CHAPTER 44 Acute Renal Failure and Chronic Kidney Disease
Renal failure occurs when approximately three fourths of the nephrons of both kidneys cease to function. Acute renal failure (ARF) results from an abrupt decline in renal function and is usually caused by an ischemic or toxic insult to the kidneys, although leptospirosis is reemerging as an important infectious cause of ARF. Ischemic or toxicant-induced injury usually results in damage to the metabolically active epithelial cells of the proximal tubules and thick ascending loop of Henle, causing impaired regulation of water and solute balance. Nephrotoxicants interfere with essential tubular cell functions and cause cellular injury, swelling, and death. Renal ischemia causes cellular hypoxia and substrate insufficiency, which leads to the depletion of adenosine triphosphate (ATP), cellular swelling, and death. Vasoconstriction secondary to toxic or ischemic tubular epithelial injury further decreases glomerular filtration. It is important to note, however, that tubular lesions and dysfunction caused by toxic and ischemic insults may be reversible. In contrast, the nephron damage associated with chronic kidney disease (CKD) is usually irreversible. Regardless of whether the underlying disease primarily affects the glomeruli, tubules, interstitium, or renal vasculature, irreversible damage to any portion of the nephron renders the entire nephron nonfunctional. Irreversibly damaged nephrons are replaced by fibrous connective tissue; therefore a specific cause is rarely determined once end-stage kidney damage is present. CKD occurs over a period of weeks, months, or years and is a leading cause of death in dogs and cats. Once advanced stage CKD has occurred, improving renal function is usually not possible. The goal of CKD treatment is threefold: (1) to identify and correct the primary disease process, (2) to monitor and slow disease progression, and (3) to alleviate patient clinical signs.
Many different and sometimes confusing terms are used to describe renal function and its deterioration (Fig. 44-1):
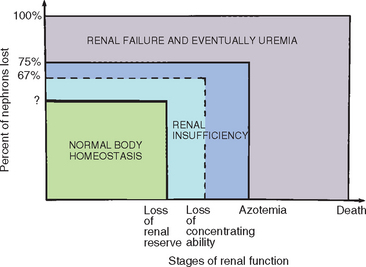
FIG 44-1 The stages of renal function.
(From Grauer G et al: Chronic renal failure in the dog, Compend Contin Educ Pract Vet 3:1009, 1981.)
ACUTE RENAL FAILURE
Etiology and Pathogenesis
The kidneys are highly susceptible to the effects of ischemia and toxicants because of their unique anatomic and physiologic features (Box 44-1). For example, the large renal blood flow (approximately 20% of the cardiac output) results in the increased delivery of blood-borne toxicants to the kidney, as compared with that to other organs. The renal cortex is especially susceptible to toxicants because it receives 90% of the renal blood flow and contains the large endothelial surface area of the glomerular capillaries. Within the renal cortex, the epithelial cells of the proximal tubule and thick ascending loop of Henle are most frequently affected by ischemia and toxicant-induced injury because of their transport functions and high metabolic rates. Toxicants disrupt the metabolic pathways that generate ATP, and ischemia can rapidly deplete cellular ATP stores. With the resulting loss of energy, the sodium-potassium pump (Na/K) fails, leading to cell swelling and death. By resorbing water and electrolytes from the glomerular filtrate, tubular epithelial cells may be exposed to increasingly higher concentrations of toxicants. Toxicants that are either secreted or resorbed by tubular epithelial cells (e.g., gentamicin) may accumulate in high concentrations within these cells. Similarly, the countercurrent multiplier system may concentrate toxicants in the medulla. Finally, the kidneys also play a role in the biotransformation of many drugs and toxicants. This usually results in the formation of metabolites that are less toxic than the parent compound; however, in some cases (e.g., the oxidation of ethylene glycol to glycolate and oxalate), the metabolites are more toxic than the parent compound.
 BOX 44-1 Factors that May Predispose the Kidney to Ischemia and Toxicant-Induced Injury
BOX 44-1 Factors that May Predispose the Kidney to Ischemia and Toxicant-Induced Injury
The kidneys receive 20% of cardiac output; the cortex receives 90% of the renal blood flow.
The glomerular capillaries have a large surface area.
Proximal tubule and thick ascending loop of Henle cells have a high metabolic rate and are susceptible to hypoxia and nutrient deficiency.
Tubular secretion and resorption may concentrate toxicants within cells.
A countercurrent multiplier system may concentrate toxicants within the medulla.
Xenobiotic metabolism within the kidney may generate toxic metabolites (e.g., metabolism of ethylene glycol).
Box 44-2 presents a partial list of potential nephrotoxicants. It should be noted that toxic insults to the kidney often can be caused by therapeutic agents, in addition to the better-known nephrotoxicants. Gentamicin and ethylene glycol are two of the most common causes of toxicant-induced ARF. Box 44-3 presents a partial list of ischemic causes of ARF. Leptospirosis is a common cause of ARF; the organisms colonize and proliferate within renal tubular epithelial cells and can lead to acute interstitial nephritis. Acute renal damage leading to failure can also occur in dogs with leptospirosis because of renal vasculitis and the development of swelling that further compromises renal blood flow. Acute renal failure has also recently been associated with ingestion of pet food containing contaminated wheat and corn gluten and rice protein concentrates. The investigation has focused on melamine and cyanuric acid as the major contaminants; however, melamine-related substances (e.g., ammelide and ammeline) may also be involved in the pathogenesis. It is thought that a chemical reaction between melamine and cyanuric acid produces insoluble crystals that form in the distal renal tubules of affected animals, compromising renal function. Clinical presentation is quite variable and ranges from severe ARF to mild azotemia associated with urine-concentrating deficits to no clinical signs. Crystalluria (round, yellow crystals with radiant striations that may resemble urate crystals) is observed in many cases. Identification of the crystals can be accomplished at veterinary diagnostic laboratories. Treatment of affected patients in largely symptomatic, long-term intravenous (IV) fluid therapy may be required for recovery in some cases.
BOX 44-2 Partial List of Potential Nephrotoxicants in Dogs and Cats
In many cases, ARF inadvertently develops in the hospital setting in conjunction with the performance of diagnostic or therapeutic procedures. For example, ARF may be caused by hypotension and decreased renal perfusion associated with anesthesia and surgery or with the use of vasodilators or nonsteroidal antiinflammatory drugs (NSAIDs). Prolonged anesthesia with inadequate fluid therapy in older dogs and cats with preexisting, subclinical renal insufficiency is a frequent cause of renal ischemia and ARF in the hospital setting. Similarly, ARF frequently occurs in animals treated with potential nephrotoxicants such as gentamicin or amphotericin. The kidneys can maintain adequate renal perfusion pressure by autoregulation as long as the mean arterial blood pressure exceeds approximately 60 to 70 mm Hg. Renal blood flow and perfusion pressure must be maintained for glomerular filtration and cellular delivery of oxygen and nutrients to occur. Cellular swelling secondary to decreased Na/K pump activity results from the osmotic extraction of water from the extracellular space, causing the amount of water in the plasma to decrease. The consequences of a decreased amount of plasma water in the renal vasculature are red blood cell aggregation and vascular congestion and stasis, which tend to potentiate and perpetuate decreased glomerular blood flow and decreased oxygen and nutrient delivery. The common result of ischemic or toxicant-induced tubular cell swelling, injury, and death is nephron dysfunction leading to a decreased glomerular filtration rate (GFR).
In ARF dysfunction and reduced glomerular filtration occur at the individual nephron level as a result of a combination of tubular obstruction, tubular backleak, renal arte riolar vasoconstriction, and decreased glomerular capillary permeability. Specifically, cellular debris within the tubule may inspissate and obstruct the flow of filtrate through the nephron. Alternatively, interstitial edema may compress and obstruct renal tubules. A backleak, or abnormal reabsorption of filtrate, occurs because of a loss of tubular cell integrity, allowing the filtrate to cross from the tubular lumen into the renal interstitium and subsequently the renal vasculature. Tubular backleak is facilitated by tubular obstruction because of the increased intratubular pressures proximal to the obstruction. The decreased resorption of solute and water by damaged proximal tubule segments results in the increased delivery of solutes and fluid to the distal nephron and macula densa in many nephrons, which causes afferent glomerular arteriole constriction. The exact mediators of this vasoconstriction are not known, but natriuretic factor, the renin-angiotensin system, and thromboxane may be involved. A decrease in the permeability of the glomerular capillary wall also leads to a reduction in glomerular filtration. For example, aminoglycosides have been shown to decrease both the number and size of fenestrae in glomerular capillary endothelial cells, thereby decreasing the surface area available for ultrafiltration. The impaired glomerular capillary permeability that occurs in ARF often persists after vasoconstriction and renal blood flow have been corrected.
Acute tubular damage leading to ARF has three distinct phases: (1) initiation, (2) maintenance, and (3) recovery. During the initiation phase, therapeutic measures that reduce the renal insult can prevent the development of established ARF. In the initiation phase, individual tubules are damaged but overall renal function remains adequate. Acute tubular damage, occurring before the development of ARF, is suggested by renal tubular epithelial cells and casts in the urine sediment (discussed in more detail later). The maintenance phase is characterized by the development of tubular lesions and nephron dysfunction (i.e., renal azotemia and urine-concentrating deficits). Although therapeutic interventions during the maintenance phase are often life saving, they usually do little to diminish the severity of existing renal lesions, improve function, or hasten recovery. In the recovery phase, renal lesions are repaired and function improves. Tubular damage may be reversible if the tubular basement membrane is intact and viable epithelial cells are present. Although new nephrons cannot be produced and irreversibly damaged nephrons cannot be repaired, the functional hypertrophy of surviving nephrons may adequately compensate for the decrease in nephron numbers. Even if renal functional recovery is incomplete, adequate function may be reestablished.
Clinical Features and Diagnosis
Clinical signs of ARF are often nonspecific and include lethargy, depression, anorexia, vomiting, diarrhea, and dehydration; occasionally, uremic breath or oral ulcers may be present. A diagnosis of ARF is suspected if azotemia develops acutely and is associated with persistent isosthenuria or minimally concentrated urine. Prerenal dehydration and azotemia superimposed on an inability to concentrate urine (e.g., Addison’s disease, hypercalcemia, overzealous use of furosemide) initially mimics renal failure; however, in these prerenal cases, volume replacement results in resolution of the azotemia.
ARF occurs within hours or days of exposure to the insult. Unique clinical signs and clinicopathologic findings associated with ARF include enlarged or swollen kidneys, hemoconcentration, good body condition, active urine sediment (e.g., granular casts, renal epithelial cells), and relatively severe hyperkalemia and metabolic acidosis (especially in the face of oliguria; see Box 41-7). Clinical signs in an animal with ARF tend to be severe compared with those seen in an animal with CKD and similar magnitude of azotemia. Renal ultrasonographic findings in dogs and cats with ARF are usually nonspecific, with diffusely normal to slightly hypoechoic renal cortices. In animals with calcium oxalate nephrosis associated with ethylene glycol ingestion, the renal cortices can be very hyperechoic (Fig. 44-2). Doppler estimation of the resistive index (RI) in renal arcuate arteries is increased in many dogs with ARF; however, this method of evaluation must be more extensively correlated with the renal histopathologic changes before firm conclusions regarding the merits of the RI can be drawn.
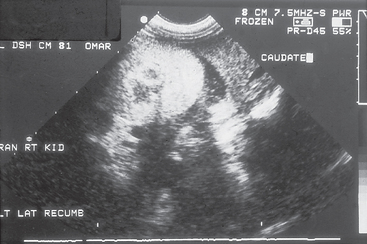
FIG 44-2 Ultrasonographic appearance of a kidney from a dog that ingested ethylene glycol. Notice the markedly increased renal cortical echogenicity.
(Courtesy Dr. Phillip Steyn, Colorado State University, Fort Collins, Colo.)
Renal biopsy specimens from dogs and cats with ARF show proximal tubular cell degeneration, ranging from cloudy swelling to necrosis, with edema and mononuclear and polymorphonuclear leukocyte infiltration in the interstitium. Ethylene glycol and melamine-associated nephrotoxicity is frequently associated with intratubular crystals. Although toxicant-induced ARF cannot be histopathologically differentiated from ARF caused by ischemia in all cases, renal histologic findings are often helpful in establishing a prognosis. Evidence of tubular regeneration (e.g., flattened, basophilic epithelial cells with irregular nuclear size; mitotic figures; high nuclear/cytoplasmic ratios) and the finding of generally intact tubular basement membranes are good prognostic findings and may be observed as early as three days after the insult. Conversely, large numbers of granular casts, extensive tubular necrosis, and interstitial mineralization and fibrosis with disrupted tubular basement membranes are poor prognostic signs. In addition to the renal histopathologic changes, the degree of functional impairment and, even more important, the response to therapy should be considered when formulating a prognosis.
RISK FACTORS FOR ACUTE RENAL DAMAGE/FAILURE
Although the prevention of trauma (e.g., being hit by car) that may lead to shock and the development of renal ischemia or exposure to nephrotoxicants outside the hospital relies on client education and environmental control, an important aspect of the prevention of hospital-acquired ARF is the identification of patients at increased risk. Several risk factors that predispose dogs to the development of gentamicin-induced ARF have been identified (Box 44-4); however, it is likely that many of these factors also predispose dogs and cats to the development of other types of toxicant-induced ARF as well as ARF induced by ischemia. In many cases a combination of decreased renal perfusion or treatment with nephrotoxic agents in the context of more chronic, preexisting risk factors is responsible for the development of ARF in the hospital setting. Once the clinician detects predisposing risk factors, he or she can assess the risk : benefit ratio in individual cases in which an elective anesthetic procedure is considered or treatment with potentially nephrotoxic drugs is indicated. In some situations, predisposing risk factors can be eliminated or corrected before any potential renal insults occur.
Major categories of risk factors include disorders affecting renal perfusion, preexisting renal disease, electrolyte disturbances, treatment with nephrotoxic drugs, and dietary influences. Poor renal perfusion increases the risk of nephrotoxic and ischemic damage to the kidney. Dehydration and volume depletion are perhaps the most common causes of decreased renal perfusion. Renal hypoperfusion can also be caused by decreased cardiac output, decreased plasma oncotic pressure, increased blood viscosity, or systemic vasodilation. In addition to decreased renal perfusion, volume depletion also leads to a decreased volume of distribution of nephrotoxic drugs and a decreased flow of tubular fluid. Decreased tubular flow, in turn, potentiates tubular resorption, which can increase the intratubular concentration of nephrotoxicants. Preexisting renal disease and advanced age, which is often associated with some degree of decreased renal function, may increase the potential for nephrotoxicity produced by several mechanisms. For example, the pharmacokinetics of potentially nephrotoxic drugs may be altered in the face of decreased renal function. Specifically, the excretion of gentamicin has been shown to be decreased in partially nephrectomized dogs with subclinical renal dysfunction. Animals with renal insufficiency or advanced age may also have reduced urine-concentrating ability and thus a decreased ability to compensate for dehydration. Preexisting renal disease may also compromise the production of vasodilatory prostaglandins. The resulting unbalanced vasoconstriction could result in decreased renal perfusion.
Studies in dogs have shown that reduced dietary potassium intake exacerbates gentamicin-induced nephrotoxicity, possibly because potassium-depleted cells are more susceptible to necrosis. It is important to note that an adverse effect of high-dose gentamicin treatment in dogs is an increase in the urinary excretion of potassium. It is possible that this could result in potassium depletion (especially if it occurs in combination with anorexia or vomiting) and thus increase the risk of gentamicin-induced nephrotoxicity. Because potassium is primarily an intracellular cation, any patient with prolonged anorexia, vomiting, or diarrhea may have whole-body potassium depletion even if serum potassium concentrations are within the normal range.
The administration of potentially nephrotoxic drugs or drugs that may enhance nephrotoxicity obviously increases the risk of ARF. For example, the concurrent use of furosemide and gentamicin in dogs is associated with an increased risk of ARF and an increased severity of ARF, should it occur. Furosemide probably potentiates gentamicin-induced nephrotoxicity by causing dehydration, reducing the volume of distribution of gentamicin, and increasing its renal cortical uptake. Fluid repletion minimizes but does not negate the additive effect of furosemide on gentamicin-induced nephrotoxicity in the dog because furosemide facilitates the tubular uptake of gentamicin independent of hemodynamic changes. By means of similar mechanisms, furosemide has been shown to enhance radiocontrast agent and cisplatin-induced nephrotoxicity in human beings.
The use of NSAIDs can also increase the risk of acute renal damage and ARF. In well-hydrated, healthy patients, NSAIDs are usually well tolerated. However, in situations associated with high renin concentration (e.g., sodium or volume depletion, hypotension, congestive heart failure, CKD) the potential for adverse effects on renal function increases. High renin states stimulate the production of angiotensin and aldosterone, which can, in turn, decrease renal blood flow and GFR. Normally, renal prostaglandins counteract this decrease in renal blood flow and GFR. However, in patients with CKD and those undergoing treatment with NSAIDs, the protective effects that prostaglandin has on renal blood flow and GFR may be compromised. Dogs appear to be particularly sensitive to NSAIDs such as ibuprofen and naproxen, which, in addition to ARF, may cause gastrointestinal tract ulceration. At one time, COX 2–specific inhibitors were thought to have less effect on renal blood flow; however, research shows that COX 2 enzymes are present or expressed in the canine kidney; therefore any NSAID, regardless of its COX specificity or sparing properties, has the potential to produce adverse renal effects. In particular, dogs express higher basal levels of COX 2 in the kidney than some other species and may be uniquely sensitive to the nephrotoxic effects of COX 2–selective drugs. There is also the concern that patients treated with angiotensin-converting enzyme inhibitors (ACEIs) may have increased risk of renal toxicity when treated with NSAIDs because some of the beneficial effects of ACEI are derived from kinin-stimulated production of prostaglandins. In one study of normal dogs treated with enalapril and tepoxalin, no alteration of GFR was noted.
Studies in healthy dogs have shown that the quantity of protein fed before a nephrotoxic insult can significantly affect the degree of renal damage and dysfunction. High-dietary-protein (27.3%) conditioning beginning 21 days before and continuing during gentamicin administration was found to reduce nephrotoxicity, enhance gentamicin clearance, and result in a larger volume of distribution compared with the findings in dogs fed medium (13.7%) or low levels of protein (9.4%). In addition, creatinine clearance and the renal elimination of gentamicin were preserved throughout 7 days of treatment in dogs fed a high-protein diet, whereas these parameters decreased during the treatment period in dogs fed a medium- or low-protein diet. Although dietary protein conditioning may not be practical in the clinical setting, it is important to realize that anorectic animals may be at increased risk for ARF as a result of decreased protein intake.
Risk factors are additive, and any complication occurring in high-risk animals increases the potential for ARF. By virtue of their diseases, animals in shock or with acidosis, sepsis, or major organ system failure are at increased risk for ARF, and these are also the animals that are likely to require anesthesia or chemotherapy, which is potentially damaging to the kidneys. For example, ARF is common in dogs with pyometra and Escherichia coli endotoxin–induced urine-concentrating defects. If fluid therapy is inadequate during anesthesia for ovariohysterectomy or during the recovery period, dehydration and decreased renal perfusion may result in ARF. Trauma, extensive burns, pancreatitis, diabetes mellitus, and multiple myeloma are examples of disorders associated with a high incidence of ARF in people. Additional clinical conditions that are thought to enhance the risk of ARF in dogs include vasculitis, fever, and prolonged anesthesia.
MONITORING PATIENTS AT RISK FOR ACUTE RENAL DAMAGE/FAILURE
The recognition and appropriate management of renal injury in the initial phase of ARF are associated with improvement in prognosis; therefore animals receiving potentially nephrotoxic drugs and high-risk animals undergoing anesthesia should be monitored closely.
Along with blood pressure, urine production is an excellent parameter to monitor during anesthesia. Ideally, urine production should be greater than 2 ml/kg/h. Increased urinary excretion of protein, glucose (normoglycemic glucosuria), or casts and/or renal tubular epitheial cells may be an early indication of renal tubular damage in animals receiving potentially nephrotoxic drugs. As an alternative to standard clinicopathologic tests, the detection and quantification of urine enzymes (enzymuria) have been used to recognize early nephrotoxicity in the dog. Inasmuch as most serum enzymes are not filtered by the glomerulus because of their large molecular weight, enzymuria may be an indication of renal tubular leakage or necrosis. Several enzymes originate from specific cellular organelles and thus can serve as markers for damage to a specific site. For example, γ-Glutamyl transpeptidase (GGT) originates from the proximal tubular brush border and N-acetyl glucosaminidase (NAG) is a lysosomal enzyme. Enzymuria usually precedes azotemia and decreased urine-concentrating ability associated with nephrotoxic proximal tubular injury by several days. The urine GGT : creatinine and NAG : creatinine ratios have been shown to accurately reflect 24-hour urine GGT and NAG excretion in dogs, if determined before the onset of azotemia. Baseline urine GGT : creatinine and NAG : creatinine ratios therefore should be determined in all dogs that are to receive potentially nephrotoxic drugs. Twofold to threefold increases in the GGT/creatinine or NAG/creatinine ratio over the baseline are suggestive of clinically relevant tubular damage. Drug therapy should be discontinued if this occurs.
Treatment
The goals of treatment of established ARF are to eliminate renal hemodynamic disorders and alleviate water and solute imbalances to give the nephrons additional time to repair and hypertrophy. A positive response to therapy is indicated by a decrease in the serum creatinine concentration and an increase in urine production. Induction of diuresis facilitates the management of ARF by decreasing serum urea nitrogen, phosphorus, and potassium concentrations and by lessening the likelihood of overhydration. Even though the GFR and renal blood flow may improve in response to diuresis, they are frequently unchanged, and the increased urine production is actually a result of decreased tubular resorption of filtrate (Table 44-1). Increased urine production alone does not indicate an improvement in GFR.
TABLE 44-1 Hypothetical Comparison of the Glomerular Filtration Rate and Urine Production in Normal and Nonoliguric Acute Renal Failure States*
| NORMAL | ACUTE RENAL (L/DAY) | FAILURE (L/DAY) |
|---|---|---|
| Glomerular filtration rate | 100 | 10 |
| Tubular resorption | 99 | 07 |
| Urine production | 1 | 03 |
* These show the effect of tubular resorption on urine production in the face of decreased glomerular filtration.
Treatment guidelines for ARF are listed in Box 44-5. Most dogs and cats with ARF are dehydrated because of gastrointestinal fluid loss (e.g., vomiting) superimposed on their inability to concentrate urine. Replacement of these volume deficits will correct the prerenal component of the ARF and help protect against any additional ischemic renal tubular damage. Once the patient is rehydrated, establishing or augmenting diuresis can facilitate excretion of solutes that are reabsorbed and secreted by renal tubular cells (e.g., urea nitrogen and potassium). Increasing tubular flow rates and volumes will hinder reabsorption and favor secretion of solutes.
 BOX 44-5 Treatment Guidelines for Dogs and Cats with Acute Renal Failure
BOX 44-5 Treatment Guidelines for Dogs and Cats with Acute Renal Failure
Discontinue all potentially nephrotoxic drugs; consider measures to decrease absorption (e.g., induction of emesis and administration of activated charcoal and sodium sulfate).
Start specific antidotal therapy if applicable (e.g., alcohol dehydrogenase inhibitors for ethylene glycol).
Identify and treat any prerenal or postrenal abnormalities.
Start intravenous fluid therapy with normal saline solution or 0.45% saline solution in 2.5% dextrose:
Assess volume of urine production.
Correct acid-base and electrolyte abnormalities; rule out hypercalcemic nephropathy.
If necessary, to increase urine production, provide mild volume expansion while monitoring urine volume, body weight, plasma total solids, hematocrit, and central venous pressure.
Administer diuretics, if necessary, to increase urine production:
Base subsequent fluid volumes on urine production plus 20 ml/kg/24 h.
Consider peritoneal dialysis if there is no response to above treatment; biopsy kidney at time of dialysis catheter placement.
Treat vomiting and gastroenteritis with:
The large volume of fluid and rapid administration rate necessary in patients with ARF require that fluids be given intravenously. Jugular catheters or other central venous lines are ideal because they facilitate frequent blood sampling and infusion of hypertonic solutions (e.g., mannitol) and allow access for central venous pressure (CVP) measurement. Deficit fluid requirements should be replaced over the first 4 to 6 hours of treatment unless the patient has a cardiac disorder that requires a slower administration rate. A fluid bolus challenge of 20 ml/kg body weight given intravenously over 10 minutes can help assess the possibility of a subsequent volume overload. The CVP should not increase by more than 2 cm of water if the patient’s cardiovascular function is normal. Because measurement of CVP is not always accurate or reproducible, results should always be interpreted in light of other paramenters (e.g., patient’s body weight, hematocrit, plasma total solids, and physical examination findings). The purpose of replacing volume deficits over the first 4 to 6 hours rather than over the normal 12 to 24 hours is to rapidly improve renal perfusion and decrease the likelihood of continued ischemic damage. Normal saline (0.9% solution) is the fluid of choice for rehydration unless the patient is hypernatremic, in which case a 0.45% saline with 2.5% dextrose solution should be used. The amount of fluid required to restore extracellular fluid deficits can be calculated by multiplying the estimated percentage of dehydration by the patient’s body weight in kilograms.
During this rapid rehydration phase the patient should be closely observed for signs of overhydration. Frequent assessment of body weight, CVP, packed cell volume, and plasma total solids will help detect early overhydration. An increase in the CVP of ≥ 5 to 7 cm of water over baseline values suggests the likelihood of overhydration. Physical manifestations of overhydration include increased bronchovesicular sounds or overt crackles and wheezes, tachycardia, restlessness, chemosis, and serous nasal discharge; however, these signs tend to be observed after the development of pulmonary edema. Overhydration in dogs and cats with oligoanuric ARF is a common complication that is extremely difficult to correct.
Urine production should be measured and electrolyte and acid-base status assessed during the period of rehydration. Urine production (ml/kg/hour) should be measured so that maintenance fluid needs can be accurately administered. Because approximately two thirds of normal maintenance fluid needs are due to fluid loss in urine, oliguric and nonoliguric patients can have large variations in their maintenance fluid needs (see Table 44-2). Metabolism cages, urinary catheters, and manual collection of voided urine are methods used to collect and measure urine volume. With regard to indwelling urinary catheters, strict aseptic technique and closed collection systems must be used. Because of the possibility of urinary tract infection, intermittent urinary bladder catheterization is usually recommended over indwelling catheterization for timed urine volume collections. In cats weighing the litter pan before and after voiding is a useful, although less accurate, method for assessing urine production. If an indwelling urinary catheter or a metabolism cage is not available, patients should be weighed in the same scale two or three times a day to assess fluid gain or loss.
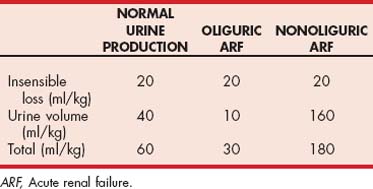
Initially, most patients with ARF have normal serum sodium and chloride concentrations on account of isonatremic fluid loss. However, hypernatremia can develop after several days of therapy with fluids containing large amounts of sodium (0.9% NaCl, lactated Ringer’s solution, and Normosol) and/or in association with sodium bicarbonate treatment of metabolic acidosis. If hypernatremia occurs, the use 0.45% NaCl with 2.5% dextrose fluids will usually correct the problem.
Disorders of calcium balance can also occasionally occur in patients with ARF. If moderate to severe hypercalcemia is observed, a primary hypercalcemic disorder (e.g., neoplasia or vitamin D3 intoxication) should be considered as the cause of the renal failure. In most cases assessment of the ionized calcium concentration is preferable to measurement of the total calcium concentration. Immediate treatment for hypercalcemia includes rehydration with 0.9% NaCl followed by diuresis induced with furosemide. Glucocorticoids will also help lower calcium concentrations by decreasing intestinal absorption and facilitating excretion, but their use may interfere with the diagnosis of the underlying disorder (e.g., lymphoma). Intravenous bisphosphonates (pamidronate-Aredia, 1 mg/kg as a constant rate infusion (CRI) in 0.9% saline solution) are effective in lowering serum calcium concentration and do not affect the clinician’s ability to diagnose the primary cause of hypercalcemia. Conversely, significant hypocalcemia can be observed in dogs and cats with ARF associated with ethylene glycol intoxication.
Oliguric ARF patients are at risk for hyperkalemia. Serum potassium concentrations greater than 6.5 to 7.0 mEq/L can cause cardiac conduction disturbances (bradycardia, atrial standstill, idioventricular rhythms, ventricular tachycardia, ventricular fibrillation, asystole) and electrocardiographic changes (peaked T waves, prolonged PR intervals, widened QRS complexes, or the loss of P waves). Mild to moderate hyperkalemia typically resolves with administration of potassium-free fluids (dilution) and improved urine flow (increased excretion). More severe hyperkalemia (>7-8 mEq/L) or hyperkalemia resulting in electrocardiographic (ECG) abnormalities should be treated with agents that rapidly decrease serum potassium concentrations or counteract the effects of hyperkalemia on cardiac conduction. Sodium bicarbonate (see discussion of dosage later in this chapter) helps correct metabolic acidosis and lower serum potassium concentration by exchanging intracellular hydrogen ions for potassium. Insulin can also be used to increase intracellular shifting of potassium. Regular insulin is administered intravenously at a dosage of 0.1 to 0.25 U/kg, followed by a glucose bolus of 1 to 2 g per unit of insulin given. Blood glucose monitoring should be maintained for several hours after administration of insulin because hypoglycemia may occur. Ten percent calcium gluconate (0.5-1.0 ml/kg administered intravenously over 10 to 15 minutes) will counteract the cardiotoxic effects of hyperkalemia without lowering the serum potassium and can be used in emergency situations. The effects of the aforementioned regimens are short-lived, and fluid and acid-base therapy to initiate and maintain a diuresis and maintain blood pH and bicarbonate within the normal range (discussed in more detail later in this chapter) are important to maintain potassium excretion and normokalemia.
Mild to moderate metabolic acidosis also generally resolves after fluid therapy, and specific treatment is usually not necessary unless the blood pH is less than 7.2 or the total CO2/CO3H is less than 12 mEq/L. Bicarbonate requirements can be calculated using the base deficit as determined from arterial blood gas, or an estimated base deficit [body weight (kg) × 0.3 × base deficit or (20 - T CO2) = mEq bicarbonate required]. Optimally, one half the calculated bicarbonate dosage should be administered intravenously over 15 to 30 minutes, and then acid-base parameters reassessed. Overzealous bicarbonate administration may result in ionized hypocalcemia, paradoxical cerebral spinal fluid (CSF) acidosis, and/or cerebral edema.
If signs of overhydration are not present and oliguria persists after apparent rehydration, mild volume expansion (3% to 5% of the patient’s body weight in fluid) may be initiated inasmuch as dehydration of this magnitude is difficult to detect clinically. If volume expansion is attempted, the possibility of inducing overhydration increases and close patient observation is necessary. Unfortunately, most patients that have oliguria will remain oliguric after rehydration and volume expansion.
In the past, diuretic therapy was frequently recommended in patients that were persistently oligoanuric despite appropriate fluid therapy. Compared with those patients with diminished urine production, polyuric ARF patients are thought to have less severe tubular injury, improved excretion of solutes that are reabsorbed or secreted (e.g., urea nitrogen and potassium), and less risk of developing overhydration and pulmonary edema. There is, however, no evidence that diuretic therapy will hasten the recovery from ARF or decrease mortality associated with ARF. In humans with established ARF, there is increasing evidence that diuretic therapy may actually be associated with increased risk of death and nonrecovery of renal function. If the choice is to use diuretics in dogs or cats with ARF, they should be used only after dehydration has been corrected and the patient has been volume expanded. Furosemide and mannitol are probably the diuretics of choice. Dopamine is not recommended because of its unpredictable effects on renal blood flow and GFR.
Furosemide blocks the reabsorption of chloride and sodium in the thick ascending limb of Henle, resulting in natriuresis and osmotic diuresis. The dose recommended for oligoanuric dogs and cats is 2 to 6 mg/kg IV q8h; however, in healthy dogs CRI of furosemide with a 0.66 mg/kg IV loading dose followed by 0.66 mg/kg/h resulted in more diuresis, natriuresis, and calciuresis and less kaliuresis than did intermittent bolus infusion.
Mannitol, in a 10% or 20% solution, has been recommended as an osmotic diuretic at a dose of 0.5 to 1.0 g/kg, given intravenously as a slow bolus over 15 to 20 minutes. Urine output should increase within 1 hour if the treatment is effective. A second bolus may be attempted, but the potential for volume overexpansion and complications such as pulmonary edema increases considerably if urine production does not increase. As an osmotic agent, mannitol may decrease tubular cell swelling, increase tubular flow, and help prevent tubular obstruction or collapse. In healthy cats the renal effects of mannitol, when used as an adjunct to fluid therapy, are superior to those of furosemide and dopamine combination. The use of mannitol is contraindicated in an overhydrated patient because the resultant increase in intravascular volume may precipitate pulmonary edema.
Whether or not diuresis can be established, fluid therapy should be tailored to match urine volume and other losses, including insensible losses (e.g., water loss caused by respiration) and continuing losses (e.g., fluid loss caused by vomiting or diarrhea). Insensible losses are estimated at 20 ml/kg/day. Urine output is quantitated for 6- to 8-hour intervals, and that amount is replaced over an equivalent subsequent time period. The volume of fluid loss resulting from vomiting and/or diarrhea is estimated, and that amount is added to the 24-hour fluid needs of the patient. Fluid losses or gains can also be indirectly estimated by weighing the patient 2 to 3 times a day on the same scale. If hypernatremia and hyperkalemia are not present and a diuresis has been established, polyionic maintenance fluids (e.g., lactated Ringer’s solution, Normosol) should be used. In the recovery phase of ARF, urine volume and electrolyte losses can be great. Potassium supplementation may be necessary, especially if the patient is vomiting or anorectic.
Control of nausea and vomiting in dogs and cats with ARF is important to facilitate caloric intake. In addition, the inability to control vomiting is discouraging to owners and may result in a hastened decision for euthanasia. (Please see the section on management of chronic kidney disease for specific recommendations for the treatment of nausea and vomiting.)
When fluid therapy is successful in inducing or maintaining diuresis, the daily volume of fluid administered to the patient will eventually need to be decreased. Indications for tapering IV fluid volume include the following: (1) significant decreases in BUN and phosphorus concentrations, (2) control of vomiting and diarrhea, and (3) improved mood and renewed interest in eating and drinking. These indications rarely occur before 5 or 6 days of intense fluid therapy/diuresis and may require 10 or more days of treatment. Gradually reducing maintenance fluid requirements by 25% each day is usually recommended for fluid tapering. If the patient loses weight or increases in packed cell volume, total protein, and BUN and/or creatinine concentrations are observed, fluid therapy tapering should be discontinued and the previous maintenance volume reinstated for at least 48 hours.
Peritoneal or hemodialysis should be considered in patients with severe, persistent uremia, acidosis, or hyperkalemia. Dialysis may also be used to treat overhydration and hasten elimination of dialyzable toxicants. Renal biopsy should be performed if the diagnosis is in doubt, if the patient does not respond to therapy within 3 to 5 days, or if dialysis is considered. The long-term prognosis for dogs or cats with ARF is usually fair to good if the patient survives the period of renal tubular regeneration and compensation; however, several weeks may be required for renal function to improve. Animals with moderate to severe renal damage may require many weeks for renal repair, and the prolonged time required for recovery results in a poor prognosis. The severity of the initial azotemia/uremia, the response to fluid therapy, and assessment of renal histopathologic lesions are the most important prognostic indicators early in the course of ARF.
CHRONIC KIDNEY DISEASE
Etiology and Pathogenesis
Unlike ARF, the cause of CKD is usually difficult to determine. Because of the interdependence of the vascular and tubular components of the nephron, the end-point of irreversible glomerular or tubular damage is the same. A morphologic heterogeneity among nephrons exists in the chronically diseased kidney, with the changes ranging from severe atrophy and fibrous connective tissue replacement to marked hypertrophy. The histopathologic changes are not process-specific, and therefore the cause is usually unknown. Nevertheless, recent studies have shown that primary glomerular disorders are a major cause of CKD in the dog. Because glomerular filtration in toto is uniformly reduced, CKD may be considered a single pathologic entity, although many diverse pathways can lead to this end-point. Potential causes of CKD are listed in Box 44-6.
 BOX 44-6 Potential Causes of Chronic Kidney Disease in Dogs and Cats
BOX 44-6 Potential Causes of Chronic Kidney Disease in Dogs and Cats
Hereditary and Congenital Disorders
The pathophysiology of CKD can be considered at both the organ and systemic level. At the level of the kidney, the fundamental pathologic change that occurs is a loss of nephrons and decreased GFR. Reduced GFR, in turn, results in increased plasma concentrations of substances that are normally eliminated from the body by renal excretion. Many substances have been shown to accumulate in the plasma of patients with CKD (Box 44-7). The constellation of clinical signs known as the uremic syndrome is thought to occur, at least in part, as a result of increasing plasma concentrations of these substances. Components of the uremic syndrome include sodium and water imbalance, anemia, carbohydrate intolerance, neurologic disturbances, gastrointestinal tract disturbances, osteodystrophy, immunologic incompetence, and metabolic acidosis.
BOX 44-7 Substances that Can Increase in Concentration in the Plasma of Dogs and Cats with Renal Failure
In addition to excreting metabolic wastes and maintaining fluid and electrolyte balance, the kidneys also function as endocrine organs and catabolize several peptide hormones. Therefore hormonal disturbances also play a role in the pathogenesis of CKD. For example, the decreased production of erythropoietin (EPO) and calcitriol in animals with CKD contributes to the development of nonregenerative anemia and hyperparathyroidism. Conversely, decreased metabolism and increased concentrations of parathyroid hormone (PTH) and gastrin contribute to the development of hyperparathyroidism and gastritis, respectively.
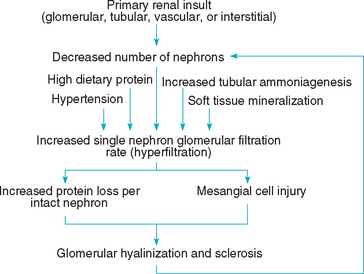
Some of the pathophysiologic changes that occur in CKD are brought about by compensatory mechanisms. The osteodystrophy of CKD occurs secondary to hyperparathyroidism, which develops in an attempt to maintain normal plasma calcium and phosphorus concentrations. Similarly, the GFR of intact hypertrophied nephrons increases in animals with CKD in an attempt to maintain adequate renal function; however, proteinuria and glomerulosclerosis in these individual nephrons, leading to additional nephron damage and loss, may be consequences of this hyperfiltration (Fig. 44-3).
Clinical Features and Diagnosis
Unlike ARF, CKD develops over a period of months or years, and its clinical signs are often relatively mild for the magni tude of the azotemia. Unique signs of CKD include a history of weight loss, polydipsia-polyuria, poor body condition, nonregenerative anemia, and small and irregularly shaped kidneys. A diagnosis of CKD is usually based on a combination of compatible historical, physical examination, and clinicopathologic findings. Plain radiographs can confirm the presence of small kidneys. Renal ultrasonography will usually show diffusely hyperechoic renal cortices with loss of the normal corticomedullary boundary. The increased cortical echogenicity results from replacement of the irreversibly damaged nephrons with fibrous connective tissue. Radiographic studies and ultrasonography can also help identify or rule out potentially treatable causes of CKD, such as pyelonephritis and renal urolithiasis. Renal biopsy is not routinely performed in animals with CKD unless the diagnosis is in question. Renal histopathologic preparations will show some combination of a loss of tubules with replacement fibrosis and mineralization, glomerulosclerosis and glomerular atrophy, and foci of mononuclear cells (small lymphocytes, plasma cells, and macrophages) within the interstitium in association with fibrous connective tissue replacement.
STAGING CHRONIC KIDNEY DISEASE
Once a diagnosis of CKD has been established and fluid therapy has resolved any prerenal azotemia, staging the disease process can help clinicians focus their diagnostic and therapeutic efforts. The International Renal Interest Society (IRIS) was created to advance the scientific understanding of kidney diseases in small animals at the Eighth Annual Congress of the European Society of Veterinary Internal Medicine in Vienna, Austria in 1998. Seventeen independent veterinary nephrologists from eight countries serve on the IRIS Board, with the mission of helping practitioners better diagnose, understand, and treat canine and feline renal disease. Table 44-3 was developed by the IRIS Board as guide to staging canine and feline CKD.

Serum creatinine concentrations must always be interpreted in light of the patient’s urine specific gravity and physical examination findings to rule out prerenal and postrenal causes of azotemia. The CKD stages are further classified by the presence or absence of proteinuria and systemic hypertension (Table 44-4).
TABLE 44-4 IRIS CKD Substaging System for Proteinuria and Hypertension
| URINE PROTEIN:CREATININE RATIO | CLASSIFICATION |
|---|---|
| <0.2 (cats and dogs) | Nonproteinuric |
| 0.2-0.4 (cats), 0.2-0.5 (dogs) | Borderline proteinuric |
| >0.4 (cats), >0.5 (dogs) | Proteinuric |
| SYSTOLIC BLOOD PRESSURE (MM HG) | CLASSIFICATION |
|---|---|
| <140 | Normotensive |
| 140-160 | Borderline hypertensive |
| >160 | Hypertensive |
IRIS, International Renal Interest Society; CKD, chronic kidney disease.
The classic diagnosis of renal failure based on renal azotemia (persistent azotemia superimposed on the inability to concentrate urine) pertains to CKD stages II through IV. Stage I CKD (nonazotemic CKD) could be diagnosed in cats and dogs with persistent proteinuria, urine-concentrating deficits, increases in serum creatinine concentration over time even if the values remain in the normal range (e.g., serum creatinine concentration that increases form 0.6 to 1.2 mg/dl could indicate a 50% reduction in GFR), or abnormal renal palpation or renal ultrasonographic findings.
Further Diagnostics and Treatment
In general, the diagnostic approach to a patient in which CKD has been identified and staged is focused on three areas: (1) characterization of the renal disease, (2) characterization of the stability of the renal disease and renal function, and (3) characterization of the patient’s problems associated with the decreased renal function (Fig. 44-4). Further definition of the renal disease (beyond a standard minimum database) could include, for example, quantification of proteinuria, measurement of blood pressure, urine culture, kidney imaging, and possibly kidney biopsy. The stability of the renal function may be assessed by serial monitoring of abnormalities identified during the initial evaluation of the renal disease. This monitoring should always include serial serum biochemistry profiles, urinalyses, quantification of proteinuria, and measurement of blood pressure, but it may also include follow-up urine cultures and ultrasonographic examinations. Characterization of the renal disease and its stability is most important in the earlier stages of CKD, when appropriate treatment has the greatest potential to improve or stabilize renal function. Characterization of the patient’s problems becomes more important in the later stages of CKD, when clinical signs tend to be more severe. In the later stages of CKD, diagnostic (and subsequent therapeutic) efforts should be directed at the anorexia, vomiting, acidosis, potassium depletion, hypertension, anemia, and related signs.
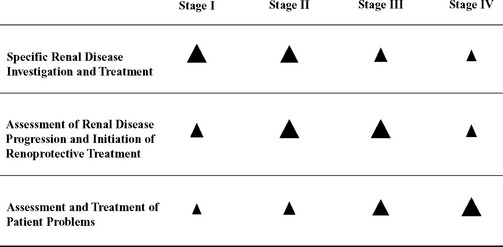
FIG 44-4 Prioritization of diagnostic and treatment efforts based on the stage of chronic kidney disease. The larger the arrowhead, the higher the priority.
Similar to the diagnostic approach to CKD, the therapeutic approach should also be tailored to fit the patient’s stage of disease. For example, disease-specific treatments for nephroliths or bacterial pyelonephritis as well as treatments designed to slow the progression of renal disease (so-called renoprotective treatments) will be of most value in the earlier stages of CKD. Examples of renoprotective treatments include dietary change designed to reduce serum phosphorus concentrations and ACEIs designed to normalize systemic and intraglomerular blood pressures and reduce proteinuria. In the later stages of CKD, treatment tends to be focused on ameliorating the patient’s clinical signs associated with the decreased renal function.
Specific treatment in patients with CKD is directed at the primary cause of the kidney disease. Although it may not be possible to identify the primary cause of the CKD, specific treatment have the potential to reduce the magnitude of subsequent renal damage. As an example, bacterial pyelonephritis can cause or complicate CKD, and the condition can be specifically treated with appropriate antibiotic therapy. The prevalence of urinary tract infection (UTI) increases in older dogs and cats, and especially dogs and cats with CKD, because the antibacterial properties of the urine decline as its concentration decreases. In a study of cats with naturally occurring CKD, 29% had occult UTI. Bacterial infection of the renal pelvis and parenchyma (i.e., pyelonephritis) can then result from an ascending lower UTI. Initially with ascending UTI, the renal cortex is not affected; however, as chronic pyelonephritis develops, the entire kidney may become involved. Pyelonephritis also can precipitate the development of renal calculi, and, conversely, renal calculi can increase the risk of pyelonephritis. Long-term antibiotic therapy based on culture and sensitivity may halt the renal damage associated with pyelonephritis; however, if renoliths are present, antibiotic therapy alone is usually ineffective. Calcium oxalate uroliths are the most common type of renoliths in older cats, and because they cannot be dissolved, surgery is necessary for stone removal. Anesthesia and surgery, however, have the potential to further compromise renal function in the cat with CKD. In most cases, the patient is closely monitored for obstructive uropathy and surgery is not performed unless an obstruction develops. Concurrent pyelonephritis that cannot be resolved with antibiotic treatment is another potential indication for surgical intervention.
Similar to bacterial pyelonephritis, hypertension (HT) can cause or complicate CKD. Gradual reduction of dietary salt intake is often recommended as the first line of treatment for HT; however, no studies document the efficacy of dietary salt reduction in lowering blood pressure in dogs or cats. In many cases vasodilators (ACEI and calcium channel blockers [CCBs]) may be necessary to control hypertension. Although ACEIs are usually recommended for HT associated with CKD in dogs, amlodipine is often recommended as the first-choice antihypertensive medication for cats. Recent studies, however, have raised the concern that amlodipine as a monotherapy in animals with renal disease may expose the glomeruli to higher pressures because of efferent arteriolar constriction caused by local increases in renin-angiotensin-aldosterone system activity. If so, cats with renal disease should benefit from therapy with both ACEIs and CCBs. Cats with CKD are mild to moderate HT should be treated with an ACEI (e.g., benazepril: 0.5 to 1.0 mg/kg q24h) because of the positive effects on intraglomerular hypertension and proteinuria. In cats with severe HT (systolic blood pressure>180 mm Hg) or cats in which HT persists despite ACEI treatment, amlodipine (0.625 to 1.25 mg/cat q24h) treatment should be initiated. Several studies have documented renoprotective effects of ACEIs in dogs and cats with naturally occurring CKD.
Direct-acting vasodilator drugs such as ACEIs and CCBs are the most successful in achieving acute reduction of blood pressure, but sympathetic nervous system–mediated increases in heart rate and aldosterone-mediated sodium and water retention may modulate the effects of the vasodilation over time. Combining antihypertensive treatments with different modes of action may block the compensatory effects caused by one medication when used alone. For example, diuretics, aldosterone antagonists, and β-blockers, which may have minimal antihypertensive effect alone, may produce additive effects when given in combination with ACEIs or CCBs. Overall, the risk of target organ damage in the eyes, brain, kidneys, and heart is thought to be minimal if systolic blood pressure is <150 mm Hg (Table 44-5).
TABLE 44-5 Risk of Target Organ Damage Associated with Hypertension in Dogs and Cats
| SYSTOLIC BLOOD PRESSURE (MM HG) | DIASTOLIC BLOOD PRESSURE (MM HG) | RISK LEVEL |
|---|---|---|
| <150 | <95 | Minimal |
| 150-159 | 95-99 | Low |
| 160-179 | 100-119 | Moderate |
| >180 | >120 | High |
In many dogs and cats with stage II to IV CKD, renal lesions progress and renal function deteriorates (see Figure 44-1). Progressive loss of function as well as the rate of decline are monitored by longitudinal measurement of serum creatinine concentrations. In addition to the antihypertensive treatment discussed previously, ACEIs (to control intraglomerular hypertension and proteinuria) and dietary phosphorus restriction are examples of so-called renoprotective treatments. Reduction of dietary phosphorus is one of the cornerstones of management of CKD and can be accomplished by feeding specifically formulated diets for CKD. From a practical standpoint, dietary phosphorus reduction is combined with dietary protein reduction (discussed in more detail later). If, after 3to 4 weeks of dietary phosphorus reduction, serum phosphorus concentrations remain high, enteric phosphate-binding gels containing calcium acetate, calcium carbonate, or aluminum hydroxide should be administered with meals (initial dosage of 30 mg/kg body weight with the dosage increased as needed to achieve normophosphatemia).
Hyperphosphatemia in patients with CKD occurs as a result of decreased renal excretion of phosphates. Concurrently, decreased renal production of the active form of vitamin D3 decreases intestinal absorption of calcium, which, in conjunction with impaired renal reabsorption of calcium, decreases plasma ionized calcium concentrations. Decreased vitamin D3 and serum calcium concentrations stimulate PTH secretion, which facilitates renal excretion of phosphorus and increases serum calcium concentrations by increasing renal calcium reabsorption and calcium absorption from bones and the gastrointestinal tract. The disadvantages of this hyperparathyroidism, however, can be severe and include osteodystrophy, bone marrow suppression, and soft tissue mineralization. Soft tissue mineralization occurs predominantly in damaged tissue, and if mineralization occurs in renal tissue, the result may be a progressive decline in renal function. If the product of the serum calcium and phosphorus concentrations is greater than 50 to 70 mg/dl, the patient is at risk for soft tissue mineralization. Studies in dogs and cats with remnant kidney CKD have shown that normal dietary phosphorus intake is associated with microscopic renal mineralization and fibrosis, and these changes were prevented by reducing dietary phosphorus. Similarly, in dogs and cats with naturally occurring CKD, feeding a diet specifically formulated to meet their needs, together with phosphate-binding drugs, if required, controls hyperphosphatemia and secondary renal hyperparathyroidism and is associated with a prolonged survival time. Physiologic doses of calcitriol may also be beneficial in dogs and cats with hyperparathyroidism and hyperphosphatemia associated with CKD. In a prospective, randomized, controlled clinical trial in dogs with spontaneous CKD (stages III and IV), calcitriol treatment (initial dose of 2.5 ng/kg/day that was adjusted within the range of 0.75 to 5.0 ng/kg/day according to serial determination of ionized calcium and PTH concentrations) resulted in decreased all-cause mortality and prolonged survival compared with placebo treatment. Calcitriol should not be administered until hyperphosphatemia has been controlled with diet and enteric binders. In addition, if the Ca X Phos product exceeds 60 to 70 mg/dl, calcitriol should not be used because of the risk of soft tissue mineralization. Serial serum calcium determinations are recommended in dogs and cats receiving calcitriol to help prevent hypercalcemia, especially if the patient is also receiving a calcium-containing enteric phosphorus binder.
Diagnosis and management of proteinuria in dogs and cats with CKD should be accomplished in a step-wise fashion. Because the specificity of the dipstick screening test for proteinuria in both dogs and cats is poor, confirmation of proteinuria should be accomplished with a more specific follow-up test, such as the sulfosalicylic acid (SSA) turbidimetric test, urine protein : creatinine ratio, or canine or feline specific albuminuria assay (see Chapter 42). The second step in assessment of proteinuria is to determine its origin. Proteinuria of renal origin can adversely affect the prognosis of dogs and cats with CKD, and therefore physiologic or benign proteinuria and prerenal and postrenal proteinuria should be ruled out. Subsequently, via serial monitoring, the clinician should determine whether the proteinuria is persistent or transient. Persistent proteinuria is defined as at least two positive tests at 2-week intervals. Relatively mild proteinuria in dogs and cats with spontaneous chronic renal failure appears to be a negative predictor of survival. In azotemic patients persistent proteinuria of renal origin with a urine protein : creatinine ratio > 0.4 (cats) or > 0.5 (dogs) should be treated with an ACEI and/or dietary protein reduction (discussed in more detail later).
Symptomatic treatment becomes a higher priority in the later stages of CKD, when the renal failure and uremia have a more pronounced effect on the patient’s quality of life. In addition to phosphorus restriction, dietary management includes protein reduction (dietary protein is reduced not restricted in these diets; restriction of any dietary component generally means feeding less than the daily requirements), salt reduction, n-3 fatty acid supplementation, and alkalinization. Feeding specifically formulated renal failure diets not only may allow the animal to live more comfortably with decreased renal function but also may significantly prolong survival. Ideally, dietary protein reduction allows all essential amino acid requirements to be met without excesses. This is accomplished by feeding smaller quantities of high biological value protein and results in a decreased need for renal clearance of urea and other nitrogenous metabolites. When feeding reduced protein diets, the clinician must remember that the energy requirements of the body have a higher priority than does protein anabolism; therefore, if the available carbohydrates and fats are insufficient to meet caloric requirements, endogenous proteins will often be used as a source of energy. Catabolism of endogenous proteins for energy increases the nitrogenous waste that the kidney must excrete and exacerbates the clinical signs of renal failure.
A good recommendation for dietary protein reduction for both dogs and cats is to feed the maximum amount of high biological value, highly digestible protein that the animal can tolerate at his/her level of renal function. A favorable response to therapy consists of stable body weight and serum creatinine and albumin concentrations and decreasing serum urea nitrogen and phosphorus concentrations. Moderate dietary protein reduction should be employed early in the course of renal failure, and use of markedly reduced protein diets should be reserved for patients that are refractory to moderate dietary protein reduction.
Most diets for CKD are alkalinizing diets; however, potassium citrate or sodium bicarbonate, given orally to effect, may be indicated if the patient remains acidemic (total CO2 < 12 mEq/L) 2 to 3 weeks after diet change. Oral potassium citrate supplementation may also prevent hypokalemia and potassium depletion in cats with CKD. Anorexia; high-protein, acidifying diets; polyuria-polydipsia; and vomiting can all contribute to potassium depletion; however, only 20% to 30% of cats with CKD have hypokalemia as an initial clinicopathologic finding. Potassium is predominantly an intracellular cation, and approximately 95% of total body potassium is present in skeletal muscle; therefore serum potassium concentrations may not accurately reflect total body potassium stores, especially in the early stages of potassium depletion. It has been documented that cats with CKD have lower muscle potassium concentrations and higher serum potassium concentrations than do normal cats. This data may suggest the need for oral potassium supplementation early in the course of CKD in cats. Generalized muscle weakness is the primary clinical sign associated with hypokalemia/potassium depletion. Muscle weakness usually resolves within 1 to 5 days after initiation of oral potassium supplementation.
Vomiting and anorexia are common in dogs and cats with CKD and can often result in decreased caloric intake. Causes of vomiting and anorexia include (1) stimulation of chemoreceptor trigger zone by uremic toxins, (2) decreased excretion of gastrin and increased gastric acid secretion (plasma gastrin concentrations in cats with chronic renal failure may be as high as 20 times the normal concentrations), and (3) gastrointestinal irritation secondary to uremia. Vomiting may be treated with metoclopramide, which blocks the chemoreceptor trigger zone. Metoclopramide also increases gastric motility and emptying without causing gastric acid secretion and is the drug of choice for vomiting associated with renal failure. H2 receptor blockers (famotidine or ranitidine) have been shown to effectively decrease gastric acid secretion, which may attenuate vomiting in CKD. Oral ulcers, stomatitis, and glossitis may occur as a result of gastritis and vomiting or the effect of uremic toxins on mucosal membranes and will often also result in anorexia. If vomiting has been controlled but anorexia persists, placement of a gastrostomy or esophagostomy tube will often facilitate the maintenance of caloric intake and hydration status. In many cases without feeding tubes, fluid therapy with polyionic solutions, given intravenously or subcutaneously in the hospital or subcutaneously by owners at home (10 to 50 ml/kg subcutaneously every 1 to 3 days), will help improve the patient’s quality of life.
The nonregenerative anemia observed in dogs and cats with CKD occurs as a result of a combination of decreased EPO production, shortened red blood cell survival, gastrointestinal tract blood loss, and the effects of uremic toxins such as PTH on erythropoiesis. In addition, nutritional deficiencies (e.g., vitamins B6 and B12, niacin, and folic acid) and iron depletion can contribute to the anemia associated with CKD. Anabolic steroids are usually of little benefit; however, treatment with recombinant human EPO in dogs and cats with CKD and anemia has generally been successful. Although not approved for use in veterinary medicine, the dosage that has been recommended is 100 U/kg of recombinant EPO given subcutaneously three times weekly. The dose interval is lengthened once a target packed cell volume is achieved (approximately 40% in dogs and 35% in cats). Usually, a dosage of 75 to 100 U/kg once or twice weekly is sufficient for maintenance. This treatment, in addition to increasing the packed cell volume, often results in increased appetite, weight gain, increased strength, and an improved sense of well-being. It should be noted, however, that antibodies may form in dogs and cats treated with human recombinant products. Studies show that antirecombinant EPO-binding antibodies will develop in approximately 25% to 30% of dogs and cats and that these antibodies may also react with endogenous EPO, making the animal transfusion dependent. Development of anti-r-HuEPO antibodies should be suspected in patients with a sudden decrease in packed cell volume. Iron deficiency; external blood loss; hemolytic disorders; and concurrent infectious, inflammatory, or neoplastic diseases should be ruled out in such patients. The absence of peripheral reticulocytes and severe erythroid hypoplasia (M : E ratio>10) on bone marrow cytology is compatible with the presence of anti-r-HuEPO antibodies. Iron supplementation (iron dextran: 10 mg/kg administered intramuscularly every 3 to 4 weeks) should be employed during recombinant EPO treatment because of the rapid initiation of erythropoiesis and marginal depletion of iron stores that occur in animals with CKD. Until canine and feline recombinant EPO become commercially available, treatment with human recombinant products should be reserved for those animals with weakness and lethargy attributable to their anemia.
Adin DB, et al. Intermittent bolus injection versus continuous infusion of furosemide in normal adult greyhound dogs. J Vet Intern Med. 2003;17:632.
Behrend EN, et al. Hospital-acquired acute renal failure in dogs: 29 cases (1983–1992). J Am Vet Med Assoc. 1996;208:537.
Brown SA. Management of chronic kidney disease. In Elliott JA, Grauer GF, editors: BSAVA manual of canine and feline nephrology and urology, ed 2, Gloucester, England: British Small Animal Veterinary Association, 2007.
Brown S, et al. Guidelines for the identification, evaluation, and management of systemic hypertension in dogs and cats. J Vet Intern Med. 2007;21:542.
Cowgill LD, Francey T. Acute uremia. In Ettinger SJ, Feldman EC, editors: Textbook of veterinary internal medicine, ed 6, St Louis: Elsevier/Saunders, 2005.
DiBartola SP. Familial renal disease in dogs and cats. In Ettinger SJ, Feldman EC, editors: Textbook of veterinary internal medicine, ed 6, St Louis: Elsevier/Saunders, 2005.
Elliott JA. Staging chronic kidney disease. In Elliott JA, Grauer GF, editors: BSAVA manual of canine and feline nephrology and urology, ed 2, Gloucester, England: British Small Animal Veterinary Association, 2007.
Fischer JR. Peritoneal and hemodialysis. In Elliott JA, Grauer GF, editors: BSAVA manual of canine and feline nephrology and urology, ed 2, Gloucester, England: British Small Animal Veterinary Association, 2007.
Grauer GF. Management of acute renal failure. In Elliott JA, Grauer GF, editors: BSAVA manual of canine and feline nephrology and urology, ed 2, Gloucester, England: British Small Animal Veterinary Association, 2007.
Jacob F, et al. Association between initial systolic blood pressure and risk of developing a uremic crisis or of dying in dogs with chronic renal failure. J Am Vet Med Assoc. 2003;222:322.
Jacob F, et al. Evaluation of the association between initial proteinuria and morbidity rate or death in dogs with naturally occurring chronic renal failure. J Am Vet Med Assoc. 2005;226:393.
Jepson RE, et al. Effect of control of systolic blood pressure on survival in cats with systemic hypertension. J Vet Intern Med. 2007;21:402.
Kerl ME. Renal tubular disease. In Ettinger SJ, Feldman EC, editors: Textbook of veterinary internal medicine, ed 6, St Louis: Elsevier/Saunders, 2005.
McCabe JR, et al. The effects of fluids and diuretic therapies on glomerular filtration rate, renal blood flow, and urine output in healthy cats (abstract). J Vet Intern Med. 2004;18:415.
Platinga EA, et al. Retrospective study of the survival of cats with acquired chronic renal insufficiency offered different commercial diets. Vet Rec. 2005;157:185.
Polzin DJ, et al. Chronic kidney disease. In Ettinger SJ, et al, editors: Textbook of veterinary internal medicine, ed 6, Philadelphia: WB Saunders, 2005.
Ross SJ, et al. A case-control study of the effects of nephrolithiasis in cats with chronic kidney disease. J Am Vet Med Assoc. 2007;230:1854.
Stepien RL, Elliott JA. Measurement of blood pressure. In Elliott JA, Grauer GF, editors: BSAVA manual of canine and feline nephrology and urology, ed 2, Gloucester, England: British Small Animal Veterinary Association, 2007.
Syme HM, et al. Survival of cats with naturally occurring chronic renal failure is related to severity of proteinuria. J Vet Intern Med. 2006;20:528.
Vaden SL, et al. Retrospective analysis of 106 dogs with acute renal failure. J Vet Intern Med. 1995;9:209.
Worwag S, et al. Retrospective, acute renal failure in cats: 25 cases (1997-2002) (abstract). J Vet Intern Med. 2004;18:416.