Chapter 2 Cancer: a Disease of Cellular Proliferation, Life Span, and Death
1. Cancer arises from genetic dysfunction in the regulation of the cell cycle, cell life span, and cell suicide.
Control of the Cell Cycle (Proliferation)
1. Cell division is the result of a clocklike cell cycle.
2. Cyclin-dependent kinases are the “engines” driving the cell cycle.
3. The CDK “engines” are controlled by both throttle (oncogene) and brake (tumor suppressor) controls.
Growth Factor Pathway: Principal Stimulator of Cell Proliferation
1. The cell cycle is stimulated by growth factors that bind to and activate receptor tyrosine kinases.
2. The ras oncogene contributes to many cancers and serves as a model for understanding “small G proteins.”
3. The MAP kinase pathway leads to the expression of cyclins and other stimulators of the cell cycle.
4. The MAP kinase pathway also mediates the stimulation of the cell cycle by cell adhesion.
Tumor Suppressors: Inhibitors of Cell Cycle
1. Checkpoints in the cell cycle are manned by tumor suppressors.
2. The retinoblastoma and p53 proteins are the main “gatekeepers” for the cell cycle.
Mechanisms Regulating Cell Suicide and Cell Life Span
1. Apoptosis is the process of cell suicide.
2. Resistance to apoptosis via the intrinsic pathway is a hallmark of cancer.
3. Cellular life span is determined by DNA sequences at the ends of chromosomes.
Traditionally, cancer was (and often still is) first detected in humans, cats, dogs, and horses by clinicians feeling for an unusual mass of cells, tumor cells. Thus, cancer is quite intuitively a disease affecting cellular growth. In the last 20 years, enormous progress has been made in understanding several normal control pathways that regulate cell growth, as well as how these “Rube Goldberg” pathways (see Chapter 1) go wrong in cancer.
The first path to be unraveled, long thought to play a major role in cancer, was the pathway controlling cellular proliferation. Cellular proliferation was known to occur by a regular clocklike cycle of chromosomal doubling followed by mitotic division, called the cell cycle. However, almost nothing was known about molecular control of the cell cycle. Progress arose from the study of cancer cells, but importantly also from the study of the proteins synthesized by fertilized sea urchin eggs, how frogs ovulate, and how yeast cells divide. Cell growth depends not only on new cells being formed by cell division, but also on cells dying. As a result of studying in detail the history and fate of every cell that arises during embryonic development to form a soil roundworm (a nematode), it was discovered that cells are programmed to commit “suicide.” That is, cells can actively kill themselves using metabolic machinery if the cell has internal damage, such as mutations or oxidative stress. This surprising discovery quickly led to the realization that not only do cancer cells divide inappropriately, but they are also resistant to programmed death and thus continue to divide despite the internal damage. The final general process affecting cellular growth is that normal cells, like the organisms they are part of, have a characteristic life span. However, cancer cells were long known to be “immortal,” being able to divide indefinitely. How cells age, or become immortal, was not understood until the process of chromosomal duplication was studied in a ciliated protozoan, similar to the familiar Paramecium of college biology laboratories.
As these examples illustrate, our understanding of cellular proliferation, cellular life span, and cell suicide came in large part from the study of problems that first seemed distant from the cancer seen in the clinic. As such, the recent progress on cancer is an unusually dramatic example of the importance of understanding basic biology to understand medicine. The vast majority of cancer studies are conducted on humans and in mice, the animal model for cancer, and using cultured cells derived from human and mouse tumors. The much smaller number of studies on domestic animals strongly indicate that the principles derived from humans and mice are generally applicable. However, it is also clear that humans and mice differ in a few aspects of cancer, and thus there are likely to be “special” aspects of cancer for each species.
Cancer Arises from Genetic Dysfunction in the Regulation of the Cell Cycle, Cell Life Span, and Cell Suicide
Cancer is a genetic disease (but not usually a hereditary disease) and a uniquely cellular disease. As shown in Figure 2-1, tumors and other cancers arise from the division of a single mutant cell whose descendants accumulate several additional mutations to become increasingly damaged with respect to control of cellular proliferation, life span, and cell death. That is, cancer is a genetic disease caused by the accumulation of mutations in body cells, such as those of the epithelia lining the lungs or the secretory epithelia of the mammary glands.
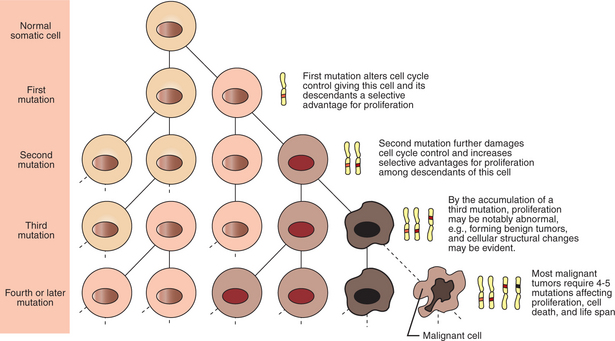
FIGURE 2-1 Clonal basis of cancer. Cancer is the result of the accumulation of mutations in a cell lineage of somatic (nongamete) cells of the body. Beginning with a normal cell, mutations occur by chance or by environmental inputs, such as radiation or cancer-causing chemicals, and accumulate to cause cancer.
All the cells of a tumor can trace their ancestry back to a single cell that developed an initial deleterious mutation. This first mutation usually occurs in a gene controlling proliferation, such that the cell produces a mutant protein that is a dysfunctional, more permissive regulator of the cell cycle. This greater “permissiveness” provides the mutant cell with more opportunity to proliferate, and it thus has a selective advantage compared with its normal neighbors. Perhaps because of this selective advantage, or because of continued exposure to mutagens (e.g., cigarette smoke, agricultural chemicals), a descendant of this cell accumulates another mutation that also affects some aspect of the cell cycle or cell death. This increases the doubly mutant cell’s selective advantage further still, and the downward spiral of increasingly abnormal, dividing cells begins to spin out of control. Scientists agree that this accumulation of mutations in individual genes is necessary for cancer to develop, but some think it is not sufficient. Rather, they argue that cancer only results when the accumulation of mutations eventually leads to large-scale genetic instability, such that whole chromosomes are gained and lost. the majority of spontaneous tumors do have cells with abnormal sets of chromosomes, a phenomenon called aneuploidy. Whether aneuploidy is necessary for cancer remains to be seen, but there is no disagreement that cancer cells are in some way badly damaged with respect to genes controlling growth.
The mutations leading to cancer are the same type as those that underlie Mendel’s familiar laws of heredity. These include base-pair changes, deletions or additions of nucleotides in the gene, and translocation of one piece of a chromosome to another. However, it is important to understand that the cells in which the mutations are occurring are different than those underlying Mendel’s laws of inheritance. Mendelian inheritance results from mutations occurring in the germ line of the organism. These are the cells that will become gametes, either sperm or eggs, and whose deoxyribonucleic acid (DNA) will be passed down to every cell of the offspring. The mutations leading to cancer are occurring in nonreproductive cells throughout the body, called somatic cells. These are passed down only to a limited number of other somatic cells by cell division, not to offspring through sexual reproduction. Thus, although cancer is a genetic disease, only about 10% of the time is it a “hereditary disease,” that is, the result of mutation inherited from a parent. In general, cancer appears to be the result of the accumulation of mutations leading to genetic instability in a particular lineage of somatic cells.
Traditionally, cancers are divided into categories based on the cell type involved. Carcinomas are cancers of epithelial cells; sarcomas are derived from connective tissue or muscle; and leukemias are cancers of blood-forming cells. There are many subdivisions based on specific cell types and location of the tumors. However, these names are traditional only; they do not reflect any fundamental differences in the biology of the cancer. Rather, it is now clear that cancers of all types share broadly similar types of dysfunctions controlling cell proliferation, cell suicide, and cell life span.
CONTROL OF THE CELL CYCLE (PROLIFERATION)
Cell Division Is the Result of a Clocklike Cell Cycle
The “Rube Goldberg device” that controls cell growth is particularly complex, with many, many more components than the “garage door opener” of Figure 1-13. To explain these pathways, we begin with the cell cycle that, like the carriage house door, is near the end of the system of control. That is, most of the control elements feed “downstream” to control the cell cycle, or intersect with some aspect of cell cycle control.
Figure 2-2 shows the classic diagram of the cell cycle in which the cell changes its state toward division, progressively going around the diagram, like the hands of a clock. For most mammalian cells, the duration of one cell cycle in culture varies between 18 and 30 hours. Two phases of the cell cycle were identified first and seemed to be where the most important events of the cell cycle occurred. One is S (synthesis) phase, during which the DNA is duplicated. The second is M (mitosis) phase, during which the duplicated chromosomes are separated to opposite sides of the cell and the cytoplasm divides. In addition to the obvious need for such events if cells are to reproduce, note that both phases must be highly precise. It is crucial for the cell that DNA synthesis produces exactly twice the original amount of DNA, no more and no less. Otherwise, there will not be two identical copies of the genetic material to pass on to two identical cells. Similarly, the machinery segregating the duplicated chromosomes during mitosis must partition exactly equal numbers and types of chromosomes to daughter cells, or the cells will be aneuploid. If DNA is not precisely replicated, or if the chromosomes are not properly aligned, the cell cycle is halted, by “checkpoints,” as described later.
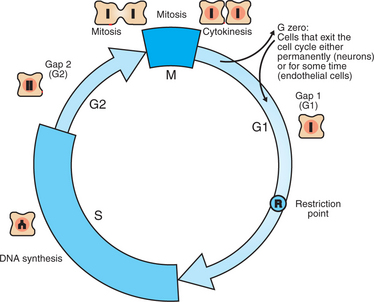
FIGURE 2-2 The mammalian cell cycle. Cell proliferation occurs by a clocklike progression of phases in which characteristic events occur. The most familiar is M phase (mitosis), during which the cytoplasm and replicated chromosomes are distributed to the daughter cells. Cells then enter G1, during which a “decision” is made whether or not to go forward with the cell cycle; this is the R (restriction) point. The events in G1 then allow S (synthesis) phase to proceed, during which the DNA is replicated to produce exactly two copies. After DNA synthesis, the cell prepares for mitosis during G1, and the cycle is complete. Although cells in culture typically go around the cycle continuously, most cells in the body divide only occasionally. These quiescent cells, as well as cells such as neurons that never divide after differentiation, are in G0, a nondividing phase. Under appropriate stimulation, cells can then exit G0 and are said to reenter the cell cycle.
However, the events during G1 (“gee-one”) and G2 phases remained a mystery. The “G” stands for gap, because of the decades-long gap in our understanding of what was happening during this time. Although it was suspected that the cell was preparing itself for DNA synthesis during G1 and preparing for mitosis during G2, the nature of these “preparations” proved difficult to determine. In the mid-1980s, work initially conducted on frog oocytes revealed that specialized protein kinases were activated during G1 and G2 to drive the cell into S phase and M phase, respectively. These special protein kinases are now called cyclin-dependent kinases (CDKs).
Cyclin-Dependent Kinases Are the “Engines” Driving the Cell Cycle
Recall from Chapter 1 that protein kinases, which are enzymes that phosphorylate other proteins, are important as elements of signaling pathways. For example, the second messenger cyclic adenosine monophosphate (cAMP) acts by activating protein kinase A (see Figure 1-18), and diacylglycerol as a second messenger activates protein kinase C (see Figure 1-19). Protein kinases play a major role in many aspects of control of the cell cycle; most importantly, CDKs, when activated, can directly cause a cell to enter either S phase or mitosis, whether the cell is ready or not.
Active CDKs are composed of two different types of protein subunits (Figure 2-3). The catalytic subunits (numbered, CDK1, CDK2, etc.) are the subunits that have enzymatic activity for hydrolyzing adenosine triphosphate (ATP) and transferring the phosphate group to a protein substrate. The other subunit is an activator of the catalytic subunit and is called a cyclin; the abundance of this protein increases and decreases during the cell cycle (i.e., the protein concentration cycles up and down during the cell cycle). Different cyclins are specific for various CDKs and for the different phases of the cell cycle. The various cyclins are identified by letters, such as cyclin A and cyclin B. Cyclins must reach a threshold concentration to activate the catalytic subunit, and the threshold is achieved as a result of protein accumulation from new synthesis during the G phases.
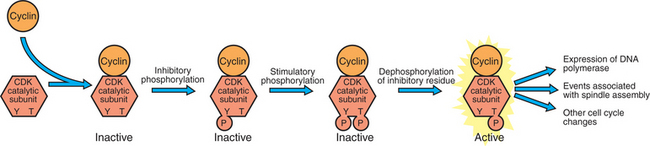
FIGURE 2-3 Activation of the cyclin-CDK “engines” of the cell cycle. Activation of cyclin-dependent kinases depends on the association of a cyclin with a catalytic subunit and then an appropriate pattern of inhibitory and stimulatory phosphorylations on the catalytic subunit.
Once the cyclins have bound to their appropriate catalytic subunit, the cyclin-CDK complex as a whole is activated by achieving a particular state of phosphorylation. There are inhibitory sites of phosphorylation around amino acid 15 of the catalytic subunit, and these must be dephosphorylated. There is also a stimulatory phosphorylation site at amino acid 167, and this must be phosphorylated for cyclin-CDK activity. Once activated, the CDK phosphorylates various substrates associated with either S phase or mitosis. For example, the cyclin-CDK complex responsible for mitosis directly phosphorylates the protein filaments that strengthen the nuclear membrane (lamins). This phosphorylation causes the filaments to disassemble, in turn allowing the nuclear membrane to dissolve, which is an early event of mitosis.
The different phases of the cell cycle are controlled by different cyclin-CDK pairs, as shown in Figure 2-4. Thus the complex of CDK1 with either cyclin B or cyclin A is the particular CDK pair responsible for driving the cell into mitosis. Cyclins E and A interacting with CDK2 play important roles in initiating and maintaining DNA synthesis in S phase. Cyclin D interacting with either CDK4 or CDK6 functions in late G1 in a “decision” by the cell to commit to DNA synthesis. This decision is called the R (restriction) point and is discussed in the later section on tumor suppressors.
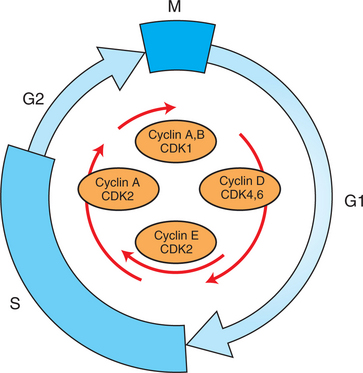
FIGURE 2-4 Cyclins and CDKs around the cell cycle. Different phases of the cell cycle are associated with and driven by different cyclin-CDK pairs, as shown here.
Given the importance of cyclins and CDKs in driving the cell cycle, one would expect they would have some connection to cancer. Overexpression of cyclin D is associated with human and mouse breast cancer, and ablation of cyclin D provides some protection against breast cancer in mice. Virtually all multiple myelomas, a type of leukemia, show overexpression of cyclin D. Overexpression of cyclin A is strongly associated with some lung cancers and with testicular cancer of humans, and overexpression of cyclin E is associated with certain human leukemias. Curiously, in contrast to the cyclin subunit, the CDK enzymatic subunit is not known to be mutant in any common cancer.
The CDK “Engines” Are Controlled by Both Throttle (Oncogene) and Brake (Tumor Suppressor) Controls
The CDK-cyclin pairs are controlled by both stimulatory and inhibitory pathways, analogous to an automobile engine controlled by throttle and brake mechanisms. The throttle mechanisms are largely the result of the cell’s environmental inputs. That is, various environmental cues, both soluble signal molecules and insoluble molecules found in tissue, are required for cells to divide. However, the pathways sending inhibitory signals to the cell cycle, the “brakes” for cell division, are largely internal and are activated by damage or stress to the cell. In general, these inhibitory signals are like the safety interlocks on an automobile. Just as one cannot start a car in gear, so the cell should not divide if DNA synthesis has not exactly duplicated all the genes and chromosomes, or if something is wrong with the mitotic spindle.
The environmental stimulatory signals for cell division can be as simple and nonspecific as availability of nutrients, to the extent that cells only divide when they have approximately doubled in size through synthetic growth. However, two more specific stimulators of the cell cycle are primarily implicated in cancer. One is the response to soluble growth factors found in the circulation and in the extracellular fluid surrounding cells (see Chapter 1). Growth factors are proteins secreted by a variety of other cell types that are required for the division, and indeed survival, of normal, noncancerous cells. Cancer cells, however, can divide and survive with little or no stimulation from growth factors because of the acquired ability to synthesize growth factors of their own, or the activation of downstream elements in the signaling pathway.
The second stimulatory pathway of general importance in cancer is cell attachment. The cells of multicellular organisms must be tightly attached to one another and to their surrounding matrix, similar to tendon; otherwise we would be jelly, juice, and bubbles on the floor. Also, however, attachment of cells to their surroundings is a source of specific and complex information to the physiology of the cell. One of the most important such messages is a “permissive” signal to divide. Normal cells must be anchored to some substrate in order to respond to other signals to divide. That is, most normal animal cells show anchorage dependence of growth. for this reason, vertebrate cells in culture are grown on the surface of a dish or flask, not in suspension the way bacteria are cultured. Once again, cancer cells have lost this normal restriction on proliferation, and many cancer cells can divide and survive in suspension. The common test for the absence of anchorage dependence is growth in soft agar: cancer cells will, but normal cells will not, divide and form colonies when suspended in soft agar. Thus, cancer cells can survive unattached while riding the circulation to relocate in a different tissue than that of the original tumor. In this way, cancer is able to spread through the body, a process called metastasis, which is ultimately the cause of death in most cases of cancer.
The “Rube Goldberg” pathways that underlie the proliferative signals of growth factors and adhesion are similar and intersect. These “throttle” contraptions begin with a soluble signal binding to a growth factor receptor and a “solid-state” signal about attachment to the surrounding tissue. However, both pathways quickly converge on the same stimulation pathway for conserved cell division. These stimulatory pathways are driven by proteins that were originally identified as being encoded by genes in viruses that caused cancer in animals. Thus these were named oncogenes, literally “cancer genes.” A major breakthrough came with the discovery that these oncogenes were actually derived from the host genome, not genes normally encoded in the virus. That is, viruses had stolen cell cycle control genes from their animal host cell. Being viruses, they did not take good care of the animal cell cycle genes they stole. The stolen genes mutated into deranged cell cycle regulators. Subsequently, the same mutant genes that were found in viruses were found to explain many spontaneous cancers in humans and in the long-used experimental tumors of mice. The finding that cancer was caused by abnormal host genes helped confirm that cancer was a somatic genetic disease due to mutations in the tumor cells.
Further analysis revealed that these oncogenes often encode normal stimulators of the cell cycle, and the mutations involved had the effect of permanently activating an element in the cell cycle pathway. You can see how this would work based on the Rube Goldberg cartoon of Figure 1-13. Note that all the elements in the garage door opener are stimulatory; if any one turns “on,” a signal is sent “downstream” to cause the garage door to open. If the fish tank of the cartoon were to “mutate” by developing a leak, an “on” signal would be sent downstream of the fish tank, regardless of whether a car had pulled into the driveway. So it is with the oncogene elements controlling the cell cycle. If one of the elements mutates to turn itself “on,” that is, acquired a gain-of-function mutation, it will stimulate cell division and contribute to cancer. To return to the automobile analogy, oncogenes represent a stuck throttle or accelerator pedal. The normal, well-behaved versions of the oncogene (a watertight fish tank before the bullet, Figure 1-13) are called proto-oncogenes. Thus, strictly speaking, “oncogenes” have their normal equivalent as “proto-oncogenes.” However, given this awkward usage, increasingly the normal versions are also informally called oncogenes, and it is usually clear from the context whether the mutant or normal version is being discussed. The molecules and molecular events of the oncogene pathway (also called the “growth factor” or MAP kinase pathway) are discussed later.
The mechanisms to stop the cell cycle, the “brakes,” are called checkpoints. Progress through the cell cycle depends on appropriate conditions being reached within the cell before a “decision” is made to go ahead with division. The first such checkpoint occurs before S phase. During G1, the cell checks itself over particularly with respect to DNA damage. The cell has sophisticated pathways to detect and repair DNA damage, such as mismatched bases detected in the double helix. for needed repairs to take place, however, DNA synthesis is delayed; the checkpoint is “engaged.” If the DNA is properly repaired, the checkpoint is disengaged, and after the delay, the cell goes ahead into S phase. However, if the DNA damage cannot be repaired, the checkpoint machinery is supposed to signal a more serious consequence. If the checkpoint is not disengaged after about a day, the cell “commits suicide.” Thus the checkpoint (or braking machinery) is tied into both the CDK engines and the processes of cell suicide, as described later. Similarly, the second checkpoint is in mitosis and checks for proper mitotic spindle assembly and correct chromosome alignment. Here again, if damage is detected, there are repair mechanisms, and a properly repaired cell will go into M phase after a delay for repair. If no repair can be made, the cell commits suicide.
The molecules and their interactions that underlie both oncogene (“throttle”) pathways and checkpoint (“brake”) pathways are now covered in greater detail, beginning with the role of growth factors.
GROWTH FACTOR PATHWAY: STIMULATOR OF CELL PROLIFERATION
The Cell Cycle Is Stimulated by Growth Factors That Bind to and Activate Receptor Tyrosine Kinases
The growth factor/oncogene pathway begins with growth factors that function in a familiar way, as discussed in Chapter 1: they bind to and activate an integral membrane protein receptor. Indeed, growth factor receptors belong to the third family of receptors for environmental signals, the receptor tyrosine kinase family. This family of signal transducers has some similarities with the G-protein–coupled receptors (GPCRs), but also some important differences. Receptor tyrosine kinases (RTKs) do not require second messengers, but they do function through protein kinase activity (as many GPCRs do). The structure of RTKs is such that binding of ligand (a growth factor) by the extracellular portion of the receptor directly activates protein kinase activity by the cytoplasmic portion of the protein. The receptor itself is an enzyme (Figure 2-5). Thus the RTK itself carries the message across the plasma membrane, without the need for a second message. RTKs specifically add a phosphate group to a tyrosine residue of the substrate protein. This differs from the protein kinases discussed in Chapter 1 (A and C), which add the phosphate to serine or threonine residues. Phosphorylation of tyrosine residues within a protein is largely (but not exclusively) specialized to control cell growth pathways, and therefore tyrosine kinase activity generally is associated with stimulation of proliferation.
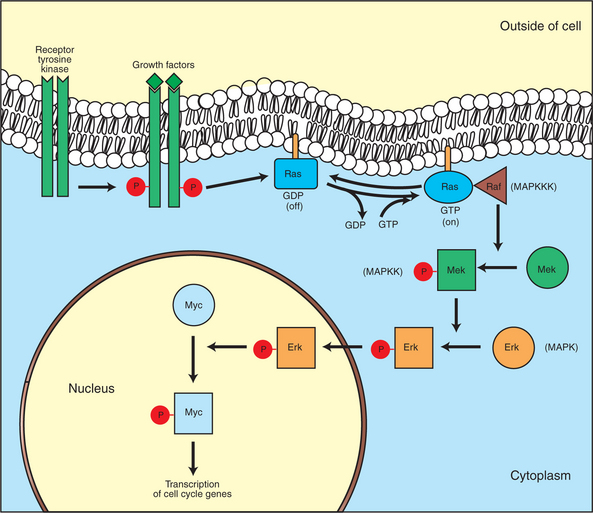
FIGURE 2-5 Growth factor/oncogene pathway. This diagram shows the normal stimulatory pathway by which growth factors lead to cell division. Growth factors bind to membrane receptors (receptor tyrosine kinases, RTKs) that are themselves protein kinases. As shown here, after activation by binding a growth factor, the first protein to be phosphorylated at tyrosine residues is the receptor protein itself. This in turn causes a “small G protein,” Ras, to exchange GDP for GTP and thus be turned “on.” The activated Ras then activates the first protein kinase in a conserved pathway of three kinases, called the MAP kinase pathway. For more detail on Ras and the MAP kinase pathway, see the text. Finally, this series of activating phosphorylations lead to the activation of transcription factors, such as Myc, in turn leading to the expression of genes directly involved in driving the cell cycle (e.g., expression of cyclin D). In this pathway, gain-of-function mutations of the RTKs, Ras, and Myc are particularly important in human cancers.
The growth factors that bind to the RTKs are too diverse to be discussed at length in this chapter. Rather, one important similarity for introductory professional students is that these factors are all poorly named, so do not judge the factor by its name. Sometimes growth factors have “growth factor” in their name; some are referred to as “cytokines”; and some are called “colony-stimulating factors” (for growth of colonies in soft agar, as previously mentioned). Further confusion arises because their names always reflect their history but rarely their broader function. Thus, “epidermal growth factor” stimulates cell division in many more types of cells than only skin cells, but it was discovered using skin cells. The other, more important similarity among growth factors is that whatever their name, as with the numerous ligands binding GPCRs and nuclear receptors, they share a conserved basic pathway and “strategy” for controlling the CDK engines of the cell cycle. Growth factor activation of RTKs stimulates a pathway involving a G-protein “on-off” molecular switch, the Ras protein introduced in Chapter 1, and uses a cascade of protein kinases, both tyrosine and serine-threonine, called the MAP kinase pathway. Ultimately, the MAP kinase pathway activates transcription factors, in turn controlling the expression of cyclins, and other direct regulators of CDKs (see Figure 2-5).
The Ras Oncogene Contributes to Many Cancers and Serves as a Model for Understanding “Small G Proteins”
After activation of the RTK, the next major step in the growth factor/oncogene pathway in normal cells is activation of the protein product of the ras proto-oncogene. Investigations of how it worked revealed that the Ras protein was an important member of the “small G-protein family” of molecular regulators, all of which have intrinsic guanosinetriphosphatase (GTPase) activity and serve as molecular “on-off switches.” These proteins control many basic cellular functions, and the heterotrimeric G protein evolved from Ras-like ancestor proteins (see Chapter 1). Indeed, in yeast it is Ras, not a heterotrimeric G protein, that controls adenyl cyclase and phospholipase C (see Figure 1-18). Figure 2-6 illustrates the duty cycle of this on-off switch and its basic similarity to the alpha subunit (Gα) of the heterotrimeric G proteins. Ras, other small G proteins, and Gα all are in the “on” state when they have guanosine triphosphate (GTP) bound to them (because of receptor activation). All are in the “off” state when the G protein hydrolyzes its GTP so that guanosine diphosphate (GDP) is now bound. You can see how this gene could be discovered as an oncogene, that is, a gene in which a gain-of-function mutation contributes to the development of cancer. If the GTPase activity is lost by mutation, this simple, enzymatic on-off switch remains trapped in the “on” position (the accelerator pedal is stuck). It continues to send an activating signal to the downstream cell cycle machinery without the presence of growth factors or the activation of RTKs. In fact, such mutations in Ras underlie its oncogenic function, and it is estimated that 30% of human cancers have activating mutations in their Ras gene.
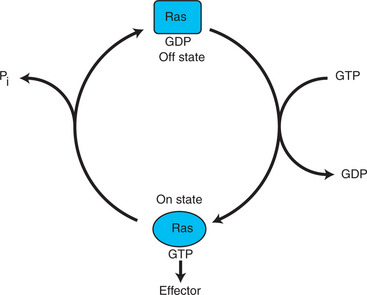
FIGURE 2-6 Duty cycle of the Ras molecular “on-off switch.” Ras serves as a model for the activity of “small G proteins,” of which there are hundreds in the cell. The molecular mechanism of Ras is similar to the alpha subunit of the heterotrimeric G protein, discussed in Chapter 1 and which evolved from Ras-like proteins. As shown here, Ras is in the “off” state when bound to GDP. Activation of RTKs leads to nucleotide exchange: GDP is lost and GTP is bound. In the GTP-bound form, Ras is in the “on” state and sends a stimulatory signal downstream, in this case to Raf in the MAP kinase pathway (see Figure 2-4). Normally, Ras rapidly returns to the off state because an intrinisc GTPase activity of the Ras protein hydrolyzes the GTP to GDP. This nucleotide-dependent on-off cycle is characteristic of all normal small G proteins.
Other small G proteins control a myriad of cellular functions, including others involved in cancer. Thus the Rho subfamily of small G proteins is directly involved in the spread of cancer because it helps regulate actin assembly and activity. As described later, the spread of cancer depends on the ability of cells to migrate through tissues. This “crawling” motility in turn depends on a musclelike mechanism based on actin and myosin (see Figure 1-4). Although the basic, on-off activity of Ras and Rho are the same as that shown in Figure 2-6, Rho is connected to actin, whereas active Ras activates the elements of the MAP kinase pathway
The MAP Kinase Pathway Leads to the Expression of Cyclins and Other Stimulators of the Cell Cycle
GTP-bound Ras causes the sequential activation of a series of protein kinases, called Raf, Mek, and Erk. That is, Raf phosphorylates and activates Mek, which in turn phosphorylates and activates Erk, as shown in Figure 2-5. This trio of kinases is called the mitogen-activated protein kinase, or MAP kinase, pathway (a mitogen is a stimulator of mitosis, e.g., a growth factor). If any of these three protein kinases should experience a gain-of-function mutation irreversibly activating the protein kinase, a stimulatory signal is sent down the remainder of the pathway. Thus, as with ras, these three kinase genes act as oncogenes. In addition, Raf, Mek, and Erk are a specific example of yet another conserved but diverse general “module” of information transduction. That is, there are MAP kinase trios other than Raf, Mek, and Erk. Although it is not worthwhile to give names to all the various specific pathways, it should be noted that these trios have a systematic set of names for their elements. Raf is a MAP kinase, kinase, kinase (a MAPKKK). Mek is a MAP kinase, kinase (MAPKK), and Erk protein is the MAP kinase (MAPK) itself. this jargon is awkward, but it is widely used and logical, as Figure 2-5 suggests.
When activated, Erk activates one or more transcription factors that control the transcription and translation of a key regulator of the cyclin-CDK engine. One of these transcription factors, Myc (“mick”), is encoded by another important oncogene/proto-oncogene. As with ras, themyc gene is mutated in a high frequency of human tumors, giving rise to an oncogenic form able to activate the cell cycle. As shown in Figure 2-5, Myc protein is involved in the transcription of a variety of cyclins and of the CDK2 catalytic subunit and plays a significant role in allowing the cell to pass from G1 to S phase. Myc is also involved in many other transcription events related to cell growth, differentiation, and cancer.
This completes the growth stimulatory pathway beginning with a growth factor binding to its RTK receptor that, through Ras, a MAP kinase cascade, and a transcription factor, eventually leads to a direct “throttling up” of a cyclin-CDK engine. This same pathway is used similarly to transduce the information about the other major stimulator of cell division, cell attachment.
The MAP Kinase Pathway Also Mediates the Stimulation of the Cell Cycle by Cell Adhesion
As noted earlier, the other major throttle mechanism to regulate the cyclin-CDK engines of the cell cycle is cell adhesion. Cell adhesion, as with growth factor stimulation, ultimately stimulates cyclin-CDK pairs through the MAP kinase pathway. Two types of cell contact are involved in normal growth and proliferation. The most obvious is cell-cell adhesion; most cells are tightly attached to their neighboring cells. The second type is cell adhesion to an extracellular matrix (ECM) of fibrous proteins. Eighty percent of human and mouse cancers arise from epithelial cells (carcinomas), and all epithelial layers are attached to an ECM. The adhesion proteins that bind to other cells or to the ECM are adhesion receptors. Adhesion receptors are responsible for the mechanical aspect of attachment, but also act similar to other receptors in transducing information across the plasma membrane. In this case, adhesion receptors communicate the information that the cell is anchored and can divide.
Both cell-cell and cell-ECM adhesion activate the MAP kinase pathway, similar to growth factors, but the Ras intermediate is less important here. Figure 2-7 shows the activation of the MAP kinase pathway as a result of cell-ECM adhesion. The adhesion receptors that bind to ECM are called integrins and these activate the MAP kinase pathway via two important intermediates that are themselves oncogenes. One is Src (“sark”), a protein tyrosine kinase and the first oncogene (src) to be discovered. Unlike the RTKs previously described, Src is not itself a receptor. However, Src is located on the inside face of the plasma membrane, where it can interact with adhesion receptors. Another important intermediate is also a protein tyrosine kinase, called Fak (focal adhesion kinase). As before, activation of Src and Fak activate the MAP kinase pathway, leading to increased cell division. Once again, mutation or overexpression ofsrc and fak sends inappropriate stimulation to the cell cycle machinery, which facilitates cancer. As mutant oncogenes,fak is associated with aggressive melanomas in humans. The src oncogene was named because of its ability to cause sarcomas in chickens.
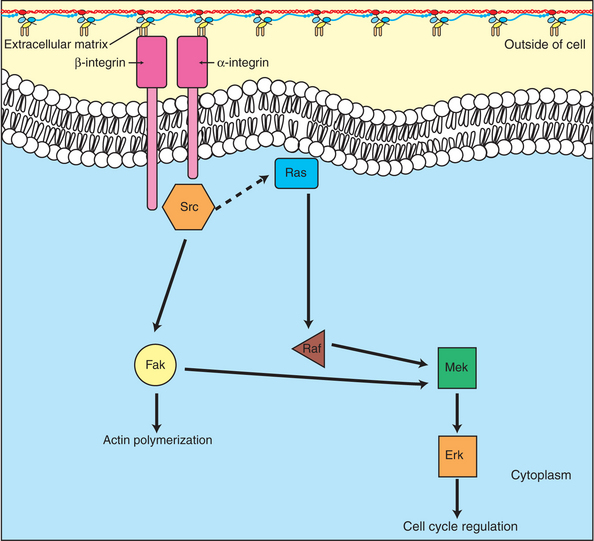
FIGURE 2-7 Cell adhesion functions through the MAP kinase pathway to stimulate cell division. In addition to the growth factor stimulation of proliferation shown in Figure 2-5, normal epithelial cells also require stimulation of the MAP kinase pathway through adhesion to the extracellular matrix. The “adhesion receptors” are integral membrane proteins called integrins, which are activated by binding proteins of the extracellular matrix. Activation of integrins leads to activation of two protein kinases, Src and focal adhesion kinase (Fak), which in turn activate the MAP kinase pathway.
Several other growth stimulatory pathways work in much the same manner as the growth factor and adhesion pathways. Most stimulatory pathways involve protein kinases and G proteins controlling the transcription of genes encoding proteins that are part of or close to the workings of the cyclin-CDK engines.
Having introduced the fundamentals of stimulatory pathways in the cell cycle, we now change our focus to consider the equally Rube Goldberg–like pathways that provide the brakes to the cell cycle.
TUMOR SUPPRESSORS: INHIBITORS OF CELL CYCLE
Checkpoints in the Cell Cycle Are Manned by Tumor Suppressors
The cell cycle machinery also has crucial “brake” mechanisms that function as checkpoints, as noted earlier. The components of the brake and checkpoint mechanisms were discovered by fusing a normal cell with a cancer cell of the same type, to form a hybrid cell with two nuclei. The resulting hybrid cell invariably showed normal regulation of growth. Apparently, a normal copy of some gene or genes present in the normal cell was able to suppress the altered activity of a mutant gene in the cancer cell. Thus these genes and their encoded proteins were called tumor suppressors.
Tumor suppressors play several different functional roles in braking and checking, and they can be divided into two broad types, gatekeepers and caretakers. Gatekeepers are genes and proteins that are involved in the actual checkpoint machinery connecting cell damage with a halt in the cell cycle. Thus, p53 (“protein of 53-kilodalton mass”) is a gatekeeper importantly involved in the pathway that detects DNA damage; it causes a halt in the cell cycle and, if the damage cannot be repaired, signals the cell to undergo programmed death. It is thought that about 50% of human cancers have a mutation in p53. Caretakers are usually proteins involved in the repair of damage or the normal maintenance of proteins crucial in the cell cycle. A human example of a caretaker gene and protein is BRCA1 (“breast cancer 1”). This protein is normally involved in the repair of nucleotide mismatches (e.g., G paired with T rather than with C in the complementary DNA strand), and its mutant gene has been found to underlie familial (hereditary) breast cancer in some families.
With these normal functions, one can see how these genes and proteins would suppress tumor activity and cell proliferation. If they are working, DNA is repaired before the cell attempts to divide; this would tend to prevent mutation or other types of genetic instability. However, loss-of-function mutation in these genes means the cell now has lost the ability to detect or repair DNA damage. For example, when p53 is nonfunctional, even a badly damaged cell may not receive an adequate signal to commit suicide, and this already-mutant cell can continue to divide. Thus, tumor suppressor genes are associated with loss-of-function mutations in cancer, not gain-of-function mutations as for oncogenes. Returning to the automobile analogy of brakes, mutant tumor suppressor genes resemble dysfunctional braking systems, or no brakes at all.
We focus on two gatekeeper-type tumor suppressors because their role and importance in cancer are clear. The role of caretakers such as BRCA1 is both more complex and more uncertain (see suggested reading on BRCA in the Bibliography).
The Retinoblastoma and p53 Proteins Are the Main “Gatekeepers” for the Cell Cycle
Retinoblastoma is a rare, hereditary, childhood cancer of the retina of the eye. Despite its rarity and that it cannot be induced in mice, retinoblastoma has played an important role in the study of cancer. A statistical study of the disease in the early 1970s provided the best evidence then available that human cancer is a genetic disease. Alfred Knudsen showed that children with retinoblastoma typically inherit one mutant copy from a parent (a “germ line mutation”), but then required a second somatic mutation in cells giving rise to the retina. Knudsen’s “two-hit hypothesis” was a forerunner to the idea that cancer develops by the accumulation of mutations in a cell lineage. (Retinoblastoma tumors do require the accumulation of additional mutations beyond the two retinoblastoma genes being mutant.) Subsequently, the retinoblastoma gene, RB, was the first tumor suppressor gene to be cloned. Study of the encoded protein, pRB, showed that it played a central role in controlling the transition from G1 to S phase of the cell cycle.
The RB protein is a repressor of a transcription factor whose activity is required for the cell to enter S phase from G1 (Figure 2-8). The transcription factor is E2F, which controls the expression of a wide variety of genes/proteins required for DNA synthesis, including cyclin A, CDK1 (see Figure 2-4), and subunits of DNA polymerase. The RB protein is a potent inhibitor of E2F only when it is bound to E2F directly, which requires the RB protein to be in an unphosphorylated state (see Figure 1-1, B, and Figure 1-17). The repression of E2F is released by phosphorylation of RB by cyclin-CDK pairs operating early in G1 in the cell cycle. As discussed, growth factor stimulation of the MAP kinase pathway leads to expression of cyclin D (see Figure 2-5), which in turn makes a pair with either CDK4 or CDK6 to make an active CDK. One of the substrates for cyclin D/CDK4,6 is the RB protein. When RB is phosphorylated by CDK4,6, it releases from E2F, allowing this transcription factor to promote RNA polymerase activity on genes with E2F promoter regions (Figure 2-8). It is this release of inhibition by CDK-mediated phosphorylation of RB that constitutes the molecular mechanism underlying the R-point “decision” to divide late in G1 mentioned earlier and shown in Figure 2-2. If both copies of RB are mutant, as in retinoblastoma, there will be no active repressor molecules to bind to E2F, and the decision will always be to divide, regardless of other conditions. E2F then promotes uncontrolled expression of S-phase genes whether or not CDK4,6 has been activated (in part) by growth factors and adhesion, thus making a contribution to unregulated growth and to cancer. Conversely, in its normal, nonmutant form, RB tends to suppress tumor formation by acting as a gatekeeper, only allowing the cell “to cross the border” between G1 and into S phase if normal growth factor and adhesion signals are received. Thus, pRB plays a crucial gatekeeper role in healthy, normal cell cycle control.
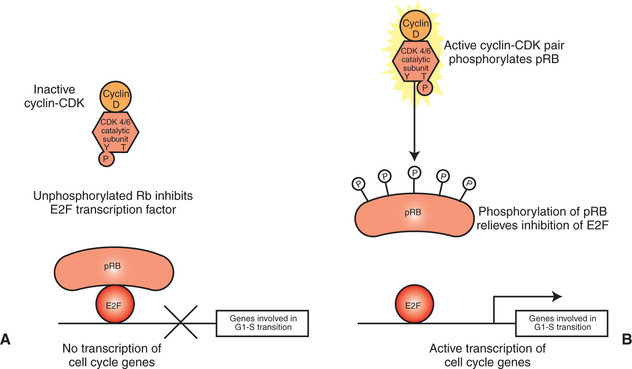
FIGURE 2-8 Retinoblastoma protein and the G1-to-S transition. A, In quiescent cells or cells early in G1, retinoblastoma protein (pRB) exists in a nonphosphorylated state that is a direct inhibitor of the E2F transcription factor. The principal CDK pair of G1, cyclin D with CDK4 or CDK6, phosphorylates pRB, releasing its inhibition of E2F. B, Activated E2F then participates in the expression of a variety of genes required for S phase, including the cyclins and CDKs of S phase and subunits of DNA polymerase.
The other crucial gatekeeper between G1 and S phase is p53. Unlike pRB, p53 does not participate in healthy cell cycles; p53 is only active in response to cell damage, usually DNA damage, or stress, such as low O2 concentration or oncogene activation (Figure 2-9). The role of p53 is to ensure that stressed/damaged cells are either repaired or, if not, commit suicide before being allowed to replicate their DNA. As a gatekeeper, p53’s mechanism is also more direct than pRB; p53 is itself a transcription factor, and p53 activation stimulates the expression of a protein that is a powerful general inhibitor of all the cyclin/CDK engines. As a transcription factor, p53 also mediates the expression of genes that encode stimulators of cell death, as discussed shortly. Whether the cell responds to p53 by cell cycle arrest to allow repair, or by committing suicide, depends on multiple factors, but presence of an oncogene is among the most important. Normally, the cell cycle arrest activity of p53 is dominant to its death-inducing activity. However, in the presence of oncogenes, including myc, suicide is favored. This illustrates clearly the normal tumor suppressor activity of p53: although a cell expressing an oncogene will tend toward increased proliferation, the same oncogene, acting through p53, activates a death pathway to prevent expansion of the mutant cell population.
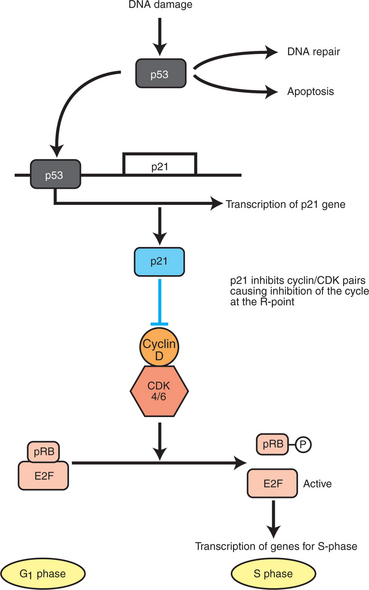
FIGURE 2-9 p53 and the response to DNA damage. Normally, p53 is maintained at low levels in the cell by continuous synthesis and breakdown. DNA damage inhibits breakdown, allowing p53 to build up to functional levels. p53 is itself a transcription factor, and its targets include p21, a potent inhibitor of all cyclin-CDK pairs. Thus, upregulation of p53 brings the cell cycle to a halt, typically by inhibiting phosphorylation of pRb, as shown here. Subsequently, if the DNA is repaired, p53 returns to low concentration. If the DNA remains damaged, p53 leads to an apoptotic response by mediating expression of pro-apoptotic proteins, as described in the text.
The activation of p53 occurs in part through mechanisms familiar from previous examples of protein control, including phosphorylation and binding with other proteins. In addition, p53 activity is also regulated simply by an increase in its concentration within the cell. That is, p53 is normally synthesized at a steady but slow rate throughout the cell cycle and is normally degraded at a similar rate. In healthy cells the half-life for a p53 molecule is about 30 minutes, but this increases threefold to sevenfold in response to DNA damage. Even one double-strand break in DNA has been shown to increase p53 concentration rapidly in some cells. Again, it is clear how p53 serves as both a gatekeeper and a tumor suppressor. Activated p53 prevents a cell with DNA damage from crossing the G1-S boundary (its gatekeeper function), which in turn prevents mutant cells from being allowed to accumulate additional mutations (its tumor suppressor function).
However, if the p53 gene suffers a loss-of-function mutation and the protein cannot act as a transcription factor, a damaged cell will indeed be able to divide, increasing the probability of accumulating further damage and leading to possible cancer. Thus, p53 is one of the most important single genes and proteins involved in human cancers; in 1993 the journal Science even named it “Molecule of the Year.” About 50% of human tumors have a mutation in p53, with most of these eliminating DNA binding, disabling its transcription factor activity. When the p53 gene was “knocked out” in mice, 74% of the animals developed cancers by 6 months of age (young adult). Among experimental mice that had one or two normal copies of the gene, only 1 in 100 animals developed a tumor by 9 months.
In addition to a checkpoint for S phase in which DNA damage provides an important regulatory signal, the other major checkpoint occurs during mitosis. This checkpoint responds to mitotic spindle abnormalities or damage and to abnormalities in the array of chromosomes within the spindle. Here again, one can easily see how mutations that disrupted such “safety interlocks” could lead to further damage, by segregating both replicated chromosomes into one daughter cell, for example, with no copy of that chromosome in the other daughter cell. This would lead directly to aneuploidy. Among human cancers, colon cancer is frequently found to have mutations in mitotic checkpoint genes.
However, we leave the topic of mitotic checkpoints at this somewhat intuitive level and do not address the molecular mechanisms in detail. Such an effort would require a lengthy background discussion of the structure, functions, and control of the microtubule-based mitotic spindle, more suitable for a course in cell biology than animal physiology. Instead, we now discuss the controls on cell growth other than proliferation and briefly summarize what is known about programmed cell death and the control of cell life span.
MECHANISMS REGULATING CELL SUICIDE AND CELL LIFE SPAN
Apoptosis Is the Process of Cell Suicide
The process of cell death by external damage, involving cellular swelling, bursting, and engagement of the inflammatory response, has been well described for more than 100 years. This form of cell death is called necrosis and is familiar from experiences as common as a cut or abrasion. A rather different process of cell death was described in the 1970s in which cells shrink, the DNA fragments in a systematic way, the plasma membrane bubbles and churns, and the cell breaks up into small pieces that are rapidly engulfed by neighboring cells (Figure 2-10). This “neater and cleaner” form of cell death was named apoptosis (a-pah-toe-sis; Greek, “falling off”). Apoptosis was largely ignored for the next 20 years, until studies of nematode development discovered genes whose only role was to control apoptosis. Further studies revealed the highly conserved mechanisms of apoptosis and its importance in normal development, immune function, and disease. Resistance to apoptosis is clearly a major contributor to cancer. (Conversely, too much apoptosis plays an important role in neurodegenerative diseases and stroke.) Particularly relevant to clinical practice, most cancer drugs and radiation therapy kill the target cells (and unfortunately many bystander cells) by stimulating apoptosis.
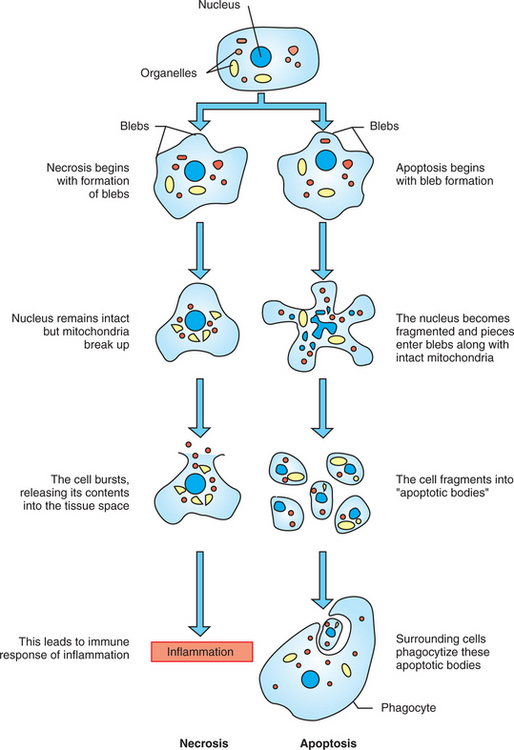
FIGURE 2-10 Necrosis vs. apoptosis. Necrosis is cell death as a result of external damage to the cell that leads to bursting of the cell and release of cell contents, leading to inflammation. Apoptosis is cell death as a result of intrinsic mechanisms in which the cell is broken down into cell fragments that then undergo phagocytosis by neighboring cells. This produces no inflammatory reaction and is so “tidy” that apoptosis is difficult to observe.
There are two broad pathways that lead to apoptosis. The intrinsic pathway of apoptosis responds to internal damage or stress from within the cell. The extrinsic pathway begins with a signal molecule binding to a “death receptor” on the cell surface (Figure 2-11). However, both pathways converge on the same “executioners.” Caspases are a family of proteolytic enzymes that have a cysteine amino acid at their active site (the “c” in caspase) and that cleave the substrate proteins at an aspartate amino acid (the “asp” in caspase). Similar to many other proteases, including digestive enzymes and blood-clotting factors, caspases are themselves activated by proteolytic cleavage. That is, as initially translated, the protease contains an inhibitory peptide that must be cleaved away to allow active proteolysis by the enzyme. In the case of the caspases, the activating protease is itself another caspase. Thus, caspases are divided into activating caspases, which respond directly to one or another element in the intrinsic or extrinsic pathway, and downstream executioner caspases, which lead to specific cleavage of cellular structures. Among other tasks, executioner caspases cleave cytoskeletal proteins, leading to cell shrinkage, and activate the DNA-degrading enzymes involved in the systematic fragmentation of DNA.
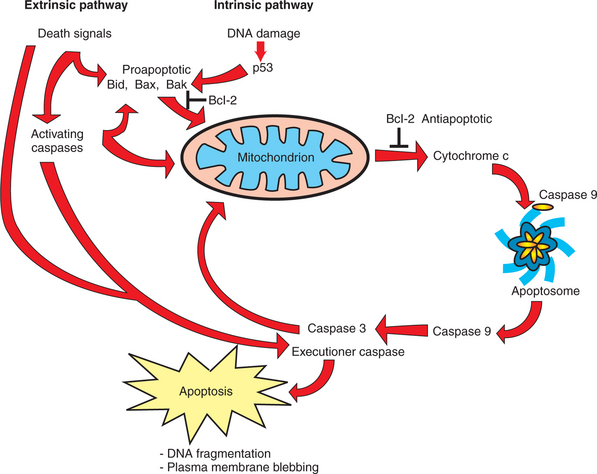
The basic extrinsic pathway of apoptosis, also called the “death receptor pathway,” is unusually short and straightforward considering the extreme and irreversible outcome. An extracellular signal, which can be either soluble or attached to the surface of another cell, binds to and activates a death receptor on the cell destined to commit suicide. The cytoplasmic domain of the death receptor recruits one or two adapter proteins that directly activate an activating caspase, which in turn activates one or more executioner caspases (see Figure 2-11). The activating caspase of the extrinsic pathway can also engage in “cross-talk” with the intrinsic pathway, described shortly, to increase the extent of caspase activation. The extrinsic pathway plays a crucial role in regulating the immune system, where the vast majority of immune cells initially generated are eliminated. The role of the extrinsic pathway in cancer is more limited. A few types of cancers overexpress “decoy receptors,” which bind to the death signals but attenuate, rather than activate, the apoptotic response. Interestingly, cancer cells are often responsive to an extrinsic pathway, including the one involved in immune cell elimination, but their normal counterparts are not. It is hoped that this differential sensitivity to extrinsic death signals can be exploited as a therapeutic cancer treatment in the future.
Resistance to Apoptosis via the Intrinsic Pathway Is a Hallmark of Cancer
Internal cellular damage or stress, including DNA damage, absence of cell anchorage, too little or too much oxygen metabolism, oncogene activation, and radiation damage, can stimulate the intrinsic pathway of apoptosis in normal cells. Most, and perhaps all, cancer cells are more resistant than normal cells to apoptosis through this pathway. Resistance to apoptosis not only increases the probability that the cell will be able to accumulate further genetic damage, but also reduces the likelihood that cancer cells can be eliminated. This is because the antitumor activity of the immune system, as well as most chemotherapy and radiation treatments, depend on apoptosis. Thus, resistance to apoptosis often means resistance to treatment.
The intrinsic pathway is considerably more complex than the extrinsic pathway, and this discussion focuses on three major elements of the pathway involved in activating caspases: p53, the mitochondrion, and the Bcl family of proteins (see Figure 2-11). This family of proteins was originally discovered in a cancer (“Bcl” is from B-cell lymphoma, a type of leukemia in which the first such protein was discovered) and includes both pro-apoptotic and anti-apoptotic members. The balance between pro- and anti-apoptotic members determines whether the cell lives or dies. The resistance of cancer cells to apoptosis arises not only from mutations, such as those already described for p53, but also from under-expression of pro-apoptotic mediators and overexpression of anti-apoptotic proteins.
We begin with the mitochondrion, familiar as the “powerhouse” of the cell responsible for generating ATP, but also the central control point for the intrinsic pathway of apoptosis. Recall that the mitochondrion has both an inner membrane, responsible for electron transport, and an outer membrane, responsible for compartmentation of this organelle. Pro-apoptotic signals cause the outer membrane of the mitochondria to become leaky, releasing several pro-apoptotic proteins not normally found in the cytoplasm. Among the most important is cytochrome c, an electron transport protein that is only loosely attached to the inner membrane. In the cytoplasm, cytochrome c stimulates the assembly of a multiprotein complex (the apoptosome) that directly stimulates the activity of an activating caspase (caspase-9), ultimately leading to the activation of executioner caspases. What then determines the extent of permeability (leakiness) of the mitochondrial outer membrane?
The Bcl family members are major regulators of mitochondrial outer membrane permeability. The pro-apoptotic members of this family, such as Bax, lead to permeabilization by assembling to form channels in the outer membrane through which cytochrome c can pass. Pro-apoptotic members of the family can also cause the channel through which ATP normally passes into the cytoplasm to open wider than usual. The anti-apoptotic members of the family, such as Bcl-2, seem to function by binding to pro-apoptotic members, inhibiting their activity. In a healthy cell, anti-apoptotic Bcl members are at high enough concentration to neutralize pro-apoptotic activity. Damage increases the amount of pro-apoptotic Bcl molecules and leads to membrane permeabilization. Thus the balance between pro- and anti-apoptotic members of the family controls the permeability state of mitochondria and the survival of the cell.
With about 20 different members of the Bcl family, the balance between pro- and anti-apoptotic Bcl molecules has multiple controls, but p53 activity is certainly a major player. Recall that when activated (e.g., by DNA damage), p53 acts as a transcription factor, and at least three different pro-apoptotic Bcl genes are transcriptionally activated by p53. These include Bax, and also the particularly powerful pro-apoptotic protein, PUMA. Downstream, p53 also activates the transcription of the activating caspase-9 gene, and the gene of a major cytoplasmic component of the apoptosome. In addition to acting as an activating transcription factor, p53 serves as an inhibitory transcription factor for some genes, including that of the anti-apoptotic Bcl-2 protein. Finally and independent of transcription, activated p53 can directly activate Bax, which is required for its ability to assemble into channel structures. With these multiple effects on apoptotic genes and proteins, p53 is regarded as a central apoptotic control point, in addition to its role in cell cycle regulation.
As noted earlier in the discussion of p53, the importance of apoptosis to tumorigenesis is that with normal apoptosis, almost all damaged cells are eliminated. Without apoptosis, damaged cells live to accumulate additional damage, which illustrates why multiple mutations and dysfunctions are required for tumors to reach a clinically significant stage. The resistance of cancer cells to apoptosis arises from many types of mutations and disruptions of normal gene expression. Perhaps most importantly, mutation of the p53 gene eliminates its DNA binding and thus transcriptional activity. Related to p53 activity is a protein engaged in p53’s normal proteolytic breakdown (see previous discussion). Overexpression of this protein (MDM2) in various cancers of soft tissues inhibits the accumulation of p53 to active levels and therefore inhibits both cell cycle arrest and apoptosis. The anti-apoptotic Bcl-2 protein is overexpressed in a variety of human cancers, including 60% of human follicular lymphomas, but also some lung cancers, melanoma, and prostate cancer. Another common apoptotic lesion seen in cancer cells is overexpression of proteins that bind to and directly inactivate caspases, as well as mutation or loss of expression of the caspases themselves.
Cellular Life Span Is Determined by DNA Sequences at the Ends of Chromosomes
The final major dysfunction of growth control found within cancer cells is the most recently discovered, but also seems to be the most common single molecular lesion in cancers: the expression of a reverse transcriptase called telomerase. (A reverse transcriptase is any enzyme that synthesizes DNA from an RNA template.) Telomerase is responsible for replicating telomeres, the specialized, noncoding regions of DNA found at the end of chromosomes. However, telomerase is normally expressed only in embryonic cells and in adult stem cells. (stem cells are specialized normal cells that do have limitless replicative potential, such as gamete-generating cells and the blood-forming cells of the bone marrow, as discussed later.) The vast majority of normal somatic cells do not express telomerase, but it is expressed in 85% to 90% of all cancers and is the major determinant of the “immortality” of cancer cells.
Telomeres are segments of highly repetitive DNA, representing hundreds of repeats of the simple nucleotide sequence TTAGGG (in vertebrates), found at the ends of chromosomes. Telomeres serve as caps at chromosomal ends, protecting them against end-to-end joining of chromosomes. Telomeres also prevent the ends of chromosomes from being recognized as sites of DNA damage (double-strand DNA breaks). Most relevant for cancer, telomeres protect against the loss of coding DNA from each chromosomal end with every round of DNA replication; this is needed because normal DNA polymerases have a serious limitation: they cannot fully replicate the end of a double-strand DNA molecule. As a result, the ends of chromosomes become shorter with each round of DNA replication. (Bacteria solve this problem by having circular DNA chromosomes.)
Telomeres are expendable DNA, at the ends of chromosomes, whose progressive shortening does not compromise the coding function of the genome. Although no coding sequence is lost, the shortening of telomeres nevertheless plays an important role in the cell. The shortening of telomeres serves as a kind of clock, measuring the number of times a cell has divided, and the length of the telomere reflects the age of the cell. Through poorly understood mechanisms, cells can detect the length of their telomeres, and when they reach a critically short length, the cell ceases to divide and is said to undergo senescence (Latin for “growing old”). As noted earlier, normal cells have a finite life span, such that a cell taken from a middle-aged human will divide 20 to 40 times in culture before senescence. When placed into culture, the number of subsequent cell divisions before senescence reflects the original length of the telomeres. Further, various degenerative diseases, including cirrhosis of the liver, have been shown to accelerate telomere shortening. In principle, senescence is a powerful block to cancer because the original damaged cell (see Figure 2-1) would be unable to divide for a sufficient number of generations to accumulate the necessary multiple mutations required to produce a tumor. Telomerase expression (and other, less common means of elongating telomeres) effectively eliminates this block to cancer development by causing the cells to become immortal.
Telomerase has both protein and RNA components. The protein provides the catalytic reverse transcriptase, allowing the enzyme to elongate the telomere sequence based on the RNA template it carries. That is, the RNA component of telomerase is complementary to the telomere DNA sequence and is used as the template for telomere DNA replication. Telomerase is not expressed in normal adult somatic cells except for stem cells, mentioned earlier. However, immortal tissue culture cells do express telomerase, as do cancer cells. Experimental expression of telomerase in human cells dramatically increases the replicative life span of the cells. Thus the observed expression of telomerase in the vast majority of human cancers permits these cells to divide indefinitely, providing yet another selective advantage for these cells to accumulate other damage over time.
In the last sections of this chapter, we turn our attention to the cancer cell in the context of a tumor, which is a population of cancer cells interacting with one another and with surrounding normal tissue. We end our discussion of the intrinsic growth controls of normal and cancer cells with an experimental result that seems to confirm the importance of the types of damage discussed thus far. This experiment showed that four genetic changes were sufficient to transform normal human kidney cells into cancer cells able to form tumors when transplanted into a mouse host (with no immune system). The four genetic changes were to “engineer” into the cells an activating mutation for the ras oncogene, inactivation of both the RB and p53 proteins, and expression of the catalytic subunit of telomerase. Thus, damage to the genes or expression of these molecules, emphasized here, reflects the minimum requirements for a normal cell to grow as a cancer.
TUMOR ORIGIN AND THE SPREAD OF CANCER
Cancer Cells May Be Related to Stem Cells
As noted in the previous section, some normal adult cells do have unlimited replicative potential. These are “stem cells,” and that term has been much in the news recently. A stem cell is a self-renewing cell of high proliferative potential that can also give rise to differentiated cells. Typically, stem cell division produces one cell that remains a stem cell while the other daughter cell differentiates into a specialized cell with the usual limited life span (Figure 2-12). The cell that continues being a stem cell does not lose any developmental capacity and can divide indefinitely, continuing to produce additional stem cells and additional differentiated cells.
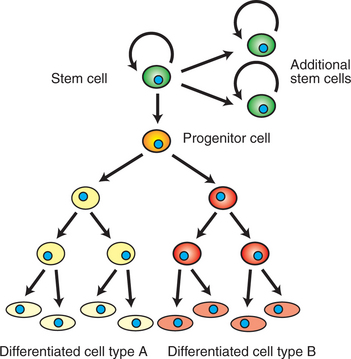
FIGURE 2-12 Stem cells. Stem cells are self-renewing cells of high, sometimes unlimited, replicative potential. Their proliferation forms both additional stem cells and “progenitor cells.” These progenitor cells divide and eventually differentiate to become one or more types of differentiated somatic cells specialized for certain tasks (e.g., erythrocytes and monocytes of blood).
Much of the recent attention in the news centers on embryonic stem cells. These are embryonic cells that can either continue to form stem cells or differentiate, in principle, to any and every cell type within the body. Even in the adult, however, the maintenance of many normal tissues is critically dependent on stem cells. Adult stem cells, however, can only differentiate into a limited array of different cell types, not every cell type in the body. Best understood is that all the various cells of the blood arise from the division of hematopoietic stem cells in the bone marrow; one daughter cell remains a stem cell in the bone marrow while the other differentiates to become one of the several types of blood cells (but the blood stem cell can only form blood cells, not nonblood cells). The cells lining the gut and skin cells also arise from a stable population of adult stem cells, some of whose descendants differentiate into specialized gut and skin cells. For this reason, chemotherapy that is intended to cause apoptosis in cancer cells typically also affects these same populations of normal stem cells; common side effects of chemotherapy include anemia, hair loss, and digestive dysfunction.
Cancer cells resemble stem cells in their immortality, but the relationship of cancer cells to stem cells may go further. Based on the presentation thus far, you may have the mental image of a tumor composed of a uniform population of badly damaged cells, any of which would be capable of forming a new tumor if transplanted. In fact, real tumors are not a homogeneous population of cells, but rather are composed of a variety of cells that differ significantly in their phenotype, despite all being clonal descendants of a single somatic cell, as shown in Figure 2-1. (Keep in mind that all somatic cells of the body are clonal descendants of the fertilized egg, so phenotypic differences arising within clonal lines is not surprising by itself.) Further, experiments with a variety of cancers show that only 1% or less of tumor cells are capable of forming another tumor, even in the same patient (or mouse). Thus, tumors may contain a small subpopulation of cancer stem cells that are responsible for producing the heterogeneous cells in the tumor and are uniquely able to continue cancer growth. This would also give tumors the capacity to adapt to their surroundings; because stem cells can differentiate in various ways, differentiated cells that allowed continued growth and survival would be selected.
This hypothesis has been persuasively supported only in leukemias, but it may apply to other cancers as well. Also for leukemias, the cancer stem cells express some marker proteins characteristic of normal hematopoietic stem cells. Further, only those leukemia stem cells expressing certain normal markers are capable of forming new cancers when transplanted. However, blood is unusual in ways other than being a fluid, not a solid tissue, and it is unknown whether other types of cancers will prove similar. Nevertheless, these results suggest that cancer therapy should be directed primarily at cancer stem cells and not the majority of cells in a tumor. Also, stem cell markers might be used for drug targeting, thus sparing the vast majority of cells in the body from side effects of the treatment.
Finally, the genetic changes summarized in this chapter might need to occur in a normal adult stem cell to produce cancer cells. This “cancer stem cell hypothesis” is controversial and remains to be proved, but it suggests that most cells of the adult are not capable of giving rise to cancer; only changes in stem cells can give rise to tumors. This is the most extreme of the controversial ideas surrounding the relationships between stem cells and cancer cells. Given the results for certain leukemias, however, and the resulting “revolution” if found to be characteristic of other cancers, it seems worthwhile to consider the emerging ideas about stem cells and cancer.
Death by Cancer Is Usually the Result of Its Spread, Not the Original Tumor
Death from cancer is often the result of the spread of the cancer from the initial tumor, the primary tumor, to various distant sites. This process of cancer cells colonizing other tissues is called metastasis. For some cancer types, including leukemias, and those of the brain, the primary tumor itself can be fatal. In contrast, the primary tumor for melanoma, a very deadly cancer, is little more than a mole on the skin that does not become life threatening until these cancer cells spread. Although metastasis is the deadliest aspect of cancer, much less is known about it than about the dysfunctions of cell growth leading to the primary tumor.
The best understood aspect of metastasis is that it occurs by a multistep process called the metastatic cascade. In this step-by-step process, cells escape from the primary tumor, breaking through tissue barriers to gain access to the circulatory system. The cells are carried until they escape the circulatory system to invade a new tissue (Figure 2-13). The steps of the metastatic cascade suggest that dysfunction of three broad types of cellular function are particularly important: cellular adhesion, cellular motility, and secretion of proteases. How these dysfunctions arise from the genetic damage of growth in the primary tumor is, once again, unknown, but mutation resulting from the genetic instability of the primary tumor is typically suggested as a link.
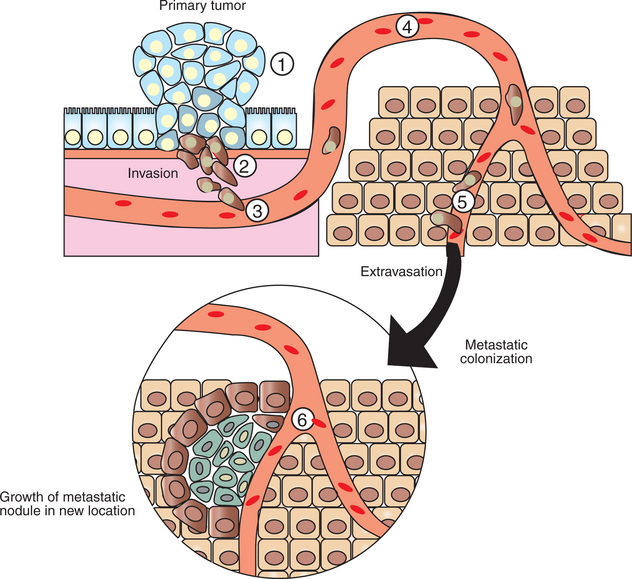
FIGURE 2-13 Metastatic cascade, the path from primary tumor to metastatic tumor. Cells of the primary tumor alter their cell adhesion and motility properties to migrate away from the primary tumor site (1). These cells secrete proteases to digest their way through the surrounding tissue (2). They then crawl into the vasculature (3), a process called intravasation, where they are then carried passively around the circulation (4). At some point, they adhere to the sides of the blood vessel and crawl out of the vasculature (5), a process called extravasation. Some metastatic cells are able to colonize the new location to form a new, deadly metastatic tumor (6).
The first step of the metastatic cascade is the loss of cell adhesion by the cancer cell, both to neighboring cells and to the extracellular matrix (ECM). Accordingly, many types of cancer cells show greatly reduced expression of a cell-cell adhesion receptor, E-cadherin, important for epithelial adhesion. Similarly, primary tumor cells show a wide variety of abnormalities in the number and type of cell-ECM adhesion receptors, integrins, they express. In addition to loosening the bonds to the primary tumor, allowing cells to escape, one hypothesis is that these changes in cell adhesion molecules underlie the curious tendency of various cancers to metastasize preferentially to certain other tissues. Melanoma, for example, has a strong tendency to metastasize to the brain and to bone. Melanoma’s particular array of abnormal (for skin) adhesion molecules may represent a “postal code” favoring delivery to a particular distant site.
Having altered its adhesion, enabling escape from the primary tumor, the metastatic cell must make its way toward the circulatory system, enter the circulation (called intravasation) to “hitch a ride” around the body. Although “circulation” typically refers to the bloodstream, cancer cells can also be disseminated by traveling within the lymphatic system, which collects extracellular tissue fluid for return to the blood. Indeed, invasion of lymph nodes, which are major collection sites for extracellular fluid and debris, is a common test for initial metastases. For either route, however, the cell’s ability to achieve intravasation depends on altering normal motility and expressing proteases. Most animal cell types are capable of “crawling” locomotion using actin and myosin mechanisms similar to muscle contraction (see Figure 1-4). This crawling locomotion is similar to the motility of amebae. Migrating breast cancer cells have been imaged directly and show solitary cells with amoeboid morphology. The entire actin and myosin system of most cancer cells is dysregulated, causing changes in cell shape and the ability and tendency to locomote. For example, normal skin cells are generally quite stationary, but melanoma cells are highly motile. The dysregulation of the actomyosin system results in part from mutations of the Rho family of small, Ras-like G proteins, mentioned briefly earlier. Mutations in rho are common among highly metastatic melanoma cells, but such mutations are rare among weakly metastatic melanoma cell lines.
Because cells in solid tissues are crowded together, increased motility appears to be helped by secretion of proteases that digest some of the cell matrix “obstacles” in the cancer cell’s path. Epithelial cells give rise to approximately 80% of human cancers. As noted earlier, all epithelial cells are attached to an ECM, which is characterized by a particular type of collagen. Proteases specific for this type of collagen are generally overexpressed by metastatic cells. The number of different proteases and the net amount of protease secreted tend to increase with increasing metastatic potential. In addition, cancer cells appear to have the capacity to cause surrounding normal cells to increase their secretion of proteases. Proteases not only aid the metastatic cell in intravasation, but also stimulate cell survival and proliferation by largely unknown mechanisms. However, not all ECM represents an obstacle to movement that must be proteolytically degraded. Some types of ECM appear to provide stimulatory pathways for the migrating cells to follow toward the circulation. Migration toward the circulation is also aided by chemoattraction; epidermal growth factor in blood vessels seems to be an attractant for breast cancer cells.
After intravasation, the metastatic cell rides around the circulation until it can attach to the vessel wall. Then, reversing the process of intravasation, the cell “climbs out” of the circulation, called extravasation. As one might expect, this also depends on changes in adhesion, motility, and protease secretion. After extravasation, the metastatic cell must be able to survive and proliferate in its new environment.
Fewer than 1 in 10,000 cells escaping the primary tumor colonize a new location successfully. It was once assumed that this high rate of failure reflected an “exceedingly rough ride” around the circulatory system. More recent evidence suggests that the limiting factor of metastasis is the survival of the cell in its new location. This represents another example of the natural selection, the “microevolution,” occurring in cancer. The foreign environment exerts a strong negative selective pressure on cancer cell arrivals, and the vast majority do not survive. Ironically, our current thinking about metastasis is similar to the “seed and soil hypothesis” first proposed in 1889. Metastasis requires a cancer cell from the primary tumor (the seed) capable of carrying out (selected for) all the steps of the metastatic cascade, and the metastatic cell must colonize a region (the soil) appropriate for its subsequent growth. Normal cells cannot survive in a new location within the same body. Different tissues have different chemical milieus (e.g., different mixes of growth factors), and these are specialized for the survival and growth of the particular cell types found in the region. The process of metastasis necessarily selects for cells capable of growing in a foreign environment. It is thought that most metastatic cells become dormant in their new location and that additional genetic changes are required and must be selected to enable uncontrolled growth. Fewer than 1% of micrometastases grow to a clinically relevant size. A key aspect of this selection process for uncontrolled growth in the foreign location is the secretion by the cancer cells and by surrounding cells of a variety of mediators to stimulate growth of new blood vessels to supply the tumor. Another key feature of selection is remodeling of the local ECM, which normally is required for proliferation. these phenomena of inducing blood supply and remodeling of the ECM are other aspects of metastasis that are poorly understood.
Indeed, it is poorly understood how metastasis actually leads to death, except that it often involves a profound and overall wasting of the body, cachexia. Cachexia is complex, but it is caused in part by generalized inflammatory reactions, which attack other aspects of the patient’s physiology. Thus, it appears that the presence of foreign cells, selected for growth in an abnormal location, causes the body’s defense mechanisms to be fully mobilized and attack itself, a nightmare of biological “friendly fire.” Presumably, the “foreignness” of the metastatic tumor explains the highly inappropriate response, which primary tumors typically do not instigate. For this reason, in part, complete removal of the primary tumor before metastasis occurs often leads to total recovery. In other cases, death from metastatic disease, similar to death from some primary tumors, is the result of cancer cells simply overwhelming a vital organ, leading to organ failure.
Growth of Solid Tumors Depends on Development of New Blood Vessels
Tumors, as with normal tissue, require blood vessels to supply them with oxygen and nutrients and to remove waste. Much attention has been focused on the development of tumor capillaries because it is a rate-limiting step in the tumor’s growth and progression. Both primary and metastatic tumors require new vessels; without them the tumor remains too small to be visible or palpable, about 1 to 2 mm in diameter. Dormant tumors of this size have been found in autopsies of people who did not die of cancer, so not all tumors develop the blood supply needed for growth. Thus the ability of tumors to stimulate new blood vessel development is a distinct and important step in tumor progression. As this suggests, it is also a relatively early step in tumor progression but is covered here after metastasis because most new vasculature arises from existing capillaries invading new regions of tissue, sharing some features with metastasis.
The discussion of Starling’s hypothesis in Chapter 1 notes that blood capillaries are composed primarily of a single layer of a specialized epithelial cell type, the endothelial cell. The first capillaries in the embryo are formed by vasculogenesis, the differentiation of precursor cells (angioblasts) to form a basic capillary network. However, most new capillaries are formed by angiogenesis, the sprouting and branching of existing capillaries to supply new tissue regions. Larger blood vessels, such as arterioles and veins, all develop from the subsequent growth of capillaries. In the adult, only angiogenesis normally occurs and depends on invasive cellular processes similar to those involved in metastasis: proliferation of existing endothelial cells; migration of the cells into the region to be supplied, involving changes in actin function and adhesion to the surrounding cells; and remodeling the surrounding ECM so the extending cells intercalate among the tissue cells, ultimately to form a hollow tube. Although the cancer cells within a tumor are abnormal, the endothelial cells composing the new capillaries are normal. Thus, tumor capillaries can arise by vasculogenesis (because of the abnormal environment of the tumor) or, and primarily, by angiogenesis. Similarly, the endothelial cells of tumor capillaries respond to the normal stimulatory and inhibitory signals for angiogenesis. Nevertheless, the pathological features of the tumor stimulate abnormal growth of blood vessels, whose pattern, composition, and function differ from normal capillaries.
In normal adult tissue, except for the female reproductive tract, endothelial cells are among the most slowly proliferating cell type. Only 1 in 10,000 adult endothelial cells are in cell division at any one time, compared with about 10% of gut epithelial cells. Normal angiogenesis is under tight regulation by both stimulatory and inhibitory influences. Stimulatory influences include injury and hypoxia which in turn lead to the secretion of angiogenic growth factors such as vascular endothelial growth factor (VEGF, “vedge-eff”). This growth factor strongly stimulates endothelial cell proliferation and migration and suppresses apoptosis. VEGF also increases permeability of existing vessels. Inhibitory influences include thrombospondin-1, which is an ECM component that inhibits endothelial cell proliferation and motility. Inhibitory influences also include soluble factors such as angiostatin, which stimulates apoptosis in proliferating endothelial cells, and endostatin, which inhibits the migration of endothelial cells. The growth, stasis, or regression of capillaries, depends on the balance between pro- and anti-angiogenic stimuli, much as cellular life and death depends on the balance between pro- and anti-apoptotic signals discussed earlier.
The relative quiescence of normal capillaries is in sharp contrast to capillaries of tumors, which have been compared to “wounds that never heal,” in that tumor capillaries undergo continuous growth and remodeling. Tumor endothelial cells divide 20 to 40 times more frequently than normal endothelial cells, and tumors typically have a much higher density of vessels than normal tissue. As a result, tumor vasculature is abnormal in structure and function. Tumor vessels can exhibit strange combinations of capillary, venous, and arteriole structures and often incorporate cancer cells as part of the vessel wall. These vessels tend to be convoluted and dilated, follow tortuous paths, and even form dead ends. As a result, blood flow is equally abnormal, with the vessels leakier than normal vessels, and in some cases the blood flows back and forth rather than circulates.
Perhaps the most important factor in this vascular pathology is the high concentration of VEGF in and around tumors. Most human tumors secrete large amounts of VEGF and also cause surrounding tissue to secrete VEGF. Much evidence from experiments on mice supports the crucial role of VEGF in tumor angiogenesis and growth. Antibodies against VEGF suppress growth of existing tumors; cancer cells engineered to be incapable of expressing VEGF were unable to form tumors; and inhibition of the VEGF receptor inhibited the growth of a variety of tumors. In part, the secretion of VEGF by tumor cells seems to be the result of the initial hypoxic conditions of the avascular tumor. Hypoxia is normally a strong inducer of VEGF production, and the centers of many solid tumors show necrotic cells indicative of death from lack of oxygen. In addition, the genetic damage to cells in their progression to a cancer cell also seems to contribute to VEGF overexpression. Mutations of Ras and overexpression of Bcl-2, the anti-apoptotic factor, have been shown to play important roles in this regard.
Tumor vessels are also substantially more permeable than normal vessels, to the point of being almost hemorrhagic, which is also thought to be caused by overexpression of VEGF (which has an alternative name of “vascular permeability factor”). The leakiness of tumor vessels has several consequences with respect to tumor physiology, spread, and treatment. The high vascular permeability of tumors is believed to aid metastasis in that metastasis requires intravasation of tumor cells into the circulation, and leakier vessels makes this more likely. Leakier vessels also disrupt capillary fluid transport, as discussed in Chapter 1. Recall that capillary filtration and reabsorption depend on the balance between hydrostatic and oncotic forces across the capillary wall. The increased fluid leaking from tumor vessels distends the interstitial space, increasing its hydrostatic pressure and thus reducing the pressure gradient across the capillary wall. The oncotic pressure gradient is also reduced because the leak of proteins into the interstitial space means that the oncotic pressure of the interstitial space approaches that of the blood. The result is uncommonly high net interstitial fluid pressure. This can cause collapse of some vessels, leading to hypoxia of the surrounding tissue and further upregulation of VEGF expression. High interstitial fluid pressure also causes poor fluid transport out of the blood into the tumor. This poor fluid exchange seems to inhibit the delivery of chemotherapeutic agents from the blood to the tumor. Studies on chemotherapy of breast cancer and melanoma show that tumors with high interstitial fluid pressure tended not to respond as well to the therapy.
As with the other insights into tumor biology, the possibility of controlling tumor angiogenesis for therapy is being actively pursued. About a dozen antiangiogenesis compounds are being tested, and bevacizumab (an antibody to VEGF) is approved as a first-line therapy for metastatic colon cancer. Unlike most cancer therapy that targets the abnormal cancer cell, antiangiogenic therapy would be targeting normal endothelial cells. These cells are not genetically unstable, and therefore development of drug resistance may be less likely (see following discussion). Also, because normal endothelial cells are unusually quiescent, inhibiting angiogenesis should produce fewer toxic side effects than standard chemotherapies. As with other cellularly targeted therapies, however, antiangiogenesis inhibitors that showed dramatic results in preclinical studies have been much less successful in treating patients.
PROSPECTIVE CANCER THERAPY
Cancer Therapy Has a Hopeful but Challenging Future
At the clinical level, the explosion of knowledge about the molecular and cellular basis of cancer has had only modest effects on the diagnosis and treatment of most types of cancer. Although new treatment trials and attempts at practical molecular diagnosis have been disappointing for different reasons, three common themes seem to account for treatment failure, reflecting the fundamental properties of cancer.
First, the accumulation of mutations, along with the differences in this process from individual to individual, means that single molecular markers have not proved very useful in refining diagnosis. For example, assessing the different mutations occurring in such important genes as ras or the p53 gene in breast cancer have had conflicting results in predicting disease outcomes. Presumably this is because these mutations have differing effects, depending on the other mutations involved in the cancer and their interaction with the normal alleles of the individual patient. As a result, it appears that multiprotein/multigene molecular “signatures” will be needed. If such signatures can be developed as body fluid or other relatively noninvasive tests, it could lead to major improvements in treatment, insofar as diagnosing cancer as early as possible is crucial for a favorable prognosis.
The second common theme is that the multiple types of genetic damage and selective processes required for cancer also function to cause resistance to treatment. That is, the unstable and abnormal genetic status of cancer cells that produce the growth abnormalities also lead to abnormal responses to drugs and other interventions. A vivid illustration of this is, ironically, one of the notable successes of targeting particular molecular processes in treating cancer. Chronic myeloid leukemia (CML) is known to begin with a specialized mutation (a particular chromosomal translocation) that disrupts the gene for a specific tyrosine kinase, Abl, so that it becomes an activated oncogene. A fairly specific inhibitor of this tyrosine kinase was developed, imatinib (Gleevec), that blocks binding of ATP, disabling kinase activity. This has had marked benefits for patients in the early, chronic stage of CML, which is debilitating but not fatal. In many patients, this drug causes complete remission of CML and has thus far prevented progression to the fatal, acute stage. However, some patients have developed resistance; in most of these cases the abl oncogene has mutated yet again such that ATP binding is restored despite Gleevec. More ominously, careful analysis of the blood of CML patients actually in remission indicates a remaining pool of leukemic cells (cancer stem cells apparently), which may subsequently lead to development of resistance in later years. Nevertheless, there are currently more than 20 “specific” protein kinase inhibitors in clinical trials, and practitioners would welcome additional drugs with the effectiveness of Gleevec, despite its limitations.
In addition to outright mutations leading to cancer, we have mentioned several examples in which changes in gene expression of normal proteins stimulate cancer development. This also applies to drug resistance, and this mechanism underlies another, broader example of the obstacle to treatment presented by the genetic status of cancer cells. Multiple-drug resistance (MDR) is a phenotype in which cells develop resistance to many current, initially effective, chemotherapeutic agents for a wide variety of cancers. This is the result of the expression, or overexpression, of a pump protein that causes the efflux of the drug from the cell. As with the selection among cancer cells for continued ability to proliferate, administration of the drug selects for those cancer cell variants that have changes in gene expression, such that the efflux pump reduces the effectiveness of the drug. Thus, new drug development must contend not only with the genetics of cancer, but also the genes and gene expression involved in drug resistance. (An interesting aspect of the drug efflux pump often expressed in cancer cells is that it is also expressed in normal stem cells!)
The third common theme identified as an obstacle to molecular cancer treatments is that, as discussed, cancers reflect physiological dysfunctions at a particularly fundamental level. It is not easy to interfere with these functions without compromising other functions, or interference engages compensatory mechanisms normally serving as “backup” to crucial functions. At the simplest level, interventions that alter these basic mechanisms of cellular life and death often prove to be too disruptive to the physiology of some normal cells to be useful. Many inhibitors in the growth factor/MAP kinase pathway (see Figure 2-5) that showed promise on cultured cells and in mice proved to be too toxic for therapeutic use.
Other results indicate that effective treatments will need to resemble the normal molecular biology of the cell very closely. Experiments attempting to target p53 are noteworthy in this respect. Because mutation of one p53 gene will predispose to cancer (if the other copy were lost, an important checkpoint would be lost), activation of the remaining normal copy might protect against cancer. Such enhanced p53 activity did protect against cancer in mice, but the mice also showed notably shortened life span and visible signs of early aging. As shown by this unexpected role of p53 in aging, the central roles played by proto-oncogenes and tumor suppressor genes means that they often have multiple roles that complicate development of therapies. In experiments in which expression of activated p53 was limited to mammary tissue, mice were again protected against cancer, but at the cost of inhibiting lactation and mammary development. The best anticancer results obtained from experimentally manipulating p53 expression has come from experiments in which whole artificial chromosomes with the p53 gene and all its normal control elements were introduced into mice. These mice showed increased resistance to chemically induced cancers with no apparent effects on aging. introducing genes with all relevant control elements, however, is a rather high hurdle for practical therapies.
Finally, the importance of these normal genes and proteins to cell function means that they often have redundant mechanisms of control. This seems to apply to that other “usual suspect” in cancer, Ras. Evidence that association with the plasma membrane via lipid “tails” was required for Ras activity (similar to the alpha subunit of the heterotrimeric G protein, see Figure 1-14) led to the development of drugs, farnesyl transferase inhibitors (FTIs), that block addition of the lipid tail. Although FTIs have proved clinically useful against some types of cancer in some patients, their effects are highly variable. One idea is that the FTIs only inhibit one pathway for Ras membrane association. Used alone, these drugs showed only modest effects on tumors, but in combination with standard chemotherapeutic drugs, FTIs worked relatively well on some cancers. However, it was puzzling that some cancers importantly involving ras mutations, such as lung cancer, were not affected by the inhibitors. Further, some ras-independent tumors were just as susceptible to FTIs. It now seems that these drugs may not be acting only through Ras membrane association.
It should be noted that standard chemotherapies and radiation therapies are highly toxic by the usual standards of clinical practice. Cancer therapy is a prime medical example of “drowning men grasping at straws.” Thus the modest clinical advances accompanying significant basic science advances in understanding cancer are widely regarded as being hopeful. The enormous success in treating infectious diseases with antibiotics and vaccines may be an unrealistic model for chronic diseases generally, and cancer in particular. Currently, much work is directed at developing “cocktails” of tumor inhibitors that would be both more specific and less toxic than current treatments.
CLINICAL CORRELATIONS
Dog That Collapsed While Running
History.
A spayed, 10-year-old female golden retriever collapsed while running outside earlier today. The dog is still very lethargic and does not want to move.
Clinical Examination.
The dog has pale mucous membranes with a normal temperature. The capillary refill time is prolonged. Heart rate and respiratory rate are increased. On palpation there appears to be fluid in the abdomen, and the dog is in pain.
Comment.
Based on this history and physical examination, there is concern that this dog has hemorrhaged into the abdomen. Hemangiosarcoma is a common tumor of older dogs and originates from a transformed endothelial cell. Dogs often present after having collapsed when the tumor, which is present in the spleen, causes internal bleeding. The dogs must often have emergency surgery to have a splenectomy (spleen removed). In some cases, dogs may show other, nonspecific clinical signs (inappetence, lethargy), so a diagnosis may be made before the dog collapses from acute bleeding. A diagnosis is often made through a combination of modalities, including radiographs, ultrasound, biopsies, histopathology, and immunohistochemistry, to determine the nature of the tumor. In many cases, by the time the diagnosis has been made, the tumor has already metastasized, usually via hematogenous route, to other organs. The lung and liver are more frequently affected, but other sites include kidney, muscle, brain, mesentery, skin, and lymph nodes. Recently it has been demonstrated that canine hemangiosarcomas express platelet-derived growth factor beta (PDGF-β). Suppression of this RTK signaling using imatinib (Gleevec) suppressed the canine cell line in a mouse model.
Treatment.
Depending on the stage at which the tumor is diagnosed, as in this case, the animal presents with shock and hemorrhage. In these cases the patient is stabilized, surgery is performed, and the spleen (in this patient) is removed. The overall prognosis for these cases is poor because the tumor has usually metastasized by the time the initial diagnosis is made. Radiation therapy is palliative for these cases, and it is sometimes used when there is a large, local unresectable mass. Chemotherapy is usually the treatment of choice, although median survival time for these dogs typically is not long. Medications often include the VAC protocol: doxorubicin, cyclophosphamide, and vincristine. Doxorubicin inhibits DNA synthesis, DNA-dependent RNA synthesis, as well as protein synthesis, and it acts throughout the cell cycle. Cyclophosphamide inhibits DNA replication as well as RNA transcription and replication. Vincristine binds to specific microtubular proteins to inhibit cell division. Complications associated with chemotherapy include myelosuppression and sepsis. Experimental treatments are still being tested and target endothelial cells, blocking adhesion factors and inhibiting growth factors associated with endothelial cell growth.
Bjerkvig R, Tysnes BB, Aboody KS, et al. The origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005;5:899.
Chan SR, Blackburn EH. Telomeres and telomerase. Phil Trans R Soc Lond B. 2004;359:109.
Engers R, Gabbert HE. Mechanisms of tumor metastasis: cell biological aspects and clinical implications. J Cancer Res Clin Oncol. 2000;126:682.
Goldman JM, Melo JV. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1084.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57.
Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:769.
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899.
Krontiris TG. Molecular medicine: oncogenes. N Engl J Med. 1995;333:303.
Tisdale MJ. Molecular pathways leading to cachexia. Physiology (Bethesda). 2005;20:340.
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171.
PRACTICE QUESTIONS
1. Which of the following is associated with normal stimulation of cellular proliferation?
2. In the growth factor pathway, the growth factor first binds to ________ which leads to activation of _______, in turn causing activation of a cascade of ___________ enzymes leading to alterations in transcription.
3. Which of the following mediate(s) apoptosis?
4. The tumor suppressor pRB is a(n) ________ and participates in regulating the cell cycles of _________ cells, whereas p53 is a(n) ______________ and participates in regulating the cell cycle of ____________ cells.
5. Normal stem cells are similar to cancer cells but differ from normal somatic cells in that normal stem cells and cancer cells both:
VOCABULARY
This chapter is unusual in that it contains a large number of vocabulary words that are specialized for cancer and related topics, words that generally will not be used in later chapters. You should be familiar with these vocabulary words, and you should be able to define them and state their role in normal cells, and whether and how they differ in cancer cells.