Cellular Adaptations, Injury, and Death
Morphologic, Biochemical, and Genetic Bases
The simple definition that pathology is “the study of disease” understates the wide range of contributions of this discipline to modern medicine. An understanding of pathology is fundamental to developing a mechanistic comprehension of how disease occurs in a chronologic sequence of events and consequently, how it can be diagnosed, treated, and prevented.
To students of medical science, pathology is the course of study that finally connects the study of normal structure and function (histology, anatomy, and physiology) to the study of clinical medicine. Pathology is fundamental to making sense of how the various causes of disease such as infectious microorganisms interact with animals and result in clinically identifiable conditions.
Pathology is also an important professional discipline that directly supports the practice of clinical medicine. Diagnostic pathologists, for example, perform postmortem examinations (necropsies), which provide clinicians with essential information on how to manage disease outbreaks in herds and how to improve management of individual cases. Surgical pathologists examine tissue removed from live animals (biopsy) and provide diagnoses that help clinicians treat animals under their care. Toxicologic pathologists test and evaluate the effects and safety of drugs and chemicals in laboratory animals. Clinical pathologists perform tests on blood and other body fluids (hematology and serum chemistry, for example) and examine cells (cytology) to provide detailed and essential information for clinicians. Experimental pathologists study the tissue, cellular, and molecular mechanisms of human and animal diseases in the fields of biomedicine and biomedical engineering.
Pathology is also an experimental science that makes essential contributions to advance our understanding of disease mechanisms through the use of a diverse variety of scientific techniques. Advanced methods of cell and molecular biology are used to unravel the complexities of how cells and animals respond to injury, so that a deeper understanding of disease processes can help improve treatment and prevention.
In summary, pathology is according to one dictionary definition (Stedman’s Medical Dictionary) “the medical science, and specialty practice, concerned with all aspects of disease, but with special reference to the essential nature, causes, and development of abnormal conditions, as well as the structural and functional changes that result from the disease processes.”
Basic Terminology
If pathology is the study of disease, what is disease? A dictionary definition (Dorland’s Medical Dictionary) states that disease is “any deviation from or interruption of the normal structure or function of any part, organ, or system (or combination thereof) of the body that is manifested by a characteristic set of symptoms and signs and whose etiology, pathology, and prognosis may be known or unknown.” Disease is not just illness or sickness but includes any departure from normal form (lesions) and function, whether it is clinically apparent or not.
Pathologists study lesions, as well as the causes (etiologic agents) of the lesions to understand the pathogenesis of a disease. Pathogenesis is the mechanism of how a disease develops from its initiation to its cellular and molecular manifestations. Understanding pathogenesis is essential to understanding how a disease is initiated and progresses, how these changes relate to clinical signs at different stages of the disease, and how appropriate clinical action can be taken.
The relationship of pathology to clinical medicine and the use of some of the basic terms discussed previously along with some additional terms are illustrated in the following clinical scenario.
In a beef feedlot, several steers and heifers are exhibiting difficult breathing, hunched posture, and depression (clinical signs). Physical examination of some of the infected animals reveals elevated temperatures, pulse rates, and respiration rates. Auscultation of the thorax reveals absence of air movement in the cranial region of the thorax along with some crackles and wheezes in other lung fields. A clinical diagnosis of bronchopneumonia is made. Some of the animals die, and a necropsy (postmortem examination) is done. The cranioventral lobes of the lungs are dark red and firm, with fibrin covering the surface (gross lesions). A gross morphologic diagnosis of severe acute fibrinopurulent cranioventral bronchopneumonia is made. Formalin fixed samples are taken for microscopic examination (histopathology), neutrophilic inflammation of airways and alveoli with fibrin are noted (microscopic lesions), and a histologic morphologic diagnosis of severe acute fibrinopurulent bronchopneumonia is made. Fresh samples of lung are taken for bacterial and viral examination, and Mannheimia haemolytica and a bovine herpes virus (etiologic agents or causes) are identified. An etiologic diagnosis of Mannheimia bronchopneumonia and a disease diagnosis of “shipping fever pneumonia” are made.
The pathogenesis of this disease might be stated in an abbreviated form like the following:
Various viruses, such as infectious rhinotracheitis virus, and environmental agents, such as dust and noxious gases, disrupt the clearance mechanisms of the airway epithelium allowing opportunistic organisms, such as the bacterium Mannheimia haemolytica, to colonize and invade the alveoli. Virulence factors of the bacteria, such as endotoxin and various exotoxins, cause necrosis and inflammation, which result in the filling of alveoli and airways with fibrin and neutrophils.
Although a diagnostic pathologist did the histologic diagnosis of the disease, the details of this pathogenesis were discovered over time by researchers in many fields, including experimental pathologists.
Types of Diagnosis
Note that various levels of diagnosis were made in the previous scenario. Diagnosis is a concise statement or conclusion concerning the nature, cause, or name of a disease. The accuracy of a diagnosis is limited by the evidence (lesions) available for study. A clinical diagnosis is based on the data obtained from the case history, clinical signs, and physical examination. It often suggests only the system involved, or it provides a list of differential diagnoses. The differential diagnosis (often termed rule outs in clinical medicine) is a list of diseases that could account for the evidence or lesions of the case. A clinical pathologic diagnosis is based on changes observed in the chemistry of fluids and the hematology, structure, and function of cells collected from the living patient. A morphologic diagnosis or lesion diagnosis is based on the predominant lesion(s) in the tissue(s) (see Chapter 3 and Fig. 3-23). It may be macroscopic (gross) or microscopic (histologic) and describes the severity, duration, distribution, location (organ or tissue), and nature (degenerative, inflammatory, neoplastic) of the lesion. An etiologic diagnosis is even more definitive and names the specific cause of the disease. A disease diagnosis is equally specific and states the common name of the disease.
One of the goals in making a diagnosis in a case is to enable a clinician to predict how the disease will progress or resolve. Prognosis is a statement of what the expected outcome of a condition is likely to be. If the lesion is expected to resolve (return to normal) with no expected lasting harm, the prognosis is good or excellent. If the outcome is uncertain—the lesion could resolve or become worse as a result of unforeseen factors—the prognosis is guarded. If the animal is not expected to recover from the lesion or disease, the prognosis is grave. Accurate determination of the prognosis demands a thorough understanding of the disease, especially pathogenesis.
As in this book, the study of pathology is often divided into two basic parts: general pathology and pathology of organ systems. General pathology is the study of basic responses of cells and tissues to insults and injuries, irrespective of the organs, systems, or species of animal involved. This area of pathology is one of the most complex and rapidly growing fields in the natural sciences, largely as the result of the availability and power of new research techniques. General pathology is studied first, so students will have a thorough understanding of the general principles of disease processes that they will encounter repeatedly in the study of diseases of body systems. Pathology of organ systems (sometimes called systemic or special pathology) involves the study of how each organ system reacts to injury associated with specific diseases.
Morphologic Changes and How They Are Detected and Evaluated
The study and practice of pathology historically have been based on the macroscopic and microscopic changes that take place in diseased cells, tissues, and organs, that is to say, the morphology of lesions. Consequently, most pathology texts tend to emphasize anatomic pathology. Morphologic techniques remain the cornerstones of pathology, but progress in our deeper understanding of the mechanisms and in the diagnosis of disease rely more and more on techniques derived from cellular and molecular biology.
The basic tools for the study and practice of pathology begin with an open and inquiring mind, skills in observation, and careful and consistent postmortem techniques. The diagnosis of many diseases can be accurately accomplished with no more than gross examination of a body. Confirmation of gross lesions and discovery and interpretation of microscopic changes typically involves observation of tissue placed on glass microscope slides. Tissues are first fixed (i.e., preserved) usually in 10% formalin, embedded in blocks of paraffin wax, microtome sectioned to about 5 µm thickness, and routinely stained with hematoxylin and eosin (H&E). H&E stained sections are the mainstay of histopathology in both postmortem and surgical pathology, and interpretation of lesions in these specimens can often lead to a final diagnosis. A simplistic explanation of the labeling characteristics of the H&E stain as applied to tissue sections is as follows: Hematoxylin stains nucleic acids (nucleus, ribosomes, mitochondria) blue, whereas eosin stains proteins, such as those found intracellularly (e.g., enzymes, actin, and myosin) or those proteins found extracellularly (e.g., collagen and extracellular matrix [ECM]), red or pink.
A variety of ancillary techniques are also used in histopathology. Histochemistry applies a variety of chemical reactions carried out on tissue sections. Glycogen, for example, can be identified in hepatocytes using periodic acid–Schiff (PAS) reaction. Suspected mast cell tumors are routinely stained to demonstrate the metachromatic mast cell granules using toluidine blue or Giemsa stains.
Increasing use in diagnostic laboratories is being made of immunohistochemistry, in which specific antigens are identified in tissue by antibodies linked to a chromogen. Detection of specific intermediate fibers by immunohistochemistry, in tumors, for example, can separate malignant striated muscle tumors from other sarcomas. Specific infectious agents, such as the coronavirus causing feline infectious peritonitis, can also be identified using immunohistochemistry.
A variety of techniques for identification of molecules or genetic sequences are now in use, with more expected. In situ hybridization, in which labeled nucleic acid probes can identify complementary strands of host or microbe deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) in intact cells and tissues, is particularly useful in the diagnosis and study of viral disease. These techniques are not as sensitive as polymerase chain reaction (PCR), in which small amounts of target DNA in biologic material are amplified and identified. Small amounts of target DNA of microbes (for example) can be identified in tissues, and RNA sequences can be identified after conversion to DNA and subsequent amplification.
The typical light microscope can magnify to about 1000× and is adequate for routine histopathology. Specialized microscopes, such as dark field, phase contrast, and fluorescence microscopes, are also used, often for identification of microbes. In both diagnostic and experimental settings, electron microscopy is used to visualize the subcellular structure of cells and microbes. Transmission electron microscopy of ultrathin sections allows resolution of ultrafine structures of less than a nanometer. Scanning electron microscopy allows the ultrafine observation of surfaces. Specialized analytical electron microscopes are also in use. Finally, laser capture microdissection allows pathologists to isolate and capture groups of similar cells from tumors or a diseased tissue. Using DNA microarrays and other genomic techniques, the genes expressed by these cells can be identified and characterized, thus providing a “genetic fingerprint” of the disease process that clinically can be used to develop therapeutic strategies and assess the outcome. Proteomics, which are molecular techniques used to reveal the protein profiles from genes in tissues and fluids, are also being increasingly used.
The Normal Cell
Components of Normal Cells and their Vulnerabilities
The early pathologists Morgagni and Bichat emphasized the importance of organs and tissues as the seat of disease. Virchow later focused on individual cells as the primary cause of abnormal function and structure associated with diseases. Before we can interpret lesions of sick cells, it is essential that we understand normal cell structure and function. The cell can be visualized simplistically as a membrane-enclosed compartment, subdivided into numerous smaller compartments (organelles) by membranes (Fig. 1-1). This vast interconnecting system of membrane-bound spaces is termed the cytocavitary network. The function of these organelles is largely determined by the type and quantity of specific enzymes associated with each membrane and in the cytoplasmic matrix.
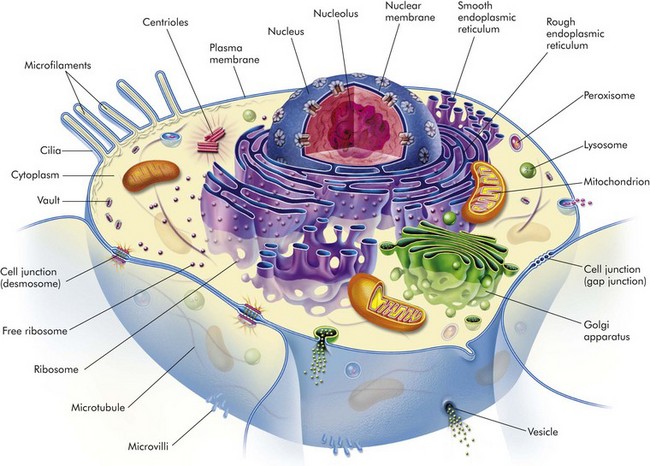
Fig. 1-1 Cell structure and the organization of organelles, cytoskeleton, and membrane enhancements. (From McCance K, Huether S: Pathophysiology: the biologic basis for disease in adults and children, ed 5, St Louis, 2006, Mosby.)
It is essential to have a clear understanding of the structure and function of the components of normal cells and how they are interrelated in a normally functioning cell. Cell membranes and organelles serve as targets for injury by microbes, harmful environmental agents, and a variety of genetic, metabolic, and toxicologic diseases discussed in greater detail in the Pathology of Organ Systems chapters of this book.
Cell Membranes
Cell membranes are a fluid phospholipid bilayer penetrated by numerous specific proteins (Fig. 1-2). The two main biologic functions of these membranes are (l) to serve as selective barriers and (2) to form a structural base for the enzymes and receptors that determine cell function. Cell membranes form the boundaries of many organelles and separate them from the cytosol.
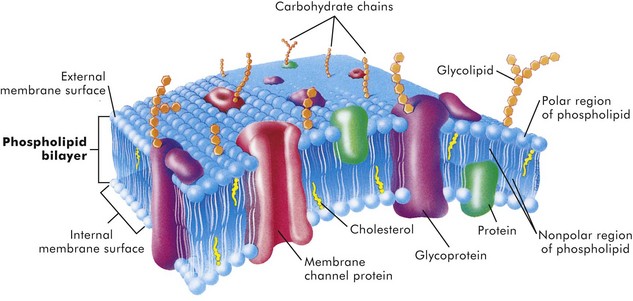
Fig. 1-2 Fluid mosaic model of cell membrane structure.
The lipid bilayer provides the basic structure and serves as a relatively impermeable barrier to most water-soluble molecules. (From McCance K, Huether S: Pathophysiology: the biologic basis for disease in adults and children, ed 4, St Louis, 2002, Mosby.)
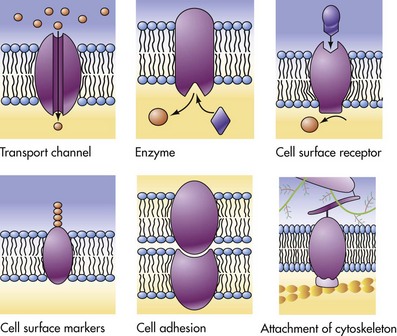
The plasma membrane is the cell’s first contact with injurious agents. Microvilli and cilia are specialized areas of the plasma membrane and are often specifically altered in disease (see Fig. 1-1). Plasma membranes separate the interior of the cell from external surfaces, neighboring cells, or surrounding matrix. Surface proteins, such as fibronectin, play a role in cell-to-cell and cell-to-ECM interactions. Transmembrane proteins embedded in the phospholipid bilayer serve in a variety of structural, transport, and enzymatic functions essential to cell viability (Fig. 1-3). It is these transmembrane proteins that are often used by infectious microbes to enter or use cell systems during their life cycles, thus initiating a process that often results in injury to the host cell.
Cytosol
The cytosol is the watery gel in which the cell’s organelles and inclusions are dispersed. Many chemical reactions occur in the cytosol mediated by “free” enzymes or macromolecular complexes such as proteasomes. The cytosol is a highly organized microtrabecular network.
Mitochondria
Mitochondria (singular = mitochondrion) are the “powerhouses” of highly specialized eukaryotic cells. They are the site of fatty acid oxidation, the citric acid cycle, and oxidative phosphorylation. Transfer of electrons from reduced cytochrome oxidase to molecular oxygen is the final and critical step culminating in these catabolic pathways. Important structural components of a mitochondrion are the outer membrane, outer compartment, inner membrane, inner compartment (matrix), cristae, and mitochondrial DNA. Damage to mitochondria results in diminished adenosine triphosphate (ATP) production and if damage is unchecked, cell death (see Fig. 1-6).
Nucleus
The nucleus is that portion of the cell responsible for storage and transmission of genetic information (see Fig. 1-1). Chains of DNA, complexed to protein, are chromatin. Areas of uncoiled chromatin (euchromatin) are active in the generation of messenger RNA (mRNA) for protein synthesis. Highly coiled chromatin (heterochromatin) is inactive in transcription. The outer nuclear membrane is continuous with that of the rough endoplasmic reticulum (RER).
Nucleolus
The nucleolus is a basic organelle of the nucleus and is composed of RNA, nucleolus-associated chromatin, and protein (see Fig. 1-1). It functions in the synthesis of ribosomal RNA (rRNA), essential in protein synthesis. The nucleolus can be basophilic or eosinophilic, and its prominence is a subjective measure of the cell’s synthetic activity.
Rough Endoplasmic Reticulum
The RER is a network of intracellular membranes studded with ribosomes (Fig. 1-4). RER is prominent in cells producing large amounts of extracellular protein (e.g., reactive fibroblasts, hepatocytes, plasma cells, and pancreatic acinar cells). The RER is responsible for the basophilia of the cytoplasm because of the numerous ribosomes, which contain acid (i.e., RNA).
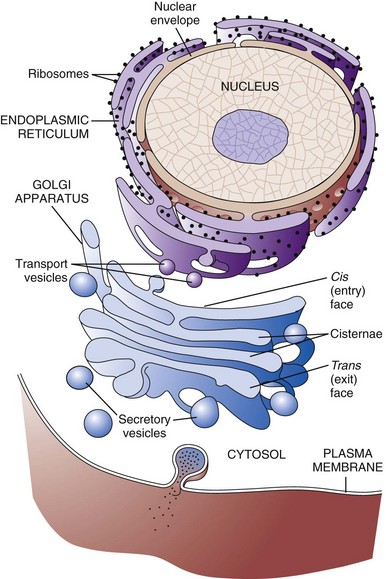
Fig. 1-4 Membrane systems.
The rough endoplasmic reticulum and Golgi apparatus are important organelles in cellular biosynthesis of proteins and glycoproteins inserted into cell membranes and used in and secreted from cells. Transcription, translation, assembly, modification, and packaging of these molecules occur in an orderly sequence from the nucleus to the cell membrane as shown. Alterations in one or more of these steps can result in cell injury and serve as the underlying pathogenesis of a disease process. (From Copstead L, Banasik J: Pathophysiology, ed 4, St Louis, 2010, Mosby.)
Smooth Endoplasmic Reticulum
Smooth endoplasmic reticulum (SER) is a tubular or vesicular form of cell membrane that lacks ribosomes (see Fig. 1-1). SER is the locus of enzymes that metabolize steroids, drugs, lipids, and glycogen. It gives the cytoplasm a pale, finely vacuolated appearance as viewed in the light microscope.
Golgi Complex
The Golgi complex consists of several lamellar stacks or flattened sacs of membranes, vesicles, and vacuoles (see Fig. 1-4). It functions in the synthesis of complex proteins by the addition of carbohydrate molecules and in the production of secretory vesicles and lysosomes.
Lysosomes
Lysosomes are small membrane-bound vesicles laden with hydrolytic enzymes essential for intracellular digestion (see Fig. 1-1). They are discussed more completely as components of phagocytic cells. Peroxisomes are similar to lysosomes but also play a role in energy metabolism.
Microfilaments, Intermediate Filaments, and Microtubules
These structures are composed of protein subunits and function in the cytoskeleton and in cell movement (Fig. 1-5). They have a prominent role in the mitotic spindle, cilia, microvilli, neurons, myocytes, and phagocytic cells. Many cell types besides muscles, for example, contain actin microfilaments.
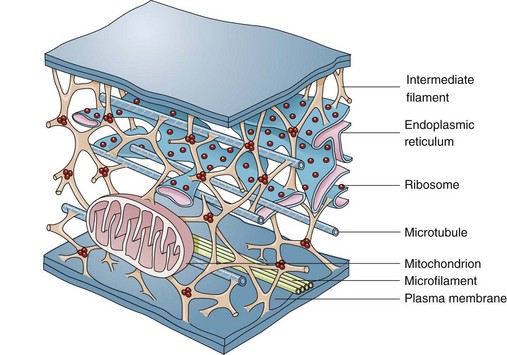
Fig. 1-5 Cytoskeleton.
The complexity of and interrelations between intermediate filaments, microtubules, endoplasmic reticulum, and other cytoplasmic organelles that can be involved in the pathogenesis of diseases are shown. (From McCance KL, Huether SE: Pathophysiology: the biologic basis for disease in adults and children, ed 5, St Louis, 2006, Mosby.)
Intermediate filaments are about 10 nm in diameter and are important in cell shape and movement. Different cell types have different intermediate filaments; for example, cytokeratins are found in epithelial cells, desmin in muscle cells, and vimentin in cells of mesenchymal origin such as fibroblasts. Intermediate filaments can be useful markers for classifying undifferentiated neoplasms.
Cellular Inclusions
Inclusions include glycogen granules, proteinaceous vacuoles, lipid debris, hemosiderin, viral particles, and calcium granules (discussed in greater detail later in this chapter). Some of these are normal, whereas others are the result of cell injury and are discussed later in this chapter in the section dealing with intracellular and extracellular accumulations.
Extracellular Matrix
Although not part of the cell itself, the ECM and its integrity influences cell health and function (see Chapter 3 and Web Figs. 3-23 and 3-24). ECM includes basement membranes and interstitial matrices composed of various collagens, proteoglycans, and adhesive glycoproteins among a variety of other molecules that interact with cells by means of various integrin molecules. Basement membrane integrity, for example, is essential for the proper structure and functioning of epithelial cells. Other components of the ECM influence how cells grow and differentiate.
Causes of Cell Injury
Causes of cell injury are numerous and can be classified in a variety of ways. Some causes, such as physical trauma, viruses, and toxins, are clearly extrinsic, whereas others, such as spontaneous genetic mutations, are clearly intrinsic. Others, such as workload imbalance, nutritional abnormalities, and immunologic dysfunctions, can have components of both extrinsic and intrinsic mechanisms. General mechanisms of injury include ATP depletion (often caused by hypoxia), membrane damage (a result of a myriad of causes, including oxygen-derived free radicals), disturbances of cellular metabolism, and genetic damage (Fig. 1-6).
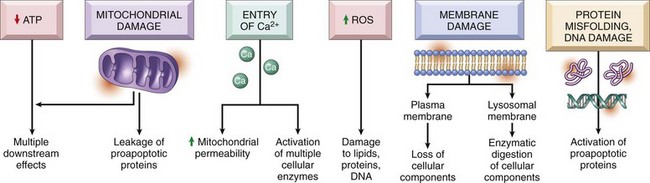
Fig. 1-6 Cellular and biochemical sites of damage in cell injury.
ATP, Adenosine triphosphate; ROS, reactive oxygen species. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Understanding disease starts with understanding the cell. Until the nineteenth century, the dominant theory of disease in western societies was humoral pathology, wherein disease was attributed to a maldistribution of body fluids or “humors.” In the mid-1800s, Rudolph Virchow, a German pathologist now considered to be the founder of modern pathology, redefined pathology and medical science with his idea of the body as an organization of cells, each suited for specific functions. He taught that disease resulted from injury to, or dysfunction of, specific populations of cells. The recent rapid advancement in medical science is owed to a great extent to Virchow’s original emphasis on cellular pathology and more recently on molecular pathology.
Cells can be injured through a large number of causes (etiologic agents). Fortunately, the types of responses of the cell to injury are not as large. The responses to injury depend on many factors, including the type of agent, the extent of injury, the duration of injury, and the cell type affected. Renal tubular cells deprived of adequate blood supply, for example, may exhibit only cell swelling, if oxygen is soon restored. Prolonged lack of adequate blood supply (ischemia) can lead to cell death. Diminished but sublethal reduction in blood supply may result in cells adapting by decreasing their metabolic rates, which could lead to recovery or if adaptation is inadequate, eventually death.
Cells respond to stimuli and stressors in a variety of ways to maintain homeostasis. Cell injury takes place when a cell can no longer maintain a steady state. Some types of cell injury, such as cell swelling, can be reversible if the extent and duration of injury are not excessive. But if the injury exceeds certain limits, cell death and irreversible change occur. Not all cell injury results in cell death. Cell injury may be sublethal and result in a variety of types of cell degenerations or accumulations and/or adaptations by the cell to the injury. In essence, cells or tissues respond to injury (or stress) in three important ways: (l) adaptation, (2) degeneration or intracellular or extracellular accumulations, and (3) death (Fig. 1-7).
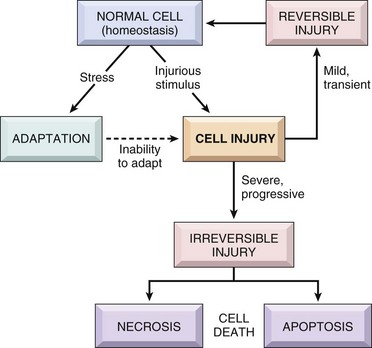
Fig. 1-7 Stages in the cellular response to stress and injurious stimuli. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Pathologically, reversible cell injury is injury from which the cell can adapt or recover and thus return to normal or nearly normal function. Irreversible cell injury results in a dead cell. This distinction seems clear-cut, but the point at which a cell transitions from reversible cell injury to irreversible cell injury (i.e., “the point of no return”) has been a major research challenge for the past few decades and remains so today (Fig. 1-8). The lesions of reversible and irreversible cell injury are discussed in greater detail in subsequent sections; however, in summary, the cytomorphologic changes characteristic of irreversible cell injury include the following:
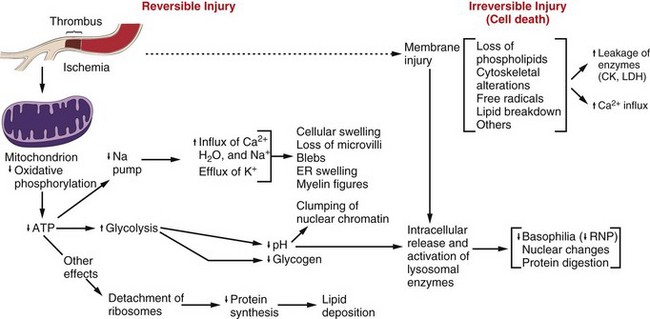
Fig. 1-8 Postulated sequence of events in reversible and irreversible ischemic cell injury.
Note that although reduced oxidative phosphorylation and adenosine triphosphate (ATP) levels have a central role, ischemia can cause direct membrane damage. ER, Endoplasmic reticulum; CK, creatine kinase; LDH, lactate dehydrogenase; RNP, ribonucleoprotein. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
• Mitochondrial swelling and vacuolization
The causes of reversible and irreversible cell injury resulting in cell death, cell adaptation and degeneration, and finally cellular accumulations are now discussed.
Oxygen Deficiency
Hypoxia is one of the most common and important causes of cell injury and death (see Fig. 1-8). Hypoxia is a partial reduction in the O2 concentration supplied to cells or tissue; a complete reduction is referred to as anoxia. Oxygen is critically important for oxidative phosphorylation, especially in highly specialized cells such as neurons, hepatocytes, cardiac myocytes, and renal tubule cells. Hypoxia can result from inadequate oxygenation of blood as a result of heart failure or respiratory failure, loss or reduction of blood supply (ischemia), reduced transport of O2 in blood (e.g., anemia or carbon monoxide toxicity), and blockage of cell respiratory enzymes (cyanide toxicosis).
Physical Agents
Trauma, extremes of heat and cold, radiation, and electrical energy may seriously injure cells. Trauma may cause direct rupture and death of large numbers of cells, or it may damage the blood supply to cells. Extreme cold impairs the blood flow, and intracellular ice crystals rupture cell membranes. Extreme heat denatures essential cell enzymes and other proteins. Excessive heat can increase the rate of metabolic reactions so that substrates, water, and pH changes reach lethal levels. Electricity generates great heat as it passes through tissue. It also alters conduction of nerves and muscle. Ionizing radiation causes ionization of cellular water with production of highly reactive “free radicals” that injure cell components. Many forms of radiation may damage genetic material, resulting in reproductive death of cells by apoptosis, genetic defects from mutations, and neoplasia.
Infectious Agents
Viruses are obligate intracellular parasites that redirect host cell enzyme systems toward synthesis of viral proteins and genetic materials to the detriment of host cells. Cell changes induced by viral agents vary from little effect to cell death or neoplastic transformation.
Injury caused by bacterial infection varies and can result from the action of potent toxins on specific host cells (clostridial infections, enterotoxigenic Escherichia coli infection) or from an overwhelming or ineffective inflammatory response to uncontrolled bacterial replication in tissue. Some bacteria, such as Lawsonia intracellularis, can result in excessive intestinal epithelial cell replication.
Mycotic agents resist destruction by the body that can lead to progressive, chronic inflammatory disease with loss of normal host tissues. Protozoal agents replicate in specific host cells, often resulting in destruction of infected cells. Metazoan parasites cause inflammation, distort tissue, and use host nutrients.
Nutritional Deficiencies and Imbalances
Dietary protein-calorie deficiencies are seen sporadically in animals and humans (known as kwashiorkor). These deficiencies require metabolic adaptation by large populations of cells. Lipolysis, catabolism of muscle protein, and glycogenolysis enable short-term survival. Calorie excess, as seen in many pets and people of affluent societies, is implicated in cardiovascular disease and several other diseases. Vitamin and mineral imbalances are common due to errors in formulating rations and hypersupplementation by well-meaning animal owners.
Genetic Derangement
A normal genetic apparatus is essential for cell homeostasis. Mutations, whatever their origin, may cause no disease, may deprive a cell of a protein (enzyme) critical for normal function, may result in neoplasia, or may be incompatible with cell survival. A few examples of genetic diseases are defects of clotting factors (hemophilia), lysosomal storage disease (mannosidosis), combined immunodeficiency of Arabian foals, and defects of collagen synthesis (dermatosparaxis). Besides causing overt disease, some genotypes cause the host to be more prone to certain types of extrinsic or intrinsic disease, a condition often termed genetic predisposition.
Workload Imbalance
Cells that are overworked may adapt to the demand or eventually become exhausted and die. Conversely, cells that are no longer stimulated to work may shrink in size and waste away. An example is the way endocrine tissues react to the presence or absence of specific trophic hormones. Muscle fibers, deprived of work or their nerve supply, will atrophy and ultimately disappear, leaving a fibrous stroma.
Chemicals, Drugs, and Toxins
Chemicals, drugs, and toxins influence cells by a multitude of mechanisms. Drugs produce their therapeutic effects by modifying the function (and morphology) of specific populations of cells. Most drugs cause these cells to adapt within a tolerable range of homeostasis. Chemicals, including drugs and toxins, can block or stimulate cell membrane receptors, alter specific enzyme systems, produce toxic free radicals, alter cell permeability, damage chromosomes, modify metabolic pathways, and damage structural components of cells.
Immunologic Dysfunction
The immune system may fail to respond to infectious agents and other antigens as a result of congenital or acquired defects of lymphoid tissue or their products (see Chapter 5). Examples of congenital defects are thymic aplasia of nude mice and combined immunodeficiency of Arabian foals. Affected animals may die at an early age from infection by opportunistic microorganisms. Acquired immunodeficiency disease may be transient and results from damage to lymphoid tissue by viral infection, chemicals, and drugs.
The immune response directed toward foreign antigens (pathogenic organisms) is usually beneficial to the host, but sometimes the response is misdirected against antigens of host cells. This large group of diseases is referred to as autoimmune disease. An inappropriate or exaggerated response to certain antigens results in immunologic disease referred to as hypersensitivity (allergy). Some examples are anaphylaxis, feline asthma, and flea allergy dermatitis. The activity of the immune system is greatly amplified by its effect on serum complement and inflammation. These reactions often lead to serious injury to the kidney, skin, and joints.
Aging
The diminished capacity of aged cells and tissue to carry out their normal functions can hardly be disputed. One can argue that aging is simply the culmination of life’s injuries inflicted by chemicals, infectious agents, work imbalances, or poor nutrition. We use the aging category for those lesions commonly found in aged animals; lesions for which we have no other defensible mechanistic explanation. Some of the lesions commonly found in older animals include nodular hyperplasia of parenchymal cells in the liver, pancreas, adrenal, spleen, and thyroid. There appear to be defects in growth control of these cell populations, but the cause is unclear. Aged cells may suffer a lifetime of damage to their DNA, or there may be accumulation of cellular debris that interferes with normal cell functions. One could argue that many cancers are caused by old age, rather than by exposure to specific chemicals, foods, viruses, or other insults.
Reversible Cell Injury
Cell swelling, also called hydropic degeneration and by other names in different organ systems (e.g., cytotoxic edema in the central nervous system and ballooning degeneration in the epidermis), is the most common and fundamental expression of cell injury (Fig. 1-9). It is manifested as increased cell size and volume resulting from an overload of water caused by a failure of the cell to maintain normal homeostasis and regulate the ingress and excretion of water. It is accompanied by modification and degeneration of organelles. Mechanisms responsible for acute cell swelling usually involve damage to cellular membranes, failure of cellular energy production, or injury to enzymes regulating ion channels of membranes. Cell swelling occurs in response to loss of the cell’s homeostasis secondary to mechanical, hypoxic, toxic, free radical, viral, bacterial, and immune-mediated injuries.
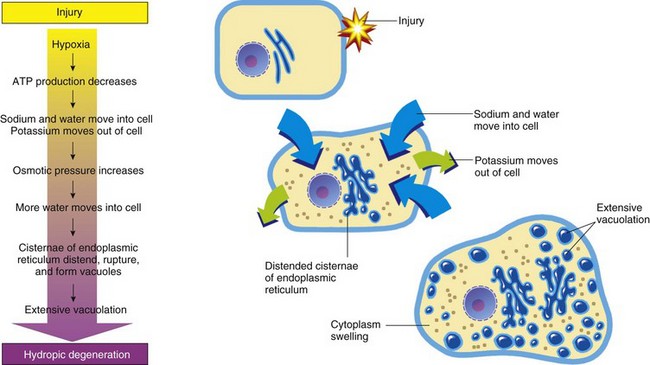
Fig. 1-9 The process of acute cell swelling (hydropic degeneration).
ATP, Adenosine triphosphate. (From Huether S, McCance K: Understanding pathophysiology, ed 3, St Louis, 2004, Mosby.)
The functional and morphologic changes begin with increased uptake of water and then to diffuse disintegration of organelles and cytoplasmic proteins. Cell swelling must be distinguished from cell enlargement (hypertrophy) that is caused by an increase of normal organelles. Organs composed of swollen cells are themselves swollen. Affected organs are larger and heavier than normal and pale in color. The parenchyma of swollen organs, such as kidney and liver, may bulge a little from beneath their capsule when incised. Because of the increase of intracellular water, the specific gravity of affected tissues is slightly less than those of normal tissues.
Normal Cell Volume Control and Mechanisms of Acute Cell Swelling
In the normal cell, energy derived from ATP drives the Na+-K+ ion pumps within cell membranes to continuously drive Na+ out of the cell in exchange for K+ moving into the cell. For each molecule of ATP used, the pump moves three Na+ out of the cell and two K+ into the cell. By this means, the ion pumps maintain the transmembrane ionic gradients required for normal nerve and muscle function. Because water moves passively across cell membranes in response to the osmotic pressure gradient generated by Na+ and proteins, the Na+-K+ pump is the key to regulation of intracellular water. The best-studied laboratory models of cell swelling are (1) hypoxia-induced failure of ATP synthesis and (2) carbon tetrachloride (CCl4)–induced membrane damage.
Hypoxic Injury Resulting in Acute Cell Swelling
Hypoxia is probably the most important fundamental cause of acute cell swelling. Hypoxia-induced cell injury results from any defect in the transport of O2, from inspired air to its role as the final acceptor of electrons from cytochrome oxidase in oxidative phosphorylation. Ischemia is reduced blood flow to a region of the body, usually because of obstruction of the blood supply. Blockage of coronary arteries by atherosclerotic plaque leads to ischemia and hypoxic injury to heart muscle, a common cause of “heart attacks” in humans. Therefore cellular hypoxia occurs with suffocation, anemia, pneumonia, shock or other damage to the circulation, and interference with mitochondrial enzymes.
In acute hypoxic injury, cell O2 is depleted in moments, aerobic oxidative phosphorylation stops, and ATP levels fall. The drop in cellular ATP stimulates phosphofructokinase, the initial regulator step of anaerobic glycolysis. The metabolic switch to anaerobic metabolism of glucose rapidly depletes the cell’s glycogen stores and leads to the accumulation of intracellular lactate and inorganic phosphates. Although the anaerobic generation of ATP is inefficient, it provides for some short-term survival. Some highly specialized cell types, such as neurons, cannot generate ATP anaerobically and thus are especially prone to hypoxic injury. Ultimately, this deficiency of ATP leads to a failure of Na+-K+ pumps and loss of cell volume control.
The cardiac glycosides of plant origin, digitalis and ouabain, specifically inhibit the action of the Na+-K+ pump. This inhibition modifies the contractility of cardiac myocytes, but it may also cause them to swell.
Cell Membrane Injury in Acute Cell Swelling
Damage to the cell membranes, both plasma membranes and organelle membranes, destroys the selective permeability barrier that retains proteins and electrolytes within the cytosol and that restricts the entry of Na+, Ca2+, and water from the extracellular space. Failure of the barrier results from chemical modification of phospholipids by free radicals, covalent binding of toxic chemicals to macromolecules, interference with ion channels, and insertion of transmembrane protein complexes (e.g., complement activation).
The hepatotoxicities of CCl4 and chloroform (CHCl3) provide classic examples of cell membrane injury (Fig. 1-10). Toxic effects of CCl4 occur when the chemical is converted to the trichloromethyl radical, CCl3•, by the mixed-function oxidase system of the SER in hepatocytes. The toxic metabolite, CCl3•, next causes progressive lipid peroxidation of unsaturated fatty acids of cellular membranes, progressing from the SER to mitochondria and other cell membranes. Chloroform is toxic to hepatocytes when it is metabolized to the electrophilic metabolite, phosgene (COCl2•). The hepatic lesions associated with these two toxins are indistinguishable, and both may result in fatty liver.
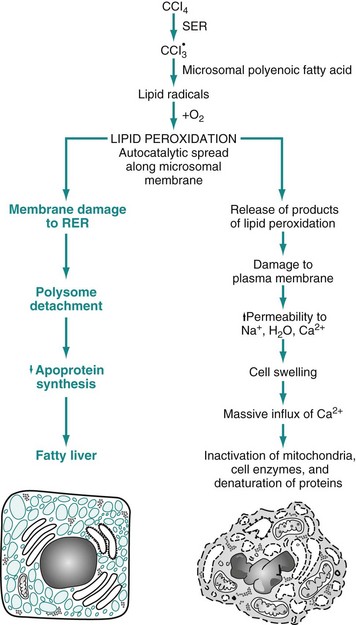
Fig. 1-10 Sequence of events leading to fatty change and cell necrosis in carbon tetrachloride (CCl4) toxicity.
RER, Rough endoplasmic reticulum; SER, smooth endoplasmic reticulum. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Besides toxins, other processes may cause cell membrane injury leading to acute cell swelling. The membrane-attack complex of serum complement (see Chapter 3) and the hemolysin of streptococci (streptolysin-O) penetrate cell membranes to form a channel for free passage of water, proteins, and electrolytes between intracellular and extracellular compartments. Affected cells are quickly lysed by water overload (hypotonic lysis). Cytotoxic effects of natural killer (NK) cells are mediated in part by the implantation of similar hollow protein-complexes into target cell membranes.
The sequence of events in acute cell swelling caused by hypoxia or ischemia (see Fig. 1-8) is as follows:
2. Decrease of oxidative phosphorylation and ATP
3. Increased glycolysis, increased intracellular lactate, and depletion of glycogen stores
4. Failure of Na+-K+ pump as the result of ATP deficiency
5. Net influx of Na+, Ca2+, and H2O with loss of intracellular K+ and Mg2+
6. Swelling of mitochondria and the cytocavitary network (RER, SER, Golgi, and outer nuclear membrane)
7. Detachment of ribosomes, clumping of nuclear chromatin, loss of microvilli, vesiculation of endoplasmic reticulum (ER), formation of membrane whorls (“myelin figures”)
8. Severe disruption of cell membranes, influx of Ca2+ into mitochondria and cytosol, overall cell enlargement, and clearing of the cytosol
When acute cell swelling results from membrane injury, the sequence of events is similar to those listed previously, except that changes start at about step 5 or 6.
Morphology of Acute Cell Swelling
Gross Appearance: Acute cell swelling is recognized as pallor, organ swelling, and decreased specific gravity. For example, the liver will be pale and somewhat turgid (Fig. 1-11, A). The parenchyma of organs with capsules may bulge when incised.
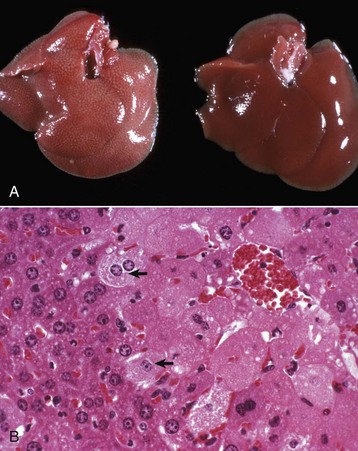
Fig. 1-11 Acute cell swelling, liver, mouse.
A, Hepatic swelling in a mouse exposed to chloroform 24 hours previously. The accentuated lobular pattern and slight pallor in the liver on the left are the result of acute cell swelling (hydropic degeneration) and necrosis of centrilobular hepatocytes. The right liver is normal. B, Liver from a mouse with chloroform toxicosis. While many hepatocytes in the centrilobular areas (at right) are necrotic, several cells at the interface of normal and necrotic (arrows) are still undergoing acute cell swelling (hydropic degeneration). H&E stain. (Courtesy Dr. L.H. Arp.)
Microscopic Appearance: The influx of water dilutes the cytoplasmic matrix and dilates organelles to give cells a pale, finely vacuolated appearance (cloudy swelling). Renal tubule epithelial cells bulge and impinge on the tubular lumen. Swollen hepatocytes and endothelial cells intrude upon and diminish vascular lumens. Although mechanisms of cell swelling are limited, variations in appearance may occur because of differences in cell type and cause of injury.
Hydropic degeneration is a common term used for the microscopic appearance of acute cell swelling (Fig. 1-11, B). It occurs in endothelium, epithelium, alveolar pneumocytes, hepatocytes, renal tubular epithelial cells, and neurons and glial cells of the brain. Cytoplasm of affected cells contains translucent vacuoles that fail to stain for fat or glycogen (two other causes of vacuolar degeneration). These vacuoles represent swollen mitochondria and dilated cisternae of the Golgi and ER. Ballooning degeneration is an extreme variant of hydropic degeneration in which cells are greatly enlarged and the cytoplasm is basically a clear space (Fig. 1-12). Ballooning degeneration is typically seen in epidermal cells infected by epitheliotropic viruses (e.g., poxvirus). This lesion frequently progresses to the formation of vesicles or bullae (blisters) from lysis of the epidermal cells. These viral infections cause both degradation of cytoplasmic proteins (cytoplasmic proteolysis) and net flux of water into the cytoplasm.
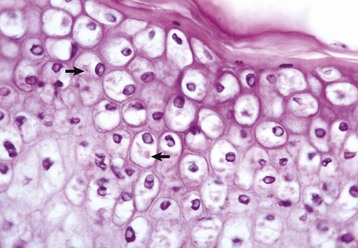
Fig. 1-12 Ballooning degeneration, papular stomatitis, oral mucosa, cow.
Cells infected by some types of virus, such as papular stomatitis virus, are unable to regulate their volume and swell at certain stages of the infection. These cells may become very large (ballooning degeneration) and eventually rupture. Some of the cells have viral inclusion bodies (arrows). H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Ultrastructural Appearance: As visualized with the electron microscope, swollen epithelial cells have lost and distorted cilia, microvilli, and attachment sites, as well as “blebbing” of cytoplasm at the cell surfaces. The cytoplasm is rarefied, and the cisternae of the ER, Golgi, and mitochondria are dilated. The cytocavitary network becomes fragmented into numerous vesicles. Proteins and Ca2+ precipitate in the cytoplasm and in organelles. Acute cell swelling in the central nervous system has other distinctive features (see section on Cerebral Edema in Chapter 14).
Significance and Fate of Acute Cell Swelling
Injured cells that can no longer regulate water and electrolytes are no better equipped to maintain other cell functions. Significance to the patient depends on the number of cells affected and the immediate importance of the lost cell function. Cells highly vulnerable to hypoxia and cell swelling include cardiac myocytes, proximal renal tubule epithelium, hepatocytes, and endothelium. In the central nervous system (CNS), besides endothelium, neurons, oligodendrocytes, and astrocytes also are swollen, and the process in the CNS is called cytotoxic edema (see Chapter 14). Swollen neurons fail to conduct nervous impulses, resulting in stupor or coma. Swollen myocardial cells contract with less force or with an abnormal rhythm. Swollen renal epithelium may not only fail to absorb and secrete but also may compress delicate interstitial blood vessels, resulting in further injury. Capillaries lined by swollen endothelium are prone to obstruction, exacerbating the lesions by worsening cellular hypoxia. Injured cells with abnormal membrane permeability may be detected by finding their specific cytoplasmic enzymes in serum.
If adequate oxygen is restored to the cells and membrane injury is repaired before a certain point is reached, the “point of no return,” most cells can be restored to normal or nearly normal function. Some cells may retain evidence of previous injury in the form of lipofuscin accumulation after autophagocytosis of damaged organelles. What happens when the stage of reversibility is passed is the topic of the next sections, beginning with cell death.
In summary, cell swelling is a manifestation of reversible, sublethal cell injury. However, unless the cause of injury to critically important cell types is removed quickly, progressive injury to these dependent cells and tissues may culminate in the death of the animal.
Irreversible Cell Injury and Cell Death
As we have just seen, major mechanisms of acute cell swelling are hypoxia, including ischemia, and membrane injury, often by toxins. Cell swelling can be reversible if the extent and duration of injury is not excessive. But if the injury exceeds certain limits (discussed shortly), cell death occurs (Fig. 1-13). Not all cell injury results in cell death. Cell injury may be sublethal and result in a variety of types of cell degenerations and/or adaptations by the cell to the injury. In essence, cells or tissues respond to injury (or stress) in three important ways: (l) adaptation (with or without accumulations or degenerative changes), (2) reversible injury (again with or without subcellular changes), and (3) death. In this section, we deal with cell death. Various types of cell adaptations, degenerations, and accumulations are addressed in subsequent sections.
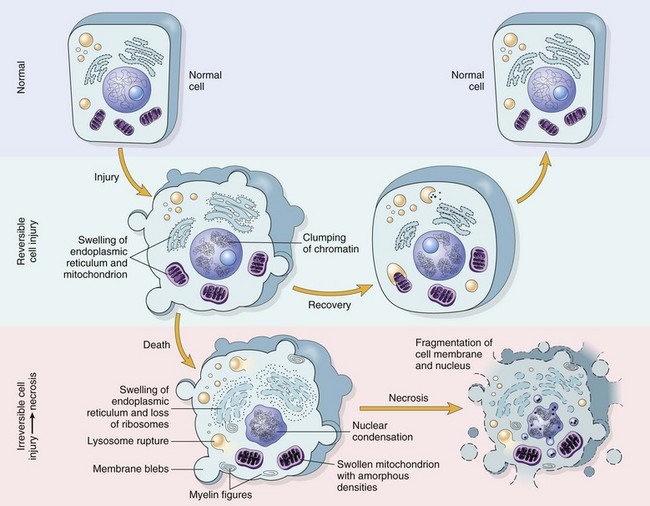
Fig. 1-13 Normal cell and the changes in reversible and irreversible cell injury.
Reversible injury is characterized by generalized swelling of the cell and its organelles, blebbing of the plasma membrane, detachment of ribosomes from the endoplasmic reticulum, and clumping of nuclear chromatin. Transition to irreversible injury is characterized by increasing swelling of the cell, swelling and disruption of lysosomes, presence of large amorphous densities in swollen mitochondria, disruption of cellular membranes, and profound nuclear changes. The latter include nuclear condensation (pyknosis), followed by fragmentation (karyorrhexis) and dissolution of the nucleus (karyolysis). Laminated structures (myelin figures) derived from damaged membranes of organelles and the plasma membrane first appear during the reversible stage and become more pronounced in irreversibly damaged cells. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Cell Death
Cell death can occur in many ways. For example, extremes of temperature or direct trauma may result in nearly instantaneous destruction or death of cells. On the other hand, death of an animal (somatic death) results in eventual death of all cells that make up the animal (postmortem autolysis). During most of the last century, cell death and necrosis were thought of as being more or less the same and in most pathologic situations; necrosis was usually thought to be preceded by cell swelling as discussed earlier. It is clear that cells die before macroscopic or histologic evidence can be detected. Although necrosis can be defined as the death of cells in a living animal, it should be understood to mean the specific morphologic changes (either macroscopic or microscopic) indicative of cell death in a living animal.
In the last few decades of the twentieth century, it became clear that cells die also by shrinkage, both under physiologic and pathologic circumstances, and this complex and now well-studied process has become known as apoptosis, a type of programmed cell death. Cell death then began to be classified in two major types: necrosis or apoptosis. Because of the long history of use of necrosis and because apoptosis, cell death with shrinkage, is distinctly different from death following swelling, the term oncosis (onco-, meaning swelling) has been proposed for what was previously termed necrosis. As in most biologic processes, it is not always possible to make the distinction between these two types of cell death based on histologic examination, and often both swelling and shrinkage are present.
How then are we to use the term necrosis? Attempts are being made by toxicologic pathologists to use the term necrosis for the histologic changes that occur after cell death by either mechanism, using the terms oncotic cell death or apoptotic cell death when a distinction needs to be made. We attempt to adhere to this distinction here, but long-used terminology does not easily change. The next sections first discuss cell death after irreversible cell injury by hypoxia and cell membrane damage (oncotic necrosis), and then apoptosis or apoptotic necrosis (Fig. 1-14).
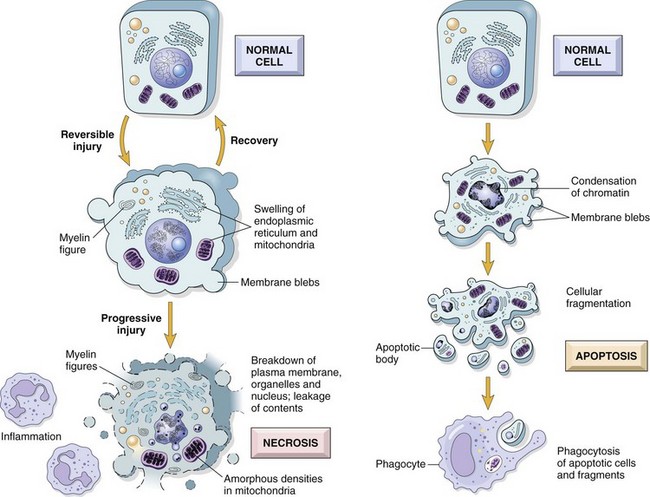
Fig. 1-14 The sequential ultrastructural changes seen in necrosis (left) and apoptosis (right).
In apoptosis, the initial changes consist of nuclear chromatin condensation and fragmentation, followed by cytoplasmic budding and phagocytosis of the extruded apoptotic bodies. Signs of cytoplasmic blebs, accumulation of myelin figures representing damaged phospholipid membranes, and digestion and leakage of cellular components characterize necrosis. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Cell Death by Oncosis (Oncotic Necrosis)
Oncosis is cell death after irreversible cell injury by hypoxia, ischemia, and direct cell membrane injury. Hypoxic injury, as discussed earlier in the section on Acute Cell Swelling, is a common cause of cell death and oncotic necrosis. Cell membrane damage caused by toxins and other substances and mechanisms can also lead to necrosis, but the resulting morphologic changes are similar. Hypoxia is often due to blockage of or markedly diminished blood supply to an area (ischemia). Ischemic injury is typically more severe than hypoxia alone because not only is the amount of oxygen lowered in the tissue, but also the inflow of metabolic substrates and nutrients is decreased and cell waste and by-products accumulate, some of which are injurious in their own right.
Acute cell swelling can result in necrosis or can be reversible. Despite much interest and research about where the exact point of no return is between injury that is reversible and where it is irreversible, resulting in necrosis, is still not clear. There is convincing evidence for the role of Ca2+ in the eventual demise of severely injured cells. Earlier work consistently identified two features of irreversible cell injury: (1) an inability to restore mitochondrial function and (2) evidence of cellular membrane damage.
Research directed at understanding coronary heart disease has led to improved understanding of the role of Ca2+. Heart muscle deprived of its blood supply (ischemia) suffers from hypoxia and substantial loss of cell volume regulation and the influx of Ca2+ because of inadequate ATP to run the ion pumps. If the blood supply is resupplied to the ischemic area, often reversal of the injury is not attained, but instead, injury is accelerated. It has been shown that restored blood flow to areas with potentially viable cells results in added membrane damage shortly after blood supply is reestablished. This phenomenon is termed ischemia-reperfusion injury.
Prevention of Ca2+ influx can reduce irreversible injury. The reactivity of free Ca2+ ion and its role as an intracellular messenger and enzyme activator are known, and these actions are thought to contribute much to the final demise of the cell in necrosis. What does Ca2+ do to cause the ultimate demise of many severely injured cells as it influxes from the extracellular space (Fig. 1-15)? At least one endogenous, membrane-bound phospholipase (phospholipase A) is activated by free Ca2+. Activated phospholipases then break down the normal phospholipids of the inner mitochondrial membrane and other cell membranes. These events then preclude any possibility for cell survival. Activation of phospholipases also generates arachidonic acid, the substrate for many lipid mediators of inflammation (to be discussed later). Therefore it is usual to see some degree of inflammation around foci of necrosis. In addition to phospholipases, Ca2+ also activates proteases that result in cytoskeleton and membrane damage, adenosine triphosphatases (ATPases) that accelerate depletion of ATP, and endonucleases that result in chromatin degradation. Irreversible injury to mitochondrial membranes appears to be the deathblow to the cell. As if this were not enough, cells injured by ischemia can also die by apoptosis because of the leakage of proapoptotic molecules from injured mitochondria.
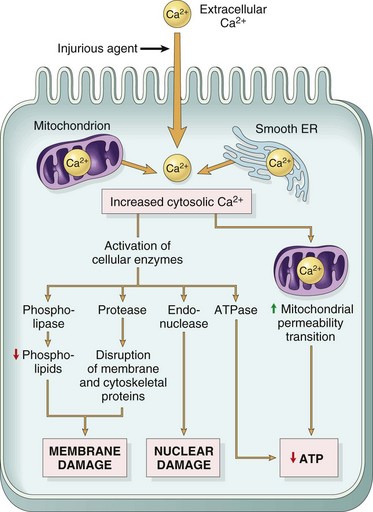
Fig. 1-15 Sources and consequences of increased cytosolic calcium in cell injury.
ER, Endoplasmic reticulum; ATP, Adenosine triphosphate. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Ischemia-reperfusion injury, for example, after restoration of blood supply caused by coronary artery blockage and myocardial ischemia (heart attack), has many components that contribute to irreversible injury. Besides the effects of Ca2+ influx, generation of free radicals in hypoxic cells combined with restored oxygen results in a variety of reactive oxygen and nitrogen species. These include production of peroxynitrite from nitric oxide (NO), a molecule of endothelial and parenchymal cells that is normally involved in vasodilation, inhibition of platelet aggregation, and prevention of leukocyte adhesion. Free radicals derived from inflammatory cells are also increased as the result of accumulation of neutrophils in reperfused areas after release of cytokines such as tumor necrosis factor-alpha (TNF-α). Reduction of reperfusion injury should theoretically be improved by agents blocking Ca2+ influx, by antioxidants such as vitamin E that decrease membrane damage because of the oxygen metabolites (free radicals), and by antiinflammatory agents that decrease the influx of inflammatory cells and impact of inflammatory mediators.
Cell Membrane Injury Leading to Cell Death: Chemical injury to cells in many cases may occur because of organelle and plasma cell membrane damage. Classically studied and referred to in the section on acute cell injury is the toxicity for hepatocytes of CCl4 (see Fig. 1-10). After ingestion and absorption by the gastrointestinal (GI) tract, CCl4 is transported via the portal vein to the liver where it enters hepatocytes. CCl4 itself is fairly innocuous, but metabolism by the cytochrome p450 system in the SER in hepatocytes results in the formation of a toxic metabolite, CCl3•. This free radical causes lipid peroxidation of organelle membranes starting from the SER and spreading to other organelles and eventually to the plasma membrane. This outcome has a variety of consequences. Injury to mitochondria results in decreased oxidative metabolism, decreased ATP production, and consequently an influx of calcium into mitochondria. This outcome results in decreased activity of the Na+-K+ pump and dysregulation of cell volume and massive intracellular increase in calcium with its lethal consequences. Direct damage to the plasma membrane itself by lipid peroxidation can have the same consequences to cell volume control and influx of calcium.
Lysosomal swelling and release of hydrolytic enzymes can result in autodigestion of cell components. Injury to RER of the hepatocyte can result in decreased protein synthesis, and this deficiency then causes insufficient production of lipoproteins required to export lipids and then results in increased fatty acid content in the cell and hepatic lipidosis (see discussion later), if the changes are not lethal.
Free Radical Injury: Injury to cell and organelle membranes can occur in many ways. One of the most common and important is free radical injury due especially to reactive oxygen species (Fig. 1-16). A free radical is any molecule that has an unpaired electron. These molecules are highly reactive, transient chemical species, generated as by-products of normal oxidative metabolism or by exposure to radiation, toxic gases, chemicals, and drugs. Most, but not all, are reactive oxygen radicals. Oxygen radicals are also produced by phagocytic cells in inflammatory lesions and account for significant damage to surrounding tissue. Antineoplastic drugs, such as doxorubicin, generate oxygen radicals that cause significant injury to cardiac myocytes. Cellular components at risk of free radical injury include proteins, membrane lipids, and nucleic acids. Lipid peroxidation of plasma membranes and organelle membranes by free radicals can have similar consequences to those described earlier from CCl4.
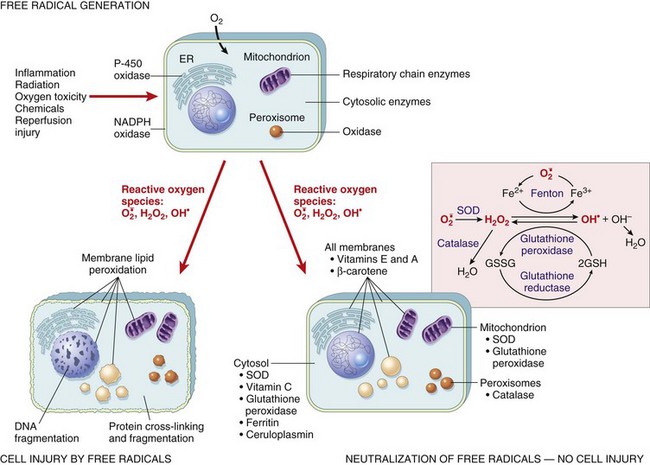
Fig. 1-16 The role of reactive oxygen species in cell injury.
Oxygen is converted to superoxide ( ) by oxidative enzymes in the endoplasmic reticulum (ER), mitochondria, plasma membrane, peroxisomes, and cytosol.
) by oxidative enzymes in the endoplasmic reticulum (ER), mitochondria, plasma membrane, peroxisomes, and cytosol.  is converted to H2O2 by dismutation and thence to OH• by the Cu2+/Fe2+-catalyzed Fenton reaction. H2O2 is also derived directly from oxidases in peroxisomes. Not shown is another potentially injurious radical, singlet oxygen. Resultant free radical damage to lipid (peroxidation), proteins, and DNA leads to various forms of cell injury. Note that superoxide catalyzes the reduction of Fe3+ to Fe2+, thus enhancing OH• generation by the Fenton reaction. The major antioxidant enzymes are superoxide dismutase (SOD), catalase, and glutathione peroxidase. GSH, Reduced glutathione; GSSG, oxidized glutathione; NADPH, reduced form of nicotinamide adenine dinucleotide phosphate. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
is converted to H2O2 by dismutation and thence to OH• by the Cu2+/Fe2+-catalyzed Fenton reaction. H2O2 is also derived directly from oxidases in peroxisomes. Not shown is another potentially injurious radical, singlet oxygen. Resultant free radical damage to lipid (peroxidation), proteins, and DNA leads to various forms of cell injury. Note that superoxide catalyzes the reduction of Fe3+ to Fe2+, thus enhancing OH• generation by the Fenton reaction. The major antioxidant enzymes are superoxide dismutase (SOD), catalase, and glutathione peroxidase. GSH, Reduced glutathione; GSSG, oxidized glutathione; NADPH, reduced form of nicotinamide adenine dinucleotide phosphate. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Free radical injury is usually controlled by intracellular antioxidants such as superoxide dismutase (SOD), glutathione peroxidase, and vitamins E and C; however, injury can be catastrophic when these antioxidative systems are defective. In many species of domestic animals, severe cellular damage occurs to heart muscle when there is a deficiency of selenium or vitamin E in the tissues. Vitamin E is one of several cytoprotective molecules that acts as an antioxidant and inhibits production of or quenches free radicals, even in normal cell metabolism. Insufficient antioxidant activity can result in severe cell injury and necrosis as a consequence of the free radicals generated. Selenium is an essential component for some glutathione peroxidases, which also inactivate some free radicals generated within cells.
Morphologic Appearance of Necrotic Cells and Tissues (Oncotic Necrosis): In contrast to postmortem autolysis, necrosis occurs in the living animal, but the degradative processes of the cells involved are similar. One challenge to veterinarians and pathologists is to distinguish necrosis (tissues that died before somatic death) from tissues that died with the rest of the animal (postmortem autolysis).
At this point, there may be some confusion about the term autolysis. Most veterinarians and pathologists use this term synonymously with postmortem changes. Technically, autolysis means the self-digestion or degradation of cells and tissues by the hydrolytic enzymes normally present in those tissues. Therefore by the strict definition, autolysis occurs in all tissues that die (and even before they die), regardless of whether cells die before or after the animal dies. Postmortem change includes both autolysis and putrefaction, which is the process by which bacteria break down tissues.
The appearance of necrotic cells varies with the tissue involved, the cause of cell death, and the duration of time. For our immediate purposes, necrosis here will for the most part be used to mean oncotic necrosis. Apoptotic necrosis will be discussed later.
Ultrastructure of Necrotic Cells (Oncotic Necrosis): Cells dying after acute cell swelling are first swollen. There is tremendous swelling of all mitochondria, ER is dilated and fragmented, chromatin is clumped, the nuclear membrane is folded, the cytoplasm is pale and structureless, and organelles are poorly visualized. As the intracellular and extracellular compartments reach equilibrium across the altered cell membrane, the cell collapses and shrinks like a hot air balloon that has lost its air. The formerly swollen cell is shrunken; cytoplasm and organelles are homogeneous, electron dense, and hard to identify. Specialized areas of the plasma membrane, such as desmosomes, microvilli, and cilia, are distorted or absent.
Histologic Changes in Necrosis (Oncotic Necrosis): Nuclear changes of dead cells are strong histologic evidence of cell death. These changes are variable and are described by the terms pyknosis, karyorrhexis, and karyolysis (Fig. 1-17). All of the following nuclear changes may be visible in necrotic cells in the same necrotic lesion. Basophilic fragments of nuclear debris can be confused with bacteria, protozoa, and calcium deposits. Histomorphology of the nucleus of a necrotic cell includes one or more of the following:
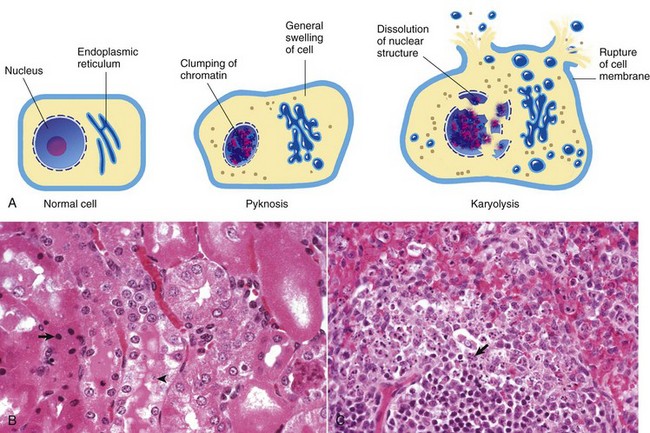
Fig. 1-17 Cytoarchitecture of cellular necrosis.
A, Schematic representation of nuclear and cytoplasmic changes in the stages of necrosis. B, Pyknosis and karyolysis, renal cortex, chloroform toxicosis, mouse. Some epithelial cells exhibit hydropic degeneration, whereas others are necrotic. Some necrotic cells exhibit pyknosis (arrow), whereas others have lost the nucleus or have a very pale nucleus (karyolysis) (arrowhead). H&E stain. C, Karyorrhexis, lymphocytes, spleen, dog. Spleen of a dog with parvovirus infection. Lymphocyte nuclei have fragmented because of the infection (arrow). H&E stain. (A from Huether S, McCance K: Understanding pathophysiology, ed 3, St Louis, 2004, Mosby; B and C courtesy Dr. L.H. Arp.)
• Pyknosis: The nucleus is shrunken, dark, homogeneous, and round, unlike the dark and dense fragmented nucleus of apoptotic cells. Pyknosis may be a sequel to chromatin clumping of early degeneration.
• Karyorrhexis: The nuclear envelope is ruptured, and dark nuclear fragments are released into the cell cytoplasm.
• Karyolysis: The nucleus is extremely pale due to dissolution of chromatin presumably by action of RNAases and DNAases.
• Absence of nucleus: This is a later stage of karyolysis in which the nucleus has been completely dissolved or lysed.
Some cell lines have a preference for a type of nuclear change in necrosis. Necrotic lymphocytes often become pyknotic, sometimes karyorrhectic, followed by release of nuclear debris. Necrotic renal proximal tubular epithelial cells often have karyolytic nuclei, but the distal tubules may have predominantly pyknotic nuclei.
Cytoplasmic changes in dead cells: Early in cell necrosis, the cytoplasm becomes homogeneous pink in H&E-stained sections (Fig. 1-18). Increased eosinophilia may reflect a loss of rRNA, which is responsible for cytoplasmic basophilia, or a consolidation of cytoplasmic components as the cell collapses. Degradation of cytoplasmic proteins eventually gives the necrotic cell a pale, ghostlike appearance. Necrotic cells usually lose their adherence to basement membranes and neighboring cells become “individualized” so they are found free in tubules, alveoli, follicles, and other lumens or on surfaces. Rupture of cells with loss of cell integrity is the most obvious evidence of cell death.
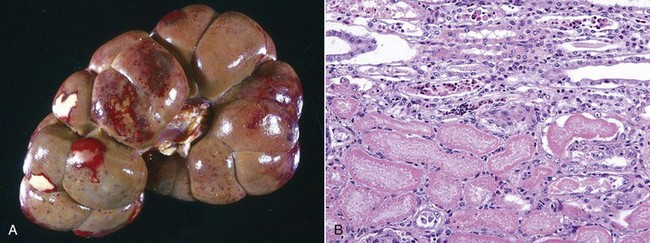
Fig. 1-18 Coagulation necrosis, infarcts, kidney, cow.
A, Note the white-yellow areas of acute coagulation necrosis surrounded by a red rim of active hyperemia and inflammation (far left). B, Acute coagulation necrosis of renal tubular epithelial cells. Necrotic cells have homogeneous eosinophilic cytoplasm and more or less retained cell outlines (lower half of figure) and nuclear changes such as pyknosis and nuclear absence (upper half of figure). H&E stain. (A courtesy Dr. D.E. Tyler, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Gross Appearance of Necrotic Tissue: Depending on the duration of injury and the type of organ (e.g., liver and kidney), necrotic tissue is usually pale, soft and friable, and sharply demarcated from viable tissue by a zone of inflammation (see Fig. 1-18, A). An exception to the pale color occurs when blood oozes into the necrotic tissue from damaged blood vessels in adjacent viable tissue as happens in renal infarcts, which are often surrounded by a narrow (1 to 3 mm) red rim (active hyperemia). A sharp line of demarcation between necrotic and viable tissue is often a reliable means to distinguish necrosis from autolysis. It must be emphasized that necrotic changes are first apparent ultrastructurally (less than 6 hours), then histologically (6 to 12 hours), and finally grossly (24 to 48 hours). Therefore, except for vascular changes, morphologic evidence of cell death is often sparse or absent in cases of peracute or acute death.
Types of Oncotic Necrosis: Foci of necrosis in tissue have a limited number of morphologic appearances, depending on the tissue involved, the cause of cell injury, and somewhat on the time since injury has occurred. Classification of necrotic lesions enables the pathologist to describe the lesion with a minimum of repetitious detail, but more than one pattern or type of necrosis may be seen in an organ or tissue. The recognized types of necrosis in most tissues are the following:
These are classically or historically derived, and although commonly used, do not always accurately describe the complexity of what has happened to the involved cells and tissues.
Coagulation necrosis: Coagulation necrosis (coagulative necrosis) is characterized by preservation of the basic cell outlines of necrotic cells (see Fig. 1-18, B). Cytoplasm is homogeneous and eosinophilic due to coagulation of cell proteins, similar to what happens to heat coagulation of proteins of a cooked egg white. Presumably the injury or subsequent cellular acidosis denatures not only structural proteins but also enzymes. This delays proteolysis of the cell. Nuclei are pyknotic, karyorrhectic, karyolytic, or absent. This form of necrosis may occur in any tissue except brain parenchyma, although it does occur initially in individual neurons. It is classically seen in kidney, liver, and muscle, and the necrotic tissue will eventually lyse within several days and be phagocytosed. The presence of coagulation necrosis suggests hypoxic cell injury as seen in local loss of blood supply or in shock. Bacterial exotoxins and chemical toxins also cause the lesion. Infarction is necrosis caused by ischemia. An infarct, for example, occurring in the human heart as a result of the blockage of a coronary artery by an atherosclerotic plaque is an area of coagulation necrosis that results from a sudden loss of blood supply to an area.
Caseation (caseous) necrosis: Caseation necrosis (caseous necrosis) implies conversion of dead cells into a granular friable mass grossly resembling cottage cheese (Fig. 1-19). The necrotic focus is composed of a coagulum of nuclear and cytoplasmic debris. Compared with coagulation necrosis, this is an older (chronic) lesion often associated with poorly degradable lipids of bacterial origin. Any tissue may be affected, and much of the necrotic debris is dead leukocytes. Dystrophic calcification commonly occurs later within the central parts of the lesion. The classic cause of this lesion is tuberculosis. Related bacteria, such as Corynebacterium, also cause this lesion in sheep. Delayed degradation of the bacterial cell wall is thought to play a role in the development of a lesion caused by these bacteria and results in a focus of caseous necrosis surrounded by granulomatous inflammatory cells and an outer fibrous connective tissue capsule. In birds and reptiles, necrotic areas are slow to liquefy and often undergo caseation necrosis, likely caused by insignificant amounts of myeloperoxidase in their heterophils, the equivalent inflammatory cell of the neutrophil in mammals.
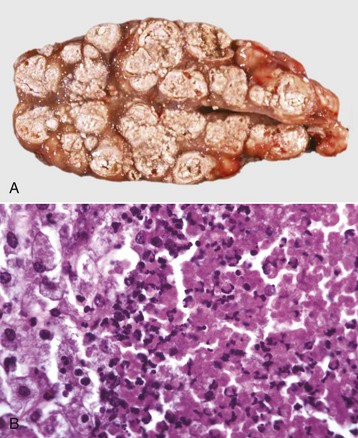
Fig. 1-19 Tuberculosis, lymph node, transverse section, ox.
A, The lymph has been replaced by a caseating granuloma. Note the caseous necrosis characterized by a pale yellow, crumbly exudate. B, Granulomatous inflammation in caseous necrosis. Cell walls are disrupted and tissue architecture is lost. Mineralization (not seen here) is common in this type of necrosis. H&E stain. (A courtesy Dr. M. Domingo, Autonomous University of Barcelona; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Liquefactive necrosis: Liquefactive necrosis is the usual type of necrosis in the CNS, although the neuron cell bodies themselves initially show coagulation necrosis, followed by liquefaction (Fig. 1-20). Hypoxic death of cells in the CNS results in rapid enzymatic dissolution of the neuropil (liquefaction), likely the result of the large amount of cell membranes present. With loss of astrocytes and because there is normally very little fibrous connective tissue in the CNS, little remains to support the tissue or fill in dead space. The result is a cavity filled with lipid debris and fluid. These cystic areas are cleared of debris by macrophages that become gitter cells (described further in Chapter 14).
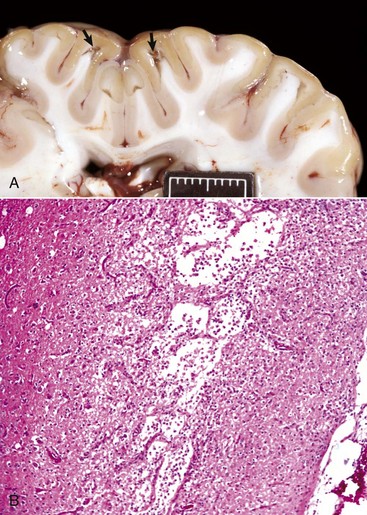
Fig. 1-20 Liquefactive necrosis.
A, Acute polioencephalomalacia, brain, goat. A thiamine deficiency has resulted in cerebrocortical malacia, which microscopically is liquefactive necrosis and varying degrees of tissue separation (arrows). Scale bar = 2 cm. B, Cortical necrosis, cerebrum, dog. The pale horizontal band in the cerebral cortex contains areas of near total loss of cells and tissue loss termed liquefactive necrosis. The cells in the spaces are gitter cells. Grossly, this band would have a fluid consistency. H&E stain. (A courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University. B courtesy Dr. L.H. Arp.)
In other tissues, focal infection by pyogenic bacteria leads to release of enzymes from accumulating leukocytes. Early in this process, heterolysis leads to a focal liquid collection of necrotic neutrophils and tissue debris (pus), and the lesion is an abscess that is also a type of liquefactive necrosis. If the abscess persists, loss of fluid or inspissation of the pus results in it becoming more caseous.
Gangrenous necrosis: The three types of gangrene are dry gangrene, moist gangrene, and gas gangrene. They are included here because the initial lesion is coagulation necrosis.
• Moist gangrene is defined as an area of necrotic tissue (usually coagulation necrosis), which is further degraded by the liquefactive action of saprophytic bacteria (defined as organisms living in dead organic matter) that usually cause putrefaction (defined as the decomposition of organic matter by microorganisms [i.e., rotten]). The initial coagulation necrosis can be caused by infarction of an extremity (too tight a bandage on a limb, penetrating damage to an artery supplying the leg by a bullet or shrapnel) or of a segment of intestine, or as in the case of the lung, by direct action of aspirated irritants such as medicaments or even ruminal fluid. The saprophytic bacteria contaminate the dead tissue from the local environment (air, skin contaminants, and soil) in the case of a limb, from inhaled air in the lung, and from the adjacent ingesta in an intestinal infarct. Grossly, tissues become soft, moist, and reddish-brown to black, and if the saprophytic bacteria produce gas, as they usually do, then gas bubbles and a putrid odor from the hydrogen sulfide, ammonia, and mercaptans may occur (Fig. 1-21, A). With time, if death does not supervene from toxemia, gangrenous tissue of the leg and udder are separated from the normal tissue by inflammation and may slough. Microscopically, initially areas of coagulation necrosis contain a few proliferating bacteria. These quickly proliferate and produce liquefaction and depending on the bacteria, gas bubbles. As the lesion progresses, most of the necrotic tissue is liquefied by saprophytic bacteria and infiltrating neutrophils.
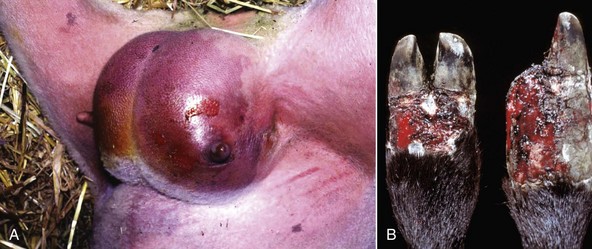
Fig. 1-21 Gangrenous necrosis.
A, Moist gangrene, udder, sheep. The surrounding tissue is well vascularized, which contributes to the wet and bloody nature of the lesion. Often saprophytic bacteria and clostridia contaminate areas of necrosis. B, Dry gangrene, fescue toxicity, digits, cow. Fescue toxicity is a disease in which the blood supply to the distal extremities is lost because of vasoconstriction from the toxic effect on vessels. The dry leathery appearance adjacent to the hooves is termed dry gangrene. There is still some blood in the skin, indicating that at least a partial blood supply has been retained or restored. Note that one of the claws (right) has been lost due to the process. (A courtesy Dr. C. Wallace, College of Veterinary Medicine, University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
• Dry gangrene is really coagulation necrosis secondary to infarction, which is followed by mummification. It involves the lower portion of an extremity (leg), tail, ears, and udder and can be caused by ingested toxins (ergot and fescue poisoning) or cold (frostbite). Ergot produces a marked peripheral arteriolar vasoconstriction and damage to capillaries, which leads to thrombosis and infarction. Fescue poisoning in cattle has a similar pathogenesis and lesions. Exposure to very cold temperatures also causes dry gangrene (frostbite). The pathogenesis involves both direct freezing and disruptions of cells by intracellular and extracellular ice crystal formation and vascular damage leading to ischemia and infarction characterized by coagulation necrosis (see Chapter 17). In dry gangrene, after necrosis, the tissues are depleted of water, for example, by low humidity, and this dehydration results in mummification. There is no proliferation of bacteria, as dry tissues do not provide an environment favorable for their proliferation and spread. Grossly, the tissue is shriveled, dry, and brown to black (Fig. 1-21, B) and affected parts may slough
• Gas gangrene is also an example of bacteria proliferating and producing toxins in necrotic tissue, but in this case the bacteria are anaerobes, usually microbes such as Clostridium perfringens and Clostridium septicum. Penetrating wounds into muscle or subcutis introduce these bacteria. The necrotic tissue then provides an anaerobic medium for growth of the clostridia. Another example, with similar lesions, is caused by Clostridium chauvoei (blackleg), which, unlike the bacteria of gas gangrene, is not introduced by a penetrating wound but from spores spread hematogenously from the intestine and lodged in muscle. Here they stay until by some mechanism, such as trauma, necrosis occurs and thus produces anaerobic conditions in which the spores can germinate and the bacteria proliferate. Grossly, affected tissues are dark red to black with gas bubbles and a fluid exudate that may contain blood. Microscopically, the lesions are characterized by coagulation necrosis of muscle, a serohemorrhagic exudate, and gas bubble formation (see Chapter 15). Some authors do not classify the lesions of blackleg as gas gangrene because it is a result of hematogenously disseminated bacterial spores and not from bacterial contamination of a wound.
Necrosis of Fat (Fat Necrosis): The three types of fat necrosis, enzymatic necrosis of fat, traumatic necrosis of fat, and necrosis of abdominal fat of cattle, are as follows:
• Enzymatic necrosis of fat, also called pancreatic necrosis of fat, refers to the destruction of fat in the abdominal cavity and usually adjacent to the pancreas by the action of activated pancreatic lipases in pancreatic fluid that has escaped from the duct system of the pancreas (Fig. 1-22).
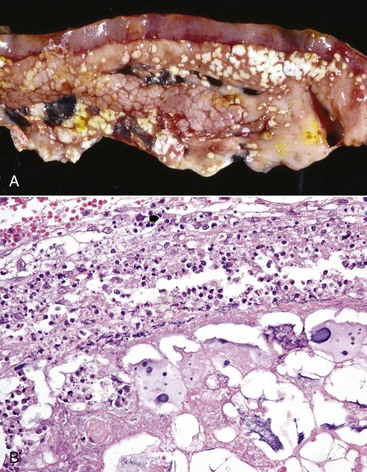
Fig. 1-22 Fat necrosis.
A, Enzymatic necrosis of fat (fat necrosis), dog with previous bouts of pancreatitis. Necrotic fat often becomes saponified and so grossly the lesion is chalky to gritty and pale white. B, Pancreas, dog. Note the large area of fat necrosis with acute inflammation and saponification (basophilic areas). H&E stain. (A courtesy Dr. J. Wright, College of Veterinary Medicine, North Carolina State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
• Traumatic necrosis of fat is seen when adipose tissue is crushed. It occurs in fat adjacent to the pelvic canal of heifers after dystocia and in subcutaneous tissue that has been injured, for example, in the subcutaneous and intramuscular fat over the sternum of recumbent cattle.
• Fat necrosis of abdominal fat of cattle is characterized by large masses of necrotic fat in the mesentery, omentum, and retroperitoneally. The cause is unknown and may not be detected until necropsy. In extreme cases, mesenteric fat may surround the intestine and cause stenosis.
Grossly affected fat is white, firm, and chalky. Histologically, the fat solvents used in the preparation of the paraffin-embedded sections do not remove necrotic fat. Necrotic adipocytes are eosinophilic but become basophilic if free fatty acids react with Ca2+ to form soap (saponification of fat).
Sequelae to Oncotic Necrosis: In contrast to apoptosis and obviously postmortem autolysis, necrosis incites a notable inflammatory reaction in most tissues except the CNS in the surrounding viable tissue. Therefore the necrotic foci are often surrounded by a well-demarcated band of white blood cells and the hyperemia of inflammation. The purpose is to digest (by heterolytic enzymes of leukocytes) and liquefy the necrotic tissue so that it can be removed by macrophages and diffusion into blood and lymph vessels and replaced by normal tissue (regeneration) or fibrous connective tissue (healing). Healing of an abscess occurs after the sequestered pus is phagocytosed and/or carried off by the lymphatics. The process is greatly hastened by drainage, either by rupture to the outside or by surgical drainage of the abscess. Material not liquified is phagocytosed by macrophages and removed via lymphatics or veins. A fragment of necrotic material, especially bone, may resist degradation and form a sequestrum. This may cause chronic irritation and delay repair.
Morphologic Appearance of Postmortem Changes: Postmortem autolysis refers to the autolysis of cells occurring after somatic death. These changes are amplified and accelerated by bacterial decomposition from bacteria that have entered the tissue shortly before death or after death (usually by either direct migration from the lumen of the gut of the dead or dying animal, or from the gut into the blood and then disseminated throughout the body by the final beats of the heart). Postmortem bacterial metabolism and dissolution of host tissues (postmortem decomposition) result in the production of color and texture changes, gas production, and odors collectively termed putrefaction.
Somatic death refers to death of the entire body; however, cell types vary greatly in time of viability after cessation of heartbeat, respiration, and brain wave activity. In somatic death, many neurons and cardiac myocytes suffer irreversible injury within minutes; kidney and liver cells may survive for an hour; and fibroblasts and bone survive much longer. Interpretation of lesions is usually clouded by changes that occur between the time of death and the time of necropsy (or fixation of tissue). Postmortem autolysis results from total diffuse hypoxia, and cells degenerate as described for hypoxic cell injury. A long death-to-fixation interval can lead to problems in histopathologic diagnosis of necrosis and other lesions; thus keeping postmortem changes to a minimum is important for accurate gross and histopathologic interpretation.
Postmortem changes vary greatly in onset and rate depending on the cause of death, environmental and body temperature, and microbial flora. Cool environmental temperatures and refrigeration (without freezing if possible because freezing induces artefacts such as intracellular and extracellular ice crystals, which disrupt cells and tissues, respectively) inhibit autolysis and delay putrefaction. Animals examined 24 hours after death, after being maintained at 5° C, have relatively few postmortem changes and artefacts to interpret versus an animal that has been maintained at room temperature for a similar time. An exception is herbivores. In the ruminant forestomach and equine cecum and ascending colon, ingesta will continue to undergo bacterial fermentation after death, with formation of heat and gas. Consequently, these animals, even if refrigerated immediately after death, show considerable intraabdominal postmortem decomposition 24 hours later. High environmental temperatures accelerate autolysis, as does elevated antemortem body temperature caused by fever, high metabolic rate, heat stroke, and exercise. Delay in cooling is especially common in fat animals and those with a heavy coat, especially wool. Young and small animals, such as neonates, cool more rapidly than large obese ones. Determining the time since death has occurred can be difficult because of the many factors just listed that influence the rate of cooling.
In summary, postmortem changes can interfere with accurate interpretation of both gross and histologic changes in tissue. Postmortem changes can be minimized by rapid cooling of the carcass and decreasing the death to tissue fixation time to a minimum. The following are examples of common postmortem changes, with some reference to their sequence of occurrence:
• Rigor mortis is the contraction of muscles occurring after death. It commences l to 6 hours after death and persists for l to 2 days. When ATP and glycogen (required to relax muscle contraction) are depleted, the contraction is irreversible except by autolysis. Muscular animals often have stronger rigor than those with less muscle mass. High heat and activity before death accelerate the onset of rigor. In animals with cachexia or extreme malnutrition, the energy stores (ATP, glycogen) in the muscles may be so depleted that no contraction of myofibers is possible, and thus these animals do not develop rigor mortis.
• Algor mortis is gradual cooling of the cadaver. Cooling of the carcass depends on temperature of the body at death (e.g., fever, environmental temperature, insulation of the carcass [fat, wool, coverings], body mass, air movement, and other factors) and is difficult to interpret precisely for establishing time of death.
• Livor mortis (hypostatic congestion) (Fig. 1-23) is the gravitational pooling of blood to the down side of the animal. In large vessels, there is clotting followed by separation of blood cells and plasma. This process begins within an hour after death, and the clotted blood can become “fixed” in place (whereby movement of the animal will not influence the distribution of the change) within 12 to 24 hours. It is often not appreciated in animals because of pigmented skin or a thick hair coat and thus is most likely to be evident in white-skinned animals with little hair (e.g., white pigs).
Fig. 1-23 Livor mortis, pig.
Note red to purple staining of the skin on the right side, the side on which the pig was lying when it died. This color change is termed livor mortis or hypostatic congestion. The pale white areas are pressure points on the down side into which blood could not flow after death. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
• Postmortem clotting (Fig. 1-24) in the heart and vessels usually occurs within several hours and can be influenced by antemortem changes in blood. Warfarin poisoning and hereditary coagulopathies, for example, will delay or cause failure of blood to clot. Before the blood clots, erythrocytes may settle to the bottom of a large vessel. This results in the clot having two portions: a bottom red mass made up chiefly of erythrocytes and an upper pale yellow mass of clotted serum. The latter type of clot is called a chicken fat clot. This separation depends upon the erythrocyte sedimentation rate (ESR) of the blood. It is high in normal horses and increased in all animals as a systemic inflammatory response. Inflammation results in increased plasma fibrinogen, which causes erythrocytes to form stacks (rouleau formation) that sediment more rapidly. Postmortem clots must be distinguished from antemortem mural thrombi and thromboemboli. Postmortem clots are unattached to vessel walls and tend to be shiny and wet and form a perfect cast of vessel lumens. Antemortem mural arterial thrombi are attached to arterial walls, tend to be dry and duller in color, and are laminated with a tail extending downstream from the point of attachment. Antemortem venous thrombi are also attached, but loosely so, and in many cases may closely resemble postmortem clots.
Fig. 1-24 Postmortem clot, dog.
The postmortem clot is pale white to yellow (chicken fat clot) in some areas and shiny red (currant jelly clot) in others. Note how it conforms to the shape of the lumen of the vessels from which it was removed. (Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
• Hemoglobin imbibition (Fig. 1-25) is a term applied to the red staining of tissue, especially evident in the endocardium and tunica intima of arteries (particularly evident in the aorta) and veins beginning some hours after death. Once the integrity of the intima is lost, hemoglobin released by lysed erythrocytes penetrates the vessel wall and extends into the adjacent tissue. Hemoglobin staining of the intima can also occur in acute intravascular hemolysis. It is usually very obvious in aborted fetuses that are retained for hours or days after their in utero deaths.
Fig. 1-25 Imbibition of hemoglobin, viscera, pig that has been dead for several hours before being necropsied.
Note the pink color on the serosal surfaces of the stomach and small intestine. This is termed imbibition of hemoglobin and is due to staining by hemoglobin that has seeped out of autolyzed red blood cells. (Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
• Bile imbibition (Fig. 1-26) occurs within hours of death. Bile in the gallbladder starts to penetrate its wall and stain adjacent tissue yellowish, and later this may become greenish brown. Tissues involved include the adjacent liver and any intestine in contact with the gallbladder. Sometimes, similar changes may be seen near the bile ducts.
Fig. 1-26 Postmortem autolysis.
Cross-sections of livers from three different pigs at different stages of postmortem autolysis. The section on the right has green staining around the bile ducts caused by leakage of bile into the surrounding parenchyma after death (bile imbibition). All of these livers are softer than normal, but the one on the left is notably softer, another characteristic of autolytic tissue. (Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
• Pseudomelanosis is the term used for the blue-green discoloration of the tissue by iron sulfide (FeS) formed by the reaction of hydrogen sulfide (H2S) generated by putrefactive bacteria and the iron from hemoglobin released from lysed erythrocytes. Because it depends on bacterial action, it usually takes a day or more to develop.
• Bloating (Fig. 1-27) is the result of postmortem bacterial gas formation in the lumen of the GI tract. Herbivores tend to bloat more rapidly and severely than carnivores. Rumen gases are produced normally in the forestomachs of live animals, but the gas is removed by eructation. In ruminants the rumen can become markedly distended by gas within hours of death, and this can be so severe as to rupture the diaphragm. The rate of gas formation depends on the diet, the substrate for the bacteria, and the temperature. Postmortem bloat can sometimes be difficult to distinguish from antemortem bloat (ruminal tympany) in ruminants that have not developed a “bloat line” (see Chapter 7). Bacteria disseminated hematogenously from the GI tract shortly before death can lodge in a variety of tissues and produce gas (postmortem emphysema).

Fig. 1-27 Postmortem bloat or emphysema.
Cow killed by lightning several hours earlier. When animals die, especially ruminants, the bacteria in the gastrointestinal tract continue to grow and produce gas. Rumen microbes may produce very large amounts of gas causing the carcass to swell tremendously. (Courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
• Organ displacement occurs after distention of the viscera; for example, distention of the rumen by gas from fermentation can cause increased intraabdominal pressure, which can result in displacement of abdominal viscera, rectal prolapse, and compression of the diaphragm, which then compresses the thoracic viscera. The latter can result in the expulsion of frothy fluid, originally in the lungs, from the mouth and nose.
• Pale foci subserosally on the liver (Fig. 1-28) can result from two causes: increased intraabdominal pressure, which squeezes blood from these areas (e.g., pressure from the overlying ribs can leave their imprints on the liver), and bacterial action. Under very hot conditions, pale areas can appear on the surface of the bovine liver within hours of death. Histologically, these areas resemble coagulation necrosis in which there are extremely numerous bacteria. Presumably these bacteria have been disseminated agonally from the gut into the portal vein.
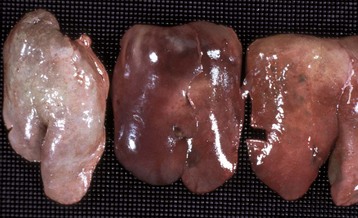
Fig. 1-28 Postmortem autolysis.
Pig livers at various intervals after death. Pale foci on the middle liver are due to blood being forced out of the parenchyma by intestinal swelling (intestinal imprints) and from pressure from the overlying ribs (rib imprints). Multiple small pale foci can sometimes be caused by colonies of postmortem bacteria and can be confused with antemortem necrosis. (Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
• Softening (see Figs. 1-26 and 1-28) of tissue results from autolysis of cells and connective tissue often aided by putrefactive bacteria.
• Mucosal sloughing occurs rapidly in the rumen, often within a few hours as a result of the enzymes within the ingesta and the low rate of cooling.
• Lens opacity (Fig. 1-29, also see Chapter 20) occurs when the carcass is very cold or frozen. The change will reverse to normal transparency on warming, but it can be confused with cataracts in cold carcasses.
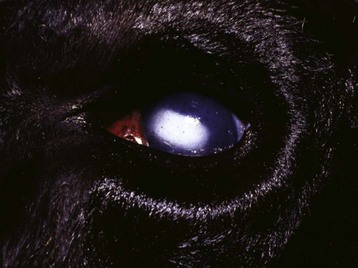
Fig. 1-29 Postmortem autolysis, eye, lens, calf.
Note that the cornea is clear. The cloudiness of the lens is due to cooling or freezing and is reversible as the carcass warms up. It should not be confused with cataracts. (Courtesy Dr. P.N. Nation, University of Alberta; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Cell Death By Apoptosis
The terms apoptosis and programmed cell death have been used virtually synonymously to refer to individual cell death in which there is initiation of a self-induced cell death process some refer to as cellular suicide. Apoptosis can be pathologic or physiologic and is only one of types of programmed cell death (others include cell death with autophagy and keratinocyte cornification). Programmed cell death is better reserved for physiologic cell death (which often involves apoptosis) that occurs, for example, in developing animals (embryogenesis and normal growth) and deletion of immune cells. For those circumstances in which pathologic cell death occurs with shrinkage first as a feature, apoptosis or apoptotic necrosis is more appropriately used. Apoptosis occurs in a variety of pathologic circumstances, including viral diseases such as yellow fever in humans, gland involution after duct blockage, immunologic damage by T lymphocytes, and as a component of injury caused by hypoxia and some chemicals and drugs.
Mechanisms of Apoptosis*: Mechanisms of programmed cell death and apoptosis have been extensively researched within the last decades. A variety of stimuli result in a self-programmed, genetically determined, energy-dependent sequence of molecular events involving initiation by cell signaling, control and integration by regulatory molecules, a common execution phase by caspase family genes, and dead cell removal. Some of these mechanisms are initiated by inflammatory mediators such as TNF and the Fas ligand (FasL). Others involve deprivation of growth factors, mitochondrial damage, DNA damage, accumulation of misfolded proteins, or immune stimulation (Fig. 1-30).
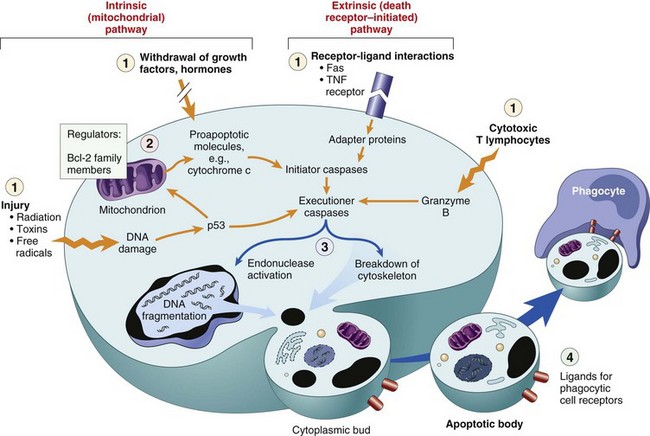
Fig. 1-30 Mechanisms of apoptosis.
Labeled (1) are some of the major inducers of apoptosis. These include specific death ligands (tumor necrosis factor [TNF] and Fas ligand), withdrawal of growth factors or hormones, and injurious agents (e.g., radiation). Some stimuli (such as cytotoxic cells) directly activate execution caspases (right). Others act by way of adapter proteins and initiator caspases, or by mitochondrial events involving cytochrome c. (2) Control and regulation are influenced by members of the Bcl-2 family of proteins, which can either inhibit or promote the cell’s death. (3) Executioner caspases activate latent cytoplasmic endonucleases and proteases that degrade nuclear and cytoskeletal proteins. This results in a cascade of intracellular degradation, including fragmentation of nuclear chromatin and breakdown of the cytoskeleton. (4) The end result is formation of apoptotic bodies containing intracellular organelles and other cytosolic components; these bodies also express new ligands for binding and uptake by phagocytic cells. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
The process of apoptosis may be divided into an initiation phase, during which caspases become catalytically active, and an execution phase, during which these enzymes act to cause cell death. Initiation of apoptosis occurs principally by signals from two distinct but convergent pathways: the extrinsic, or receptor-initiated, pathway and the intrinsic, or mitochondrial, pathway. Both pathways converge to activate caspases. These two pathways are described separately because they involve largely distinct molecular interactions, but it is important to remember that they may be interconnected at numerous steps.
The Extrinsic (Death Receptor–Initiated) Pathway: The extrinsic pathway is initiated by engagement of cell surface death receptors on a variety of cells (see Fig. 1-30). Death receptors are members of the TNF receptor family that contain a cytoplasmic domain involved in protein-protein interactions that is called the death domain because it is essential for delivering apoptotic signals. (Some TNF receptor family members do not contain cytoplasmic death domains; their role in triggering apoptosis is much less established.) The best-known death receptors are the type 1 TNF receptor (TNFR1) and a related protein called Fas (CD95), but several others have been described. The mechanism of apoptosis induced by these death receptors is well illustrated by Fas (Fig. 1-31). When Fas is cross-linked by its ligand, membrane-bound FasL, three or more molecules of Fas come together, and their cytoplasmic death domains form a binding site for an adapter protein that also contains a death domain and is called Fas-associated death domain (FADD). FADD is attached to the death receptors, which in turn bind an inactive form of caspase-8 (and in humans, caspase-10), again via a death domain. Multiple procaspase-8 molecules are thus brought into proximity, and they cleave one another to generate active caspase-8. The enzyme then triggers a cascade of caspase activation by cleaving and thereby activating other procaspases, and the active enzymes mediate the execution phase of apoptosis (discussed later). This pathway of apoptosis can be inhibited by a protein called FLIP, which binds to procaspase-8 but cannot cleave and activate the enzyme because it lacks enzymatic activity. Some viruses and normal cells produce FLIP and use this inhibitor to protect infected and normal cells from Fas-mediated apoptosis. The sphingolipid ceramide has been implicated as an intermediate between death receptors and caspase activation, but the role of this pathway is unclear and remains controversial.
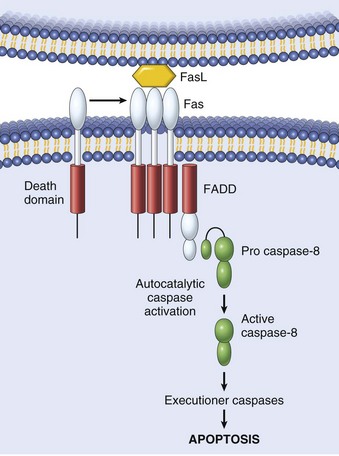
The Intrinsic (Mitochondrial) Pathway: The intrinsic pathway of apoptosis is the result of increased mitochondrial permeability and release of proapoptotic molecules into the cytoplasm, without a role for death receptors (see Fig. 1-30). Growth factors and other survival signals stimulate the production of antiapoptotic members of the Bcl-2 family of proteins. This family is named after Bcl-2, which was identified as an oncogene in a B lymphocyte lymphoma and is homologous to the Caenorhabditis elegans protein, Ced-9. There are more than 20 proteins in this family, all of which function to regulate apoptosis; the two main antiapoptotic ones are Bcl-2 and Bcl-x. These antiapoptotic proteins normally reside in mitochondrial membranes and the cytoplasm. When cells are deprived of survival signals or subjected to stress, including misfolded proteins and ER stress, or when DNA is damaged, Bcl-2 and/or Bcl-x are lost from the mitochondrial membrane and are replaced by proapoptotic members of the family such as Bak, Bax, and Bim. When Bcl-2/Bcl-x levels decrease, the permeability of the mitochondrial membrane increases, and several proteins that can activate the caspase cascade leak out (Fig. 1-32). One of these proteins is cytochrome c, well known for its role in mitochondrial respiration. In the cytosol, cytochrome c binds to a protein called apoptosis activating factor-1 (Apaf-1, homologous to Ced-4 in Caenorhabditis elegans), and the complex activates caspase-9. (Bcl-2 and Bcl-x may also directly inhibit Apaf-1 activation, and their loss from cells may permit activation of Apaf-1.) Other mitochondrial proteins, such as apoptosis-inducing factor (AIF), enter the cytoplasm, where they bind to and neutralize various inhibitors of apoptosis, whose normal function is to block caspase activation. The net result is the initiation of a caspase cascade. Thus the essence of this intrinsic pathway is a balance between proapoptotic and protective molecules that regulate mitochondrial permeability and the release of death inducers that are normally sequestered within the mitochondria. There is evidence that the intrinsic pathway of apoptosis can be triggered without a role for mitochondria. Apoptosis may be initiated by caspase activation upstream of mitochondria, and the subsequent increase in mitochondrial permeability and release of proapoptotic molecules amplify the death signal. However, these pathways of apoptosis involving mitochondria-independent initiation are not well defined. We have described the extrinsic and intrinsic pathways for initiating apoptosis as distinct, but there may be overlaps between them. For instance, in hepatocytes, Fas signaling activates a proapoptotic member of the Bcl family called Bid, which then activates the mitochondrial pathway. It is not known if such cooperative interactions between apoptosis pathways are active in most other cell types.
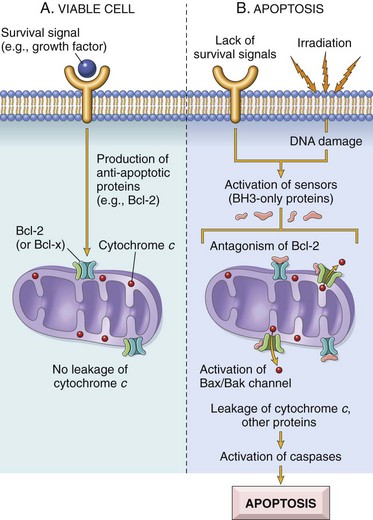
Fig. 1-32 The intrinsic (mitochondrial) pathway of apoptosis.
A, Cell viability is maintained by the induction of antiapoptotic proteins such as Bcl-2 by survival signals. These proteins maintain the integrity of mitochondrial membranes and prevent leakage of mitochondrial proteins. B, Loss of survival signals, DNA damage, and other insults activate sensors that antagonize the antiapoptotic proteins and activate the proapoptotic proteins Bax and Bak, which form channels in the mitochondrial membrane. The subsequent leakage of cytochrome c (and other proteins, not shown) leads to caspase activation and apoptosis. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
The Execution Phase: This final phase of apoptosis is mediated by a proteolytic cascade, toward which the various initiating mechanisms converge. The proteases that mediate the execution phase are highly conserved across species and belong to the caspase family, as previously mentioned. They are mammalian homologues of the Ced-3 gene in Caenorhabditis elegans. The term caspase is based on two properties of this family of enzymes: The “c” refers to a cysteine protease (i.e., an enzyme with cysteine in its active site), and “aspase” refers to the unique ability of these enzymes to cleave aspartic acid residues. The caspase family, now including more than 10 members, can be divided functionally into two basic groups: initiator and executioner, depending on the order in which they are activated during apoptosis. Initiator caspases, as we have seen, include caspase-8 and caspase-9. Several caspases, including caspase-3 and caspase-6, serve as executioners. Like many proteases, caspases exist as inactive proenzymes, or zymogens, and must undergo an activating cleavage for apoptosis to be initiated. Caspases have their own cleavage sites that can be hydrolyzed not only by other caspases but also autocatalytically. After an initiator caspase is cleaved to generate its active form, the enzymatic death program is set in motion by rapid and sequential activation of other caspases. Execution caspases act on many cellular components. They cleave cytoskeletal and nuclear matrix proteins and thus disrupt the cytoskeleton and lead to breakdown of the nucleus. In the nucleus, the targets of caspase activation include proteins involved in transcription, DNA replication, and DNA repair. In particular, caspase-3 activation converts a cytoplasmic DNAase into an active form by cleaving an inhibitor of the enzyme; this DNAase induces the characteristic internucleosomal cleavage of DNA.
Morphologic Appearance of Apoptosis: Morphologically, apoptotic cells have condensed chromatin and cytoplasm, and fragments of them are often found in adjacent or phagocytic cells as apoptotic bodies (Fig. 1-33). Because single cells are dead, gross changes (and even microscopic changes) are usually not obvious. In addition, because the cell fragments into membrane-bound particles, phagocytosis occurs without the inflammation that is so often seen in necrosis.
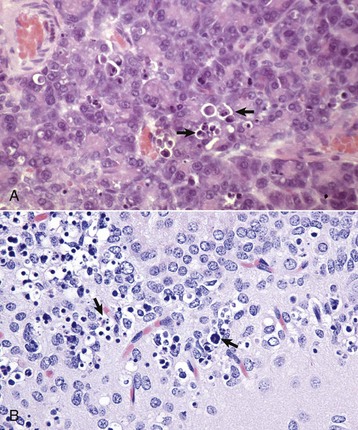
Fig. 1-33 Apoptosis, cytoarchitecture of cells.
A, Pancreas, rat. Individual acinar cells are shrunken and their chromatin condensed and fragmented (arrows). Cytoplasmic buds are found as apoptotic bodies in adjacent cells. Inflammation is absent. H&E stain. B, Hippocampal formation, mouse. Individual neurons are shrunken and their chromatin condensed and fragmented (arrows). (A courtesy Dr. M.A. Wallig, College of Veterinary Medicine, University of Illinois. B courtesy Drs. V.E. Valli and J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Although typically discussed separately, necrosis by oncosis and apoptosis can be seen within the same tissue as the result of the same agent (Fig. 1-34). Cell injury by a chemical, for example, that injures mitochondria may release cytochrome c and initiate the apoptosis program. Cells with more severely affected mitochondria may die from swelling or oncosis.
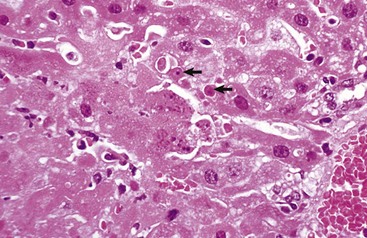
Fig. 1-34 Necrosis and apoptosis, mouse hepatitis virus infection, liver, mouse.
This disease causes hepatocyte death, typically by oncotic necrosis but sometimes by apoptosis. Note areas of coagulation necrosis in the lower left and apoptotic bodies in the center, some of which have been taken up by adjacent hepatocytes (arrows). H&E stain. (Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
The histopathologic characteristics of apoptosis are as follows:
Chronic Cell Injury and Cell Adaptation
As mentioned previously in the discussion of cell swelling and necrosis, cells respond to injury (or stress) in three possible major ways: (l) adaptation, (2) reversible injury with or without degeneration, and (3) death. Sublethal injury to a cell over time can lead to a variety of cell alterations. Cells may adapt by producing more cells (hyperplasia) or by producing more organelles, leading to an increase in size (hypertrophy); in some cases, adaptation results in fewer organelles and a decrease in cell and tissue size (atrophy). Cells may degenerate in a variety of ways, some of which involve the accumulation of excess normal or abnormal substances. Impaired function may result, and morphologic changes in the cell and tissue may give a clue as to the cause of the cell injury.
Sequelae to Sublethal Injury and Subcellular Changes
Autophagocytosis is the process by which cells with sublethal injury remove damaged and effete organelles. Cells with sublethal injury often have various amounts of damaged organelles. As in organized societies, the cell has a system to clean up after a “storm.” In autophagy, portions of the cytoplasmic matrix and its damaged organelles are enveloped by cell membranes to form autophagosomes, which subsequently fuse with lysosomes (Fig. 1-35). When phagocytic white cells ingest dead or dying cells, the process is very similar and termed heterophagy. Autophagy is a common reaction of sublethally injured cells, epithelial cells such as endometrium undergoing cyclic physiologic regression, and in atrophy as the result of many causes. Recent evidence suggests that autophagocytosis pathways may result in a distinct type of cell death termed autophagic cell death.
Fig. 1-35 Autophagy and heterophagy.
Schematic representation of heterophagy (left) and autophagy (right). The mechanisms are similar for processing cell debris, both from intrinsic sources and extrinsic sources (heterophagy). (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
By light microscopy, autophagic vacuoles may appear as eosinophilic inclusions (see section on Intracellular Hyaline Proteins) and are common in the liver and kidney. As digestion progresses, electron-dense and lamellar debris is formed. Some vacuoles are evicted from the cell by exocytosis; others remain as residual bodies, and the contents form lipofuscin, the so-called wear-and-tear pigment.
Misfolded proteins or those otherwise altered occur in a variety of circumstances within the cell, both normally and in disease states. These proteins may be repaired by chaperones, or they may be degraded by the ubiquitin-proteasome pathway. The targeted proteins are conjugated to ubiquitin (one of several heat shock proteins) that through a cascade results in polyubiquitination and direction of the protein into a proteasome, a multisubunit complex with a catalytic core that degrades the protein for removal (see Fig. 1-48). Removal of all sorts of proteins, including cell-signaling molecules, allows proper control of cell function, growth, and replication. This pathway also plays a role in both activation and inhibition of apoptosis as well as in sublethal injury.
Adaptive Changes Leading to Change in Cell Size, Number, or Appearance
Adaptive changes to cell stress or injury can lead to an increase in the size of a tissue or organ (by hyperplasia and/or hypertrophy), a decrease in tissue and cell size (atrophy), or a change to a different cell type (metaplasia) (Fig. 1-36). Hypertrophy is an increase in the size of cells or organs. Hyperplasia is an increase in the number of cells in a tissue or organ. The two often occur together as an adaptive change and are considered positive responses to injury or stress.
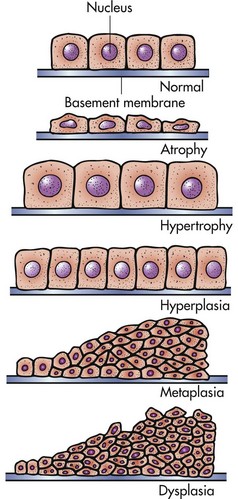
Fig. 1-36 Adaptive changes in epithelium. (From McCance KL, Huether SE, Brashers VL, et al: Pathophysiology: the biologic basis for disease in adults and children, ed 6, St. Louis, 2010, Mosby.)
Hypertrophy
In simple cellular hypertrophy, the number of cells in an organ or tissue does not increase. Cells synthesize more organelles, and cell enlargement occurs. The histologic architecture of the organ is normal, but cells are bigger. Hypertrophy can occur in most organs and tissues but tends to occur in cells that undergo little replication (i.e., stable or permanent cells). It is most common in striated muscle. In response to increased workload, smooth muscle may undergo hypertrophy and hyperplasia. Causes of hypertrophy usually involve demands for increased function (e.g., the increased workload on a muscle and resultant hypertrophy of that muscle in weight lifters).
The size and configuration of organelles reflect the work requirements of the cell. Chronic exposure to drugs, such as phenobarbital, Dilantin, and alcohol, leads to enlargement of the SER in hepatocytes. SER contains the mixed oxidase enzyme systems that function to catabolize these substances. The increased size of the Golgi complex and RER are a reflection of a need for synthesis of extracellular proteins (e.g., immunoglobulin, collagen, and secretions). These organelles increase in size by duplication of membranes. The number of mitochondria adjusts to the ATP requirements of the cell. The size of nucleoli and proportion of euchromatin also reflect the synthetic activity of the cell.
Physiologic hypertrophy is common and expected after work. Compensatory hypertrophy is a response to the loss of a part of an organ or one of the paired organs or from obstruction of the lumen of a hollow muscular organ. For example, hypertrophy occurs in one kidney after the loss of the opposite kidney. The kidney enlarges because of the increased length of nephrons and not the increased numbers of nephrons. Functional capacity increases with the increased size. Hypertrophy of the right ventricle of the heart caused by stenosis of the pulmonary outflow tract is another example of compensatory hypertrophy (Fig. 1-37).
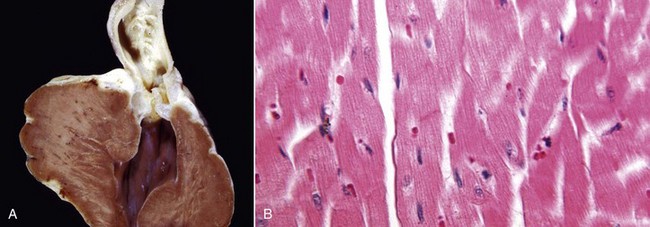
Fig. 1-37 Hypertrophy, heart, dog.
A, Narrowing of the pulmonary outflow tract caused by pulmonic valve stenosis has forced the right ventricle to contract with much more pressure. This increased workload has caused hypertrophy of the wall of the right ventricle, which is much thicker here than it would normally be. B, Note the increased size (hypertrophy) of myocytes in the overworked heart muscle. (Courtesy Dr. L. Miller, Atlantic Veterinary College, University of Prince Edward Island; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Hypertrophy is common, protective, limited, and reversible and may rarely cause harm to adjacent structures. Hypertrophy may not always be useful. In myocardial hypertrophy, enlargement of myofibers may occur with a corresponding increase in intercellular stroma, making the myocardium stiff. In addition, the blood supply may not increase adequately to serve the needs of the increased mass of myocytes, and this results in hypoxic injury. The term hypertrophy is used in gross pathology to describe lesions that involve gross enlargement of an organ regardless of cause.
Cellular mechanisms leading to hypertrophy vary by tissue and cause, and details are lacking for most entities. Growth factors likely play a role in altering gene expression in many circumstances, whereas in myofiber hypertrophy, the type of mechanical stress can influence the way the muscle enlarges, for example with increased mitochondria, required for oxidative metabolism in endurance training. Muscular hypertrophy of the uterus results from binding of estrogen to cytosolic estrogen-receptors that in turn activate genes leading to muscle protein production. These specific changes and others all are likely due to activation of specific genes.
Hyperplasia
Because hyperplasia is an increase in the number of cells, increased mitotic division is implied. Hyperplasia increases the size of a tissue, an organ, or part of an organ and may appear grossly as hypertrophy. It is a common change. Microscopically, cells resemble normal cells but are increased in numbers. Hyperplastic cells may also be increased in size (i.e., cellular hypertrophy).
The ability of different adult cell types to undergo hyperplasia varies. Labile cells—those that routinely proliferate in normal circumstances, such as those of the epidermis, intestinal epithelium, and bone marrow cells—readily become hyperplastic. Permanent cells, such as neurons and cardiac and skeletal muscle myocytes, have very little capacity to regenerate or become hyperplastic in most situations. Stable cells, such as bone, cartilage, and smooth muscle, are intermediate in their ability to become hyperplastic.
Hyperplasia is traditionally divided into physiologic hyperplasia and pathologic hyperplasia, as follows:
• Physiologic hyperplasia is usually either hormonal or compensatory. Hormonal hyperplasia includes conditions such as increased mammary gland epithelial proliferation before lactation and enlargement of the pregnant uterus. Compensatory hyperplasia, or regeneration, occurs after a portion of an organ is lost. For example, if the skin is abraded, the basal layer of the epidermis undergoes mitosis to regenerate superficial layers. Removal of part of the liver can cause mitosis in the remaining hepatocytes resulting in the restoration of the liver to its normal size but not necessarily its normal shape. This regenerative process takes as little as 2 weeks in rats after partial hepatectomy.
• Pathologic hyperplasia is often caused by excessive hormonal stimulation of target cells or chronic irritation. Cystic endometrial hyperplasia of the canine uterus as a result of prolonged progesterone influence is common. Microscopically, there is folding of increased numbers of epithelial cells into glands and onto the lumen surface. The mucosa thickens and may trap or impair secretions, causing dilation of glands and cyst formation within the mucosa. The process is reversible if the stimulus is removed.
Pathologic hyperplasia may cause diffuse enlargement of an organ, such as in benign prostatic hyperplasia in dogs and in goiter (hyperplasia of the thyroid gland) (Fig. 1-38), or be localized as nodular hyperplasia. Nodular hyperplasia may occur without known cause and occurs in the spleen, liver, and pancreas of old dogs. One must differentiate hyperplasia, particularly nodular hyperplasia, from neoplasia.
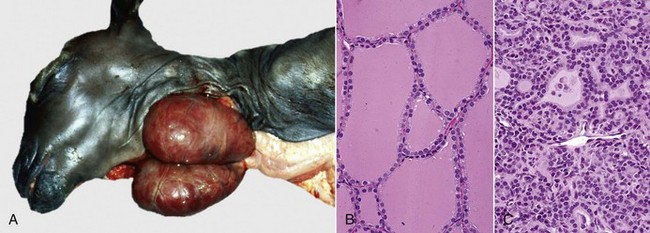
Fig. 1-38 Hyperplasia, thyroid goiter, goat.
A, Deficiency of maternal dietary iodine during pregnancy has resulted in hyperplasia (and hypertrophy) of thyroid follicular epithelial cells in this neonatal goat and thus results in a symmetric enlargement of the glands (goiter). B, Thyroid follicular epithelial cells from a normal thyroid gland. H&E stain. C, Thyroid follicular epithelial cells from a case of thyroid goiter. Note the increased number (and size) of the follicular epithelial cells. H&E stain. (A courtesy Dr. O. Hedstrom, College of Veterinary Medicine, Oregon State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B and C courtesy Dr. B. Harmon, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
The significance of hyperplasia usually lies in determining its cause. If it is hormonal in origin, the disturbance in the source organ should be determined. If chronic irritation is the cause, determining the agent is often important. Hyperplasia is usually induced by known stimuli. It is a controlled process that stops when the stimulus ceases, can serve a useful purpose (e.g., repair defects, compensate for tissue loss, prepare for increased function, and aid in protection), and is subject to regular growth controls. These features are not part of neoplastic processes, which otherwise may be similar to hyperplastic changes in appearance and behavior.
Cellular mechanisms of hyperplasia vary in details, depending on the cell affected and the cause. There are multiple controls as to whether or not a cell enters the replication cycle. In some circumstances hormones trigger cell replication, whereas in others growth factors, increased receptors for growth factors, and activation of cell signaling pathways may all have a role. In some circumstances, cytokines are important. Ultimately, transcription factors may influence the expression of a new cadre of genes leading to cell proliferation. In regeneration for restitution of parenchyma to normal amounts (see later discussion), stem cells turn on and lead to appropriate cell replication.
Metaplasia
Metaplasia is a potentially reversible change in which one adult cell type is replaced by another adult cell type of the same germ line (Fig. 1-39). Usually, specialized epithelium is replaced by less-specialized epithelium. One adult cell type does not transform into another type of adult cell. It is the less-differentiated reserve or stem cells that differentiate along a different line. For example, in cigarette smokers, chronic irritation of the normal columnar ciliated epithelium of the trachea and bronchial tree causes it to be replaced by focal or diffuse areas of stratified squamous epithelium. The squamous cells are more resistant to injury but are less protective to the lung, and, as they lack cilia, there is decreased clearance of mucus.
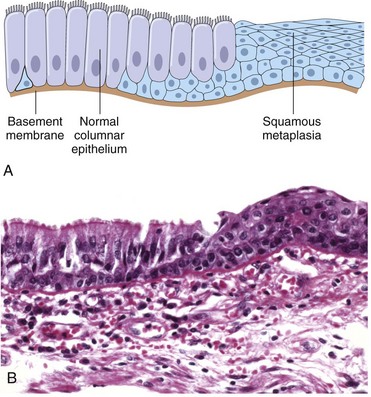
Fig. 1-39 Metaplasia to squamous epithelium.
A, Schematic diagram of columnar to squamous metaplasia. B, Metaplasia of columnar epithelium (left) to squamous epithelium (right) in a bronchus. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Metaplasia is often but not always an adaptive change to withstand adverse environmental conditions and is reversible if the cause is removed. Epithelial metaplasia is commonly a change to squamous epithelium and is usually a result of chronic irritation, but it can have other causes (e.g., avitaminosis A). Metaplasia in mesenchymal tissue is less clearly adaptive and is usually a response to change of microenvironment of cells, such as oxygen tension. One type of mesenchymal tissue changes to another, for example, fibrous tissue changes to cartilage or bone.
The following are some examples and causes of metaplasia:
• Chronic irritation from particles and chemicals in the lungs of smokers may cause the normal cuboidal and columnar epithelium of airways to become stratified squamous.
• Vitamin A deficiency causes squamous metaplasia of the transitional epithelium of the urinary tract, cuboidal and columnar epithelial cells lining the ducts within the salivary glands, and the epithelium of the mucous glands of esophageal mucosa in birds (Fig. 1-40).
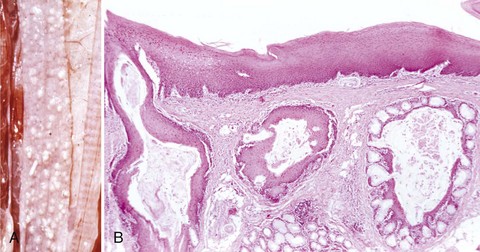
Fig. 1-40 Squamous metaplasia, esophagus, parrot.
A, The esophageal mucosa has multiple white raised nodules from squamous metaplasia of mucosal glands. Metaplasia arose from the lack of dietary vitamin A (avitaminosis A). B, Note the squamous metaplasia of the esophageal glands. Vitamin A is necessary for maintenance of the normal epithelium. Avitaminosis A results in the replacement of normal mucosal epithelium and goblet cells in the glands by keratinized stratified squamous epithelium. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
• Estrogen toxicity, among other things, causes squamous metaplasia of the urinary tract and prostate.
• Healing of glandular epithelium after mastitis may at first be squamous.
• Squamous metaplasia of salivary, biliary, and pancreatic ducts can occur if they are blocked by stones in the lumen.
• Osseous metaplasia (metaplastic bone) occasionally occurs in injured soft tissue.
• Myeloid metaplasia (extramedullary hematopoiesis) in adult spleens and livers occurs commonly after bone marrow injury or insufficiency.
• Metaplasia occurs in some tumors such as mixed mammary gland tumors of dogs.
Metaplasia is reversible (usually) if the cause is withdrawn. It may, however, be preneoplastic—for example, in the lungs of smokers where it appears before transformation to squamous cell carcinoma.
Cellular mechanisms leading to metaplasia vary. Vitamin A is important in normal differentiation of mucus-secreting epithelium by yet unspecified mechanisms. When vitamin A is deficient, these cells differentiate along squamous lines. Estrogen causes differentiation along squamous lines in specific sex-hormone responsive epithelia. Growth factors and other trophic substances presumably can influence differentiation along certain pathways from stem cells, and ECM can play an important role. How these metaplastic changes take place in response to injury is less clear.
Atrophy
Atrophy is the decrease in size or amount of a cell, tissue, or organ after normal growth has been reached (Fig. 1-41). It is caused by the decreased number and/or size of cells. It may affect virtually any organ or part of an organ. It is a regressive change usually caused by gradual and continuous injury. Some causes and examples of atrophy are as follows:
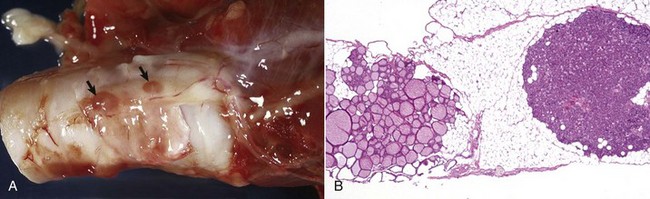
Fig. 1-41 Atrophy, right thyroid gland on trachea, dog.
A, The right thyroid gland is extremely small and difficult to discern. Only small pieces of thyroid tissue remain (arrows). B, The thyroid gland is extremely small, follicles are atrophic and of varied sizes, and colloid has a low concentration of thyroglobulin protein (pale pink color). Note that supporting stroma has been replaced by fat cells. The parathyroid gland (right) is of normal size. H&E stain. (A courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy College of Veterinary Medicine, University of Illinois.)
• Deficient nutritive supply. Starvation and especially a decreased blood supply. For example, liver atrophy results from decreased blood flow through the portal vein (Fig. 1-42).
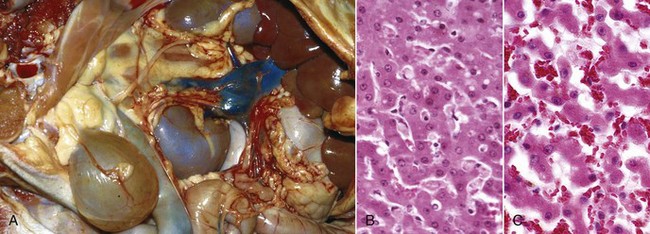
Fig. 1-42 Atrophy, liver, dog.
A, Note the small size (up under the rib cage) but normal color of the liver in this dog and the anomalous size of the caudal vena cava in the mesentery (i.e., shunt between the portal vein and the systemic circulation). This shunt caused bypassing of blood from the liver. The reduction in blood flow to the liver causes decreased nutrients (hepatocyte trophic factors) to the hepatocytes and therefore decreased size of hepatocytes. B, Normal liver. H&E stain. C, Liver, atrophy. Hepatocytes are smaller and narrower than those in the normal liver (B). As a consequence, the sinusoids are correspondingly wider. H&E stain. (A courtesy Dr. J. Sagartz, College of Veterinary Medicine, The Ohio State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. C courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
• Decreased workload. For example, muscle fibers atrophy in sedentary people.
• Disuse. Muscles in a limb in a cast atrophy.
• Denervation. Muscle fibers decrease in size rapidly if their motor neuron is severed.
• Pressure. Atrophy, degeneration, and necrosis occur adjacent to tumors because of pressure and compromised blood supply.
• Loss of endocrine stimulation. Atrophy of the zona fasciculata of the adrenal follows prolonged steroid therapy.
Involution is the decrease in size of a tissue caused by reduction in the number of cells (usually by apoptosis) and is usually used to refer to physiologic processes. For example, the thymus involutes with age, and many tissues become smaller because of senile involution. The uterus involutes after parturition, and its smooth muscle cells decrease notably in size and number.
The pathogenesis of atrophy implies an adverse environment. Cells regress to a smaller cell size and survive, but with decreased function. The general cause is inadequate cellular nutrition for any reason. Synthesis of proteins is exceeded by degradation or loss.
Autophagocytosis, lysosomes, and the ubiquitin proteasome pathway (see the section on Sublethal Injury and Subcellular Changes) may all play a role in decreasing the organelle and protein content of a cell. The actual triggers and cellular mechanisms are unclear for many circumstances. Atrophy may resolve if the cause is removed. It may persist as is, with or without harm to the organism, or it may progress.
Atrophied organs grossly have a decreased weight and volume, may have a loose covering membrane (e.g., wrinkled skin), have tortuous blood vessels too large for the volume of tissue, and often are firmer because of fibrosis or condensation of remaining collagen. Microscopically, cells are smaller and/or reduced in number. Ultrastructurally, there are fewer mitochondria, ER, and myofilaments (muscle) and often an increase in autophagic vacuoles and maybe lipofuscin.
Serous atrophy of fat is a very important necropsy finding because it may indicate starvation. Grossly, fat deposits are completely or partially depleted, and a clear or yellowish gelatinous material remains. Histologically, adipocytes are smaller, and interstitial hyaluronic acid mucopolysaccharides are increased. It is most evident in the epicardial and perirenal fat but may affect any fat depot, including bone marrow. The cause of starvation may be virtually anything: malnutrition, malabsorption, chronic infection, parasitism, neoplasia, and so forth. It is common in neonates, often the result of mismothering.
Intracellular Accumulations*
One of the manifestations of metabolic derangements in cells is the intracellular accumulation of abnormal amounts of various substances (Fig. 1-43). The stockpiled substances fall into three categories: (1) a normal cellular constituent accumulated in excess, such as water, lipids, proteins, and carbohydrates; (2) an abnormal substance, either exogenous, such as a mineral or products of infectious agents, or endogenous, such as a product of abnormal synthesis or metabolism; or (3) a pigment. These substances may accumulate either transiently or permanently, and they may be harmless to the cells, but on occasion they are severely toxic. The substance may be located in either the cytoplasm (frequently within phagolysosomes) or the nucleus. In some instances, the cell may be producing the abnormal substance, whereas other cells may be merely storing products of pathologic processes occurring elsewhere in the body.
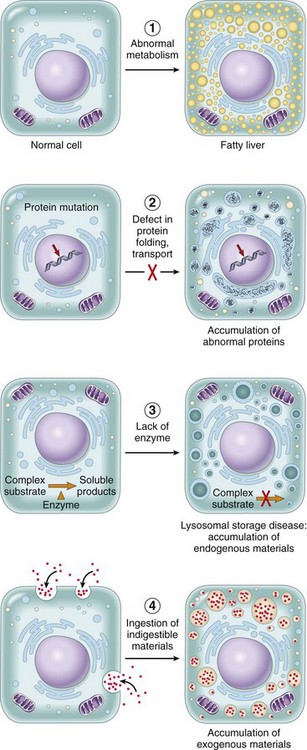
Fig. 1-43 Mechanisms of intracellular accumulations.
A, Abnormal metabolism, as in fatty change in the liver. B, Mutations causing alterations in protein folding and transport, as in α1-antitrypsin deficiency. C, Deficiency of critical enzymes that prevent breakdown of substrates that accumulate in lysosomes, as in lysosomal storage diseases;. D, Inability to degrade phagocytosed particles, as in hemosiderosis and carbon pigment accumulation. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Many processes result in abnormal intracellular accumulations, but most accumulations are attributable to the following three types of abnormalities.
1. A normal endogenous substance is produced at a normal or increased rate, but the rate of metabolism is inadequate to remove it. An example of this type of process is fatty change in the liver because of intracellular accumulation of triglycerides (described in a later section). Another example is the appearance of reabsorption protein droplets in the epithelial cells of renal proximal tubules because of increased leakage of protein from the glomerulus.
2. A normal or abnormal endogenous substance accumulates because of genetic or acquired defects in the metabolism, packaging, transport, or secretion of these substances. One example is the group of conditions caused by genetic defects of specific enzymes involved in the metabolism of lipid and carbohydrates resulting in intracellular deposition of these substances, largely in lysosomes in so-called storage diseases. Another is α1-antitrypsin deficiency, in which a single amino acid substitution in the enzyme results in defects in protein folding and accumulation of the enzyme in the ER of the liver in the form of globular eosinophilic inclusions.
3. An abnormal exogenous substance is deposited and accumulates because the cell has neither the enzymatic machinery to degrade the substance nor the ability to transport it to other sites. Accumulations of carbon particles and nonmetabolizable chemicals, such as silica particles, are examples of this type of alteration.
Whatever the nature and origin of the intracellular accumulation, it implies the storage of some product by individual cells. If the overload is due to a systemic derangement and can be brought under control, the accumulation is reversible. In genetic storage diseases, accumulation is progressive, and the cells may become so overloaded as to cause secondary injury, leading in some instances to death of the tissue and the patient.
Lipids
Hepatic Lipidosis (Fatty Liver, Fatty Change, Hepatic Steatosis): All major classes of lipids can accumulate in cells: triglycerides, cholesterol/cholesterol esters, and phospholipids. Phospholipids are components of the myelin figures found in necrotic cells. In addition, abnormal complexes of lipids and carbohydrates accumulate in the lysosomal storage diseases.
Lipidosis is the accumulation of triglycerides and other lipid metabolites (neutral fats and cholesterol) within parenchymal cells. Although it occurs in heart muscle, skeletal muscle, and the kidney, clinical manifestations are most commonly detected as alterations in liver function (elevated liver enzymes, icterus) because the liver is the organ most central to lipid metabolism.
Hepatic lipidosis, the prototype example of this type of cellular degeneration, can occur as the result of one of the following five mechanisms:
1. Excessive delivery of free fatty acids either from the gut or from adipose tissue
2. Decreased β-oxidation of fatty acids to ketones and other substances because of mitochondrial injury (toxins, hypoxia)
3. Impaired synthesis of apoprotein (CCl4 toxicity, aflatoxicosis)
4. Impaired combination of triglycerides and protein to form lipoprotein (uncommon)
5. Impaired release (secretion) of lipoproteins from the hepatocyte (uncommon)
The underlying pathogenesis of hepatic lipidosis centers on the biochemical pathways of free fatty acid formation and metabolism. Free fatty acids, derived from triglycerides, provide a large component of the basal energy needs for parenchymal cells. They are obtained directly from the diet through digestive processes, from chylomicrons in the blood, or from adipose cells in body fat stores (adipose tissue). Chylomicrons transport dietary lipids consisting predominately of triglycerides from the alimentary system to the liver, muscle, and adipose tissue. Lipoprotein lipase and other proteins act synergistically within the chylomicron to free fatty acids from triglycerides for their use as an energy source. In the liver, free fatty acids are esterified to triglycerides, converted into cholesterol or phospholipids, or oxidized to ketones. Triglycerides can only be transported out of hepatocytes if apolipoprotein converts them to lipoproteins (Fig. 1-44). Alterations in one or more of these biochemical processes can result in the accumulation of triglycerides and other lipid metabolites, resulting in hepatic lipidosis.
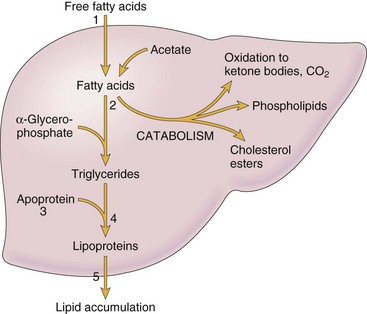
Fig. 1-44 Fatty liver.
Schematic of typical hepatic lipid metabolism (uptake, catabolism, and secretion) and possible mechanisms resulting in lipid accumulation. 1, Excessive delivery of free fatty acids (FFA) from fat stores or diet. 2, Decreased oxidation or use of FFAs. 3, Impaired synthesis of apoprotein. 4, Impaired combination of protein and triglycerides to form lipoproteins. 5, Impaired release of lipoproteins from hepatocytes. (Modified from Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
In domestic animals, hepatic lipidosis most commonly arises from conditions that cause increased mobilization of body fat stores. Such conditions usually occur when there is increased demand for energy over a short duration, as in late pregnancy and early lactation in dairy cows (pregnancy toxemia and ketosis, respectively). Hepatic lipidosis is also observed with nutritional disorders, including obesity (increased transport of dietary lipids or mobilization from adipose tissue), protein-calorie malnutrition (impaired apolipoprotein synthesis), and starvation (increased mobilization of triglycerides), but it also can occur secondarily in inherited storage diseases, such as Wilson’s disease, and in endocrine disease, such as diabetes mellitus (increased mobilization of triglycerides). Poisoning by chemicals, such as CCl4 (used in industrial applications) and yellow phosphorus (used in manufacturing of incendiary munitions and once in the manufacture of matches), can also induce hepatic lipidosis via decreased oxidation of free fatty acids. These chemicals are strictly regulated and diseases caused by them are rarely seen in clinical medical practice today. In some disorders, such as feline hepatic lipidosis (feline fatty liver syndrome) and fat cow syndrome, the cause of hepatic lipidosis is unclear.
Grossly, mild fatty change may not be detectable, but livers with notable lipidosis are enlarged, yellow, soft and friable, and the edges of the lobes are rounded and broad instead of sharp and flat (Fig. 1-45, A). When incised, the cut surface of severely affected livers can bulge and the hepatic parenchyma is soft and friable and has a greasy texture attributable to lipid within hepatocytes. In addition, a 1-cm-thick transverse section from a liver lobe may float in formalin, again indicative of lipid within hepatocytes.
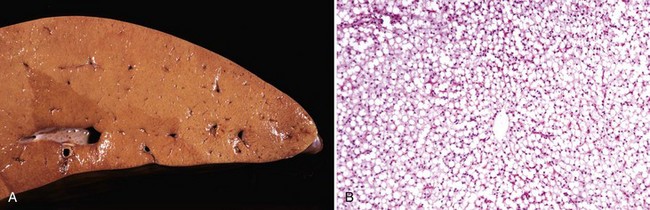
Fig. 1-45 Steatosis (fatty liver, fatty change, hepatic lipidosis), liver, ox.
A, Note the uniformly pale yellow surface. The liver is usually enlarged and the edges rounded. The cut surface bulges on incision and may feel greasy. B, In this severely affected liver, all hepatocytes are vacuolated and their nuclei have been displaced to the side. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
It is important to distinguish these gross lesions from the lesions present in glucocorticoid (steroid) hepatopathy in dogs. The liver in glucocorticoid hepatopathy is also enlarged and has rounded edges, but it is pale beige to tan-white, firm, and nongreasy (Fig. 1-46, A). Cut sections do not float in formalin. These gross lesions are attributable to the accumulation of glycogen and water in the cytoplasm of hepatocytes (see Chapter 8).
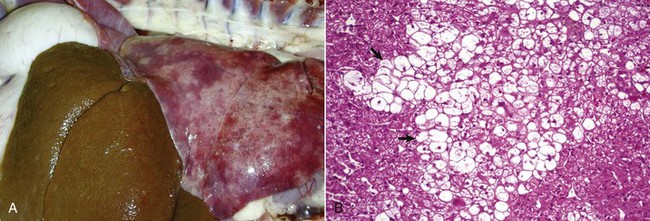
Fig. 1-46 Glucocorticoid hepatopathy, liver, dog.
A, Extensive accumulation of glycogen in hepatocytes leads to an enlarged and pale brown to beige liver in dogs with glucocorticoid excess from endogenous (Cushing’s disease) or exogenous sources. The liver is usually enlarged and the edges rounded. This cut surface would bulge on incision and not be greasy. B, Note the swollen hepatocytes (arrows) with extensive cytoplasmic vacuolation. H&E stain. (A courtesy Dr. K. Bailey, College of Veterinary Medicine, University of Illinois. B courtesy Dr. J. M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Microscopically, hepatocytes with lipidosis are vacuolated, with the extent of the vacuolation depending on the severity of the lipidosis. Initially, there are a few small clear vacuoles that increase in size and number and eventually coalesce into larger vacuoles. These vacuoles have sharply delineated borders (Fig. 1-45, B), which are attributed to the hydrophobic interface between water and lipid in the cell’s cytoplasm and should be compared with the vacuoles that result from glycogen accumulation (Fig. 1-46, B). In hepatocytes with large amounts of fat, the nucleus can be displaced to the periphery, and the cell can resemble an adipocyte. In extremely affected livers in which all of the hepatocytes are filled with lipid, the liver can resemble fat and can be identifiable only by the presence of portal areas.
Vacuoles in hepatocytes may be due to fat accumulation but can also occur as the result of intracellular accumulation of glycogen or water. Fat is confirmed by special stains, but as alcohol and clearing agents used during the processing of paraffin-embedded sections dissolve fat, formalin-fixed frozen sections—properly stained for fat—must be used to confirm its presence in hepatocytes. Fat stains, which are alcoholic solutions of fat-soluble dyes, include Sudan III, Scharlach R, and Oil-Red-O. Glycogen is confirmed by the PAS and PAS-diastase reactions described later (see section on Glycogen). Vacuoles that do not stain with either fat or PAS are presumed to be a result of the accumulation of water (hydropic degeneration).
Fatty Infiltration: Fatty infiltration should not be confused with fatty change or steatosis, in which the lipid is intracellular (see previous discussion). Adipocytes are normally present in connective tissue and depending on the nutritional state of the animal in limited numbers between fasciculi of skeletal muscle and subepicardially between cardiac myocytes. When increased lipid is to be stored, adipocytes increase in number, and the process is called fatty infiltration. It occurs in old age and in obesity in which there is hyperplasia of adipocytes by means of proliferation of preadipocytes. When myocytes of skeletal muscle atrophy and disappear, the lost myocytes may be replaced by adipocytes (see Chapter 15 and Fig. 15-9).
Glycogen
Variable amounts of glycogen are normally stored in hepatocytes and myocytes (the amount in the liver depends on the interval between sampling and the last meal). Hepatocytes of starved animals are usually devoid of glycogen. Excessive amounts of glycogen are present in animals in which glucose or glycogen metabolism is abnormal, such as in diabetes mellitus, in genetic disorders such as glycogen storage diseases (type Ia and type III), and in animals that have received excess amounts of corticosteroids. Large amounts of glycogen can be found in the livers of young growing animals and in animals that are well nourished and on diets of commercially produced feeds.
In diabetes, glycogen is found not only in hepatocytes but also in the epithelial cells of renal proximal tubules and in B cells of the islets of Langerhans. Hepatocytes are highly permeable to glucose, and hyperglycemia leads to increased glycogen concentration in these cells. Also in diabetes, large amounts of glucose are passed out in the glomerular filtrate and exceed the resorptive capacity of the renal tubule epithelial cells. These cells, when overloaded with glucose, convert it into glycogen, which accumulates intracellularly.
Grossly, physiologic deposits of glycogen cannot be detected, but in steroid-induced hepatopathy, in which massive amounts of glycogen are stored, the liver may be enlarged and pale (see Fig. 1-46).
Microscopically, the amount of glycogen demonstrated in hepatocytes is chiefly a function of the original concentration in the cell, the delay between death and fixation during which time the glycogen is metabolized, and the type of fixation. Despite frequent statements that glycogen is best preserved by fixing tissue in an alcoholic fixative (e.g., absolute alcohol or 10% formalin in absolute alcohol), glycogen can be well preserved by fixation in an ordinary 10% buffered neutral formalin solution at 4° C in a refrigerator during the period of fixation (Fig. 1-47, A). This procedure retains most of the glycogen but avoids the excessive shrinkage and distortion of tissues fixed in alcoholic fixatives and also avoids “polarization,” the phenomenon whereby the glycogen is displaced to the side of the cell away from the surface. Polarization is seen in fixation at room temperature but is worst with alcoholic fixatives (Fig. 1-47, B).
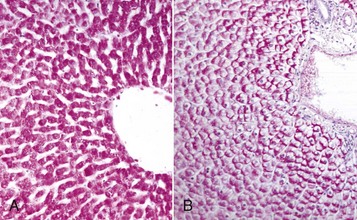
Fig. 1-47 Glycogen, liver, dog.
A, Ten-percent buffered neutral formalin fixation at 4° C. Glycogen (purplish-red) is uniformly dispersed throughout the cytoplasm of all hepatocytes. Periodic acid-Schiff technique. B, Absolute alcohol (ethanol) fixation at room temperature. The glycogen in each hepatocyte has been pushed to the side of the cell, so-called polarization of glycogen. Periodic acid-Schiff technique. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Histologically, glycogen is demonstrated specifically by the PAS reaction using two serial tissue sections mounted on glass slides. The tissue section on the first slide is pretreated with diastase, which digests the glycogen in the tissue, and the tissue section on the second glass slide is untreated. Thus the deposits digested by diastase are glycogen. The PAS reaction breaks 1,2-glycol linkages to form aldehydes, which are revealed by Schiff’s reagent. These linkages occur in substances other than glycogen, hence the use of two slides, including one pretreated with diastase, to specifically identify glycogen. Microscopically, glycogen appears as clear vacuoles in the cytoplasm of the cell. In contrast to intracellular fat whose vacuoles are rounded and sharply delineated, glycogen forms irregular clear spaces with indistinct outlines. Usually the nucleus remains centrally located in the hepatocyte. However, if very large amounts of glycogen are stored in hepatocytes, as in steroid-induced hepatopathy, hepatocyte nuclei may be displaced peripherally.
In glycogen storage diseases (glycogenoses), glycogen accumulates, sometimes in massive amounts in cells as a result of a defective enzyme. Exactly which cells store glycogen depends on the defective enzyme, but skeletal muscle is frequently involved (see Chapters 14 and 15 for more detail).
Proteins
In histologic sections, intracellular protein accumulations are of several types and include rounded eosinophilic droplets, vacuoles, and aggregates in cells. The causes of these accumulations vary widely.
Hyaline Change: The adjective “hyaline” is defined by Dorland’s Medical Dictionary as “glassy and transparent or nearly so,” and the noun “hyalin” as a “translucent albuminoid substance.” However, histologically the term has come to mean having a homogeneous, eosinophilic, and glassy (translucent) appearance. Some pathologists also add “amorphous,” and the lesion has been termed both a change and a degeneration, but the term hyaline is purely descriptive and rather loosely applied to a variety of changes, none of which is a true cellular degeneration. Hyaline substances may be intracellular or extracellular.
Intracellular Hyaline Proteins: Intracellular hyaline proteins include resorption droplets in the proximal tubule epithelial cells, Russell bodies in plasma cells, and those aggregates caused by defects in protein folding.
Resorption droplets in the epithelial cells of renal proximal tubules: There is normally very little protein in the filtrate from the glomerulus, and the proximal tubule epithelial cells resorb what is present. When the protein concentration of the filtrate is high, as for example in a proteinuria from glomerular damage, this protein is taken up by the proximal tubule epithelial cells into vesicles where, in H&E stained sections, they appear as hyaline droplets in the cytoplasm (Fig. 1-48, A). The vesicles fuse with the lysosomes to form phagolysosomes, where the protein is metabolized. If the proteinuria ceases, the formation of hyaline droplets also ceases. This condition was once called hyaline droplet degeneration. It is not a degeneration but an exaggeration of a normal process. Also, similar droplets are seen in the intestinal epithelium of neonatal pigs and calves that have recently ingested colostrum.
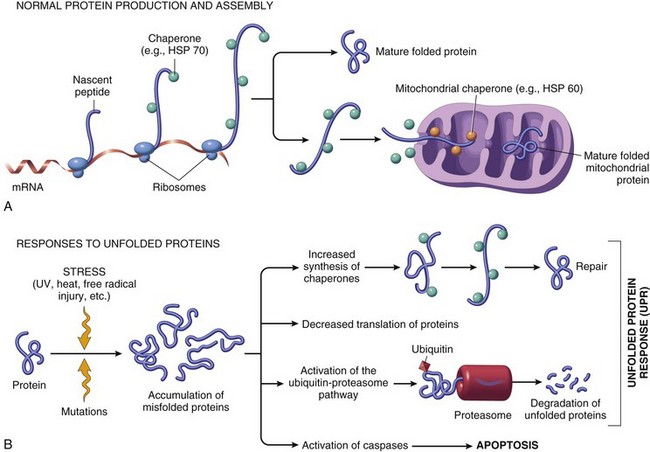
Fig. 1-49 Mechanisms of protein folding and the unfolded protein response.
A, Chaperones, such as heat shock proteins (HSP), protect unfolded proteins from degradation and guide proteins into organelles. B, Misfolded proteins trigger a protective unfolded protein response (UPR). If this response is inadequate to cope with the level of misfolded proteins, it induces apoptosis. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Excessive production of normal protein: Hyaline bodies called Russell bodies are seen in the cytoplasm of some plasma cells (Mott cells). These bodies are large, eosinophilic, homogeneous, and amorphous and consist of immunoglobulin (γ-globulin). Russell bodies have been described as “manifestations of cellular indigestion” in the ER.
Defects in protein folding: During protein synthesis on ribosomes, proper folding of the protein is essential for its transport in the cell’s organelles. Normally, if there is a defect in folding, the protein is eliminated by the proteasome complex (Fig. 1-49). On occasion, these folded proteins accumulate in cells as is seen in some of the human neurodegenerative diseases, including Alzheimer’s disease. Sometimes folded proteins may accumulate in tissue, and some types of amyloidosis are examples of this process.
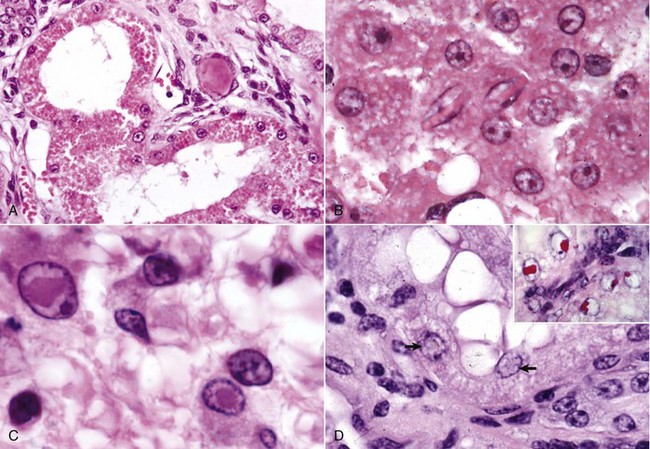
Fig. 1-48 Cell droplets and inclusion bodies.
A, Resorption droplets, proteinuria, kidney, dog. The cytoplasm of the proximal tubule epithelial cells is filled with eosinophilic homogeneous droplets—protein that has been resorbed by the cells from the glomerular filtrate. H&E stain. B, Crystalloids, hepatocytes, dog. Note the elongated crystals in the nuclei of the hepatocytes. C, Viral inclusion bodies, canine distemper, brain, dog. Note the intranuclear eosinophilic inclusion bodies in glial cells. H&E stain. D, Lead inclusion bodies, kidney, dog. The inclusions in the nuclei of these renal tubular epithelial cells are difficult to see with an H&E stain (arrows). Inset, An acid-fast stain is useful in identifying lead inclusions, which stain red. Ziehl-Neelsen stain. (A and C courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. D.D. Harrington, College of Veterinary Medicine, Purdue University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. D courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. Inset courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Other Intracellular Inclusions
Autophagic Vacuoles: Autophagic vacuoles are large eosinophilic intracytoplasmic inclusions, which ultrastructurally are autophagosomes (see Fig. 1-35). They are a common response to injury in cells with sublethal damage, particularly hepatocytes, and are a mechanism by which the cell rids itself of damaged or senescent organelles. A portion of the cell membrane invaginates and envelops the affected organelles to form an autophagosome, which then fuses with a lysosome to cause degradation of the contents. Digestion of the material in autophagic vacuoles may leave some lamellar debris, and this debris may either be exocytosed from the cell or remain intracellularly to form lipofuscin (see later discussion of pigments).
Crystalline Protein Inclusion Bodies: Crystalline protein inclusion bodies, sometimes known as crystalloids, occur in normal hepatocytes and renal tubular epithelial cells, particularly in old dogs. They are large, eosinophilic, and rhomboidal and may be so large as to distort the nucleus or the cell (Fig. 1-48, B). Except for being age related, their significance is unknown. In fact, an increased incidence of these inclusions is the most consistent age-related change in canine hepatocytes.
Viral Inclusion Bodies: Infection of host cells by some types of viruses results in the formation of characteristic inclusion bodies, which may be intranuclear, intracytoplasmic, or both. They contain viral proteins usually mixed with other proteins and are useful diagnostically to confirm a specific viral disease.
DNA viruses, such as herpesviruses, adenoviruses, and parvoviruses, tend to produce only intranuclear inclusions. These inclusions are round to oval and can be eosinophilic (herpesviruses), basophilic, or amphophilic (adenoviruses). Poxviruses are also DNA viruses, but they produce large distinct eosinophilic intracytoplasmic inclusion bodies in infected cells.
A few RNA viruses produce intracytoplasmic inclusions. Examples are the distinctive cytoplasmic neuronal inclusions of rabies (Negri bodies) and the epithelial inclusions of canine distemper. Distemper causes both intranuclear and intracytoplasmic inclusions in nervous tissue (Fig. 1-48, C). Viral inclusions are usually surrounded by a clear halo, particularly in the nucleus. Cells with inclusion bodies and adjacent cells usually have signs of degeneration or cell death. Many of these viral inclusion bodies are discussed in the systemic pathology chapters of this book.
Lead Inclusion Bodies: In lead poisoning, irregularly shaped intranuclear inclusion bodies that are acid-fast may be present in renal tubular epithelial cells (Fig. 1-48, D). They contain both lead and protein. When they are present, they are helpful in the diagnosis of lead poisoning, but are not present in all cases. In dogs, they must be distinguished from the protein crystalline protein inclusions described previously.
Extracellular Accumulations
Extracellular hyaline substances include the following:
1. Hyaline casts in the lumens of renal tubules in a proteinuria.
2. Serum or plasma in blood vessels.
3. Plasma proteins in vessel walls (e.g., in porcine edema disease). These substances are subendothelial hyaline deposits, primarily seen in arterioles of the brainstem in pigs with porcine edema disease (see Fig. 1-52).
4. Old scars. With age, the number of nuclei in collagen deposits decreases as the result of cell senescence, and the collagen fibers condense and become hyalinized.
5. Thickened basement membranes (e.g., in glomerulonephritis and in the capillaries of the choroid plexus with old age).
6. Hyaline membranes of the alveolar walls (see Chapter 9).
7. Hyaline microthrombi (e.g., platelet microthrombi) in disseminated intravascular coagulation (DIC); often visible in glomerular capillaries and pulmonary alveolar capillaries.
Amyloid: The name amyloid is given to a chemically diverse group of chiefly extracellular proteinaceous substances that appear histologically and ultrastructurally similar. The name means “starchlike” and was applied to these proteins because when the surface of an affected organ was treated with an iodine solution and then with dilute sulfuric acid, it turned blue, a positive test for starch (Fig. 1-50).
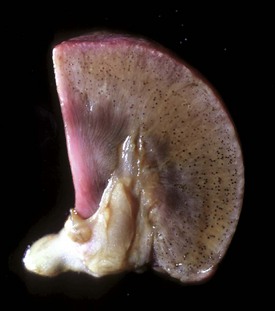
Fig. 1-50 Amyloidosis, kidney, cross section, dog.
Note the blue-black foci, which are glomeruli-containing amyloid stained by Lugol’s iodine. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Histologically, amyloid is an eosinophilic amorphous hyaline substance (Fig. 1-51, A) and needs to be differentiated from other hyaline substances on histologic examination. It is extracellular and compresses adjacent parenchymal cells, causing atrophy or death from compression and/or ischemia. This outcome is most evident in hepatic amyloidosis, in which the protein is deposited in the space of Disse. Here it compresses the adjacent hepatocytes and interferes with the hepatocytes’ access to blood and nutrients in the sinusoids.
Fig. 1-51 Amyloidosis, kidney, dog.
A, The renal glomerulus contains large amounts of pale homogeneous eosinophilic material, which is amyloid. H&E stain. B, The amyloid in the glomeruli stains orange. Congo red stain. (A and B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
The most frequently used special stain for amyloid is Congo red. It stains amyloid orange to orange red (Fig. 1-51, B) and under polarized light imparts a light green, the so-called apple green fluorescence (see Chapter 11). Congo red staining is not absolutely specific, and immunohistochemistry or transmission electron microscopy to identify 7.5- to 10-nm filaments may be necessary.
Chemically, amyloid is not one substance. It is a diverse group of glycoproteins whose protein component is configured in a β-pleated sheet pattern, which is responsible for the characteristic staining with Congo red. In humans, there are three major and several minor forms. In animals, there are two major and two minor forms, which are chemically distinct but appear similar histologically.
AL amyloid consists of immunoglobulin light chains, is monoclonal, and is secreted by plasma cells in immunocyte dyscrasias (B lymphocyte proliferative disorders). AA amyloid is not an immunoglobulin but is synthesized from a precursor protein, serum amyloid A (SAA), secreted by the liver. SAA concentration is increased in inflammatory states in response to IL-1 and IL-6, but this increase does not necessarily lead to amyloid deposition in all cases. A minor form of amyloidosis is hereditary amyloidosis found in Shar-Pei dogs and in Abyssinian cats.
β-Amyloid found in Alzheimer’s disease in humans has been detected in the brains of aged dogs.
Classification of Amyloidosis: Amyloidosis is discussed in detail in Chapter 5.
Amyloidosis has been classified in several ways (i.e., primary versus secondary, systemic [generalized] versus localized, and a combination of these categories). Systemic amyloidosis (generalized) is also divided into primary amyloidosis (immunocyte dyscrasia) and secondary amyloidosis (reactive systemic amyloidosis).
• Immunocyte dyscrasia is the most common form of amyloidosis in humans but not in animals. The amyloid consists of amyloid light chains and is indicative of a plasma cell dyscrasia. These cells also secrete large amounts of γ- and κ-light chains into blood and urine (Bence Jones proteins), which are diagnostically important.
• Reactive systemic amyloidosis was initially called secondary amyloidosis because it was secondary to chronic inflammatory conditions, particularly those causing a chronic antigenic stimulation with protracted breakdown of cells. It is the most common form of amyloidosis in animals, and the amyloid is deposited in kidney, liver, spleen, and lymph nodes. Functionally, and most often in old dogs, amyloid deposits in the kidney are the most important because they are located in the mesangium and basement membranes of renal glomeruli and cause a proteinuria. The spleen is the most frequent site in reactive systemic amyloidosis, and amyloid is deposited in the periarteriolar lymphoid sheaths and red pulp. The space of Disse of the liver is the usual site for amyloidosis in birds.
• Localized amyloidosis involves a single organ or tissue. Such localized lesions are in the nasal vestibule or rostral portion of the nasal septum and turbinates in horses and in the pancreatic islets in cats.
• β-Amyloidosis. Extracellular accumulation of amyloid-β (Aβ) protein is characteristic of Alzheimer’s disease in humans. This type of amyloid has also been identified in the brains of aged dogs, the highest concentration being in the frontal cortex. Dogs older than 13 years had Aβ plaques.
Location of Amyloid Deposits in Animals: The kidney (glomeruli in most animals and medullae in cats), liver (space of Disse in cattle, horses, dogs, and cats), and spleen (germinal centers) are common sites. Other organs affected include the stomach, intestine (lamina propria), thyroid (C-cell tumor), skin (dermis and subcutis of horses), lymph node (germinal centers), adrenal cortex, pancreas (islets of Langerhans in cats), nasal septum and turbinates (walls of submucosal vessels and basement membranes of mucosal glands of horses), and meningeal and cerebral vessels of older dogs. See the appropriate organ chapters for more detail.