Chapter 34 Endocrine Glands and Their Function
1. The thyroid hormones are synthesized from two connected tyrosine molecules that contain three or four iodine molecules.
2. Thyroid hormones are stored outside the cell and attached to thyroglobulin in the form of colloid.
3. The release of thyroid hormones involves transport of thyroglobulin with attached thyroid hormones into the cell, cleavage of the thyroid hormones from thyroxine-binding globulin, and release into the interstitial tissues.
4. Thyroid hormones are transported in the plasma attached to plasma proteins.
5. The main routes of metabolism of thyroid hormones are through deiodination or the formation of glucuronides and sulfates via hepatic mechanisms.
6. Thyroid hormones are the primary factors for the control of basal metabolism.
7. The ingestion of compounds that inhibit the uptake or organic binding of iodine blocks the thyroid’s ability to secrete thyroid hormones and causes goiter.
1. The adrenal glands are composed of two organs: the outer gland (cortex) and the inner gland (medulla).
1. The adrenal cortex has three zones: the zona glomerulosa, which secretes mineralocorticoids, and the zona fasciculata and the zona reticularis, which secrete glucocorticoids and sex steroids.
2. Adrenal corticoids are synthesized from cholesterol; the critical difference in the activity of these corticoids is related to the hydroxyl group on C-17 of glucocorticoids.
3. Adrenocortical hormones are carried in plasma in association with specific binding globulins (corticosteroid-binding globulin).
4. The metabolism of adrenocortical hormones involves the reduction of double bonds and conjugation of the steroids to glucuronides and sulfates.
5. One of the most important functions of glucocorticoids is control of metabolism, in particular the stimulation of hepatic gluconeogenesis.
6. Corticotropin is the pituitary hormone that regulates glucocorticoid synthesis by the adrenal cortex.
7. 7.One of the most important clinical uses of glucocorticoids is the suppression of the inflammatory response.
1. The synthesis of catecholamines is from tyrosine; the main catecholamine synthesized by the adrenal medulla is epinephrine.
2. The primary actions of catecholamines are on metabolism, especially effects that increase the concentration of glucose.
3. The main factors that stimulate catecholamine secretion are hypoglycemia and conditions that produce stress.
1. The synthesis of insulin is biphasic: an acute phase involves the release of preformed insulin, and a chronic phase involves the synthesis of protein.
2. The metabolism of insulin involves splitting the A and B chains and reducing the chains to amino acids and peptides.
3. The main metabolic functions of insulin are anabolic.
4. Insulin deficiency produces diabetes mellitus, which can culminate in diabetic ketoacidosis.
5. Dietary management is an important consideration in therapy for feline type 2 diabetes.
6. The most important functions of glucagon are to decrease glycogen synthesis, increase glycogenolysis, and increase gluconeogenesis.
7. Glucagon synthesis is stimulated by decreased glucose concentrations in the blood.
8. The main functions of somatostatin are to inhibit the secretion of hormones produced by the pancreas (insulin, glucagon, pancreatic polypeptide).
Calcium and Phosphate Metabolism
1. Calcium is important for many intracellular reactions, including muscle contraction, nerve cell activity, release of hormones through exocytosis, and activation of enzymes.
2. Phosphorus is important for the structure of bone and teeth, and organic phosphate serves as part of the cell membrane and several intracellular components.
3. The most important body pool of calcium involved in homeostasis is the extracellular fluid component.
THE THYROID GLAND
In most mammals the thyroid gland is located caudal to the trachea at the level of the first or second tracheal ring. The thyroid gland is composed of two lobes lying on either side of the trachea and connected by a narrow piece of tissue called the isthmus.
The thyroid gland is the most important endocrine gland for metabolic regulation. The glandular tissue has cells formed in a circular arrangement called a follicle (Figure 34-1). The follicles are filled with a homogeneous-staining substance called colloid, which is the main storage form of the thyroid hormones. The follicular cells are cuboidal when the secretion is basal and are elongated when the cells are stimulated to release hormone. Another important endocrine cell, the parafollicular cell, or C cell, is located outside the follicles. This cell secretes calcitonin, a hormone important for the regulation of calcium. The activity of this hormone is discussed in the section on calcium metabolism.
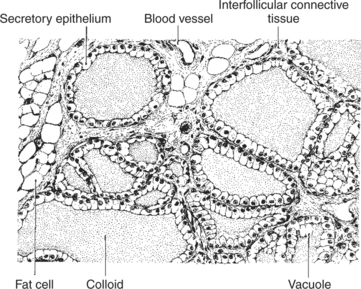
FIGURE 34-1 Histological features of the normal thyroid gland of the rat. All normal thyroid glands are structurally similar, although slight variations occur with age, diet, habitation, and sexual status (neutered or intact). The normal animals of the colony to which this rat belonged were maintained on a high-protein ration, which probably accounts for the slight hypertrophic condition of the secretory epithelium.
(From Turner CD, Bagnara JT: General endocrinology, ed 6, Philadelphia, 1976, Saunders.)
The Thyroid Hormones Are Synthesized from Two Connected Tyrosine Molecules That Contain Three or Four Iodine Molecules
The synthesis of thyroid hormone is unusual because a large amount of the active hormone is stored as a colloid outside the follicle cells, within the lumen (or acinus) created by the circular arrangement of glandular cells. Two molecules are important for thyroid hormone synthesis: tyrosine and iodine. Tyrosine is a part of a large molecule (molecular weight, 660,000D) called thyroglobulin that is formed within the follicle cell and secreted into the lumen of the follicle. Iodine is converted to iodide in the intestinal tract and then is transported to the thyroid, where the follicle cells effectively trap the iodide through an active transport process. This allows intracellular iodide concentrations to be 25 to 200 times higher than extracellular concentrations.
As iodide passes through the apical wall of the cell, it attaches to the ring structures of the tyrosine molecules, which are part of the thyroglobulin amino acid sequence. The tyrosyl ring can accommodate two iodide molecules; if one iodide molecule attaches, it is called monoiodotyrosine, and if two attach, it is called diiodotyrosine. The coupling of two iodinated tyrosine molecules results in the formation of the main thyroid hormones; two diiodotyrosine molecules form tetraiodothyronine, or thyronine (T4), and one monoiodotyrosine and one diiodotyrosine molecule form triiodothyronine (T3) (Figure 34-2). A key enzyme in the biosynthesis of thyroid hormones is thyroperoxidase (which works in concert with an oxidant, hydrogen peroxide). Thyroperoxidase catalyzes the iodination of the tyrosyl residues of thyroxine-binding globulin (TBG) and the formation of T3 and T4. In addition to the unusual molecular storage form of the hormone, thyroid hormones are also unique in that they are the only hormones that contain a halide (i.e., iodine).
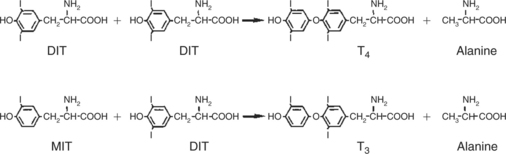
FIGURE 34-2 Production of tetraiodothyronine (thyronine, T4) and triiodothyronine (T3) by the coupling of iodinated tyrosyl residues with thyroglobulin molecule. DIT, Diiodotyrosine; MIT, monoiodotyrosine.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Thyroid Hormones Are Stored Outside the Cell and Attached to Thyroglobulin in the Form of Colloid
Once thyroid hormones are synthesized, they remain in the extracellular acinar lumen until release. This extracellular storage of hormone within an endocrine gland is a unique storage arrangement. It allows the thyroid gland to have a large reserve of hormone. From a teleologic standpoint, thyroid hormone is the most important hormone of metabolism; it allows mammals to withstand periods of iodine deprivation without an immediate effect on the production of thyroid hormones.
The Release of Thyroid Hormones Involves Transport of Thyroglobulin with Attached Thyroid Hormones into the Cell, Cleavage of the Thyroid Hormones from Thyroxine-Binding Globulin, and Release into the Interstitial Tissues
In order for thyroid hormones to be released from the thyroid gland, thyroglobulin with its attached monoiodotyrosine, diiodotyrosine, T3, and T4 molecules must be translocated into the follicle cell, and the hormones must be cleaved from thyroglobulin (Figure 34-3). Key enzymes in this transfer are found in the lysosomes. On entering the cell, the TBG molecules fuse with lysosomes, and lysosomal enzymes cleave both the iodinated tyrosine molecules and the iodinated thyronines from the thyroglobulin molecule. The thyronines are released through the basal cell membrane (they freely pass through the cell membrane); monoiodotyrosine and diiodotyrosine are deiodinated by an enzyme called iodotyrosine dehalogenase; and both the iodide and the remaining tyrosine molecules are recycled to form new hormone in association with thyroglobulin.
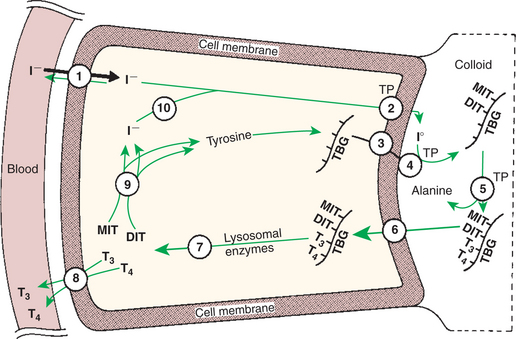
FIGURE 34-3 Depiction of follicular cell showing steps in the synthesis and release of triiodothyronine (T3) and thyronine (T4). The numbers identify the major steps: 1, trapping of iodide; 2, oxidation of iodide; 3, exocytosis of thyroglobulin; 4, iodination of thyroglobulin; 5, coupling of iodotyrosines; 6, endocytosis of thyroglobulin; 7, hydrolysis of thyroglobulin; 8, release of T3 and T4; 9, deiodination of monoiodotyrosine (MIT) and diiodotyrosine (DIT); and 10, recycling of iodide. TBG, Thyroxine-binding globulin; TP, thyroperoxidase.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
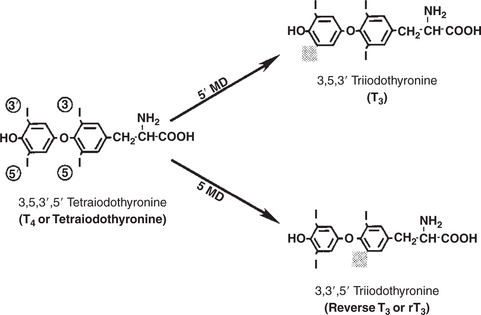
The majority of T3 formation occurs outside the thyroid gland by deiodination of T4. Tissues that have the highest concentration of deiodinating enzymes are those of the liver and kidneys, although muscle tissue produces more T3 on the basis of relative size. The enzyme that is involved in the removal of iodide from the outer phenolic ring of T4 in the formation of T3 is called 5′-monodeiodinase (Figure 34-4). Another type of T3 in which an iodide molecule is removed from the inner phenolic ring of T4, a compound called reverse T3, is also formed. Reverse T3 has little of the biological effects of thyroid hormones and is formed only by the action of extrathyroidal deiodinating enzymes and not by activity of the thyroid gland.
Thyroid Hormones Are Transported in the Plasma Attached to Plasma Proteins
As indicated in Chapter 33, lipid-soluble hormones are transported in the vascular system through association with specific binding plasma proteins. There is considerable species variation in the proteins that bind thyroid hormones. The most important carrier protein is TBG, which has high affinity for T4, although it also has low capacity because of its low concentration. TBG also is an important carrier protein for T3. TBG has been reported in all domestic animals except the cat. Albumin is also involved in the transport of thyroid hormones; however, albumin has low affinity for T3 and T4 but high capacity because of its high concentration in plasma. In the absence of TBG, albumin is the most important carrier of thyroid hormones. All species have a third plasma protein, thyroxine-binding prealbumin, which is specific for T4 and has a specificity and capacity that are intermediate between those of TBG and albumin. The term prealbumin refers to the migration of the protein during electrophoresis, not to synthesis of the molecule.
As with all lipid-soluble hormones that are transported in plasma, most of the T3 and T4 is bound; little is free to interact with receptors on the cells of the target tissues. The amount of thyroid hormone that is free in plasma is remarkably low (e.g., in humans, 0.03% of T4 and 0.3% of T3). In dogs the amount of free hormone is somewhat greater (slightly less than 1.0% for T4 and slightly more than 1.0% for T3) because of less affinity between plasma-binding proteins and thyroid hormones in canine plasma than in human plasma. The equilibrium between free and bound hormone is easily shifted because of physiological or pharmacological situations, such as the increase in estrogen concentrations that occurs during pregnancy. Estrogens cause increased synthesis of TBG by the liver, resulting in a shift toward the bound form. Adjustments to maintain a normal amount of free hormone occur rapidly, with a decline in the rate of metabolism or with stimulation of thyroid hormone production through the release of thyroid-stimulating hormone (TSH).
The Main Routes of Metabolism of Thyroid Hormones Are Through Deiodination or the Formation of Glucuronides and Sulfates via Hepatic Mechanisms
The main form of metabolism of thyroid hormones involves the removal of iodide molecules. Except for the T3 formed from T4, none of the deiodinated thyronine derivatives has any significant metabolic activity. The two enzymes involved in T3 and reverse T3 synthesis, 5′-deiodinase and 5-deiodinase, are also involved in the catabolism of thyroid hormones. Only these two enzymes are needed for catabolism because they do not differentiate between the 3 and 5 positions of the phenolic rings of the thyronines. Skeletal muscle, liver, and kidney tissues are important tissues involved in the catabolism of thyroid hormones through deiodination. The formation of thyroid hormone conjugates represents another form of inactivation; sulfates and glucuronides are formed mainly in the liver and kidneys. Conjugation is less common than deiodination as a means of metabolism of thyroid hormones. Another form of metabolism involves modification of the alanine moiety of the thyronines by either transamination or decarboxylation. The deiodinated and conjugated forms of the thyronines are eliminated primarily in the urine; unmetabolized thyronines are excreted with feces through bile secretion. Degradation of the conjugate forms in the feces results in the production of iodide molecules, which are reabsorbed as part of the enterohepatic cycle. Humans are more efficient than dogs in recovery of iodide both intrathyroidally and enterohepatically.
One of the striking aspects of thyroid hormones is their long half-lives in humans; T3 has a half-life of 1 day and T4 of 6 to 7 days, whereas most other hormones have half-lives of seconds or minutes. One reason for these long half-lives is the large percentage of the circulating thyronines that are bound to the plasma proteins, which protects them from degradation. The difference in half-lives between T3 and T4 results from the tighter T4 protein binding compared with T3 and the resultant reduction in free circulating hormone. In contrast, the half-life for T4 is relatively short in certain domestic species; dogs and cats exhibit a T4 half-life of less than 24 hours.
Thyroid Hormones Are the Primary Factors for the Control of Basal Metabolism
The mechanism of action of thyroid hormones at the cellular level is based on their ability to penetrate the cell membrane even though they are amino acids; in essence, they are lipophilic. Although it is thought that thyroid hormones interact directly with the nucleus to initiate the transcription of messenger ribonucleic acid (mRNA) (Figure 34-5), the presence of T3 receptors has been reported on mitochondria.
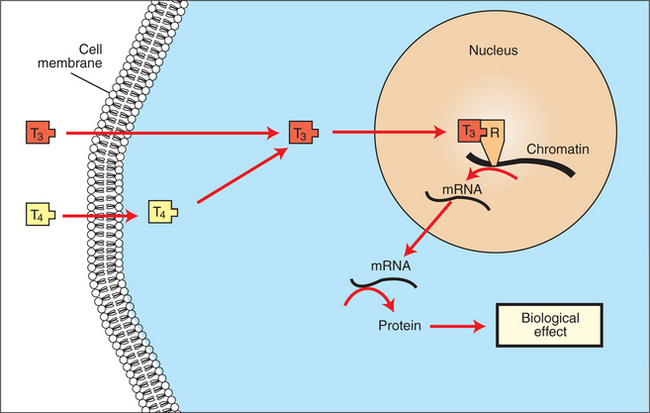
FIGURE 34-5 Proposed subcellular mechanism of thyroid hormone action. mRNA, Messenger ribonucleic acid; R, receptor.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Thyroid hormones are likely the primary determinants of basal metabolism. It is difficult to define their precise physiological effects, however, because many of the effects of thyroid hormones have been demonstrated through the creation of hypothyroid or hyperthyroid states. Nevertheless, it has long been recognized that thyroid hormones increase oxygen consumption of tissues and, as a result, heat production. This effect is known as the calorigenic effect. One site of action of the calorigenic effect of thyroid hormones is within the mitochondrion.
Thyroid hormones affect carbohydrate metabolism in several ways, including increasing intestinal glucose absorption and facilitating the movement of glucose into both fat and muscle. Furthermore, thyroid hormones facilitate insulin-mediated glucose uptake by cells. Glycogen formation is facilitated by small amounts of thyroid hormones; however, glycogenolysis occurs after larger dosages.
Thyroid hormones in concert with growth hormone are essential for normal growth and development. This is accomplished in part by the enhancement of amino acid uptake by tissues and enzyme systems that are involved in protein synthesis.
Whereas thyroid hormones affect all aspects of lipid metabolism, the emphasis is placed on lipolysis. One particular effect of thyroid hormones is the tendency to reduce plasma cholesterol levels. This appears to involve both increased cell uptake of low-density lipoproteins (LDLs) with associated cholesterol molecules and a tendency for increased degradation of both cholesterol and LDL. These effects on lipid metabolism are usually seen in pathophysiological situations involving hypersecretion of thyroid hormone or in thyroid deficiency states in which hypercholesterolemia is a hallmark of thyroid deficiency. In this same context, the effects of thyroid hormones on metabolic processes, including carbohydrate, protein, and lipid metabolism, are often described as catabolic.
Thyroid hormones have noteworthy effects on the nervous and cardiovascular systems. The effects of the sympathetic nervous system are enhanced by the presence of thyroid hormones. This is thought to occur through thyroid stimulation of β-adrenergic receptors in tissues that are targets for the catecholamines, such as epinephrine and norepinephrine. In the central nervous system (CNS), thyroid hormones are important for normal development of tissues in the fetus and neonate; inhibition of mental activity occurs when thyroid hormone exposure is inadequate. In humans, persons with hypothyroid activity are mentally dull and lethargic, which suggests that normal CNS function in the adult depends on the presence of adequate amounts of thyroid hormone.
Thyroid hormones increase the heart rate and force of contraction, probably through their interaction with the catecholamines. This interaction is caused by an increase in tissue responsiveness through the induction of catecholaminergic β receptors by thyroid hormones. Blood pressure is elevated because of increased systolic pressure, with no change in diastolic pressure; the end result is an increase in cardiac output. These responses are most easily observed in situations of increased thyroid activity. In regard to the effect of thyroid hormones on cardiovascular activity, it may be concluded that they are important for maintaining normal contractile activity of cardiac muscle, including the transmission of nerve impulses.
Thyroid hormone was used in classic experiments involving the metamorphosis of amphibian larvae. Thyroxine administration causes the differentiation of tadpoles into frogs, whereas thyroidectomy results in development into large tadpoles. Thyroid-induced metamorphosis is limited to amphibians, but thyroid hormones are important for many (subtle) aspects of differentiation in other classes of animals.
Thyroid hormone activity is usually defined in terms of tissue or organ responses to inadequate or excessive amounts of hormone. A more balanced view is that thyroid hormones are important for the normal metabolic activity of all tissues.
TSH, or thyrotropin, is the most important regulator of thyroid activity. It acts through the initiation of cyclic adenosine 3′,5′-monophosphate (cAMP) formation and the phosphorylation of protein kinases. Thyrotropin secretion is regulated by thyroid hormones through negative-feedback inhibition of the synthesis of thyrotropin-releasing hormone (TRH) at the level of the hypothalamus and by inhibition of TSH activity at the level of the pituitary gland (Figure 34-6).
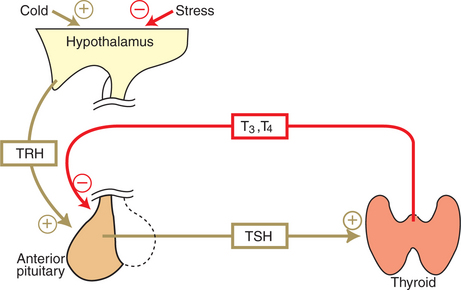
FIGURE 34-6 Hypothalamopituitary-thyroid axis. Plus signs indicate stimulation; minus signs indicate inhibition. T3, Triiodothyronine; T4, thyronine; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The Ingestion of Compounds That Inhibit the Uptake or Organic Binding of Iodine Blocks the Thyroid’s Ability to Secrete Thyroid Hormones and Causes Goiter
An inability to secrete adequate amounts of thyroid hormone often leads to the enlargement of the thyroid gland, a condition known as goiter. In many places in the world, this condition is, or has been, caused by a deficiency of iodine in the diet. This has largely been corrected through the use of iodized salt. Certain plants, such as cruciferous plants (e.g., cabbage, kale, rutabaga, turnip, rapeseed), contain a potent antithyroid compound called progoitrin, which is converted into goitrin within the digestive tract. Goitrin interferes with the organic binding of iodine. Many of the goitrogenic feeds also contain thiocyanates, which interfere with the trapping of iodine by the thyroid gland. The feeding of excess iodine can sometimes overcome the effects of thiocyanate but has less influence on overcoming the effects of goitrin. Studies of these phenomena have led to the development of compounds for the treatment of hyperthyroidism, the most potent being the thiocarbamides, thiourea and thiouracil. Other antithyroid drugs include sulfonamides, p-aminosalicylic acid, phenylbutazone, and chlorpromazine.
Hypothyroidism in Dogs
Hypothyroidism is most common in the dog, and the usual etiology of primary hypothyroidism is lymphocytic thyroiditis. Congenital hypothyroidism may be caused by thyroid dysgenesis, dyshormonogenesis, T4 transport defects, goitrogens, or in rare cases, iodine deficiency. Secondary hypothyroidism may be a secondary effect of pituitary tumors, radiation therapy, or ingestion of endogenous or exogenous glucocorticoids. Tertiary hypothyroidism can be acquired, as in the case of hypothalamic tumors, or can be congenital as a result of defective TRH or TRH receptor defects.
The signalment of hypothyroid dogs carries a distinct breed predisposition; high-risk breeds manifest symptoms as early as 2 to 3 years of age, and low-risk breeds manifest symptoms at a slightly older age (4-6 years). Breeds predisposed to hypothyroidism include golden retrievers, Doberman pinschers, dachshunds, Irish setters, miniature schnauzers, Great Danes, miniature poodles, boxers, Shetland sheepdogs, Newfoundlands, chow chows, English bulldogs, Airedale terriers, cocker spaniels, Irish wolfhounds, giant schnauzers, Scottish deerhounds, and Afghan hounds.
Clinical signs of hypothyroidism are gradual and subtle in onset; lethargy and obesity are most common. Dermatological evidence of hypothyroidism is the next most common clinical finding. Symmetric truncal or tail head alopecia is a classic finding in hypothyroid dogs. The skin is often thickened because of myxedematous accumulations in the dermis. Common hair coat changes seen in the hypothyroid dog include dull dry hair, poor hair regrowth after clipping, and presence or retention of puppy hair.
Cardiovascular signs of hypothyroidism include bradycardia, decreased cardiac contractility, and atherosclerosis, but these are uncommon presenting complaints. Neuromuscular signs such as myopathies and megaesophagus are also uncommon manifestations of canine hypothyroidism. Neuropathies, including bilateral or unilateral facial nerve paralysis, vestibular disease, and lower motor neuron disorders, are occasionally seen in hypothyroid dogs. Myxedema coma is an unusual finding in hypothyroid dogs and is secondary to myxedematous fluid accumulations in the brain and severe hyponatremia. Less common signs of hypothyroidism include reproductive disorders in female dogs, such as prolonged interestrous intervals, silent heat, and delivery of weak or stillborn puppies. Corneal lipid deposits and gastrointestinal problems such as constipation are occasionally observed in hypothyroid dogs.
Clinicopathological findings, such as anemia resulting from erythropoietin deficiency, decreased bone marrow activity, and decreased serum iron and iron-binding capacity, are observed in about 25% to 30% of hypothyroid dogs. Hypercholesterolemia is seen in approximately 75% of hypothyroid dogs because of altered lipid metabolism, decreased fecal excretion of cholesterol, and decreased conversion of lipids to bile acids. Hyponatremia, a common finding in humans with hypothyroidism, is observed as a mild decrease in serum sodium in about 30% of hypothyroid dogs in one study. Hyponatremia is caused by an increase in total body water as a result of impaired renal excretion of water and by retention of water by hydrophilic deposits in tissues. An unusual clinicopathological feature of hypothyroidism is increased serum creatine phosphokinase levels, possibly as a result of hypothyroid myopathy.
Diagnosis is based on measurement of serum basal total thyroxine (T4) and triiodothyronine (T3) concentrations, serum free T4 and T3 concentrations, and endogenous canine serum thyrotropin (TSH) levels (Table 34-1) and/or results of dynamic thyroid function tests, including the TRH and TSH stimulation tests. The many variables that affect T4 include age, breed, environmental and body temperature, diurnal rhythm, obesity, and malnutrition. Specifically, affected greyhounds have approximately half the normal total thyroxine (TT4) and free thyroxine (unbound) (FT4) concentrations of normal dogs. Obese dogs have mild increases in serum TT4 concentrations. In puppies the serum TT4 concentration is two to five times higher than in adult dogs. Furthermore, there is an age-related decline in serum TT4 concentrations and response to TSH stimulation in dogs. Euthyroid sick syndrome is characterized by a decrease in serum TT4 and increase in reverse T3. Concurrent illnesses such as diabetes mellitus, chronic renal failure, hepatic insufficiency, and infections can cause euthyroid sick syndrome, resulting in decreases in serum TT4 concentrations. Drugs such as anesthetics, phenobarbital, primidone, diazepam, trimethoprim-sulfa, quinidine, phenylbutazone, salicylates, and glucocorticoids can also decrease serum basal TT4 concentrations.
Table 34-1 Serum T4 and T3 Values by Radioimmunoassay
| Species* | T4 (mg/dL) | T3 (ng/dL) |
|---|---|---|
| Equine | ||
| M ± SD | 1.63 ± 0.51 | 77.1 ± 45.75 |
| Range | 0.95-2.38 | 31-153 |
| Bovine | ||
| M ± SD | 6.22 ± 2.03 | 92.50 ± 53.61 |
| Range | 3.60-8.9 | 41-170 |
| Caprine | ||
| M ± SD | 3.45 ± 0.47 | 145.9 ± 29.32 |
| Range | 3.0-4.23 | 88-190 |
| Ovine | ||
| M ± SD | 4.41 ± 1.13 | 99.6 ± 27.34 |
| Range | 2.95-6.15 | 63-150 |
| Porcine | ||
| M ± SD | 3.32 ± 0.80 | 89.8 ± 36.7 |
| Range | 1.70-4.68 | 43-140 |
| Canine | ||
| M ± SD | 1.15 ± 0.38 | 96.2 ± 21.39 |
| Range | 0.70-2.18 | 63-130 |
| Feline | ||
| M ± SD | 2.02 ± 0.61 | 64.7 ± 20.62 |
| Range | 1.18-2.95 | 39-112 |
* N = 10, for all species listed.
T3, Triiodothyronines; T4, thyronine; M ± SD, median plus/minus standard deviation.
From McDonald LE, Pineda MG, editors: Veterinary endocrinology and reproduction, ed 4, Philadelphia, 1989, Lea & Febiger.
Free thyroid hormone concentrations, or unbound T4 and T3, are used in human medicine to differentiate between euthyroid sick syndrome and true hypothyroidism. In humans the diagnostic accuracy of a single FT4 measurement is approximately 90%. Measurement of FT4 concentrations is achieved by equilibrium dialysis (“gold standard”) or analogue immunoassays. Theoretically, FT4 is not subject to spontaneous or drug-induced changes that occur with TT4. Results of early studies, classifying dogs as hypothyroid on the basis of TSH stimulation tests, indicated that FT4 measurements by equilibrium dialysis were 90% accurate, whereas other FT4 assays (analogue assays) were no better than TT4. Glucocorticoids decrease both FT4 fraction and TT4 in dogs.
With the advent of the endogenous canine TSH assay, veterinarians now have a method of assessing the thyroid-pituitary axis in dogs without dynamic testing. With thyroid gland failure, decreases in serum FT4 and TT4 are sensed by the pituitary gland, resulting in an increase in serum endogenous TSH concentration. Initial studies in dogs with experimentally induced hypothyroidism have been encouraging. In humans, when endogenous TSH concentrations are increased and FT4 concentrations are decreased, diagnostic accuracy for primary hypothyroidism approaches 100%. As FT4 concentration falls, there is a logarithmic increase in serum endogenous TSH concentration, which makes the TSH assay the most sensitive test for the detection of early hypothyroidism. The use of endogenous TSH alone is not recommended as a method of assessing thyroid function.
The antithyroglobulin autoantibody test (ATAA) appears promising on the basis of initial study results. The presence of antithyroglobulin antibodies theoretically presages the onset of hypothyroidism in dogs with autoimmune thyroiditis. It is hoped that this test will identify dogs with hereditary thyroid disease before breeding. However, no large studies of dogs with naturally occurring thyroid disease have been performed to evaluate this assay.
For many years the TSH stimulation test was considered the gold standard for diagnosis of hypothyroidism in dogs. Unfortunately, this test does not differentiate between early hypothyroidism and euthyroid sick syndrome and does not identify dogs with secondary or tertiary hypothyroidism. Furthermore, exogenous bovine TSH is no longer commercially available. Other thyroid function tests include the TRH stimulation test, thyroid scan, and thyroid biopsy. However, each test has drawbacks (expense, inaccuracy, or invasiveness).
In summary, diagnosis of canine hypothyroidism is based on signalment, historical findings, physical examination findings, clinicopathological features, and confirmation with a battery of thyroid function tests. The author uses TT4 and endogenous TSH (eTSH) initially, followed by FT4 by dialysis. If all measurements are abnormal, the dog is hypothyroid. If two of the three are abnormal, secondary hypothyroidism (low FT4, low TSH) or early primary hypothyroidism (high TSH, low FT4) is possible. If only one of the three thyroid measurements is abnormal, the dog should be reevaluated in 3 to 6 months.
Hyperthyroidism in Cats
Hyperthyroidism is the most common endocrinopathy of cats and is caused by adenomatous hyperplasia of the thyroid gland. Middle-aged to older cats are typically affected, and there is no predilection for breed or gender. Hyperthyroidism is characterized by hypermetabolism; therefore, polyphagia, weight loss, polydipsia, and polyuria are the most prominent features of the disease. Activation of the sympathetic nervous system is also seen; hyperactivity, tachycardia, pupillary dilation, and behavioral changes are characteristic of the disease in cats. Long-standing hyperthyroidism leads to hypertrophic cardiomyopathy, high-output heart failure, and cachexia, which may lead to death.
Clinicopathological features of hyperthyroidism include erythrocytosis and an excitement leukogram (neutrophilia, lymphocytosis) caused by increased circulating catecholamine concentrations. Increased catabolism of muscle tissue in hyperthyroid cats may result in increased levels of blood urea nitrogen (BUN) but not creatinine. In fact, glomerular filtration rate (GFR) is increased in hyperthyroid cats, and this increase may mask underlying renal insufficiency. Although hyperthyroidism increases GFR, the effect of thyroid hormone excess on the urinalysis is variable. Most cats, however, have decreased urine specific gravity, particularly if they are exhibiting polyuria as a clinical sign. Increased metabolic rate results in liver hypermetabolism; therefore, serum activities of liver enzymes (alanine aminotransferase, aspartate aminotransferase) increase in 80% to 90% of hyperthyroid cats. Serum cholesterol decreases, not because of decreased synthesis, but rather because of increased hepatic clearance mediated by thyroid hormone excess.
Feline hyperthyroidism is diagnosed through measurement of TT4; TT3 measurement is generally noncontributory to a diagnosis. Because the disease has become more common and recognized in its early stages, FT4 concentrations have been shown to be more diagnostic of early or “occult” hyperthyroidism. However, FT4 concentrations should be interpreted in light of the TT4 because nonthyroidal illness (chronic renal failure) can result in spurious elevations of FT4 as well. Free triiodothyronine (FT3) concentrations do not provide any further advantage over FT4.
THE ADRENAL GLANDS
The Adrenal Glands Are Composed of Two Organs: the Outer Gland (Cortex) and the Inner Gland (Medulla)
The adrenal glands are two bilaterally symmetric endocrine organs located just anterior to the kidneys. Each gland is divided into two separate entities, a medulla and a cortex (Figure 34-7), each of which produces different types of hormones. These adrenal tissues have different embryonic origins. The medulla arises from the neuroectoderm and produces amines such as norepinephrine and epinephrine. The cortex arises from the mesodermal coelomic epithelium and produces steroid hormones such as cortisol, corticosterone, sex steroids, and aldosterone. The utility of placing two such disparate tissues together is not apparent. The one common factor is that both sets of hormones are important for adaptation to adverse environmental conditions (i.e., stress).
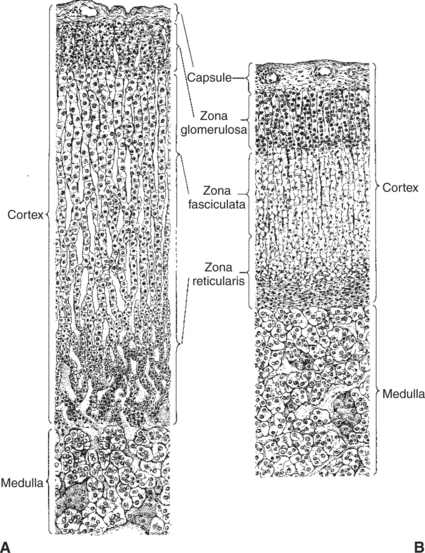
FIGURE 34-7 Depiction of comparable sections through the adrenal glands of A, normal rats, and B, hypophysectomized rats. Because the functional capacity of the adrenal cortex is conditioned by the release of corticotropin, hypophysectomy results in tremendous shrinkage of the cortex. The medulla is not influenced by hypophysectomy. Both sections are drawn to scale.
(From Turner CD, Bagnara JT: General endocrinology, ed 6, Philadelphia, 1976, Saunders.)
Interest in the function of the adrenal cortex was heightened in the 1930s because of the research of Hans Selye. He published a series of papers on the effects of adrenalectomy and the ability of the surgically treated animal to defend itself against injury. Selye’s hypothesis was termed the general adaptation syndrome, which he divided into three parts: the alarm reaction, the stage of resistance, and the stage of exhaustion. The critical aspect of this theory was that in addition to specific responses to injury, animals responded in nonspecific ways to combat injury, and the adrenal cortex was the most important organ in leading the nonspecific response. One example of the beneficial effects of glucocorticoids in a situation of injury is the mobilization of glucose, a readily usable source of energy for running away or healing injury. The adaptation of animals to stressful environments is often accompanied by enlargement of the adrenal cortex, such as in domestic chickens raised in crowded conditions and wild animals living in relatively high density.
THE ADRENAL CORTEX
The Adrenal Cortex Has Three Zones: the Zona Glomerulosa, Which Secretes Mineralocorticoids, and the Zona Fasciculata and the Zona Reticularis, Which Secrete Glucocorticoids and Sex Steroids
The adrenal cortex is organized into three zones in mammals (see Figure 34-7). The outer zone, the zona glomerulosa, is relatively narrow, and its cells are organized in a whorl-type arrangement. The middle zone, the zona fasciculata, is relatively wide, and its cells are organized in columns. In the cow and sheep, the zona fasciculata is further divided into inner and outer layers. The inner zone of the adrenal cortex, the zona reticularis, which is adjacent to the adrenal medulla, is intermediate in size, and cells are more randomly organized.
All the cells of the adrenal cortex have intracellular features characteristic of steroid hormone synthesis: an abundance of lipid droplets (containing cholesterol esters), mitochondria, and smooth endoplasmic reticulum. Human adrenal glands have an additional zone, the fetal zone, that is present during fetal life and for the first year of life. The fetal zone participates with the placenta in the production of estrogen during gestation. Immature mice and rabbits have an inner X zone that becomes the zona reticularis at puberty.
The adrenal cortex produces two major types of steroid hormones: the mineralocorticoids and the glucocorticoids. These hormones have distinctly different functions. The mineralocorticoids, produced by the zona glomerulosa, play an important role in electrolyte balance and therefore are important in the regulation of blood pressure (see later discussion). The major mineralocorticoid is aldosterone. The glucocorticoids, produced by the zona fasciculata (which accounts for the majority of glucocorticoid production) and zona reticularis, are important in the regulation of all aspects of metabolism, either directly or through an interaction with other hormones. The major glucocorticoid is cortisol.
Adrenal Corticoids Are Synthesized from Cholesterol; the Critical Difference in the Activity of These Corticoids Is Related to the Hydroxyl Group on C-17 of Glucocorticoids
The synthesis of adrenal steroids involves the classic pathways for steroid biosynthesis. As indicated previously, cholesterol is the major starting material for the synthesis of steroid hormones. Cholesterol is readily available to the steroid-synthesizing cells because it is stored in large quantities in ester form within lipid droplets in these cells. One of the initial steps in steroid formation is the hydrolysis of the ester. The first step in steroid synthesis involves an enzyme that cleaves the carbon side chain from the steroid molecule, leaving a C-21 steroid known as pregnenolone. This step occurs within the mitochondrion (Figure 34-8). The synthesis of all steroid hormones, regardless of their form, utilizes pregnenolone in the synthetic pathway (see Figure 33-5).
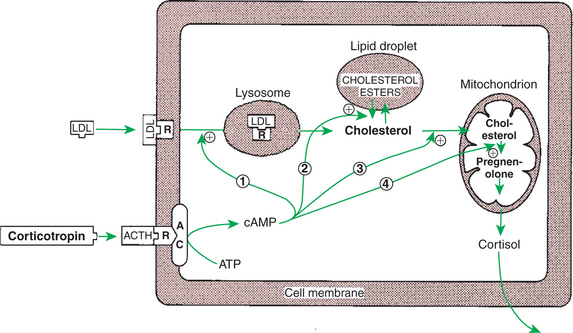
FIGURE 34-8 Mechanism of action of corticotropin (adrenocorticotropic hormone, ACTH) on adrenocortical steroidogenesis. The numbers indicate the processes stimulated (indicated by plus signs) by corticotropin as follows: 1, stimulation of the uptake of low-density lipoproteins (LDL), which are further processed to free cholesterol; 2, stimulation of the hydrolysis of stored cholesterol esters to generate free cholesterol; 3, stimulation of the transport of cholesterol into mitochondria, where cleavage of the cholesterol side chain occurs; and 4, promotion of the binding of cholesterol to the enzyme. AC, Adenyl cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; R, receptor.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The critical aspect of adrenal corticoid synthesis, which differentiates adrenal corticoids from the progesterone family of steroids, is a hydroxylation step at C-21 (directed by a C-21 hydroxylase). The difference between the mineralocorticoids (aldosterone) and the glucocorticoids (cortisol) is a hydroxyl group on C-17, which is part of the glucocorticoid molecule. As expected, cells of the zona fasciculata and zona reticularis have the hydroxylating enzyme for C-17 (17α-hydroxylase), whereas cells of the zona glomerulosa do not have this enzyme. Both aldosterone and cortisol have hydroxyl groups on C-11. Because of the marked difference in biological activity of the mineralocorticoids and glucocorticoids, it is useful to view the zona glomerulosa as an endocrine organ that is distinct from the zona fasciculata and zona reticularis.
Two intermediate compounds in the synthesis of aldosterone have significant adrenocortical activity. 11-Deoxycorticosterone has significant mineralocorticoid activity, although it is secreted in relatively small amounts. Corticosterone, the immediate precursor to aldosterone, is a relatively important glucocorticoid in animals, although its potency is less than that of cortisol.
In adrenal cortical cells, biosynthetic pathways allow some synthesis of androgens and estrogens. Although the amount of sex steroids produced by the adrenal cortex under normal conditions is low, significant amounts can be synthesized under pathological conditions.
Adrenocortical Hormones Are Carried in Plasma in Association with Specific Binding Globulins (Corticosteroid-Binding Globulin)
Steroid hormones, as indicated previously, are lipids and depend on binding to plasma proteins for transport in the blood. A specific globulin that has a high affinity for cortisol has been identified: corticosteroid-binding globulin, or transcortin. Of the cortisol carried in plasma, 75% is bound to transcortin and 15% to albumin, leaving 10% in the unbound, or free, state. This amount of free hormone is large compared with thyroid hormones: less than 0.1% of T4 is free. The transport of aldosterone is mainly associated with albumin (50%), and only 10% is associated with transcortin, leaving a very large amount (40%) in the free state.
Changes in physiological or pathophysiological states can influence the amount of binding proteins present in plasma. Estrogen produced in increasing amounts by the fetoplacental unit during pregnancy results in an increase in hepatic synthesis of transcortin, whereas liver dysfunction can result in lower concentrations of transcortin. The large pool of hormone present in the bound state during pregnancy gives animals a good reserve from which to make appropriate adjustments in the amount of free hormone available for influencing biological activity. Because the total amount of glucocorticoid is determined in the assay of plasma concentrations, the veterinary clinician needs to be aware that total concentrations not only reflect secretion rate, but also can be influenced by the amount of glucocorticoid-binding plasma proteins.
The Metabolism of Adrenocortical Hormones Involves the Reduction of Double Bonds and Conjugation of the Steroids to Glucuronides and Sulfates
The clearance half-life of cortisol is about 60 minutes, and that of aldosterone is about 20 minutes. This difference is attributable to the observed difference in protein binding of these hormones within the plasma. In general, metabolism of mineralocorticoid and glucocorticoid hormones involves the reduction of double bonds and ketone configurations, which reduces the biological activity of the molecules. The liver, an organ important for modification of these hormones, is also an important site for the conjugation of these steroids with sulfates and glucuronides; this reduces their biological potency and renders them water soluble for passage in the urine.
One of the Most Important Functions of Glucocorticoids Is Control of Metabolism, in Particular the Stimulation of Hepatic Gluconeogenesis
The mechanism of action of adrenal hormones is similar to that of other lipophilic hormones: they are able to penetrate the cell membrane and interact in the cytoplasm with specific cytosolic receptors. This complex is transferred to the nucleus, resulting in transcription of certain genes and the synthesis of specific proteins that affect the biological action of the adrenal hormones.
As emphasized previously, adrenocortical hormones are classified as either glucocorticoid or mineralocorticoid in their activity. Before the biological actions of each class are discussed, it is important to realize that there is overlap of activity (Table 34-2). For example, whereas cortisol is the dominant glucocorticoid hormone, it also has mineralocorticoid effects, although at a reduced potency.
Table 34-2 Relative Glucocorticoid and Mineralocorticoid Potencies of Various Steroids
| Steroid | Glucocorticoid potency (relative to cortisol) | Mineralocorticoid potency |
|---|---|---|
| Cortisol | 1 | 1 |
| Aldosterone | 0.1 | 400 |
| Corticosterone | 0.2 | 2 |
| 11-Deoxycorticosterone | <0.1 | 20 |
| Dexamethasone | 30 | 2 |
| Fludrocortisone | 10 | 400 |
| Prednisone | 4 | 0.7 |
| Triamcinolone | 5 | <0.1 |
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
The glucocorticoid hormones are important mediators of intermediary metabolism. One of the important specific effects of glucocorticoids is the stimulation of hepatic gluconeogenesis, which involves the conversion of amino acids to carbohydrates. The net result is an increase in hepatic glycogen and a tendency to increase blood glucose levels. These effects on glycogen metabolism are observed mainly in animals that have excessive glucocorticoid secretion (hyperadrenocorticism) or an insulin deficiency. The effect of glucocorticoids on carbohydrate metabolism is “permissive”; that is, their presence is required for the gluconeogenic and glycogenolytic actions of glucagon and epinephrine, respectively.
Whereas glucocorticoids and insulin have similar effects on liver glycogen metabolism, their effects on the peripheral use of glucose are different. Glucocorticoids inhibit glucose uptake and metabolism in the peripheral tissues, particularly in muscle and adipose cells. This effect has been termed the anti-insulin effect. The chronic administration of glucocorticoids can lead to the development of a syndrome called steroid diabetes because of the hyperglycemic effect produced at the level of the liver; use of glucose decreases in the peripheral tissues because of insulin antagonism.
Whereas the actions of glucocorticoids on fat metabolism tend to be complex, the direct effect on adipose tissue is to increase the rate of lipolysis and to redistribute fat into the liver and abdomen. This fat redistribution leads to the classic “potbelly” appearance of animals and humans with hyperadrenocorticism.
Protein synthesis is inhibited by glucocorticoids; in fact, protein catabolism is enhanced, with an accompanying release of amino acids. This process supports hepatic gluconeogenesis. Two tissues, cardiac and brain, are spared from the effect of glucocorticoids on protein catabolism. Chronic administration of glucocorticoids results in muscle wasting and the weakening of bone. The mobilization and incorporation of amino acids into glycogen result in an increase in urinary excretion of nitrogen and a negative nitrogen balance.
Glucocorticoids play a role in water diuresis (i.e., the enhancement of water excretion). Whereas glucocorticoids inhibit vasopressin activity in the distal tubule, the most important effect is to increase the GFR. Table 34-3 summarizes the effects of glucocorticoids.
Table 34-3 Glucocorticoid Effects and Target Tissues
| Effect | Site of action |
|---|---|
| Stimulates gluconeogenesis | Liver |
| Increases hepatic glycogen | Liver |
| Increases blood glucose | Liver |
| Facilitates lipolysis | Adipose tissue |
| Is catabolic (negative nitrogen balance) | Muscle, liver |
| Inhibits corticotropin secretion | Hypothalamus, anterior pituitary gland |
| Facilitates water excretion | Kidney |
| Blocks inflammatory response | Multiple sites |
| Suppresses immune system | Macrophages, lymphocytes |
| Stimulates gastric acid secretion | Stomach |
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
Corticotropin Is the Pituitary Hormone That Regulates Glucocorticoid Synthesis by the Adrenal Cortex
The control of the secretion of the glucocorticoids by the zona fasciculata and zona reticularis is by the tropic hormone (corticotropin) (Figure 34-9). A negative-feedback system exists, whereby glucocorticoids inhibit the release of hypothalamic corticotropin-releasing hormone, which in turn results in decreased corticotropin secretion by the pituitary gland. Some evidence indicates that glucocorticoids also have a negative-feedback effect at the level of the pituitary gland. The potency of a glucocorticoid in negative-feedback inhibition of corticotropin is directly related to its glucocorticoid potency; for example, cortisol has more potent negative-feedback effects than corticosterone and has more potent glucocorticoid effects.
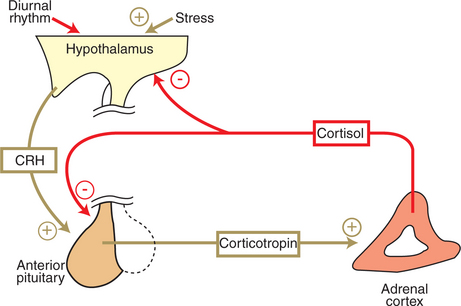
FIGURE 34-9 Regulation of cortisol secretion by the hypothalamopituitary axis. Plus signs indicate stimulation; minus signs indicate inhibition. CRH, Corticotropin-releasing hormone.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The negative-feedback control system that exists for the secretion of glucocorticoids does not result in the maintenance of uniform hormone concentrations in blood throughout the day. Sleep and activity patterns are superimposed on the negative-feedback system, so a predictable circadian rhythm occurs in which concentrations of glucocorticoids are lowest late at night and highest in the early-morning hours (Figure 34-10).
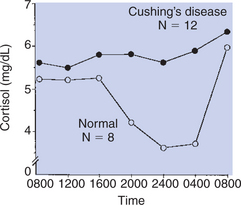
FIGURE 34-10 Circadian changes in cortisol secretion in normal horses (open circles), in comparison with no circadian change in horses with equine Cushing’s disease (solid circles).
(From Dybdal N: Studies on equine Cushing’s disease, Davis, University of California–Davis, 1990 [PhD thesis].)
Another factor that can modify the negative-feedback control of glucocorticoids is stress. Stress can result from physical or psychological stimuli that are harmful to the individual. The effects of stress, as with the factors that influence circadian rhythms of glucocorticoid secretion, are mediated through the CNS. The glucocorticoid response to stress is immediate: concentrations of cortisol increase rapidly to reach, within minutes, values that are several-fold higher than normal. The glucocorticoid response is proportional to the severity of the stress; that is, lower levels of stress result in less cortisol production than do higher levels of stress.
One of the Most Important Clinical Uses of Glucocorticoids Is the Suppression of the Inflammatory Response
Glucocorticoids have valuable clinical effects, particularly the inhibition of the inflammatory response, including the prevention of capillary dilation, extravasation of fluid into tissue spaces, leukocyte migration, fibrin deposition, and connective tissue synthesis. Whereas the process of inflammation is important for the walling off and destruction of systemic noxious agents, the end response is often the replacement of functional tissue with fibrous connective tissue, with a resultant loss of function. For example, inflammatory processes in the mammary gland often result in the isolation of the injurious agent by the laying down of connective tissue as a part of the defense mechanism; however, the gland may lose much of its functional capacity as a result. Administration of glucocorticoids, in conjunction with antibiotic therapy, can help reduce the loss of functional tissue by inhibiting the development of connective tissue. Figure 34-11 shows the chemical structures of some of the synthetic glucocorticoids used in clinical practice.
FIGURE 34-11 Chemical structures of some clinically useful glucocorticoid analogues.
(From Martin CR: Endocrine physiology, London, 1985, Oxford University Press.)
One of the ways in which glucocorticoids inhibit the inflammatory response is through the inhibition of the formation of substances that promote inflammation. Glucocorticoids inhibit the synthesis of inflammatory mediators, such as prostaglandins, thromboxanes, and leukotrienes, that arise as a result of arachidonic acid metabolism. This effect is mediated through the stabilization of lysosomal membranes and the prevention of phospholipase A2 activation. Glucocorticoids are also used to inhibit allergic reactions. This action occurs through the inhibition of the release of certain biogenic amines, such as histamine, from the granules of mast cells.
Hyperadrenocorticism
Hyperadrenocorticism (Cushing’s syndrome) in the dog may be caused by a pituitary tumor, pituitary hyperplasia, adrenal tumors, adrenal hyperplasia, or nonendocrine tumors (usually of the lung), or it may be iatrogenic. Approximately 85% of dogs with hyperadrenocorticism have pituitary gland–dependent disease, whereas 15% exhibit adrenal tumors. Hyperadrenocorticism is a disease of middle-aged and older dogs (7-12 years). Breeds typically affected by pituitary-dependent hyperadrenocorticism include miniature poodles, dachshunds, boxers, Boston terriers, and beagles. Adrenal tumors are seen more frequently in large-breed dogs, and there is a predilection for females (3:1 ratio with males). Hyperadrenocorticism is a rare endocrine disorder of cats and is usually pituitary in origin in that species.
The most common clinical signs associated with canine hyperadrenocorticism are polydipsia, polyuria, polyphagia, heat intolerance, lethargy, abdominal enlargement or “potbelly,” panting, obesity, muscle weakness, and recurrent urinary tract infections (UTIs). Dermatological manifestations of canine hyperadrenocorticism can include alopecia (especially truncal), thin skin, phlebectasias, comedones, bruising, cutaneous hyperpigmentation, calcinosis cutis, pyoderma, dermal atrophy (especially around scars), seborrhea, and secondary demodicosis. Thin skin is the hallmark of feline hyperadrenocorticism. Cats with Cushing’s syndrome develop such severe thinning of the epidermis that they may incur open wounds just by grooming themselves.
Attempts to diagnose hyperadrenocorticism can be challenging. Uncommon clinical manifestations of hyperadrenocorticism in dogs can include signs such as hypertension, congestive heart failure, bronchial calcification, pulmonary thromboembolism, polyneuropathy, polymyopathy, pseudomyotonia, behavioral changes, and blindness. Evidence of increased collagenase activity caused by hypercortisolemia may result in nonhealing corneal ulceration and bilateral cranial cruciate rupture (in small dogs). Unusual reproductive signs may include testicular atrophy, prostatomegaly in castrated male dogs, clitoral hypertrophy, and perianal adenoma in females or castrated males.
Serum chemistry abnormalities associated with hypercortisolemia in dogs include increased serum activities of alkaline phosphatase and alanine aminotransferase, hypercholesterolemia, hyperglycemia, and decreased BUN. The hemogram is often characterized by evidence of erythroid regeneration (nucleated red blood cells) and a classic “stress leukogram.” Basophilia is occasionally observed. Many dogs with hyperadrenocorticism have evidence of UTI without pyuria. Proteinuria resulting from glomerulosclerosis is also common. Urine specific gravity is usually decreased and may be hyposthenuric. Thyroid status is often affected in animals with hyperadrenocorticism, as evidenced by (1) decreases in TT4 and TT3 caused by euthyroid sick syndrome and (2) a response to TSH stimulation that is attenuated as a result of overcrowding of pituitary thyrotrophs by adrenocorticotrophs. Overt diabetes mellitus may result from the insulin antagonism caused by hypercortisolemia in about 15% of dogs with hyperadrenocorticism and 85% of cats with hyperadrenocorticism. Conversely, hyperadrenocorticism can be a cause of insulin resistance and poor glycemic control in diabetic animals.
The diagnosis of hyperadrenocorticism should be based on suggestive clinical signs and supporting minimal database abnormalities (e.g., high serum cholesterol, increased serum alkaline phosphatase activity) and confirmed by an appropriate screening test. If screening test results are inconclusive, the dog should be retested at a later date (3-6 months) rather than be subjected to treatment without a definitive diagnosis.
Screening tests for hyperadrenocorticism, such as the low-dose dexamethasone suppression (LDDS) test and the corticotropin stimulation test, work on the principle of suppression or stimulation of the pituitary-adrenal axis. In the case of the LDDS test, dexamethasone is administered at a low dosage to cause negative feedback to the pituitary gland. In a normal animal, this negative feedback results in a decrease in endogenous corticotropin secretion and a resultant decrease in circulating cortisol concentrations. Dexamethasone is the only synthetic corticosteroid that does not cross-react with the cortisol assay. The corticotropin stimulation is used to determine the extent of adrenal enlargement. Adrenal glands that are enlarged because of chronic pituitary stimulation by corticotropin or that are neoplastic show an exaggerated response to exogenous corticotropin.
The LDDS test has traditionally been the screening test of choice for canine hyperadrenocorticism. It is sensitive (92%–95%); only 5% to 8% of dogs with pituitary dependent hyperadrenocorticism exhibit suppressed cortisol concentrations at 8 hours (i.e., 5%–10% false-negative results). In addition, 30% of dogs with pituitary-dependent hyperadrenocorticism exhibit suppression at 3 or 4 hours, followed by “escape” of suppression at 8 hours; this pattern is diagnostic for pituitary-dependent disease and makes further testing unnecessary. The major disadvantage of the LDDS test is the lack of specificity in dogs with nonadrenal illness. It is recommended that a dog be allowed to recover from the nonadrenal illness before being assessed for hyperadrenocorticism with the LDDS test.
Mineralocorticoids
The mineralocorticoids, produced in the outer zone (zona glomerulosa) of the adrenal cortex, have surprisingly different functions compared with glucocorticoids; the functions are surprising because both types of hormones are produced by tissues that are part of the same gland. As indicated previously, electrolyte balance and blood pressure homeostasis represent the principal physiological effects of mineralocorticoids (Table 34-4). These actions are carried out at the level of the distal tubules in the kidney. The effect of the mineralocorticoids is to promote retention of sodium and secretion of potassium and hydrogen. The cellular response to mineralocorticoids is to synthesize a protein that increases the permeability of the luminal cell surface to sodium influx from the renal filtrate and increases sodium/potassium-adenosinetriphosphatase (Na+,K+-ATPase) activity in the contraluminal cell surface, which allows movement of Na+ out of the cell into the interstitial tissue (Figure 34-12).
Table 34-4 Mineralocorticoid Effects and Target Tissues
| Effect | Site of action |
|---|---|
| Stimulates Na+ reabsorption | Kidney, salivary glands, sweat glands |
| Stimulates K+ excretion | Kidney, salivary glands, sweat glands |
| Stimulates H+ excretion | Kidney |
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
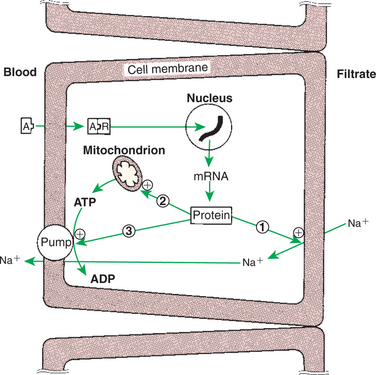
FIGURE 34-12 Mechanisms of action of aldosterone on sodium transport in the renal tubular cell. The numbered arrows indicate the three putative sites of action of aldosterone: 1, increasing the permeability of the luminal membrane to sodium; 2, increasing mitochondrial adenosine triphosphate (ATP) production; and (3) increasing Na+,K+-ATPase activity in the contraluminal membrane. Plus signs indicate stimulation. A, Aldosterone; ADP, adenosine diphosphate; mRNA, messenger ribonucleic acid; R, receptor.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The control of secretion of K+ by mineralocorticoids is passive in the sense that K+ is retained in the renal filtrate to maintain the osmolality of urine. However, evidence suggests that mineralocorticoids have an effect on Na+ secretion that is independent of Na+ retention. The secretion of K+ continues to be influenced by mineralocorticoids after mineralocorticoid administration, whereas Na+ retention decreases within a few days.
In situations of excessive mineralocorticoid production, the effects of increased Na+ retention are to increase the extracellular fluid volume and to cause hypertension; conversely, low blood pressure (hypotension) occurs as a result of inadequate secretion of mineralocorticoids. Hypersecretion of mineralocorticoids can also lead to excessive hydrogen ion (H+) loss and metabolic alkalosis, whereas hyposecretion can result in increased retention of H+ and metabolic acidosis.
The regulation of mineralocorticoid secretion, in contrast to glucocorticoid secretion, is not controlled by tropic hormones from the pituitary gland (Figure 34-13). In the case of mineralocorticoids, the main controlling factors are produced in the target organ, the kidney. Cells in the juxtaglomerular apparatus of the kidney produce an enzyme, renin, in response to decreases in blood pressure. Renin acts on angiotensinogen, an α2 globulin produced by the liver and present in the circulation, and this results in the production of angiotensin I, a decapeptide. Angiotensin I is further hydrolyzed to angiotensin II, an octapeptide, by angiotensin-converting enzyme. Angiotensin II stimulates the zona glomerulosa to produce mineralocorticoids. Angiotensin II also increases peripheral resistance of the blood vascular system by causing vasoconstriction of smooth muscle of the blood vessels. Angiotensin II, if present on a long-term basis, also increases the size of the zona glomerulosa.
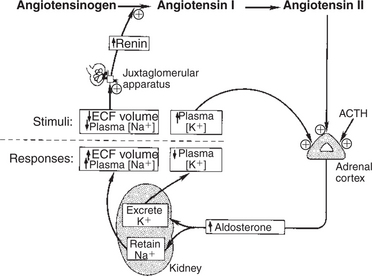
FIGURE 34-13 Regulation of aldosterone secretion by the zona glomerulosa of the adrenal cortex. Plus signs indicate stimulation. ACTH, Corticotropin (adrenocorticotropic hormone); ECF, extracellular fluid.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Evidence indicates that cells of the macula densa, groups of specialized cells located at the origin of the kidney’s distal tubule (Figure 34-14), exert control on the renin-angiotensin system. This is done through the sensing of changes in Na+ concentrations in tissue fluids; increased Na+ results in decreased renin release, and decreased Na+ results in increased renin release. In either case, the change produced tends to restore the mineralocorticoid concentrations to normal. In addition to the effect of sodium, the macula densa may control changes in the renin-angiotensin system through the sensing of changes in chloride ion (Cl−) concentrations in tissue fluids.
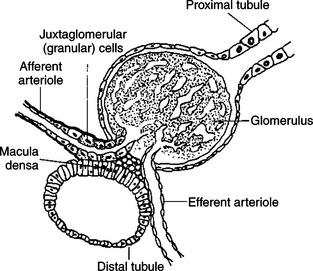
FIGURE 34-14 Diagrammatic representation of juxtaglomerular apparatus.
(From Martin CR: Endocrine physiology, London, 1985, Oxford University Press.)
Another major regulatory factor in the control of mineralocorticoid secretion is the blood potassium concentration. An increase in K+ concentration stimulates the zona glomerulosa to secrete mineralocorticoids, whereas a decline in K+ has the opposite effect. This stimulation is independent of the renin-angiotensin system.
It has been thought that corticotropin has minimal effect on control of the zona glomerulosa, because experimental studies showed that hypophysectomy has little effect on the zona glomerulosa. More recent studies have shown that cells of the zona glomerulosa have receptors for corticotropin, and corticotropin may play some role, although minor, in the control of mineralocorticoid secretion.
In contrast to the sodium-conserving effect of mineralocorticoids, the 28–amino acid atrial natriuretic peptide (ANP) reduces Na+ retention by the kidneys. ANP also causes peripheral vasodilation and thus a lowering of blood pressure. ANP may inhibit the production of mineralocorticoids and renin as well. ANP is produced by cells of the cardiac atria, but it is also produced in other sites, including the brain.
Hypoadrenocorticism
Hypoadrenocorticism, caused by lack of mineralocorticoids and glucocorticoids, is most often diagnosed in young female dogs and usually has an immune-mediated etiology. Certain breeds, such as Leonbergers, standard poodles, and Portuguese water dogs, are at increased risk for the disease; however, hypoadrenocorticism may be diagnosed in any breed. Historical findings compatible with hypoadrenocorticism include intermittent vomiting, diarrhea, weight loss, lethargy, anorexia, and weakness. These symptoms often resolve with fluid therapy and corticosteroid treatment. Physical examination of animals in an acute hypoadrenal crisis reveals weak pulse, bradycardia, prolonged capillary refill time, severe mental slowness, and profound muscle weakness. Clinical features of hypoadrenocorticism that should raise suspicion include a normal or slow heart rate in the presence of circulatory shock and the “waxing and waning” course of disease before collapse.
Electrolyte abnormalities consisting of severe hyponatremia and hypochloremia associated with hyperkalemia are the hallmarks of hypoadrenocorticism. Azotemia and hyperphosphatemia also accompany primary hypoadrenocorticism, which makes it difficult to differentiate it from acute renal failure. Azotemia may be prerenal as a result of dehydration and hypovolemia, or increase in BUN may be caused by gastrointestinal hemorrhage. Hematological abnormalities consist of eosinophilia and lymphocytosis, or eosinophil and lymphocyte counts may be normal in the presence of severe metabolic stress. The anemia of hypoadrenocorticism has classically been attributed to lack of glucocorticoid effects on the bone marrow. However, more recent studies have suggested that hemorrhagic gastroenteritis contributes significantly to the anemia. Although hypoglycemia is more common with secondary or atypical hypoadrenocorticism, it is rarely seen with typical hypoadrenocorticism.
Urine specific gravity is frequently low, attributable to medullary washout (inadequate medullary gradient caused by sodium depletion) and decreased medullary blood flow. Dilute urine in the presence of azotemia and hyperkalemia may easily be mistaken for acute renal failure. Hormonal assays are necessary to confirm the presence or absence of adrenal disease and to differentiate between hypoadrenocorticism and renal failure.
Diagnosis of primary hypoadrenocorticism is based on clinical signs, classic electrolyte imbalances, and confirmation with a corticotropin response test. The baseline cortisol sample should be collected with the initial blood work, and synthetic corticotropin (cosyntropin [Cortrosyn], 0.25 mg) should be administered intravenously during the initial fluid therapy. A 1-hour, postcorticotropin sample may then be drawn, and glucocorticoids may be administered after the 1-hour sample is taken. Intramuscular injection of corticotropin (gel or synthetic) may not be absorbed in animals in circulatory shock; therefore, intravenous administration of synthetic corticotropin is preferred. If glucocorticoids must be administered before the measurement of cortisol, dexamethasone sodium phosphate is preferred because it does not interfere with the cortisol assay. Endogenous plasma corticotropin may be measured to determine whether the hypoadrenocorticism is primary or secondary.
Dogs and cats with primary hypoadrenocorticism exhibit a subnormal response to corticotropin administration. Both the baseline and the postcorticotropin cortisol concentrations are usually low or undetectable. Endogenous plasma corticotropin concentrations are dramatically increased in animals with primary hypoadrenocorticism as a result of loss of negative feedback to the pituitary gland, caused by decreased serum cortisol concentrations. In the case of secondary hypoadrenocorticism, caused by a pituitary deficiency of corticotropin, the endogenous corticotropin concentrations are typically decreased (<20 pg/mL). The response to exogenous corticotropin is diminished, but not as dramatically as for primary hypoadrenocorticism. Baseline and postcorticotropin cortisol concentrations may be in the normal range.
THE ADRENAL MEDULLA
The adrenal medulla, as its name indicates, occupies the central portion of the adrenal gland (see Figure 34-7). A stimulatory effect of adrenal medullary extracts on cardiac activity was first recognized by Oliver and Schafer in 1894. Thereafter, the main hormone of the adrenal medulla, epinephrine, became the first hormone to be isolated (by Abel in 1898), crystallized (by Takamine and Aldrich in 1901), and synthesized (by Stolz in 1904). Theories on the importance of the adrenal medulla include that of Cannon, who in 1932 proposed the “fight or flight” hypothesis, in which the adrenal medulla is activated to aid in combating situations of extreme stress. Others advocated the “tonus” theory, which stated that cells of the adrenal medulla are constantly in a state of readiness. In fact, the adrenal medulla has a constant output of catecholamines that can be accentuated dramatically if the need arises.
It was recognized early in this research that cells of the adrenal medulla are the equivalent of postganglionic cells of the sympathetic nervous system. Therefore, it was assumed that epinephrine was the mediator of postganglionic activity of the sympathetic nervous system. It was later recognized that another catecholamine, norepinephrine, is the neurotransmitter of the sympathetic nervous system. Both epinephrine and norepinephrine are released when preganglionic nerve fibers to the adrenal medulla are stimulated; in fact, most of the norepinephrine found in plasma originates from the adrenal medulla. However, epinephrine is the major catecholamine secreted by the adrenal medulla of most mammals. Exceptions to this generalization include the dominance of norepinephrine over epinephrine in whales and chickens and in the fetal tissues of all species.
The Synthesis of Catecholamines Is from Tyrosine; the Main Catecholamine Synthesized by the Adrenal Medulla Is Epinephrine
The cells of the adrenal medulla that synthesize catecholamines are classified as chromaffin cells. This classification is based on the histochemical reaction of the cells when exposed to potassium dichromate, that is, a darkening of the cells as a result of the formation of colored pigments in conjunction with the oxidation of the catecholamines. The cells that produce epinephrine are different from those that synthesize norepinephrine; accordingly, the type of chromaffin granule present is different for each cell type. In cattle the epinephrine-secreting cells tend to be on the outer edge of the medulla. Acetylcholine release from the preganglionic nerve fibers initiates the synthesis of the catecholamines by the medullary cells (Figure 34-15). Acetylcholine also stimulates the release of catecholamines from chromaffin granules, a phenomenon called stimulus-secretion coupling.
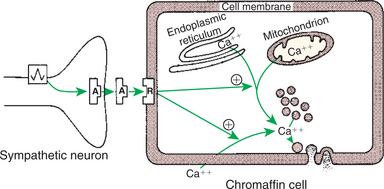
FIGURE 34-15 Stimulus-secretion coupling in the adrenal chromaffin cell. Note that cytosolic calcium may be derived from intracellular or extracellular sources. Circled plus signs indicate stimulation. A, Acetylcholine; R, receptor.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The synthesis of the catecholamines begins with either of the amino acids phenylalanine or tyrosine. However, tyrosine is a naturally occurring amino acid, and most synthesis of catecholamines begins with it (Figure 34-16). The initial step in the biosynthetic pathway begins with the conversion of tyrosine to 3,4-dihydroxyphenylalanine, or dopa. Tyrosine hydroxylase, the enzyme responsible for the conversion of tyrosine, is the rate-limiting enzyme in the formation of catecholamines. The end products of tyrosine metabolism, including dopa, dopamine, norepinephrine, and epinephrine, inhibit the activity of tyrosine hydroxylase. Dopa is converted to dopamine through the enzymatic activity of aromatic-l-amino acid decarboxylase (dopa decarboxylase). To this point, the biochemical transformations have occurred in the cytosol. The conversion of dopamine to norepinephrine occurs within the chromaffin granule because the key enzyme, dopamine-β-hydroxylase, is localized within the granule (Figure 34-17).
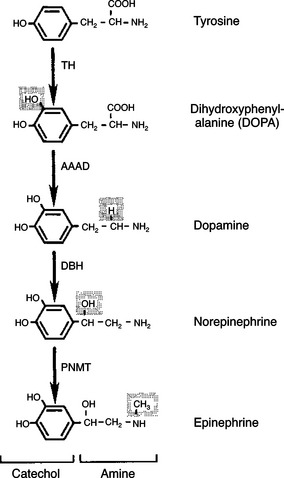
FIGURE 34-16 Pathway of catecholamine synthesis in the adrenal medulla. Shaded areas denote the structural changes occurring at each step. AAAD, Aromatic-l-amino acid decarboxylase; DBH, dopamine-β-hydroxylase; PNMT, phenylethanolamine-N-methyltransferase; TH, tyrosine hydroxylase.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
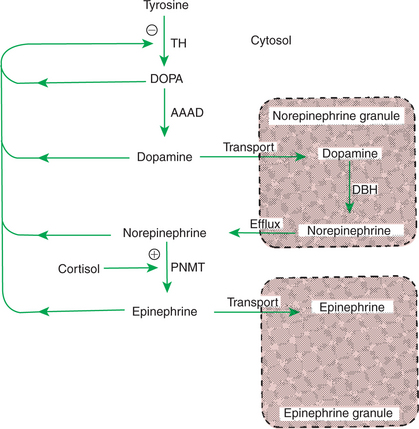
FIGURE 34-17 Regulation of catecholamine biosynthesis in the adrenal medulla. Plus sign indicates stimulation; minus sign indicates inhibition. AAAD, Aromatic-l-amino acid decarboxylase; DBH, dopamine-β-hydroxylase; DOPA, dihydroxyphenylalanine; PNMT, phenylethanolamine-N-methyltransferase; TH, tyrosine hydroxylase.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
If the cell secretes norepinephrine, the biochemical pathway is ended, and the hormone remains in the norepinephrine granule, ready for secretion. If the cell secretes epinephrine, norepinephrine moves back into the cytosol, where it is converted to epinephrine through the activity of phenylethanolamine-N-methyltransferase (PMNT). Epinephrine then moves into an epinephrine granule for storage before its release. The metabolism of catecholamines is rapid (2 minutes for norepinephrine, less for epinephrine) and is accomplished mainly by the liver and kidneys.
The importance of the anatomical association of the adrenal cortex and medulla may be related to the fact that cortisol is important for the activity of the enzyme PMNT. The chromaffin cells are located close to the venous sinuses that drain the adrenal cortex and therefore are exposed to venous effluent that contains high concentrations of cortisol.
The Primary Actions of Catecholamines Are on Metabolism, Especially Effects That Increase the Concentration of Glucose
The actions of the catecholamines involve the regulation of intermediary metabolism, as well as responses that allow animals to adjust to situations involving acute stress. The actions of catecholamines are mediated through adrenergic receptors located on target tissues (Figure 34-18). There are two major types of adrenergic receptors, alpha (α) and beta (β), which are subdivided into α1, α2, β1, and β2. The α-adrenergic receptors control catecholamine release from sympathetic nerve endings, with α1 affecting postsynaptic nerve endings and α2 affecting presynaptic terminals. The β1 receptors affect mainly the heart, and β2 receptors affect smooth muscle contraction and intermediary metabolism. Whereas all adrenergic receptors are responsive to both epinephrine and norepinephrine, the responses to the two catecholamines are different. In addition, the receptor types on various tissues vary in number, which, together with the different responses of adrenergic receptors on tissues, results in variable adrenergic responses being produced by a particular catecholamine.
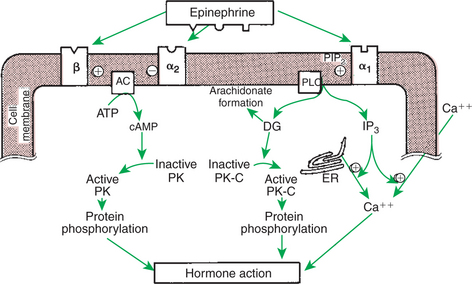
FIGURE 34-18 Mechanisms of action of epinephrine in target cells mediated by β-, α2-, and α1-adrenergic receptors. Plus signs indicate stimulation; minus sign indicates inhibition. AC, Adenyl cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; DG, diacylglycerol; ER, endoplasmic reticulum; IP3, inositol 1,4,5-triphosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; PK, protein kinase; PK-C, protein kinase C; PLC, phospholipase C.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The metabolic effects of catecholamines are mediated mainly by β2 receptors. Because epinephrine is 10 times more potent than norepinephrine with β2 receptors, epinephrine plays a much more important role in the control of intermediary metabolism than does norepinephrine. The effects of epinephrine on glucose metabolism are similar to those of glucagon and opposite to those of insulin. Epinephrine increases blood glucose concentrations, with the effect mainly in the liver; that is, epinephrine promotes both hepatic glycogenolysis and gluconeogenesis. Epinephrine also stimulates glycogenolysis in skeletal muscle, which in this situation is in contrast with the action of glucagon. Because glucose-6-phosphatase is not present in skeletal muscle, lactate is produced instead of glucose; the liver takes up lactate and converts it to glucose. Additional effects on glucose metabolism include the inhibition of insulin secretion (through α receptors) and stimulation of glucagon secretion by the pancreas; both actions increase blood glucose concentrations.
Epinephrine promotes lipolysis through interaction with two receptors on adipose cells. Activation of a lipase enzyme results in an increase in free fatty acids in the blood. Glucocorticoids potentiate the effect of epinephrine on lipolysis.
Catecholamines stimulate cardiac function. Both epinephrine and norepinephrine interact with β1 receptors to increase both the force of contraction and the heart rate, the latter resulting from the promotion of a shorter period of diastolic depolarization. Whereas both catecholamines promote arteriolar constriction through interaction with α receptors, epinephrine, through its high affinity for β2 receptors, causes the dilation of blood vessels both in the heart and in skeletal muscle. The end result is that total peripheral resistance is decreased by the action of epinephrine, with a concomitant decline in diastolic pressure; however, blood pressure is minimally changed, and cardiac output increases because of the increase in heart rate. The action of epinephrine to increase cardiac output is an obvious beneficial effect in situations that are described as “fight or flight.”
Catecholamines affect smooth muscle. Epinephrine causes relaxation of bronchial smooth muscle, particularly when the muscle is in a contracted state. Because the action is mediated through β2 receptors, norepinephrine has little effect on bronchial smooth muscle. Epinephrine causes relaxation of the smooth muscle of the gastrointestinal (GI) tract through interaction with β2 receptors. Catecholamine stimulation of β-adrenergic receptors results in contraction of uterine smooth muscle, and stimulation of β2 receptors results in relaxation. Because of its dominant effect on β2 receptors, epinephrine causes relaxation of the uterus, whereas both epinephrine and norepinephrine interact with α receptors to cause contraction.
The effects of the catecholamines on bladder smooth muscle depend on different locations of α and β receptors; α-adrenergic receptors are located within the neck of the bladder, and β-adrenergic receptors are located within the body of the bladder. Epinephrine relaxes the body and contracts the neck of the bladder; norepinephrine contracts the neck of the bladder. The net effect is retention of urine.
Although the parasympathetic nervous system is the principal system involved in penile erection, the sympathetic nervous system may also play a role. Epinephrine promotes erection through vasodilation of the vasculature mediated by β receptors. Higher concentrations of epinephrine (and norepinephrine) can cause ejaculation through α-receptor interaction and vasoconstriction.
In the eye, epinephrine causes relaxation of the lens through stimulation of β receptors on the ciliary muscles. It causes dilation of the pupil through stimulation of α receptors, with resultant contraction of the radial muscle of the iris.
The effects of epinephrine on the CNS are excitatory. Drugs that affect the CNS probably do so by modulation of catecholamine concentrations, whereby sedation is associated with lower values of epinephrine. Other effects of catecholamine include the promotion of sweating and piloerection. Epinephrine also increases renin production by the renal juxtaglomerular cells. Table 34-5 summarizes the effects of catecholamines.
Table 34-5 Responses of Target Tissues to Catecholamines
| Target tissue | Receptor type | Responses |
|---|---|---|
| Liver | β2 | Glycogenolysis, lipolysis, gluconeogenesis |
| Adipose tissue | β2 | Lipolysis |
| Skeletal muscle | β2 | Glycogenolysis |
| Pancreas | α2 | Decreased insulin secretion |
| β2 | Increased insulin secretion | |
| Cardiovascular system | β1 | Increased heart rate, increased contractility, increased conduction velocity |
| α2 | Vasoconstriction | |
| β2 | Vasodilation in skeletal muscle arterioles, coronary arteries, and all veins | |
| Bronchial muscle | β2 | Relaxation |
| Gastrointestinal tract | β2 | Decreased contractility |
| Urinary bladder | α2 | Sphincter contraction |
| β2 | Detrusor relaxation | |
| Uterus | α2 | Contraction |
| β2 | Relaxation | |
| Male sex organs | α2 | Ejaculation, detumescence |
| β2 | Erection? | |
| Eye | α1 | Radial muscle contraction |
| β2 | Ciliary muscle relaxation | |
| Central nervous system | α2 | Stimulation |
| Skin | α2 | Piloerection, sweat production |
| Renin secretion | β1 | Stimulation |
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
The Main Factors That Stimulate Catecholamine Secretion Are Hypoglycemia and Conditions That Produce Stress
Any factor that increases sympathetic nervous system stimulation of the adrenal medulla results in the immediate secretion of catecholamines. The main physiological factor that influences catecholamine secretion is hypoglycemia. In this situation, epinephrine secretion is stimulated by decreases in blood glucose concentrations that are within normal physiological limits. In contrast, other parts of the sympathetic nervous system are depressed by decreases in blood glucose levels. Factors that elicit a massive release of catecholamines are categorized as “stressful,” particularly those that are acute. Catecholamines are especially important for the mainte-nance of blood pressure in conjunction with severe blood loss; decreased blood pressure stimulates epinephrine secretion. Catecholamines are also important for adaptation to cold exposure in terms of increased heat production; decreased temperature increases epinephrine secretion. The response to acute stress can be particularly marked, because each preganglionic sympathetic neuron that supplies the adrenal medulla affects a number of chromaffin cells; that is, the signal is greatly amplified.
HORMONES OF THE PANCREAS
The pancreas has important endocrine and nonendocrine functions. The nonendocrine functions result from activity of the exocrine part of the pancreas and are involved in GI function. The endocrine portion of the pancreas is organized as discrete islets (islets of Langerhans) that contain four cell types, each of which produces a different hormone (Figure 34-19). The most numerous of the islet cells are β cells, which produce insulin; α cells produce glucagon; D cells produce somatostatin; and F or PP cells produce pancreatic polypeptide (Figure 34-20). Although these hormones have different functions, all are involved in the control of metabolism and, more specifically, in glucose homeostasis.
FIGURE 34-19 Depiction of section through the pancreas of the rat. The islet of Langerhans is a gland of internal secretion, whereas the surrounding acinar tissue forms an exocrine gland.
(From Turner CD, Bagnara JT: General endocrinology, ed 6, Philadelphia, 1976, Saunders.)
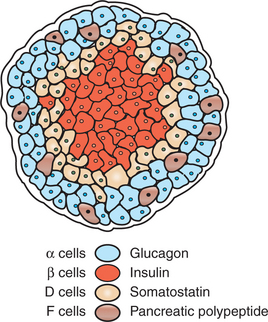
FIGURE 34-20 Depiction of the pancreatic islet.
(From McDonald LE: Veterinary endocrinology and reproduction, ed 4, Philadelphia, 1989, Lea & Febiger.)
Insulin
The first studies that associated the pancreas with carbohydrate metabolism were done by von Mering and Minkowski in 1889, when they showed that pancreatectomy of dogs resulted in signs similar to those characteristic of diabetes mellitus. Later, Banting and Best were able to show that injection of pancreatic extracts could alleviate the signs of diabetes mellitus in dogs and humans. Able was the first to crystallize insulin, and its structure was elucidated by Sanger in 1960.
Insulin is a protein consisting of two chains, designated A (21 amino acids) and B (30 amino acids), that are connected by two disulfide bridges. The monomer form of the hormone is thought to be the active form; insulin also exists in dimer and hexamer forms, the latter complexed with two zinc molecules. Although there are some differences in amino acid composition among species, the differences are small; for example, cattle, sheep, horses, dogs, and whales differ only in positions 8, 9, and 10 of the A chain. As a result, the biological activities of insulin are not highly species specific. Of the domestic species, feline insulin is most similar to bovine insulin, and canine insulin is identical to porcine insulin in its amino acid structure.
The Synthesis of Insulin Is Biphasic: an Acute Phase Involves the Release of Preformed Insulin, and a Chronic Phase Involves the Synthesis of Protein
The synthesis of insulin, similar to that of other peptide hormones, begins with the formation of a linear polypeptide preproinsulin within the rough endoplasmic reticulum. A small peptide fragment is removed to form proinsulin. Proinsulin is coiled, and the end fragments are joined by disulfide bonds. Proinsulin is transferred to the Golgi apparatus, where it is further processed and packaged into granules that contain both insulin and the connecting or C peptide (33 amino acids in length).
The secretion of insulin follows biphasic kinetics in response to appropriate stimuli (Figure 34-21). The initial, acute release of insulin involves the exocytosis of preformed insulin from secretion granules. After the acute phase, a chronic phase of secretion occurs that involves the synthesis of protein and thus probably the synthesis of insulin.
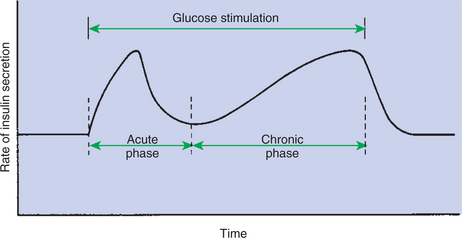
The Metabolism of Insulin Involves Splitting the A and B Chains and Reducing the Chains to Amino Acids and Peptides
Insulin is metabolized mainly by the liver and kidneys. Enzymes that are present reduce the disulfide bonds that link the A and B chains, and the chains are then subjected to protease activity, which reduces them to peptides and amino acids. The half-life of insulin is about 10 minutes.
The Main Metabolic Functions of Insulin Are Anabolic
Insulin acts at a number of sites within the metabolic pathways of carbohydrates, fats, and proteins (Figure 34-22). It is important to realize that the liver is an especially important target organ, in part because the pancreatic venous effluent passes directly to the liver. The net effect of the actions of insulin is to lower blood concentrations of glucose, fatty acids, and amino acids and to promote intracellular conversion of these compounds to their storage forms: glycogen, triglycerides, and protein, respectively (Table 34-6). Glucose does not readily penetrate cell membranes except in a few tissues, such as brain, liver, and red and white blood cells, all of which must have continual access to glucose. The presence of insulin is critical to the movement of glucose through the plasma membrane into the cell.
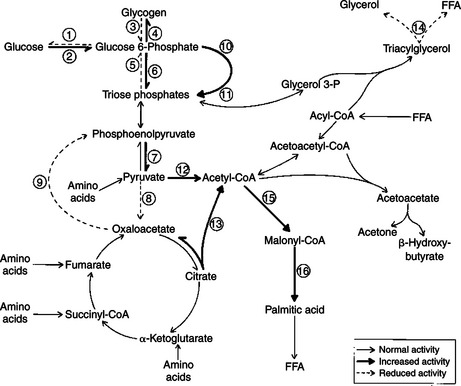
FIGURE 34-22 Metabolic pathways affected by insulin. The numbers correspond to each of the following enzymes: 1, glucose-6-phosphatase; 2, glucokinase; 3, phosphorylase; 4, glycogen synthase; 5, fructose-1,6-bisphosphate aldolase; 6, 6-phosphofructokinase; 7, pyruvate kinase; 8, pyruvate carboxylase; 9, phosphoenolpyruvate carboxykinase; 10, glucose-6-phosphate-dehydrogenase; 11, 6-phosphogluconate dehydrogenase; 12, pyruvate dehydrogenase; 13, adenosine triphosphate (ATP)–citrate lyase; 14, hormone-sensitive lipase; 15, acetyl–coenzyme A (CoA) carboxylase; 16, fatty-acid synthase. FFA, Free fatty acid.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
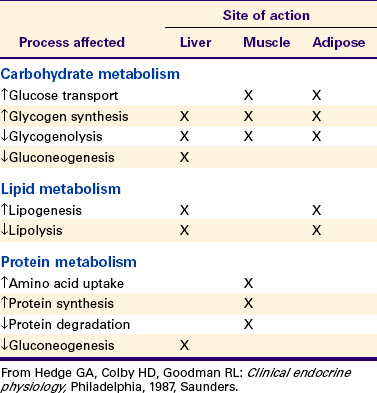
Insulin has profound effects on carbohydrate metabolism. Insulin facilitates the use of glucose: namely, glycolysis, which involves the oxidation of glucose to pyruvate and lactate through the induction of enzymes, such as glucokinase, phosphofructokinase, and pyruvate kinase. Insulin promotes glycogen production in the liver, in adipose tissue, and in skeletal muscle by increasing glycogen synthetase activity with a concomitant decrease in glycogen phosphorylase activity. Gluconeogenesis is decreased by insulin because of the promotion of protein synthesis in peripheral tissues, thereby decreasing the amount of amino acids available for gluconeogenesis. In addition, insulin decreases the activities of hepatic enzymes (fructose 1,6-bisphosphate aldolase, pyruvate carboxylase, phosphoenolpyruvate carboxylase, and glucose-6-phosphatase) that are involved in the conversion of amino acids to glucose.
In adipose tissue, insulin promotes the synthesis of triglycerides. Insulin facilitates the intracellular use of glucose, which results in increased levels of pyruvate, a precursor of acetyl–coenzyme A (CoA) (in turn, a precursor of fatty acids) and increased glycerol 3-phosphate for the esterification of fatty acids. Insulin activates the enzymes pyruvate dehydrogenase and acetyl-CoA carboxylase, which promote the synthesis of fatty acids from acetyl-CoA. Insulin also increases the activity of lipoprotein lipase located in the endothelium of capillaries of extrahepatic tissues, which promotes the movement of fatty acids into adipose tissue. Finally, insulin decreases lipolysis in adipose tissue.
With protein metabolism, insulin promotes uptake of amino acids by most tissues, including skeletal muscle, but not liver. Insulin promotes protein synthesis and inhibits protein degradation. Therefore, insulin promotes the maintenance of a positive nitrogen balance. With insulin deficiency, protein catabolism increases, with increased amounts of amino acids available for hepatic gluconeogenesis and a resultant increase in the blood glucose concentrations.
The most important factor in the control of insulin secretion is the concentration of blood glucose. Increased concentrations of blood glucose initiate the synthesis and release of insulin by the β cells of the pancreatic islets (Figure 34-23). Two theories explain the mechanism of cellular induction of insulin synthesis and release. In the first, insulin exists within the plasma membrane, whereby glucose interacts with a membrane receptor protein that directs intracellular events toward the synthesis and release of insulin. In the second theory, insulin occurs at the intracellular level, whereby the metabolism of glucose produces the signal for insulin synthesis and release. Glucose control of insulin secretion is a positive-feedback system in which increased concentrations of glucose lead to increased concentrations of insulin.
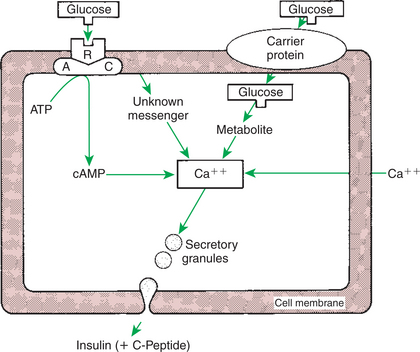
FIGURE 34-23 Proposed mechanisms of action of glucose on insulin secretion by the β cell. AC, Adenyl cyclase; ATP, adenosine triphosphate; R, receptor; cAMP, cyclic adenosine monophosphate.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Because the oral administration of glucose produces a greater insulin response than systemic administration, factors from the intestinal tract were thought to affect insulin secretion. It is now known that a number of GI hormones stimulate insulin secretion, including gastrin, cholecystokinin, secretin, and gastric inhibitory peptide. The presence of amino acids and fatty acids in the intestinal tract also stimulates the release of insulin, although with less potency than that of glucose (Box 34-1).
Box 34-1 Factors Affecting Insulin Secretion
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
Hormones other than those from the GI tract are important for the control of insulin secretion. Glucagon from the α cells of the pancreas has a direct stimulatory effect on the β cells to secrete insulin. Conversely, somatostatin inhibits the secretion of insulin. Both hormones work through the adenyl cyclase system, glucagon being stimulatory and somatostatin being inhibitory. Catecholamines tend to decrease insulin secretion through an interaction with the α-adrenergic receptors on the β cells. Whereas epinephrine is the main circulating catecholamine that affects insulin secretion, norepinephrine also influences insulin secretion because the pancreas has adrenergic innervation by the autonomic nervous system. The pancreas also has cholinergic innervation by the autonomic nervous system, and in contrast to adrenergic stimulation, cholinergic activity increases insulin secretion through the release of acetylcholine.
Insulin Deficiency Produces Diabetes Mellitus, Which Can Culminate in Diabetic Ketoacidosis
A lack or deficiency of insulin produces a syndrome called diabetes mellitus (DM). DM may be type 1, which is most common in dogs, or type 2, which is most common in cats. Type 1 DM is caused by an autoimmune destruction of the β cells of the pancreas; it leads to absolute insulin deficiency and a propensity to develop ketosis. Diabetic ketoacidosis (DKA) is the culmination of DM that results in unrestrained ketone body formation in the liver, metabolic acidosis, severe dehydration, shock, and possibly death. Hepatic lipid metabolism becomes deranged with insulin deficiency, and nonesterified fatty acids are converted to acetyl-CoA rather than incorporated into triglycerides. Acetyl-CoA accumulates in the liver and is converted into acetoacetyl-CoA, then ultimately to ketones, including acetoacetic acid, β-hydroxybutyrate (primary ketone in dogs and cats), and acetone.
As insulin deficiency culminates in DKA, accumulation of ketones and lactic acid in the blood and loss of electrolytes and water in the urine result in profound dehydration, hypovolemia, metabolic acidosis, and shock. Ketonuria and osmotic diuresis, resulting from glycosuria, cause sodium and potassium loss in the urine, exacerbating hypovolemia and dehydration. Nausea, anorexia, and vomiting, caused by stimulation of the chemoreceptor trigger zone through ketonemia and hyperglycemia, contribute to the dehydration caused by osmotic diuresis. Dehydration leads to further accumulation of glucose and ketones in the blood. Stress hormones such as cortisol and epinephrine contribute to the hyperglycemia in a vicious cycle. Eventually, severe dehydration may result in hyperviscosity, thromboembolism, severe metabolic acidosis, renal failure, and finally death.
Historical Findings. Most dogs and cats with DKA present with a previous history of uncomplicated diabetes, including polyuria, polydipsia, and dramatic rapid weight loss in the presence of a good or even ravenous appetite. More recent historical findings include anorexia, weakness, depression, vomiting, and diarrhea. Occasionally, owners fail to notice the significance of the classic signs of DM, and the animals are presented solely with an acute history of DKA. DKA may also develop in previously well-controlled, treated diabetic patients.
Physical Examination Findings. The most common examination findings in DKA are lethargy and depression, dehydration, unkempt hair coat, and muscle wasting. Hepatomegaly is common in both diabetic cats and diabetic dogs. Cataracts are often observed in diabetic dogs. A plantigrade rear limb stance resulting from diabetic neuropathy is often observed in diabetic cats. Other findings include tachypnea, dehydration, weakness, vomiting, and occasionally a strong acetone breath odor. Cats may present recumbent or comatose, which may be a manifestation of mixed ketotic-hyperosmolar syndrome. Icterus can develop from the complicating factors of hemolysis, hepatic lipidosis, and acute pancreatitis.
Laboratory Findings. Glucose level is greatly elevated. The average blood glucose concentration in patients with DKA is 25 mmol/L. Values can range from 10 mmol/L to greater than 50 mmol/L, but the latter is more characteristic of hyperosmolar coma. Although portable glucose meters are typically used to monitor glucose concentrations in DKA, caution is advised in relying on these monitors for baseline glucose concentrations because of inaccuracies in animals with severe hyperglycemia. All DKA patients have a relative or absolute deficiency of insulin and excessive hepatic production of glucose, resulting in hyperglycemia. Hyperglycemia is further exacerbated by dehydration and the corresponding reduction in GFR, and these factors are important determinants of its severity. This is supported by the findings that (1) glucose concentrations should exceed 25 mmol/L only when dehydration is severe enough to reduce GFR, and thus the ability of the kidneys to excrete glucose, and (2) fluid administration alone can significantly reduce blood glucose concentrations.
Osmolality usually is mildly to extremely increased in the DKA patient as a result of hyperglycemia, but this may not be detected, in part because of concurrent hyponatremia. Sodium (and to a lesser extent potassium), glucose, and urea concentrations are the determinants of the calculated serum osmolality. Reference values for serum osmolality in dogs and cats are approximately 290 to 310 mOsm/kg. Hyperosmolality is generally mild enough to resolve with intravenous fluid and insulin therapies.
Nonketotic hyperosmolar diabetes is defined by extreme hyperglycemia (>30 mmol/L), hyperosmolality (>350 mOsm/L), severe dehydration, and CNS depression, without ketone formation and with absent or mild metabolic acidosis. Affected patients are more likely to have underlying renal or cardiovascular disease and to be non–insulin dependent. Although this specific syndrome, as defined in humans, is infrequently encountered in veterinary medicine, ketotic or nonketotic diabetic cats may be seen, with significant hyperosmolality and CNS alterations.
Most patients with DKA have a total body K+ deficit caused by urinary (osmotic diuresis), anorexia, and GI losses (vomiting, anorexia). The metabolic acidosis, relative or absolute insulin deficiency, and serum hypertonicity combine to cause a K+ shift from the intracellular to the extracellular compartment. This is capable of masking the severity of total-body hypokalemia when plasma concentrations are measured. Insulin therapy, as well as correction of the acid-base disturbance with fluids and bicarbonate, will drive serum K+ intracellularly, potentially causing marked circulating hypokalemia. Polyuric patients are predisposed to severe hypokalemia, whereas oliguric or anuric patients are predisposed to severe hyperkalemia.
In general, DKA causes significant total-body Na+ deficits. Excessive urinary loss of Na+ results from the osmotic diuresis induced by high glucose and ketone concentrations and the lack of insulin, which usually aids in reabsorption of Na+ from the distal nephron. Hyperglucagonemia, vomiting, and diarrhea also contribute to the total-body Na+ loss. Hyperosmolality may contribute to a low Na+ concentration because as osmolality increases, water is drawn from the interstitium into the vascular space, thus diluting plasma Na+ and Cl−.
Phosphorus is the major intracellular anion and is important for energy production and for maintenance of cell membranes. Concentrations are regulated by dietary intake, renal elimination, factors promoting its movement into and out of cells, and vitamin D and parathyroid interactions. In DKA, circulating concentrations are usually within reference range or increased initially because of dehydration and renal disease. Phosphorus may also be low at presentation because of urinary loss from osmotic diuresis. As long as renal function is not compromised, a significant decrease in phosphorus should be anticipated with therapy. After insulin administration, phosphorus shifts to the intracellular compartment with glucose. Clinical signs of hypophosphatemia, such as hemolytic anemia (also seen with Heinz bodies in DKA), lethargy, depression, and diarrhea, may develop once concentrations reach 0.32 mmol/L. Oversupplementation of phosphorus should be avoided because hypocalcemia or metastatic calcification may result.
Magnesium (total serum) is not usually measured routinely, but concentrations may be abnormal in DKA. A recent study in cats demonstrated high total serum magnesium concentrations at presentation in those with DKA; after 48 hours of therapy, however, total serum magnesium concentrations were significantly decreased. Magnesium deficiency may be caused by poor oral intake, decreased intestinal absorption, increased renal loss, or changes in distribution because it is the second most abundant intracellular cation. Clinical signs of hypomagnesemia include neuromuscular weakness and cardiac arrhythmias, signs that can be seen with other electrolyte alterations. Hypomagnesemia can also cause decreases in other electrolytes, such as potassium and calcium. Correction of deficits may resolve electrolyte disturbances and may improve clinical outcome in the severely deficient patient.
Liver enzyme elevations are common in DM patients. Further increases potentially occur in DKA. Alanine aminotransferase and aspartate aminotransferase are most often affected, increasing secondary to hypovolemia and poor hepatic blood flow, with subsequent hepatocellular damage. Further increases in serum alkaline phosphatase concentration may occur if pancreatitis and secondary cholestasis ensue. Cholesterol and triglycerides may be elevated secondary to derangements of lipid metabolism resulting from decreased insulin.
Metabolic acidosis is one of the most prominent features of DKA. As ketone bodies accumulate in the blood and overwhelm the body’s buffering capabilities, there is an increase in hydrogen ions and a decrease in bicarbonate. As dehydration worsens, blood flow to peripheral tissues decreases, and the resulting lactic acidosis may contribute to the acid-base disturbance. Acidosis may manifest as lethargy, vomiting, hyperventilation, decreased myocardial contractility, peripheral vasodilation, stupor, and coma. Initiation of insulin therapy (to stop ketogenesis) and fluid therapy (to correct dehydration) will help improve the metabolic acidosis in most patients. Bicarbonate supplementation should be pursued with caution and is generally not recommended unless the patient’s blood pH is less than 7.1 or serum bicarbonate is less than 12 mmol/L.
Anion gap may be normal or elevated. An elevated value further characterizes the metabolic acidosis caused by DKA. The anion gap is a representation of the circulating anions not routinely measured on biochemical analyses. The normal anion gap ranges from 10 to 20 and is calculated by the following equation:
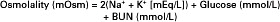
In DKA the ketones become unmeasured anions as they dissociate from ketoacids. However, if significant dehydration is present secondary to the osmotic diuresis and vomiting, lactic acidosis secondary to tissue hypoxia may contribute to the unmeasured anions, thus increasing the anion gap.
Circulating urea and creatinine concentrations may be within reference range or high. These values are high in most patients because of severe dehydration, but renal insufficiency or failure is also a possible cause. Increases in urea and creatinine must be evaluated in light of the urine specific gravity. A low urine specific gravity at presentation does not always guarantee a diagnosis of renal insufficiency or failure, because osmotic diuresis and chronic hypokalemia can contribute to low specific gravities in DM patients. Therefore, reevaluation of urea, creatinine, and urine specific gravity must be done after treatment of the crisis. If urea and creatinine are initially elevated and remain static or increase with appropriate therapy, concurrent renal disease is strongly suspected.
The most important part of urinalysis is measurement of glucose and ketones. A strongly positive glucose result confirms DM, and a positive result for ketones confirms DKA. However, a negative ketone result does not definitively rule out ketosis. The nitroprusside reagent used in urine sticks detects only acetoacetate and acetone. It is not as sensitive for β-hydroxybutyrate, the most prevalent ketone body, and therefore may be negative in the presence of ketosis. A recent study reported that β-hydroxybutyrate concentrations greater than 1.9 mmol/L were the most sensitive indicator of DKA, and values greater than 4.8 mmol/L were highly specific for its diagnosis. Using a cutoff value of 3.8 mmol/L was associated with the best combination of specificity (95%) and sensitivity (72%) for DKA.
The presence of pyuria and hematuria on urinalysis, along with confirmation by examination of urine sediment, supports the presence of UTI. However, urine culture should be performed regardless of urine sediment activity.
The hemogram may be normal at presentation but usually reveals a leukocytosis with a mature neutrophilia (common in cats) or a stress leukogram. There may be a regenerative or degenerative left shift suggestive of a severe inflammatory and infectious process. The red blood cell count and hematocrit may be elevated as a result of dehydration. Heinz bodies, with or without anemia, may be noted in cats because feline hemoglobin is uniquely susceptible to oxidative damage.
Concurrent Disease. Frequently, an underlying stressful event precipitates the shift from DM to DKA or nonketotic hyperosmolar DM. Impaired immune function secondary to DM increases the risk of infections. The precipitating event may be a UTI or other viral or bacterial infection or inflammatory disorder, such as pancreatitis, pyelonephritis, cholangiohepatitis, inflammatory bowel disease (IBD), eosinophilic granuloma complex, prostatitis, pyometra, upper respiratory infection, or pneumonia. Other concurrent diseases may include renal insufficiency or failure, hepatic lipidosis, neoplasia, and congestive heart failure. Recent drug therapy may also precipitate a crisis, especially administration of corticosteroids or progestagens. Therefore, further diagnostic testing of the diabetic patient that presents in a crisis is essential, particularly abdominal radiography or ultrasonography, as well as thoracic radiography and echocardiography if indicated. Additional testing for concurrent endocrine diseases, such as hyperthyroidism in cats and hypothyroidism and hyperadrenocorticism in dogs, may also be indicated but should be postponed until some control of the DM is achieved, because uncontrolled disease may affect the results of the tests.
Ancillary tests for pancreatitis include abdominocentesis or diagnostic peritoneal lavage if pancreatitis is suspected. Serum amylase and lipase, if determined on presentation, may be elevated in the absence of pancreatitis, secondary to severe dehydration or renal insufficiency, and demonstration of an elevated circulating concentration of trypsin-like immunoreactivity (TLI) may be preferable. Cats and dogs with acute necrotizing pancreatitis usually present with vomiting, abdominal pain, and concurrent DKA. Physical examination findings include icterus, cranial abdominal pain, and abdominal effusion. Radiographs may reveal a “ground glass” appearance of the abdomen, and abdominal ultrasound usually shows enlargement and hypoechogenicity of the pancreas. Concurrent hepatopathies are often present in patients with DKA, but evaluation is complicated by the effect of both DM and DKA on liver enzymes and liver function tests. Ultrasonography may be more useful in these cases. Diagnostic peritoneal lavage is usually necessary to demonstrate inflammatory, nonseptic peritonitis, and abdominal lipase is usually increased dramatically in affected cats and dogs.
Dietary Management Is an Important Consideration in Therapy for Feline Type 2 Diabetes
Type 2 DM is caused by insulin resistance and secondary β-cell failure. Type 2 diabetes may be managed using oral hypoglycemic agents, diet, or insulin. DM is one of the most common feline endocrinopathies, affecting 1 in 300 cats. The pathogenesis of type 2 DM in cats is reviewed earlier. Diagnosis of DM can be challenging, particularly in the early stages when the cats are non–insulin dependent. However, once clinical signs of diabetes are observed (polyuria, polydipsia, neuropathy), many cats may still benefit from alternatives to insulin therapy. In general, the primary abnormalities associated with type 2 DM, such as obesity and insulin resistance, are reversible. Insulin secretory ability, however, may be reversible (glucose toxicity) or irreversible (pancreatic amyloid deposition). In cats the differentiation of insulin-dependent (type 1) DM and non–insulin-dependent (type 2) DM is virtually impossible before treatment; therefore the clinician may have to rely on the response to oral hypoglycemic agents as a guide to whether the cat has sufficient β-cell function to be managed with oral hypoglycemic agents.
Goals of therapy for DM include restoration of normal fasting serum glucose concentrations, normalization of serum fructosamine, and reversal or attenuation of chronic complications, such as diabetic neuropathy and nephropathy. As in human patients with type 2 DM, the best approach to cats is a stepwise progression from dietary management to oral hypoglycemics and finally to insulin therapy when “islet burnout” occurs.
Exercise and diet is the cornerstone of therapy in human patients with type 2 DM. In most diabetic cats, exercise is not a reasonable option. One mechanism by which cats may be encouraged to exercise is by feeding the cat multiple small meals hidden in various places within the house. For example, an obese diabetic cat might be encouraged to jump up on the refrigerator or counter to find small amounts of food and then have to hunt for the rest of the food at the opposite end of the house.
In human diabetic patients, fiber supplementation is beneficial in the management of the disease. In humans and dogs, increased amounts of fiber slow the rate of glucose absorption from the intestine and minimize the postprandial fluctuations in blood glucose. This allows better glycemic control and correction of obesity; however, the data in cats are less compelling. In the only study of high-fiber diets in cats, 9 of 13 diabetic cats showed significant improvement in glycemic control with consumption of a high-fiber diet. Examples of high-fiber diets include prescription diet w/d and r/d, Science Diet Maintenance Light, Purina OM, and Iams Less Active. Because many cats find high-fiber diets unpalatable, soluble fiber such as psyllium can be mixed into the cat’s regular food, and glycemic control may still be enhanced. If the cat’s weight is normal at the start of therapy, the diet should be fed at a maintenance level of 60 to 70 kcal/kg/day. If the patient is obese, caloric intake should be limited to 70% to 75% of the energy needs for the cat’s optimal weight.
The cat is an obligate carnivore and as such is unique among mammals in its insulin response to dietary carbohydrates, protein, and fat. The feline liver exhibits normal hexokinase activity, but glucokinase activity is virtually absent. Glucokinase converts glucose to glycogen for storage in the liver and is important in “mopping up” excess postprandial glucose. Normal cats are similar to diabetic humans because glucokinase levels drop precipitously with persistent hyperglycemia in humans with type 2 DM. Amino acids, rather than glucose, are the signal for insulin release in cats. In fact, a recent study demonstrated more effective assessment of insulin reserve in cats using the arginine response test rather than a glucose tolerance test. Another unusual aspect of feline metabolism is the increase in hepatic gluconeogenesis seen after a normal meal. Normal cats maintain essential glucose requirements from gluconeogenic precursors (i.e., amino acids) rather than from dietary carbohydrates. As a result, cats can maintain normal blood glucose concentrations even when deprived of food for over 72 hours; furthermore, feeding has negligible effect on blood glucose concentrations in normal cats. In summary, the cat is uniquely adapted to a carnivorous diet (mice) and is not metabolically adapted to ingestion of excess carbohydrate.
When type 2 DM occurs in cats, the metabolic adaptations to a carnivorous diet become even more deleterious, leading to severe protein catabolism; feeding a diet rich in carbohydrates may exacerbate hyperglycemia and protein wasting in these diabetic cats. In humans with type 2 diabetes, the first recommendation is to restrict excess dietary carbohydrates such as potatoes and bread and to control obesity by caloric restriction. Furthermore, human patients with type 2 DM have improved glycemic control and nitrogen turnover during weight loss when a low-energy diet (high protein) is combined with oral hypoglycemic therapy.
The authors have found high-protein diets to be beneficial in increasing lean body mass and reducing postprandial hyperglycemia. Caution should be used when high-protein, restricted-carbohydrate diets are used in cats also treated with insulin because the insulin requirement may decrease. Usually the insulin dose is decreased by 25% in cats given high-protein diets and insulin. On the other hand, high-protein diets and oral hypoglycemic agents appear to be complementary treatments in cats that are underweight.
Glucagon
Glucagon is a protein hormone produced by the α cells of the islets of Langerhans. It has a close relationship with insulin in the control of glucose metabolism.
Glucagon is a polypeptide consisting of a single chain composed of 29 amino acids. There is considerable homology in amino acid composition among species. Glucagon is produced in other sites besides the pancreas; the stomach produces a molecule called gut glucagon that is identical to the pancreatic glucagon molecule, and the small intestine produces an immunologically similar molecule called glicentin. As with other polypeptide hormones, glucagon is first synthesized in the endoplasmic reticulum as part of a precursor molecule, packaged in the Golgi apparatus, and final processing occurs in the secretory granules. Glucagon is released by exocytosis. Glucagon is metabolized mainly by the liver and kidneys. It has a half-life in plasma of about 5 minutes.
The Most Important Functions of Glucagon Are to Decrease Glycogen Synthesis, Increase Glycogenolysis, and Increase Gluconeogenesis
The physiological actions of glucagon are the opposite of those of insulin; the main effect of glucagon is centered in the liver. Glucagon increases cAMP production in the liver, which leads to decreased glycogen synthesis, increased glycogenolysis, and increased gluconeogenesis, the last related to the effects of glucagon on protein metabolism (Figure 34-24). The net result is an increase in glucose concentrations in the blood.
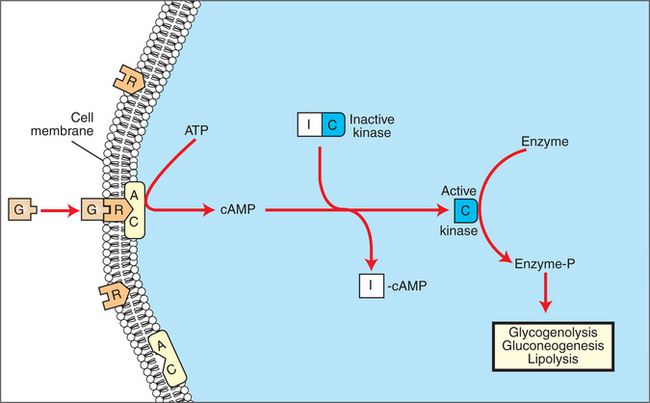
FIGURE 34-24 Mechanism of action of glucagon (G) on its target cells. AC, Adenyl cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; I and C, inhibitory and catalytic subunits of the kinase, respectively; R, receptor.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Changes in glucagon secretion counterbalance the effects of insulin in association with the daily ingestion of food and the intervals between food intake periods. After the consumption of food, the initial response of the metabolic system is increased insulin secretion, which results in conservation of energy through the formation of storage forms of carbohydrates, fats, and proteins. Glucagon secretion, which begins with the ingestion of food, increases as the interval from food ingestion lengthens and blood glucose concentrations begin to decline. This secretion allows the individual to mobilize energy stores for the maintenance of glucose homeostasis (i.e., to prevent postprandial hypoglycemia) (Figure 34-25).
FIGURE 34-25 Effects of hyperglycemia and hypoglycemia on the secretion of insulin and glucagon by the pancreatic β cells and α cells, respectively. Plus signs indicate stimulation; minus signs indicate inhibition.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Glucagon Synthesis Is Stimulated by Decreased Glucose Concentrations in the Blood
The main factor that regulates glucagon secretion is plasma glucose concentration. In contrast to insulin synthesis, decreased glucose concentrations stimulate glucagon synthesis and release, a relationship that represents a negative-feedback system. It must be remembered that glucagon regulation works in tandem with insulin regulation to maintain glucose concentrations within the physiological range. In fact, if glucagon were not secreted to maintain blood glucose concentrations, the individual would die of hypoglycemic shock. Because the α cells require insulin for glucose entry into the cells (as do most cells), in clinical syndromes involving insulin insufficiency (diabetes mellitus), glucose entry into the α cells is reduced, and plasma glucagon concentrations are paradoxically elevated. Glucagon promotes lipolysis and an increase in fatty acids, which has a negative-feedback effect on glucagon secretion.
Protein ingestion represents an exception to the rule of opposite responses of glucagon and insulin. The release of both insulin and glucagon in response to protein ingestion appears logical; increased insulin secretion, in response to increased plasma amino acid levels, leads to lower glucose concentrations, and increased glucagon would counteract this through increased hepatic gluconeogenesis, resulting in maintenance of blood glucose within normal limits. The complementary responses of insulin and glucagon allow growth to occur in animals fed a diet only of protein and fat.
Intestinal hormones, with the exception of secretin, stimulate both glucagon and insulin secretion. A similar (inhibitory) response to somatostatin is observed for both glucagon and insulin. Both sympathetic and parasympathetic stimulation of the autonomic nervous system induce secretion of glucagon (Figure 34-26).
FIGURE 34-26 Regulation of insulin and glucagon secretion by the autonomic nervous system. Plus signs indicate stimulation; minus sign indicates inhibition. NS, Nervous system.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Some birds have a predominance of glucagon in the pancreas, which suggests that glucagon may have a more important role in carbohydrate metabolism of avian species than in mammals.
Somatostatin
As indicated in Chapter 33, somatostatin was first described in the brain as a 14–amino acid peptide that inhibits growth hormone secretion by the pars distalis. The molecule has since been identified in a number of tissues, including other areas of the brain, the GI tract, and the D cells of the pancreatic islets. Its synthesis and secretion are similar to those observed for other protein hormones. The metabolism of somatostatin is rapid (about 5 minutes) and occurs mainly in the liver and kidneys.
The Main Functions of Somatostatin Are to Inhibit the Secretion of Hormones Produced by the Pancreas (Insulin, Glucagon, Pancreatic Polypeptide)
The actions of somatostatin can be classified as inhibitory. Pancreatic somatostatin inhibits the digestive processes by decreasing nutritive absorption and digestion. The motility and secretory activity of the GI tract are decreased by somatostatin. One of the most important physiological functions of pancreatic somatostatin is the regulation of the endocrine cells of the pancreas (Figure 34-27). Somatostatin inhibits secretion of all endocrine cell types of the islets of Langerhans, including the D cells. The α cells are more affected by the inhibitory action of somatostatin than are β cells; therefore, glucagon secretion is more affected by somatostatin than is insulin secretion.
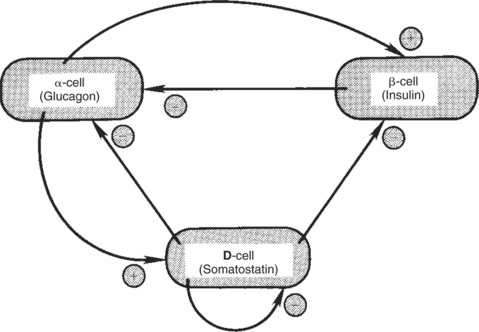
FIGURE 34-27 Possible cell-to-cell interactions in the pancreatic islets. Plus signs indicate stimulation; minus signs indicate inhibition.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Somatostatin secretion is increased by nutrients (e.g., glucose, amino acids) and by the neurotransmitters of the autonomic nervous system (epinephrine, norepinephrine, acetylcholine). Of the hormones produced by the pancreas, only glucagon stimulates somatostatin secretion.
Pancreatic Polypeptide
Pancreatic polypeptide, a 36–amino acid polypeptide, is produced by the F cells of the pancreas (see Figure 34-20). In contrast to somatostatin secretion, pancreatic polypeptide secretion is limited to the pancreas.
The effects of pancreatic polypeptide are directed toward the GI tract. The secretion of pancreatic enzymes and the contraction of the gallbladder are inhibited by the actions of this hormone. Both gut motility and gastric emptying are increased by the action of pancreatic polypeptide.
The secretion of pancreatic polypeptide is stimulated by intestinal hormones, including cholecystokinin, secretin, and gastrin. Stimulation of the vagus nerve also promotes pancreatic polypeptide secretion. The ingestion of protein is stimulatory for secretion, whereas carbohydrates and fats have little effect. As indicated previously, somatostatin inhibits pancreatic polypeptide secretion.
CALCIUM AND PHOSPHATE METABOLISM
Calcium Is Important for Intracellular Reactions, Including Muscle Contraction, Nerve Cell Activity, Release of Hormones Through Exocytosis, and Activation of Enzymes
The control of calcium and phosphate metabolism is important because these ions play a major role in physiological processes. Calcium homeostasis is tightly controlled; adjustments are made within a range of 5% of normal. Calcium is important for a number of intracellular reactions, including muscle contraction, nerve cell activity, the release of hormones through the process of exocytosis, and the activation of several enzymes. Calcium is important for coagulation of blood and for maintaining the stability of cell membranes and the linkage between cells. On a less acute basis, calcium is important for the structural integrity of bone and teeth.
Phosphate Is Important for the Structure of Bone and Teeth, and Organic Phosphate Serves as Part of the Cell Membrane and Several Intracellular Components
Phosphate concentrations are controlled by the same systems that control calcium concentrations. Inorganic phosphate in blood serves as the source of phosphate, which is important for the structure of bone and teeth. Inorganic phosphate also functions as an important H+ buffering system in blood. Organic phosphate is an important part of the cell, including the plasma membrane and intracellular components, such as nucleic acids, adenosine triphosphate, and adenosine monophosphate.
The Most Important Body Pool of Calcium Involved in Homeostasis Is the Extracellular Fluid Component
Almost all calcium (99%) in the body is in bone in the form of hydroxyapatite crystals, which contain calcium, phosphate, and water. The next largest pool of calcium is intracellular calcium. As stated previously, calcium is important for the response of cells in carrying out their physiological activities, including the secretion of hormones. In the inactive cell state, calcium concentrations are relatively low in the cytosol; calcium is bound to proteins or contained within the mitochondria or granules of the endoplasmic reticulum. Increased intracellular calcium concentrations are indicative of increased cell activity.
The smallest pool of calcium, which resides in the extracellular fluid (ECF), is the most important pool for physiological control of calcium concentrations in the blood. This component comprises interstitial calcium, blood calcium, and a small (0.5%) but important part of the bone calcium pool, which exists as amorphous crystals or in solution. The soluble bone calcium pool allows access to the large reserve of calcium that resides in bone.
The regulation of calcium levels involves control of the movement of calcium between the ECF and three body organs: bone, GI tract, and kidneys. The exchange of calcium ions between the ECF and intracellular fluid occurs in conjunction with the control of intracellular metabolism, with little effect on plasma concentrations of calcium.
The absorption of calcium from the GI tract is by passive diffusion and active transport. The passive diffusion of calcium across the intestinal mucosa occurs in the presence of high concentrations and, as such, is not an important aspect of calcium absorption. Active transport involves the movement of calcium into the intestinal cell down a concentration gradient, which is facilitated by carrier proteins located on the luminal side of the mucosal cell. Calcium is moved through the serosal side of the mucosal cell into the interstitial fluid through a calcium pump system. The active transport system adjusts according to the amount of calcium in the diet, becoming more active when calcium concentrations in the diet are lower and less active when calcium concentrations are higher. Calcium excretion into the GI tract is not affected by calcium uptake, and this can exacerbate conditions involving hypocalcemia. The GI tract serves as the source of calcium for the body, even though both absorption and excretion of calcium occur through the tract. As discussed later, vitamin D plays an important role in the absorption of calcium from the GI tract.
The kidneys serve as the route of excretion of calcium. Most of the calcium that passes into the kidneys is reabsorbed, with a net loss of only about 2%. This amount is matched by net absorption of calcium by the GI tract. Most of the calcium filtered by the kidneys is reabsorbed in the proximal tubules; the next largest amount is absorbed by the distal tubules, and a lesser amount, by the ascending loop of Henle. The distal tubules are under hormonal control and therefore are the sites of regulation of calcium in the kidneys.
The most important regulation of calcium metabolism between bone and ECF involves the soluble portion of bone. Amorphous crystals and soluble calcium, which form the source of ready exchange of ions with the blood, are located between the osteoblasts, which line the blood vessel channels, and the osteocytes, which are deeper in the bone (Figure 34-28). These two cell types have cytoplasmic projections that interact intimately through the presence of tight cell junctions. For labile bone calcium to reach the blood, calcium must cross the membrane barrier created by the osteoblasts and osteocytes. Movement of calcium from stable bone into the ECF also occurs but has little impact on the acute regulation of calcium concentrations. The process of remodeling bone, which occurs on a continuous basis, involves the breakdown of hydroxyapatite crystals by osteoclasts, a laying down of organic matrix by osteoblasts in the tunnels made by the osteoclasts, and finally the mineralization of the organic matrix by hydroxyapatite crystals. If an animal is subjected to prolonged changes involving calcium metabolism, the slowness of bone calcium exchange can have a significant impact on calcium metabolism.
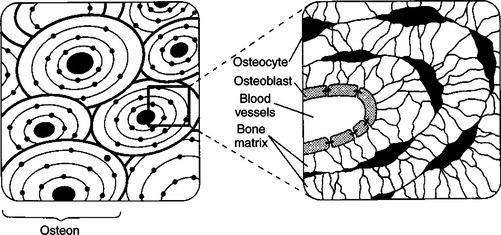
FIGURE 34-28 Structure of the osteon, the functional unit of bone, depicted in cross section at two magnifications.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Parathyroid Hormone
The main organ involved in the control of calcium and phosphate metabolism is the parathyroid gland (Figure 34-29). Most domestic animals have four pairs of parathyroid glands that are generally located at the poles of the two lobes of the thyroid gland; the pig has only one pair of parathyroid glands, which lie anterior to the thyroid. The cranial pair of parathyroid glands in dogs and cats are at the craniolateral poles of the thyroid, and those of ruminants and horses are anterior to the thyroid. The caudal pair of parathyroid glands in dogs, cats, and ruminants are located within the medial surface of the thyroid, whereas in the horse they lie near the bifurcation of the carotid trunk. The parathyroid cells that are in the active process of hormone secretion are called chief cells, whereas inactive, or degenerate, cells are called oxyphil cells.
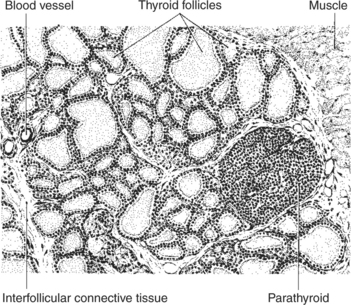
FIGURE 34-29 Depiction of a section of the thyroid and parathyroid glands of the rat as seen under low power of the microscope. Notice that the parathyroid gland lies near the surface of the thyroid gland and is surrounded on three sides by the thyroid follicles.
(From Turner CD, Bagnara JT: General endocrinology, ed 6, Philadelphia, 1976, Saunders.)
The synthesis of parathyroid hormone (PTH) is similar to the synthesis of other protein hormones; a prepro-PTH of 115 amino acids is synthesized in the rough endoplasmic reticulum and then cleaved by 25 amino acids to form pro-PTH. A 6–amino acid “pro” portion is removed by the Golgi apparatus; the resulting PTH has 84 amino acids. PTH is secreted by the process of exocytosis. PTH is rapidly metabolized by the liver and kidneys and has a relatively short half-life (5-10 minutes) in blood.
The effect of PTH is to increase calcium and decrease phosphate concentrations in ECFs. PTH has direct effects on bone and kidney metabolism of calcium and indirect effects on GI metabolism of calcium. The initial effect of PTH on bone is to promote the transfer of calcium across the osteoblast-osteocyte membrane. This level of action occurs without the movement of phosphate and therefore has no effect on phosphate concentrations in blood. PTH has additional effects on stable bone, which results in the resorption of the bone. This effect involves increased osteoclast activity and an inhibition of osteoblast activity. The effect of PTH on stable bone results in the release of both calcium and phosphate.
PTH acts on the distal convoluted tubules of the kidneys to increase absorption of calcium and decrease renal phosphate reabsorption through an effect on the proximal tubules. PTH is involved also in the activation of vitamin D at the kidney level. PTH mediates the absorption of calcium from the gut indirectly through its effect on vitamin D.
PTH secretion is controlled by free (ionized) calcium concentrations in blood; decreases in calcium levels stimulate PTH secretion, and increases in calcium turn off secretion (Figure 34-30). Both actions are mediated by an effect on cAMP metabolism. Epinephrine stimulates PTH secretion through stimulation of β-adrenergic receptors. Magnesium affects PTH secretion in the same manner as calcium, but its physiological impact is much less. Sleep affects the secretion of PTH; values are highest immediately after waking.
FIGURE 34-30 Changes in plasma immunoreactive parathyroid hormone (PTH) levels in response to hypercalcemia induced by calcium infusion; in response to hypocalcemia produced by ethylenediaminetetraacetic acid (EDTA) infusion; and in response to hyperphosphatemia with normocalcemia in a cow.
(From Capen CC: The calcium regulating hormones: parathyroid hormone, calcitonin, and cholecalciferol. In McDonald LE, Pineda MH, editors: Veterinary endocrinology and reproduction, Philadelphia, 1989, Lea & Febiger.)
Calcitonin
Calcitonin, a hormone produced by cells in the thyroid gland, also affects calcium metabolism. Cells of the type involved in the synthesis of calcitonin—parafollicular, or C, cells—are scattered throughout the thyroid gland and are distinctly different from the cells that synthesize thyroid hormones. During the early studies of calcitonin in animal classes such as fish, amphibians, reptiles, and birds, which have separate thyroid and ultimobranchial glands, it was found that all the calcitonin activity was in the ultimobranchial glands. Therefore the calcitonin cells represent ultimobranchial gland tissue that has been incorporated into the thyroid during embryonic development.
Calcitonin, synthesized as a preprohormone, has 32 amino acids; a ring structure at the amino terminus contains a disulfide link that bridges between amino acids 1 and 7. The processing of the molecule is interesting because calcitonin is located in the middle of procalcitonin, so an additional enzyme cleavage is required for the formation of the active molecule. The secretion of calcitonin is by exocytosis from granules.
Calcitonin acts as a counterbalance to PTH because it causes hypocalcemia and hypophosphatemia. The effect of calcitonin on mineral metabolism is mainly on bone (Figure 34-31). Calcitonin decreases the movement of calcium from the labile bone calcium pool (behind the osteoblast-osteocyte barrier) to the ECF and decreases bone resorption through an inhibitory effect on osteoclasts. Whereas the inhibition of bone resorption explains one aspect of the hypophosphatemic effects of calcitonin, calcitonin also increases movement of phosphate from the ECF into bone. Calcitonin decreases GI activity directly by inhibiting gastric acid secretion and indirectly by inhibiting gastrin secretion. The physiological importance of this is not known. Calcitonin also increases renal excretion of calcium and phosphate.
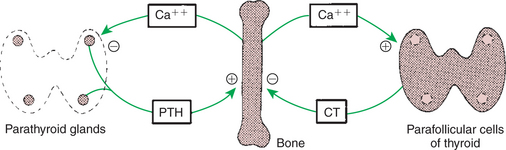
FIGURE 34-31 Negative-feedback loops controlling parathyroid hormone (PTH) and calcitonin (CT) secretion. Plus signs indicate stimulation; minus signs indicate inhibition.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The control of calcitonin secretion is by calcium; increased calcium concentrations cause increased secretion of calcitonin. The physiological control of calcium metabolism by calcitonin operates in situations of hypercalcemia with increased secretion of calcitonin and concomitant inhibition of PTH secretion. During hypocalcemic conditions (Figure 34-31), calcitonin synthesis is inhibited, and PTH becomes responsible for reestablishing normal calcium concentrations in the ECFs. GI hormones, including gastrin, cholecystokinin, secretin, and glucagon, stimulate the secretion of calcitonin, with gastrin the most potent. These hormones limit postprandial hypercalcemia.
Vitamin D
Vitamin D is important for the absorption of calcium from the gut. It is a steroidlike molecule, and because it is produced in one tissue and transported by the blood to a distant site of action, it should probably be called a hormone instead of a vitamin. All the vitamin D produced by the body is produced in the skin. Epithelial cells of the skin synthesize the immediate precursor of vitamin D, 7-dehydrocholesterol, from acetate. Exposure of the skin to ultraviolet light results in cleavage of the C-9 and C-10 bonds of 7-dehydrocholesterol, which results in the formation of vitamin D (Figure 34-32). The vitamin D molecule, as such, is inactive and must be transformed by both the liver and the kidney before the molecule is biologically activated. The liver first hydroxylates the molecule at the C-25 position, and the kidney subsequently hydroxylates the molecule at C-1 to produce the active compound, 1,25-(OH)2-vitamin D (1,25-vitamin D).
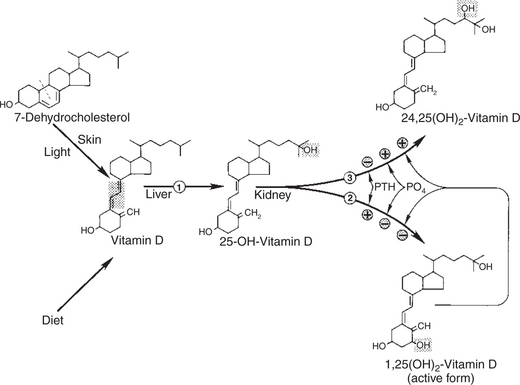
FIGURE 34-32 Synthesis and metabolism of vitamin D. The position of hydroxylation of 25-OH-vitamin D in the kidney is controlled by parathyroid hormone (PTH), phosphate (PO4), and 1,25-(OH)2-vitamin D. Shading indicates structural change at each step; dashed line indicates the position of cleavage of 7-dehydrocholesterol to produce vitamin D. Enzymes: (1) 25-hydroxylase, (2) 1α-hydroxylase, (3) 24-hydroxylase. Plus signs indicate stimulation; minus signs indicate inhibition.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Control of the C-1 hydroxylase in the kidney by PTH is the most important control linkage for the synthesis of 1,25-vitamin D. Decreases in calcium concentrations stimulate PTH secretion, which in turn favors the synthesis of active vitamin D and increased intestinal absorption of calcium. Phosphate also regulates vitamin D metabolism. Increased serum phosphate concentrations stimulate an enzyme that promotes hydroxylation of C-24 (instead of C-1) by the kidney, which leads to the formation of 24,25-(OH)2-vitamin D, an inactive molecule. The active molecule, 1,25-vitamin D, also regulates itself by decreasing C-1 hydroxylase and increasing C-24 hydroxylase activity; decreased amounts of active vitamin D is the result.
Because of its lipid nature, 1,25-vitamin D is transported by binding to proteins in the plasma. Most of vitamin D is carried in association with a specific α globulin called transcalciferin, a molecule synthesized by the liver.
The most important effects of vitamin D involve increased absorption of calcium by the GI tract. Vitamin D stimulates the synthesis of protein within the mucosal cells, which aids the rate-limiting step in calcium absorption: movement of calcium into the mucosal cell (Figure 34-33). Because the intestinal effect of vitamin D depends on the activation of protein synthesis by mucosal cells, the effect on calcium absorption usually requires several hours. Although the stimulation of protein synthesis relates mostly to active transport of calcium, vitamin D also stimulates passive transfer of calcium. Vitamin D also has effects on bone, promoting the movement of calcium ions from the labile pool into ECFs and the resorption of bone, as well as enhancing the effects of PTH on bone metabolism of calcium.
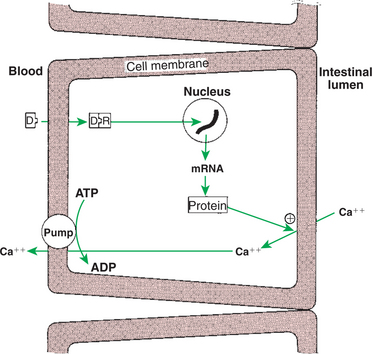
FIGURE 34-33 Mechanism of action of 1,25-(OH)2-vitamin D (D) to increase calcium absorption in the intestine. Plus sign indicates stimulation. R, receptor.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
The control of 1,25-vitamin D synthesis is by PTH and phosphate. A decrease in calcium concentrations results in increased PTH secretion and increased formation of 1,25-vitamin D through enhancement of C-1 hydroxylation (Figure 34-34). This action leads to the correction of hypocalcemia by increasing absorption of calcium by the gut. A decline in phosphate concentrations results in decreased inhibition of the C-1 hydroxylation, which indirectly results in increased 1,25-vitamin D production and increased absorption of phosphate. Some evidence suggests that hormones associated with pregnancy, such as growth hormone and prolactin, increase 1,25-vitamin D production by stimulating C-1 hydroxylation.
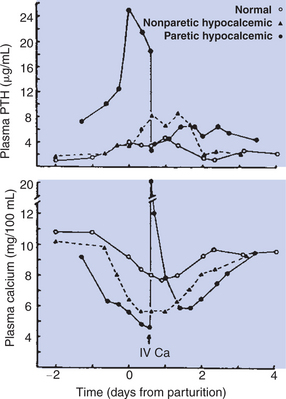
FIGURE 34-34 Development of varying degrees of hypocalcemia in cows near parturition, with corresponding increases in plasma parathyroid hormone (PTH) levels. The cow developing severe hypocalcemia (<5 mg/100 mL) had a considerably greater increase in plasma PTH levels than the moderate rise detected in the nonparetic hypocalcemic and the normal cow. Note that PTH levels declined rapidly after treatment of the paretic cow with intravenous calcium (IV Ca).
(From Mayer CP: The roles of parathyroid hormone and thyrocalcitonin in parturient paresis. In Anderson JJB, editor: Parturient hypocalcemia, New York, 1970, Academic Press.)
In the overall control of calcium metabolism, PTH is primarily responsible for the maintenance of calcium homeostasis. The primary target tissue for PTH in calcium homeostasis is the labile pool in bone; changes in renal absorption of calcium are also important. In the case of long-term calcium deficit in the diet, both PTH and 1,25-vitamin D are important for correction of the deficit. Decreased dietary calcium leads to decreased concentrations of calcium in the ECFs and the release of PTH. PTH affects resorption of calcium by the kidneys, but most importantly for long-term correction of the problem, it causes increased 1,25-vitamin D secretion with increased absorption of dietary calcium. PTH also contributes to the overall calcium pool through its effect on stable bone, that is, the promotion of resorption.
Hypercalcemia has a variety of causes, including malignancy, hyperparathyroidism, fungal disease, osteoporosis, hypoadrenocorticism, chronic renal disease, and hypervitaminosis D. The initial signs of hypercalcemia are polydipsia and polyuria resulting from impaired response of distal renal tubules to antidiuretic hormone. Listlessness, depression, and muscle weakness result from depressed excitability of neuromuscular tissue. Mild GI signs of hypercalcemia include inappetence, vomiting, and constipation. Persistent mild elevations in serum calcium (12-14 mg/dL) can cause uroliths and signs of urinary tract disease such as hematuria and stranguria. On the other hand, severe hypercalcemia (>14 mg/dL) can progress rapidly to acute renal failure when the calcium-phosphorus product (Ca × PO4) exceeds 60 to 80 mg/dL because of mineralization of renal tissue.
The diagnostic approach to hypercalcemia consists of ruling out the most common cause: hypercalcemia of malignancy. A thorough history and physical examination, including lymph node and rectal examination (for anal sac adenocarcinoma), complete blood cell count, urinalysis, serum chemistry profile, and chest and abdominal radiographs, are necessary to search for underlying neoplastic processes. If lymphoma is not detected on the minimum database, a bone marrow examination and survey skeletal radiographs may be necessary. Once a diagnosis of neoplasia has been excluded, the next primary differential for hypercalcemia is chronic renal failure. This is the most difficult differential to exclude because other causes of hypercalcemia may result in renal damage because of soft tissue mineralization of the kidneys. Therefore an animal with hypercalcemia, azotemia, and hyperphosphatemia could have primary hyperparathyroidism, primary renal failure with secondary renal hyperparathyroidism, or vitamin D intoxication. Furthermore, patients with hypercalcemia secondary to renal disease may also exhibit elevations in intact PTH. Diagnosis of primary hyperparathyroidism is based on the findings of hypercalcemia (preferably ionized), hypophosphatemia (unless azotemic), high-normal to elevated serum PTH concentrations, and a mass in the cervical region. Intact PTH, demonstrated by a “sandwich” assay validated for use in the dog and cat, should be measured. A normal PTH concentration in the presence of elevated total and/or ionized calcium is considered inappropriate for the calcium level and would be considered diagnostic for primary hyperparathyroidism. For suspected cases of hypercalcemia of malignancy in which the diagnostic approach has failed to identify a neoplastic process, PTH-related protein (PTH-rp) concentrations may be measured.
The classic biochemical findings in animals with hypoparathyroidism are hypocalcemia (both total and ionized) and hyperphosphatemia. Other causes of hypocalcemia include iatrogenic (post-thyroidectomy) hypoparathyroidism, chronic and acute renal failure, acute pancreatitis, hypoalbuminemia, puerperal tetany (eclampsia), ethylene glycol intoxication, intestinal malabsorption, and nutritional secondary hyperparathyroidism. Early signs of hypocalcemia are nonspecific and include anorexia, facial rubbing, nervousness, and a stiff, stilted gait. Later signs progress to paresthesias, hyperventilation, and finally generalized tetany and seizures.
Primary hypoparathyroidism is diagnosed by means of an intact PTH assay. Serum or plasma PTH concentrations should be measured on a freshly drawn morning sample in a fasting animal. Handling of the sample is crucial to appropriate diagnosis because PTH may degrade if subjected to warm temperatures. Intact PTH refers to the entire 85–amino acid sequence of PTH; this is measured in a double-antibody “sandwich” assay in most endocrine laboratories that perform PTH measurement. For the diagnosis of primary hypoparathyroidism, the sample should be analyzed for both ionized calcium and intact PTH. Low ionized calcium and undetectable intact PTH concentrations are diagnostic for hypoparathyroidism.
CLINICAL CORRELATIONS
Diabetes Mellitus
History.
You are presented with a 10-year-old, intact, female poodle whose owner is upset because the dog urinates in the house. In addition, the owner has noticed that the animal now drinks larger amounts of water than in the past. Although the owner indicates the dog has a good appetite, it appears to have lost weight over the past few months.
Clinical Examination.
During the examination you check the dog’s breath and detect a sweet odor. Among the organ systems you check are the eyes, and you find developing cataract formation. Because you have seen this dog many times, you check its weight and find that it has lost 2 pounds since its last admittance a year ago. You are able to run a blood glucose determination in your hospital and tell the owner that the glucose concentration is 278 mg/dL.
Comment.
The findings in diabetes mellitus (DM) are all attributable to inadequate availability of insulin. Glycogen synthesis decreases in tissues, whereas glycogenolysis and gluconeogenesis increase, contributing to the high concentrations of glucose found in blood. When glucose concentrations exceed the reabsorption capacity in the tubular cells of the kidney, glucose appears in the urine. The loss of glucose in urine causes an osmotic diuresis (polyuria), and the dog compensates for this by drinking additional amounts of water. The sweetness of the breath is caused by the presence of ketone bodies. These form as the result of decreased triglyceride synthesis in adipose tissue, which stimulates lipase activity and the release of free fatty acids. These fatty acids are metabolized to ketone bodies (acetoacetate, acetone, β-hydroxybutyrate) by the liver in a situation of excess fatty acids. The end result is both a ketonemia and a ketonuria. Protein metabolism shifts toward catabolism during DM, with decreased protein synthesis and increased protein degradation by muscle cells. This process increases the circulating concentrations of amino acids available to the liver for gluconeogenesis. The end result is nitrogen loss and a decrease in the muscle mass of the animal. The changes noted in the lenses of the eyes represent only one of a number of changes that occur in the presence of DM as a result of glycosylation of proteins, including lens proteins and hemoglobin.
Treatment.
Insulin administration is essential in the treatment of insulin-dependent (type 1) DM. During the initial stages of treatment, considerable care must be taken to ensure that the dosage is correct. The goal of treatment is to maintain glucose concentrations between a low of 80 mg/dL and a high of 200 mg/dL, with a serum fructosamine less than 400 μmol/L and one or two insulin injections every 24 hours. Too much insulin has the potential for producing a hypoglycemic coma. Two other important aspects of treatment include feeding the animal a diet high in soluble fiber in conjunction with insulin administration and adequate exercise. Finally, the owner needs to be educated and prepared for the necessity of intensive involvement in the management of the disease.
Eiler H. Endocrine glands. Reece WO, ed. Dukes’ physiology of domestic animals, ed 12, Ithaca, NY: Cornell University Press, 2004.
Feldman EC, Nelson RW. Canine and feline endocrinology and reproduction, ed 3, Philadelphia: Saunders, 2004.
Foster DW, Kronenberg HM, Larsen PR, Wilson JD. Williams textbook of endocrinology, ed 9. Philadelphia: Saunders, 1998.
Hedge GA, Colby HD, Goodman RL. Clinical endocrine physiology. Philadelphia: Saunders, 1987.
Martin R. Endocrine physiology. New York: Oxford University Press, 1985.
Pineda MH, Dooley MP. McDonald’s veterinary endocrinology and reproduction, ed 5, Ames: Iowa State University Press, 2003.
Tepperman J, Tepperman M. Metabolic and endocrine physiology, ed 5. Chicago: Year Book Medical Publishers, 1987.
PRACTICE QUESTIONS
1. The other main hormone secreted by the thyroid gland, in addition to tetraiodothyronine and triiodothyronine, is:
2. The most important function of mineralocorticoids is control of:
3. The pancreas has four types of cells, each of which produce a specific hormone. For example, the α cells of the pancreas produce:
4. Two hormones play an important role in calcium homeostasis. The two hormones, __________ and _________________, cause an increase and a decrease in calcium concentrations, respectively:
5. The main functions of the catecholamines are to allow rapid body responses to acute stimuli, which include the mobilization of glucose. The catecholamines are secreted by the sympathetic portion of the autonomic nervous system. The _____________ hormone is the main neurotransmitter of the sympathetic nervous system, whereas __________________ is the main hormone produced by the postganglionic fibers of the adrenal medulla.