Neoplasia and Tumor Biology*
Despite the relatively short lifespan of most animals, neoplasia is an important concern for veterinary practitioners, diagnosticians, and researchers. Tumor diagnosis and treatment for individual animals is becoming an increasingly prominent part of small animal practice. In food animals, infectious and environmental causes of cancer can have a major impact on herd or flock health. Furthermore, animal models provide important insights into the cause and treatment of human cancer.
Definitions
A neoplasm is a “new growth” composed of cells, originally derived from normal tissues, that have undergone heritable genetic changes allowing them to become relatively unresponsive to normal growth controls and to expand beyond their normal anatomic boundaries. Other common terms for neoplasms describe their clinical appearance or behavior: tumor (“swelling”) and cancer (“crab”). Although the terms neoplasm and tumor may refer to benign or malignant growths, the term cancer always denotes a malignant growth. Oncology is the study of neoplasia; the term is derived from the Greek word oncos (“tumor”).
Benign tumors do not invade surrounding tissue or spread to new anatomic locations within the body; thus these tumors are usually curable and rarely responsible for the death of the host. Malignant tumors, if left untreated, invade locally, may spread by metastasis (“change of place”), and ultimately kill the host. Interestingly, tumors of the nervous system very rarely metastasize; however, many of these tumors are notably invasive and kill their hosts, thus the tumors are malignant.
With the recognition that tumor development is a stepwise process, potentially preneoplastic changes have assumed new diagnostic and clinical significance. These changes include hyperplasia (increased cell number in a tissue), metaplasia (transformation of one differentiated cell type into another), and dysplasia (abnormal pattern of tissue growth) (Fig. 6-1). Hyperplasia, which is an increase in the number of cells in a tissue, should be distinguished from hypertrophy, which is an increase in individual cell size rather than number. Metaplasia is seen most commonly in epithelial tissue. In several species of animals, vitamin A deficiency is characterized by squamous metaplasia of respiratory and digestive epithelium. Dysplasia usually refers to disorderly arrangement of cells within epithelium. In general, preneoplastic changes are reversible. They arise in response to physiologic demands, injury, or irritation and resolve with the removal of the inciting factor. For example, epidermal hyperplasia is a normal part of wound repair, and skeletal muscle hypertrophy is an adaptive response to increased workload. Preneoplastic changes often indicate an increased risk for neoplasia in the affected tissue, and preneoplastic lesions may progress to neoplasia. The terms dysplasia and metaplasia can be applied to tumors to describe changes that persist during the transition from preneoplasia to neoplasia; however, the terms hyperplasia and hypertrophy are not appropriate in descriptions of true neoplasms.
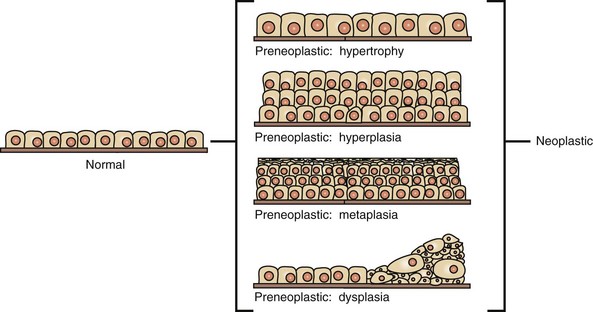
Fig. 6-1 Preneoplastic changes that may precede tumor emergence.
Preneoplastic changes in tissues include alterations in cell number, size, and organization. In this example, preneoplastic changes are illustrated in simple cuboidal epithelium, although such changes may also occur in other epithelial and mesenchymal cell types. (Redrawn with permission from Dr. D.F. Kusewitt, College of Veterinary Medicine, The Ohio State University.)
Nomenclature
Most tumors appear to consist of a single cell type, and the name of the neoplasm reflects the cell type (mesenchymal or epithelial) from which the tumor is presumed to arise.
Mesenchymal Tumors
Mesenchymal tumors arise in cells of embryonic mesodermal origin. Benign tumors originating from mesenchymal cells are usually named by adding the suffix -oma to the name of the cell of origin. Thus a lipoma is a benign tumor derived from a lipocyte (“fat cell”) (Fig. 6-2, A), and a fibroma is a benign tumor of fibroblast origin. A malignant tumor of mesenchymal origin is a sarcoma (“fleshy growth”). A prefix or modifier indicates the tissue of origin. For example, a liposarcoma is a malignant tumor of lipocyte origin (Fig. 6-2, B), and a fibrosarcoma is a tumor composed of malignant fibroblasts. The cells of the hematopoietic system are mesenchymal. Tumors arising from circulating blood cells or their precursors are termed leukemias (“white blood”); neoplastic hematopoietic cells are usually found in large numbers in the bloodstream (Fig. 6-3), although they may also form solid tumor masses.
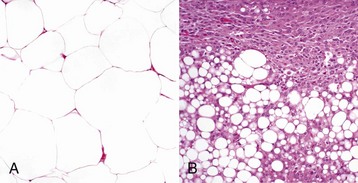
Fig. 6-2 Comparison of benign and malignant tumors of lipocyte origin, dog.
A, The benign lipoma is composed of mature fat cells indistinguishable from normal cells. H&E stain. B, The liposarcoma consists of poorly differentiated cells, many of which do not have the morphologic features characteristic of lipocytes. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
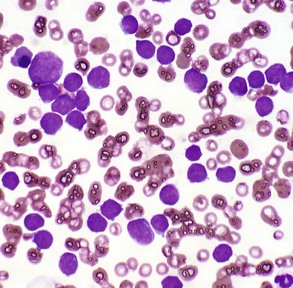
Fig. 6-3 Acute lymphoid leukemia, peripheral blood, dog.
The peripheral blood smear contains numerous neoplastic large lymphocytes. Flow cytometry identified these cells as B lymphocytes. The white blood cell count of this animal was 293,000 leukocytes/µL. Wright’s stain. (Courtesy Dr. M.L. Wellman, College of Veterinary Medicine, The Ohio State University.)
Epithelial Tumors
All of the three embryonic cell layers, endoderm, mesoderm, and ectoderm, can give rise to epithelial tissue and to tumors derived from this tissue. Terms for both benign and malignant epithelial tumors are frequently modified by prefixes or adjectives describing their appearance or the response they elicit in surrounding tissue. For instance, the adjective “squamous” is applied to an epithelial neoplasm that demonstrates squamous differentiation.
Benign tumors that arise from glandular epithelium are called adenomas, regardless of their microscopic appearance. However, the term is also applied to many tumors that are derived from nonglandular epithelial tissues but that have a tubular appearance such as renal adenomas. The term papilloma refers to a benign exophytic growth arising from an epithelial surface, whereas a polyp is a grossly visible, benign epithelial tumor projecting from a mucosal surface (Fig. 6-4).
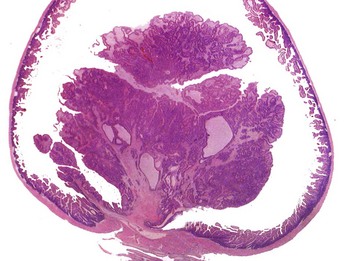
Fig. 6-4 Polyp, small intestine, mouse.
The neoplastic growth arises from the mucosa and extends into the lumen of the intestine. There is no invasion of the intestinal wall. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
All malignant tumors of epithelial origin are termed carcinomas (“cancers”). The general term carcinoma may be further modified to indicate the organ of origin, as in hepatocellular carcinoma. The prefix adeno- indicates a glandular pattern of tumor growth. Adenocarcinomas may be described as papillary, tubular, or cystic. Carcinomas and adenocarcinomas that stimulate the formation of abundant collagen in surrounding connective tissue (desmoplasia) may be termed scirrhous. The neoplastic epithelial cells of mucinous carcinomas and adenocarcinomas produce abundant mucin. Carcinoma in situ is a preinvasive form of carcinoma that remains within the epithelial structure from which it arises and does not penetrate the basement membrane to enter the stroma.
Undifferentiated Tumors
The appearance of some malignant tumors gives no clue to their cell of origin; thus they are termed undifferentiated neoplasms.
Mixed Tumors
Mixed tumors contain multiple cell types derived from a single or multiple germ layers. Mixed tumors are believed to arise from a single pluripotent or totipotent cell capable of differentiating into a variety of more mature cell types. Teratomas and teratocarcinomas arise from totipotential germ cells; thus they contain tissue derived from all embryonic cell layers and consist of a bizarre mixture of adult and embryonic tissue types. The mixed mammary gland tumor of dogs is generally considered a mixed tumor. A mixed mammary tumor is composed of a variable admixture of neoplastic epithelial elements (luminal epithelium and myoepithelium) and mesenchymal elements (fibrous connective tissue, fat, cartilage, and bone) (Fig. 6-5).
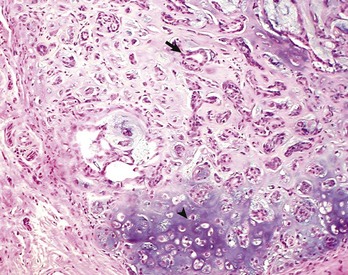
Tumorlike Lesions
Hamartomas are disorganized but mature mesenchymal or epithelial tissues found in their normal anatomic location. Many of the hamartomas identified in animals consist of abnormal proliferations of blood vessels. Hamartomas apparently represent the result of aberrant differentiation rather than true neoplasia, and their behavior is completely benign. Choristomas are composed of normal mature tissue located at an ectopic site. An example is the dermoid, a mass consisting of mature skin and its appendages, which may be found in a variety of unusual sites, including the cornea.
Veterinary Nomenclature
In Web Table 6-1, the names of common benign neoplasms in animals and their malignant counterparts are shown. The names given are those commonly employed in veterinary medicine. The terms used by veterinary pathologists to describe tumors in animals may differ from the terms used by medical pathologists to describe human tumors. This is partly because conventional usage plays an important role in tumor nomenclature; thus tumor nomenclature may be dictated by historic precedent rather than by logic. Moreover, attempts to standardize diagnostic terms for tumors in veterinary medicine have lagged far behind such efforts in the medical arena. A significant difference between veterinary and human nomenclature is that a benign tumor arising from melanocytes is termed a benign melanoma or melanocytoma by veterinary pathologists and a nevus by medical pathologists. Medical pathologists reserve the term melanoma for a malignant tumor of melanocyte origin, whereas veterinary pathologists term such tumors malignant melanomas.
Tumor Characteristics
Benign Versus Malignant Tumors
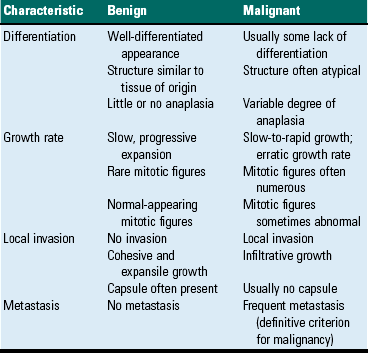
The most important distinction between benign and malignant tumors is that malignant tumors are able to invade locally and metastasize systemically, but benign tumors are not. The invasive capabilities of malignant tumors are associated with enhanced tumor cell motility, increased production of proteases, and altered tumor cell adhesion characteristics. Although benign tumors are ultimately distinguished from their malignant counterparts based on invasiveness, a variety of morphologic and behavioral features are generally considered to predict the potential for malignant behavior (Table 6-1). Although both benign and malignant tumors are composed of proliferating cells, malignant tumors have essentially unlimited replicative potential. The tumors are relatively independent of exogenous growth stimulatory molecules and are insensitive to growth inhibitory signals from their environment. Moreover, malignant cells are better able than benign cells to evade programmed cell death (apoptosis) and to escape the host’s cytotoxic immune response. Compared with benign tumors, malignant tumors stimulate marked angiogenesis (the formation of new blood vessels), thus assuring adequate tumor nutrition.
Because some benign tumors evolve into malignant neoplasms and some malignant tumors develop increasingly aggressive behavior over time (a process termed malignant progression), tumors may be graded to reflect where they lie on the continuum from benign to highly malignant and/or staged to indicate the extent of tumor spread. Together, the grade and stage of the tumor indicate the risk the tumor poses to the host and help determine therapeutic strategy. It should be noted, however, that many benign tumors, such as equine sarcoids, have little or no malignant potential and rarely if ever evolve into malignant tumors.
Differentiation
Morphology: Each normal, fully differentiated, mature tissue type has a characteristic gross and microscopic appearance that varies little from individual to individual of a species. Neoplastic tissues lose these differentiated features of cellular morphology and organization to a variable extent. In general, malignant tumors appear less differentiated than benign tumors. Loss of morphologic hallmarks of tissue maturity is often accompanied by loss of functional capacity and development of aggressive behavior.
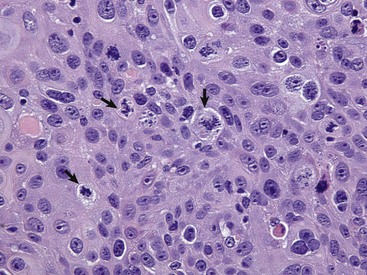
Neoplastic cells often show considerable morphologic variability compared with the normal tissue from which they are derived. Tumor cells, especially malignant tumor cells, may exhibit anaplasia (cellular atypia). Anaplastic cells are poorly differentiated cells that exhibit notable cellular and nuclear pleomorphism (variation in size and shape). In some tumors, bizarre tumor giant cells are seen (Fig. 6-6). Nuclei may exhibit extreme variability in number, size, shape, chromatin distribution, and nucleolar size and number (Fig. 6-7). Anaplastic nuclei are often hyperchromatic (darkly staining) because of increased DNA content; are disproportionately large relative to cell size, resulting in an increased nuclear : cytoplasmic ratio; and have prominent nucleoli. Mitotic figures in tumor cells may be numerous. Many of the nuclear changes seen in neoplastic cells reflect the frequent cell division, chromosomal abnormalities, and active metabolic state that characterize these cells.
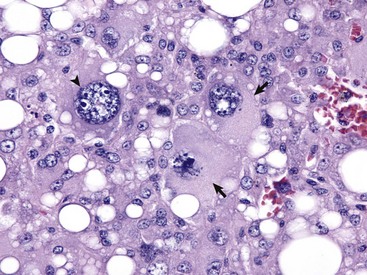
Fig. 6-6 Anaplastic liposarcoma, subcutis, dog.
Anaplastic tumors of epithelial or mesenchymal cell origin often contain bizarre tumor giant cells such as the cells indicated by the arrows. Also, note the large nuclei with abundant coarsely aggregated chromatin and multiple nucleoli (arrowhead). H&E stain. (Courtesy College of Veterinary Medicine, University of Illinois.)
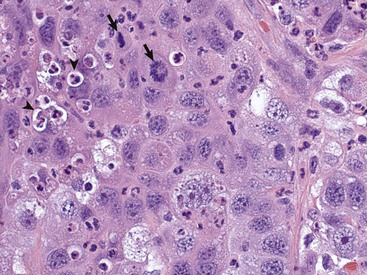
Fig. 6-7 Anaplastic bronchioalveolar carcinoma, dog.
This tumor exhibits marked nuclear pleomorphism and has a fairly high mitotic index. Note the prominent mitotic figures (arrows) and phagocytosis of neutrophils by the tumor cells (emperipolesis) (arrowheads). H&E stain. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Neoplastic cells often exhibit loss of characteristic cytoplasmic and nuclear features. For example, poorly differentiated mast cell tumors often lack the prominent cytoplasmic granules that are a hallmark of normal mast cells (Fig. 6-8). Special stains or immunohistochemistry may be able to highlight some characteristic morphologic feature retained in at least a subpopulation of tumor cells. As an example, characteristic granules may be revealed in some cells of feline and canine mast cell tumors by staining with toluidine blue or Giemsa. Many tumor cells have noticeably basophilic cytoplasm as a result of the presence of large numbers of ribosomes required for rapid cell growth and frequent cell division.
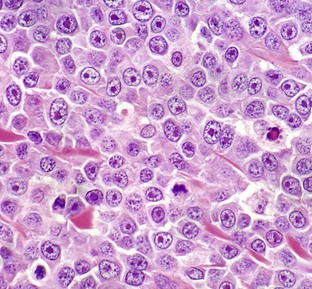
Fig. 6-8 Mast cell tumor, skin, cat.
With H&E staining, hallmark mast cell granules are not visible. To see these granules, the section must be stained with a metachromatic stain such as toluidine blue or Giemsa. Note the very large single nucleolus and marginated chromatin in the neoplastic cells. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
In tumors, normal tissue organization is usually lost to some extent. Increasing loss of normal architecture in tumors correlates with increasing independence of tumor cells from their surrounding tissue. As an example, lymphomas arising in lymph nodes often consist of solid sheets of neoplastic cells that partially or completely efface normal lymph node architecture (Fig. 6-9). In tissue that normally undergoes continual renewal, such as the skin and oral mucosa, the normal maturation sequence may be altered. Thus in squamous cell carcinomas, the orderly morphologic progression from basal cell layer to fully keratinized stratum corneum may not be seen (Fig. 6-10).
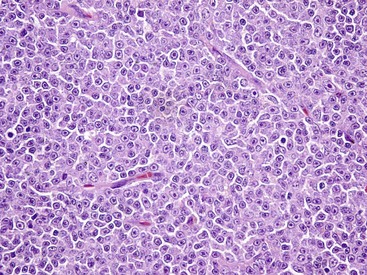
Fig. 6-9 Lymphoma (lymphosarcoma), lymph node, dog.
The normal lymph node architecture has been completely effaced by solid sheets of neoplastic lymphocytes that are relatively uniform in morphology. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
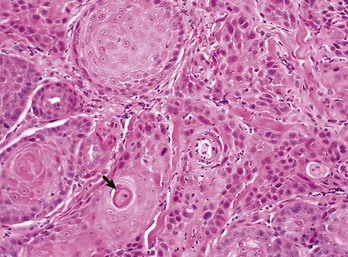
Fig. 6-10 Squamous cell carcinoma, tongue, cat.
The orderly pattern of epidermal maturation seen in normal oral mucosa is absent from this squamous cell carcinoma. An occasional “keratin pearl” (arrow) reveals the tissue of origin for this tumor. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
Function: Loss of differentiated function frequently accompanies loss of differentiated morphology in tumors. Thus neoplastic cells arising from alveolar lining cells of the lung generally fail to perform normal respiratory functions and tumors of primitive germ cell origin do not form normal sperm or ova. Some aspects of normal function may be retained. Thyroid adenomas may continue to produce thyroid hormones, and plasma cell tumors may secrete immunoglobulins. However, in the majority of cases, these functions are no longer regulated appropriately because the neoplastic cells have lost responsiveness to and dependence on normal regulatory pathways. Thus thyroid adenomas may produce clinical hyperthyroidism, and plasma cell tumors may cause hypergammaglobulinemia.
Behavior: Benign tumors are generally expansile and may compress adjacent tissue, whereas malignant tumors have invasive and in many instances metastatic capabilities. In malignant tumors, alterations in adhesion, motility, and protease production allow tumor cells to leave the tumor mass and penetrate surrounding tissue. Moreover, for malignant cells to invade and ultimately metastasize, they must become completely independent of local growth regulatory controls and acquire an independent blood supply. Acquisition of these features allows tumors to spread well beyond their ordinary anatomic niches.
Stem Cells and Differentiation
Most tumors are composed of cells that lack fully differentiated morphologic, functional, and behavioral characteristics. Furthermore, many neoplastic cells share some features with the embryonic cells that gave rise to the mature tissue in which the tumor originated. This similarity between embryonic and neoplastic cells may be accounted for in two different ways. First, normal mature cells may undergo dedifferentiation as they evolve into tumor cells, leading to the reemergence of more primitive characteristics. Second, tumors may arise from the small population of stem cells found in all adult tissue; such stem cells are required for normal tissue renewal. The appearance and behavior of the tumor that develops from a neoplastic stem cell is determined by the stage of differentiation at which the malignant phenotype is manifested; the neoplastic stem cell is said to have undergone maturation arrest at that stage of its development. The diversity of cell types that can arise from a single progenitor stem cell is limited by the differentiation potential of that cell.
Totipotent stem cells, such as embryonic stem cells, can give rise to all tissues of the body, whereas multipotent or pluripotent stem cells can give rise to a smaller variety of tissue types. The plasticity of most adult stem cells is generally considered to be relatively restricted. Leukemias provide excellent examples of neoplasms arising from stem cells. A leukemia almost always arises from a single hematopoietic stem cell that has undergone heritable genetic change. The progeny of this stem cell all exhibit the same genetic change, although the cell type and degree of differentiation of the progeny may vary. Thus in myelogenous leukemia, a neoplastic multipotential stem cell may give rise to a combination of leukemic cells of the granulocytic, monocytic, and erythroid series (Fig. 6-11). The concept of a stem cell origin for cancer explains not only the embryonic characteristics of neoplastic cells but also the success of treatment strategies that use differentiating agents such as retinoids (vitamin A derivatives used to induce maturation of some human leukemia cells).
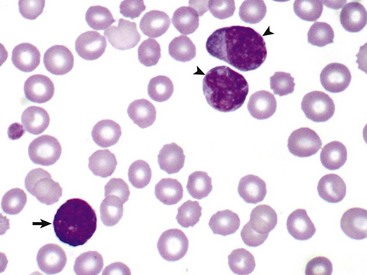
Fig. 6-11 Myelomonocytic leukemia, peripheral blood, dog.
In this unusual case, leukemic cells of both monocytic (arrowheads) and granulocytic (basophil) (arrow) origin were present in peripheral blood. The animal had a marked leukocytosis (103,000 white blood cells/µL) and thrombocytopenia. Wright’s stain. (Courtesy Dr. M.J. Burkhard, College of Veterinary Medicine, The Ohio State University.)
Proliferation
The cell cycle consists of G1 (presynthetic), S (DNA synthesis), G2 (premitotic), and M (mitotic) phases (Fig. 6-12). Quiescent cells are in a physiologic state called G0. In adult tissue, many cells reside in G0 and are unable to enter the cell cycle at all or do so only when stimulated by extrinsic factors. Moreover, in response to DNA damage, even actively dividing normal cells undergo cell-cycle arrest, usually at one of several cell-cycle checkpoints. Cell-cycle arrest is initiated by the multifunctional tumor suppressor gene product p53 and gives the cell time to repair DNA damage.
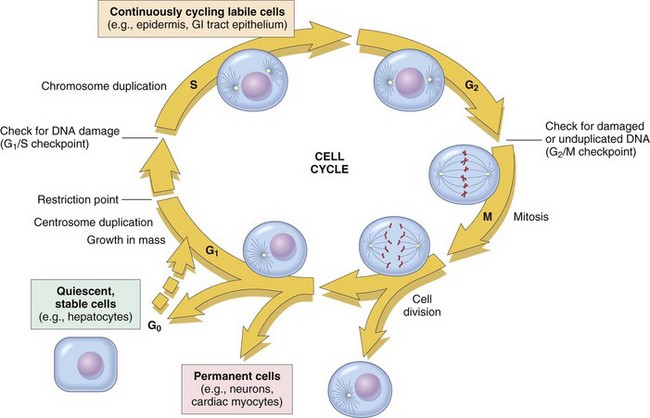
Fig. 6-12 Cell-cycle landmarks.
The figure shows the cell-cycle phases (G0, G1, G2, S, and M), the location of the G1 restriction point, and the G1/S and G2/M cell-cycle checkpoints. Cells from labile tissues, such as the epidermis and the gastrointestinal (GI) tract, may cycle continuously; stable cells, such as hepatocytes, are quiescent but can enter the cell cycle; permanent cells, such as neurons and cardiac myocytes, have lost the capacity to proliferate. (Modified from Pollard TD, Earnshaw WC: Cell biology, Philadelphia, 2002, Saunders.)
Proliferative Activity in Nonneoplastic Tissue
Although composed primarily of quiescent cells in G0, most mature tissue contains some combination of continuously dividing cells, terminally differentiated cells, stem cells, and quiescent cells that can enter the cell cycle. The tissues of the body may be divided into three groups on the basis of their proliferative activity.
Continuously Dividing Tissues (Labile Tissues): In continuously dividing tissues (also called labile tissues), cells proliferate throughout life, replacing those that are lost. These tissues include surface epithelia, such as stratified squamous surfaces of the skin, oral cavity, vagina, and cervix; the lining mucosa of all the excretory ducts of the glands of the body (e.g., salivary glands, pancreas, and biliary tract); the columnar epithelium of the gastrointestinal tract and uterus; the transitional epithelium of the urinary tract; and cells of the bone marrow and hematopoietic tissue. In most of these tissues, mature cells are derived from stem cells, which have an unlimited capacity to proliferate and whose progeny may differentiate into a variety of mature cell types.
Quiescent Tissues (Stable Tissues): Quiescent (or stable) tissues normally have a low level of replication; however, cells from these tissues can undergo rapid division in response to stimuli and are thus capable of reconstituting the tissue of origin. They are considered to be in the G0 stage of the cell cycle but can be stimulated to enter G1. This category includes the parenchymal cells of the liver, kidneys, and pancreas; mesenchymal cells, such as fibroblasts and smooth muscle; vascular endothelial cells; and resting lymphocytes and other leukocytes. The regenerative capacity of stable cells is best exemplified by the ability of the liver to regenerate after partial hepatectomy and after acute chemical injury. Fibroblasts, endothelial cells, smooth muscle cells, chondrocytes, and osteocytes are quiescent in adult mammals but proliferate in response to injury. Fibroblasts in particular may proliferate extensively.
Nondividing Tissues (Permanent Tissues): Nondividing (permanent) tissues contain cells that have left the cell cycle and cannot undergo mitotic division in postnatal life. Neurons and skeletal and cardiac muscle cells belong to this group. If neurons in the central nervous system (CNS) are destroyed, the tissue is generally replaced by the proliferation of the CNS supportive elements, the glial cells. However, recent results demonstrate that limited neurogenesis from stem cells may occur in adult brains. Although mature skeletal muscle cells do not divide, skeletal muscle does have some regenerative capacity, through the differentiation of the satellite cells that are attached to the endomysial sheaths. If the ends of severed muscle fibers are closely juxtaposed, muscle regeneration in mammals can be excellent, but this is a condition that can rarely be attained under practical conditions. Cardiac muscle has very limited, if any, regenerative capacity, and extensive injury to the heart muscle, as may occur in myocardial infarction, is followed by scar formation.
Normal Tissue Growth
In adult tissues, the size of a cell population is determined by the relative rates of cell proliferation, differentiation, and death. Fig. 6-13 depicts these relationships and shows that increased cell numbers may result from either increased proliferation or decreased cell death.
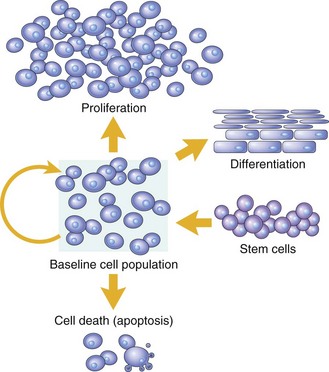
Fig. 6-13 Mechanisms regulating cell populations.
Cell numbers can be altered by increased or decreased rates of stem cell input, by cell death due to apoptosis, or by changes in the rates of proliferation or differentiation. (Modified from McCarthy NJ, Smith CA, Williams GT: Cancer Metastasis Rev 11:157-178, 1992.)
Proliferation: Cell proliferation is largely controlled by signals (soluble or contact-dependent) from the microenvironment that either stimulate or inhibit cell proliferation. An excess of stimulators or a deficiency of inhibitors leads to net growth. Although accelerated growth can be accomplished by shortening the cell cycle, the most important mechanism of growth is the conversion of resting or quiescent cells into proliferating cells by making the cells enter the cell cycle. Both the recruitment of quiescent cells into the cycle and cell-cycle progression require stimulatory signals to overcome normal physiologic blocks to cell proliferation. Cell proliferation can be stimulated under both physiologic and pathologic conditions. The proliferation of mammary epithelium under hormonal stimulation during lactation is an example of physiologic proliferation. Pathologic conditions, such as tissue injury, cell death, and mechanical alterations, also stimulate cell proliferation. Excessive physiologic stimulation may create pathologic conditions, such as enlargement of the thyroid, as a consequence of increased serum levels of thyroid-stimulating hormone.
Differentiation: Differentiation also impacts the size of a cell population and its proliferative potential. For example, myocytes and neurons are terminally differentiated cells (i.e., they are at an end stage of differentiation and are not capable of replicating). In some adult tissues, such as liver and kidney, differentiated cells are normally quiescent but are able to proliferate when necessary. In proliferative tissue, such as bone marrow and the epithelia of the skin and gut, the mature cells are terminally differentiated, short-lived, and incapable of replication, but they may be replaced by new cells arising from stem cells. Thus in such tissues there is a homeostatic equilibrium between the proliferation of stem cells, their differentiation, and the death of fully differentiated cells.
Cell Death: A variety of cell death mechanisms, including senescence, apoptosis, and autophagy, eliminate irreversibly damaged or effete cells to maintain normal tissue homeostasis. In response to DNA damage, oxidative stress, and telomere shortening, proliferating cells may undergo a permanent arrest in the G1 phase of the cell cycle termed cellular senescence. Senescence is mediated by activation of the p53 or retinoblastoma pathways of cell cycle arrest. Senescent cells often express senescence-associated β-galactosidase.
Apoptosis is a form of “programmed cell death” that serves both as a normal physiologic process and as a response to pathologic stimuli. In proliferative tissue, such as gut epithelium, terminally differentiated cells undergo apoptosis and are thus removed from the cell population. Apoptosis may occur in response to withdrawal of survival factors from the cell environment or by binding of death factors, such as Fas ligand and tumor necrosis factor-α (TNF-α) to cell surface receptors. Hypoxia and lack of essential nutrients may end in apoptosis. DNA damage may also induce apoptosis; in this case, apoptosis is triggered by p53. Apoptosis may be stimulated by the activity of cytotoxic immune cells, including T lymphocytes and natural killer (NK) cells. Signals for apoptosis activate a variety of signaling pathways, many of which ultimately result in the release of cytochrome C from mitochondria. The final effectors of apoptosis are the caspases, intracellular proteases that selectively destroy cellular organelles and degrade genomic DNA into nucleosome-sized fragments. The morphologic hallmarks of apoptosis include margination of chromatin, condensation and fragmentation of the nucleus, and condensation of the cell with preservation of organelles. Ultimately, the cell breaks into membrane-bound apoptotic bodies that are engulfed by surrounding cells without stimulating an inflammatory response (Fig. 6-14).
Fig. 6-14 Lymphoma, apoptosis, lymph node, horse.
The light microscopic appearance of apoptosis is characterized by condensation and fragmentation of nuclei (arrows), cell shrinkage, engulfment of apoptotic bodies by surrounding cells, and lack of inflammation. H&E stain. (Courtesy Dr. R. Tan, College of Veterinary Medicine, University of Illinois.)
Autophagy refers to degradation of a cell’s own organelles within autophagosomes. Autophagy can be a mechanism for cell survival in the face of nutrient deprivation, as it salvages important cellular components for reuse; however, extensive autophagy can also lead to a form of programmed cell death. The kinase mammalian target of rapamycin (mTOR) is the major cellular inhibitor of autophagy.
Tumor Growth
Essentially unlimited proliferative potential is a hallmark of neoplasia, especially of malignant neoplasms. Unlike normal cells, many tumor cells are immortal. This immortality is due to a combination of the alterations discussed later. In general, neoplastic cells escape normal limits on cell division, become independent of external growth stimulatory and inhibitory factors, and lose their susceptibility to apoptotic signals. This results in an imbalance between cell production and cell loss and a net increase in tumor size. However, it should be noted that the growth of a tumor is not completely exponential. A proportion of tumor cells is continually lost from the replicative pool because of irreversible cell-cycle arrest, differentiation, and death (Fig. 6-15).
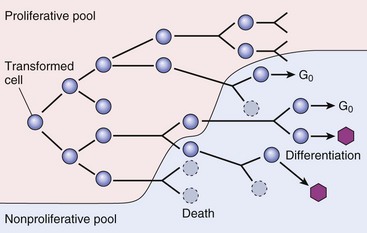
Fig. 6-15 Schematic representation of tumor growth.
As the cell population expands, a progressively higher percentage of tumor cells leave the replicative pool by reversion to G0, differentiation, and death. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Latency: As illustrated in Fig. 6-16, the latent period for a tumor is the time before a tumor becomes clinically detectable. The smallest clinically detectable mass is about 1 cm in diameter and contains about 109 cells. To form a tumor that size, a single transformed cell must undergo about 30 rounds of cell division, if all the progeny remain viable and capable of replication. Thus, by the time most tumors become clinically evident, they have probably been developing in the host for many years. However, once tumors reach a clinically detectable size, their growth may appear to be very rapid, because only 10 doubling cycles are required to convert a 1-g tumor into a 1-kg tumor. In fact, volume doubling times for tumors vary considerably, depending on the rate at which tumor cells divide, the fraction of tumor cells that are replicatively competent, and the rate at which tumor cells die. In general, benign neoplasms grow more slowly than malignant tumors, although there is considerable variation among tumors. Moreover, tumors may grow erratically, depending on their blood supply, the effect of extrinsic growth-regulating factors such as hormones, the efficacy of the host immune response, and the emergence of subpopulations of particularly aggressive tumor cells.
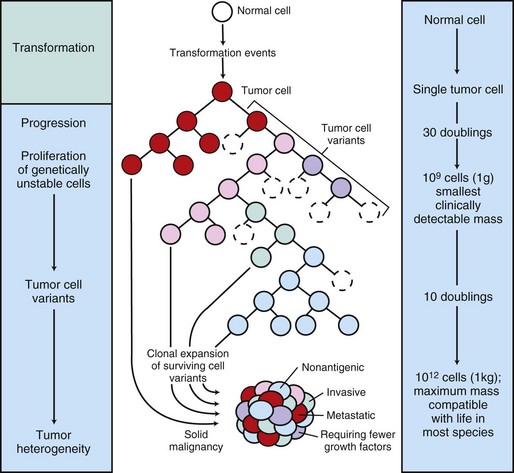
Fig. 6-16 Biology of solid tumor growth.
The center panel illustrates clonal evolution of a tumor and generation of tumor-cell heterogeneity. New subclones arise from descendents of the original transformed cell. With progressive growth, the tumor mass becomes enriched for those variants that are more adept at evading host defenses and are likely to be more aggressive. The left panel shows the corresponding stages of tumor progression, and the right panel depicts minimal estimates of tumor-cell doublings that precede the formation of a clinically detectable tumor mass. It is evident that by the time a solid tumor is detected, it has already completed a major portion of its life cycle, as measured by population doublings. The maximum tumor size compatible with life depends to some extent on the species affected. (Modified from Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Proliferation: Many neoplastic cells no longer respond to extrinsic or intrinsic signals directing them into G0 and no longer express functional p53. Thus the cells move continuously through the cell cycle. Moreover, because the tumor cells do not undergo cell-cycle arrest after DNA damage, they progressively accumulate potentially mutagenic DNA damage (Fig. 6-17). For homeostasis to be maintained, normal cells must engage in a continual dialogue with their environment. There is a constant exchange of information among cells via soluble mediators, including growth stimulatory factors, growth inhibitory factors, and hormones. These soluble mediators tightly control the growth of nonneoplastic cells. Neoplastic cells, on the other hand, often lose both their dependence on extrinsic growth stimulatory substances and their susceptibility to growth inhibitory signals from their environment. The mechanisms by which this occurs are discussed later. The end result is that tumor cells are no longer responsive to the needs of the organism as a whole and develop the capacity to drive their own replication.
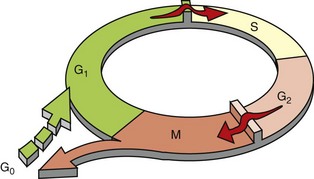
Fig. 6-17 Schematic representation of the cell cycle.
Many normal cells reside in G0, a nonreplicative state. When they do enter the proliferative cycle, they can arrest at cell-cycle checkpoints at the G1-S and G2-M boundaries in response to a variety of stimuli, including DNA damage. In contrast, tumor cells spend little time in G0 and often do not undergo cell-cycle arrest in response to DNA damage or lack of extrinsic growth stimuli. (Modified from Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
The mitotic index is usually defined as the number of tumor cells in a microscopic field that contain condensed chromosomes and lack nuclear membranes (Fig. 6-18). Such cells are interpreted as being actively dividing, and the mitotic index of a tumor is considered to indicate its malignant potential. However, the mitotic index can be misleading. The fraction of tumor cells observed to be in mitosis depends not only on the number of cells undergoing mitosis but also on the length of time required to complete the process. In tumor cells, the time required for completion of the cell cycle is generally as long as or even longer than for normal cells. Mitotic figures may persist in cells unable to complete cell division and abnormal mitotic figures may be seen.
Differentiation: As discussed previously, tumor cells are generally less differentiated than normal cells. In some instances, however, some tumor cells can be forced to differentiate into more mature, near-normal cells. Leukemia cells are particularly susceptible to differentiation therapy, and retinoids are routinely employed to treat acute promyelocytic leukemia in human patients. Other differentiating agents, including vitamin D compounds and cytokines, have been less effective in this disease. Vitamin D compounds are showing some promise in differentiation therapy of human epithelial tumors, and compounds that epigenetically alter tumor cells by modifying the histones in chromatin may also enhance differentiation of tumor cells (discussed later). A common assumption underlying differentiation therapies is that more differentiated tumor cells will have a less stem cell-like phenotype and will thus have reduced proliferative potential.
Cell Death: Because the DNA replication machinery is unable to duplicate the extreme ends of DNA templates, the telomeres that form the ends of chromosomes are shortened at each cell division. Embryonic cells express telomerase, a riboprotein enzyme that allows telomeres to be replicated and even expanded; however, most adult cells do not express this protein and their telomeres shrink with each round of cell division. Very short telomeres are incompatible with continued cell division and trigger cellular senescence in normal cells. However, many neoplastic cells regain the ability to produce telomerase and thus to replicate their telomeres. Reexpression of telomerase appears to play an important role in the escape of tumor cells from senescence and their consequent immortality.
Although virtually all normal cells in the body can undergo apoptosis in response to appropriate physiologic signals, many cancer cells acquire resistance to apoptosis. This blocks a major route of tumor cell loss and enhances the overall growth rate of the tumor. Many tumor cells circumvent apoptosis by functional inactivation of the p53 gene, thus removing a key proapoptotic molecule. Additionally, tumor cells may constitutively activate survival signaling pathways, rendering the cells independent of exogenous survival factors. Finally, tumor cells may develop mechanisms for inactivating death factor signaling pathways, thus evading apoptosis in response to homeostatic signals from the cellular environment.
Autophagy plays a poorly understood and somewhat paradoxic role in tumor growth. In many tumors, authophagy is suppressed, thus presumably preventing autophagic tumor cell death. However, increased autophagy may also enhance tumor cell survival under the conditions of reduced nutrient availability that arise during therapy.
Genomic Instability
Evolving genomic instability is a hallmark of cancer. Many tumor cells fail to undergo cell-cycle arrest or apoptosis in response to DNA damage. They produce long and unstable telomeres subject to breakage, they lose the ability to carry out effective DNA repair, they demonstrate aberrant DNA methylation, and they exhibit increased rates of gene amplification, recombination, conversion, and transposition. These factors contribute to an increased rate of mutation and chromosomal aberration in neoplastic cells. Ultimately, this genomic instability results in aneuploidy, a chromosome complement that is not a simple multiple of the haploid chromosome content, or polyploidy, a chromosome complement more than twice the haploid chromosome number. The karyotypes of cancer cells may thus be notably abnormal and unstable. As a rule, increasing aneuploidy is correlated with increasingly malignant behavior. Genomic instability is discussed in more detail later in the chapter.
Tumor Evolution
Neoplasms develop as the result of multiple genetic and epigenetic changes that occur over a relatively long time course. It is the cumulative effect of these alterations that creates a tumor. The stepwise evolution of tumors has been studied most thoroughly in carcinomas. There are several types of carcinoma that develop in an orderly and predictable fashion. For instance, squamous cell carcinoma arises from the epithelium of the eyelid in many species of animals, including cattle, horses, cats, and dogs. In all species, these tumors develop through the same sequence of steps: epidermal hyperplasia, carcinoma in situ, and invasive carcinoma. Extensive studies of experimentally induced squamous cell carcinomas in the skin of mice have revealed a similar morphologic pattern of tumor evolution (Fig. 6-19) and have led to a detailed model of stepwise carcinoma development as described in the next section (Fig. 6-20).
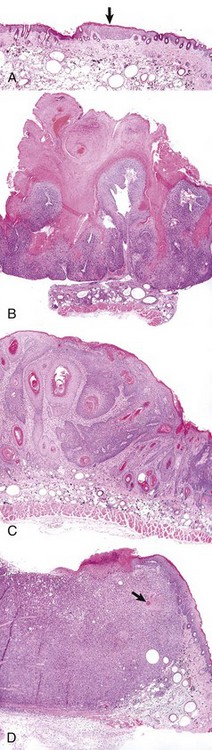
Fig. 6-19 Development of squamous cell carcinoma in the skin of a hairless mouse exposed to ultraviolet (UV) radiation.
A, A focus of epidermal hyperplasia (arrow) is the earliest lesion seen. B, This develops into a papilloma, a benign exophytic papillary growth that is highly keratinized and does not penetrate into the dermis. C, As the papilloma undergoes conversion into a malignant squamous cell carcinoma, it begins to invade the dermis and to lose the regular pattern of epithelial differentiation. D, A fully developed squamous cell carcinoma has lost most differentiated characteristics and extends deep into the dermis. Only a few keratin “pearls” (arrow) indicate the origin of this tumor from the epidermis of the skin. All figures were taken at the same magnification. H&E stain. (Courtesy Dr. T.M. Oberyszyn, The Ohio State University.)
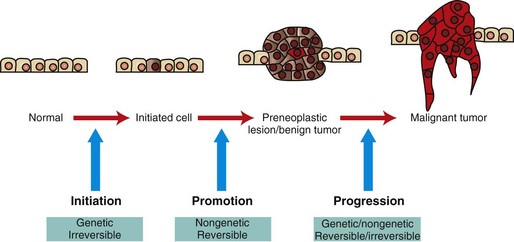
Fig. 6-20 Illustration of stepwise tumor development.
Initiated cells have irreversible genetic damage. In the presence of a promoter, these initiated cells expand to form a preneoplastic lesion or benign tumor. With further genetic and epigenetic alterations, a malignant tumor emerges from a subclone of cells within the benign precursor lesion. (Redrawn with permission from Dr. D.F. Kusewitt, College of Veterinary Medicine, The Ohio State University.)
Initiation
The first step is initiation, the introduction of irreversible genetic change into basal cells of the skin by the action of a mutagenic initiating agent or initiator. Initiators are chemical or physical carcinogens that damage DNA. Mutation induction requires not only the introduction of a DNA lesion, but also mispairing of the DNA lesion during subsequent DNA replication to produce an altered complementary DNA strand. Thus at least a single round of DNA replication is necessary for fixation of the genetic change to occur. Initiated cells appear morphologically normal and may remain quiescent for many years. However, these cells harbor mutations that provide them with a growth advantage under special conditions. For example, the initiated cells may respond more vigorously to mitogenic signals or be more resistant to apoptosis-inducing stimuli than their neighbors.
Promotion
The second stage of tumor development is promotion. Promotion refers to the outgrowth of initiated cells in response to selective stimuli. Most of these selective stimuli, termed promoting agents or promoters, drive proliferation. For example, the skin irritant croton oil is an effective skin tumor promoter. In general, promoters are not mutagenic; instead, they alter gene expression in initiated and uninitiated cells to create an environment in which initiated cells have a growth advantage. Because promoters are nonmutagenic, their effects are usually reversible, and some papillomas can actually undergo regression. What emerges at the end of the promotion phase of tumor development is a papilloma, which is a benign tumor.
Progression
The final stage of tumor development, progression, includes the conversion of a benign tumor to an increasingly malignant tumor and ultimately to a metastatic tumor. Malignant conversion represents an irreversible change in the nature of the developing tumor. Progression is a complex and poorly understood process involving both genetic and epigenetic changes in tumor cells and their environment that select for increasingly malignant clones of tumor cells. Karyotypic instability in tumor cells and increasing tumor cell heterogeneity are hallmarks of progression.
Tumor Heterogeneity and Clonal Selection
Most tumors are believed to be of clonal origin (i.e., they are ultimately derived from a single transformed cell). Tumor cell heterogeneity is generated during the course of tumor growth by the progressive accumulation of heritable changes in tumor cells (see Fig. 6-16). With each new genetic alteration, the progeny of the genetically altered tumor cell constitute a subclone of tumor cells. The generation of subclones is fostered by the marked genetic instability of tumor cells compared with normal cells. Successful subclones are those that have a high proliferative rate, are able to evade the host immune response, can stimulate the development of an independent blood supply, are independent of exogenous growth factors, and are able to escape from the primary tumor and spread to distant sites. These characteristics give successful subclones a selective advantage over other subclones of cells within the tumor. A tumor subclone with a selective advantage will eventually predominate.
Molecular Changes Underlying Tumor Progression
In a few well-studied tumor types, such as chemically induced skin tumors in mice and colonic carcinomas in humans, the stepwise molecular changes that underlie morphologic changes in the tumors have been determined. Many of these genetic changes are associated with proliferation, DNA repair, angiogenesis, and invasiveness, as detailed later.
Tumor Spread
Malignant tumors are often highly invasive. They do not respect anatomic boundaries, and they infiltrate adjacent normal tissue. Benign tumors, on the other hand, are generally expansile rather than infiltrative. The border between a benign tumor and adjacent tissue is usually distinct, and benign tumors of epithelial origin are often encapsulated (surrounded by a connective tissue capsule). Metastasis occurs when colonies of tumor cells take up residence at some distance from the parent tumor. Metastasis is the single most reliable hallmark of malignancy. Benign tumors do not metastasize. However, some malignant tumors, notably those of the CNS, are also nonmetastatic. Metastatic disease is believed to be responsible for 90% of human cancer deaths. Moreover, it is estimated that approximately 30% of solid cancers in humans have already metastasized by the time of initial diagnosis, greatly reducing the possibility of successful therapy. Cancer may metastasize by seeding the body cavities and surfaces (transcoelomic spread), by lymphatic spread, or by hematogenous spread.
Pathways of Tumor Metastasis
When cancers arise on the surface of an abdominal or thoracic structure, they encounter few anatomic barriers to spread. Thus mesotheliomas may be confined to the abdominal or pleural cavities, but the tumor cells within these cavities readily spread to cover all visceral and parietal surfaces (Fig. 6-21). In both humans and dogs, ovarian adenocarcinomas preferentially spread transcoelomically. Although such tumors are rare in dogs, they are commonly encountered in women. Even in the absence of invasion into the underlying organs, tumors such as mesotheliomas and ovarian adenocarcinomas are extremely difficult to treat and are generally fatal.
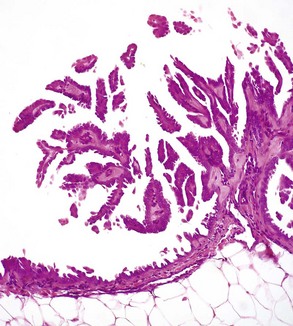
Fig. 6-21 Mesothelioma, peritoneum of the abdominal cavity, dog.
Mesotheliomas spread extensively within body cavities, but rarely metastasize by lymphatic or hematogenous routes. Note in the figure that neoplastic mesothelial cells cover the serosal surfaces and form papillary fronds, but they do not infiltrate underlying tissue. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
Lymphatic
In general, most carcinomas metastasize via the lymphatic system, although sarcomas may also employ this route of spread. The pattern of lymph node involvement is usually dictated by preexisting routes of regional lymphatic drainage. The lymph nodes closest to the tumor are usually colonized earliest and develop the largest metastatic tumor masses (Fig. 6-22). Thus adenocarcinomas of the intestine in all species usually metastasize first to the mesenteric lymph nodes and later to other lymph nodes within and outside the abdominal cavity. For many years, it was assumed that cancers spread in a stepwise manner from the primary site to regional lymph nodes, then to distant sites, such as the lung, and that regional lymph nodes actually represented a mechanical barrier to the spread of cancer. Thus removal of regional lymph nodes containing tumor tissue was believed to protect the patient from further spread of the tumor. However, regional lymph nodes may be bypassed as a result of natural, tumor-related, or treatment-induced anomalies in lymphatic drainage. More recent studies suggest that lymphatic spread does not occur in an orderly fashion and that metastasis to regional lymph nodes indicates that neoplastic disease has become widely systemic.
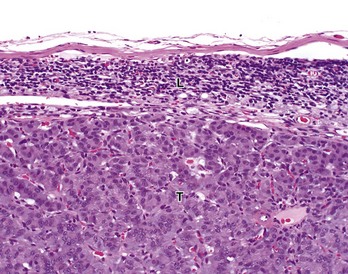
Fig. 6-22 Pancreatic carcinoma, metastatic, hepatic (portal) lymph node, dog.
Tumor cells (T) have almost completely replaced the normal architecture of the lymph node, except for a thin subcapsular rim of lymphocytes (L). The tumor was confirmed by immunohistochemistry to be a functional pancreatic islet β-cell carcinoma. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
Hematogenous
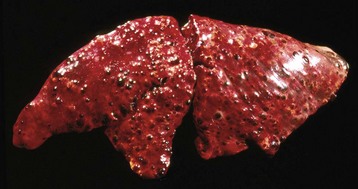
Because lymphatic vessels connect with the vascular system, the distinction between lymphatic and hematogenous spread is somewhat artificial. However, sarcomas do tend to use the hematogenous route of spread more frequently than carcinomas. Tumors generally invade veins rather than arteries because arterial walls are much thicker and more difficult to penetrate. Tumors that enter veins ultimately enter the vena cava and lodge in the lungs (Fig. 6-23) or enter the portal system and lodge in the liver. Neoplasms metastatic to the lungs may secondarily enter the arterial circulation. Some tumors have a notable predilection for veins. Pheochromocytomas of many species frequently enter the adrenal veins, where they may form large tumor masses extending into the vena cava.
Mechanisms of Invasion and Metastasis
Metastasis is a complex process requiring invasion of the extracellular matrix (ECM), entry into blood vascular or lymphatic vessels, extravasation of tumor cells, and colonization of the metastatic site (Fig. 6-24). These activities require many coordinated alterations in cell-cell and cell-matrix adhesion, motility, and invasiveness. Because of the complexity of the metastatic process and the heterogeneity of tumor cells, metastasis is inefficient. Of the many tumor cells that enter the circulation, only a few are able to produce metastases. Local invasion is a prerequisite for metastasis; thus the two processes share many features.
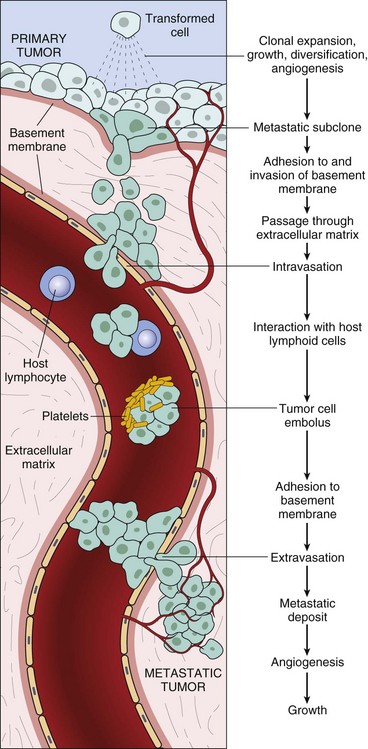
Fig. 6-24 The metastatic cascade.
This figure illustrates the sequential steps involved in the hematogenous spread of a tumor. Similar events occur during lymphatic spread. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Adhesion
As a first event in invasion and metastasis, tumor cells must detach from the main tumor mass, penetrate the basement membrane, and enter the ECM. For cells to separate from each other, intercellular adhesion structures, including desmosomes and adherens junctions, must be dismantled. In many tumor cells of epithelial origin, this occurs because of the loss of cadherin or catenin function. These molecules are essential structural elements of intercellular junctions. At the same time that tumor cells detach from each other, they must also establish contacts with ECM components. Integrins and other specific receptors on the tumor cell membrane recognize and bind to a variety of ECM components such as fibronectin, laminin, collagen, and vitronectin. During invasion and metastasis, carcinoma cells often express increased numbers of these receptors. Moreover, instead of being localized to the basal surface of the tumor cell, the receptors are redistributed to cover the entire cell membrane. Tumor cells also appear to be able to modulate the types of ECM receptors that they express, allowing the cells to adapt to different microenvironments.
Invasion
Nonneoplastic epithelial cells generally rest on a specialized extracellular structure called the basement membrane, to which they are firmly attached by hemidesmosomes. In benign epithelial tumors, the basement membrane usually remains intact, whereas in malignant tumors the neoplastic epithelial cells penetrate the basement membrane to invade surrounding tissue. Tumor cells actively degrade basement membrane and ECM components by increasing the net protease activity in their vicinity (Fig. 6-25). Net protease activity is determined by a variety of interacting factors, including the rate of protease synthesis, the rate at which proteases are activated, and the rate at which protease inhibitors are produced. Proteases and antiproteases may be produced and activated by the tumor cells themselves, or tumor cells may induce nonneoplastic stromal cells to produce these enzymes. Proteases that appear to play an important role in tumor metastasis include matrix metalloproteinases, such as type IV collagenase, and urokinase, a serine protease.
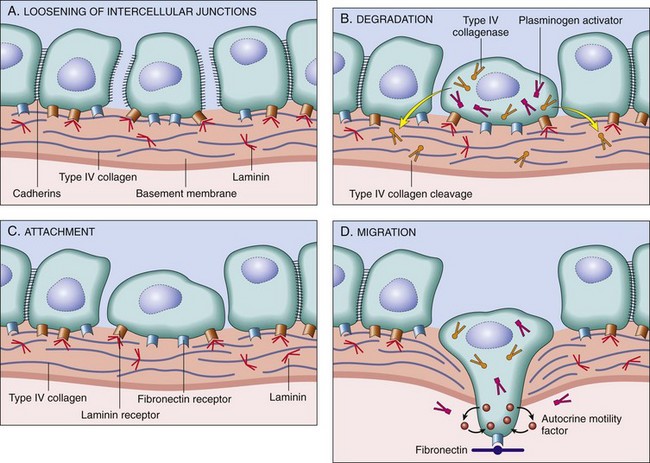
Fig. 6-25 Schematic illustration of the sequence of events in the invasion of epithelial basement membranes by tumor cells.
Tumor cells detach from each other because of reduced adhesiveness (A). Cells then attach to the basement membrane via laminin receptors (B) and secrete proteolytic enzymes, including type IV collagenase and plasminogen activator. Degradation of the basement membrane (C) and tumor cell migration (D) follow. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Migration
At many points during invasion and metastasis, tumor cells migrate actively. This migration appears to be mediated by coordinated alterations in the cytoskeleton and the cellular adhesion structures with which they are intimately associated. Tumor cell migration is stimulated by autocrine growth factors, such as hepatocyte growth factor, also called “scatter factor,” and by cleavage products of ECM components, including fragments of collagen.
Tumor Emboli
The processes by which tumor cells invade blood and lymphatic vessels, extravasate from these vessels, and invade the ECM at a metastatic site are variations on those discussed earlier. Once inside a lymphatic or blood vessel, tumor cells tend to associate to form small emboli. In vessels, tumor cells may be recognized and attacked by host lymphocytes or may be surrounded by platelets. Interestingly, platelets may actually protect the tumor embolus and enhance tumor metastasis. The site at which tumor cells exit the blood vascular or lymphatic system is determined both by the pattern of lymphatic or vascular drainage of the primary tumor and by the ability of tumor cells to interact with adhesion molecules on endothelial cells. In addition, metastatic sites must provide a suitable microenvironment for tumor cell growth. Some tumors preferentially metastasize to specific sites. For example, prostate carcinomas in both humans and dogs frequently spread to bone (Fig. 6-26).
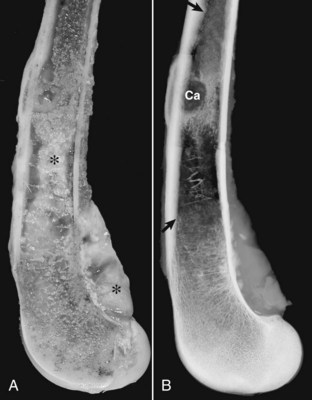
Fig. 6-26 Prostate carcinoma, metastasis, femur, dog.
A, The gross photograph of a sectioned femur reveals metastatic prostate carcinoma (*). B, The radiograph illustrates an osteolytic bone metastasis (Ca). In the regions between the arrows, extensive proliferation of new bone has occurred in response to the tumor. (Modified from Rosol TJ, Tannehill-Gregg SH, LeRoy BE, et al: Animal models of bone metastasis. In Keller ET, Chung LWK, editors: Cancer treatment and research, Boston, 2004, Kluwer Academic Publishers.)
Metastasis Suppression
A wide variety of genetic and epigenetic changes affect tumor cell adhesion, motility, and protease production. Thus metastatic potential is probably the cumulative effect of many different genetic alterations, and it seems unlikely that any individual genetic change single-handedly makes a tumor metastatic. On the other hand, a small number of genes have been identified that seem to function to suppress metastasis effectively. A good example of a candidate metastasis suppressor gene is the gene encoding E-cadherin, a component of adherens junctions, because loss of intercellular junctions appears to be essential for tumor metastasis.
Tumor Stroma
A tumor consists of tumor cells proper, the tumor parenchyma, and nonneoplastic supporting structures, the stroma (Fig. 6-27, A). The stroma is composed largely of extracellular connective tissue and consists of proteins and glycoproteins, such as collagen, embedded in a complex matrix of proteoglycans. The stroma contains the blood vessels that supply nutrients to the tumor, fibroblasts, and a variety of inflammatory and immune cells. The amount of stroma associated with tumors can vary considerably. The extracellular material in the stroma of epithelial tumors is produced primarily by surrounding nonneoplastic mesenchymal cells (Fig. 6-27, B), whereas many mesenchymal tumors can produce the ECM in their stroma. For example, many osteosarcomas produce bone, a specialized form of connective tissue stroma. Stromal tissue may form a connective tissue capsule around epithelial tumors that limit neoplastic spread. In general, encapsulated epithelial tumors are considered to have a better prognosis than unencapsulated tumors (Fig. 6-28).
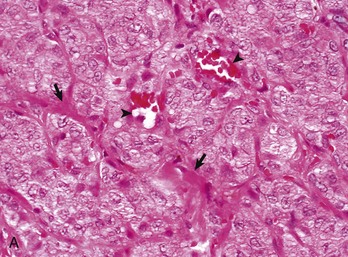
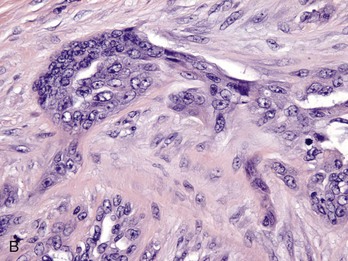
Fig. 6-27 Tumor stroma.
Tumor cells and the stroma in which they are embedded interact in a variety of ways that serve to modulate the growth rate, differentiation state, and behavior of both components. A, Pancreatic adenocarcinoma, pancreas, dog. The tumor parenchyma is divided into incomplete lobules by the tumor stroma (arrows) composed of collagen and extracellular matrix components in which blood vessels (arrowheads), fibroblasts, and inflammatory and immune cells are embedded. H&E stain. B, Scirrhous carcinoma, mammary gland, dog. Carcinomas and adenocarcinomas that stimulate the formation of abundant collagen in surrounding connective tissue (desmoplasia) may be termed scirrhous. Tumor-associated fibroblasts may secrete a fetal type of extracellular matrix (ECM) and co-evolve with adjacent tumor cells. Note how there is intimate interaction between the anaplastic malignant epithelial cells and the scirrhous connective tissue in which they are embedded, thus tumor stroma may both enhance and/or limit tumor development and spread. H&E stain. (A courtesy College of Veterinary Medicine, The Ohio State University. B courtesy of Dr. L. Borst, College of Veterinary Medicine, North Carolina State University.)
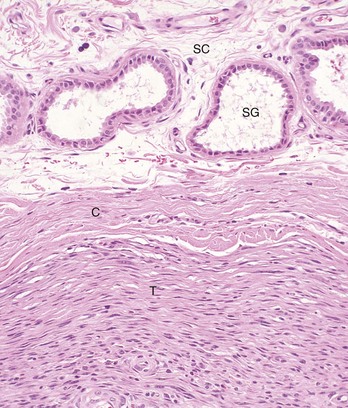
Fig. 6-28 Adnexal tumor, benign, skin, dog.
This tumor (T) lies within the subcutis (SC) and is surrounded by a thick connective tissue capsule (C), which is more frequently associated with benign than with malignant tumors. SG, Normal apocrine sweat glands of the subcutis. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
In the stroma of rare tumors, both mesenchymal and epithelial, there is an amorphous eosinophilic substance termed amyloid. Amyloid consists of one of a variety of abnormal proteins arranged in β-pleated fibrils. The proteins that form amyloid are usually secreted by the tumor cells themselves; for instance, λ-light chain protein secreted by neoplastic plasma cells forms the amyloid sometimes seen in the extramedullary plasmacytomas of various species.
Tumor-Stromal Interactions
Tumor cells interact with their stroma in a complex fashion, exchanging a wide variety of signaling molecules, including growth factors, cytokines, hormones, and inflammatory mediators (Fig. 6-29). These exchanges modulate the growth rate, differentiation state, and behavior of both stromal and tumor cells. Platelet-derived growth factor (PDGF) released by tumor cells stimulates tumor-associated fibroblasts to increase the production of collagen. In some cases, this process leads to an extensive fibrous reaction, termed a scirrhous or desmoplastic response, in the stroma. Transforming growth factor-α (TGF-α) of tumor origin can stimulate tumor-associated fibroblasts to differentiate into myofibroblasts, which are fibroblasts with contractile capabilities. Tumor-associated fibroblasts may acquire special characteristics that distinguish them from normal fibroblasts. Such fibroblasts may secrete a fetal type of ECM. In some tumors, tumor-associated fibroblasts acquire heritable genetic and epigenetic changes that allow them to co-evolve with adjacent tumor cells. Tumor cells may induce surrounding stromal cells to produce cytokines that promote tumor cell proliferation and motility. Furthermore, growth factors are sequestered in the ECM of the stroma, where they bind to proteoglycans. This interaction controls the bioavailability of these factors, which can be released from the ECM by the activity of proteases secreted by tumor cells, stromal fibroblasts, or inflammatory cells.
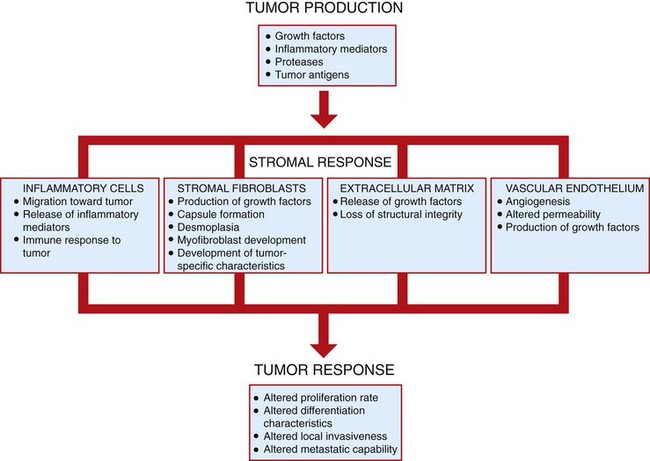
Fig. 6-29 Tumor-stromal interactions.
Tumor cells and the stroma in which they are embedded interact in a variety of ways that serve to modify the growth and behavior of both elements. Tumor stroma may both enhance and limit tumor development and spread. (Redrawn with permission from Dr. D.F. Kusewitt, College of Veterinary Medicine, The Ohio State University.)
Angiogenesis
Continued growth of solid tumors absolutely depends on an adequate blood supply to provide oxygen and nutrients to tumor cells. Without the development of new blood vessels, a process termed angiogenesis, tumors are limited to a maximum diameter of 1 to 2 mm. At some point during tumor development, an angiogenic switch occurs that confers on tumor cells the ability to induce and sustain new tumor vasculature. Angiogenesis is a complex process involving recruitment of endothelial cells from preexisting blood vessels, endothelial cell proliferation, directed migration of endothelial cells through the ECM, and maturation and differentiation of the capillary sprout. Angiogenesis is controlled by the balance between a plethora of angiogenesis-stimulating and angiogenesis-inhibiting factors. Tumors induce angiogenesis by the production and release of angiogenic factors, such as vascular endothelial growth factor (VEGF), or by downregulating production of antiangiogenic factors, such as thrombospondin. In addition, angiogenic and antiangiogenic factors bound to ECM components can be released and activated by tumor protease activity. VEGF and acidic and basic fibroblast growth factors are among the most potent angiogenic factors produced by tumors. The tumor blood vessels that develop in response to angiogenic signals are usually more dilated, more tortuous, and more permeable than normal blood vessels (Fig. 6-30).
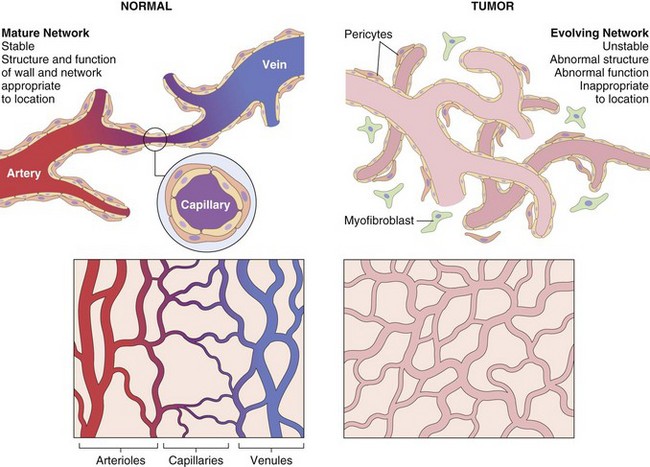
Fig. 6-30 Tumor angiogenesis.
Compared with normal vessels (left panels), tumor vessels are tortuous and irregularly shaped (right panels). The tumor vasculature is formed from circulating endothelial precursor cells and existing host vessels; myofibroblasts give rise to pericytes that surround vessels. In contrast to the stable vessel network of normal tissue, the networks formed by tumor vessels are unstable and leaky. Arterioles, capillaries, and venules are clearly distinguishable in normal vasculature; in the tumor, vessels are disorganized and specific vessel types cannot be identified. (Modified from Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
In addition to supplying nutrients, tumor vasculature plays other roles in tumor development. Vessel leakiness allows perivascular deposition of a fibrin network that promotes formation of collagenous tumor stroma. The endothelial cells of tumor blood vessels produce growth factors, such as PDGF and interleukin-1 (IL-1), that can stimulate the growth of tumor cells. Moreover, without access to the circulatory system, tumors cannot metastasize. Because solid tumor growth absolutely depends on an adequate blood supply, therapeutic strategies to inhibit angiogenesis are being developed.
Investigation of the development of lymphatic vasculature of tumors has demonstrated that tumor lymphangiogenesis shares many features with tumor angiogenesis. Tumor-associated lymphatic vessels sprout from preexisting lymphatic vessels in response to tumor-secreted factors such as VEGF. Tumor-associated lymphatics appear to be essential for metastasis of solid tumors to regional lymph nodes. In human tumors, there is a strong correlation between the levels of VEGF expression and lymphatic metastasis; and in genetically engineered mice that do not express VEGF, lymphatic metastasis does not occur.
Inflammation
Many tumors are heavily infiltrated with neutrophils, eosinophils, mast cells, lymphocytes, histiocytes, or a combination of these cells. Inflammatory cells are attracted to tumors by chemokines and cytokines released by the tumor cells themselves or by mononuclear and polymorphonuclear cells infiltrating the tumor. Inflammatory cells serve as a source of prostaglandins, leukotrienes, and reactive oxygen species. In general, inflammation does not appear to protect against tumors. In fact, many chronic inflammatory conditions increase the risk of cancer in affected organs. As an example, the development of postvaccinal sarcomas in cats is clearly linked to the presence of inflammation at sites of inoculation. Moreover, in humans, epidemiologic studies suggest that nonsteroidal antiinflammatory drugs (NSAIDs) reduce the incidence of some cancers.
Tumor Immunity
Tumor antigens are proteins, glycoproteins, glycolipids, or carbohydrates expressed on the tumor cell surface (Fig. 6-31). They include both tumor-specific antigens restricted to tumor cells and tumor-associated antigens present on both tumor cells and normal cells. Tumor antigens can be exploited both for diagnostic and therapeutic purposes. Tumor antigens released into the bloodstream allow noninvasive detection of tumors and monitoring tumor response to treatment. In combination with sophisticated imaging techniques, antibodies against tumor-restricted antigens can be used to localize tumors and detect metastases. Some tumor antigens can serve as the targets of effective immunosurveillance. However, many tumor antigens are not appropriate therapeutic targets. The antigens may not be restricted to tumor cells or may not elicit a strong cytotoxic response. Therapeutic strategies to enhance the immune response to tumor antigens are currently the subject of intense investigation.
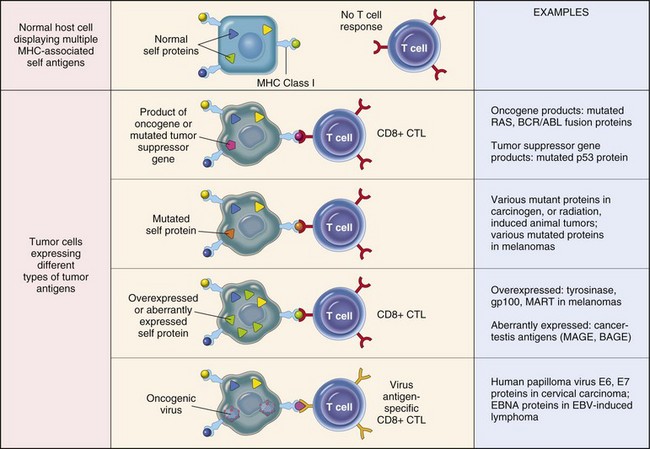
Fig. 6-31 Tumor antigens recognized by CD8+ T lymphocytes.
Note that all tumor antigens are presented to CD8+ T lymphocytes by major histocompatibility (MHC) class I molecules. CTL, Cytotoxic T lymphocyte. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Some tumor-specific antigens are newly expressed molecules, such as antigens derived from oncogenic viruses, or altered cellular products encoded by mutated genes. In these instances, productive viral infection or gene mutation is restricted to tumor cells and their progeny. Embryonic antigens or oncofetal antigens, normally not expressed in adult tissue but reexpressed in tumor tissue, may also behave as tumor antigens. For example, the developmental antigens, carcinoembryonic antigen and α-fetoprotein, are reexpressed in some tumors in a variety of species and may be released into the circulation. Serology for these antigens is widely used to test for recurrence of liver and intestinal tumors in humans. Tumor-specific shared antigens are encoded by genes that have very limited expression in adult tissue but that are expressed by many types of tumor tissue. A striking example of useful tumor-specific shared antigens is the MAGE family of proteins, found in humans and other species. These antigens are not present on the surface of normal adult cells; however, they are expressed by a wide variety of tumor types and are promising candidates for antitumor immunotherapy. Tissue-specific antigens are shared by tumors and the normal tissues from which they arise. In some cases, these antigens are expressed only at specific stages of differentiation in the normal tissue and are thus termed differentiation antigens. When tissue-specific or differentiation antigens are expressed at considerably higher levels on tumor cells than normal cells, they may function much like tumor-specific antigens.
Immunosurveillance
It is likely that the vertebrate immune system evolved primarily for the purpose of recognizing and destroying infectious organisms and the host cells they infect. However, the immune system also attacks tissue transplanted from genetically dissimilar animals of the same or different species. In special instances, the immune system can respond to self-antigens. It has been postulated that the immune system is capable of recognizing self-antigens on tumor cells as foreign and attacking antigen-bearing tumor cells as if they were microbially infected cells or foreign cells. This process is termed immunosurveillance. It is argued that effective immunosurveillance suppresses tumor development and that a failure of immunosurveillance allows tumors to emerge. The immunosurveillance hypothesis is supported by the dramatically increased tumor susceptibility of immunosuppressed transplant recipients. The inability of these individuals to mount effective antitumor responses apparently allows the emergence of many tumors usually eliminated by immunosurveillance. The presence of lymphocyte and macrophage infiltrates within and around tumors of many types in many species also suggests that tumors can elicit an immune response. Finally, there is abundant experimental evidence that mice can mount an effective immune response against chemically induced tumors. Much of our knowledge of tumor immunity comes from experimental studies employing mice.
Antitumor Effector Mechanisms
The body may mount a variety of immune responses against tumor antigens (Fig. 6-32). The type of immune response and its effectiveness against tumor cells is largely determined by the inherent immune responsiveness of the host and the characteristics of the tumor antigen under attack. The least specific immune response to tumor cells is carried out by the innate immune system. The innate immune response is believed to be the first line of defense against cancer cells. Antitumor effectors of the innate immune system, including NK cells and macrophages, do not require antigen-specific priming by dendritic cells. More specific immune responses are undertaken by the adaptive immune system, consisting of both cell-mediated and humoral components. The cell-mediated immune response is believed to mount the most effective antitumor defenses. Any adaptive antitumor immune response requires that tumor antigens be presented to immune cells in a recognizable context. Dendritic cells capture antigens that are secreted by viable tumor cells or released from dying tumor cells. The dendritic cells ingest these antigens, fragment them to a suitable size, link them to class I or class II major histocompatibility (MHC) antigens, and present them on the cell surface in association with appropriate co-stimulatory molecules. A dendritic cell can then interact with many different lymphocytes to prime their response to the specific tumor antigen presented by the dendritic cell. Antigen-activated CD8+ and CD4+ T lymphocytes develop into tumor-specific cytotoxic and T helper (TH) lymphocytes, respectively, whereas B lymphocytes develop into immunoglobulin-secreting plasma cells. CD8+ lymphocytes recognize tumor antigens in the context of MHC class I antigens, whereas CD4+ cells recognize these antigens only in association with MHC class II molecules.
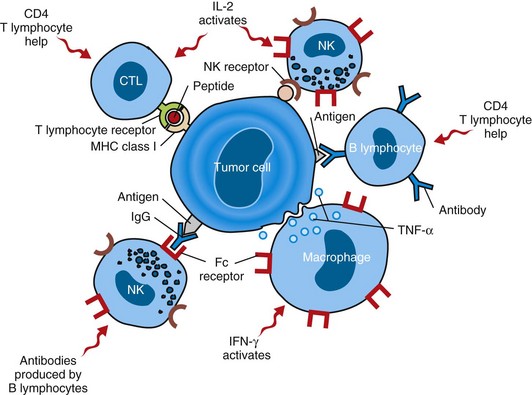
Fig. 6-32 Cells involved in immunosurveillance against tumors.
Antitumor responses involve cytotoxic CD8+ and helper CD4+ lymphocytes, natural killer (NK) cells, macrophages, B lymphocytes, and a variety of immunomodulatory cytokines. CTL, Cytotoxic T lymphocyte; IFN-γ, interferon-γ; IgG, immunoglobulin G; IL-2, interleukin-2; MHC, major histocompatibility complex; TNF-α, tumor necrosis factor-α. (Modified from Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Natural Killer Cells
NK cells are lymphocytes that lack many of the usual markers of T or B lymphocytes. NK cells display a variety of receptors, both inhibitory and activating, that recognize MHC molecules and stress-induced ligands on tumor cells. NK cells can kill a wide variety of neoplastic and virally infected cells. Cells that express MHC class I molecules are preferentially spared by NK cells, whereas cells lacking MHC molecules are specifically targeted. After binding to a tumor cell, the NK cell releases lytic granules that activate apoptosis in the target cell. This mechanism of cell killing or cytolysis is shared with T lymphocytes and is discussed in more detail later.
Macrophages
Macrophages are migratory phagocytic cells capable of killing tumor cells by releasing reactive oxygen intermediates, lysosomal enzymes, nitric oxide, and tumor necrosis factor. Their antitumor activity is stimulated by interferon-γ (IFN-γ), which is produced by both T lymphocytes and NK cells. Macrophage-mediated tumor cell killing is independent of MHC antigens, tumor-specific antigens, and the type of transformed cell being targeted, but direct contact between the macrophage and tumor cell is required.
T Lymphocytes
Cytotoxic T lymphocytes (CTLs) are the primary effector cells of the adaptive antitumor immune response. Most CTLs are CD8+ T lymphocytes that have been primed by dendritic cells to recognize and engage tumor antigens on the surface of tumor cells. When a CTL attaches to its target cell, a well-organized immunologic synapse is rapidly formed at the site of cell-to-cell contact and persists for more than an hour. Into this synapse, the CTL releases lytic granules containing perforin, a pore-forming protein, and granzymes, which are serine proteases. Perforin mediates the entry of granzymes into the target cell. Once inside the target cell, granzymes initiate both caspase-dependent and caspase-independent apoptosis. CD4+ T lymphocytes do not appear to be essential for the generation or maintenance of a CTL response; instead, CD4+ lymphocytes are generally considered to function as TH lymphocytes that enhance the function of CD8+ CTLs and antigen-producing B lymphocytes. CD4+ TH lymphocyte activities are largely mediated through secretion of cytokines, such as IL-2, which drives CD8+ cell proliferation, and IFN-γ, which stimulates CD8+ T lymphocyte differentiation.
B Lymphocytes
Many tumor antigens can incite both cell-mediated and humoral immune responses. Antibody-producing B lymphocytes mediate the humoral immune response to tumors. Antibodies that recognize tumor antigens kill tumor cells by binding to the cells and activating a local complement cascade. Activation of the complement cascade generates a membrane attack complex that induces loss of tumor cell membrane integrity and rapid cell death with the morphologic hallmarks of necrosis. In addition, antitumor antibodies may be bound by their constant regions to NK cells or macrophages, leaving the variable regions of the immunoglobulins available for specific recognition of tumor antigens. This allows the effector cells to recognize, attach to, and kill tumor cells by the mechanism of antibody-dependent cell-mediated cytotoxicity (ADCC).
Evasion of the Immune Response
Many tumors are able successfully to evade immunosurveillance, using a variety of mechanisms (Fig. 6-33).
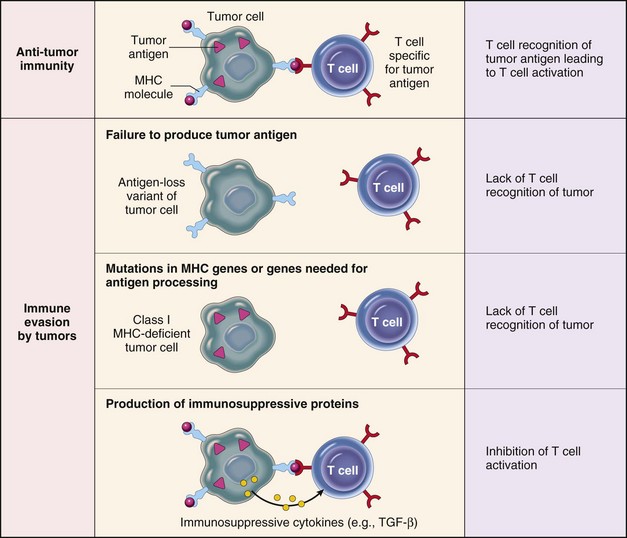
Fig. 6-33 Mechanisms by which tumors evade the immune system.
Tumor cells employ a variety of mechanisms to avoid attack by cytolytic T lymphocytes. MHC, Major histocompatibility complex; TGF-β, transforming growth factor-β. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Altered Major Histocompatibility Complex (MHC) Expression
CTLs recognize tumor antigens only on tumor cells that display the antigens in the context of MHC class I molecules. Thus tumor cells that lose or downregulate expression of class I MHC antigens may have a distinct selective advantage. However, tumors that fail to express class I antigens are more susceptible to NK cell killing. Tumors may also downregulate expression of class II MHC antigens. Class II antigens are required for activation of the TH lymphocytes that stimulate CTL differentiation, and loss of these antigens prevents the generation of an optimal antitumor CTL response.
Antigen Masking
Tumors may become invisible to the immune system by losing or masking tumor antigens. The outgrowth of clonal tumor variants that do not express tumor antigens is favored during tumor evolution. Tumor antigens on the cell surface may be hidden from the immune system if they are complexed with glycocalyx molecules, fibrin, or even antibodies. Some humoral responses to tumor antigens may therefore promote tumor survival by protecting tumor antigens from recognition by CTLs.
Tolerance
Although the immune system responds vigorously to non–self-antigens, it is tolerant to self-antigens. Thus tumor antigens shared with normal tissue usually are not able to evoke an immune response because the body has already been “tolerized” to the antigen. Tolerance can also result from presentation of non–self-antigens in a “tolerogenic” context (i.e., in the absence of co-stimulatory molecules required for effective T lymphocyte activation).
Immunosuppression
Tumor cells or their products may be immunosuppressive. Many tumors produce TGF-α, which inhibits the proliferation and function of lymphocytes and macrophages. Tumors may also produce Fas ligand. Fas ligand expressed by tumor cells binds to Fas receptors on nearby T lymphocytes and triggers their apoptosis. By this mechanism, T lymphocyte clones that recognize a tumor may be specifically deleted. Finally, tumor cells release tumor antigens into the circulation that form immune complexes with antibodies, and these immune complexes may be immunosuppressive.
Tumor Immunotherapy
The fact that the antitumor immune response attacks only the tumor cells and not normal tissue makes it an attractive candidate as a therapeutic modality. Moreover, effective immunotherapy would reduce or eliminate the need to use highly cytotoxic chemotherapeutic agents that indiscriminately target dividing cells. These drugs can cause significant morbidity and mortality in cancer patients. To date, immunotherapy results have been variable and disappointing; however, many new approaches are presently under investigation, some of which show promise.
In general, immunotherapeutic strategies are aimed at (1) providing the patient with mature effector cells or antibodies that recognize and destroy tumors (passive immunotherapy) or (2) stimulating the immune response of the host against the tumor (active immunotherapy).
Administration of monoclonal antibodies raised against tumor antigens or tumor-specific effector lymphocytes generated in vitro generates rapid but short-lived passive tumor immunity. Monoclonal antibodies raised in other species have a limited usefulness because the tumor host may develop an immune response to these antibodies that abrogates their effectiveness. However, coupling toxins to monoclonal antibodies may allow targeted delivery of therapeutic agents to tumor cells. Antitumor lymphocytes are generated by removing lymphocytes from the host or tumor and expanding them in vitro by incubation with IL-2; these autologous immune cells are then readministered to the patient. Many approaches to stimulate the active immunity of patients against their tumors have been attempted. These include vaccination with tumor cells or tumor antigens to generate antitumor CTLs, administration of cytokines to increase effector cell number and function, and nonspecific stimulation of the immune system by treatment with proinflammatory substances, such as bacterial products.
Systemic Effects on the Host
Direct Effects Versus Paraneoplastic Syndromes
Tumors directly compromise the function of the organs in which they arise by replacing normal tissue and by disrupting normal anatomic relationships of affected organs. In both the tissue of origin and in metastatic sites, expanding tumor tissue may compress surrounding normal tissue or the blood vessels that supply this tissue, resulting in pressure atrophy or frank necrosis. This is a particularly severe problem in organs encased in bone, such as the brain, that can expand to only a very limited extent. In the brain, even benign tumors that are not surgically accessible may prove fatal. Seizure activity is a common manifestation of brain tumors. Tumor invasion into the wall of a hollow organ, such as the stomach, may lead to organ rupture. Tumors may erode blood vessel walls, causing acute hemorrhage, whereas tumor extension into blood vessels may lead to the formation of tumor emboli that produce infarcts at distant sites.
In addition to direct effects, tumors may cause a variety of systemic clinical signs termed paraneoplastic syndromes. Paraneoplastic disorders are indirect and usually remote effects that are caused by tumor cell products rather than by the primary tumor or its metastases. Approximately 75% of human cancer patients develop paraneoplastic syndromes, but the incidence in veterinary cancer patients is unknown. These syndromes are best described for the dog, although some affecting the cat and the horse have been reported (Web Table 6-2). Recognition of paraneoplastic syndromes is important for the following reasons: (1) paraneoplastic syndromes may appear early in the course of tumor development and may be associated with specific tumor types, thus recognition of these syndromes can facilitate early tumor diagnosis; (2) treatment of the metabolic abnormalities associated with paraneoplastic syndromes and antitumor therapy may be required to assure effective cancer management; and (3) the severity of paraneoplastic abnormalities reflects the tumor burden; thus monitoring such abnormalities may be useful in determining tumor response to therapy and identifying tumor recurrence or spread.
Cachexia
Many animals with cancer show notable weight loss and debility. In cancer cachexia, both muscle and fat are lost, whereas in simple starvation fat is lost preferentially. Also, in starvation, there is a compensatory decrease in basal metabolic rate not seen in cancer cachexia. The etiology of cancer cachexia is complex. Contributing factors include anorexia, impaired digestion, nutritional demands of tumor tissue, nutrient loss in cancer-related effusions or exudates, and a variety of metabolic and endocrine derangements. Extra calories do not reverse the catabolic state of cancer cachexia. Many humoral factors, including cytokines and hormones, contribute to the development of cachexia. Among the best characterized of these factors are TNF-α, IL-1, IL-6, and prostaglandins.
Web Fig. 6-1 Apocrine gland carcinoma, anal sac, dog.
This tumor is composed of the typical solid lobules of malignant epithelial cells that characterize apocrine gland adenocarcinomas. Apocrine gland carcinoma, lymphoma, and multiple myeloma may be associated with hypercalcemia of malignancy. H&E stain. (Courtesy College of Veterinary Medicine, The Ohio State University.)
Endocrinopathies
A functioning neoplasm of an endocrine tissue produces the normal hormonal products of the tissue of origin, but the host is usually able to regulate this hormone production only to a very limited extent. Frequently, the result is an overproduction of hormone as the result of increased numbers of hormone-producing tumor cells, increased production of hormone by individual neoplastic cells, or both. In endocrine glands with more than one cell type, such as the pancreatic islet, the anterior pituitary, the thyroid, and the adrenal, generally only a single cell type becomes neoplastic. Thus a pancreatic islet cell adenoma probably produces only a single hormone, such as insulin, glucagon, gastrin, or somatostatin, and not a combination of hormones. Several clinically significant endocrinopathies are seen fairly commonly in veterinary medicine. These include hyperthyroidism, caused by thyroid neoplasia in the cat, and Cushing’s disease (hyperadrenocorticism), resulting from pituitary tumors in several species. A variety of nonendocrine neoplasms may also produce hormonally active substances not normally found in the tissue of tumor origin. This is termed ectopic hormone production. The hormone produced may be identical to the normally produced hormone, may be a modified form of the normal hormone, or may be the product of a gene that encodes a protein related to but not identical with the true hormone.
Probably the two most frequently observed metabolic derangements resulting from cancer-associated endocrine abnormalities in animals are hypercalcemia of malignancy and hypoglycemia. Hypercalcemia is seen with functional tumors of the parathyroid gland that produce excess parathyroid hormone, the major regulator of calcium levels in the body. In the great majority of cases, however, hypercalcemia of malignancy is due to ectopic production of parathyroid hormone and parathyroid hormone-related protein by neoplastic tissue, including a wide variety of carcinomas and sarcomas. In dogs, hypercalcemia is seen most frequently with adenocarcinoma of the anal sac (≈90% of cases) (Web Fig. 6-1), lymphoma (≈20% of cases), and multiple myeloma (≈15% of cases). Hypercalcemia of malignancy in cats appears to be relatively rare. Parathyroid hormone and parathyroid-related hormone are encoded by two separate genes. The genes are regulated independently and produce proteins of somewhat different size and sequence. However, the amino terminal 34 amino acids of the two proteins are functionally equivalent. The hormones increase serum calcium by increasing calcium release from bones, enhancing reabsorption of calcium in the kidneys, and stimulating absorption of calcium in the intestine. Clinical signs of hypercalcemia include muscle weakness, cardiac arrhythmia, anorexia, vomiting, and renal failure. Hypercalcemia may also occur as a result of tumor metastasis to bone and resultant bone resorption; however, this is not a true paraneoplastic disorder because it is a direct effect of the tumor.
Hypoglycemia is seen primarily with insulinomas, functioning tumors of the pancreatic islet b cells. However, profound hypoglycemia of unknown origin may also occur with other tumor types. Because of the absolute dependence of the nervous system on glucose for energy, clinical signs of hypoglycemia are mostly neurologic and may include lethargy, incoordination, muscle weakness, and seizures.
Web Fig. 6-2 Hypertrophic osteopathy, forelimbs, dog with a pulmonary tumor.
On this radiograph, the arrows indicate newly deposited bone that is less dense than normal cortical bone. Note that multiple bones on both limbs are affected and that new bone deposits are located primarily in the diaphyseal region of the long bones. (Courtesy Dr. J. Mattoon, College of Veterinary Medicine, The Ohio State University.)
Skeletal Syndromes
Hypertrophic osteopathy occurs in both cats and dogs. It usually presents clinically as a symmetric lameness. Radiographically, there is evidence of extensive periosteal new bone growth (Web Fig. 6-2). Hypertrophic osteopathy occurs with a variety of tumor types, although there is a strong association with space-occupying thoracic lesions, both neoplastic and nonneoplastic. The cause of this condition is not known, although abnormalities of growth hormone production are suspected. Myelofibrosis results from overgrowth of nonneoplastic fibroblasts in the bone marrow. It may be associated with myeloproliferative disease or with distant tumors. The cause is unknown.
Vascular and Hematologic Syndromes
Nonhematopoietic cancer in animals may result in a variety of vascular and hematologic syndromes, including eosinophilia and neutrophilic leukocytosis. The etiology of these conditions is unclear, but circulating cytokines are likely to be involved. Anemia is commonly seen in veterinary cancer patients. There are numerous potential causes for anemia in these animals, including anemia of chronic disease, bone marrow invasion, blood loss, and hemolysis. Polycythemia associated with ectopic production of erythropoietin has been reported. Thrombocytopenia is seen in approximately one third of all dogs with cancer. Disseminated intravascular coagulation (DIC) may be secondary to any large tumor. Anemia and DIC are frequently seen in dogs with hemangiosarcoma, a tumor that accounts for 7% of canine malignancies. Excessive immunoglobulin production by tumors, particularly monoclonal gammopathies caused by multiple myeloma, can result in hyperviscosity syndrome, manifested as altered neurologic function, congestive heart failure, or bleeding disorders.
Neurologic Syndromes
Paraneoplastic neurologic disease in veterinary cancer patients is usually related to hypercalcemia, hypoglycemia, or hyperviscosity and is often manifested as seizure activity. Primary peripheral nervous system disease has also been reported. In many dogs with cancer, there is microscopic evidence of peripheral neuropathy; however, clinical signs of disease, such as areflexia, muscle weakness, reduced muscle tone, or paralysis, are much less common. Myasthenia gravis is occasionally seen in veterinary cancer patients, usually in association with a mediastinal tumor such as thymoma. The underlying defect in myasthenia gravis is a failure of nerve impulse transmission at neuromuscular junctions. Clinical signs include muscle weakness, reduced exercise tolerance, and dysphagia. Many neurologic paraneoplastic syndromes in humans are immune mediated, and this is likely to be the case in animals as well.
Cutaneous Syndromes
There are only a few reports of cutaneous manifestations of paraneoplastic disease in dogs and cats. Clinical signs of flushing, alopecia, or necrolytic dermatitis have been associated with a variety of tumor types. The syndrome of nodular dermatofibrosis in German shepherd dogs is a heritable disorder characterized by multiple benign-appearing fibrous nodules in the skin in conjunction with bilateral renal cystadenocarcinomas.
Miscellaneous Syndromes
Gastrin-secreting tumors have been reported in dogs and cats. These tumors can cause gastroduodenal ulceration, abdominal pain, vomiting, and blood loss. Mast cell tumors are very common in dogs. Neoplastic mast cells produce a wide variety of biologic mediators, including histamine, heparin, platelet-activating factor (PAF), TNF-α, prostaglandins, and proteases. Release of these compounds, particularly histamine, is responsible for such paraneoplastic disorders as gastrointestinal ulceration and hemorrhage. Although widespread mastocytosis is frequently associated with clinical signs, cutaneous mast cell tumors rarely produce systemic signs.
Genetics and Cancer
Cancer is ultimately caused by heritable changes in the DNA of the tumor cells. These changes are manifested in enhanced expression of normal proteins, decreased or absent expression of normal proteins, and expression of abnormal proteins. For instance, tumor suppressor proteins, such as those encoded by p53, may fail to be expressed or may be expressed in an inactive form. Oncogene-encoded proteins may be overexpressed or expressed in constitutively active form. The altered protein repertoire of the tumor cell determines its phenotype. Such changes create cells with the features of neoplasia, including unlimited proliferative potential, growth factor independence, resistance to apoptosis, and invasive capabilities.
Genetic alterations in the coding region of a gene clearly may result in the production of an altered protein as a result of an altered coding sequence or premature protein termination. However, changes in other regions of the genome may also affect the sequence of the encoded protein and the level at which it is expressed. An alteration in splice site sequence can yield incorrectly spliced mRNA that is translated into an aberrant protein. Because the 5′ untranslated region of a gene generally contains its promoter and the 3′ untranslated region often contains messenger RNA (mRNA) stabilization motifs, sequence alterations in these regions of the genome may also have profound effects on gene expression level.
Stepwise Accumulation of Changes
Cancer is associated with a progressive accumulation of genetic and epigenetic abnormalities. These defects lead to disruption of cell growth, cell death, apoptosis, differentiation, DNA repair, and other critical pathways. Some model systems demonstrate an orderly morphologic progression through premalignant to malignant to invasive and metastatic disease, and molecular genetic investigations of these various stages have made important contributions to our understanding of cancer biology. The molecular events that occur in the development of familial adenomatous polyposis, a form of human colorectal cancer, provide an excellent example of the genetic evolution that underlies progressive morphologic changes in cancer (Fig. 6-34). The initiating event is loss or mutation of the adenomatous polyposis coli (APC) tumor suppressor gene, leading to adenoma formation. This is followed by an activating mutation of a RAS oncogene and loss of genetic material harboring additional tumor suppressor genes. Ultimately, a malignant carcinoma emerges.
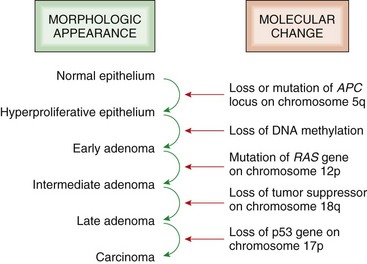
Fig. 6-34 Molecular model for the evolution of human colorectal cancers through the adenoma-carcinoma sequence.
Although adenomatous polyposis coli (APC) mutation is an early event and loss of p53 occurs late in the process of tumorigenesis, the timing for the other changes may show variations. Note also that individual tumors may not have all of the changes listed. (Adapted from Vogelstein B, Kinzler KW: Colorectal tumors. In Vogelstein B, Kinzler KW, editors: The genetic basis of human cancer, New York, 2002, McGraw-Hill.)
As illustrated in Fig. 6-35, DNA is susceptible to many types of chemical and physical alterations. Some of these changes are caused by injurious endogenous and exogenous agents. In addition, DNA alterations occur as part of normal processes of genome replication, repair, and rearrangement. Under some circumstances, these normal processes can lead to changes in DNA structure that contribute to the neoplastic phenotype. Furthermore, as tumors evolve, they often demonstrate reduced DNA repair capabilities and increased genomic instability. These developments accelerate the accumulation of genetic alterations in tumor cells.
Fig. 6-35 Heritable alterations contributing to carcinogenesis.
Many genetic changes caused by extrinsic and intrinsic DNA-damaging agents, normal physiologic processes, and aging alter the amino acid sequences of encoded proteins and the levels at which these proteins are expressed. These changes in turn are responsible for the neoplastic phenotype. (Redrawn with permission from Dr. D.F. Kusewitt, College of Veterinary Medicine, The Ohio State University.)
Why do we study the molecular basis of cancer? Cancer is often treated using cytotoxic drugs and/or radiation therapy, neither of which discriminates between normal and tumor cells. Nonselective cell killing is responsible for many of the deleterious side effects of cancer treatment. More rational therapies would specifically target molecularly altered tumor cells, leaving nontumor cells lacking the molecular defect(s) relatively unharmed. Examples of molecularly targeted therapies used in humans include Gleevec, which is active against the BCR/ABL chromosomal translocation in chronic myeloid leukemia, and Iressa, which targets the epidermal growth factor receptor (EGF-R) in lung cancer. Specific molecular defects can also be exploited for early detection and/or early intervention at a stage when the tumor may be more responsive to treatment. Mutations in the BRCA1 gene are associated with a high risk for development of breast and/or ovarian cancer in women. Identification of the carrier status gives women the option of prophylactic mastectomy and/or oophorectomy. Mutations and other molecular defects can also be used to stratify patients for treatment and/or prognostic purposes.
Genetic Changes in Cancer
DNA damage, per se, does not constitute mutation. Mutation occurs during DNA replication, when the DNA strand containing unrepaired or misrepaired DNA damage is used as a template for the synthesis of a complementary DNA strand (see later discussion). At this time, unrepaired DNA lesions are misread by DNA polymerases, which insert incorrect bases in the newly synthesized DNA strand, and incorrectly repaired DNA lesions are reproduced in the daughter DNA strand. This process is known as mutation fixation; at least one and sometimes two rounds of replication are required for mutations to become fully fixed in the genome.
A chemical or physical change in an individual base may lead to the substitution of one base for another. The point mutation thus created can lead to altered protein sequence, if it is located in an exon or at a splice site, and to altered levels of expression, if it is located in the promoter or in the mRNA stabilization motif of a gene. Single- and double-strand breaks in DNA are caused by physical and chemical agents and viruses; they may also occur during normal physiologic processes such as recombination of immunoglobulin genes and T lymphocyte receptor genes. Although single-strand breaks are usually readily repaired, they can in some circumstances lead to gene conversion, the replacement of a gene or part of a gene by DNA derived from a closely related gene. Gene conversion is one mechanism by which organisms routinely generate diversity in large families of related genes, for example, MHC antigen genes. Double-strand breaks produce unprotected, recombinogenic DNA ends and often lead to major chromosomal anomalies, including deletions and translocations. Clearly, such large-scale chromosomal changes have the potential to alter the gene expression repertoire of a cell in a dramatic fashion. Insertions of DNA bases into the genome may be as small as a single base or larger than a viral genome. Insertion of individual bases by the enzyme terminal deoxynucleotide transferase is part of the system by which antibody diversity is normally generated. Retroviral genomes replicate only after insertion into the host genome, and these large insertions can interrupt the coding sequence of host genes, abrogating their expression. On the other hand, juxtaposition of viral promoter elements adjacent to cellular coding sequences can lead to dysregulated, often markedly increased, host gene expression. Unscheduled amplification of DNA segments is a poorly understood process by which multiple rounds of localized DNA replication produce hundreds or thousands of copies of DNA segments up to several megabases in length. Such amplified DNA segments may be visible in cytogenetic preparations as homogeneously staining regions in chromosomes or as episomal double minutes. Amplification of oncogenes or drug resistance genes may have significant effects on the life history of a tumor. Expansion or contraction of a small region of tandemly repeated DNA sequences can occur as the result of DNA polymerase slippage during replication. This process can introduce sequence alterations into protein encoding genes.
Chromosomal Instability
The karyotypes of many tumor cells are extremely abnormal because of changes in chromosome number and configuration. These alterations are the result of chromosomal instability. In tumors with notable chromosomal instability, each cell may have a different karyotype and exhibit a remarkable array of duplications, deletions, and reciprocal and nonreciprocal translocations. Such abnormalities can dramatically affect the levels of gene expression by altering the numbers of gene copies present and by juxtaposing regulatory and structural elements of unrelated genes. In some cases, specific chromosomal abnormalities are associated with specific disease entities. Probably the best studied example of such an association is the relationship between chronic myeloid leukemia in humans and a reciprocal translocation between chromosomes 9 and 22 that yields an abnormal chromosome called the Philadelphia chromosome. This translocation fuses portions of the BCR and ABL genes; the fusion gene thus produced encodes an abnormal protein that appears to be responsible for neoplastic transformation of myeloid cells.
Types of Genetic Alterations in Cancer Cells
Aneuploidy: The numeric aberrations in cancer can involve many different chromosomes or be limited to a single chromosome. Cytogenetic analysis can determine the copy number of each chromosome. Monosomy is the term used when only one copy of a chromosome exists. Trisomy is the term used when three copies of a chromosome exist. For example, one quarter of canine lymphomas show trisomy of chromosome 13. In mice, trisomy of chromosome 15 occurs in almost all T lymphocyte lymphomas and leukemias, suggesting that overexpression of a gene or genes on this chromosome plays an important role in tumor causation. Alterations in chromosome number (aneuploidy) are largely the result of mistakes in chromosome segregation caused by multipolar spindles, centrosome amplification, kinetochore malfunction, or abnormal cytokinesis.
Translocation: Translocations often occur when the cell response to DNA damage is dysregulated. When cells respond appropriately to DNA damage, multiple signaling cascades such as the p53 pathway are activated, leading to cell-cycle arrest, enhanced DNA repair, and apoptosis in the face of excessive DNA damage. However, if these pathways are no longer functional, chromosomal instability results. Dysfunctional telomeres also contribute to chromosomal instability (Fig. 6-36). The precise mechanisms by which an intact DNA damage response and normal telomerase activity maintain chromosomal integrity are unclear.
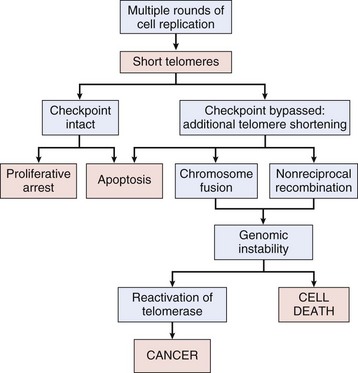
Mutation: In addition to abnormal numbers of chromosomes, mutations (heritable change in the DNA sequence) are also present in most tumors. Mutations can result in loss or inactivation of a protein. On the other hand, some mutations cause uncontrolled activation of a protein. One example is a point mutation of the RAS oncogene. This mutation causes constitutive and unregulated activation of the protein and gives the affected cell a growth advantage over surrounding normal cells. Another type of mutation is a dominant-negative mutation, in which the mutant gene product causes inactivation of the wild-type product, effectively eliminating the cellular contribution of the normal gene. A missense mutation that changes a codon for one amino acid into a codon for a different amino acid has been identified in the Birt-Hogg-Dubé- (BHD) gene in German shepherd dogs with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis (see later discussion).
Deletion: Deletions involve loss of DNA from a chromosome and can be as small as one base pair or as large as an entire arm of a chromosome. Heterozygous deletions occur on only one chromosome, whereas homozygous deletions occur on both chromosomes. Small deletions of one or two base pair cause a shift in the reading frame during protein synthesis (frameshift mutation). One possible consequence is that the mutated sequence codes for a premature stop codon, resulting in a truncated protein. Large deletions often encompass chromosome regions that harbor tumor suppressor genes.
Amplification: Genomic amplifications result in the presence of more than one diploid copy of a DNA sequence. The amplified region can involve large segments of a chromosome and encompass millions of base pairs. Alternatively, the amplified region may be very small and contained within a portion of a single gene such as the internal tandem duplication (ITD) of the c-kit gene in canine mast cell tumors. Regions of genomic amplification are useful for identifying potential oncogenes.
Epigenetic Mechanisms of Altered Gene Expression
Definition of Epigenetic: Epigenetic alterations have recently come to light as being major players in tumor biology. The term epigenetic refers to a heritable change in gene expression in somatic cells resulting from something other than a change in the DNA sequence. The most frequently studied epigenetic changes are DNA cytosine methylation and histone modifications. These epigenetic modifications can enhance or suppress gene expression and can be transmitted to daughter cells during cell division. Although DNA methylation and histone modifications are carried out by normal cellular enzymes, the activity and specificity of these enzymes can be altered by exogenous agents acting through a variety of oncogenic pathways. And although epigenetic changes are stable for the most part, they can be modulated with various pharmacologic agents. This quality makes them attractive targets for therapeutic intervention designed to restore gene expression to its normal state.
Types of Epigenetic Changes in Cancer Cells:
DNA Methylation: DNA methylation involves the addition of a methyl group to carbon 5 of cytosine, in cytosines located immediately 5′ to guanine (CpG dinucleotide). The methylation process is carried out by various methyltransferase enzymes. Methylation is essential for regulating gene expression in normal cells. Cancer cells, however, are characterized by an overall decrease in 5-methylcytosine in the bulk of the genome (global hypomethylation), with a paradoxic increase in gene-specific methylation of clusters of CpG sites located in the promoter and/or exon 1 of genes (CpG islands). In general, hypomethylation of genes, particularly of promoter regions, leads to gene activation, whereas hypermethylation results in gene silencing. Promoter methylation has been found in every type of human cancer studied (Fig. 6-37).
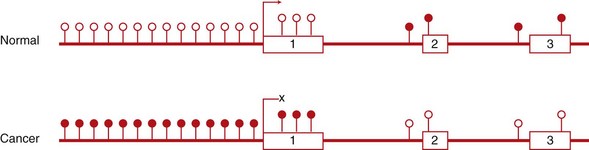
Fig. 6-37 CpG island methylation.
In most normal tissues, the dense clusters of CpG sites in the 5′ regions of genes (CpG islands) are unmethylated (open lollipops), whereas those in the body of the gene are methylated (filled lollipops). The reverse is often seen in cancer where 5′ CpG islands become hypermethylated, and there is concurrent hypomethylation of CpG sites in the body of the gene. Unmethylated 5′ CpG islands are associated with active transcription (arrow), whereas methylated 5′ CpG islands are associated with transcriptional repression (x). (Redrawn with permission from Dr. L.J. Rush, College of Veterinary Medicine, The Ohio State University.)
Imprinting: Another situation in which methylation plays a role is in imprinting. Genomic imprinting refers to the parent-of-origin, allele-specific expression of certain genes whereby only the maternal or paternal allele is expressed. This monoallelic expression is controlled in part by DNA methylation, but this regulation is sometimes lost in cancer. The loss of imprinting can allow a double dose of a growth-promoting gene product. For example, insulin-like growth factor-2 (IGF-2) is an imprinted gene that is expressed from only the paternal allele in most normal tissue. If a cancer cell undergoes relaxation of imprinting, the methylation-mediated silencing of one allele is lost, enabling biallelic expression and higher than normal levels of this growth-promoting gene product.
Histone Modification: In addition to DNA methylation, a second type of epigenetic regulation involves histone modification. DNA is wound around histones to form chromatin. Chromatin can exist in an open configuration in which DNA is accessible to transcription factors (euchromatin) or in a closed, compact configuration in which DNA is inaccessible to transcription factors (heterochromatin). Posttranslational modifications, such as acetylation, methylation, and phosphorylation, take place on the histone tails. In the aggregate, these modifications form the “histone code,” which plays an important role in determining gene expression. For example, the addition of a negatively charged acetyl group (CH3CO−) to certain lysine residues in a histone tail results in a weaker bond between the DNA and the histone. This allows a relaxed chromatin configuration and increases transcription of the associated gene (Fig. 6-38).
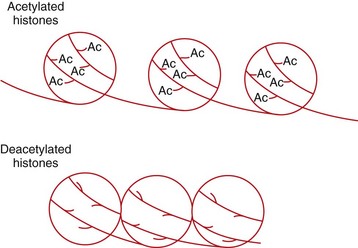
Fig. 6-38 Histone acetylation.
DNA is wound around histones. The presence of acetyl groups (Ac) on histone tails is associated with relaxed chromatin, which is permissive to gene transcription. Removal of acetyl groups by histone deacetylases results in a closed chromatin configuration that is less permissive to the transcription machinery. (Redrawn with permission from Dr. L.J. Rush, College of Veterinary Medicine, The Ohio State University.)
Posttranscriptional Regulation of Gene Expression by MicroRNAs
MicroRNAs (miRNAs) are small noncoding RNA molecules that posttranscriptionally regulate the expression of other genes. MiRNA genes are found throughout the genome, both within other genes and in intergenic regions. MiRNA genes are transcribed into large precursor RNAs that undergo extensive enzymatic processing within the nucleus and after export into the cytoplasm. Mature miRNAs are 18 to 25 nucleotides in length and bind to their target mRNAs as the result of sequence complementarity. Once bound, miRNAs either trigger degradation of their target mRNAs or prevent translation of these mRNAs into proteins.
Approximately 1000 miRNA genes are present in the human genome, and each miRNA can regulate translation of about 200 target mRNA species. Overall, miRNAs control translation of perhaps one-third of all protein-coding genes in the human genome. The pattern of miRNA expression is extensively dysregulated in cancers by a wide variety of genetic and epigenetic mechanisms. Altered patterns of miRNA expression in turn create extensive changes in cellular processes related to neoplasia, including proliferation, apoptosis, invasiveness, and genomic stability. Current research is focused on understanding how altered patterns of miRNA expression can be exploited for diagnostic and therapeutic purposes.
Cancer Etiology
The genetic alterations that contribute to cancer development include both heritable changes in germline sequences present in all cells within an organism and somatic changes that accumulate in individual cells and tissues of the body over time. Human families and genetically related animals that exhibit distinct Mendelian inheritance of specific types of cancer are said to have cancer syndromes. In these cases, a single germline mutation is generally responsible for tumor development. In contrast, sporadic tumors occur randomly in the population and are not associated with specific germline characteristics.
Germline Mutations and Cancer Syndromes
Heritable genetic lesions undergo germline transmission from one generation to the next, and the affected individual is born with one defective copy of a gene in each cell. Heritable mutations may be linked to familial cancer syndromes. Characteristics of heritable familial cancers include an early age of onset compared with sporadic cases, the formation of bilateral tumors in paired organs, the occurrence of multiple primary tumors in nonpaired organs, and a family history of cancer. Although many of the familial cancer syndromes are the result of mutations in recessive tumor suppressor genes, the syndromes generally show a predominantly autosomal dominant pattern of inheritance. This apparent paradox is explained later in the discussion of hereditary retinoblastoma. Some cancer syndromes show a recessive mode of inheritance. In such syndromes, the affected individual must inherit the genetic defect from both parents. For instance, the mutant ter gene carried by strain 129/Sv-ter mice confers high susceptibility to testicular teratoma when present in the homozygous state but not when carried in the heterozygous condition. Interestingly, the role of germline mutations in oncogenes in hereditary cancer syndromes remains unclear.
The seminal work that identified the retinoblastoma (RB) tumor suppressor gene came about through the study of such inherited cancer syndromes (see later discussion). Other well-known inherited cancer syndromes in humans include germline mutations of p53 in Li-Fraumeni syndrome, mutations of NF1 and NF2 that lead to neurofibromatosis, mutations of BRCA1 and BRCA2 associated with breast and ovarian cancers, and mutations in MEN1 and RET, which are associated with multiple endocrine neoplasia. A well-documented veterinary cancer syndrome is the disease with the unwieldy name “hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis” that occurs in the German shepherd dog. This disease is characterized by bilateral and multifocal renal tumors, uterine leiomyomas, and nodules in the skin (dermatofibrosis). The gene responsible has been mapped to a locus homologous to the human BHD locus; mutations in BHD cause a phenotypically similar human disease.
Although the inherited cancer syndromes account for less than 10% of all human cancers, they have led to the identification of a number of important tumor suppressor genes that also play a significant role in sporadic tumors. In dominant cancer syndromes, tumor risk is very markedly increased because of changes in a single gene; however, there are also a variety of tumor susceptibility genes that alter cancer incidence to a considerably lesser extent. A variety of cancer susceptibility patterns have been identified in the dog as shown in Table 6-2. The tumor susceptibility genes identified to date generally encode proteins involved in carcinogen metabolism or DNA repair. Their effects are substantially modified by interactions with each other and with the environment. For instance, the lack of pigmentation in white cats contributes to their susceptibility to squamous cell carcinoma of the ears; however, sun exposure makes a major contribution to tumor incidence in these animals.
TABLE 6-2
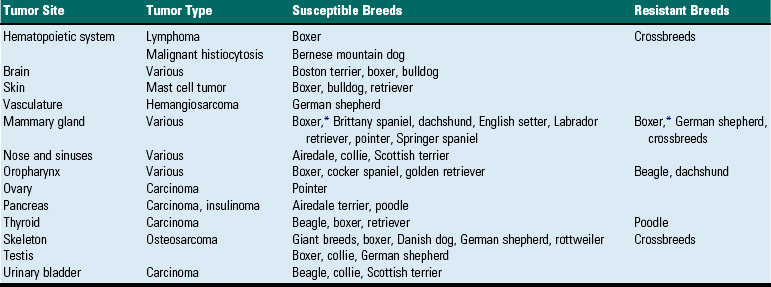
*Information obtained from two independent studies.
Modified from McCullen JM, Page R, Misdorp W: An overview of cancer pathogenesis, diagnosis, and management. In Meuten DJ, editor: Tumors in domestic animals, Ames, 2002, Iowa State Press.
Acquired Somatic Mutations
In contrast to germline mutations, acquired mutations occur in somatic cells and are restricted to individual cells and their progeny. These mutations are not passed through the germline but are required in the specific cells that give rise to the tumor. Somatic genetic alterations are caused both by intrinsic and extrinsic agents. These changes accumulate randomly over time, thus aging dramatically increases the risk of cancer. The graph shown in Fig. 6-39 illustrates the increased risk of cancer with age in the human population, for which solid epidemiologic data are available. Somatic mutations are responsible for the majority of cancers in humans and probably in animals.
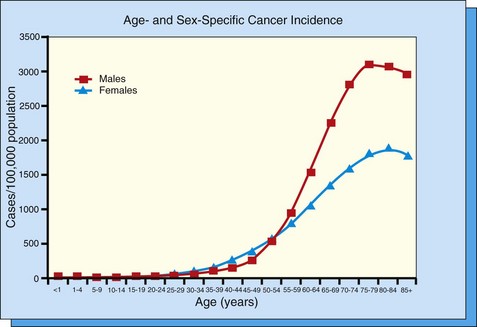
Fig. 6-39 Cancer incidence by age in the human population of the United States (US) for the year 2001.
Cancer incidence is shown as the number of cases of invasive cancer per 100,000 persons. Data for all races and all cancer sites are combined. Basal and squamous cell carcinomas of sun-exposed skin and all in situ cancers except urinary bladder cancers are excluded. Data are from selected state and metropolitan area cancer registries and cover approximately 92% of the US population. (Compiled by Dr. D.F. Kusewitt from data released by the Centers for Disease Control and Prevention.)
Intrinsic Factors
As a by-product of ordinary metabolism, a variety of DNA damaging agents are produced, including reactive oxygen species. Moreover, DNA is susceptible to ordinary hydrolytic attack. These processes induce a variety of DNA lesions that can result in mutation. Additionally, in the course of many rounds of replication, DNA changes are introduced as a result of DNA polymerase errors. Illegitimate recombination, inappropriate nucleotide addition, and altered DNA methylation, activities carried out by normal cellular enzymes, can lead to the accumulation of DNA alterations. Chromosomal abnormalities also arise as a result of decreased telomere length, altered telomerase activity, and mistakes in chromosome segregation.
Extrinsic Factors
Extrinsic factors that interact with DNA to cause cancer include chemical, physical, and infectious agents. Mutagens are agents that create DNA damage that gives rise to mutations, whereas carcinogens are agents that cause cancer. Many mutagens are also carcinogens. However, there are carcinogens with unknown mechanisms of action; such carcinogens may or may not be mutagens.
Chemicals: Chemical carcinogens are widespread in the environment. All animals are exposed to low levels of carcinogens in air, water, food, and medications; accidental exposure to very high levels of carcinogens occasionally occurs. One notable chemical cause of cancer in animals is the bracken fern plant, the toxin of which causes urinary bladder cancer in cattle grazing pastures containing the plant. Unlike humans, however, animals do not voluntarily expose themselves to potent carcinogens such as tobacco smoke.
Radiation: Natural sources of radiation include the sunlight and cosmic rays that bombard the earth and naturally occurring radioisotopes, such as radon, in the soil. Medical procedures, including diagnostic radiographs and radiation therapy, can provide substantial radiation exposure. Radiant energy with wavelengths greater than 5 × 10−5 cm (visible light, infrared radiation, microwaves, radio waves, and electrical waves) is generally considered to be noncarcinogenic. However, shorter wavelength γ-rays, x-rays, and ultraviolet (UV) radiation can cause notable DNA damage and resultant mutation. In addition, high-energy particles, including α-particles (helium nuclei), β-particles (electrons), and neutrons, can be carcinogenic. Ionizing radiation includes those forms of radiation with enough energy to eject electrons from target molecules (γ-rays, x-rays, α-particles, β-particles, and neutrons). Because most animals have hair, UV radiation is generally not an important veterinary carcinogen. There are, however, two instances in which the UV radiation in sunlight poses a significant risk to animals: squamous cell carcinoma of the ears in white cats and ocular squamous cell carcinoma in Hereford cattle with nonpigmented eyelids.
Viruses as a cause of cancer were first identified and have been most thoroughly studied in animals. Virally induced tumors in animals often affect a relatively large number of animals in a flock or herd, frequently arise in relatively young animals, and generally show a pattern of occurrence consistent with an infectious cause. There are many types of oncogenic viruses that induce cancer by a large variety of mechanisms. Web Table 6-3 summarizes those viruses of significance to veterinary medicine. Oncogenic retroviruses are small RNA viruses that cause many neoplastic diseases of importance in animals, including avian leukosis, feline leukemia, and bovine leukosis. Oncogenic DNA viruses include papillomaviruses that cause skin cancer, hepadnaviruses associated with liver cancer, and herpesviruses that induce adenocarcinomas, lymphomas, and leukemias. Species of Helicobacter bacteria have been recently demonstrated to play a role in gastric carcinoma and gastric lymphoma in ferrets and mice and in hepatocellular carcinoma in mice.
WEB TABLE 6-3
Modified from Wyke J: Viruses and cancer. In Franks LM, Reich NM, editors: Cellular and molecular biology of cancer, New York, 2003, Oxford University Press.
Mechanisms of Carcinogenesis
A very wide variety of chemicals can cause cancer. Direct-acting chemical carcinogens are effective in the form in which they enter the body, but most carcinogens are procarcinogens that require metabolic activation by cellular enzymes to form ultimate carcinogens. These are thus termed indirect-acting carcinogens. Despite their varied composition, the effective form of most carcinogens is an electrophilic compound that binds covalently to DNA to form DNA adducts. Fig. 6-40 illustrates the general mechanism of chemical carcinogenesis.
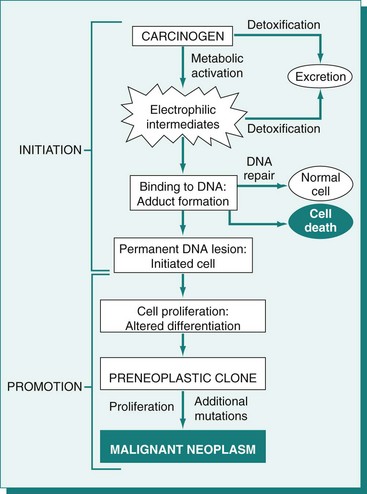
Fig. 6-40 General schema of events in chemical carcinogenesis.
Note that promoters cause clonal expansion of the initiated cell, thus producing a preneoplastic clone. Further proliferation induced by the promoter leads to formation of a benign neoplasm. Accumulation of additional mutations during the phase of tumor progression results in emergence of a malignant tumor. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
As discussed previously, experimental carcinogenesis studies have been critical in elucidating the stepwise development of cancer. Moreover, these studies have clearly defined the contribution of initiating versus promoting agents to cancer development (Fig. 6-41). In multistage tumorigenesis models, such as the skin carcinogenesis model in mice, the initiator must be administered before the promoting agent. The initiator is ineffective without subsequent application of a promoter. Moreover, multiple, closely spaced promoter treatments are required to drive tumor emergence.
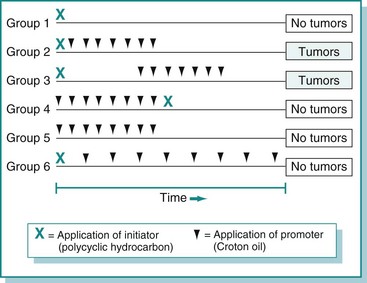
Fig. 6-41 Experiments demonstrating the initiation and promotion phases of chemical skin carcinogenesis in mice.
Tumors arose only if application of an initiator was followed by multiple applications of a promoter. For group 2, application of the promoter was repeated at twice-weekly intervals for several months. For group 3, application of the promoter was delayed for several months and the promoter was then applied twice weekly. When the promoter was applied at monthly rather than twice-weekly intervals (group 6), it did not effectively promote tumor emergence. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Radiation
Unlike chemicals, all forms of radiation are complete carcinogens (i.e., they are able both to initiate and with continued exposure, to promote tumorigenesis). Direct DNA damage caused by ionizing radiation consists primarily of single- and double-strand breaks and base elimination. However, ionizing radiation also generates free radicals from many cellular molecules. The interactions of these highly reactive molecules result in many forms of DNA damage, including altered bases and DNA-protein cross-links. Absorption of UV radiation by DNA results in the formation of hallmark DNA adducts, pyrimidine dimers, which are potentially mutagenic. To a lesser extent, UV radiation generates reactive oxygen species, leading to indirect DNA damage like strand breaks and DNA-protein cross-links.
Viral
Oncogenic viruses employ a remarkable array of direct and indirect mechanisms to induce cancer.
Dominant Oncogene: The genomes of many rapidly transforming viruses include a dominant oncogene that drives tumor development. Oncogenes may be of ancient or more recent host cell origin such as the fes, fgr, abl, fms, and kit genes transduced by oncogenic retroviruses of cats. Viruses may also contain oncogenes not of host cell origin. For example, papillomavirus genomes include the E6 and E7 genes that encode inhibitors of the p53 and pRb tumor suppressor proteins, respectively; some viruses contain genes that encode growth stimulatory molecules such as the small T antigen of SV40.
Insertional Mutagenesis: Viruses that do not possess their own oncogenes can instead activate expression of cellular oncogenes by a process of insertional mutagenesis. For example, most tumors caused by the avian leukosis virus exhibit a similar site of viral insertion, suggesting that virally induced alterations in genes at that site in the host genome are essential for tumorigenesis.
Hit and Run Mechanism: In the two mechanisms discussed previously, the viral genome or portions of the genome persist in the host cell. However, some viruses also cause tumors merely by transient residence in target cells. The precise mechanism by which this might occur has not been elucidated. Bovine papillomavirus uses such a hit and run mechanism of cell transformation.
Indirect Mechanisms: Viruses may also stimulate tumorigenesis by suppression of the host immune system or stimulation of target cell proliferation. The herpesvirus that causes Marek’s disease is an example of a virus that suppresses the ability of the host to eliminate transformed cells, whereas hepatitis viruses exemplify viruses that stimulate cell proliferation.
Molecular Determinants of Cancer
Fig. 6-42 illustrates the key steps in the molecular basis of cancer.
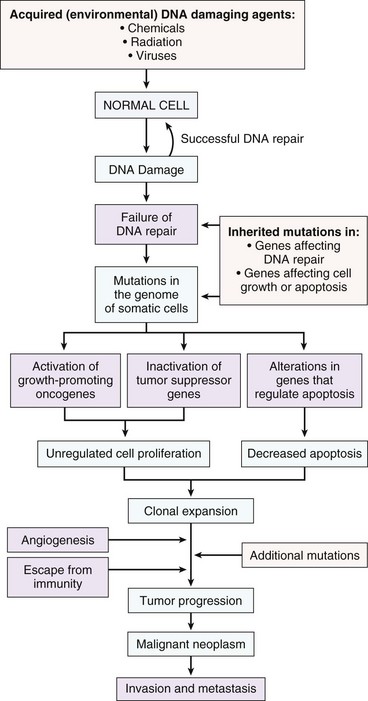
Fig. 6-42 Schematic diagram illustrating the molecular basis of cancer. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Tumor Suppressor Genes
Cells receive signals that both stimulate and inhibit cell growth and must maintain the proper balance between these stimuli. The designation of tumor suppressor gene was originally given to genes that inhibited cell growth. Over time, the class of tumor suppressor genes has expanded to include many different types of cancer-related genes that, when inactivated through genetic and/or epigenetic means, allow uncontrolled proliferation or tumor growth. This includes genes that control cell cycle, apoptosis, DNA repair, and other fundamental pathways.
The pivotal concept of tumor suppressor genes was posited by Alfred Knudson in 1971, based on his observations of children with familial and sporadic retinoblastoma, an uncommon tumor arising in retinoblasts. According to Knudson’s “two-hit” hypothesis, both alleles of a tumor suppressor gene must undergo mutation (i.e., a genetic “hit”) for cancer to develop. In inherited cancer syndromes, the person is born with a mutation in one allele of all cells in the body (Web Fig. 6-3). The second hit is acquired as a somatic mutation of the remaining tumor suppressor allele in a single cell. In contrast, development of a sporadic tumor in those born with two normal tumor suppressor alleles requires that a cell sustain two hits, one on each allele of the tumor suppressor gene. Loss of both alleles in a single cell is a rare event; however, loss of only one allele occurs much more frequently. In cells with only a single copy of a tumor suppressor gene, loss of the second allele can occur by a variety of mechanisms, including point mutation in the allele, deletion of the allele or the chromosomal segment where it resides, deletion of the entire chromosome containing the allele, or mitotic recombination resulting in replacement of the normal allele by the mutant allele. When the normal allele has been completely lost, the cell and its progeny contain only a single abnormal allele. This condition is termed loss of heterozygosity.
Web Fig. 6-3 Schematic illustration of the pathogenesis of retinoblastomas (RBs) and the “two hit” hypothesis.
Two mutations of the RB gene lead to neoplastic proliferation of the retinal cells. In the familial form (left), all somatic cells have one mutant copy of the RB gene inherited from a carrier parent. The second mutation affects the RB locus in one of the retinal cells after birth. In the sporadic form (right), both RB mutations are acquired by the retinal cells after birth. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Tumor suppressor genes are frequently located in areas of the genome that are deleted in cancer. The areas of chromosomal loss can be limited to small deletions of a few hundred base pairs or encompass a whole chromosome arm or even an entire chromosome. By examining a large number of tumors, the minimal region of loss can be defined as the common region deleted in all tumors of the type under investigation. Candidate tumor suppressor genes within the boundaries of this area can then be further interrogated for silencing of the remaining allele. In addition to loss of genetic material and inactivating mutations, it is now widely recognized that DNA methylation, as discussed earlier, is an alternative method of silencing tumor suppressor genes.
Although the classic definition of a tumor suppressor gene dictates that both alleles must be inactivated, recent evidence suggests that for certain genes inactivation of only one copy is sufficient for tumor growth (haploinsufficiency). Haploinsufficiency can contribute to tumor development by a number of mechanisms. One mechanism is a simple gene-dosage effect in which half the normal amount of a protein is insufficient to maintain the normal homeostatic balance in the cell. Alternatively, a mutation in one allele can give rise to a dominant-negative protein whereby the abnormal protein blocks the function of the normal protein produced by the remaining wild-type allele. A third mechanism occurs when a mutation in one allele of a tumor suppressor gene decreases the amount of normal protein in a cell, which may in turn influence other pathways that may have concurrent abnormalities.
Many tumor suppressor genes are key components of the cell cycle. One of the most widely studied tumor suppressor genes is p53, and it is inactivated most commonly by mutation in more than half of human cancers. It is a DNA-binding protein that regulates transcription of numerous genes and plays a critical role in cell-cycle arrest and induction of apoptosis after DNA damage. For these reasons, it has been entitled the “guardian of the genome.” Levels of p53 are rapidly elevated in response to DNA damage. This leads to increased transcription of p53 target genes, such as p21 and GADD45, which function in cell-cycle arrest and DNA repair, respectively. The half-life of p53 is short (about 20 minutes) and it is rapidly degraded after completion of DNA repair. If the DNA repair is unsuccessful, p53 directs cell death by activating BCL2-associated X protein (BAX), a key component of the apoptotic cascade (Fig. 6-43). It is clear then that loss of functional p53 can have devastating consequences for maintaining integrity of the genome. Without p53, mutagenic DNA damage goes unrepaired, the cell proceeds through division, and the mutation becomes fixed in the genome.
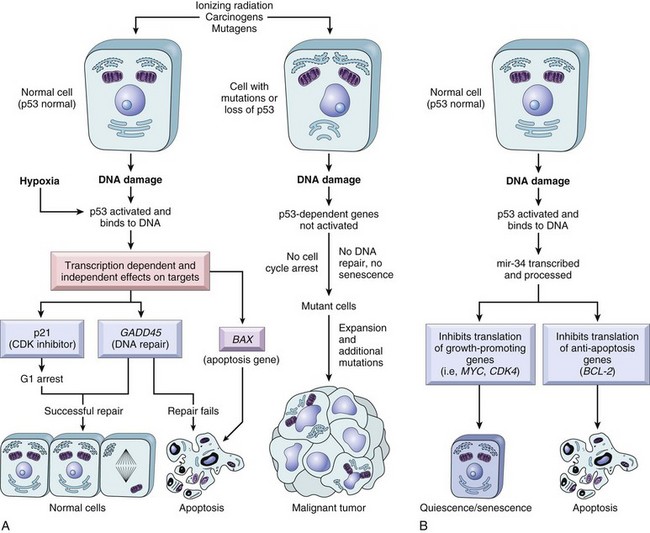
Fig. 6-43 The role of p53 in maintaining integrity of the genome.
DNA damage or hypoxia activate normal p53, leading to G1 arrest and induction of DNA repair by upregulation of p21 and GADD45, respectively. Successful DNA repair allows cells to proceed through the cell cycle. If DNA repair fails, BCL2-associated X protein (BAX) promotes apoptosis. In cells with loss or mutation of p53, DNA damage does not induce cell-cycle arrest or DNA repair. The genetically damaged cells proliferate and may eventually give rise to malignant neoplasms. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Another widely studied tumor suppressor gene is the retinoblastoma gene. As described earlier, RB is a target for inactivation both in germline and somatic cells and like p53, plays an important role in cell-cycle control. In quiescent cells, RB is hypophosphorylated and active. When RB is hyperphosphorylated and inactive, the cell can proceed through the G1/S transition. Control of proper RB function and phosphorylation status involves a host of other genes, including p16INK4a, cyclin D, and CDK4. Therefore a defect in any one of these gene products disrupts the RB pathway and leads to unregulated progression through the cell cycle.
Oncogenes
Oncogenes are derived from proto-oncogenes, normal cellular genes that regulate cell growth and differentiation. Proto-oncogenes encode products involved in diverse pathways and include molecules such as growth factors and their receptors, cell-cycle regulators, DNA-binding proteins, transcription factors, protein kinases, and others. Oncogenes were first identified as RNA tumor virus proteins. Later it was discovered that normal cells have the equivalent gene products.
There are a number of ways in which oncogenes can be abnormally activated to promote tumor growth. The genomic region containing the gene can be amplified, resulting in dramatically increased copy number. For example, the N-MYC oncogene can be amplified up to 100 times in human neuroblastomas. Oncogenes can undergo mutations that cause constitutive activation of the gene. In these cases, the gene is always “turned on” and is unresponsive to inhibitory signals. This scenario is often the case for cell surface receptors such as the EGF-R in carcinomas. Many tyrosine kinase receptors have activating mutations that result in constitutive activation even in the absence of ligands. Tumor cells can also produce large amounts of stimulatory molecules along with their cognate receptors, forming a growth-promoting autocrine loop. All of these mechanisms serve to drive proliferation and render the cell unresponsive to normal inhibitory signals.
The prototype of signal transduction oncogenes is the RAS family of guanine triphosphate (GTP)-binding proteins (G proteins). There are several members of this family, including HRAS, KRAS, and NRAS. In normal cells, RAS transmits growth stimulatory signals from outside the cell to the nucleus, ultimately activating transcription of genes for cell-cycle progression. RAS alternates between an active and inactive state in its normal location on the cytoplasmic side of the cell membrane where it is closely associated with farnesyl transferase. Inactive RAS binds guanine diphosphate (GDP). Upon receiving a stimulatory signal from a growth factor, RAS exchanges GDP for GTP. RAS bound to GTP is the active form, which triggers the RAS–RAF–mitogen-activated protein kinase (MAPK) pathway and results in transcription of genes that promote cell division (Fig. 6-44). The activation of RAS is normally short lived, as RAS has an intrinsic GTPase that hydrolyzes GTP to GDP and converts RAS to its inactive state. In many cancers, RAS is constitutively activated either by mutations in RAS itself or by failure of the GTPase to inactivate RAS. RAS family members, the farnesyl transferase anchor to the plasma membrane, and other components of the signal transduction pathway are all attractive molecular targets for therapeutic intervention in cancer patients.
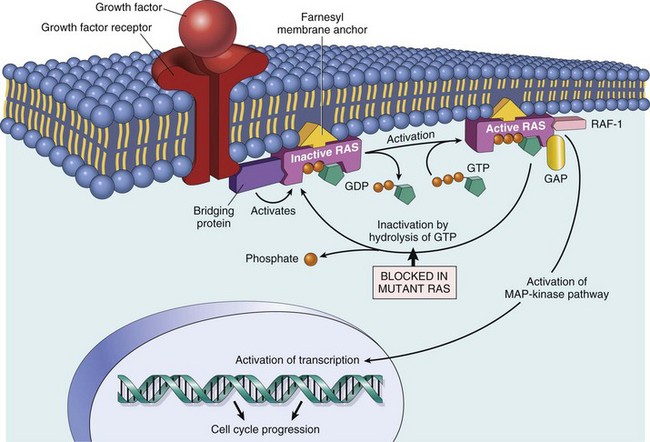
Fig. 6-44 Model for action of RAS genes.
When a normal cell is stimulated through a growth factor receptor, inactive (guanine diphosphate [GDP]-bound) RAS is activated to a guanine triphosphate (GTP)-bound state. Activated RAS recruits RAF and stimulates the mitogen-activated protein kinase (MAPK) pathway to transmit growth-promoting signals to the nucleus. In cancer, mutated RAS is permanently activated because of the inability to hydrolyze GTP, leading to continuous stimulation of cells in the absence of an external trigger. The anchoring of RAS to the cell membrane by the farnesyl moiety is essential for its action. GAP, GTPase-activating protein. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Defects in Dna Repair
Defects in DNA repair enzymes are one cause of mutations and genomic instability. If mutations inactivate tumor suppressor genes and activate oncogenes, the cell has a selective growth advantage. Specific types of DNA repair mechanisms have evolved to repair specific DNA lesions. Mismatch repair enzymes, such as MLH1 and MSH2 proofread DNA, much like the “spell check” function on a computer, to locate and fix single nucleotide mismatches that occur on a regular basis. For example, if an adenine is mistakenly paired with a guanine during DNA replication, this error will be recognized and corrected.
It is well recognized that exposure to UV light leads to cross-linking of pyrimidine residues and formation of pyrimidine dimers. These lesions require a large cohort of DNA repair proteins to perform a nucleotide excision repair procedure. This process is similar to the “cut and paste” function of a computer in which the dimer is excised and the correct nucleotides are replaced and ligated in its stead. As discussed earlier, p53 levels rise in response to DNA damage from any number of agents and give the cell time to carry out the repair processes.
Additional DNA repair genes include ATM, BRCA1, and BRCA2 in humans. Failure of DNA repair of any type can lead to the mutation becoming fixed in the genome and passed on with subsequent rounds of cell division. When the DNA repair gene itself is defective, either through mutation, promoter methylation, or genomic loss, the result is a myriad of mutations throughout the genome with widespread genomic instability and increased cancer susceptibility.
Animals and Cancer
Animal models have been and remain critically important tools for understanding the cause of human cancer and for testing cancer therapeutic agents. Animal models of cancer include both experimentally induced and naturally occurring. In experimentally induced cancer models, administration of carcinogenic substances or transplantation of human cancer cells results in de novo development of cancer in test animals. Naturally occurring models of cancer rely on the spontaneous development of tumors in the test animal.
Experimentally Induced Tumors
A major advantage of experimental model systems is the rapid and reproducible induction of cancer in a very large proportion of experimental animals. Rodents, mice in particular, are often used for such studies. Mice are small and relatively inexpensive to maintain. Moreover, mice reproduce rapidly and their genetics are highly defined and readily manipulated. The mouse genome has been sequenced, essentially in its entirety, and detailed comparative maps of the human and mouse genomes have been developed. Many inbred mouse strains, each consisting of genetically identical or syngeneic individuals, are available. Syngeneic mice readily accept tissue grafts from each other. Genetic homogeneity of syngeneic mice tends to standardize responses and thus reduce the numbers of animals required for studies. A number of inbred mouse strains have been developed that are particularly suited to specific needs in cancer research. For instance, Sencar mice are highly susceptible to tumors of keratinocyte origin and have thus been employed for many skin carcinogenesis studies. Nude mice, which are profoundly immunodeficient, accept tumor or normal tissue grafts from other species and provide an environment in which these xenografts can be maintained, manipulated, and studied. Differences in strain susceptibility to different experimentally induced cancers have been exploited to identify modifier genes that dramatically affect tumor incidence. With the advent of effective means for creating genetically engineered mice, specific genes of interest can be introduced into or inactivated in the mouse genome. A gene introduced into the mouse genome is generally termed a transgene. A mouse lacking a functional normal gene is a knockout for that gene. Moreover, the timing, location, and level of gene expression in genetically engineered mice can now be precisely controlled, thus allowing gene expression to be turned on or off in particular tissues as required for specific studies. Gene expression modulated in this fashion is termed conditional gene expression.
Conventional inbred mice have been used extensively to determine the carcinogenicity of chemical and physical agents and to test the safety and efficacy of anticancer therapeutics. Studies in mice, particularly studies of chemically induced skin cancer, have been critical in defining the stages of carcinoma progression. Genetically engineered mice have been essential for identifying the mechanisms by which specific genes act to retard or enhance tumor development, growth, and spread. However, mice have several inherent shortcomings as models of human cancer. The genetic homogeneity of inbred mice does not reflect the high degree of genetic diversity within the general human population. The spectrum of experimentally induced tumors in mice differs from the spectrum of naturally occurring tumors in humans. Moreover, experimentally induced tumors in mice rarely metastasize; in humans, on the other hand, tumor metastasis is of overwhelming importance. Finally, the short lifespan and small size of mice make them less than ideal for long-term testing of tumor therapies.
Naturally Occurring Tumors
Several naturally occurring cancers in animals, including avian leukosis, bovine lymphoma, and feline leukemia, have provided invaluable information about the cause, transmission, and prevention of virally induced cancers. However, virally induced cancers do not appear to account for a large proportion of human cancers. Recently, the dog has become the focus of increasing attention as a useful animal model of nonviral human cancers. Sequencing of the canine genome and comparative alignment with human and murine genomes enhance the usefulness of dogs as a naturally occurring cancer model.
The age-adjusted cancer incidence in dogs is 381 per 100,000; this is comparable with the cancer incidence in humans. With the large number of pet dogs in this country, many cancer cases are thus available for entry into clinical trials. Like humans, dogs are outbred. Moreover, dogs share a common environment with humans and are exposed to many of the same carcinogens. As in humans, many canine tumors metastasize widely. Because tumors in dogs progress more rapidly than human tumors, studies can be completed within a reasonable time frame. On the other hand, the time course of tumor development is sufficiently long to allow meaningful comparison of response times in different treatment groups. Because dogs are relatively large, they provide abundant tumor tissue for diagnostic and experimental purposes. In addition, many therapeutic approaches that are difficult to test in small rodents can readily be examined using larger dogs. Clinical trials in dogs are much easier to instigate and much cheaper to carry out than comparable studies in humans. Many dog owners are enthusiastic participants in clinical trials that may benefit their pets. Tumor types for which dogs are particularly good models of human cancer include osteosarcoma and lymphoma.
Clinical Oncology
Histopathologic Diagnosis: A definitive diagnosis of cancer is frequently obtained by standard histopathology and/or cytology from incisional or excisional biopsies or aspirates. Biopsy specimens are analyzed by routine hematoxylin and eosin (H&E) staining for histopathologic evaluation and by Wright’s stain or Diff-Quik for aspirates. Cells are scrutinized for features of malignancy, including abnormal morphology, evidence of invasion and/or metastasis, high mitotic index, presence of abnormal mitoses, high nuclear : cytoplasmic ratio, and absence of encapsulation. The degree of differentiation is evaluated. Malignant neoplasms are frequently poorly to moderately differentiated, and some may be so anaplastic that the cell of origin cannot be determined. Abnormal structures or cellular products may also provide clues to the presence of malignancy.
Special staining techniques may be used to aid in the diagnosis of some tumors. For example, immunohistochemistry can establish if a lymphoma originates from B or T lymphocytes (Web Fig. 6-4). This knowledge may be useful to the clinician in designing treatment or delivering a prognosis. The type of intermediate filaments present in an undifferentiated malignancy can indicate if the tumor is of epithelial (positive for cytokeratin staining) or mesenchymal (positive for vimentin staining) origin. Some neoplasms, such as mesotheliomas and synovial cell sarcomas, are often positive for both cytokeratin and vimentin. Squamous cell carcinomas that have undergone epithelial-to-mesenchymal transformation will have areas positive for one stain or the other, and the transition areas may be positive for both. An exhaustive list of antibodies is beyond the scope of this chapter, but immunohistochemical staining is becoming a widely used tool that assists the pathologist in providing a more complete diagnosis in cases in which routine H&E evaluation is limited.
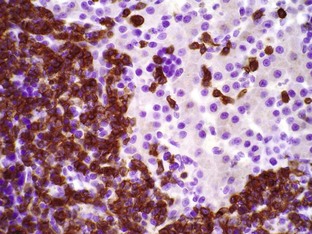
Web Fig. 6-4 Lymphoma, liver, dog.
Note the neoplastic lymphocytes invading the liver. The malignant cells are immunohistochemically positive for CD3 (brown color), indicating a T lymphocyte neoplasm. The surrounding hepatocytes are not labeled. Immunohistochemistry. (Courtesy Dr. D.F. Kusewitt, College of Veterinary Medicine, The Ohio State University.)
Histochemical stains can also aid in diagnosis. Poorly differentiated canine mast cells may have granules that are not clearly visible by H&E staining. Staining with toluidine blue often highlights the granules and confirms the diagnosis in otherwise challenging cases.
Web Fig. 6-5 Diagram depicting the use of X-linked isoenzyme markers to assess monoclonality of tumors.
Because of random X-inactivation, all females are mosaics with two cell populations (G6PD isoenzyme A or B in this case). Monoclonal neoplasms are composed of cells that contain the active maternal (XA) or the paternal (XB) X chromosome but not both. These assays can only be used in females who are heterozygous at the X-linked marker being tested. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Clonality Assays
Although cells can exhibit some degree of heterogeneity within a tumor as a result of clonal selection (see previous discussion), all of the neoplastic cells originated from expansion of a single transformed cell. Clonality can be assessed using the human androgen receptor assay (HUMARA) method in females who are polymorphic at the X-linked androgen receptor locus (Web Fig. 6-5). Other X-linked genes that can be interrogated include glucose-6-phosphate dehydrogenase (G6PD) or phosphoglycerate kinase (PGK). Because females have random inactivation of one X chromosome in each cell, clonality can be demonstrated by showing that each cell within a tumor population has the same X chromosome inactivated (thus the need to use polymorphic markers). If the tumor population has equal distribution of inactivated X chromosomes, it is not monoclonal. These assays have not been well established in companion animals.
Clonality can be determined by a different method in lymphoid malignancies. Sometimes it is difficult to distinguish benign lymphoid hyperplasia from lymphoma by morphology alone. Establishing that the lymphocyte population is clonal gives more weight to the diagnosis of a malignancy. This can be done by analyzing the lymphocytes for T or B lymphocyte receptor rearrangement using the polymerase chain reaction (PCR). If the entire lymphocyte population has a single rearrangement, this is indicative of a clonal population. Conversely, if each lymphocyte has a different receptor rearrangement, this indicates a nonclonal population, which is more consistent with a reactive specimen. It must be remembered that the presence of a clonal population does not, by itself, mean that the specimen is a malignancy. Some nonneoplastic conditions, such as canine ehrlichiosis, can give rise to clonal lymphocyte populations. Therefore results need to be interpreted in conjunction with clinical signs and other clinicopathologic data.
Other Molecular Diagnostic Techniques
A full description of molecular techniques used in cancer diagnosis is beyond the scope of this chapter. However, it is certain that their use will become more widespread and commonplace in veterinary medicine. Cytogenetic analysis can be a useful tool for diagnosis, determining the presence of residual disease after treatment, and stratification of high- and low-risk patients. The discovery of recurrent chromosomal abnormalities and translocations, particularly in leukemias and lymphomas, will aid in diagnosis and understanding the pathogenesis of these diseases.
The increasing use of microarrays for global gene expression analysis in the research setting will undoubtedly lead to identification of panels of specific biomarkers that can be used for diagnosis in the future. Microarrays, which allow the measurement of thousands of genes simultaneously in a single sample, are already available for mice, rats, dogs, chickens, pigs, cows, and horses, and it is anticipated that other species will be added to the list. Large-scale studies using carefully selected tumor samples will be necessary to identify significant diagnostic and prognostic changes in gene expression patterns, which can be applied to individual tumors to provide important information to the clinician. Likewise, the advancement of proteomics for the large-scale interrogation of protein expression in tumors will add to our diagnostic armamentarium.
Finally, identification of genes involved in inherited cancers can be accomplished through the detailed analysis of well-described pedigrees, particularly in certain cancer-prone breeds. As is the case in humans, the elucidation of these genes is important not only in the diagnosis and screening of high-risk animals but also in providing insight into the pathogenesis of sporadic tumors.
Grading
A tumor grade is assigned by the pathologist to provide some indication of how similar or dissimilar the neoplastic cells are to their normal counterparts. The underlying assumption is that this provides some indication about biologic behavior. This is not universally true, however, and experience has demonstrated that tumor stage (see next section) is sometimes a more useful parameter. Grading schemes vary depending on the tumor type. The ideal scheme incorporates criteria that are easily identified on an H&E stained section, and the grade is strongly linked to prognosis or response to therapy.
All grading schemes evaluate the degree of differentiation of tumor cells. The tumor grade classifications usually include well-differentiated (cells appear very similar to normal cells), moderately differentiated (somewhat similar to normal cells), and poorly differentiated (anaplastic) cells. These categories translate to low, medium, and high grade, or grades I, II, and III, respectively. Other criteria that may be included in grading schemes include the mitotic rate (i.e., the number of mitotic figures in ten 40× fields), the degree of necrosis, location and invasiveness of the tumor, and overall cellularity. To be biologically relevant, the grading system for a particular tumor type should be based on objective criteria that can be assessed on routine sections with high concordance between different evaluators and provide the clinician with useful information. As such, criteria need to be periodically reevaluated in light of new discoveries and diagnostic capabilities.
Staging
Tumor stage gives an indication of the extent of tumor growth and spread. In general, staging guides the clinician in developing a therapeutic plan and offering an estimate of prognosis to the client. One of the most widely used schemes is the TNM system, which is based on the size of the primary tumor (T), degree of lymph node involvement (N), and extent of metastasis (M). Within each category, a number is assigned based on clinical, diagnostic, and histopathologic evaluation. A designation of T0 is given to carcinoma in situ, whereas T1 to T4 indicate increasing size of the primary tumor. N0 indicates the absence of detectable lymph node involvement, whereas N1 to N3 indicate progressive involvement. Similarly, M0 signifies no detectable metastasis, whereas M1 and M2 indicate metastasis to one and two organs, respectively.
Overall, TNM staging provides a standard measurement by which the natural course of disease and impact of treatment modalities can be compared. Increased availability of more sophisticated diagnostic testing, such as computerized tomography (CT) and magnetic resonance imaging (MRI), as well as more sensitive histologic detection, such as the use of immunohistochemistry for cytokeratin to detect micrometastases in lymph nodes of patients with carcinoma, may influence comparisons of studies among investigators.
Surgical Margins
Typically, excisional biopsies are performed with the intent of complete removal of the tumor mass to effect a cure or greatly reduce tumor burden. Histologic evaluation of surgical margins has long been a valuable service provided by the diagnostic pathologist. Margins of the tumor are evaluated microscopically for the presence or absence of tumor cells to confirm that the tumor was completely excised (see Figs. 17-32, 17-33, and 17-34). Residual malignant cells at the surgical site may warrant a second surgical procedure. However, evaluation of margins is not always straightforward. Sometimes it is difficult for the pathologist to properly orient the gross specimen with respect to lateral, deep, and superficial margins. At other times it may be difficult to distinguish true surgical margins from those produced at trimming. It is important to realize that having clean surgical margins on a histologic slide does not guarantee that the patient is free of tumor. Neoplasms are three-dimensional lesions and only a portion of the mass is examined in any one section. In addition, multifocal or multicentric lesions may not be submitted to the pathologist. Lastly, lymphatic or hematogenous spread may not be evident on the section or sections examined.
Taken together, the tumor type, grade, stage, and completeness of excision are used by the clinician to develop the most appropriate treatment plan for the patient. As we learn more about the molecular pathogenesis of certain tumors, the need for specialized diagnostic tests will certainly increase. Targeted molecular therapies will be effective only if the target is present in the patient’s tumor.
Knowles, M, Selby, P. Introduction to the cellular and molecular biology of cancer, ed 4. Oxford: Oxford University Press; 2005.
Hanahan, D, Weinberg, RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Herman, JG, Baylin, SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054.
Hong, WK, Bast, RD, Hait, WN, et al. Holland Frei cancer medicine, ed 8. Shelton, CT: PMPH-USA; 2010.
Meuten, DJ. Tumors in domestic animals, ed 4. Ames: Iowa State Press; 2002.
Paoloni, MC, Khanna, C. Comparative oncology today. Vet Clin North Am Small Anim Pract. 2007;37(6):1023–1032.
Payne, SR, Kemp, CJ. Tumor suppressor genetics. Carcinogenesis. 2005;26(12):2031–2045.
Pitot, HC. Fundamentals of oncology, ed 4. New York: Marcel Dekker; 2002.
Sell, S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. 2004;51(1):1–28.
*Dr. Laura J. Rush, Department of Veterinary Biosciences, The Ohio State University, made contributions to this chapter in the fourth edition.