Management of cleft lip and palate
Nicky Kilpatrick

Introduction
Epidemiology
Clefts of the lip and cleft palate are fusion disorders that affect the midfacial skeleton. They are one of the most common congenital anomalies with a worldwide incidence of between 0.8 and 2.7 cases per 1000 live births. The incidence of clefting varies by region, gender, ethnicity and maternal characteristics. Racial differences are apparent with Native Americans having the highest incidence (followed by Maoris, Chinese, Anglo Saxons) and African-Americans the lowest.
Aetiology
Despite being common, the aetiology of many clefts of the lip/palate remains obscure. In addition to chromosomal abnormalities and teratogenic effects, there are over 500 recognized Mendelian syndromes associated with a cleft of the lip/palate. In many instances, specific genetic mutations have been identified, e.g. van de Woude syndrome (IRF6) and Treacher Collins (TCOF1). However, over 70% of clefts of the lip and palate and up to 50% of clefts of the palate only are apparently isolated anomalies. The aetiology of these non-syndromic forms of cleft is multifactorial and the relative genetic and environmental contributions remain unclear. Consequently, while it is understood that offspring and siblings of individuals with a cleft have a higher risk of having a cleft themselves this lack of clarity makes it difficult to give families accurate information regarding the recurrence risks or to offer informed genetic counselling.
Embryology
Orofacial clefts result from a failure of complete fusion between any of the independently derived facial primordia that form the orofacial complex. Development of the human face begins around day 22 with the migration of cranial neural crest cells (ectomesenchyme) from the dorsal region of the anterior neural tube into the facial region. On either side of the foregut, ectodermal swellings form five pairs of branchial (pharyngeal) arches that are derived from primitive gill or visceral arches of fish. These facial primordial establish during the 4th week of gestation, consisting of five distinct prominences:
• A pair of mandibular prominences develop into the lower jaw.
• An unpaired frontonasal prominence that gives rise to a pair of lateral and medial nasal processes during the 5th week. During the following 2 weeks, the medial nasal processes enlarge and merge with each other and the flanking maxillary prominences, completing the formation of the upper lip.
• A pair of bilateral maxillary prominences that result in the upper jaw.
The secondary palate protrudes as a medial outgrowth from the maxillary prominences and consists of neural crest-derived mesenchymal cells. The shelves grow vertically (downwards) initially from the bilateral maxillary processes during the 6th week. Growth of the embryo causes rotation and extension of the head, dropping the tongue that was lying in the nasal cavity into the mouth (stomodeum). At the same time, the palatal shelves elevate to the horizontal position above the tongue during the 7th week of gestation. Following elevation, the palatal shelves continue to grow towards each other during the 8th week until the epithelial surfaces covering the opposing palatal shelves meet and fuse at the midline to form the secondary palate. The epithelium breaks down allowing mesenchymal consolidation.
Events that disrupt the fusion may result in clefting:
• Clefts of the primary palate (i.e. lip, alveolus and palate anterior to the incisive foramen) are caused by disruption of fusion of the medial nasal processes and maxillary prominences around Week 6
• Clefts of the secondary palate (i.e. hard and/or soft palate) are caused by disruption of the elevation and/or fusion of the palatal shelves around week 9.
Anatomy
Clefts of the lip and palate may be unilateral or bilateral, complete or incomplete. The presentation of clefting conditions can be categorized into the following groups:
Clefts of the lip and alveolus (primary palate)
The primary palate forms anterior to the incisive foramen. A cleft of the primary palate may vary from an incomplete cleft (forme fruste) to a minimal defect involving just the vermilion border, to a complete defect extending from the vermilion border to the floor of the nose with clefting of the alveolus. Even in the absence of a frank cleft of the bony alveolus there may be evidence of clefting in the form of a dental anomaly such as a missing or microdont lateral incisor (see Chapter 11). Clefts of the primary palate may be unilateral or bilateral.
Cleft lip and palate (CL/P)
Varying degrees of clefting of the lip and palate can also exist; complete to incomplete, in a wide range of combinations; uni- and bilateral, symmetrical and asymmetrical. In complete clefts, a direct communication exists between the oral and nasal cavities on the cleft side (Figure 15.1A). There can be substantial variation in the degree of palatal shelf separation and while the maxillary arch form generally appears normal at birth, medial collapse of the maxillary segments occurs soon after.
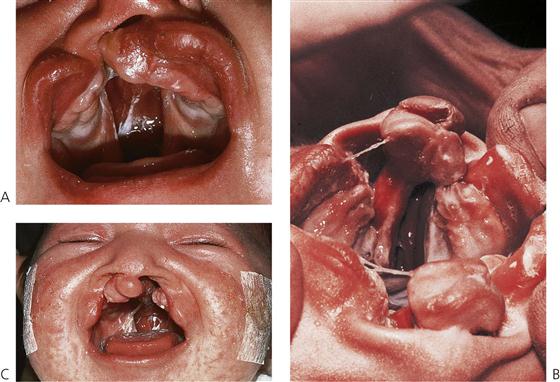
Figure 15.1 (A) Unilateral complete cleft lip and palate, showing the extent of the malformation in the palate. (B) Bilateral complete cleft lip and palate. The premaxillary segment is clearly visible as an extension of the nasal septum. The central incisors are contained within this process. (C) Anterior view of a child with a bilateral complete cleft of the lip and palate. The columella and philtrum are extremely short and there is a wide defect between the segments.
Bilateral clefts of the lip and palate can be either symmetrical or asymmetrical. In the bilateral complete cleft lip and palate, both nasal chambers are in direct communication with the oral cavity. The palatal processes are divided into two equal parts and the turbinates are clearly visible within both nasal cavities. The nasal septum forms a midline structure that is firmly attached to the base of the skull but is fairly mobile anteriorly, where it supports the premaxilla and columella (Figure 15.1B). In these clefts, the premaxilla protrudes considerably forward of the facial profile and is attached to a stalk-like vomer and the nasal septum. The ‘lip’ component of the medial segment contains only collagenous connective tissue. It is, therefore, grossly deficient in bulk and lacks the features normally produced by muscle (Figure 15.1C).
Cleft palate
A cleft of the palate may involve only the soft palate, both soft and hard palates, but almost never the hard palate alone. Deficiency of mucosa and bone are the main features of clefts of the hard palate (Figure 15.2). The cleft may extend forward from the uvula to varying degrees, from a bifid uvula (Figure 15.3) to a ‘V’-shaped cleft extending through the hard palate to the incisive foramen. Some babies have a ‘U’-shaped cleft of the palate that is described in ‘Robin sequence’ (Figure 15.2). Robin sequence is characterized by upper airway problems associated with a very small mandible and glossoptosis and is thought to be secondary to mandibular hypoplasia in the 1st trimester, which causes the tongue to sit high in the mouth and prevent fusion of the palatal shelves.
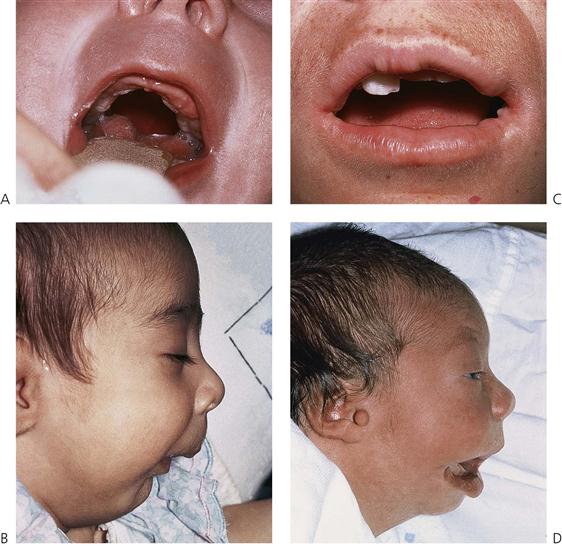
Figure 15.2 (A) Cleft of the palate with an intact lip and alveolus. This U-shaped cleft was associated with the Robin sequence and resulted from a failure of embryonic head rotation. This maintained the tongue in the oral cavity and the palatal shelves subsequently formed around the tongue, giving rise to the characteristic cleft shape. (B) Another manifestation of this condition is extreme micrognathia. The Robin sequence may also be associated with transverse clefts of the face. These transverse clefts may be rather subtle as demonstrated in (C), or severe as in (D). This latter patient has a first and second branchial arch deformity associated with Goldenhar’s syndrome. Note the preauricular skin tag.
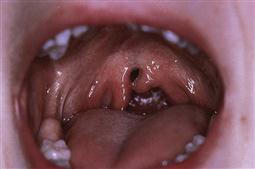
Figure 15.3 A bifid uvula associated with a submucous cleft palate. The fibres of tensor palati are not joined, although the epithelium is intact. These children may present with nasal air escape due to a shortened palate and velopharyngeal incompetence. There is often a notch felt at the posterior border of the hard palate and a bluish median line extends to the uvula.
Submucous cleft palate
Clefting of the velum or soft palate can be ‘submucous’ where the mucous membrane remains intact, despite clefting of the underlying musculature. Submucous clefts of the palate occur in around 1 in 1200 live births, with only half of the cases having clinically significant symptoms.
Diagnosis
Clefting disorders are now increasingly diagnosed prenatally as part of routine ultrasound screening between 18 and 20 weeks in utero. The reported diagnostic accuracy of routine 2D ultrasonography in a low risk population varies between 9–100% for complete clefts of lip and palate but is much lower (0–22%) for clefts of the palate only (Maarse et al 2010). Much higher accuracy is achieved using 3D ultrasound but again isolated clefts of the palate often remain undetected. Prenatal screening provides knowledge of the impending birth of a baby with a cleft, which allows both the family and appropriate clinicians to prepare for this event.
Management of individuals with clefts of the lip and/or palate
Although surgery can correct many of the structural defects, affected individuals can still face a lifetime of functional, social and aesthetic challenges. In addition, there is growing evidence that individuals with CL/P may also have subtle neuropsychological and developmental deficits, which are associated with learning difficulties and other educational challenges. The care of these infants often starts antenatally and continues from birth through to adulthood and involves a large multidisciplinary team of clinicians. The specialties involved in this team vary greatly but usually include:
In addition, genetics, ophthalmology, neurosurgery, periodontics and implantology may be involved at different times. Such orofacial clefting places enormous burdens, both financial and psychosocial on, not just the individual themselves, but also their family and society more broadly.
While there is a lot of variation (and very little high-level evidence) in the sequencing and timing of treatment and the clinical techniques and interventions used, there are some commonly accepted aims and principles of treatment. The appropriate specialists (usually a specialist nurse and/or primary surgeon) within a cleft team aim to provide a rapid initial evaluation, often within hours of birth. Subsequent regular contact through either home visits or attendance at specialist cleft review clinics ensures that families are supported appropriately and social and psychological issues identified and resolved early. Treatment plans can then be formulated and implemented collaboratively by the multidisciplinary team in partnership with the families.
The goals of the treatment of a child with a cleft of the lip and/or palate are to restore both form (appearance) and function (especially speech and mastication), while optimizing an individual’s general health and well-being.
Management in the neonatal period
Feeding
Efficient feeding is important for growth and development in infancy. A baby with a cleft of any kind may experience feeding difficulties. However, babies with cleft palate or combined cleft lip and palate usually have more problems than those with cleft lip. A cleft palate prevents the baby from sealing the oral cavity and generating the negative pressure necessary for efficient feeding. Babies with large clefts of the lip and palate may also have difficulty generating positive pressure (compression), which is also important for feeding. The early involvement of specialist nurses with experience of cleft care and in particular the feeding problems associated with clefting is essential. While there is no doubt that breast-feeding is best for babies and should be encouraged, it is usually unsuccessful for infants with a cleft of the lip and palate or palate alone, therefore bottle feeding (preferably using expressed breast milk) is often the most appropriate approach. Once a child is feeding well, and is progressively gaining weight, they will be able to cope with surgery.
Presurgical orthopaedics (Figure 15.4)
Presurgical orthopaedics (PSO) takes advantage of the plasticity of the nasal cartilage in the neonate by using orthodontic-like plates, with or without tapes and nasal stents to reposition the maxillary segments and the nasal structures. PSO needs to be started as soon after birth as possible to maximize the chances of an infant tolerating the appliances. It is technically complex and demanding, not least because taking a maxillary impression in a neonate is particularly challenging with the associated risks to the airway. PSO should only be done by appropriately trained and experienced clinicians. Its use remains controversial, not least because of the associated financial burden coupled with the lack of strong evidence of effectiveness.
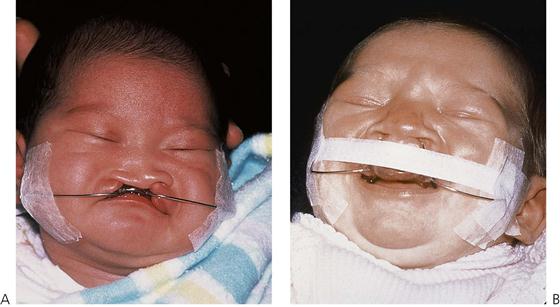
Figure 15.4 (A) Presurgical orthopaedic appliance. The plate is held by micropore tape to the cheeks. (B) Strapping aids in the positioning of the labial segments, especially in cases of bilateral complete clefts. Depending on the institution, these appliances are used in cases of bilateral cleft lip and those with very wide unilateral clefts as shown in (A).
Counselling and parent support
Careful counselling by an appropriately informed cleft specialist is often needed by parents. In many cleft teams, this is provided by the specialist cleft nurses who may also carry out home visits to provide additional support. Parents not only have many questions but are also often confused and anxious and may seek advice about the intermediate and long-term implications of the cleft defect and the necessary surgery. They also often appreciate, and benefit from, the assistance available from members of a parent support group such as CleftPALs (Australia), CLAPA (UK) and American Cleft Palate-Craniofacial Association.
Primary surgery
Many surgical techniques have been described for the primary closure of cleft lip and palate. However, there is still controversy regarding the timing of surgery and which is the most reliable technique consistent with ensuring optimal growth of the face and development of speech.
• Lip repair is less controversial than is palate repair and is generally undertaken around 10–12 weeks and almost certainly by 6 months of age, provided the infant is otherwise developing well. The aim of the lip repair is to restore the continuity of the orbicularis oris muscle of the lip, and with it, the appearance of the upper lip (Figure 15.5).
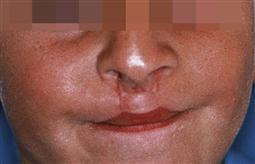
Figure 15.5 Surgical repair of a bilateral complete cleft lip with columella lengthening. The alar base is symmetrical, although there is an accentuation of the cupid’s bow and eversion of the vermillion border.
• Palate repair aims to reconstruct the abnormally inserted musculature of the soft palate to normalize movements of the soft palate and permit the development of normal speech. The extent and timing of palatal surgery is one of the major and continuing controversies in cleft management. This relates to the perceived balance between the benefits of good speech development (which is promoted by early closure) versus the deleterious effects on midfacial growth through surgical trauma and associated scarring (which can be minimized by delaying the surgery).
To date, there is no strong evidence regarding the best approach to primary surgery. Attempts to address this gap in the evidence are being made through large multi-centre international clinical trials.
Management during childhood
Speech and language and ENT problems
Children with clefts and other craniofacial malformations are at an increased risk of speech and language difficulties. Regular assessments are required to monitor the speech and language acquisition process, to assist in making decisions about the need for either further surgery or speech therapy or both.
During childhood, care needs to be exercised to ensure hearing is optimal for speech and language development. Otitis media is common in infants and children with cleft palate and some may also have sensorineural loss. Suboptimal hearing can affect speech and language development so regular audiology assessments for all infants with clefts of the lip and palate is important.
Velopharyngeal inadequacy (VPI)
Children with clefts are at increased risk of VPI. Normal speech requires that the muscles that make up the velopharyngeal sphincter (predominantly the muscles of the soft palate and nasopharynx) work in a coordinated fashion. Defects in any aspect of the nasopharyngeal anatomy and/or physiology may lead to VPI. It is uncommon for otherwise healthy individuals to have VPI. However, for individuals with anomalies of their velopharyngeal sphincter such as a cleft of the palate (hard and/or soft, repaired or unrepaired) VPI can, if severe, compromise speech production rendering a child incomprehensible to others.
• The speech pathologist’s assessment of speech production is critical in the identification and management of VPI and they work closely with the primary cleft surgeons in deciding what, if any, intervention is required to optimize speech.
• If significant VPI is identified, then surgery is usually needed. There are a number of different approaches to the surgical management of VPI, including narrowing the velopharyngeal opening (pharyngoplasty), lengthening the soft palate (intravelar veloplasty) and bulking up the posterior pharyngeal wall (fat augmentation). Although surgery can be expected to improve structural obstacles which hinder the development of normal speech, postoperative speech therapy may still be needed (before and after surgery) to correct poor speech habits that have developed because of the structural malformation.
• Occasionally, surgical correction of VPI is not possible or does not bring resolution. In this case, a palatal obturator or a speech bulb may reduce the velopharyngeal space sufficiently for normal speech to develop.
Orthodontics
Children with clefts of the lip and palate and those with cleft of the hard-palate are at risk of compromised growth of the maxilla as a result of post-surgical scarring. If severe, the growth restriction of the maxilla which affects all three dimensions can lead to the development of a skeletal class III relationship and an associated buccal crossbite. Individuals with complete clefts of the lip and palate may require extensive orthodontic treatment.
The first comprehensive orthodontic evaluation is generally made around the age of 8 years of age although occasionally, there may be a need for earlier review. Orthodontic management at this age can be subdivided into three main components:
Mixed dentition treatment (Figure 15.6)
• Interceptive treatment such as minor tooth alignment to facilitate anterior alignment or correction of an anterior incisal cross-bite.
• Palatal expansion prior to alveolar bone grafting comprises a relatively short period of fixed appliance therapy aimed at achieving an optimal arch shape to improve access for surgery. Bone grafting is usually undertaken when the cleft-side permanent canine (cuspid) tooth shows between half and two-thirds of root development (Figure 15.7). Where necessary, pre-bone graft expansion is commenced approximately 12 months prior to surgery and is completed using an upper fixed appliance (Figure 15.6B). The appliance may then be maintained for 4–6 months post-graft, for stability.
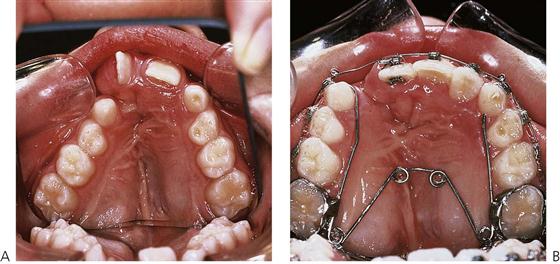
Figure 15.6 (A) One of the effects of early surgery and scarring is collapse of the palatal segments due to an inhibition of growth. Rotation of the central incisor adjacent the cleft can also be seen. (B) Palatal expansion with a quad helix appliance to correct the posterior cross-bite and open space for the bone graft.
Secondary alveolar bone grafting (Figure 15.7)
Alveolar bone grafting should be timed to precede the eruption of the permanent canine tooth on the cleft side, which usually occurs at approximately 11 years of age. The aim of bone grafting is to:
• restore the bony contour of the alveolus.
• stabilize the maxillary expansion.
• provide a bony matrix through which teeth (especially the canine) may erupt.
• allow the teeth to have a healthy supporting periodontium.
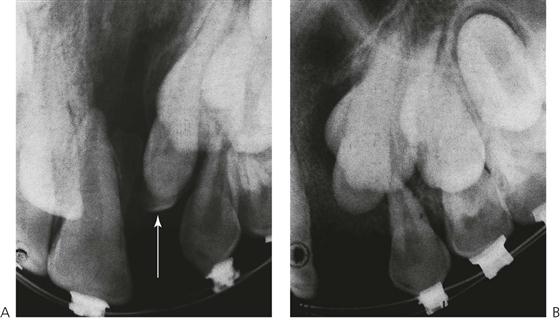
Figure 15.7 (A) Periapical radiograph of a cleft prior to bone grafting. The lateral incisor is present on the palatal aspect of the canine tooth. A small supernumerary tooth (arrowed) is also present, and was removed at the time of surgery. (B) The cleft has been filled with cancellous bone, harvested from the iliac crest. After 3 months, there is already movement of the lateral and canine and both these teeth erupted through the graft to the mouth, where they were aligned orthodontically.
Surgery
• Presurgical palatal expansion (if required) with fixed appliances such as a quad helix (Figure 15.6B).
• Preparation of recipient area may be prepared by extraction of retained primary teeth several weeks prior to surgery.
• Bone harvest for the graft is usually autologous iliac crest cortico-cancellous bone, but may be obtained from the mandibular symphysis, tibia, rib, or calvarium.
Cleft management in adolescence and early adulthood
Orthodontics
Full permanent dentition correction
In the mixed dentition, very little active orthodontic treatment if any, is done, leaving the bulk of the orthodontic treatment to be carried out in the shortest possible time, when the full permanent dentition is present. Once the full permanent dentition is established (excluding the third molars), final correction of the malocclusion and tooth alignment is undertaken. The reasons for postponing the definitive orthodontic management to mid to late adolescence include:
• In the event of a significant skeletal discrepancy orthognathic surgery may be required in conjunction with orthodontic treatment the planning for which cannot occur until growth is complete.
• At 12–16 years dental suitability and motivation to cooperate with orthodontic treatment is assessed prior to the commencement of full fixed appliance treatment.
• Rapid palatal expansion and/or fixed appliance orthodontic treatment is undertaken at the optimum time – occasionally elective extraction of teeth for orthodontic reasons is indicated.
Orthognathic surgery
If a maxillary/mandibular skeletal discrepancy exists at the time of physical maturity, an osteotomy may be required along with any further soft-tissue revision which is also undertaken at the same time, if needed.
If necessary, further aesthetic surgery such as a rhinoplasty can be undertaken as a final surgical procedure. In some instances, functional lip and nose revisions are combined with alveolar bone grafting at an earlier stage to reduce the impact of the aesthetic deformity on the growing child but in general, such procedures are postponed until growth has ceased and orthodontic therapy completed.
The above procedures are only some of the many that may be needed by a patient with cleft lip and palate.
Importance of dental care in overall management
Children with cleft conditions require extensive interdisciplinary care throughout early life and the highest standard of oral health must be maintained because the presence of dental disease can severely compromise both surgical and orthodontic success. The paediatric dentist plays an essential role in coordinating the oral healthcare for these individuals, in order to ensure they reach adulthood with a healthy dentition and positive attitude to dental care. The paediatric dentist will generally coordinate all dental care aspects with those of other disciplines and as such, it is important for them, or their representative, to regularly review the child with a cleft lip and palate.
The first appointment
Many specialist paediatric dental academies support the notion that all infants should be seen by a dentist within 6 months of eruption of the first tooth (i.e. before the age of 1 year). This is even more important in infants born with a cleft.
The purpose of this initial consultation is to explain:
• The dental aspects of the clefting process.
• The likely course of dental management. The involvement of different specialties including restorative, radiological, orthodontic and possible later oral surgical care.
• The probability of the absence of the normal tooth in the region of the cleft, and conversely, the possibility of one or more supernumerary teeth in the cleft region should be mentioned (Figure 15.8).
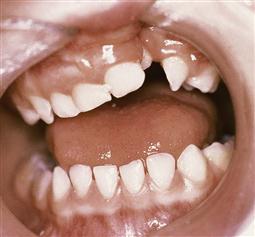
Figure 15.8 A tooth erupting in the cleft. Such teeth often become carious, but should be repaired and retained until bone grafting is performed. Where permanent teeth are congenitally absent, especially the lateral incisor, the supernumerary may be used if of adequate size.
• The likelihood of the presence of crown and/or root morphological abnormalities and enamel hypoplasia of the incisor and canine teeth adjacent to the cleft should be indicated, with the positive reassurance that these can be treated relatively simply soon after they appear.
• The absolute importance of sound preventive care and regular dental visits should be emphasized.
It will almost certainly be necessary to reinforce the introductory information and advice given at this visit at many subsequent outpatient visits over the years of treatment. Many cleft palate clinics produce parent handbooks that are particularly useful.
Babies born with a cleft of the lip and palate not uncommonly have a tooth present at, or erupting soon after, birth – a natal tooth. This can worry parents and may contribute to the existing anxiety around feeding. Such teeth often sit in the premaxillary region and may be very mobile. In most instances, they can be extracted simply using topical anaesthetic cream. Care should be taken to protect the airway from the extracted tooth.
Preventive dental care
Children with clefts of the lip and/or palate may be at increased risk of developing dental caries for a number of reasons, however the evidence is fairly weak. Reasons include:
• PSO (if carried out) may predispose infant oral cavity to colonization by mutans streptococci and hence, to the early development of dental caries.
• Early infant feeding problems may lead to prolonged and more cariogenic feeding habits.
• Presence of developmental defects of enamel particularly around the cleft site.
• Presence of dental crowding, malocclusion and orthodontic appliances.
• Existence of other comorbidities, which may increase caries risk, e.g. reduced salivary flow associated with velocardiofacial syndrome.
• Risk of being ‘spoilt’ with sugary treats to compensate for hospitalizations and other healthcare exposures.
Preventive dental care is essential for these patients, using all known techniques including:
• Early and regular exposure to a fluoridated toothpaste.
• Oral hygiene technique instruction – for parents and later for the individuals themselves.
• Home application of topical fluoride agents.
• Fissure sealing of both primary and permanent teeth.
• Dietary advice to child and parents by paediatric dentist (or dietitian if necessary).
• Appropriate use of bitewing radiographs (in-line with national standards) to ensure early identification and treatment of carious lesions.
The prevention of dental caries and periodontal disease will help with cooperation for ultimate definitive orthodontic treatment, by reducing unpleasant visits for treatment in early childhood. Motivation is especially important for later orthodontic treatment in these patients. This should be assessed early, and enhanced during preventive visits during childhood, so as to ensure compliance.
In many countries, it is the role of the general dental practitioner (GDP) to provide routine dental care to all children. Despite being at increased risk of dental disease, there is no reason why children with craniofacial anomalies cannot receive such care from their local GDP. In many cases, this will be geographically more convenient. However, it is important that the GDP establishes early contact with the cleft palate team or surgeon and maintains a frequent dialogue.
Dental extractions and minor oral surgery
• Except in an emergency, dental extractions should not be performed for these children by the general dentist without first checking and clearing this with the supervising paediatric dentist or orthodontist.
• Primary molars should be retained by pulpotomy, or the space maintained after extraction as advised by the paediatric dentist.
• Erupted supernumerary teeth should be retained until 6–7 years of age, unless impossible to clean, resulting in progressive dental caries, gingival or mucosal inflammation.
Extraction of such teeth should then, in most cases, be carried out in the supervising hospital under either local or general anaesthetic. If a general anaesthetic is required for restorative treatment or other purpose, superficial or obstructing unerupted supernumerary teeth may be removed at the same time but only after discussion with the coordinating paediatric dentist. If a bone graft is planned for the alveolar cleft, it may be necessary to retain primary teeth that are adjacent to the graft site. This, if done 3–4 weeks prior to the grafting procedure, ensures that the healing of the bone graft site is not compromised by leaving a deficiency where the tooth has been extracted at the time of grafting. If the child’s behaviour does not permit the extraction of teeth prior to grafting, then they may be removed by the maxillofacial or plastic surgeon at the time of bone grafting.
Dental anomalies (see also Chapter 11)
Dental anomalies are extremely common in children with orofacial clefting. The most commonly affected tooth is the maxillary lateral incisor on the cleft side. This is due, in part, to disruption of the dental lamina. Anomalies may include:
• Concurrent agenesis and supernumerary teeth within or adjacent to the cleft.
• Disorders of morphogenesis (size and shape).
• Supernumerary teeth may occur in either the medial or distal segment and much less frequently in both segments (Figure 15.8).
Cosmetic restoration of malformed anterior teeth and alveolar cleft
The appearance of the teeth can be improved at any time, depending on the child’s perceptions and wishes.
• Composite resin restorations may be placed for hypoplastic or morphologically abnormal permanent incisor teeth adjacent to the cleft(s) soon after eruption, however, enamel-bonded crowns or veneers should be reserved until after passive eruption and establishment of the gingival margin at the cementoenamel junction.
• As a temporary measure, and in suitable cases, an adhesive retained bridge may be placed to replace missing incisor teeth. This form of prosthesis is a superior alternative to a removable partial denture, however an osseointegrated implant remains the ultimate treatment solution when space cannot be closed orthodontically.
Further reading
1. American Cleft Palate Association. Parameters for the evaluation and treatment of patients with cleft lip/palate and other craniofacial anomalies. In: www.acpa-cpf.org; 2009.
2. Bergland O, Semb G, Abyholm FE. Elimination of the residual alveolar cleft by secondary bone grafting and subsequent orthodontic treatment. Cleft Palate Journal. 1986;23:175–205.
3. Bessell A, Hooper L, Shaw WC, et al. Feeding interventions for growth and development in infants with cleft lip, cleft palate and cleft lip and palate (review). Cochrane Database of Systematic Reviews 2011.
4. Cheng LL, Moor SL, Ho CTC. Predisposing factors to dental caries in children with cleft lip and palate: a review and strategies for early prevention. Cleft Palate-Craniofacial Journal. 2007;44:67–72.
5. Dixon M, Marazita M, Beaty T, et al. Cleft lip and palate: understanding the genetic and environmental influences. Nature Reviews: Genetics. 2011;12:167–178.
6. Eppley BL, van Aalst JA, Robey A, et al. The spectrum of orofacial clefting. Plastic and Reconstructive Surgery. 2005;115:101e–114e.
7. Guo J, Li C, Zhang Q, et al. Secondary bone grafting for alveolar cleft in children with cleft lip or cleft lip and palate (review). Cochrane Database for Systematic Reviews 2011.
8. Hunt O, Burden D, Hepper P, et al. The psychosocial effects of cleft lip and palate: a systematic review. European Journal of Orthodontics. 2005;27:274–285.
9. Levy-Bercowski D, Abreu A, DeLeon E, et al. Complications and solutions in presurgical nasoalveolar moulding. Cleft Palate-Craniofacial Journal. 2009;46:521–528.
10. Liao Y-F, Mars M. Hard palate repair timing and facial growth in cleft lip and palate: a systematic review. Cleft Palate-Craniofacial Journal. 2006;43:563–570.
11. Maarse W, Berge SJ, Pistorius L, et al. Diagnostic accuracy of transabdominal ultrasound in detecting prenatal cleft lip and palate: a systematic review. Ultrasound in Obstetrics and Gynecology. 2010;35:495–502.
12. Nelson P, Glenny AM, Kirk S, et al. Parents experience of caring for a child with a cleft lip and/or palate: a review of the literature. Child: Care, Health and Development. 2012;38:6–20.
13. Tannure PN, Oliveira CA, Maia LC, et al. Prevalence of dental anomalies in nonsyndromic individuals with cleft lip and palate: a systematic review and meta-analysis. Cleft Palate-Craniofacial Journal. 2012;49:194–200.