig. 2-1). A network of lymphatic vessels that parallel the veins also contributes to circulation by draining fluid from extravascular spaces into the blood vascular system.
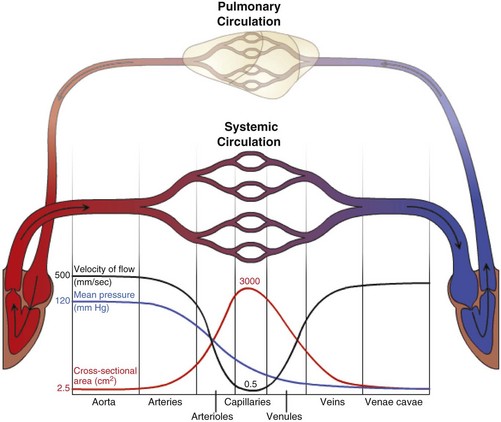
Fig. 2-1 The vascular system.
Blood travels from the left to the right side of the heart via the systemic circulation, and from the right to the left side via the pulmonary circulation. Blood flow rate and pressure in the systemic arterial circulation decrease in conjunction with increased arterial cross-sectional area. In the venous systemic circulation, blood flow rate, but not pressure, increases in conjunction with decreased venous cross-sectional area. The flow, pressure, and cross-sectional area relationships are similar but reversed (i.e., veins deliver blood and arteries collect blood) in the pulmonary circulation. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
The heart provides the driving force for blood distribution. Equal volumes of blood are normally distributed to the pulmonary circulation by the right side of the heart and the systemic circulation by the left side of the heart. The volume of blood pumped by each half of the heart per minute (cardiac output) is determined by the beats per minute (heart rate) and the volume of blood pumped per beat by the ventricle (stroke volume). Typically, each half of the heart pumps the equivalent of the entire blood volume of the animal per minute.
Arteries have relatively large diameter lumens to facilitate rapid blood flow with minimal resistance. Artery walls are thick and consist predominantly of smooth muscle fibers for tensile strength and elastic fibers for elasticity (Web Fig. 2-1). Elastic fibers allow arteries to act as pressure reservoirs, expanding to hold blood ejected from the heart during contraction and passively recoiling to provide continuous flow and pressure to arterioles between heart contractions.
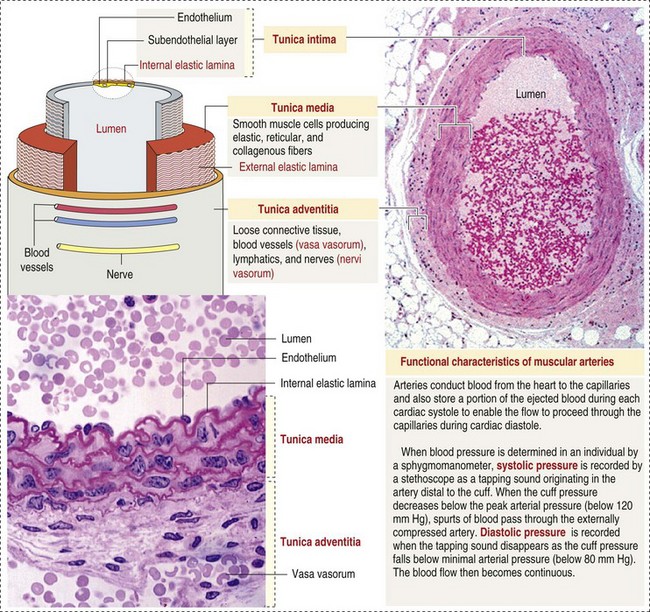
Web Fig. 2-1 Structure of a muscular artery. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby.)
Arterioles are the major resistance vessels within the circulatory system; intravascular pressure can fall by nearly half after blood passes through an arteriole. Arterioles have relatively narrow lumens, the diameter of which is controlled by the smooth muscle cells that are the major component of their walls. Extrinsic sympathetic innervation and local intrinsic stimuli regulate the degree of arteriolar smooth muscle contraction, causing arterioles to dilate or constrict to selectively distribute blood to the areas of greatest need.
Capillaries are the site of nutrient and waste product exchange between the blood and tissue. Capillaries are the most numerous vessel in the circulatory system, with a total cross-sectional area nearly 1300 times that of the aorta. However, they normally contain only about 5% of the total blood volume. The velocity of blood flow through the capillaries is very slow, and red blood cells generally move through a capillary in single file to further facilitate the diffusion of nutrients and wastes. Capillaries have narrow lumens (approximately 8 µm) and thin walls (approximately 1 µm) consisting of a single epithelial cell layer (endothelium). At the junctions between capillary endothelia are interendothelial pores, which make the capillary semipermeable to facilitate diffusion of nutrients and waste products between the blood and tissues. There are three types of capillaries: continuous, fenestrated, and discontinuous. The basic functions and tissue locations of these types of capillaries are illustrated in Fig. 2-2. They are discussed in greater detail in the chapters covering the diseases of organ systems.
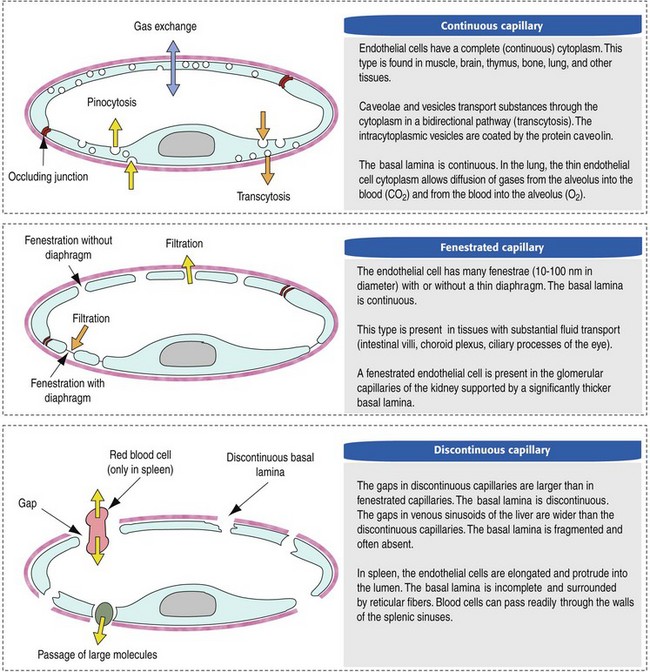
Fig. 2-2 Types of endothelium lining capillaries. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, St Louis, 2002, Mosby.)
The return trip of blood to the heart begins in the postcapillary venules. Venules have a composition similar to capillaries but may have thin layers of muscle as they become more distant from the capillary bed. Veins are composed mainly of collagen with smaller amounts of elastin and smooth muscle (Web Fig. 2-2). Venules and veins provide a low resistance pathway for the return of blood to the heart. Because of their distensibility, they can store large amounts of blood; nearly 65% of total blood volume is normally present within the systemic veins. Pressure and velocity of flow are low within venules and veins. Therefore other factors are necessary to help move venous blood toward the heart such as venous valves to prevent backflow of blood, skeletal muscle contraction, venous vasoconstriction, an increased pressure gradient due to decreased pressure in the heart during filling (cardiac-suction effect), and decreased pressure in the thoracic veins due to negative pressure within the thoracic cavity (respiratory pump).
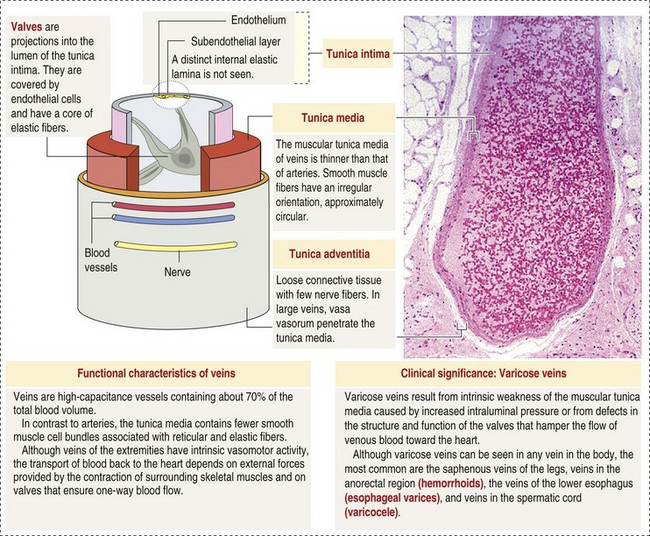
Web Fig. 2-2 The structure of a vein. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby.)
The lymphatic system originates as blind-ended lymphatic capillaries, which permeate the tissue surrounding the microcirculation (arterioles, metarterioles, capillaries, and postcapillary venules [Fig. 2-3]).Lymphatic capillaries have overlapping endothelial cells and large interendothelial gaps so that external pressure allows movement of fluid and molecules into the vessel. However, intravascular lymphatic pressure forces these overlapping edges together to prevent the flow of lymph out of the vessel. Lymphatic capillary gaps are much larger than those between blood capillary endothelium, so they can accommodate movement of larger particles and substances. Lymphatic capillaries converge into progressively larger lymph vessels that drain into lymph nodes and then ultimately empty into the venous system. Similar to venous vessels, lymphatics are distensible, low-pressure vessels that require lymphatic valves and contraction of surrounding muscles to facilitate return of fluid to the blood.
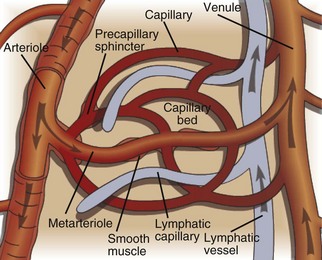
Fig. 2-3 The microcirculation.
The microcirculation consists of arterioles (small arteries proximal to a capillary bed), metarterioles (arterial capillaries), capillaries (thin, semipermeable vessels that connect arterioles and venules), and postcapillary venules (small vessels that merge to form veins after collecting blood from a capillary network). Smooth muscle of the arterioles and metarterioles regulates flow of blood into the capillary bed. There is a dramatic drop in pressure and flow rate from the arterial to the venous side of the microcirculation, facilitating interactions between capillary blood and interstitial fluid. Blind-ended lymphatic vessels that originate near capillary beds interact intimately with the microcirculation. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
All components of the circulatory system are lined by a single layer of endothelium. Endothelium forms a dynamic interface between blood and tissue and is a critical participant in fluid distribution, inflammation, immunity, angiogenesis, and hemostasis (Fig. 2-4). Normal endothelium is antithrombotic and profibrinolytic and helps maintain blood in a fluid state, but when injured, endothelium becomes prothrombotic and antifibrinolytic. Endothelial activation by oxidative stress, hypoxia, inflammation, infectious agents, tissue injury, or similar events results in the production and release of numerous substances with wide-ranging roles in physiology and pathology (Fig. 2-5 and Box 2-1). Endothelial activation is typically localized to restrict a host response to a specific area, while not affecting the normal function of endothelium and flow of blood in other parts of the body.
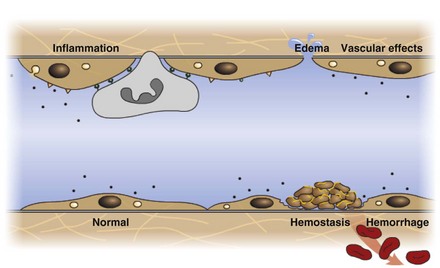
Fig. 2-4 Structure and function of the endothelium.
Endothelium is a physical barrier between intravascular and extravascular spaces, and it is an important mediator of fluid distribution, hemostasis, inflammation, and healing. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
Microcirculation, Interstitium, and Cells
The exchange of fluid, nutrients, and waste products between blood and cells takes place through the interstitium, the space between cells, and the microcirculation. The interstitium is composed of structural, adhesive, and absorptive components collectively referred to as the extracellular matrix (ECM). Type I collagen is the major structural component of the ECM and forms the framework in which cells reside. This is intimately associated with type IV collagen of cell basement membranes. Adhesive glycoproteins provide sites of attachment for structural components and also serve as receptors for cells, such as phagocytes and lymphocytes, which move through the interstitium. Absorptive disaccharide complexes (glycosaminoglycans) and protein-disaccharide polymer complexes (proteoglycans) are hydrophilic and can bind large amounts of water and other soluble molecules. In most cases, no more than 1.0 mm of interstitial space separates a cell from a capillary.
Fluid Distribution and Homeostasis
Water comprises approximately 60% of body weight, of which about 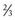 is intracellular and
is intracellular and 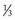 is extracellular (80% of which is in the interstitium and 20% is in the plasma). The distribution of fluid-nutrients, and waste products between the blood, interstitium, and cells is controlled by physical barriers, as well as pressure and concentration gradients between each compartment. The cell’s plasma membrane is a selective barrier that separates interstitial and intracellular compartments. Nonpolar (uncharged) lipid-soluble substances, such as O2, CO2, and fatty acids, move relatively freely across the plasma membrane based on concentration gradients. Polar (charged) lipid-insoluble particles and molecules, such as electrolytes, calcium, glucose, and amino acids, enter the cell by carrier-mediated transport. Water readily moves across the plasma membrane down its concentration gradient. Although approximately 100 times the volume of water in a cell crosses the plasma membrane in 1 second, cell fluid content remains relatively stable because of the activity of energy-dependent membrane pumps (e.g., Na+/K+-adenosine triphosphatase [ATPase] pump) and the balance between osmotic pressures exerted by interstitial and intracellular solutes.
is extracellular (80% of which is in the interstitium and 20% is in the plasma). The distribution of fluid-nutrients, and waste products between the blood, interstitium, and cells is controlled by physical barriers, as well as pressure and concentration gradients between each compartment. The cell’s plasma membrane is a selective barrier that separates interstitial and intracellular compartments. Nonpolar (uncharged) lipid-soluble substances, such as O2, CO2, and fatty acids, move relatively freely across the plasma membrane based on concentration gradients. Polar (charged) lipid-insoluble particles and molecules, such as electrolytes, calcium, glucose, and amino acids, enter the cell by carrier-mediated transport. Water readily moves across the plasma membrane down its concentration gradient. Although approximately 100 times the volume of water in a cell crosses the plasma membrane in 1 second, cell fluid content remains relatively stable because of the activity of energy-dependent membrane pumps (e.g., Na+/K+-adenosine triphosphatase [ATPase] pump) and the balance between osmotic pressures exerted by interstitial and intracellular solutes.
The capillary wall is a semipermeable barrier that influences the movement of fluid, nutrients, and waste products between the blood and interstitium. Lipid-soluble substances can pass through capillary endothelium by dissolving in the membrane lipid bilayer, and large proteins can move through the cell by transport within vesicles. Most importantly, water and polar molecules move through interendothelial pores. Normally, these pores are large enough to allow the passage of water, small nutrients (ions, glucose, amino acids), and waste products, yet small enough to prevent the movement of cells and large proteins (albumin and other plasma proteins such as complement, kinin, and coagulation proteins). Local stimuli, such as inflammation, can cause endothelial cells to contract to widen interendothelial pores and allow the passage of larger molecules. Under normal conditions, the composition of plasma and interstitial fluid is very similar, with the exception of the large plasma proteins.
Movement of substances through interendothelial pores and cell membranes is generally passive in response to concentration and pressure gradients. Nutrient-rich arterial blood contains O2, glucose, and amino acids that move down their concentration gradients into the interstitium, where they are available for use by cells. CO2 and waste products generated by cells accumulate in the interstitium and move down their gradient into the venous blood. These gradients become larger in areas where cells are metabolically active.
Water distribution between the plasma and interstitium is determined mainly by osmotic and hydrostatic pressure differentials between the compartments and is described by the following formula (Fig. 2-6):
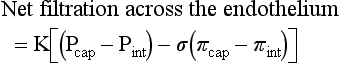
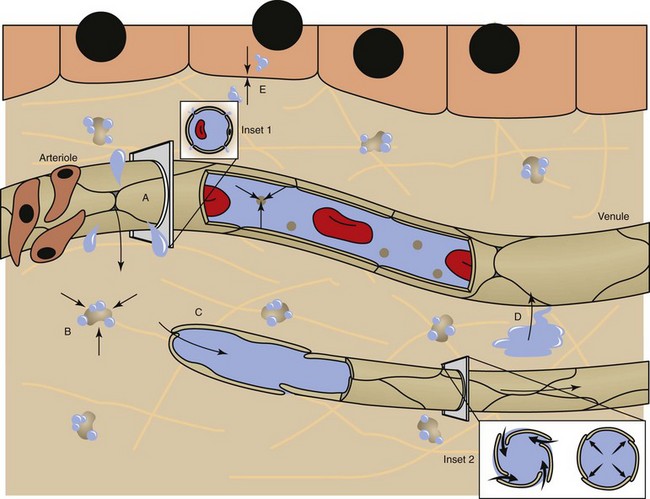
Fig. 2-6 Factors affecting fluid balance in the microcirculation.
Fluid distribution is determined by physical characteristics of the microcirculation and lymphatics and osmotic and hydrostatic forces within the blood and interstitial fluid. Intercellular gaps between endothelium allow movement of fluid and small molecules between the blood and interstitial fluid (insets 1 and 2). A, High arteriolar hydrostatic pressure forces fluid into the interstitium. B, Plasma proteins (e.g., albumin) and molecules within the ECM exert an osmotic effect to attract and retain water. C, Interstitial hydrostatic pressure forces interstitial fluid into lower pressure venules. D, The slight excess of interstitial fluid not returned to the venules enters the lymphatics to be drained from the area. E, Exchange of intracellular and interstitial fluid is balanced by osmotic forces and concentration gradients of electrolytes and other molecules across the cell plasma membrane. Inset 1, Cross-section of a blood vessel capillary showing interendothelial junctions. Endothelium forms end-to-end junctions for movement of fluid and small molecules. Inset 2, Cross-section of a lymphatic capillary showing the interendothelial junctions. Endothelium overlaps to allow movement of larger particles and closure when intravascular pressure forces overlapping endothelium together. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
Although sodium and chloride account for approximately 84% of the total osmolality of plasma, free movement of these electrolytes through interendothelial pores balances their concentrations in the plasma and interstitium, so their contribution to differences in osmotic pressure between these compartments is minimal. In contrast, nonpermeable, suspended plasma proteins make up less than 1% of the total osmolality of plasma. However, because these proteins (particularly albumin) do not readily move through interendothelial pores, they exert a colloidal osmotic pressure that is responsible for the majority of the difference in osmotic pressure between the plasma and interstitium.
In the microcirculation, intravascular and interstitial osmotic pressures and interstitial hydrostatic forces remain relatively constant and favor intravascular retention of fluid. However, high hydrostatic pressures within the arteriolar end of the capillary bed result in a net filtration of fluid into the interstitium. Lower hydrostatic pressures in the venular end of the capillary bed result in a net absorption pressure and reentry of fluid into the microvasculature. Alternatively, filtration and absorption may not occur because of a drop in hydrostatic pressure across individual capillary beds. Instead, filtration may occur across the entire length of capillary beds with open precapillary sphincters and high rates of blood flow, whereas absorption may occur across the entire length of capillary beds with closed precapillary sphincters and low blood flow rates. The slight excess of fluid that is retained in the interstitium and any plasma proteins that have escaped the vasculature enter lymphatic capillaries to be drained from the area.
The constant flow of fluid between the microcirculation and interstitium allows exchange of nutrients and waste products between these two fluid compartments to support cell functions. Additionally, the interstitium provides a fluid buffer to either increase or decrease the plasma volume to assure effective circulatory function. Excessive fluid intake will expand plasma volume and increase hydrostatic pressure, resulting in greater filtration into the interstitium to maintain a relatively constant plasma volume. Reduced fluid intake will decrease plasma volume, shifting the movement of water from the interstitium into the plasma to increase circulating fluid volume.
Abnormal Fluid Distribution
Alteration in any of the factors that regulate normal fluid distribution between the plasma, interstitium, and cells can lead to pathologic imbalances between these compartments.
Imbalance Between Intracellular and Interstitial Compartments
Distribution of fluid between the interstitium and cells is generally dynamic but stable. This stability is necessary to maintain a relatively constant intracellular environment for cell function. Generalized conditions (e.g., alterations in plasma volume) and local stimuli (e.g., inflammation) can result in slight and usually transient shifts in fluid distribution between the interstitium and cells. Excess plasma volume (hypervolemia) results in movement of additional water into the interstitium and ultimately into the cell along both osmotic and hydrostatic gradients, causing cell swelling. In contrast, reduced plasma volume (hypovolemia) can result in a flow of water in the opposite direction resulting in cell shrinkage and decreased interstitial volume. Increased interstitial volume will also cause a slight flow of fluid into cells in the affected region.
Disruption of any of the mechanisms that maintain proper fluid distribution between the cell and interstitium can have serious consequences for the cell. Failure to maintain proper osmotic balance as a result of cell membrane damage or failure of the energy-dependent plasma membrane pumps results in cell swelling, which if not quickly corrected can lead to cell death by osmotic lysis.
Imbalance Between Intravascular and Interstitial Compartments (EDEMA)
Changes in distribution of fluid between the plasma and interstitium are most commonly manifested as edema, which is an accumulation of excess interstitial fluid. Edema occurs by four major mechanisms: (1) increased microvascular permeability, (2) increased intravascular hydrostatic pressure, (3) decreased intravascular osmotic pressure, and (4) decreased lymphatic drainage (Box 2-2).
Mechanisms of Edema Formation
Increased Microvascular Permeability
Increased microvascular permeability is most commonly associated with the initial microvascular reaction to inflammatory or immunologic stimuli. These stimuli induce localized release of mediators that cause vasodilation and increased microvascular permeability. Immediate increases in permeability are induced by mediators such as histamine, bradykinin, leukotrienes, and substance P, which cause endothelial cell contraction and widening of interendothelial gaps. Subsequent release of cytokines such as interleukin-1 (IL-1), tumor necrosis factor (TNF), and γ-interferon induces cytoskeletal rearrangements within endothelial cells that result in endothelial cell retraction and more persistent widening of interendothelial gaps. Movement of intravascular fluid through these gaps into the interstitium results in localized edema that can dilute an inflammatory agent. The reaction terminates as localized edema and regresses when the stimulus is mild. However, most cases progress to the leakage of plasma proteins and emigration of leukocytes as early events in the formation of an acute inflammatory exudate.
Increased Intravascular Hydrostatic Pressure
Increased intravascular hydrostatic pressure is most often due to increased blood volume in the microvasculature. This can be the result of an active increased flow of blood into the microvasculature (hyperemia), such as occurs with acute inflammation. But more commonly, it results from passive accumulation of blood (congestion), often caused by heart failure or localized venous compression or obstruction. Increased microvascular volume and pressure cause increased filtration and reduced or even reversed fluid absorption back into the vessel. When increased hydrostatic pressure affects a localized portion of microvasculature, the edema is localized. In the case of heart failure, congestion and increased hydrostatic pressure can occur in the portal venous system (right heart failure) causing ascites; in the pulmonary venous system (left heart failure) causing pulmonary edema; or in both venous systems (generalized heart failure) causing generalized edema. Generalized edema can result in a reduction of circulating plasma volume and renal hypoperfusion, which activate a variety of volume-regulating compensatory responses. Plasma volume is increased through sodium retention induced by activation of the renin-angiotensin-aldosterone pathways, and water retention mediated by antidiuretic hormone (ADH) release following activation of intravascular volume and pressure receptors. The resulting intravascular volume overload further complicates the dynamics of fluid distribution that accompany heart failure.
Decreased Intravascular Osmotic Pressure
Decreased intravascular osmotic pressure most commonly results from decreased concentrations of plasma proteins, particularly albumin. Hypoalbuminemia reduces the intravascular colloidal osmotic pressure, resulting in increased fluid filtration and decreased absorption and culminating in edema. Hypoalbuminemia is caused by either decreased production of albumin by the liver or excessive loss from the plasma. Decreased hepatic production most commonly occurs because of a lack of adequate protein for the synthetic pathway as a result of malnutrition or intestinal malabsorption of protein. Less often, severe liver disease with decreased hepatocyte mass or impaired hepatocyte function can result in inadequate albumin production. Loss of albumin from the plasma can occur in gastrointestinal diseases characterized by severe blood loss, such as that caused by parasitism. Renal disease, in which glomerular and/or tubular function is impaired, can result in loss of albumin into the urine and dilution of remaining albumin caused by sodium retention and expanded intravascular fluid volume (e.g., nephrotic syndrome). Plasma exudation accompanying severe burns is a less frequent cause of albumin loss. Because of the systemic nature of hypoalbuminemia, edema caused by decreased intravascular osmotic pressure tends to be generalized.
Decreased Lymphatic Drainage
Decreased lymphatic drainage reduces the ability of the lymphatic system to remove the slight excess of fluid that normally accumulates in the interstitium during fluid exchange between the plasma and interstitium. This can occur because of lymph vessel compression by a neoplastic or inflammatory swelling, lymph vessel constriction caused by fibrosis, or internal blockage of a lymph vessel by a thrombus. Edema occurs once the capacity of the damaged lymphatics is exceeded and is localized to the area served by the affected lymphatic vessels.
Morphologic Characteristics of Edema
Edema is morphologically characterized by clear to slightly yellow fluid that generally contains a small amount of protein (transudate), which thickens and expands affected interstitium (Fig. 2-7). When edema occurs in tissues adjacent to body cavities or open spaces, such as alveolar lumens, the increased interstitial pressure often forces fluid into these cavities and spaces. The result can be fluid within alveolar lumens (pulmonary edema; Fig. 2-8), the thoracic cavity (hydrothorax), the pericardial sac (hydropericardium), or the abdominal cavity (ascites or hydroperitoneum; Fig. 2-9). Histologically, edema is an amorphous, pale eosinophilic fluid (hematoxylin and eosin [H&E] stain) because of its protein content (Fig. 2-10). The clinical significance of edema is variable, depending mainly on its location. Subcutaneous edema results in doughy to fluctuant skin and subcutis that is often cooler than adjacent unaffected tissue, but alone has minimal clinical impact (Fig. 2-11). Likewise, ascites does not generally have an impact on the function of abdominal organs. In contrast, edema of a tissue within a confined space, such as the brain in the cranial vault, can result in pressure within the organ that results in serious organ dysfunction. Similarly, filling a confined space with fluid, such as in hydrothorax or hydropericardium, can have a substantial impact on the function of the lungs and heart, respectively. In these situations, edema can have immediate and life-threatening implications.
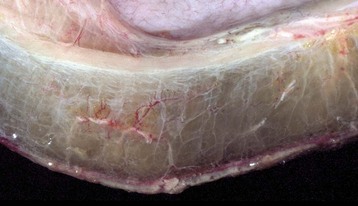
Fig. 2-7 Edema, intestine, submucosa, horse.
Note the clear to slightly yellow fluid (that generally contains a small amount of protein [transudate]), which thickens and expands the affected submucosa. (Courtesy Department of Veterinary Biosciences, The Ohio State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
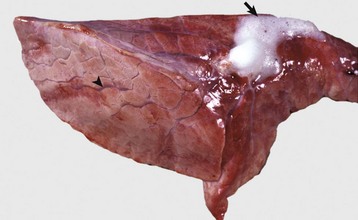
Fig. 2-8 Pulmonary edema, lung, pig.
The lung failed to collapse and is heavy and firm due to edema fluid in alveoli and the interstitium. Note the prominent interlobular septa caused by edema (arrowhead) and the frothy edema fluid exuding from the bronchus (arrow). (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
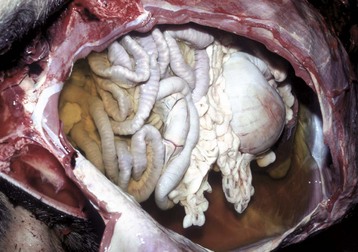
Fig. 2-9 Ascites (hydroperitoneum), peritoneal cavity, dog.
Slightly yellow fluid is present in the peritoneal cavity. When edema occurs in tissue adjacent to body cavities, the increased interstitial pressure forces the edema fluid, which is usually clear to slightly yellow (transudate), into these cavities. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
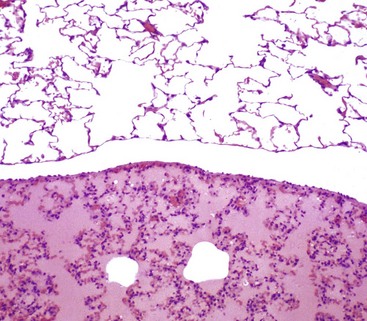
Fig. 2-10 Pulmonary edema, lung, rat.
There is eosinophilic (pink staining) fluid distending the alveoli in the lower specimen. Histologically, edema is an amorphous, pale eosinophilic fluid, and the depth of the eosinophilia is proportional to its protein content. The fluid in this specimen has a high protein content. The upper specimen is normal rat lung. H&E stain. (Courtesy Dr. A. López, Atlantic Veterinary College; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
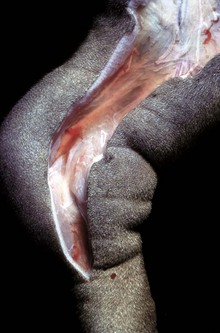
Fig. 2-11 Subcutaneous edema, congenital lymphedema, skin, dog.
This form of edema results in doughy to fluctuant skin and subcutis. Edematous skin is often cooler than adjacent unaffected skin. In congenital lymphedema, the lymph vessels are hypoplastic or aplastic. (Courtesy Dr. H. Liepold, College of Veterinary Medicine, Kansas State University.)
Hemostasis
Hemostasis is the arrest of bleeding. It is a physiologic response to vascular damage and provides a mechanism to seal an injured vessel to prevent blood loss (hemo = blood, stasis = halt, slow). Hemostasis is a finely regulated process that predominantly involves interactions between endothelium, platelets, and coagulation factors. It normally occurs only at the site of vascular injury, without affecting fluidity and flow of blood in normal undamaged vasculature. Disruption of the delicate balance of hemostasis can result in the pathologic states of blood loss (hemorrhage) or inappropriate thrombus formation (thrombosis).
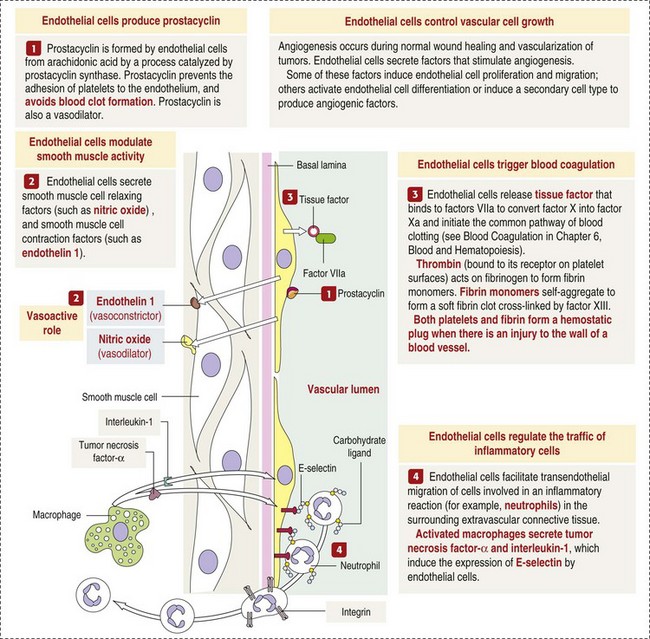
Normal endothelium provides a surface that promotes the smooth, nonturbulent flow of blood. It produces and responds to mediators that enhance vasodilation, and inhibit platelet activation and coagulation. In contrast, after injury or activation, endothelium produces or responds to mediators that induce vasoconstriction, enhance platelet adhesion and aggregation, and stimulate coagulation (Box 2-3).
Platelets are anucleate cell fragments derived from megakaryocytes. Their major role in hemostasis is to form the initial plug that covers and seals a small area of vascular damage. After vascular damage, platelets adhere to subendothelial collagen and other ECM components (e.g., fibronectin, adhesive glycoproteins, and proteoglycans). Adhered platelets express receptors that promote aggregation of additional platelets and become activated to release the products of their cytoplasmic granules and produce other mediators of coagulation (e.g., thromboxane; Box 2-4). The phospholipid surfaces of aggregated platelet membranes also provide a surface to concentrate activated coagulation factors to promote coagulation.
Coagulation factors are plasma proteins produced mainly by the liver. Their purpose in hemostasis is to form fibrin. Coagulation factors are divided into (1) a structurally related and functionally interdependent contact group (prekallikrein, high molecular weight kininogen [HMWK], and factors XII and XI); (2) a vitamin K–dependent group (factors II, VII, IX, and X); and (3) a highly labile fibrinogen group (factors I, V, VIII, and XIII). Circulating coagulation factors are activated in a cascade fashion by hydrolysis of arginine- or lysine-containing peptides to convert them to enzymatically active serine proteases (except for factor XIII, which has cysteine-rich active sites). The contact group factors are activated by contact with collagen or subendothelial components to initiate coagulation by the intrinsic pathway. The extrinsic pathway of coagulation is activated by release of tissue factor (TF, factor III) from damaged cells. The vitamin K–dependent coagulation factors play an important role in localizing coagulation by γ-carboxylating glutamic acid residues of N-terminal ends of precursor factors, so they can bind calcium to form calcium bridges with platelet phospholipids.
Hemostatic Process
The sequence of events that contribute to hemostasis are (1) transient vasoconstriction and platelet aggregation to form a platelet plug at the site of damage (primary hemostasis), (2) coagulation to form a meshwork of fibrin (secondary hemostasis), (3) fibrinolysis to remove the platelet/fibrin plug (thrombus retraction), and (4) tissue repair at the damaged site (Fig. 2-12).
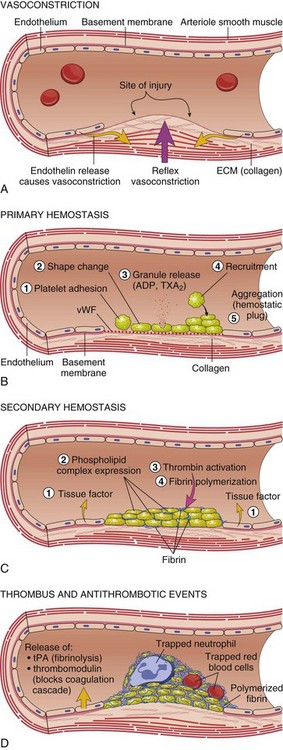
Fig. 2-12 Diagrammatic representation of the normal hemostatic process.
A, After vascular injury, local neurohumoral factors induce a transient vasoconstriction. B, After endothelial injury and disruption that exposes the subendothelial extracellular matrix (ECM), platelets adhere to the ECM via von Willebrand’s factor (vWF) and are activated, undergoing a shape change and granule release; released adenosine diphosphate (ADP) and thromboxane A2 (TXA2) lead to further platelet aggregation to form the primary hemostatic plug. C, Local activation of the coagulation cascade (involving tissue factor and platelet phospholipids) results in fibrin polymerization, “cementing” the platelets into a definitive secondary hemostatic plug. D, Counter-regulatory mechanisms, such as release of tissue plasminogen activator (tPA) (fibrinolytic) and thrombomodulin (interfering with the coagulation cascade), limit the hemostatic process to the site of injury. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Primary Hemostasis
Primary hemostasis includes the initial vascular and platelet response to injury. Neurogenic stimuli and mediators released locally by endothelium and platelets cause vasoconstriction immediately after damage (Fig. 2-12, A). The nature and effectiveness of vasoconstriction is partially determined by the size of the affected vessel, the amount of smooth muscle it contains, and endothelial integrity. Narrowing of the vessel lumen allows opposing endothelial surfaces to come into contact with and sometimes adhere to each other to reduce the volume of blood flowing through the damaged area. Platelets can directly adhere to the exposed subendothelial matrix of collagen, fibronectin, and other glycoproteins and proteoglycans (Fig. 2-12, B). However, more efficient adhesion occurs when von Willebrand’s factor released by local activated endothelium coats subendothelial collagen to form a specific bridge between collagen and the glycoprotein platelet receptor GPIb. At this stage and without further stimulation, adhered and aggregated platelets may disaggregate. Otherwise, platelets within the aggregate secrete the contents of their dense bodies and α-granules and produce substances such as thromboxane to accelerate hemostasis. Adenosine diphosphate (ADP) released from dense granules triggers the binding of fibrinogen to platelet receptor GPIIb-IIIa, resulting in the formation of fibrinogen bridges that link platelets into a loose aggregate. Platelet contraction consolidates this loose aggregate into a dense plug, which covers the damaged area. When vascular injury is minimal, platelet plugs alone may resolve the damage. If not, the exposed collagen and aggregated platelet phospholipids promote secondary hemostasis at the site.
Secondary Hemostasis
In most cases of vascular damage, the formation of fibrin is important for the prevention of blood loss. Fibrin is the end-product of a series of enzymatic reactions involving coagulation factors, nonenzymatic cofactors, calcium, and phospholipid membranes derived mainly from platelets (Fig. 2-12, C). Three coagulation pathways have been classically used to describe the coagulation process and formation of fibrin in vitro. These pathways provide a useful starting point for understanding coagulation (Figs. 2-13 and 2-14). However, integrated models of coagulation more adequately emphasize the interrelatedness of these pathways, the multiple positive and negative control loops within the system, and amplification of the process on affected cell surfaces.
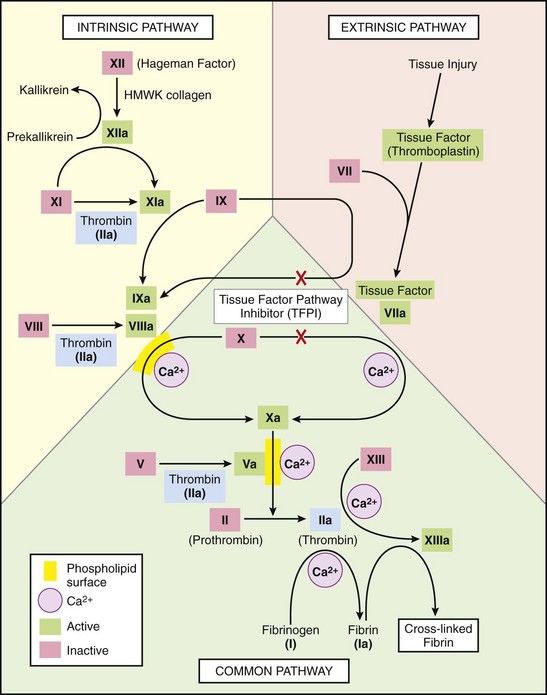
Fig. 2-13 The coagulation cascade.
Note the common link between the intrinsic and extrinsic pathways at the level of factor IX activation. Factors in red boxes represent inactive molecules; activated factors are indicated with a lower case “a” and a green box. HMWK, High molecular weight kininogen. Not shown are the anticoagulant inhibitory pathways. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
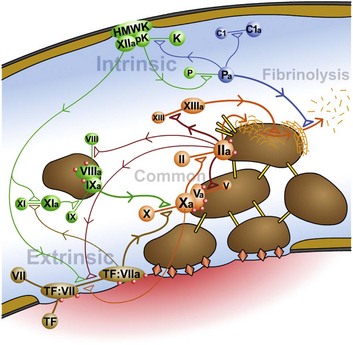
Fig. 2-14 Coagulation, fibrinolysis, and other pathways are highly integrated to balance the host response to injury.
Intrinsic coagulation is initiated by binding of high molecular weight kininogen (HMWK), factor XII, and prekallikrein (pK) to altered endothelial surfaces. Activated products of this reaction (factor XIIa and kallikrein [K]) directly or indirectly result in the formation of factor XIa (intrinsic coagulation pathway), factor VIIa (extrinsic coagulation pathway), plasmin (Pa) (fibrinolysis), and complement fragments C3a, C3b, C5a (complement cascade). Extrinsic coagulation is initiated by release of tissue factor (TF) from areas of damaged endothelium, with subsequent binding of TF to factor VII. The TF:VII complex can be activated by a wide variety of agents. Activation of factor X initiates the common cascade to ultimately result in cleavage of fibrinogen into fibrin. In addition to its role in the common pathway, factor IIa (thrombin) also influences both the intrinsic (factors XI and VIII) and extrinsic (TF:VII) coagulation pathways. Additional interactions between these factors, which are not shown in the figure, are described in the text. Specific effects of kallikrein include cleavage and activation of factors XII, IX, and VII, plasminogen, HMWK, and complement fragment C5. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
Intrinsic Pathway: Intrinsic coagulation is a complex and highly interrelated process that is initiated by the contact group of coagulation factors (see Figs. 2-13 and 2-14). Prekallikrein and factor XI normally circulate bound to HMWK, which acts as a catalytic factor for their activation. After vascular injury, circulating prekallikrein-HMWK and factor XII form a complex on negatively charged endothelial or subendothelial surfaces, which results in activation of factor XII (factor XIIa). Factor XIIa initiates a series of reactions that affect coagulation, as well as kinin formation, complement activation, and fibrinolysis. Factor XIIa activates factor XI (XIa), interacts with prekallikrein to form kallikrein, and interacts with HMWK to form kinins. Cleavage of factor XIIa by kallikrein, plasmin, and other proteolytic enzymes forms fragments (factor XIIf), which have activity that is similar to but much weaker than factor XIIa. Both kallikrein and factor XIa in the presence of Ca2+ can activate factor IX (factor IXa). Factor IXa then binds to platelet phospholipids in a complex with Ca2+ and factor VIII. After modification of factor VIII by thrombin into factor VIIIa, the complex of factor VIIIa-factor IXa/Ca2+-phospholipid (tenase) activates factor X to initiate the common coagulation pathway.
Extrinsic Pathway: Release of factor III (TF) from cells underlying damaged endothelium, or from activated endothelium, initiates the extrinsic pathway (see Figs. 2-13 and 2-14). TF is a high molecular weight phospholipid-containing glycoprotein found in the plasma membrane of many cells, including activated, but not resting, endothelium. Endothelial cell production of TF is stimulated by substances such as endotoxin, TNF, IL-1, transforming growth factor-β (TGF-β), and thrombin. When circulating factor VII comes into contact with TF, it forms a Ca2+-dependent TF:VII complex on the TF-expressing surface. Although this complex may have some enzymatic activity, activation of factor VII by substances such as factors XIIa, XIIf, IXa, Xa, IIa, and kallikrein results in the much more active TF:VIIa complex. This complex along with Ca2+ activates factor X to initiate the common pathway.
Common Pathway: The intrinsic and extrinsic pathways merge with the activation of factor X (see Figs. 2-13 and 2-14). Factor Xa is bound to endothelial or platelet membrane phospholipids where it can directly convert factor II into factor IIa (thrombin). However, when factor Xa is combined with factor Va and Ca2+ (prothrombinase complex), this reaction occurs much more rapidly. Thrombin is a multifunctional mediator whose major function is to cleave fibrinopeptides A and B from factor I (fibrinogen) to form fibrin monomers (Fig. 2-15). Removal of these fibrinopeptides reduces intermolecular repulsive forces so that fibrin monomers spontaneously form weak H+ bonds and self-polymerize into soluble fibrin polymers. Factor XIIIa, formed by the action of factors Xa and IIa on factor XIII in the presence of Ca2+, catalyzes the formation of covalent bonds that cross-link adjacent fibrin molecules to make the polymer insoluble. Cross-linking of the fibrin network, along with concurrent platelet contraction and the presence of abundant calcium, thrombin, and adenosine triphosphate (ATP), causes retraction of the fibrin-platelet thrombus. Retraction reduces the size of the thrombus to allow blood flow to continue and to pull damaged vessel edges closer together for efficient healing.
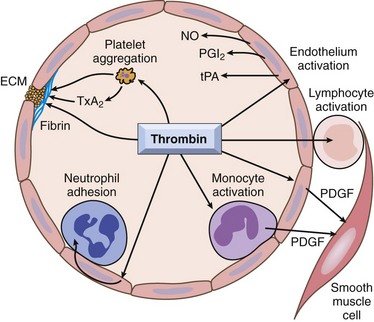
Fig. 2-15 The central roles of thrombin in hemostasis and cellular activation.
In addition to a critical function in generating cross-linked fibrin (via cleavage of fibrinogen to fibrin and activation of factor XIII), thrombin also directly induces platelet aggregation and secretion (e.g., TXA2). Thrombin also activates endothelium to generate leukocyte adhesion molecules and a variety of fibrinolytic (tPA), vasoactive (NO, PGI2), or cytokine (PDGF) mediators. Likewise, mononuclear inflammatory cells may be activated by the direct actions of thrombin. ECM, Extracellular matrix; NO, nitric oxide; PDGF, platelet-derived growth factor; PGI2, prostacyclin; TXA2, thromboxane A2; tPA, tissue plasminogen activator. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Integrated Model of Coagulation
In vivo coagulation is more like an integrated web rather than a series of independent cascades. The major stimulus for coagulation in vivo is exposure of plasma to TF and subsequent extrinsic coagulation. Two important events occur after the formation of the TF/factor VIIa complex on damaged, TF-expressing surfaces. Factor X is activated as described for the common pathway, and factor IX is activated to allow a bypass of the contact phase of classical intrinsic coagulation. Factor Xa remains localized on the damaged cell surface to initiate the formation of a small amount of thrombin. Although the amount of thrombin generated is insufficient to convert significant amounts of fibrinogen into fibrin, it does activate platelets and factors V, VIII, XI, and XIII on TF-expressing surfaces. Factor IXa can bind to the surface of activated platelets in the area to initiate the formation of tenase complexes, which activates additional factor X of the common pathway. Thrombin-activated or intrinsically activated factor XIa can also participate by activating additional factor XI on platelet surfaces. The thrombin-initiated activation of these different factors provides an amplification of the critical reactions necessary to generate large amounts of thrombin for the subsequent conversion of fibrinogen into fibrin. Other, probably less important links between the pathways also exist. For example, intrinsic factors XIIa, XIIf, and IXa and kallikrein can activate extrinsic factor VII to provide additional amplification of this pathway.
The interrelatedness of coagulation pathways also extends to anticoagulant reactions. When excessive levels of thrombin are generated, thrombin destroys rather than activates factors V and VIII. When thrombin binds to thrombomodulin on endothelial surfaces, it activates protein C, a potent anticoagulant (see section on Coagulation Inhibitors). Intrinsic pathway factors XIIa, XIIf, and XIa and kallikrein not only participate in fibrin formation but also initiate fibrinolysis by cleaving plasminogen into plasmin (see section on Thrombus Dissolution).
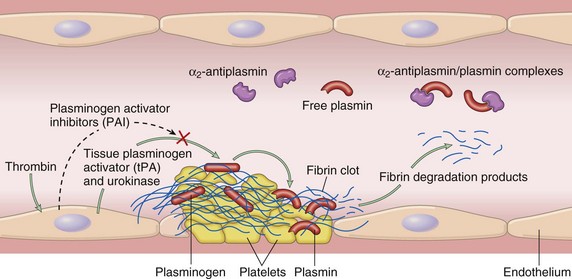
Thrombus Dissolution
The purpose of a fibrin-platelet thrombus is to form a temporary patch that is dissolved after healing of the vessel (thrombolysis). The rate of dissolution must be balanced so that it does not occur so quickly that bleeding returns but is not prolonged so that vessel occlusion may occur (Fig. 2-16). Fibrin dissolution (fibrinolysis) is initiated immediately on vessel injury by the cleavage of the plasma protein plasminogen into plasmin (Fig. 2-17). Plasminogen is activated by a wide variety of proteases, including activated contact group coagulation factors, plasminogen activators present within endothelium and other tissues (tissue plasminogen activator [tPA]), and activators present in secretions and fluids. Plasminogen adsorbs to fibrin within a thrombus, so that on activation the plasmin is localized to the site of the thrombus. The presence of fibrin increases the efficiency of tPA-dependent plasmin generation by nearly twofold. Additionally, by binding to fibrin, plasmin is protected from its major inhibitor (α2-antiplasmin). The bound plasmin restricts thrombus size by degrading both cross-linked (insoluble) fibrin within the thrombus and fibrinogen, so that additional fibrin formation is inhibited. Dissolution of insoluble, but not soluble, fibrin by plasmin results in the formation of fibrin degradation products (FDPs). FDPs are various-sized fragments of fibrin and fibrinogen that can impair hemostasis. Collectively, FDPs inhibit thrombin, interfere with fibrin polymerization, and can coat platelet membranes to inhibit platelet aggregation.
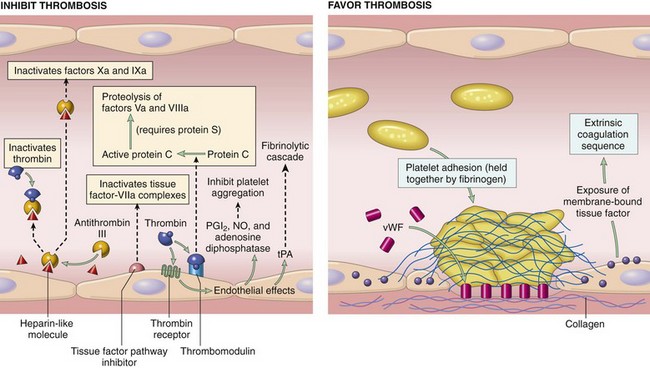
Fig. 2-16 Schematic illustration of some of the procoagulant and anticoagulant activities of endothelial cells.
Not shown are the profibrinolytic and antifibrinolytic properties. vWF, von Willebrand’s factor; PGI2, prostacyclin; NO, nitric oxide; tPA, tissue plasminogen activator. Thrombin receptor is referred to as protease-activated receptor (PAR). (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Regulation of Hemostasis
The potent biologic effects of hemostatic products must be finely regulated to achieve appropriate hemostasis, without creating detrimental effects associated with too little or too much activity. Coagulation factors are continuously activated at a low, basal level to keep the system primed for a rapid response to an injurious stimulus. Proteins that inhibit or degrade activated hemostatic products are present in the plasma or are locally produced at the site of hemostasis (see Fig. 2-17). These products help confine hemostasis to a site of vascular damage and inhibit hemostatic reactions in normal vasculature. Regulation is also achieved by simple dilution of activated agents as blood removes them from the area, and the factors are removed from the circulation by the liver and spleen.
Coagulation Inhibitors
The major anticoagulant-antithrombotic systems on endothelial cells are the protein C-protein S-thrombomodulin system and endothelial heparan sulfate to which antithrombin III (ATIII) and tissue factor pathway inhibitor (TFPI) are bound. ATIII is the most potent and clinically significant of the coagulation inhibitors, accounting for nearly 80% of the thrombin-inhibitory activity of plasma. ATIII is a circulating serine protease produced by endothelium and hepatocytes that will degrade all activated coagulation factors except for factor VIIa. However, its most important action is the neutralization of thrombin and factor Xa. ATIII can bind heparan sulfate present on the surface of normal endothelium and platelets to localize it to the site where it is most needed to inactivate thrombin and factor Xa. Through this binding, heparin accelerates the rate of ATIII-induced serine protease inactivation by 2000- to 10,000-fold. ATIII also inhibits fibrinolysis (by inhibiting plasmin and kallikrein), kinin formation, and complement activation. Although the major role of heparin is to bind and enhance the activity of ATIII, it also inhibits coagulation by enhancing the release of TFPI from endothelial cells and interfering with binding of platelet receptors to von Willebrand’s factor.
The protein C pathway also plays a critical role in preventing thrombosis. Proteins C and S are vitamin K–dependent glycoproteins that, when complexed together on phospholipid surfaces, potently inhibit coagulation by destroying factors Va and VIIIa. An important step in this process is the activation of protein C by thrombin, a reaction that normally occurs at low levels but that increases nearly 20,000-fold after the binding of thrombin to the endothelial receptor thrombomodulin. This reaction is further enhanced by the presence of a protein C receptor on the surface of endothelial cells. Protein S, in addition to serving as a nonenzymatic cofactor with protein C, can independently inhibit factors VIIIa, Xa, and Va. Binding of thrombin to thrombomodulin also results in the loss of the procoagulant functions of thrombin. The protein C-S complex may also enhance fibrinolysis by neutralizing plasminogen activator inhibitors.
TFPI is a significant inhibitor of extrinsic coagulation, which functions synergistically with protein C and ATIII to suppress thrombin formation. TFPI is a plasma protein derived mainly from endothelium and smooth muscle cells that forms a complex with factor Xa on the endothelial-bound TF:VIIa molecule to inhibit subsequent factor X activation. TFPI can interact with VIIa without Xa but at a slow rate. Therefore TFPI does not substantially inhibit extrinsic coagulation until factor Xa levels increase, after which TFPI provides negative feedback for further generation of Xa by the TF:VIIa complex.
Fibrinolytic Inhibitors
Major inhibitors of fibrinolytic agents include plasminogen activator inhibitor-1 (PAI-1) and antiplasmins, which include α2-antiplasmin, α2-macroglobulin, α1-antitrypsin, antithrombin III, and C-1 inactivator. PAI-1 inhibits tPA and urokinase, therefore inhibiting fibrinolysis and promoting fibrin stabilization. PAI-1 also inactivates activated protein C, plasmin, and thrombin. The antiplasmins function in a cooperative fashion to prevent excessive plasmin activity so that a thrombus can dissolve at a slow and appropriate rate. α2-Antiplasmin is the first to bind and neutralize plasmin. When its binding capacity is saturated, excess plasmin is taken up by α2-macroglobulin. α2-Macroglobulin also binds to certain activated factors, such as thrombin, and physically entraps but does not degrade their active sites. When α2-macroglobulin is saturated, plasmin binds to α1-antitrypsin. α1-Antitrypsin is a weak inhibitor of fibrinolysis, but a potent inhibitor of factor XIa. In addition to their fibrinolytic roles, α1-antitrypsin and α2-macroglobulin are the major plasma inhibitors of activated protein C.
Hemostasis and Other Host Responses
Hemostatic pathways are highly integrated, and many factors within the pathways have multiple roles. Thrombin has a major procoagulant role to cleave factor I to yield fibrin monomers. Thrombin also activates factors V, VIII, XI, and XIII and is a potent activator of platelets. In contrast, high concentrations of thrombin destroy, rather than activate, factors V and VIII. When thrombin binds to thrombomodulin on endothelial surfaces, it activates protein C, a potent anticoagulant.
An important link between intrinsic and extrinsic pathways is the TF/factor VIIa complex. This complex is the major component of extrinsic coagulation, but it can also activate factor IX to allow a bypass of the contact phase of intrinsic coagulation. In turn, intrinsic factors XIIa, XIIf, and IXa and kallikrein can activate factor VII, which greatly increases the efficiency of extrinsic coagulation. These features give the TF/factor VIIa complex a central role in efficient hemostasis. Extrinsic coagulation and the TF/factor VIIa complex are probably the most important mechanisms for in vivo hemostasis because bleeding tendencies are not usually associated with factor XII, prekallikrein, and HMWK deficiencies and some factor XI deficiencies in humans and animals. Although the intrinsic pathway does not appear to be essential for in vivo hemostasis, it does appear to play an essential role in the formation of pathologic thrombi in response to major vascular damage.
Hemostasis is closely tied to inflammation and other host responses. A prothrombotic environment is also proinflammatory. Inflammatory stimuli, such as IL-1 and TNF, activate endothelium to produce TF and to increase their expression of leukocyte adhesion molecules. Thrombin and histamine released by degranulating mast cells also stimulate the expression of the adhesin P-selectin. In early stages of inflammation, leukocytes can loosely attach and roll along endothelium or adhered platelets by interacting with endothelial or platelet P-selectin. During this interaction, the neutrophil αMβ2 integrin may localize neutrophils to fibrinogen on the surface of activated platelets to promote the conversion of fibrinogen into fibrin. An enhanced prothrombotic environment during inflammation also occurs because of decreased thrombomodulin function in response to inflammatory products such as endotoxin, IL-1, TNF, and TGF-β. Additionally, adhered or migrating neutrophils and platelets can release lysosomal proteases (e.g., elastase, collagenase, and acid hydrolases), which cleave many products on endothelial or platelet surfaces. The conversion of prekallikrein to kallikrein during the contact phase of intrinsic coagulation is another source of integration between hemostatic, fibrinolytic, and inflammatory pathways. Kallikrein is chemotactic, can directly cleave C5 to C5a and C5b, can cleave HMWK to form bradykinin, and can convert plasminogen into plasmin. Plasmin also influences other host responses by cleaving C3 to generate C3a and C3b. Mitogenic factors produced by activated endothelium and platelets (e.g., platelet-derived growth factor [PDGF], TGF-β, and vascular endothelial growth factor [VEGF]) contribute to healing of damaged tissue. Some hemostatic reactions initiate pathways that have multiple and sometimes opposite outcomes. Intrinsic pathway factors XIIa, XIIf, and XIa and kallikrein not only initiate the formation of fibrin but also initiate fibrinolysis by cleaving plasminogen into plasmin. Factor XIIa not only participates directly in intrinsic coagulation and fibrinolysis but also indirectly initiates kinin formation and complement activation by converting prekallikrein to kallikrein. Additionally, both kallikrein and plasmin can directly activate factor XII to result in autoamplification of all factor XIIa pathways. Not all stimuli that activate factor XII lead to fibrin formation. Certain stimuli (e.g., misfolded proteins such as amyloid) activate inflammatory factor XII pathways (kallikrein-kinin) without concurrent activation of coagulation (via subsequent activation of factor XI). Other hemostatic products that influence other host systems include factor Xa, thrombin, and fibrinopeptides, all of which have inflammatory and coagulation functions. These interactions indicate the fine balance within the hemostatic system and the interrelatedness of hemostasis, inflammation, and other host responses.
Disorders of Hemostasis: Hemorrhage and Thrombosis
The purpose of hemostasis is to prevent blood loss after vascular damage, while at the same time maintaining blood in a fluid state so that it flows freely through a normal vasculature. Failure of hemostasis can result in the extravascular loss of blood (hemorrhage) or the inappropriate formation of intravascular clots (thrombosis).
Hemorrhage
Hemorrhage occurs because of abnormal function or integrity of one or more of the major factors that influence hemostasis—the endothelium and blood vessels, platelets, or coagulation factors.
Abnormalities in blood vessels can result from various inherited or acquired problems. Trauma can physically disrupt a vessel and cause hemorrhage by rhexis (rhexis = breaking forth, bursting). Hemorrhage by rhexis can also occur following vascular erosion by inflammatory reactions or invasive neoplasms. Certain fungi commonly invade and damage blood vessels to cause extensive local hemorrhage (e.g., internal carotid artery erosion secondary to guttural pouch mycosis in horses). More commonly, minor defects in otherwise intact blood vessels allow small numbers of erythrocytes to escape by diapedesis (dia = through, pedian = leap). Endotoxemia is a common cause of endothelial injury that results in small widespread hemorrhages (Fig. 2-18). Infectious agents, such as canine adenovirus-1, or chemicals, such as uremic toxins, can also damage endothelium. Similarly, immune complexes can become entrapped between endothelial cells and activate complement and neutrophil influx to result in damage to the endothelium and vessel wall (type III hypersensitivity reaction). Developmental collagen disorders, such as the Ehlers-Danlos syndrome, are sometimes accompanied by hemorrhage. Affected blood vessels contain abnormal collagen in their basement membranes and surrounding supportive tissue, resulting in vascular fragility and predisposition to leakage or damage. Similar hemorrhages occur due to collagen defects in guinea pigs or primates with vitamin C deficiency.
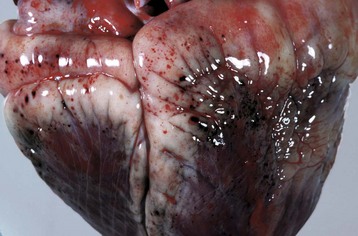
Fig. 2-18 Hemorrhage, endotoxemia, heart, cow.
Note the epicardial and subepicardial hemorrhages in the fat of the coronary groove (a common site), from injury to the endothelium from endotoxin (component of the cell wall of Gram-negative bacteria). The smaller, pinpoint hemorrhages (1 to 2 mm) are petechiae. The larger, blotchy hemorrhages (3 to 5 mm) are ecchymoses. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Decreased platelet numbers (thrombocytopenia) or abnormal platelet function (thrombocytopathy) can cause hemorrhage. Thrombocytopenia can result from decreased production, increased destruction, or increased use of platelets. Decreased production generally occurs following megakaryocyte damage or destruction as a result of causes such as radiation injury, estrogen toxicity, cytotoxic drugs, and viral or other infectious diseases (e.g., feline and canine parvoviruses). Increased platelet destruction is often immune-mediated. Autoimmune destruction due to antibody production against platelet membrane components, such as GPIIb and GPIIIa, can occur after immune dysregulation (e.g., systemic lupus erythematosus). Alteration of platelet membranes by drugs or infectious agents may also stimulate immune-mediated destruction or removal of platelets from the circulation. Isoimmune destruction of platelets in neonatal pigs has occurred after ingestion of colostrum-containing antiplatelet antibodies. Viral diseases (e.g., equine infectious anemia and feline immunodeficiency syndrome) and arthropod-borne agents are often associated with platelet destruction and their removal by the spleen. The most common cause of increased platelet use is diffuse endothelial damage or generalized platelet activation, which initiates disseminated intravascular coagulation (DIC). With DIC there is widespread intravascular coagulation and platelet activation, which rapidly results in consumption of platelets and coagulation factors (see section on Thrombosis). This results in progressive thrombocytopenia and hemorrhage as the syndrome escalates. Another platelet consumption disease that is not accompanied by coagulation is thrombotic thrombocytopenic purpura. In this condition, platelet aggregates form in the microvasculature, possibly the result of increased release of proagglutinating substances by normal or damaged endothelium.
Decreased platelet function is usually associated with an inability to adhere or aggregate at a site of vascular injury. Inherited problems of platelet function in humans include deficiency of GPIb on the platelet surface (Bernard-Soulier syndrome), deficient or defective GPIIb and GPIIIa on the platelet surface (Glanzmann’s thrombasthenia), and deficient release of platelet granule content (“storage pool disease”). Glanzmann’s thrombasthenia is a rare disease that has been reported in Otterhound and Great Pyrenees dogs. In these dogs, there is prolonged bleeding and hematoma formation from minor injury and spontaneous epistaxis because of a mutation affecting a Ca2+-binding domain of the extracellular portion of GPIIb. Abnormal synthesis or release of platelet granule content has been reported in Simmental cattle, dogs (Spitz, Basset hound, and American foxhounds), cats, and fawn-hooded rats. Defective platelet storage of ADP occurs in the Chédiak-Higashi syndrome (Aleutian mink, cattle, Persian cats, and killer whales). Acquired platelet inhibition and dysfunction is most often associated with administration of nonsteroidal antiinflammatory drugs such as aspirin. Aspirin inhibits the cyclooxygenase pathway of arachidonic acid metabolism, thus decreasing thromboxane production to result in reduced platelet aggregation. Platelet function is also inhibited by uremia because of renal failure. Secondary platelet dysfunction can also occur because of deficiencies of factors necessary for normal platelet function. In von Willebrand’s disease, or in autoimmune or myeloproliferative disorders in which autoantibodies against von Willebrand’s factor are produced, the amount of functional von Willebrand’s factor is decreased. This results in decreased platelet adhesion following vascular damage with either subclinical or severe hemorrhage.
Decreased concentrations or function of coagulation factors can also result in hemorrhage. Inherited deficiencies in coagulation factors have been recognized in many different breeds of dogs and less often other species (Box 2-5). These conditions are characterized by hemorrhage that can range from subclinical to severe. In many cases, the coagulation factor deficiency is recognized because of prolonged bleeding after venipuncture or surgery, but otherwise has minimal significance to the animal. Other inherited deficiencies are characterized by severe episodes of hemorrhage that begin soon after birth.
Acquired defects in coagulation can be caused by decreased production or increased use of coagulation factors. Severe liver disease results in decreased synthesis of most coagulation factors. Production of coagulation factors II, VII, IX, X and proteins C and S is reduced by vitamin K deficiency. Decreased vitamin K production, absorption, or function will reduce conversion of glutamic acid residues into γ-carboxyglutamic acid on these factors. Common substances that competitively inhibit this conversion include dicumarol in moldy sweet clover (Melilotus alba), warfarin-containing rodenticides, and sulfaquinoxaline (Fig. 2-19). An inherited deficiency of binding of γ-glutamyl-carboxylase with vitamin K has been reported in British Devon Rex cats. The most common acquired cause of decreased coagulation factors is increased consumption associated with DIC.
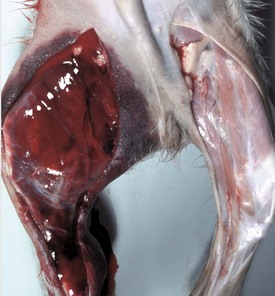
Fig. 2-19 Hemorrhage, anticoagulant (warfarin-containing) rodenticide toxicosis, skin and subcutis, medial aspect of the right hindleg, dog.
There is a large area of extensive hemorrhage in the subcutis. This lesion was attributed to decreased production of coagulation factors II, VII, IX, and X and proteins C and S resulting from a deficiency of vitamin K induced by warfarin. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
The appearance of hemorrhage depends on its cause, location, and severity. Hemorrhage within tissue is often characterized based on size. A petechia (pl. petechiae) is a pinpoint (1 to 2 mm) hemorrhage that occurs mainly because of diapedesis associated with minor vascular damage (see Fig. 2-18). An ecchymosis (pl. ecchymoses) is a larger (up to 2 to 3 cm in diameter) hemorrhage that occurs with more extensive vascular damage (Fig. 2-20), whereas suffusive hemorrhage affects larger contiguous areas of tissue than the other two types (Fig. 2-21). Hemorrhage that occurs in a focal, confined space forms a hematoma. Hematomas are most common in the ears of long-eared dogs or pigs and in the spleen after trauma to the vasculature (Fig. 2-22). The hematoma grows in size until the pressure exerted by the extravascular blood matches that within the injured vessel or the vessel seals internally by hemostasis. Hemorrhage into body cavities results in pooling of coagulated or noncoagulated blood within the cavity and is classified by terms such as hemoperitoneum (blood in the peritoneal cavity), hemothorax (blood in the thoracic cavity), and hemopericardium (blood in the pericardial sac) (Fig. 2-23).

Fig. 2-20 Ecchymotic hemorrhages (ecchymoses), subcutis, rabbit.
Ecchymoses result from moderate injury to endothelial cells in the capillary beds. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
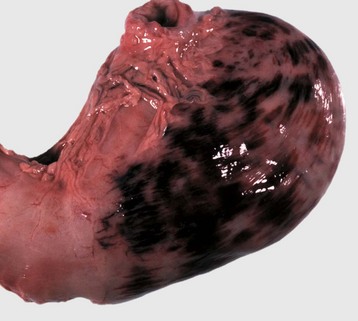
Fig. 2-21 Suffusive hemorrhage, serosa, stomach, dog.
Suffusive hemorrhage results from severe injury to endothelial cells in the capillary beds. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
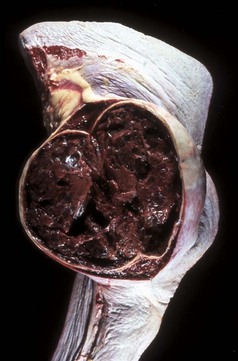
Fig. 2-22 Organizing hematoma, spleen, horse.
Trauma to the spleen has caused damage to the splenic red pulp and its vessels, resulting in bleeding into the splenic parenchyma, forming a hematoma. Note that this hematoma is not acute but is several days old because the blood clot is being degraded. The hematoma is contained by the splenic capsule. (Courtesy Dr. H.B. Gelberg, College of Veterinary Medicine, Oregon State University.)
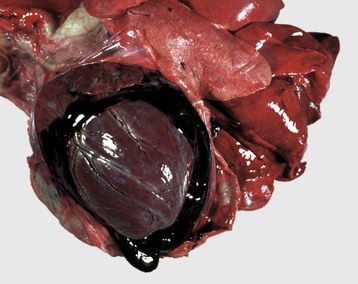
Fig. 2-23 Hemopericardium, pericardial sac, dog.
Hemorrhage into the pericardial sac has caused its distention. Extensive hemopericardium can interfere with the dilatation and contraction of the ventricles, causing cardiac tamponade. Both coagulated and noncoagulated blood are present in the pericardial sac. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
The significance of hemorrhage depends mainly on the amount, rate, and location of the blood loss. In most cases, blood loss occurs locally and is quickly stopped by hemostatic processes that seal the damaged vessel. In more severe cases, blood loss continues until local tissue pressure matches intravascular pressure and ends the hemorrhage (such as occurs with hematoma formation). When these mechanisms fail to stop blood loss, significant hemorrhage can occur externally or internally into body cavities. Rapid loss of substantial amounts of blood, such as occurs because of traumatic injury of a large vessel, can lead to hypovolemia, decreased tissue perfusion, and hypovolemic shock (see later discussion in this chapter). In contrast, slow rates of blood loss can be totally or partially compensated for by increased hematopoiesis. Many cases of gastric ulceration and hemorrhage are characterized by persistent but slow rates of blood loss. Some hemorrhages can create pressure that interferes with tissue function. This is most significant in vital organs or in tissue with little room to expand in response to the pressure, such as the brain and heart.
Thrombosis
Thrombosis is characterized by the formation of an inappropriate clot of fibrin and/or platelets along with other blood elements (thrombus; pl. thrombi) on the wall of a blood or lymphatic vessel or heart (mural thrombus), or free in their lumens (thromboembolus). Major determinants of thrombosis are historically referred to as Virchow’s triad and include the endothelium and blood vessels (vascular injury), coagulation factor and platelet activity (coagulability), and the dynamics of blood flow (stasis or turbulence) (Fig. 2-24 and Box 2-6).
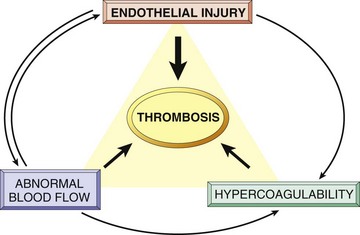
Fig. 2-24 Virchow triad in thrombosis.
Endothelial integrity is the single most important factor. Note that injury to endothelial cells can affect local blood flow and/or coagulability; abnormal blood flow (stasis or turbulence) can, in turn, cause endothelial injury. The elements of the triad may act independently or may combine to cause thrombus formation. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Alterations in the endothelium are the most important factor in thrombosis and can result in increased production of procoagulant substances and decreased production of anticoagulant substances. Endothelial injury and exposure of TF and subendothelial components, such as collagen and fibronectin, are potent stimuli for platelet aggregation and coagulation. Causes of injury are widely varied in their severity and cause and include trauma, vasculitis caused by infection or immunologic reactions, metabolic disorders, neoplasia, and toxins. Additionally, loss of anticoagulant properties of normal endothelium combined with local release of procoagulant substances can result in fibrin formation. Platelets may also adhere to intact endothelium by interacting with altered proteoglycans in the endothelial glycocalyx. Reduced prostacyclin synthesis may also increase platelet adhesion to endothelium.
Abnormal blood flow increases the risk of thrombosis. Reduced blood flow may occur systemically with heart failure or in a local region of congestion caused by vascular obstruction or vascular dilation. Reduced blood flow is most important in veins, in which the slow flow rate favors accumulation of activated coagulation factors and contact of platelets with the endothelium. Venous thrombosis is common in horses with occlusion of intestinal veins secondary to intestinal torsion. Inactivity can also lead to venous stasis and thrombosis in the limbs, a common problem in humans but not in animals. Dilated heart chambers (e.g., dilatative cardiomyopathy) or dilated vessels (e.g., aneurysms) are also areas in which reduced blood flow predisposes to thrombosis.
Turbulent blood flow also enhances the potential for thrombosis. Turbulence disrupts laminar blood flow so the thin layer of plasma that normally separates the endothelium from cellular elements, particularly platelets, is disrupted, and platelets interact more readily with the endothelium. Similarly, turbulence results in mixing of the blood, which provides greater opportunity for interactions between coagulation factors. Turbulence can also physically damage endothelium, creating a strong stimulus for platelet adhesion and coagulation. Turbulence, along with increased risk of thrombosis, is usually greatest in areas in which vessels branch, there is narrowing of the vessel lumen, or at sites of venous or lymphatic valves.
Increased coagulability of blood (hypercoagulability) is another factor that predisposes to thrombosis. Hypercoagulability usually reflects an increase or decrease in the concentration of activated hemostatic proteins (e.g., coagulation factors and coagulation or fibrinolytic inhibitors) caused by enhanced activation or decreased degradation of these proteins. Less often, an alteration in hemostatic protein function may influence coagulability. Activity of coagulation and fibrinolytic proteins can increase in certain conditions such as inflammation, stress, surgery, neoplasia, pregnancy, and renal disease (e.g., the nephrotic syndrome). Inflammation is the most common cause of hypercoagulability, resulting in a variety of changes such as increased TF, increased platelet reactivity, increased fibrinogen, increased levels of phosphatidylserine, increased PAI-1, and decreased thrombomodulin. Transient increases in fibrinogen can also occur with stress and tissue necrosis. Factor I and factor VIII are elevated by trauma, acute illness, surgery, and increased metabolism that accompanies hyperthyroidism. Deficiency of ATIII, a major inhibitor of thrombin, occurs relatively often in dogs with the nephrotic syndrome. In this syndrome, ATIII is depleted because of loss through damaged glomeruli. In affected dogs, there is an increased incidence of venous thrombosis and pulmonary embolism. Increased platelet activation (e.g., heartworm disease, nephrotic syndrome, and neoplasia) can also contribute to hypercoagulability of blood.
The appearance of a thrombus depends on its underlying cause, location, and composition (relative proportions of platelets, fibrin, and erythrocytes). Thrombi composed predominantly of platelets and fibrin tend to be pale, whereas those containing many erythrocytes are red. Cardiac and arterial thrombi are usually initiated by endothelial damage. This damage provides a site for firm platelet attachment and subsequent incorporation of fibrin. Rapid blood flow in these arteries and arterioles inhibits passive incorporation of erythrocytes into the thrombus (Fig. 2-25). Cardiac and arterial thrombi are dull, usually firmly attached to the vessel wall, and red-gray (pale thrombi) (Fig. 2-26). The thrombus may or may not occlude the vessel lumen, and large thrombi tend to have tails that extend downstream from the point of endothelial attachment. Cardiac and larger arterial thrombi often have a laminated appearance created by rapid blood flow and characterized by alternating layers of platelets, interspersed by fibrin intermixed with erythrocytes and leukocytes (lines of Zahn) (Fig. 2-27).
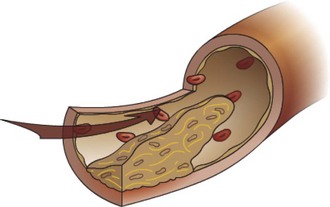
Fig. 2-25 Thrombus (mural), artery.
Thrombus formation is usually initiated by endothelial damage, forming a site of attachment for the thrombus. Growth of the thrombus is downstream, resulting in a tail that is not attached to the vessel wall. Portions of the tail can break off to form thromboemboli. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
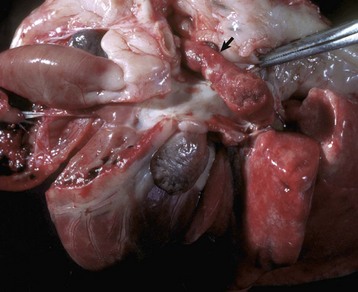
Fig. 2-26 Arterial thrombus, pulmonary artery, dog.
Arterial thrombi are composed primarily of platelets and fibrin because of the rapid flow of blood, which tends to exclude erythrocytes from the thrombus; thus they are usually tan to gray (arrow). (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
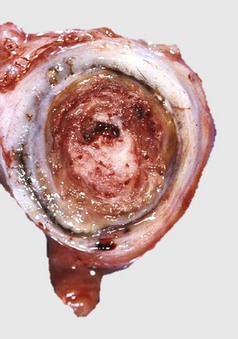
Fig. 2-27 Arterial thrombus, lines of Zahn, cranial mesenteric artery, horse.
Cardiac and larger arterial thrombi often have a laminated appearance characterized by alternating layers of platelets (white-gray) and fibrin (white) intermixed with erythrocytes and leukocytes (lines of Zahn). These lines are the result of rapid blood flow in the heart and arteries/arterioles that favors the deposition of fibrin and platelets and the exclusion of erythrocytes from the thrombus. This horse had verminous arteritis (Strongylus vulgaris fourth stage larvae) in the affected artery. (Courtesy Dr. P.N. Nation, University of Alberta; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Venous thrombi often occur in areas of stasis. Because of the slow blood flow and reduced clearance rate of activated clotting factors in these areas, erythrocytes are commonly incorporated into a loose meshwork of fibrin and platelets (Fig. 2-28). Venous thrombi are typically gelatinous, soft, glistening, and dark red (red thrombi) (Fig. 2-29). They are almost always occlusive and molded to the vessel lumen and often extend for a considerable distance upstream from their point of origin. They commonly have points of attachment to the vessel wall, but these are often very loose and difficult to discern. Venous thrombi are morphologically similar to postmortem clots (see Fig. 1-24). Compared with venous thrombi, postmortem clots are softer and do not have a point of vascular attachment. In larger vessels or in the heart, erythrocytes may settle to the bottom of the clot, leaving a yellow upper layer (chicken fat clot) indicative of postmortem formation. The presence or absence of associated lesions is often a major factor in distinguishing between an antemortem venous thrombus and a postmortem clot.
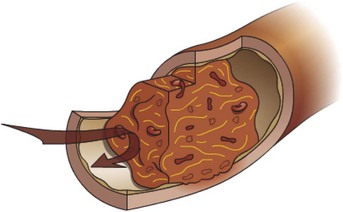
Fig. 2-28 Venous thrombus.
Thrombus formation often occurs in areas of slow blood flow or stasis. Venous thrombi are dark red and gelatinous as a result of large numbers of erythrocytes that are loosely incorporated into the thrombus because of the slow blood flow. Most venous thrombi are occlusive. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
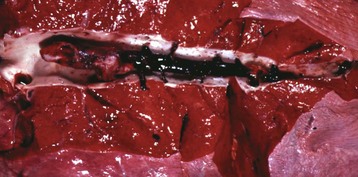
Fig. 2-29 Venous thrombi, pulmonary vein, lung, horse.
Venous thrombi become molded to the shape of the lumen of the vein and grow upstream from the site of initiation. (Courtesy Dr. J. King, College of Veterinary Medicine, Cornell University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
The significance of a thrombus is determined by its location and its ability to disrupt perfusion in a dependent tissue. Disruption of tissue perfusion is influenced mainly by the size of the thrombus, its rate of formation, and its method of resolution or repair. In general, thrombi that rapidly develop are more detrimental than those that slowly develop. A slowly developing thrombus creates progressive narrowing of the vessel lumen, but the slow rate of development provides opportunity for collateral blood flow to increase into the affected area. Small thrombi are usually less damaging than large thrombi. Small thrombi are more easily removed by thrombolysis with little residual vessel damage or tissue compromise. In contrast, large thrombi substantially narrow the vessel lumen to restrict blood flow, are often occlusive, and are less readily dissolved by thrombolysis (Fig. 2-30). Occlusive thrombi block blood flow either into (occlusive arterial thrombus) or out of (occlusive venous thrombus) an area and often result in ischemia (decreased oxygenation of tissue) or infarction (necrosis of tissue caused by lack of oxygen).
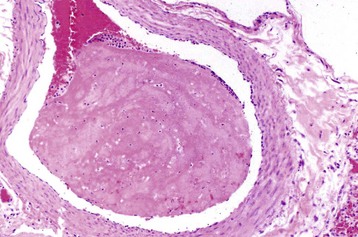
Fig. 2-30 Large thrombus, pulmonary artery, cow.
Large thrombi are less readily dissolved by thrombolysis and therefore heal by other methods. This thrombus consists of a large coagulum of fibrin that has undergone little to no resolution. H&E stain. (Courtesy Dr. M.A. Miller, College of Veterinary Medicine, University of Missouri; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Under most circumstances and after removal of the injurious stimulus, the well-regulated cascade of events in thrombosis results in the return to normal function of the endothelium and subendothelial collagen (Fig. 2-31, A). However, blood flow through a vessel containing a chronic large or occlusive thrombus can change over time. The thrombus provides an ongoing stimulus for platelet adhesion and coagulation, so thrombus propagation can result in progressive narrowing and possible occlusion of the vessel lumen. A thrombus can also be incorporated into the wall of the vessel by a process similar to that used to replace irreversibly damaged tissue. Products of the aggregated platelets stimulate permanent healing of the damaged area by recruiting fibroblasts to the damaged area. Thrombotic debris is removed by macrophages, and granulation tissue and subsequent fibrosis (organization) occur at the site of the thrombus. Concurrently, there is regrowth of endothelium over the surface of the scar. Although there is a permanent narrowing of the vessel lumen, the regrowth of endothelium over the healed thrombus decreases the stimulus for continued thrombosis (Fig. 2-31, B). In occlusive and some large thrombi, this healing process may be accompanied by invasion and growth of endothelial-lined blood channels through the fibrosed area (recanalization) (Fig. 2-31, C, and Fig. 2-32). This provides alternate routes for blood flow to reestablish through or around the original thrombus. Although reestablishment of blood flow increases tissue perfusion, the permanent vascular narrowing and altered, more turbulent blood flow at the site of a healed thrombus result in an increased risk for subsequent thrombosis at the site.
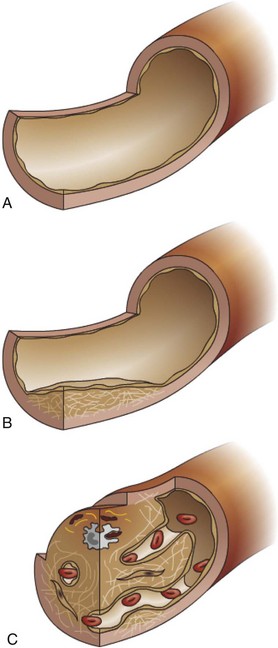
Fig. 2-31 Thrombus resolution.
A, Small thrombi are removed by thrombolysis, and the blood vessel returns to normal structure and function. B, Larger, more persistent thrombi are resolved by removal of thrombotic debris by phagocytes with subsequent granulation tissue formation and fibrosis with regrowth of endothelium over the surface to incorporate the affected area into the vessel wall. C, In large mural or occlusive thrombi that are not removed by thrombolysis or phagocytosis of the thrombotic debris, the thrombus is organized by the invasion of fibroblasts and later by the formation of new vascular channels (recanalization), which provides alternate routes for blood flow through and around the site of the original thrombus. (A, B, and C courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
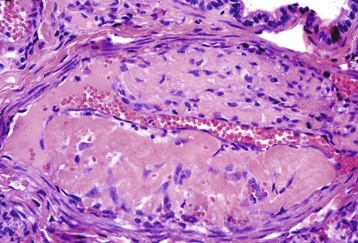
Fig. 2-32 Occlusive mural thrombus, recanalization, cat.
In occlusive and large thrombi, the healing process may occur by fibrosis and the invasion and growth of endothelial-lined vascular channels through the fibrosed area (recanalization). Note the vascular channel, horizontally in the middle of the thrombus. This provides alternate routes for blood flow to reestablish through or around the original thrombus. The permanent vascular narrowing and altered, more turbulent blood flow at the site of a healed thrombus result in an increased risk for subsequent thrombosis at the site. H&E stain. (Courtesy Dr. B.C. Ward, College of Veterinary Medicine, University of Mississippi; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
In some cases, a thrombus or portions of a thrombus can break loose and enter the circulation as an embolus (pl. emboli), a piece of free-floating foreign material within the blood. Thromboemboli (emboli derived from fragments of a thrombus) eventually become lodged in a smaller-sized vessel as the vessel diameter reaches a size that prevents the passage of the embolus, a process called embolization. Venous thromboemboli typically lodge in the pulmonary circulation where they can cause pulmonary infarcts or right-sided heart failure. Arterial thromboemboli typically lodge within a smaller artery downstream from the site of the thrombus, often near sites of vascular bifurcation. Arterial emboli frequently result in infarction of dependent tissue, depending on the tissue and nature of its vascular supply. Cardiac thromboemboli usually lodge at the bifurcation of the external iliac arteries with a portion of the thromboembolus entering each iliac vessel to form a saddle thrombus (Fig. 2-33).
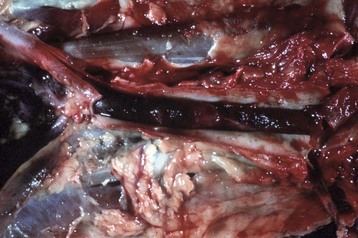
Fig. 2-33 Saddle thrombus, iliac-aortic bifurcation, cat.
Cardiac thromboemboli usually lodge at the bifurcation of the aorta into the external iliac arteries with a portion of the thromboembolus entering each iliac vessel to form a saddle thrombus. A saddle thrombus is not attached to the wall of the aorta or iliac arteries and is easily removed at necropsy. The thromboembolus is composed of layers of platelets and fibrin in which there are enmeshed erythrocytes. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Emboli can also originate from substances other than thrombi. Fat from the bone marrow can be released into the circulation after a fracture of a long bone. Most fat emboli lodge in the pulmonary circulation. Fibrocartilaginous emboli consist of portions of an intervertebral disk, which are released after rupture of a degenerative disk. These can result in occlusion of local vessels and sometimes cause localized spinal cord infarction. Bacteria from inflammatory lesions, such as vegetative valvular endocarditis, or abscesses can enter the blood to form bacterial emboli. When these lodge within vessels, they may cause infarction and secondary sites of infection. Intravascular parasites, such as heartworms (e.g., Dirofilaria), or flukes (e.g., schistosomes) can form parasitic emboli. Malignant neoplasms that invade a vessel result in the formation of neoplastic emboli composed of neoplastic cells. Less common sources of emboli include hematopoietic cells from the bone marrow, amniotic fluid, agglutinated erythrocytes, or clumps of other cells, such as hepatocytes, released after tissue trauma. In any case, the significance of these emboli is their potential to occlude a vessel and inhibit blood flow to dependent tissue.
A serious manifestation of abnormal coagulation is DIC. This is a severe dyshomeostasis caused by the generation of excess thrombin. There are many causes, including diffuse vascular damage (e.g., trauma, vasculitis, and burns), which results in exposure of blood to TF. Intravascular generation of TF by endothelial cells and monocytes can also occur in response to bacteremia, other systemic infections, or any other stimuli that activate the release of inflammatory mediators. The result is TF-induced activation of extrinsic coagulation to produce thrombin. Thrombin causes platelet aggregation and activation of coagulation factors V, VIII, and I to form fibrin, resulting in widespread microvascular clots. Concurrently, the high levels of thrombin stimulate clot dissolution by binding to thrombomodulin to activate protein C, by converting plasminogen into plasmin, and by binding to ATIII to become inactivated. The widespread nature of the coagulation response results in the consumption of these and other factors, resulting in widespread hemorrhages. This combination of microthrombosis with concurrent or rapidly sequential hemorrhage represents one of the most profound and dramatic examples of dyshomeostasis in animals.
Normal Blood Flow, Distribution, and Perfusion
The heart provides the driving pressure for blood distribution. Baroreceptors in the carotid sinus and aortic arch signal the cardiovascular control center in the medulla to balance sympathetic and parasympathetic output to maintain appropriate blood pressure. Left atrial volume receptors and hypothalamic osmoreceptors also help regulate pressure by altering water volume and sodium balance. Sodium concentration is an important contributor to blood volume, osmolality, and pressure and is controlled by the renin-angiotensin-aldosterone system. Secretion of ADH by the hypothalamus in response to a water deficit increases renal tubular reabsorption of water to help maintain blood volume.
Distribution of blood within the circulatory system is highly variable. Organs that alter or recondition blood (e.g., lungs, gastrointestinal tract, kidney, and liver) receive substantially greater blood flow than is required for their metabolic needs. O2 and CO2 are exchanged in the lungs, nutrients are obtained from the gastrointestinal tract and processed by the liver, wastes are removed and electrolytes are balanced by the kidneys, heat is dissipated in the skin, and regulatory hormones enter from endocrine tissues. Systemic neural and hormonal influences can cause general changes in blood distribution. Blood vessel β2 receptors, most abundant in cardiac and skeletal muscle, cause vasodilation and increased flow when stimulated by epinephrine. In contrast, vessel α-receptors, notably absent in the brain, induce vasoconstriction and reduced flow in most organs on stimulation with norepinephrine. Local intrinsic controls alter arteriolar diameter to adjust the blood flow to a tissue based on that tissue’s metabolic needs. These local controls generally override any central controls to maintain adequate blood flow to support normal cell function. At rest, more than 60% of the circulating blood volume is in the veins, providing a storage pool that can be quickly returned to the heart during periods of increased tissue need. In contrast, most capillary beds are closed at any given time; blood flows through only about 10% of the total capillaries of resting skeletal muscle. The orchestration of central pressure, blood composition, and blood distribution is critical to meet the varying perfusion needs of all the cells in the body despite constantly changing conditions.
Alterations in Blood Flow and Perfusion
Hyperemia is an active engorgement of vascular beds with a normal or decreased outflow of blood. It occurs because of increased metabolic activity of tissue that results in localized increased concentrations of CO2, acid, and other metabolites. These cause a local stimulus for vasodilation and increased flow (hyperemia). Hyperemia can occur as a physiologic mechanism within the skin to dissipate heat. It also occurs because of increased need such as increased blood flow to the gastrointestinal tract after a meal. Hyperemia is also one of the first vascular changes that occur in response to an inflammatory stimulus (Fig. 2-34). Neurogenic reflexes and release of vasoactive substances, such as histamine and prostaglandins, mediate the change to promote delivery of inflammatory mediators to the site. Tissues with hyperemic vessels are bright red and warm, and there is engorgement of the arterioles and capillaries.
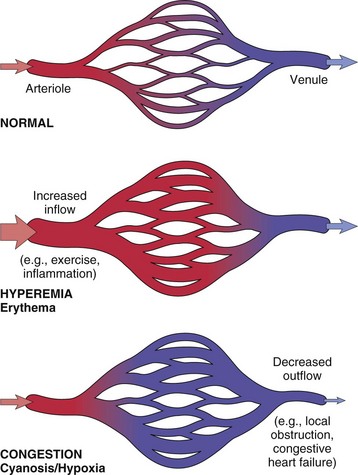
Fig. 2-34 Hyperemia versus congestion.
In both cases, there is an increased volume and pressure of blood in a given tissue with associated capillary dilation and a potential for fluid extravasation. In hyperemia, there is increased inflow leading to engorgement with oxygenated blood. In congestion, diminished outflow leads to a capillary bed swollen with deoxygenated venous blood resulting in cyanosis. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Decreased Blood Flow
Congestion is the passive engorgement of a vascular bed generally caused by a decreased outflow with a normal or increased inflow of blood (see Fig. 2-34). Passive congestion can occur acutely (acute passive congestion) or chronically (chronic passive congestion). Acute passive congestion can occur in the liver and lungs in response to acute heart failure (Fig. 2-35), after euthanasia, or in organs in which relaxation of smooth muscle from barbiturate anesthesia/euthanasia results in dilation of the vasculature and vascular sinusoids such as in the spleen. Most passive congestion is recognized clinically as chronic passive congestion. It can occur locally because of the obstruction of venous outflow caused by a neoplastic or inflammatory mass, displacement of an organ, or fibrosis resulting from healed injury. Generalized passive congestion occurs because of decreased passage of blood either through the heart or the lungs. This is most often caused by heart failure or conditions (e.g., pulmonary fibrosis) that inhibit the flow of blood through the lungs. Right-sided heart failure causes portal vein and hepatic congestion (Fig. 2-36). Left-sided heart failure results in pulmonary congestion (Fig. 2-37). Chronically, there may be fibrosis caused by the hypoxia and cell injury that accompanies congestion (e.g., chronic hepatic congestion). Congested tissues are dark red, swollen (edema), and cooler than normal. The microvasculature is engorged with blood, and there is often surrounding edema and sometimes hemorrhage caused by diapedesis.
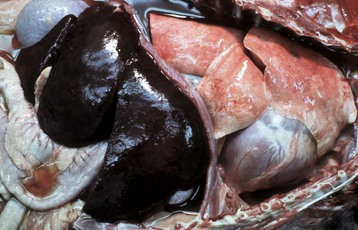
Fig. 2-35 Acute passive congestion, liver, dog.
The liver is enlarged and dark red. Acute passive congestion occurs in the vascular system and dependent organs (heart, lungs, portal system) when there is a sudden interruption of the return of blood to the heart, as occurs in heart failure resulting from arrhythmias and after euthanasia. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
Fig. 2-36 Chronic passive congestion (nutmeg liver), liver, cut surface, dog.
The cut surface has a repeating pattern of red and tan mottling (an accentuated lobular pattern). Chronic passive congestion leads to persistent hypoxia in centrilobular areas and atrophy, degeneration, and/or eventually necrosis of centrilobular hepatocytes. The red areas are dilated central veins and adjacent areas of sinusoidal dilation and congestion caused by centrilobular hepatic necrosis. The tan areas are normal, uncongested parenchyma. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
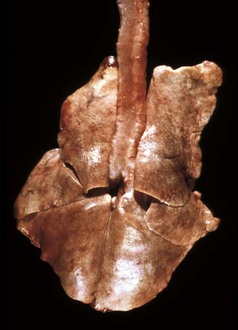
Fig. 2-37 Chronic passive congestion, lung, dog.
The lungs are moderately firm and yellow-brown because of alveolar macrophages containing hemosiderin. Inflammatory mediators produced by these macrophages also induce fibroplasia, thus there is extensive formation of interstitial collagen in the long term. This collagen is the reason the lungs fail to collapse after loss of negative pressure in the pleural cavity when the diaphragm is incised at necropsy. (Courtesy College of Veterinary Medicine, University of Illinois.)
Decreased Tissue Perfusion
Reduced blood flow to an area is usually caused by a local obstruction of a vessel, local congestion, or decreased cardiac output. Local obstruction results in either reduced blood flow into an area or inadequate blood flow out of an area. Ischemia occurs when the perfusion of tissue in the affected area becomes inadequate to meet the metabolic needs of the tissue. Ischemia caused by arterial disease is most commonly the result of incomplete luminal blockage by a thrombus or embolus. The result is a decreased flow of oxygenated blood into the area. Arteriolar vasoconstriction, if prolonged, can also result in ischemia. Ischemia resulting from venous lesions can be caused by intraluminal obstruction such as a venous thrombus. However, external pressure that occludes the vein, such as inflammatory or neoplastic masses, is a common cause. Venous obstruction leads to congestion characterized by slowing and stagnation of blood flow, with loss of tissue oxygenation, local increased hydrostatic pressure, and leakage of fluid into the interstitium (edema). Increased interstitial pressure may partially inhibit arterial inflow into the area to compound the problem. Capillaries can also become occluded by thrombi or external pressure. The severity of ischemia is determined by the local vascular anatomy and degree of anastomoses and collateral circulation, the number of microcirculatory vessels and degree of resistance of the arteriole supplying the capillaries, the extent of the decreased perfusion, the rate at which the occlusion occurred, and the metabolic needs of the tissue. Ischemia can be tolerated to different levels by different tissues. The brain and heart are most susceptible because of a high need for O2 and nutrients, combined with poor collateral circulation. In contrast, organs that recondition blood (e.g., lungs, gastrointestinal tract, kidneys, and skin) can tolerate substantial reductions in flow because they already receive more blood than necessary for their metabolic needs. Other tissues receive blood based on their immediate needs (e.g., skeletal muscle during physical activity). Rapid and complete occlusion that affects large areas of tissue is generally more severe because collateral circulation may not be able to reestablish flow to certain areas quickly enough to prevent tissue injury.
In tissue in which there has been a return of blood flow after brief ischemia, the tissue often returns to normal. The ATP of ischemic tissue is degraded to adenosine, a potent vasodilator, which relieves the ischemia and allows ATP production to resume. However, after prolonged ischemia, the return of blood flow can result in a variety of detrimental effects. Reflow results in fluid loss to the interstitium, resulting in high tissue pressure, which compresses veins and inhibits local venous return. The congested capillaries hemorrhage, TF is released, and vessels are occluded by thrombi. In ischemic cells, a breakdown product of ATP is hypoxanthine. In the absence of oxygen, this is nonreactive. However, on the return of oxygen, xanthine oxidase converts hypoxanthine into urates, hydrogen peroxide, and superoxide anions. Subsequent reaction of superoxide results in the formation of additional reactive oxygen species such as hydroxyl radicals. Collectively, these oxygen-free radicals formed during reperfusion can induce damage, in addition to that caused by ischemia and energy depletion of the cell.
An infarct is a local area of peracute ischemia that undergoes coagulative necrosis. Infarction is caused by the same events that result in ischemia and is most common secondary to thrombosis or thromboembolism. The characteristics of an infarct are variable based on the type and size of vessel that was occluded (artery or vein), the duration of the occlusion, the tissue in which it occurs, and the prior perfusion and vitality of the tissue. Complete arterial blockage usually results in immediate infarction (Fig. 2-38). In contrast, when venous obstruction occurs, such as from torsions or displacements of the bowel, there is extensive congestion and edema of the affected bowel that precedes and promotes infarction. Concurrent disease, decreased cardiovascular function, anemia, or decreased tissue vitality will increase the likelihood of localized areas of ischemia progressing to infarction. In tissue with a single blood supply and minimal anastomoses (e.g., brain, heart, kidney, and spleen), occlusion of nearly any sized vessel typically results in infarction of the dependent tissue (Fig. 2-39). In tissue with parallel blood supplies that have numerous anastomoses (e.g., skeletal muscle and gastrointestinal tract), occlusion is less serious unless it occurs in a large vessel. Tissues with dual blood supplies (e.g., liver and lung) are not commonly susceptible to infarction unless concurrent underlying disease compromises the overall blood supply.
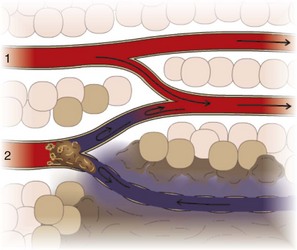
Fig. 2-38 Infarction due to arterial obstruction.
Arterial obstruction results in loss of blood flow to downstream tissue, resulting in abrupt coagulative necrosis. The amount of necrosis depends on factors such as the type and prior health of the tissue affected, its metabolic rate (neurons versus myocytes and fibroblasts), and amount of collateral circulation or alternative blood supply. 1, Normal arterial flow; 2, arterial flow obstructed by an arterial thrombus. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
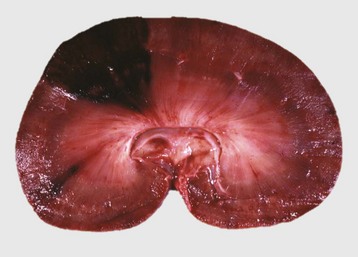
Fig. 2-39 Acute hemorrhagic infarct, kidney, dog.
There is a focal wedge-shaped hemorrhagic area of cortical necrosis. The capsular surface of the infarct bulges above that of the adjacent normal kidney, indicating acute cell swelling and hemorrhage. (Courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Most infarcts are dark red soon after their occurrence because of hemorrhage from damaged vessels in the infarcted area and backflow of blood into the area from surrounding vessels (see Fig. 2-39). As cells undergo necrosis, there is swelling of the affected area, which can force blood out of the infarcted region, giving it a pale appearance (Fig. 2-40). Additionally, hemolysis of erythrocytes and degradation and diffusion of hemoglobin give the infarct a progressively paler appearance. This change in color can occur within 1 to 5 days depending on the tissue and extent of the infarction. Certain types of tissue that have a loose (spongy) consistency, such as the lungs and storage-type spleens (e.g., dogs and pigs), usually remain red because the interstitial areas are expandable and necrosis-induced pressure does not build up to force blood out of the infarcted region (Figs. 2-41 and 2-42). Parenchymal tissues with a less expansible interstitium (e.g., kidney) generally become pale over time because of the pressure that forces blood from the necrotic area. Inflammation occurs at the periphery of the dead tissue so that leukocytes, then macrophages, enter the area to clear the necrotic debris, and subsequently neovascularization and granulation occur to replace the necrotic region with fibrous tissue. This process can occur over a period of weeks or months depending on the extent of the damage. In contrast to the coagulative necrosis caused by infarction in most tissue, infarction in the brain and nervous tissue is characterized by liquefactive necrosis. Subsequently, there is glial cell removal of damaged tissue and astrocytic production of glial fibers (astrogliosis) to replace the affected area.
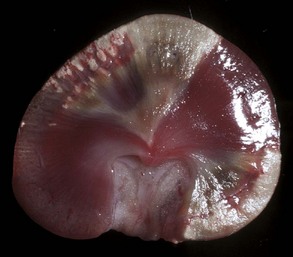
Fig. 2-40 Acute pale infarcts, kidney, rabbit.
Multiple, pale white to tan pyramidal-shaped infarcts extend from the renal cortex to the medulla. The infarcts bulge above the capsular surface (center top), indicative of acute cell swelling. The glistening areas on the right are highlights from the photographic lamps. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
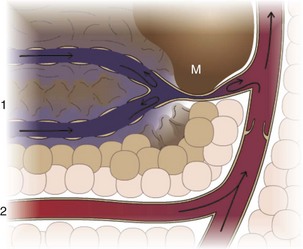
Fig. 2-41 Infarction due to venous obstruction.
Venous obstruction results in stagnation of blood flow and reduction or loss of venous return. There is progressive ischemia and ultimately coagulative necrosis of the tissue upstream of the site of vessel obstruction. The amount of necrosis depends on factors such as the type and prior health of the tissue affected, metabolic rate, and amount of collateral circulation or alternative blood supply. 1, Venous return to a larger vein (note the valve) obstructed by a mass (M); 2, normal venous return to a larger vein. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
Fig. 2-42 Venous infarction, small intestinal volvulus, pig.
Note the intensely congested loops of small intestine undergoing early venous infarction. The veins have been compressed by a volvulus that has compressed the veins but not the arteries, thus preventing the venous return. If the volvulus had rotated further, it would also have compressed the arteries. (Courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University.)
Shock
Shock (cardiovascular collapse) is a circulatory dyshomeostasis associated with loss of circulating blood volume, reduced cardiac output, and/or inappropriate peripheral vascular resistance. Although causes can be diverse (e.g., severe hemorrhage or diarrhea, burns, tissue trauma, endotoxemia), the underlying events of shock are similar. Hypotension results in impaired tissue perfusion, cellular hypoxia and a shift to anaerobic metabolism, cellular degeneration, and cell death (Fig. 2-43). Although the cellular effects of hypoperfusion are initially reversible, persistence of shock results in irreversible cell and tissue injury. Shock is rapidly progressive and life threatening when compensatory responses are inadequate. Shock can be classified into three different types based on the fundamental underlying problem: (1) cardiogenic, (2) hypovolemic, and (3) blood maldistribution. Shock attributed to blood maldistribution can be further divided into septic shock, anaphylactic shock, and neurogenic shock.
Fig. 2-43 Shock.
In hypovolemic shock, there is initially compensation characterized by increased cardiac rate and output, vasoconstriction of nonessential vascular beds, and predominantly oxidative metabolism by morphologically normal cells. With progression, cardiac output falls as peripheral vasodilation occurs, and cell metabolism shifts to glycolysis with progressive morphologic deterioration of cells. (Courtesy Dr. D.A. Mosier and L. Schooley, College of Veterinary Medicine, Kansas State University.)
Cardiogenic Shock
Cardiogenic shock results from failure of the heart to adequately pump blood. Cardiac failure can occur due to myocardial infarction, ventricular tachycardia, fibrillation or other arrhythmias, dilatative or hypertrophic cardiomyopathy, obstruction of blood flow from the heart (e.g., pulmonary embolism and pulmonary or aortic stenosis), or other cardiac dysfunctions. In all cases, there is a decrease in both stroke volume and cardiac output. Major compensatory mechanisms (e.g., sympathetic stimulation of the heart), which increase heart contractility, stroke volume, total cardiac output, and heart rate, are only variably successful depending on the nature of the cardiac damage and the ability of the damaged heart to respond. Unsuccessful compensation leads to stagnation of blood and progressive tissue hypoperfusion.
Hypovolemic Shock
Hypovolemic shock arises from reduced circulating blood volume as the result of blood loss caused by hemorrhage or the result of fluid loss secondary to vomiting, diarrhea, or burns. Reduced circulating blood volume leads to decreased vascular pressure and tissue hypoperfusion. Immediate compensatory mechanisms (e.g., peripheral vasoconstriction and fluid movement into the plasma) act to increase vascular pressure and maintain blood flow to critical tissues such as the heart, brain, and kidney. Increased pressure provides an adequate driving force on which local mechanisms can draw on to increase blood flow based on their needs. When the insult is mild, compensation is generally successful and the animal returns to homeostasis. Loss of about 10% of blood volume can occur without a decrease in blood pressure or cardiac output. However, if greater volumes are lost, adequate pressure and perfusion cannot be maintained and there is insufficient blood flow to meet the needs of the tissues. When blood loss approaches 35% to 45%, blood pressure and cardiac output can fall dramatically.
Blood Maldistribution
Blood maldistribution is characterized by decreased peripheral vascular resistance and pooling of blood in peripheral tissues. This is caused by neural or cytokine-induced vasodilation that can result from situations such as trauma, emotional stress, systemic hypersensitivity to allergens, or endotoxemia. Systemic vasodilation results in a dramatically increased microvascular area, and although the blood volume is normal, the effective circulating blood volume is decreased. Unless compensatory mechanisms can override the stimulus for vasodilation, there is pooling and stagnation of blood with subsequent tissue hypoperfusion. The three major types of shock caused by blood maldistribution are anaphylactic, neurogenic, and septic shock.
Anaphylactic shock is a generalized type I hypersensitivity. Common causes include exposure to insect or plant allergens, drugs, or vaccines. The interaction of the inciting substance with immunoglobulin E bound to mast cells results in widespread mast cell degranulation and the release of histamine and other vasoactive mediators. Subsequently, there is systemic vasodilation and increased vascular permeability, causing hypotension and tissue hypoperfusion.
Neurogenic shock may be induced by trauma, particularly trauma to the nervous system; electrocution, such as by lightning strike; fear; or emotional stress. In contrast to anaphylactic and endotoxic shock, cytokine release is not a major factor in the initial peripheral vasodilation. Instead, there are autonomic discharges that result in peripheral vasodilation, followed by venous pooling of blood and tissue hypoperfusion.
Septic shock is the most common type of shock associated with blood maldistribution. In septic shock, peripheral vasodilation is caused by components of bacteria or fungi that induce the release of excessive amounts of vascular and inflammatory mediators. The most common cause of septic shock is endotoxin, a lipopolysaccharide (LPS) complex within the cell wall of Gram-negative bacteria. Less often, peptidoglycans and lipoteichoic acids of Gram-positive organisms initiate shock. Local release of LPS from degenerating bacteria is a potent stimulus for many of the host responses induced by the infectious agent. LPS often gains entry from microflora of the bowel, entering the circulation into the reticuloendothelial system, then accumulating in the liver, spleen, alveoli, and leukocytes. LPS activates cells (mainly endothelium and leukocytes) through a series of reactions involving LPS-binding protein (an acute phase protein), CD14 (a cell membrane protein and soluble plasma protein) and Toll-like receptor 4 (TLR4, a signal-transducing protein). Endothelial activation by LPS inhibits production of anticoagulant substances (e.g., TFPI and thrombomodulin). Activation of monocytes and macrophages by LPS induces the direct or indirect release of TNF and IL-1 and other cytokines (e.g., IL-6, IL-8, chemokines). LPS directly activates factor XII to initiate intrinsic coagulation and other factor XIIa–related pathways (kinins, fibrinolysis, complement). LPS can also directly activate the complement cascade to generate the anaphylatoxins C3a and C5a. Although these events are important for enhancing the inflammatory response to control localized infections associated with relatively low concentrations of LPS, they can be detrimental if the response becomes more pronounced. This may occur with overwhelming infections by bacteria (generating large concentrations of LPS), or when prolonged intestinal ischemia as the result of other types of shock results in breakdown of the mucosal integrity and leakage of bacteria and toxins into the blood. These higher concentrations of LPS induce even more production of TNF, IL-1, and other cytokines, and the secondary effects of these cytokines become more prominent. TNF and IL-1 induce TF expression and endothelial activation of extrinsic coagulation and enhance the expression of endothelial leukocyte adhesion molecules. IL-1 also stimulates the release of platelet-activating factor (PAF) and PAI to enhance platelet aggregation and coagulation. PAF released from leukocytes, platelets, and endothelium can cause platelet aggregation and thrombosis, increased vascular permeability, and similar to TNF and IL-1, stimulation of arachidonic acid metabolite production (particularly prostacyclin [PGI2] and thromboxane). TNF and IL-1 induce nitric oxide production, which also contributes to vasodilation and hypotension. Neutrophils become activated by TNF and IL-1 to enhance their adhesion to endothelium, which further interferes with blood flow through the microvasculature. The end result of the activation of these myriad vascular, proinflammatory, and procoagulant alterations is the profound systemic vasodilation, hypotension, and tissue hypoperfusion characteristic of septic shock.
Stages and Progression of Shock
Regardless of the underlying cause, shock generally progresses through three different stages: (1) a nonprogressive stage, (2) a progressive stage, and (3) an irreversible stage.
Nonprogressive shock is characterized by compensatory mechanisms that counteract reduced functional circulating blood volume and decreased vascular pressure. Baroreceptors respond to decreased pressure by increasing medullary sympathetic nervous output and epinephrine/norepinephrine release, which increases cardiac output and causes arteriolar vasoconstriction (increased peripheral resistance) in most tissues in an attempt to raise vascular pressure. Notable exceptions are critical tissues, such as the heart, brain, and kidney, to which the blood flow is preserved. Left atrial volume receptors and hypothalamic osmoreceptors help regulate pressure by altering water and sodium balance. Reduced plasma volume stimulates ADH release and water retention and activates angiotensin II production by the renin-angiotensin system to result in aldosterone release and sodium retention. ADH and angiotensin II are also vasoconstrictors and help contribute to increased peripheral resistance. Vasoconstriction also results from endothelial release of endothelin, cold, increased O2, or decreased CO2. Decreased microvascular pressure results in a shift in fluid movement from the interstitium into the plasma to also help increase blood volume. The results of these and other responses are increased heart rate and cardiac output, as well as increased vascular pressure. This provides an adequate driving force on which local mechanisms can draw on to increase blood flow based on their needs. When the insult is mild, compensation is generally successful and the animal returns to homeostasis.
In the case of severe or prolonged hypovolemia or cardiac damage that inhibits the ability of the heart to increase output, compensatory mechanisms are inadequate and shock enters the progressive stage. In this stage, there is blood pooling, tissue hypoperfusion, and progressive cell injury. Cellular metabolism becomes less efficient and shifts from aerobic to anaerobic with pyruvate converted to lactate without entering the Krebs cycle. The deficient production of ATP and overproduction of lactic acid inhibits normal cell functions and results in cellular and systemic acidosis. Metabolic products (e.g., adenosine and potassium), increased local osmolarity, local hypoxia, and increased CO2 eventually result in arteriolar relaxation and dilation. In the case of septic shock, these events exacerbate preexisting cytokine- and mediator-induced vasodilation of the microvasculature. In hypovolemic and cardiogenic shock, the decreased vascular resistance initiates pooling and stagnation of blood within previously closed vascular beds. Widespread arteriolar dilation caused by local influences overrides systemic controls and dramatically contributes to further decreases in vascular plasma volume and pressure. When oxygen and energy stores of the cell are depleted, membrane transport mechanisms are impaired, lysosomal enzymes are released, structural integrity is lost, and cell necrosis occurs. In addition to the detrimental metabolic effects of deficient oxygenation, cell and tissue injury occur in response to the dramatic accumulation of mediators that is characteristic of progressive shock, regardless of its underlying cause. These include histamine, kinins, PAF, complement fragments, and a wide variety of cytokines (e.g., TNF, IL-1, IL-8). These mediators are associated with inappropriate systemic inflammation and systemic activation of complement, coagulation, fibrinolysis, and kinin pathways.
The exact point in which shock enters the irreversible stage is not clear. At the cellular level, metabolic acidosis that results from anaerobic metabolism inhibits enzyme systems needed for energy production. Decreased metabolic efficiency allows vasodilatory substances to accumulate in the ischemic cells and tissues. Once these local products and reflexes override centrally mediated vasoconstriction to produce vasodilation, it is unlikely that shock will be reversed. The fall in peripheral resistance as the result of widespread peripheral vasodilation decreases vascular pressure even more. Irreversibility is generally assured when shock progresses into the syndrome of multiple organ dysfunction. As each organ system fails, particularly the lung, liver, intestine, kidney, and heart, there is a reduction in the metabolic support each system provides to the others. Vicious cycles occur in which the failing function of one organ or tissue contributes to the failure of another (e.g., decreased cardiac output causes renal and pancreatic ischemia; electrolyte imbalances caused by renal ischemia then result in cardiac arrhythmias and myocardial depressant factor released by the ischemic pancreas contribute to even greater reductions in cardiac output). The endpoint of irreversible shock is often manifested as DIC, which is the profound and paradoxic dysfunction of hemostasis.
Clinical and Morphologic Features of Shock
Clinical features of shock are rapidly progressive and include hypotension, weak pulse, tachycardia, hyperventilation with pulmonary rales, reduced urine output, and hypothermia. Organ and system failure occurs in later stages, each manifesting with signs specific to that organ or tissue.
The lesions of shock are variable and depend on the nature and severity of the initiating stimulus and the stage of progression of shock. Characteristically, there are vascular changes accompanied by cell degeneration and necrosis. Generalized congestion and pooling of blood are present in most cases, unless there has been substantial blood loss. Edema, hemorrhage (petechial and ecchymotic), and thrombosis may be present as reflections of the vascular deterioration that accompanies shock. Thrombosis and platelet plugging of capillaries can be prominent in septic shock. Vascular abnormalities are most obvious in those cases that progress to DIC. Cell degeneration and necrosis are most prominent in those cells that are most susceptible to hypoxia, such as neurons and cardiac myocytes, and cells that do not obtain adequate preferential blood flow during shock. Hepatocytes, renal tubular epithelium, adrenal cortical epithelium, and gastrointestinal epithelium are often affected. With the exception of loss of neurons and myocytes, virtually all of these tissue changes can revert to normal if the animal survives. Specific changes may include severe pulmonary congestion, edema, and hemorrhage with alveolar epithelial necrosis, fibrin exudation, and hyaline membrane formation. Passive congestion and centrilobular hepatic necrosis, as well as renal tubular necrosis, are often present in these metabolically important organs. Intestinal congestion, edema, and hemorrhage with mucosal necrosis may occur. In the heart, myofibril coagulation is caused by hypercontraction of sarcomeres and is most likely a response to high sarcoplasmic calcium levels as the result of lack of energy and membrane damage. Cerebral edema and in some cases cerebrocortical laminar necrosis as a result of cerebral ischemia may be present.
Behling-Kelly, E, Czuprynski, CJ. Endothelial cells as active participants in veterinary infections and inflammatory disorders. Anim Health Res Rev. 2007;8:47–58.
Boudreaux, MK. Platelet structure. In Weiss D, Wardrop KJ, eds.: Schalm’s veterinary hematology, ed 6, Ames, IA: Wiley-Blackwell, 2010.
Boudreaux, MK, Catalfamo, JL. Platelet biochemistry, signal transduction, and function. In Weiss D, Wardrop KJ, eds.: Schalm’s veterinary hematology, ed 6, Ames, IA: Wiley-Blackwell, 2010.
Esmon, CT. Inflammation and thrombosis. J Thromb Haemost. 2003;7:1343–1348.
Furie, B, Furie, BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–949.
Gailani, D, Renné, T. Intrinsic pathway of coagulation and arterial thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:2507–2513.
Gentry, PA. Comparative aspects of blood coagulation. Vet J. 2004;168:238–251.
Hajjar, KA, Francis, CW. Fibrinolysis and thrombolysis. In Lichtman MA, Beutler E, Kipps TJ, et al, eds.: Williams hematology, ed 7, New York: McGraw-Hill, 2006.
Hajjar, KA, Esmon, NL, Marcus, AJ, Muller, W. Vascular function in hemostasis. In Lichtman MA, Beutler E, Kipps TJ, et al, eds.: Williams hematology, ed 7, New York: McGraw-Hill, 2006.
Hopper, K, Bateman, S. An updated view of hemostasis: mechanisms of hemostatic dysfunction associated with sepsis. J Vet Emerg Crit Care. 2005;15:83–91.
Lip, GY, Blann, AD. Thrombogenesis, atherogenesis and angiogenesis in vascular disease: a new “vascular triad,”. Ann Med. 2004;36:119–125.
Loscalzo, J, Schafer, AI. Thrombosis and hemorrhage, ed 3. Philadelphia: Lippincott Williams & Wilkins; 2003.
Majno, G, Joris, I. Vascular disturbances. In Majno G, Joris I, eds.: Cells, tissues, and disease: principles of general pathology, ed 2, New York: Oxford University Press, 2004.
McManus, B, Allard, MF, Yanagawa, R. Hemodynamic disorders. In Rubin R, Strayer DS, eds.: Rubin’s pathology: clinicopathologic foundations of medicine, ed 5, Philadelphia: Lippincott Williams & Wilkins, 2008.
Michiels, C. Endothelial cell functions. J Cell Physiol. 2003;196(3):430–443.
Mitchell, RN. Hemodynamic disorders, thromboembolic disease, and shock. In Kumar V, Abbas AK, Fausto N, eds.: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia: Saunders, 2009.
Parise, LV, Smyth, SS, Shet, AS, Coller, BS. Platelet morphology, biochemistry and function. In Lichtman MA, Beutler E, Kipps TJ, et al, eds.: Williams hematology, ed 7, New York: McGraw-Hill, 2006.
Roberts, HR, Monroe, DM, Hoffman, M. Molecular biology and biochemistry of the coagulation factors and pathways of hemostasis. In Lichtman MA, Beutler E, Kipps TJ, et al, eds.: Williams hematology, ed 7, New York: McGraw-Hill, 2006.
Ruggeri, ZM, Mendolicchio, GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685.
Ruggeri, ZM. Platelet adhesion under flow. Microcirculation. 2009;16:58–83.
Seligsohn, U, Hoots, WK. Disseminated intravascular coagulation. In Lichtman MA, Beutler E, Kipps TJ, et al, eds.: Williams hematology, ed 7, New York: McGraw-Hill, 2006.
Shebuski, RJ, Kilgore, KS. Role of inflammatory mediators in thrombogenesis. J Pharmacol Exp Ther. 2002;300(3):729–735.
Shen, GX. Impact and mechanism for oxidized and glycated lipoproteins on generation of fibrinolytic regulators from vascular endothelial cells. Mol Cell Biochem. 2003;246(1-2):69–74.
Smith, SA. Overview of hemostasis. In Weiss D, Wardrop KJ, eds.: Schalm’s veterinary hematology, ed 6, Ames, IA: Wiley-Blackwell, 2010.
Steffel, J, Lüscher, T. Progress and challenges in transfusion medicine, hemostasis, and hemotherapy. In: Scharf RE, ed. Endothelium and hemostasis in health and disease. Basel, Switzerland: S Karger AG, 2008.
Stockham, SL, Scott, MA. Hemostasis. In: Stockham SL, Scott MA, eds. Fundamentals of veterinary clinical pathology. Ames, IA: Blackwell Publishing, 2008.
Stokhof, AA. The extracardiac peripheral circulation and shock. In: Dunlop RH, Malbert CH, eds. Veterinary pathophysiology. Ames, IA: Blackwell Publishing, 2004.
Stokol, T, Brooks, M, Rush, JE, et al. Hypercoagulability in cats with cardiomyopathy. J Vet Intern Med. 2008;22:546–552.
Varga-Szabo, D, Pleines, I, Nieswandt, B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403–412.
Zarbock, A, Polanowska-Grabowska, R, Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2006;21:99–111.