ALTERATIONS OF PULMONARY FUNCTION IN CHILDREN


Alterations of respiratory function in children are influenced by age, development, gender, race, genetic dominance, and environmental conditions. Newborns are especially vulnerable to a variety of upper and lower airway infections caused by immunologic immaturity. Structural differences in infants and children also render them less competent to tolerate conditions that cause increased work of breathing. Access to healthcare and timeliness of immunizations influence the incidence and severity of pulmonary disorders.
STRUCTURE AND FUNCTION
A number of structural characteristics of the pulmonary system influence the way in which infants and children respond to respiratory disturbances. These include structural characteristics of the upper and lower respiratory tracts, chest wall and lung dynamics, metabolic requirements, immunologic immaturity, and physiologic control of respiration.
Upper Airway
All conducting airways (the portions of airway that do not participate in gas exchange) are present at birth and change only in size throughout childhood. Branching of the bronchial tree is in fact complete by the sixteenth week of fetal life.
Because infants and children naturally have smaller-diameter airways than do adults, they suffer exponentially more obstruction for a given degree of mucosal edema or secretion accumulation. The relative sizes of tonsils, adenoids, and epiglottis likewise are proportionately greater in the young child and with swelling can impose a significant site of obstruction. Infants up to 2 to 3 months of age are “obligatory nose breathers” and are unable to breathe in through their mouths. Nasal congestion is therefore a serious threat to a young infant.
Lower Airways and Lung Parenchyma
During fetal development the lung is transformed from a somewhat dense organ to one that is more delicately structured to facilitate air exchange. Beginning in the second trimester, there is loss of interstitial (mesenchymal) tissue with concomitant expansion of the future air spaces. Capillaries grow into the distal respiratory units that keep subdividing (alveolarization) to maximize surface area for gas exchange. In fact the number of alveoli continues to increase during the first 5 to 8 years of life, after which the alveoli increase in size and complexity. In addition to the structural development of the lung in utero there is accompanying functional maturation, and specialized cell types, such as type II cells, become manifest (Figure 34-1).
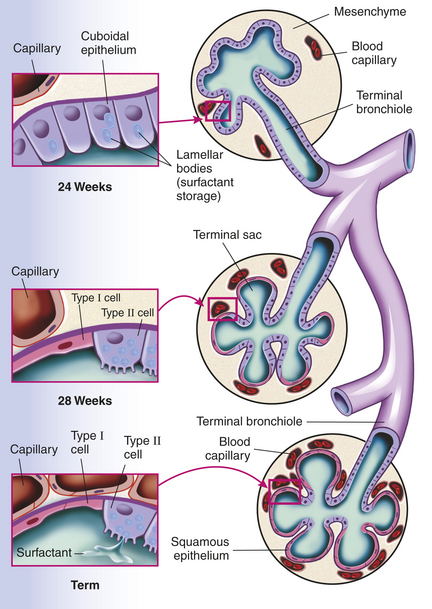
Figure 34-1 Prenatal development of the alveolar unit. Epithelial cells differentiate into type II and type I cells. Mature type II cells are cuboidal, have apical microvilli, and contain lamellar bodies for surfactant storage and secretion. Type I cells are derived from type II cells and consist of flattened epithelium overlying capillaries, thus forming part of the desired thin air-blood barrier. During fetal development the pulmonary capillaries initially are randomly distributed in mesenchyme. They progressively arrange around the epithelial tubes and establish close contacts to the lining epithelium. Overall the volume of mesenchyme decreases and that of the potential air space increases.
Surfactant is a lipid-protein mix that is produced by type II cells and is critical for maintaining alveolar expansion (thus allowing normal gas exchange). It lines alveoli and reduces surface tension, preventing alveolar collapse at the end of each exhalation. Without surfactant the alveoli tend to stay closed, demanding greater inspiratory force and work of breathing to reexpand the alveoli on the next breath. Deficiency of surfactant is often seen in premature infants and causes respiratory distress syndrome (RDS), also known as hyaline membrane disease. Thus surfactant deficiency reflects developmental immaturity. Surfactant lipid is produced by 20 to 24 weeks of gestation and is secreted into the fetal airways by 30 weeks. The more premature the infant, the higher the risk of RDS.
Chest Wall Dynamics
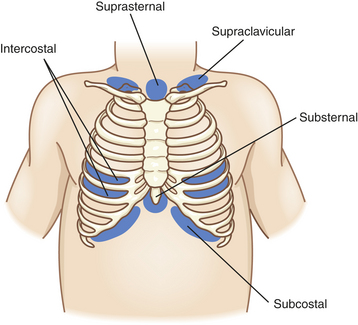
Chest wall compliance is high in infants, particularly premature infants. The cartilaginous structures of the thoracic cage are not yet well ossified (ossification continues to occur throughout childhood), and the chest wall is easily collapsible. During inspiration in the young child, air is drawn in by the downward movement of the diaphragm, but the resulting negative pressure causes the “soft” chest wall to be drawn inward (Figure 34-2); this produces so-called paradoxic breathing, or diaphragmatic breathing. Paradoxic breathing is especially seen during rapid eye movement (REM) sleep of premature infants. With pulmonary compromise the accessory muscles also are drawn inward, creating retraction of the intercostal and supraclavicular spaces (Figure 34-3).
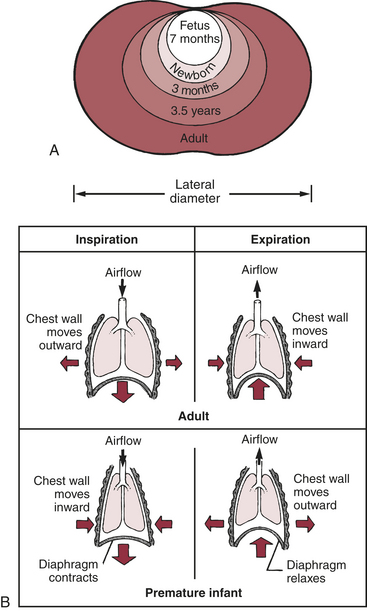
Figure 34-2 Developmental differences in the chest wall and lung mechanics. A, Changes in chest wall shape with age. B, Differences in lung mechanics caused by differences in chest wall compliance (degree of rigidity) in premature infants and adults. (Arrows indicate direction of airflow, chest wall movement, and diaphragm movement.)
Resting lung volume, or functional residual capacity (FRC), represents the balance point between the natural elastic recoil of the lungs (to collapse) and the elastic recoil of the chest wall (to expand). In the face of an overly compliant chest wall, infants up to about 1 year of age are thought to maintain their FRC and avoid atelectasis by muscular “braking” of their expiration. This may occur either by active glottic narrowing or by increased activity of the inspiratory intercostal muscles.
Metabolic Characteristics
The basal metabolic rate of a child is greater than that of an adult, and thus oxygen consumption (VO2) is greater per unit of body weight. The VO2 of the child’s normal breathing accounts for up to 25% of the total VO2. The work of breathing increases VO2 exponentially with respiratory distress. Children have less muscle glycogen reserve, which limits the efficiency of accessory muscles such that fatigue with lactic acidosis can occur quickly. Children also have a high proportion of extracellular fluid and therefore more quickly lose fluid and become dehydrated as a result of fever, environmental heat, or in association with tachypnea (which causes evaporation from the respiratory tract).
Immunologic Incompetence
Passive immunity with immunoglobulin G (IgG) is normally conveyed transplacentally from the mother to the fetus beginning at 20 weeks of gestation; thus IgG levels are lower in preterm than term infants. Breast-feeding allows transfer of secretory IgA, IgG, and IgM after birth. Because IgG has a half-life of approximately 21 days, the placentally transferred antibodies are gone after just a few months. Babies are able to make IgG, IgM, and IgA, and levels of these increase slowly with age. Cell-mediated immunity is also not fully developed in the neonate, which creates a situation of enhanced susceptibility to viral and fungal infections.
Physiologic Control of Respiration
For up to 3 weeks, the newborn has a blunted ventilatory response to hypoxia compared with older children and adults. The mechanisms for this are not well understood but may reflect reduced activity of the peripheral chemoreceptors (in the carotid body) and nonadaptive responses in the respiratory center (in the brainstem). Ventilatory response to hypercarbia is normal in term infants but may be reduced in premature infants. Congenital or acquired lesions of the central nervous system may cause hypoventilation or apnea.
PULMONARY DISORDERS
Pulmonary dysfunction can be categorized into disorders of either the upper airway or lower airway. Signs of acute respiratory failure, however, are the same regardless of etiology. These include the following:
• Increased respiratory effort with retractions (see Figure 34-3) or gasping (apnea in some conditions)
• Decreased level of consciousness
• Cardiovascular signs: tachycardia, mottled color, or bradycardia
• Physiologic compromise reflected by hemoglobin desaturation, hypoxemia, hypercarbia, and acidosis
Disorders of the Upper Airways
The crucial issue in the upper airways is patency. The most common causes of acute-onset upper airway obstruction (UAO) in children are infections, foreign body aspiration, angioedema, and trauma. Chronic UAO has many etiologies, including congenital malformations affecting the airway, cartilaginous weakness, vocal cord paralysis, and subglottic stenosis. Chronic upper airway symptoms should prompt referral to a pediatric pulmonologist or an otolaryngologist because specialized diagnostic studies may be needed. A list of causes of pediatric UAO can be found in Box 34-1.
The site and nature of the obstruction are often discernible by assessing the noise associated with breathing, the quality of the voice or cry, and presence of feeding difficulties. This assessment often can be made without even touching the patient. Likewise, the severity of the problem can to a great extent be judged by simple visual observation of signs, including retractions, nasal flaring, gasping or obstructed breaths, anxiety, restlessness, or need to maintain a specific head or body position. Agitation should be regarded as a likely sign of hypoxemia or obstruction. In acute UAO, increasing the child’s anxiety by excessive physical examination can worsen the condition. The child should be kept as calm as possible. The clinician should never attempt a pharyngeal examination if there is any suspicion of epiglottitis or retropharyngeal abscess because this maneuver may precipitate acute obstruction of the airway.
The sounds of the child’s breathing can provide key clues (Figure 34-4). A sonorous, snoring noise is typical for nasopharyngeal obstruction, such as adenotonsillar hypertrophy. A common sign of pediatric UAO is stridor, a harsh, vibratory sound of variable pitch caused by turbulent flow through the partially obstructed airway. A diagnostic approach to stridor is outlined in Figure 34-5. Whether it is present in inspiration, expiration, or both reflects the site of the problem. In general, inspiratory stridor is generated with obstruction of the extrathoracic airway (above the thoracic inlet), which includes the supraglottic structures, the larynx, the subglottic space, and the upper trachea. Expiratory stridor or a monophonic wheeze may be generated by an obstruction in the intrathoracic airway (the mid- to lower trachea and central bronchi). Biphasic stridor typically reflects obstruction at the glottis (e.g., vocal cord paralysis) itself or a fixed rather than a dynamic lesion in the subglottic space (e.g., hemangioma or subglottic stenosis). Biphasic noise may sometimes mean abnormalities of both extrathoracic and intrathoracic trachea (long-segment stenosis or malacia).
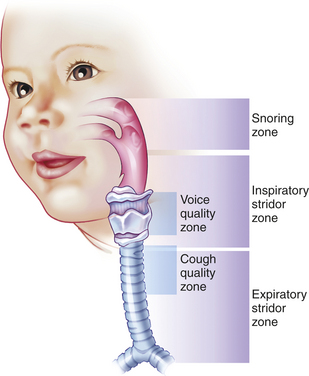
Figure 34-4 Listening can help locate the site of airway obstruction. A loud, gasping snore suggests enlarged tonsils or adenoids. Stridor during inspiration suggests the airway is compromised at the level of the supralaryngeal structures (epiglottis and arytenoid cartilages), vocal cords, subglottic region, or upper trachea. With forced inspiration, intrathoracic pressure becomes quite negative and is less than atmospheric pressure, promoting collapse at or just above the site of obstruction. Expiratory stridor or central wheeze results from narrowing or collapse of the lower trachea or bronchi. During forced exhalation, rising pleural pressure may exceed intratracheal pressure. Airway noise during both inspiration and expiration often represents a fixed obstruction of the vocal cords or subglottic space. Hoarseness or a weak cry is a byproduct of obstruction at the vocal cords. If a cough is croupy or low pitched, suspect tracheal pathology. (Redrawn from Eavey RD: Contemp Pediatr 3(6):78, 1986; used with permission; original illustration by Paul Singh-Roy.)
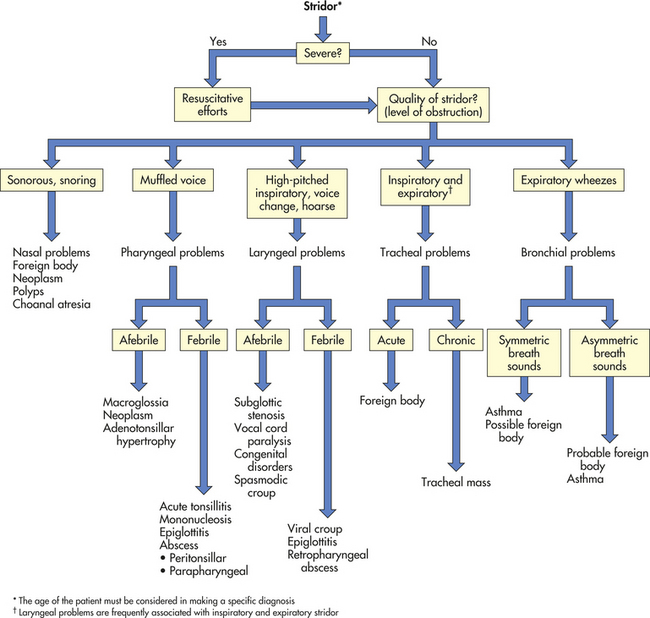
Figure 34-5 Diagnostic approach to stridor. (Adapted from Handler SD: Stridor. In Fleisher GR, Ludwig S, editors: Textbook of pediatric emergency medicine, Baltimore, 1993, Williams & Wilkins.)
Abnormalities of voice or cry (weak or hoarse) suggest problems at the larynx, such as vocal cord paralysis. Muffling of the voice, especially in an acute condition, suggests supralaryngeal obstruction, such as epiglottitis or retropharyngeal abscess. Pronounced cough may be an irritative symptom, such as that produced by an aspirated foreign body, or may be a sign of tracheal obstruction. The cough associated with croup or tracheal foreign body is usually harsh and barking.
Airway obstruction occurs sooner in infants than in older children. Obviously, airway luminal size is smaller in accordance with smaller body size, but any decrease in luminal diameter will be much more significant. This is because airway resistance is proportional to the inverse of the fourth power of the radius; thus a decrease to half the original diameter increases resistance 16-fold. Furthermore, an infant’s cartilaginous structures are more collapsible and thus are prone to creating or contributing to a situation of UAO.
Infections
Infections of the upper airway (Table 34-1) are common in children; some have the potential to cause life-threatening emergencies. Recognition and rapid evaluation of these problems are crucial pediatric care skills.
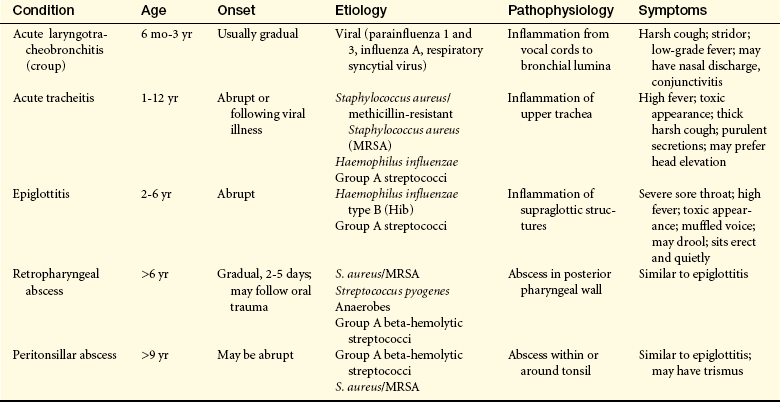
Other Acute Upper Airway Infections:
Bacterial Tracheitis: Bacterial tracheitis can cause rapidly fatal airway obstruction. It accounts for 5% to 14% of UAO in children requiring intensive care.1 The epidemiology of this illness has changed dramatically secondary to immunization against Haemophilus influenzae. Bacterial tracheitis is most often caused by Staphylococcus aureus (including methicillin-resistant Staphylococcus aureus [MRSA] strains), H. influenzae or group A beta-hemolytic Streptococcus (GABHS).2 A virus or a fungus is more likely to be seen as the source of tracheitis in immunocompromised children.1 Treatment of viral croup with corticosteroids has increased the risk for serious bacterial tracheitis (especially by GABHS) placing mortality rates between 18% and 40%.2 This makes it the most common potentially life-threatening upper airway infection in children. The presence of airway edema and copious purulent secretions leads to airway obstruction that can be worsened by the formation of a tracheal pseudomembrane and mucosal sloughing. Increased morbidity can occur because of respiratory and cardiopulmonary arrest, respiratory failure, pneumonia, septic shock, toxic shock syndrome, acute respiratory distress syndrome (ARDS), and multiple organ dysfunction syndrome (MODS). The onset of symptoms may be sudden or may be preceded by a preexisting viral upper respiratory infection or croup. The acute clinical presentation frequently includes tachypnea, stridor, hoarse voice, fever, cough, and/or increased secretions from the mouth and nose. There also may be evidence of concurrent infections, such as sinusitis, otitis, pneumonia or pharyngitis. Children with chronic gastroesophageal reflux are more likely to experience difficulty with these types of concurrent infections.1 Management requires the rapid administration of broad-spectrum intravenous antibiotics. The majority of children with tracheitis require endotracheal intubation in order to prevent airway obstruction. Corticosteroids (parenteral and inhaled) are used to decrease tracheal inflammation. Many children recover adequately to be extubated within 72 to 96 hours.
Retropharyngeal Abscess: Retropharyngeal abscess can be caused by aerobic, anaerobic, or polymicrobial infection. A change in the epidemiologic pattern has been noted in the past several years that is likely related to the use of corticosteroids in the treatment of influenza and croup. There also has been an increase in GABHS strains associated with this condition, and the emergence of MRSA as the offending microorganism is increasingly noted.3–5 Retropharyngeal abscess usually occurs in children younger than 2 years of age and as a consequence of either nasopharyngeal infection or penetrating local injury. Clinical signs include fever, dysphagia, drooling, stridor, respiratory distress, and stiff neck. This condition requires intravenous antibiotics targeted at the suspected microorganism, and sometimes incision and drainage.
Tonsillar Infections: Tonsillar infections (tonsillitis) are occasionally severe enough to cause UAO.6 As with other infections of the upper airway, the incidence of tonsillitis secondary to GABHS (group A streptococci) and MRSA has risen notably in the past 15 years. A classic example of UAO secondary to tonsillitis, now rare because of routine immunization, is diphtheria, which causes sore throat and dysphagia along with fever, malaise, headache, and nausea. Significant swelling of the tonsils and pharynx occurs, and a tenacious membrane may cover the mucosa. UAO because of tonsillitis is a well-known complication of infectious mononucleosis, especially in a young child. The development of UAO in tonsillar infections requires the use of appropriately selected antibiotics and may require the use of corticosteroids, especially in the case of mononucleosis.5,7
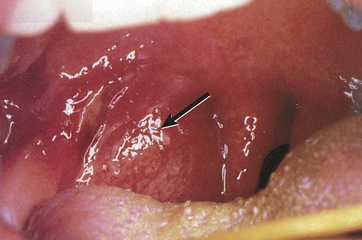
Peritonsillar abscess is usually unilateral and is most often a complication of acute tonsillitis.7 The most common causative microorganism is GABHS. Children have fever, sore throat, dysphagia, trismus, pooling of saliva, and muffled voice. Peritonsillar bulging (Figure 34-6) and cervical adenopathy on the same side are usually visible. The abscess must be drained and the child given antibiotics. Death can occur from spontaneous abscess rupture with aspiration or airway obstruction.8
Croup
Classic croup is an acute laryngotracheobronchitis and is the most common cause of acute upper respiratory obstruction in young children.9 It occurs most often in children from 6 months to 5 years of age, with peak incidence in the second year of life.10 In 85% of cases, croup is caused by a virus, most commonly parainfluenza; however, other viruses such as influenza A or respiratory syncytial virus (RSV) also can cause croup.9 Rhinovirus, adenovirus, measles, and the atypical bacteria Mycoplasma pneumoniae also have been associated with causation. The incidence of croup is highest in late autumn and winter, corresponding to the parainfluenza and RSV seasons, respectively. Croup is more common in boys than girls. In a significant portion of affected children, croup is a recurrent problem during childhood, and there is a family history of croup in about 15% of cases.
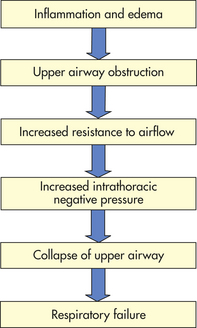
PATHOPHYSIOLOGY The pathophysiology of viral croup is caused primarily by subglottic edema from the infection. The mucous membranes of the larynx are tightly adherent to the underlying cartilage, whereas those of the subglottic space are looser and thus allow accumulation of mucosal and submucosal edema (Figure 34-7). Furthermore, the cricoid cartilage is structurally the narrowest point of the airway, making edema in this area critical. As illustrated in Figure 34-8, increased resistance to airflow leads to increased work of breathing, which generates more negative intrathoracic pressure, which in turn may exacerbate dynamic collapse of the upper airway.
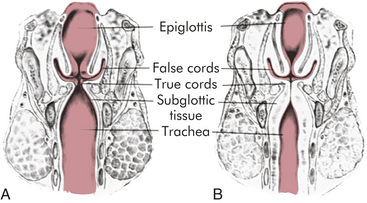
Figure 34-7 The larynx and subglottic trachea. A, Normal. B, Narrowing and obstruction from edema caused by croup. (From Hockenberry MJ et al: Wong’s nursing care of infants and children, ed 8, St Louis, 2007, Mosby.)
CLINICAL MANIFESTATIONS Typically there is a prodrome of rhinorrhea, sore throat, and low-grade fever for a few days. The child then develops the characteristic harsh (seal-like) barking cough, hoarse voice, and inspiratory stridor. Most cases are mild and resolve spontaneously after several more days. Occasionally, however, UAO becomes severe and requires urgent management.
Spasmodic croup is another clinical entity that is characterized by similar hoarseness, barking cough, and stridor but is of sudden onset, usually at night and without viral prodrome. It often resolves as quickly as it develops. The etiology is unknown although an association with a history of atopy has been observed.10
EVALUATION AND TREATMENT The degree of symptoms determines the level of treatment. Most children have a barking cough and viral symptoms and may need no specific treatment. However, the presence of stridor (especially at rest), retractions, or agitation suggests a sicker child. A number of clinical tools are used to assess the severity of croup in children. The tool most often used is the Westley croup score, which provides a cumulative score for the degree of stridor, retractions, air entry, cyanosis, dyspnea, and level of consciousness in the child.9 Severity also is classified into mild, moderate, and severe.
Croup therapy has been the subject of debate for years. Nonpharmacologic treatment options include steam inhalation, ice masks, and oxygen. The first two, however, lack scientific studies to support or refute their benefit. The consensus from numerous controlled studies is that oral, intravenous, or nebulized corticosteroids have a significant effect on croup-related hospitalizations and are cost effective.9 Symptoms improve faster, less sleep is lost by children, less stress is experienced by parents, and fewer children have a need for return healthcare visits when corticosteroids are used.11 The emergent use of nebulized epinephrine is indicated when significant respiratory distress is present. Epinephrine stimulates α- and β-adrenergic receptors and is thought to decrease airway secretions and mucosal edema. However, its effect lasts only 2 hours and should be considered a temporizing measure until concomitantly given steroids begin to take effect. Thus children who are given nebulized epinephrine should be observed for 2 to 3 hours to ensure that they will remain stable, and close follow-up is mandatory. Heliox (helium-oxygen mixture of 80:20 or 70:30) can be used for severe cases of croup, although this is not considered part of the routine treatment regimen.9,10
Acute Epiglottitis
Acute epiglottitis is a severe, life-threatening, rapidly progressive infection of the structures above the insertion point of the glottis, which include the epiglottis, aryepiglottic folds, arytenoid soft tissue, and the uvula. Historically, cases were nearly always caused by H. influenzae type B (Hib). Since the advent of Hib immunization, the overall incidence of acute epiglottitis has decreased to only 10% to 20% of previous levels.9,12 Current pediatric cases usually represent vaccine failures or are caused by alternative pathogens, such as groups A, B, C, F, and G streptococci, Streptococcus pneumoniae, Candida species. S. aureus, and viral pathogens.12 Hib still accounts for approximately 25% of the cases seen in children.12 Thermal injuries, trauma, and posttransplant lymphoproliferative disorder also have been reported as causes of epiglottitis.12
CLINICAL MANIFESTATIONS In the classic form of the disease, a child between 2 and 6 years of age suddenly develops high fever, irritability, sore throat, a “hot potato voice,” inspiratory stridor, and severe respiratory distress. The child appears ill and classically will adopt a forward-leaning position (tripod position) with drooling and dysphagia (inability to swallow). Examination of the throat may trigger laryngospasm and cause respiratory collapse. Death may occur in a few hours. Pneumonia, cervical lymph node inflammation, otitis, and, rarely, meningitis or septic arthritis may occur during the course of epiglottitis.
EVALUATION AND TREATMENT Despite its decreasing incidence, all pediatric practitioners must be familiar with epiglottitis and understand it is a life-threatening emergency. The essentials are recognition, avoidance of disturbing the child (which could worsen the obstruction), and securing the airway. Tracheal intubation should be accomplished by the most experienced personnel (usually an anesthesiologist and/or otolaryngologist) using fiberoptic laryngoscopy. Subsequent culture of the airway is obtained and intravenous broad-spectrum antibiotics are administered promptly. Therapy is reevaluated after culture results return. Corticosteroids also are generally used in treatment regimens although there are no published randomized trials to support this practice.9,12,13 Despite the severe presentation of epiglottitis, resolution with treatment is usually rapid, with intubation rarely needed for more than a couple of days. When Hib epiglottitis is diagnosed, the American Academy of Pediatrics (AAP) recommends that postexposure prophylaxis with rifampin be administered to household contacts (specific to certain ages of children present).13 When caused by microorganisms other than H. influenzae, as is now the usual situation, epiglottitis may present in ages outside the typical range and with more gradual rather than fulminant onset, thus making diagnosis less obvious. Such cases also may respond more slowly to treatment.
Aspiration of Foreign Bodies
Most children who aspirate a foreign object (foreign body aspiration) are between 1 and 3 years of age. More than 100,000 cases occur each year.14 Often the aspiration either is not witnessed or does not seem significant to the parent, thus medical care is often not pursued until after the first 24 hours. At the time of aspiration, the child may cough, choke, gag, or wheeze, and stridor or cyanosis occasionally occurs. This may be followed by a quiescent interval of minutes to even weeks or months before symptoms reappear from resulting local irritation, granulation, bronchial obstruction, or infection (pneumonia or bronchiectasis). Pronounced inspiratory stridor, cough, and wheezing are typical symptoms that prompt the parents to seek medical attention. Examples of common aspirated objects include nuts, sunflower seeds, hot dog chunks, popcorn, coins, and small toys or toy parts. Meat or food impactions are more common in adolescents. Items of particular concern are batteries and multiple magnets. In general, foreign bodies require early intervention secondary to their propensity to cause respiratory symptoms and complications, including esophageal erosions or aortoesophageal fistula.
The symptom history is often the most critical aid in diagnosis.14,15 Symptoms are determined by the size of the object and the site in which it is located, as well as the child’s age and size (see Figure 34-4). Foreign bodies lodged in the upper trachea typically produce inspiratory stridor, whereas those located in the lower intrathoracic airways more commonly produce wheezing. About 75% of aspirated foreign bodies lodge in a bronchus. Children with an unexplained persistent cough and refractory parenchymal infiltrates also should be considered for unrecognized foreign body aspiration.16 Many objects are not radiopaque; however, if the object has completely occluded a lung segment, atelectasis will be visible on a chest radiograph. Occasionally, air will accumulate distal to the obstruction if the object is causing a ball-valve effect. This effect can sometimes be documented by inspiratory and expiratory chest films (Figure 34-9). In a younger child, bilateral decubitus films may show failure to compress the obstructed lung when in the “down” position.
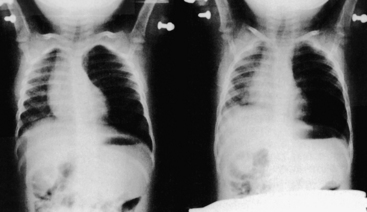
Figure 34-9 Foreign body aspiration. Inspiratory (left) and expiratory (right) chest radiographs of a child who aspirated a portion of a potato into the left mainstem bronchus. Left lung field is hyperaerated and the mediastinum is shifted to the right on expiration because of left-sided obstructive emphysema. (From Kenna MA, Bluestone CD: Pediatr Rev 10[1]:25, 1988.)
Most foreign bodies can be removed by bronchoscopy and only rarely is a pulmonary lobectomy required. Soft particles such as food as well as hard objects must be removed because infectious processes will otherwise occur. Objects lodged in the laryngeal or subglottic regions are particularly dangerous because of their potential for complete or near-complete airway occlusion.
Other Causes of Upper Airway Compromise
Angioedema: Angioedema is a localized edema involving the deep, subcutaneous layers of skin or mucous membranes. Generally, angioedema causes facial swelling first, particularly around the eyes and lips, and may progress to airway swelling.17 Angioedema is usually secondary to allergic phenomena. If airway compromise is apparent, standard treatment includes epinephrine (subcutaneous), antihistamines, and corticosteroids. An occasional cause of pediatric angioedema is use of angiotensin-converting enzyme inhibitors for treatment of hypertension or heart disease. Increased levels of bradykinin appear to mediate this adverse effect by causing vasodilation, increased vascular permeability, and histamine release.18
An inherited deficiency of the plasma protein C-1 inhibitor (C-1 INH), causes hereditary angioneurotic edema (HAE), a rare but serious problem in children. This autosomal dominant trait has an estimated prevalence of 1 in 10,000 to 50,000 births, and a family history is positive in 75% of cases.17 The mean age of onset of initial symptoms is 8 to 12 years but it also may occur as early as the first year of life. This condition is characterized by recurring attacks of angioedema involving subcutaneous tissues (especially limbs, genitalia, and face); abdominal and pelvic viscera; and, much less often, the airway. Laryngeal attacks in these individuals may be life threatening and do not respond reliably to standard measures for airway edema. The mortality of undiagnosed HAE can be as high as 50%. The mainstays of supportive care are airway monitoring, hydration, pain relief, and control of nausea.19 Concentrates of C-1 INH appear to produce rapid improvement within 15 to 60 minutes. Short- and long-term prophylaxis can be instituted using antifibrinolytic agents, attenuated androgens, and C-1 INH concentrates.20
Subglottic Stenosis: Congenital subglottic stenosis is the third most common laryngeal anomaly and is defined as a subglottic airway diameter of less than 4 mm at the cricoid region in a full-term infant, and less than 3 mm in a premature infant.21 Incomplete recanalization of the laryngotracheal tube during the third month of gestation results in this defect. Subglottic stenosis also is associated with eosinophilic esophagitis, Wegener granulomatosis, and neurofibromatosis.22–24 Traumatic injury to the upper airway with development of subglottic stenosis is a well-described complication of endotracheal intubation.25 Factors that contribute to subglottic stenosis include long-term assisted ventilation, use of an endotracheal tube that is too large, excessive movement of the tube, and individual susceptibility.26 Neonates can tolerate long periods of endotracheal intubation; the overall rate of symptomatic subglottic stenosis in neonates is 0.2%.26 The occurrence of subglottic stenosis can be minimized by ensuring that the tube size allows a small air leak during inspiration (at a peak inspiratory force of approximately 25 mmHg) and that the tube is securely taped. Sedation is generally required to reduce head movement for children who are intubated. Because of the rapid growth of the lumen of the trachea and cricoid cartilage in the first year (which triples in size), infants may outgrow the obstruction, particularly if mild or moderate.27 Clinical trials are underway to evaluate topical mitomycin C (an antineoplastic agent) as an adjunct in reducing scarring of the airways; this has met with mixed results in the adult population.28 For significant subglottic stenosis, tracheostomy or tracheal reconstructive surgery may be needed.
Laryngomalacia and Tracheomalacia: Laryngomalacia is the most common cause of chronic stridor in infants. Boys are twice as likely to present with symptoms than girls. In laryngomalacia, the epiglottis or arytenoids, or both, fold inward with inspiration partially covering the glottis (Figure 34-10). The pathophysiology of these abnormalities is still not completely understood. Two primary hypotheses are anatomic or neuromuscular.29 Anatomically there may be foreshortened or tight aryepiglottic folds or there may be redundant soft tissue in the supraglottis. The neuromuscular hypothesis suggests that there is an abnormality of the sensorimotor integrative function of the brainstem and peripheral reflexes are responsible for laryngeal tone and airway patency.30 Laryngomalacia is frequently associated with gastroesophageal reflux disease (GERD).27 Typical signs of laryngomalacia include inspiratory stridor beginning in the first days or weeks of life, accentuated with activity, and sometimes with positional changes (worse in supine or head-flexed positions). Feeding difficulties may be noted, but they are usually mild. Cry is normal. Laryngoscopy is used to confirm the diagnosis. Laryngomalacia is usually mild and improves spontaneously over the first year of life as the supralaryngeal cartilage structures stiffen, thus most cases are managed with watchful waiting. A late-onset variant of this disease has been noted in the literature and should be suspected if the following occurs: potential cause of feeding difficulties in toddlers, sleep apnea in children, and exercise intolerance in teenagers.31,32
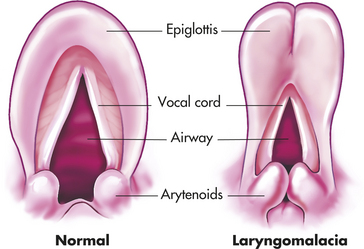
Figure 34-10 Laryngomalacia. In the normal larynx (left), supralaryngeal structures maintain their upright orientation during inspiration. In contrast, in infants with laryngomalacia (right), there is inward prolapse of the arytenoid masses, which include the prominent cuneiform tubercles and the arytenoid cartilages. The glottis becomes partially covered, and airflow is impeded. Sometimes the edges of the epiglottis curl inward, further exacerbating the obstruction. In expiration, these structures are “blown” aside passively.
In tracheomalacia, or tracheobronchomalacia, the tracheobronchial cartilages tend to collapse during the respiratory cycle. This may be classified as primary (idiopathic) or secondary. When malacia is caused by a secondary source, it is usually related to extrinsic compression of the trachea from a vascular malformation.27 Tracheobronchomalacia presents clinically as a spectrum of respiratory illnesses that range from life-threatening conditions to chronic cough and wheeze conditions.33 In some cases symptoms may be more subtle than in laryngomalacia. Low-pitched inspiratory stridor may be a sign of malacia of the upper trachea or centrally located, single-pitch (monophonic) wheeze may be present in malacia of the mid- to distal trachea. Tracheomalacia can be suspected clinically and confirmed by bronchoscopy. Depending on the type and severity of the lesion, surgical approaches for repair may be indicated.
Vocal Cord Paralysis: The vocal cords should move apart to facilitate inspiration and move together to facilitate vocalization. Paralysis of one or both vocal cords may affect breathing, swallowing, and speech. Although it is classified as the second most common congenital laryngeal anomaly, vocal cord paralysis is a relatively uncommon condition. The etiology of the congenital abnormality is unclear but may be caused by immaturity of the vagus nerve or brainstem or both.27 Iatrogenic injury is frequently cited as the major secondary cause of vocal cord paralysis, such as surgical trauma to the recurrent laryngeal nerve during cardiac surgery.34 Other secondary causes include Arnold-Chiari malformation (the region of the brainstem in which the nucleus ambiguus acts as the “relay station” for laryngeal function), cerebral palsy, hydrocephalus, myelomeningocele, spina bifida, or hypoxia.21 Other associations include infectious and neoplastic causes, trauma, and inflammatory conditions.34 In older children and adolescents, exercise has been known to precipitate vocal cord dysfunction (VCD).35
Clinical findings of vocal cord paralysis in children less than 1 year include dysphonia, glottic incompetence, GERD, and stridor.34 It sometimes resolves spontaneously (most often during the first year of life) or with correction of the underlying problem. Flexible laryngoscopy and chest x-ray are common evaluative tools that may help determine the cause. Medical therapy may include use of corticosteroids, proton pump inhibitors, and speech therapy. Recurrent pulmonary infections secondary to aspiration may occur and require treatment until the cords are repaired.21 Severe cases may necessitate endotracheal intubation and tracheostomy. Tracheostomy may be used until the vocal cords are surgically repaired or can be used as a permanent measure for bilateral vocal cord paralysis.21
Congenital Malformations: Congenital malformations of the trachea and bronchial tree cause airway obstruction. Lesions include laryngeal atresias and webs, cysts, clefts, and subglottic hemangiomas. Webs and atresias are caused by failure of the larynx to recanalize during embryogenesis. Most of these disorders are in the area of the glottis with extension into the subglottis.21 Structural abnormalities involving the great vessels also can result in tracheal compression, for example, absent pulmonary valve syndrome dilates the pulmonary artery, which can compress the trachea and bronchi.36 Tracheal or bronchial compression results in airway symptoms or feeding difficulties, or both, ranging from dysphagia, recurrent respiratory infections, wheezing, and stridor to acute respiratory distress or “dying spells.” Many older children are first thought to have gastroesophageal reflux or asthma as the principal problem. Surgical management is usually required for these conditions, and some infants may require mechanical ventilation while awaiting surgery.36
Obstructive Sleep Apnea
Obstructive sleep apnea syndrome (OSAS) is a breathing disorder defined by prolonged partial and/or intermittent complete UAO during sleep with disruption of normal ventilation and normal sleep patterns.37 Childhood OSAS is common with an estimated prevalence of 2% to 3% among middle-school children and as many as 13% of children ages 3 to 6 years. Prevalence is estimated to be two to four times higher in vulnerable populations (blacks, Hispanics, and preterm infants).38 Unlike adults, OSAS in children occurs equally among males and females. Possible influences early in life may include passive smoke exposure, socioeconomic status, and snoring together with genetic modifiers such as those that promote airway inflammation.
PATHOPHYSIOLOGY The pathophysiology of childhood OSAS is likely to be multifactorial in origin. In otherwise healthy children, the most common predisposing factor is adenotonsillar hypertrophy, which causes physical impingement on the nasopharyngeal airway. OSAS often occurs in overweight or obese children as well as in those with orthodontic/craniofacial anomalies or neurologic disorders. Allergy and asthma also may contribute to this condition. In addition to physical narrowing, other mechanisms have been suggested, such as abnormalities in the motor tone of the upper airways (frequently an issue in neurologically impaired children) or abnormal arousal mechanisms.39 Recent studies have documented that children with sleep disordered breathing (SDB) have increased inflammation in the upper airway and elevated serum levels of C-reactive protein that are relationally proportional to the severity and frequency of UAO.40 Lastly, genetic susceptibility likely plays a role in neurocognitive dysfunction associated with this condition.
CLINICAL MANIFESTATIONS OSAS may present with a history of snoring and labored breathing, restlessness, and sweating during sleep, which can be continuous or intermittent. There may be episodes of increased respiratory effort but no audible airflow, often terminated by snorting, gasping, repositioning, or arousal. Affected children are often chronic mouth breathers and have large tonsils. They also may exhibit nocturnal enuresis and intrusive nap habits.41 Unlike adults, no correlation between OSAS and sleep position has been noted in children. Similar to adults, it appears that there also may be a correlation between OSAS and elevated blood pressure. This is further linked to increased body mass index (BMI) and episodes of desaturation and apnea/hypopnea.42 Children who are overweight or obese often have severe OSAS and adopt the prone sleeping position to facilitate improved airway patency and are at a 4.6-fold increase for sleep apnea compared with healthy children.38
OSAS can result in chronic hypoxemia and hypercapnia affecting multiple organ systems. Significant morbidity is associated with OSAS including cognitive, neurobehavioral (inattention, hyperactivity, aggression, conduct problems, attention deficit/hyperactivity disorder [ADHD]/emotional [mood]) impairment, excessive daytime sleepiness, impaired school performance, and poor quality of life.38 Left untreated it also can cause cardiovascular disease, particularly left ventricular hypertrophy, and insulin resistance, as well as pulmonary complications (upper and lower respiratory tract infections) and reduced somatic growth.38,41,43–45
EVALUATION AND TREATMENT All parents should be asked if their child exhibits snoring, a symptom that is often not spontaneously reported to a healthcare provider. The history and physical examination are the most effective means of diagnosis.46 Screening tools and sleep questionnaires may be helpful in evaluating the presence of SDB.41 Radiographic image of the upper airway may reveal upper airway narrowing caused by adenoidal hypertrophy and magnetic resonance imaging (MRI) and acoustic reflectometry may detect reduced upper airway dimensions.38,41 The most definitive evaluation (“gold standard”) is the polysomnographic sleep study, which documents obstructed breathing and physiologic impairment. If obstructive sleep apnea caused by tonsillar enlargement is documented or strongly suspected clinically, children are most often referred for tonsillectomy and adenoidectomy (T&A). For severely affected children who do not respond to T&A or who have different problems, such as obesity that cannot be rapidly remedied, continuous positive airway pressure (CPAP) delivered through a tight-fitting nasal mask may be used during sleep. Treatment is important to prevent associated morbidities. Successful medical or surgical treatment results in improvement in physical, behavioral, and emotional difficulties as well as quality of life.41,47
Disorders of the Lower Airways
Lower airway disease is one of the leading causes of morbidity in the first year of life and continues to be an important component of other illnesses. Pulmonary disorders commonly observed include perinatal conditions such as neonatal RDS, congenital malformations, asthma, cystic fibrosis, infections, aspiration syndrome, and ARDS.
Neonatal Respiratory Distress Syndrome
Respiratory distress syndrome (RDS) of the newborn, also known as hyaline membrane disease (HMD), is a major cause of morbidity and mortality in premature newborns.48 The epidemiology, pathophysiology, and clinical presentation of RDS are outlined in Box 34-2. The major predisposing factor is prematurity because the immature lung is not well structured for gas exchange and has not yet developed adequate surfactant production and secretion. Occasionally RDS is seen in other situations, most notably infants of diabetic mothers. An additional factor that increases risk is cesarean delivery. It is more common in boys than girls and in whites than nonwhites. The incidence of RDS (in the absence of preventive treatment) is approximately 50% to 60% at 29 weeks of gestation and decreases significantly by 36 weeks. Preterm births account for up to 12% of all births49 and approximately 10% of newborns who require some assistance to begin breathing at birth.50 Antenatal stress on the fetus may accelerate lung maturation and decrease RDS risk. In special circumstances, such as elective early delivery (e.g., for maternal health reasons), RDS risk is assessed by sampling amniotic fluid for quantification of secreted surfactant lipids, the basis of the lecithin/sphingomyelin (L/S) ratio (value of 2 or greater predicts low risk). Another common test looks for presence of the lipid phosphatidylglycerol, which also reflects lung maturity.
PATHOPHYSIOLOGY RDS is a state of pulmonary insufficiency that in its natural course commences at or shortly after birth. Severity tends to increase over the first 2 days of life.51 It is caused primarily by surfactant deficiency and, secondarily, by a deficiency in alveolar surface area for gas exchange. Premature infants are born with many underdeveloped and small alveoli that are difficult to inflate. Those that are available for gas exchange do not have adequate surfactant, which is necessary at the air interface to maintain alveolar distention at end-expiration. The net effect is atelectasis (see Figure 34-11), which causes significant hypoxemia, and is difficult for the neonate to overcome because it requires a significant negative inspiratory pressure to open the alveoli with each breath. The chest wall is weak and highly compliant, making it difficult to overcome this increased work of breathing. This results in a decrease in tidal volume causing alveolar hypoventilation and hypercapnia. Hypoxia and hypercapnia cause pulmonary vasoconstriction, which increases intrapulmonary resistance and shunting (Figure 34-12). To make the situation more complex, prolonged hypoxemia activates anaerobic glycolysis, which produces lactic acid and thus causes metabolic acidosis. Alveolar hypoventilation makes it impossible to get rid of excess carbon dioxide (CO2), and combined metabolic and respiratory acidosis develops. Lowered pH causes further vasoconstriction. This results in hypoperfusion of the lung and a decrease in effective pulmonary blood flow. Increased pulmonary vascular resistance causes a partial return to fetal circulation, with right-to-left shunting of blood through the ductus arteriosus and foramen ovale (see Figures 34-11 and 34-12). With inadequate pulmonary circulation and alveolar perfusion, the oxygen content of the blood continues to decrease, pH decreases, and materials needed for surfactant production are not circulated to the alveoli. Capillary permeability increases, resulting in the leakage of plasma proteins. Fibrin deposits in the air spaces create the appearance of hyaline membranes for which the disorder is named. The plasma proteins leaked into the air space have the additional adverse effect of interfering with the function of surfactant that may be present. The pathogenesis of RDS is summarized in Figure 34-12.
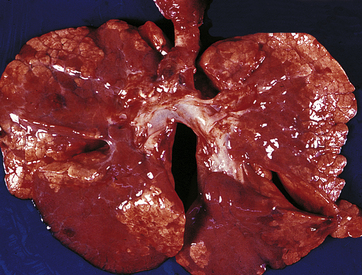
Figure 34-11 Patchy atelectasis of neonatal lungs with respiratory distress syndrome (RDS). (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
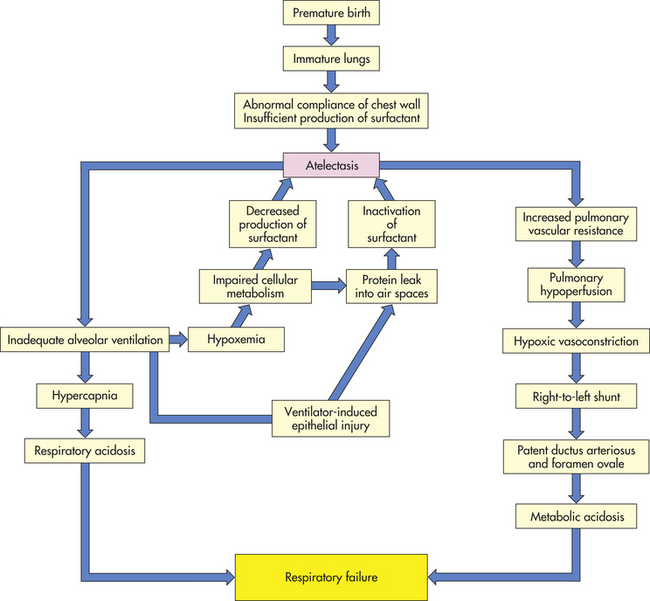
Figure 34-12 Pathogenesis of respiratory distress syndrome (RDS) of the newborn. RDS is also known as hyaline membrane disease.
CLINICAL MANIFESTATIONS Signs of RDS appear within minutes of birth. Some neonates require immediate resuscitation because of asphyxia or initial severe respiratory distress. Tachypnea (respiratory rate more than 60 breaths per minute), expiratory grunting or whining, intercostal and subcostal retractions, nasal flaring, and poor color are the most striking clinical manifestations of RDS. The natural course is characterized by progressive hypoxemia and dyspnea. Apnea and irregular respirations occur as the infant tires. The severity of the hypoxemia and the difficulty in providing adequate supplemental oxygenation give rise to the Vermont Oxford Neonatal Network definition of RDS as a PaO2 less than 50 mmHg in room air, central cyanosis in room air, or a need for supplemental oxygen to maintain PaO2 greater than 50 mmHg, as well as the classic chest film appearance.51 Within the first 6 hours of life, a chest radiograph will reveal air-filled bronchi (air bronchograms) silhouetted against lung fields that have a “ground glass” appearance associated with alveolar consolidation. RDS can progress to death in severe cases, but in most cases the clinical manifestations reach a peak within 3 days, after which there is gradual improvement with appropriate treatment.
EVALUATION AND TREATMENT Diagnosis is made on the basis of clinical manifestations, chest radiographs, and, occasionally, confirmatory analysis (e.g., L/S ratio) of amniotic fluid or tracheal aspirates. The ultimate treatment for RDS would be prevention of premature birth, but in the meantime other significant advances in treatment have been made.
The first is antenatal treatment with glucocorticoids for women in preterm labor. Glucocorticoids induce a significant and rapid acceleration of lung maturation and stimulation of surfactant production in the fetus, and there is extensive evidence that maternal steroid therapy significantly reduces the incidence of RDS, central nervous system hemorrhage, and neonatal mortality.49,52 This treatment is currently recommended in the setting of preterm labor at 24 to 34 weeks of gestation unless delivery is imminent; ideally, dosing continues for 48 hours while attempts are made to halt labor. It remains unclear whether repeated courses of steroids are safe in this setting.51
The second major advance in RDS treatment has been exogenous surfactant, either synthetic or purified from animal sources and instilled down an endotracheal tube (ETT). This may be administered in prophylactic or rescue protocol. Unfortunately, liquid dosing down the ETT may result in peridosing adverse events like hypoxia, hypercapnia, and changes in cerebral blood flow. There has been recent progress in administering surfactant by less invasive methods such as through nebulization or nasal CPAP. These modalities result in more uniform distribution of the drug than through the ETT.53 Current protocols recommend prophylactic administration of surfactant to infants weighing less than 1000 g beginning within 15 to 30 minutes of birth, after the infant is stabilized. Repeat dosing is usually given every 12 hours during the first few days. There is usually a dramatic improvement in oxygenation. For infants weighing more than 1000 g, surfactant replacement is based on clinical need. Because of concerns about intervention-induced lung injury, guidelines for surfactant administration are being reconsidered (see What’s New? Pulmonary Resuscitation of the Newborn—Setting the Stage for Injury?).
Systematic reviews of randomized, controlled trials have confirmed that surfactant replacement improves oxygenation as well as reduces the incidence of RDS, death, pneumothorax, and pulmonary interstitial emphysema.54 Therapy with surfactant should be considered complementary to antenatal glucocorticoids, which promote not only accelerated surfactant synthesis but also enhanced structural development of the lung and beneficial effects on mechanisms of fluid clearance from the lung. These two therapies together appear to have an additive effect on improving lung function. Supplemental inositol also may promote maturation of surfactant and prevent adverse neonatal outcomes in preterm infants.55
The third advance in RDS treatment has been in supportive care. Newborns with RDS need oxygen and often ventilatory support such as CPAP or mechanical ventilation. Strategies that are lung protective, such as greater reliance on nasal CPAP, permissive hypercapnia, lower oxygen saturation targets, modulation of tidal volume (VT) settings, use of nitric oxide, and use of high-frequency oscillation, are being evaluated.56 Nitric oxide has found acceptance for treatment of persistent pulmonary hypertension of the newborn and for hypoxic respiratory failure in term and near-term infants although the precise mechanisms of how it may improve lung function are yet unclear.57,58 The use of nitric oxide in moderately ill, ventilated premature infants appears safe in infants between 1000 and 1250 g birth weight. Benefits include decreased oxygen use and fewer days of ventilation, prevention of bronchopulmonary dysplasia (BPD), and neurologic protection. Additional therapies, such as antioxidants, late surfactant doses, caffeine, and improved ventilatory strategies (including steroids surrounding the time of extubation), can provide additional benefit in these vulnerable infants.59,60 Other key components of supportive care include prophylactic antibiotics, temperature control, fluid and nutritional management, maintenance of blood pressure, and management of patent ductus arteriosus.51
The extremely preterm lung is particularly vulnerable to injury. Mechanical ventilation may interfere with alveolarization and surfactant metabolism and may aggravate the proinflammatory state (as reflected by abnormal cytokine profiles) that is believed to accompany premature birth and RDS. Injury from oxygen toxicity also is a concern and is mediated through reactive oxygen species.61 This combination of factors may lead to subsequent development of chronic lung disease or bronchopulmonary dysplasia.62,63 The use of CPAP rather than intubation may reduce these complications.64 Most infants with RDS survive with treatment. However, the incidence of subsequent chronic lung disease is significant among very low-birth-weight infants.
Bronchopulmonary Dysplasia
Bronchopulmonary dysplasia (BPD), often used synonymously with chronic lung disease of infancy, is the most common chronic lung disease of infancy in the United States. It is the term used for persisting lung disease following premature birth and perinatal respiratory support. When originally described by Northway and colleagues in 1967, the term was applied to premature infants (30 to 37 weeks’ gestation) who had survived acute RDS but continued to have pulmonary dysfunction and oxygen dependence, which was attributed to injury from postnatal mechanical ventilation and oxygen therapy.65 The terms of definition and evolution of disease have changed notably since the original description of the disease. Premature infants are now consistently surviving at 23 to 26 weeks and have different mechanisms of lung injury.66
In the current era of neonatology, the widespread use of antenatal glucocorticoids and postnatal surfactant has lessened the incidence and severity of RDS, and BPD is occurring almost exclusively in the smallest premature infants (23 to 28 weeks’ gestation) who have received mechanical ventilation. Surprisingly, some of these tiny infants who develop BPD have had few or no clinical signs of RDS at birth or have initially received only low levels of supplemental oxygen or ventilatory support.67 Nevertheless, a highly significant predictor of subsequent BPD remains the need for mechanical ventilation on the day of birth. The presence of antenatal chorioamnionitis, postnatal sepsis, or a patent ductus arteriosus may confer additive risk of developing BPD.
The reported incidence of BPD is widely variable because of the lack of consistent diagnostic criteria, but is most common in infants delivered at gestational ages less than 30 weeks or who have birth weights of less than 1500 g. There are approximately 60,000 infants born at less than 1500 g in the United States on an annual basis. About 20% to 30% of these infants develop BPD. Because BPD is a multisystem condition, it also is associated with developmental disorders in other systems, such as growth retardation, pulmonary hypertension, neurodevelopmental delays (e.g., cerebral palsy), hearing defects, and retinopathy of prematurity.66,68
PATHOPHYSIOLOGY In preterm infants born at less than 28 weeks of gestation, the fetal lung is in the canalicular stage of development (16 to 28 weeks), a critical period during which type II epithelial cells appear, capillaries grow into the future distal alveolar regions, and the interstitium begins to condense. Ultimately the alveoli must have a very thin interface between the air space and the capillary for appropriate gas exchange. The extensive network of alveoli develop by septation within the terminal respiratory unit, beginning in the saccular stage, which starts at approximately 26 to 28 weeks.
Prior to the widespread use of surfactant therapy, BPD was a disease characterized by airway injury, inflammation, and parenchymal fibrosis. Now, after the initiation of surfactant therapy, what is called new BPD is most often a form of arrested lung development.69 The characteristic pathologic changes seen in new BPD are fewer and larger alveoli with less functional surface area, and reduced and dysplastic capillary ingrowth to the alveolar region. There may be accompanying pulmonary hypertensive changes, interstitial fibrosis, and smooth muscle hyperplasia, but certainly to a much lesser degree than that associated with classic BPD. Airway epithelial lesions are negligible. The pathophysiology of BPD is diagrammed in Figure 34-13.
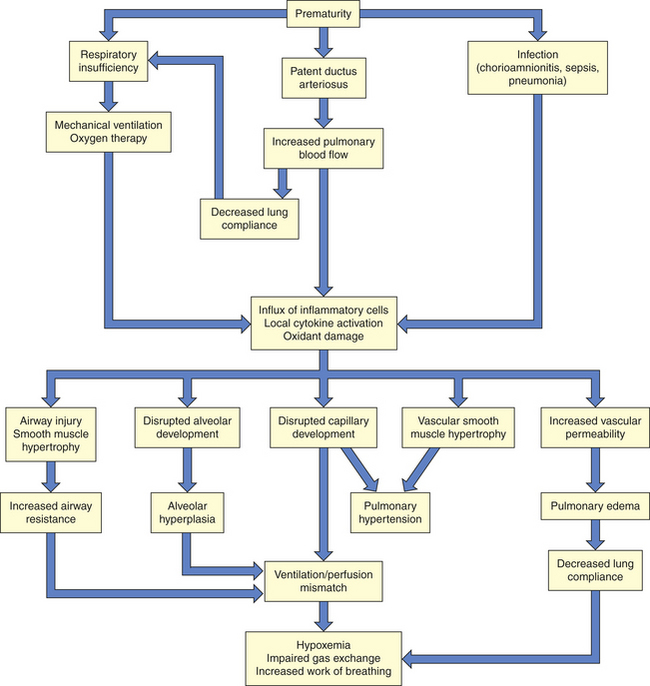
Genetic susceptibility and hereditary influences on gene expression that are pivotal for surfactant synthesis, vascular development, and inflammatory regulation have been documented as being associated with new BPD.66 To a significant extent, cytokines may mediate the abnormal alveolarization and injury response that lead to BPD, although this has not been directly proven. In the case of intrauterine infection, inflammatory mediators may prime the lung for an exaggerated path of injury after birth. Proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), IL-6, and IL-8, are elevated in the amniotic fluid or tracheal aspirates of preterm infants who later develop BPD. Interestingly, the predominant mediators of new BPD are profibrotic and angiogenic cytokines rather than proinflammatory cytokines.70 Antenatal and postnatal exposures also play into the progression of this “developmental disorder.” In the antenatal period, the use of steroids, chorioamnionitis, or the presence of intrauterine growth restriction influences the development and progression of BPD. Postnatally, administration of steroids, nutritional issues, as well as mechanical ventilator damage and pulmonary edema, influence progression of BPD.
Ventilation-perfusion matching is compromised as a result of structural underdevelopment, pulmonary hypertension, increased lung fluid content, airway injury, and smooth muscle hypertrophy, as well as adverse chest wall dynamics. Thus infants with BPD exhibit an increased oxygen requirement, increased work of breathing, and in the most severe cases, right-sided heart failure.
CLINICAL MANIFESTATIONS The current clinical definition of BPD includes the need for supplemental oxygen at 36 weeks’ postmenstrual age, and for at least 28 days after birth, with a graded severity dependent on required respiratory support at term (divided into mild, moderate, and severe based on oxygen requirements and ventilatory needs). The affected infant exhibits hypoxemia caused by ventilation-perfusion mismatch and diffusion defects. Work of breathing is elevated, resulting in hypercapnia. The ability to feed may be impaired. Intermittent bronchospasm with wheezing, mucous plugging, and pulmonary hypertension characterize the clinical course of the most severely affected babies. Dusky spells may occur with agitation or reflux because of several contributing factors, including nonhomogeneous ventilation, air trapping, bronchospasm, laryngospasm, sudden increases in pulmonary vascular resistance, or occasionally, pneumothorax.
Infants with severe BPD require prolonged, assisted ventilation with cautious weaning. Prevention of lung damage with “gentle ventilation” (see lung protective strategies of ventilation in RDS section, p. 1335) or early nasal CPAP, or both, are used in clinical situations when permitted. Use of CPAP has resulted in fewer days of oxygen and ventilator requirement by reducing the amount of lung injury as compared with mechanical ventilation.71,72 Additionally, oxygen supplementation at lower than previously accepted values (89% to 94% saturations) is a means to reduce oxidant injury to the lungs and retinal vasculature.71 Inhaled nitric oxide is also a treatment option in some circumstances and has been associated with improved neurologic outcomes in preterm infants; however, routine use of this therapy needs further follow-up for long-term effects on the pulmonary and developmental systems.73–75 Diuretics are used to control pulmonary edema. Bronchodilators are used to reduce airway resistance. Early anti-inflammatory therapies, such as steroids, may facilitate weaning but introduce significant risks, such as abnormal neurodevelopment. The current recommendation is to avoid using steroids when clinical situations permit until further studies can identify which individuals are most likely to benefit without serious steroid-associated complications.75,76 Azithromycin also may have a role in controlling the inflammatory portion of this disease process as it does in other pulmonary diseases; however, research in this area is ongoing.77 Caffeine citrate is commonly used and has been associated with decreased rates of BPD. Other therapies in the early stages of investigation include targeted cytokine and anticytokine therapies, antioxidants, and antiproteinases. Infection is a constant threat because of invasive lines, the endotracheal tube, and a compromised immune system. Nutritional needs are high and must be met to promote growth and healing. Most infants can be fed enterally. Early supplemental vitamin A and/or amino acids, which play a role in normal lung development, may be required in low-birth-weight infants and have resulted in as much as a 12% reduction in development of BPD.71,75,78
Death from BPD is usually caused by infection or respiratory failure. Recurrent cough and wheezing are frequent in survivors with BPD, as well as a high susceptibility to repeated respiratory infections and their complications. Measures to prevent viral infections and avoid environmental exposures are critical to the care and treatment of these children. Infants who survive are often discharged with home oxygen therapy (some on ventilators). In addition to respiratory management, good nutrition is essential to recovery. Gradual improvement is usually noted in the first 2 years, but pulmonary function may remain abnormal for many years, and there is an increased incidence of asthma during childhood. Characteristic abnormalities on pulmonary function testing include expiratory airflow obstruction and air trapping. For some children, improvement is seen from school age onward, with gradually diminishing clinical symptoms and improved pulmonary function.66
Respiratory Infections
Infections may be localized to the bronchioles and bronchi, alveoli, interstitium, or pleura. The cause and site of infections are related to the age of the child, seasonal variables, and environmental exposures. Infants and young children tend to have more viral infections, especially during late autumn to early spring. Environmental factors may play a role such as the presence of siblings and daycare exposure.
Bronchiolitis: Bronchiolitis is a common viral-induced lower respiratory tract infection that occurs almost exclusively in infants and young toddlers. The most common associated pathogen is RSV, but it also may be associated with adenovirus, rhinovirus, influenza, parainfluenza virus (PIV), and M. pneumoniae.79 RSV infects nearly 100% of children in the United States by 2 to 3 years of age, with the peak incidence between 2 and 6 months.79 Bronchiolitis cases in the United States and Europe average 30 per 1000 for children younger than 1 year of age.80 Bronchiolitis has a peak incidence during winter (late December), a spike in February, and then tapers off in the spring, paralleling the RSV season. There are distinct regional differences in the United States depending on variation in the viral season based on geography. Other viruses, especially human metapneumovirus (hMPV), also follow a seasonal pattern and result in coinfection with RSV.80 It is a major reason for hospital admission of children younger than 1 year, particularly children of lower socioeconomic status. Healthy infants usually make a full recovery from RSV bronchiolitis, but infants who are premature or who have underlying lung disease, heart disease, or immune deficiency may have a much more severe or even deadly course. Certain types of PIV and adenovirus are associated with more severe disease that can progress to bronchiolitis obliterans. Nonetheless, mortality rates are low at 2 per 100,000 live births.80
PATHOPHYSIOLOGY Viral infection causes necrosis of the bronchial epithelium and destruction of ciliated epithelial cells. There is infiltration with lymphocytes around the bronchioles and a cell-mediated hypersensitivity to viral antigens with release of lymphokines causing inflammation, as well as activation of eosinophils, neutrophils, and monocytes. The inflammatory process extends from the respiratory tract to the eustachian tubes and the middle ear. The submucosa becomes edematous, and cellular debris and fibrin form plugs within the bronchioles. Edema of the bronchiolar wall, accumulation of mucus and cellular debris, and possibly bronchospasm narrow or occlude many peripheral airways. Resultant uneven ventilation and atelectasis lead to perfusion mismatch and hypoxemia.
The mechanics of breathing are disrupted by bronchiolitis. Airway narrowing causes obstruction of airflow that is worse with expiration. This leads to air trapping, hyperinflation, and an increase in FRC. Airway resistance and hyperinflation result in a decrease in lung compliance and an increased work of breathing. This increased work of breathing leads to a decrease in alveolar ventilation with resultant hypercapnia.
CLINICAL MANIFESTATIONS Children with bronchiolitis generally have several of the following signs and symptoms, although there is no standard definition of the condition. They may have tachypnea, expiratory wheezing, cough, rhinorrhea, mild fever, and varying grades of respiratory distress. Mild conjunctivitis occurs in up to 33% of cases, and otitis media occurs in 16% to 50%.79,80 Infants may have difficulty with apnea (8% to 20%). Chest radiographs often reveal hyperexpanded lungs, patchy or peribronchial infiltrates, and atelectasis. Severely affected infants appear anxious and distressed because of dyspnea or hypoxemia. The thoracic cage is overexpanded, particularly in its anteroposterior diameter. The infant takes rapid, short breaths, and wheezing and rales are often heard on auscultation. With overexpansion of the lungs, the diaphragm is flattened, causing downward displacement of the liver and spleen. Abdominal distention results from air swallowing. Some individuals have persistent high airway resistance and airway hyperresponsiveness, including increased risk for asthma, long after resolution of the viral process.81 Genetic tendencies have been noted that correlate RSV bronchiolitis and long-term pulmonary sequelae.77
EVALUATION AND TREATMENT Diagnosis is made by review of signs and symptoms (e.g., rhinitis, cough, wheezing, crackles, chest retractions and/or hyperinflation, tachypnea) and radiologic examination. Nasal washings/swabbings may be tested for specific viral agents, such as RSV. RSV swabs are positive in 70% of cases of bronchiolitis. Routine chest films have fallen out of favor because they often reveal nonspecific findings (hyperinflation and patchy atelectasis) and are associated with an increase in antibiotic use that is unwarranted.82 Treatment is determined by the severity of the disease and age of the child. Infants younger than 1 year are most at risk for acute respiratory failure and may require assisted ventilation. Supplemental oxygen is given as needed, and adequate hydration should be maintained. The use of nasal CPAP and heliox (mixture of helium and oxygen) is being explored, and studies to prove effectiveness are ongoing.83 Bronchodilators have not been scientifically validated as consistently providing significant benefit, but are widely tried on an empiric basis. Likewise, steroids are not of proven benefit but have been associated with small decreases in length of stay and improved symptoms in some cases.82,84 Racemic epinephrine has shown promising results in small subsets of children.82 Antiviral agents (ribavirin) for RSV are no longer widely used because of high cost and unclear efficacy; however, several new antiviral agents are being investigated for treatment and prophylaxis. Prophylactic treatment with RSV-specific monoclonal antibody is recommended for high-risk infants younger than 2 years old, although high cost is sometimes a barrier.85 For those requiring hospitalization, length of stay is generally 3 to 4 days.
Pneumonia: Pneumonia is a process that results from infection and resultant inflammation in the terminal airways and alveoli. Community-acquired pneumonia (CAP) is one of the most common global infections in the pediatric age group, as well as one of the leading causes of hospital admission. Identification of the etiologic agent is often challenging because the range of pathogens is quite large.86 The most common agents are viral, followed by bacteria and atypical microorganisms. The incidence of viral and bacterial pneumonia varies according to age, time of year, and geographic location. In children, fungal and anaerobic pneumonias are rare, and opportunistic infections occur only in the immunocompromised child (these unusual forms of pneumonia are not discussed further in this chapter).
The incidence of CAP in the developed world is 21 to 36 per 1000 with 40% of these cases requiring hospitalization.87 Pneumonia is most common in children younger than the age of 2, with the highest frequency between 6 and 12 months.88 Risk factors for bacterial and viral pneumonia include age younger than 2 years, overcrowded living conditions, winter season, recent antibiotic treatment, attendance at daycare centers, and passive smoke exposure. Nutritional status, age, and underlying disease process influence morbidity and mortality rates related to CAP.
PATHOPHYSIOLOGY Bacterial pneumonia in young children beyond the neonatal period is most commonly the result of infection with streptococcal and staphylococcal microorganisms (Table 34-2). S. pneumoniae (pneumococcal) pneumonia is the most common causative microorganism and manifests acutely and with variable severity. Pneumococci and many of the other bacteria that commonly cause pneumonia have specific virulence factors (such as capsules) that increase their survival and proliferation while causing insult to the host.89 Infection usually begins with inhalation of microbes dispersed in ambient air or in secretion droplets (person-to-person spread) or by aspiration of one’s own nasopharyngeal bacteria into the trachea. A preceding viral infection sometimes sets the stage for bacterial infection by causing epithelial damage and reduced mucociliary clearance in the trachea and major bronchi. Colonization of the trachea then ensues, with the microorganism, host, and environment all playing a role in the development of pneumonia. Once in the alveolar region, bacteria encounter local host defenses, such as antibodies, complement, phagocytes, and cytokines, that prepare bacteria for ingestion by alveolar macrophages. If these mechanisms fail, neutrophils will be recruited and an intense cytokine-mediated inflammation will ensue. Vascular engorgement, edema, and a fibrinopurulent exudate with tissue damage occur. Alveolar filling precludes gas exchange and, if extensive, can lead to respiratory failure. If sepsis occurs at the same time, shock and end-organ hypoperfusion may develop. Staphylococcal and group A streptococcal pneumonia can be particularly fulminant and necrotizing, with a high incidence of accompanying empyema, pneumatoceles, and sepsis. Empyema is increasingly associated with pneumococcal infections and with MRSA.90,91
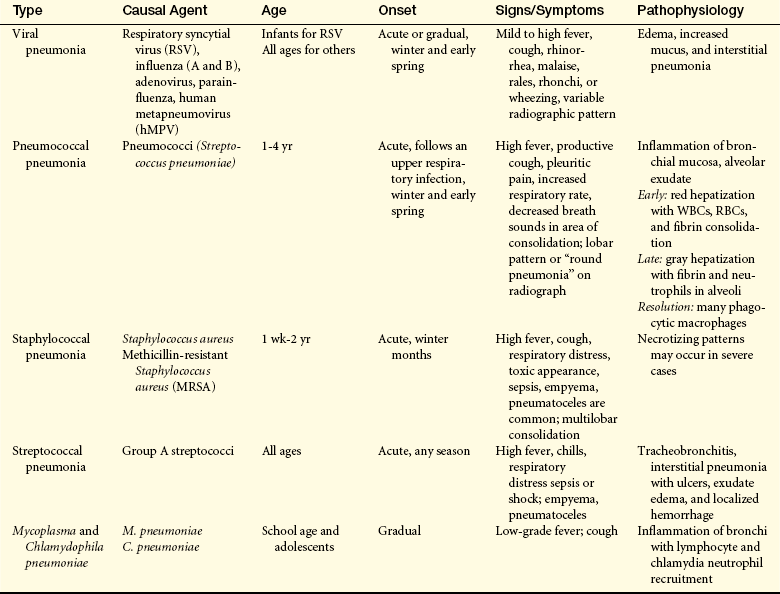
The clinical presentation of bacterial pneumonia, particularly pneumococcal, may include a preceding viral illness followed by fever with chills and rigors, shortness of breath, and an increasingly productive cough. Occasionally there is blood streaking of the sputum. Respiratory rate and oxygen saturation also are important clinical indicators. Auscultation usually reveals such abnormalities as crackles or decreased breath sounds. Other less specific findings may include malaise, emesis, abdominal pain, and chest pain. Chest film will usually present with a lobar pattern in older children and adolescents but may appear patchier with a bronchopneumonic pattern in younger children.88
Viral pneumonia is more common than bacterial pneumonia and is acquired by direct contact, droplet transmission, or aerosol. Children are two to three times more likely than adults to become infected with respiratory viruses, with the majority occurring in otherwise healthy children. Although mortality from respiratory viruses in developed countries is rare, they are a major source of morbidity. In less developed countries viral pneumonia results in nearly 5 million deaths in children younger than the age of 5 each year.79 Viral infections often occur in epidemics, whereas others occur endemically; however, most tend to follow a seasonal pattern. The most common cause of viral pneumonia in infants and young children is RSV,92 occurring most often in winter to early spring. A number of other viruses are important, including parainfluenza, influenza A and B, coronaviruses, rhinoviruses, enteroviruses, human metapneumovirus (hMPV), bocavirus, and adenoviruses.
Viral infection of the lower respiratory tract results in destruction of ciliated epithelium of the distal airway, with sloughing of cellular material. A mononuclear-predominant inflammatory response occurs first in the interstitium and may later involve the alveoli. Certain serotypes of adenovirus can cause necrotizing disease, sometimes leading to obliterative bronchiolitis and significant lung disability.
Early in the course of illness, it is often difficult to determine whether the pneumonia is of viral or bacterial origin. Differences that may be noted are elevated temperatures, absolute neutrophil counts, and percent of bands are consistently higher in bacterial pneumonias than with those of viral etiology.93 Diagnosis of a viral etiology requires laboratory confirmation (immunofluorescence tests). Development of safe agents to treat viral pneumonias continues to be a priority, as is development of more effective vaccines.79
Atypical pneumonia Chlamydophila pneumoniae (previously Chlamydia pneumoniae) is clinically indistinguishable from, and is typically grouped with, M. pneumoniae as “atypical” pneumonia.94,95 These microorganisms are the most common cause of CAP for school-age children (ages 5 and older) and young adults, accounting for nearly one fourth of all cases of bacterial pneumonia.89 Mycoplasma is known to cause a wide spectrum of disease and has more extensive complications than previously recognized. Studies reveal that it is seen increasingly in infants and younger children.96,97 Children experiencing recurrent respiratory tract infections often have been found to be infected with atypical bacteria.
Transmission of atypical microorganisms is person to person, with a 2- to 3-week incubation period. Mycoplasma microorganisms lack cell walls but have a limiting membrane and a specialized tip for attaching to ciliated respiratory epithelial cells. Local sloughing of cells occurs. Peribronchial lymphocytic infiltration develops, along with neutrophil recruitment to the airway lumen. The pattern resembles bronchitis or bronchopneumonia.
Onset of symptoms is usually gradual, resembling a typical upper respiratory infection with low-grade fever and prominent cough. There may be accompanying sore throat, myalgia, and headache. Cases are not usually clinically severe, and full recovery should be expected without complications. When complications do occur, they can include bronchopneumonia, parapneumonic effusions, and necrotizing pneumonitis.96
EVALUATION AND TREATMENT Diagnosis of pneumonia is based on clinical, laboratory and chest radiograph findings. Guidelines have been developed to improve and aid assessment and management, although consensus has not been reached in their clinical application. Identifying pathogens is very difficult in children, especially since there is often overlap between bacterial and viral pathogens. Some newer studies are recommending use of highly sensitive C-reactive protein (hs-CRP) as a tool to help discern between viral and bacterial pneumonias. hs-CRP in combination with clinical signs and symptoms and chest film may correlate to help the clinician more accurately diagnose the etiology of the pneumonia.86 Other laboratory tests that may be helpful include a white cell–granulocyte count, procalcitonin, or erythrocyte sedimentation rate (ESR) but they do not indicate a specific etiology. Several microbiologic tests are available including polymerase chain reaction (PCR) and nucleic acid amplification tests (NAAT). On chest radiography, a bacterial pneumonia initially produces an alveolar infiltrate and later causes a segmental or lobar disease. A viral infection is more likely to be associated with an interstitial pattern.
Most pneumonias may be treated on an outpatient basis; however, some children require oxygen supplementation and, occasionally, assisted ventilation. This is particularly true with infants who have a viral interstitial pneumonia, such as RSV. In addition, adequate hydration, nutrition, and supportive pulmonary therapy are required to reduce the duration and severity of illness. Many hospitalized infants are markedly tachypneic and unable to coordinate their breathing with swallowing such that they may require enteral feeding. Aspiration is always a risk with infants in respiratory distress.
Appropriate antibiotic administration, whether oral or intravenous (IV), for bacterial pneumonias is usually instituted for a minimum of 10 days, although studies suggest that shorter courses of antibiotics may be adequate in children with mild to moderate to severe cases.98 Local patterns of drug resistance must be considered as there is 20% to 40% pneumococcal resistance to penicillin and up to 40% resistance for macrolides in the United States. New antibacterials are under development for treatment of antibiotic-resistant pathogens.96,99 Use of the heptavalent pneumococcal conjugate vaccine has led to a decrease in invasive infections.100
Aspiration Pneumonitis
Aspiration pneumonitis is caused by a foreign substance, such as food material, secretions (e.g., saliva or gastric contents), or chemical compounds entering the lung and causing inflammation. The aspiration of meconium from amniotic fluid can occur at birth. Meconium contains bile salts from the fetal intestinal tract that cause inflammation. Neurologically compromised children or children undergoing sedation or anesthesia may aspirate oral secretions (containing anaerobic bacteria) or stomach contents. Children with neurologic compromise, other underlying diseases such as chronic lung disease/BPD, anatomic tracheoesophageal, or craniofacial abnormalities may present with chronic pulmonary aspiration (CPA). Left unaddressed, CPA can result in progressive lung disease, bronchiectasis, and respiratory failure and is the leading cause of death in children with severe neurologic compromise.101 The severity of lung injury after an acute aspiration incident is determined by the amount of material aspirated, the pH of the aspirated material, and the presence of pathogenic bacteria. Very low pH or very high pH causes a significant inflammatory response. With hydrocarbon ingestions, lung injury is determined by the volatility and viscosity of the aspirated substance. A low-viscosity substance, such as gasoline or lighter fluid, is the most toxic; high-viscosity hydrocarbons, such as petroleum jelly or mineral oil, are much less likely to cause pneumonitis. Treatment for aspiration pneumonitis depends on the material aspirated but generally includes broad-spectrum antibiotic therapy. Strategies for prevention of aspiration are an important part of the therapeutic plan at every well-child visit.
Bronchiolitis Obliterans
Bronchiolitis obliterans (BO), a relatively rare diagnosis in children, is fibrotic obstruction of the respiratory bronchioles and alveolar ducts secondary to intense inflammation. The inflammation leads to narrowing, complete obliteration, or both of the airway lumen. Pathologically, there are two forms: proliferative and constrictive; the latter results in the obliteration of the lumen and is the more common form. Most cases of bronchiolitis obliterans in children are associated with viral pulmonary infections, such as influenza, adenoviral infection, pertussis (whooping cough), measles, parainfluenza, RSV, human immunodeficiency virus (HIV) or M. pneumoniae. BO may occur after allograft transplantation (lung, heart-lung, and bone marrow) as a manifestation of graft-versus-host disease. It also is associated with collagen vascular disease, toxic fume inhalation, chronic hypersensitivity pneumonitis, Crohn disease, and Stevens-Johnson syndrome.102,103 For reasons that are unclear, BO seems to occur more frequently in the Southern Hemisphere, although it is certainly noted in other parts of the world as well. Genetic factors influence susceptibility; however, these factors have yet to be clarified.103 Initially cough, respiratory distress, and cyanosis occur, followed by a brief period of improvement. The progression of disease is then reflected by tachypnea, sputum production, increased anterior/posterior diameter, crackles, wheezing, and hypoxemia.104
There is no specific treatment for BO. Supportive care is usually given with mechanical ventilation, although there is evidence that this also may contribute to the progression of the disease. Use of antiviral agents may be warranted in managing those with viral infection. For transplant recipients, augmentation of immunosuppressive therapies and treatment with anti-inflammatory agents are showing promise in reducing airway inflammation, thus improving pulmonary function.105 Clinical progression can be quite variable depending on the predisposing condition. Some children experience partial recovery, whereas others follow a course of steady decline in lung function.
Asthma
Asthma is an obstructive airway disease characterized by reversible airflow obstruction, bronchial hyperreactivity, and inflammation. The onset of asthma generally occurs early in life and is associated with identifiable risk factors.106 The prevalence of asthma is significantly higher in children than adults and is the most prevalent chronic disease in childhood, affecting 5% to 13% of all children. In the prepubertal years, more boys than girls are affected. Although statistics suggest that asthma has become more prevalent in the past two decades, some of this increase may be related to improved awareness of the diagnosis of the condition.107 Populations most affected include those living in an urban setting, ethnic minorities, and those of low-socioeconomic status.108 There also may be associations between those children living with a single mother and having multiple siblings that result in reduction in treatment and worsened outcomes from asthma.109 Asthma-related deaths almost always occur outside the hospital setting. The severity and persistence of asthma are influenced by age at disease onset, genetics, behavior, atopy, air pollution, level of allergen exposure, environmental tobacco smoke, gastroesophageal reflux, and respiratory infections.106 Additional confounding variables that affect disparity in asthma morbidity and therapy are lack of health insurance, poor access to asthma specialists, inappropriate utilization of healthcare resources, and inadequate medical care.108 Inner-city black and Hispanic children have higher morbidity and mortality rates than white children.108 After decades of rising mortality rates, there has been a decrease in the number of children dying of this disease in recent years.110
The wide spectrum of clinical disease in asthma probably reflects a complex interaction between genetic susceptibility and environmental factors, including allergens (e.g., air pollution, dust mite and cockroach allergen, tobacco smoke) and infections, particularly viral respiratory infections (e.g., rhinovirus).108,111 The genetics of asthma are complex. Population genomic screening has led to the identification of many candidate genes or chromosomal regions that are associated with asthma. Included in the long list of asthma-associated genes are those that code for increased levels of immune and inflammatory mediators (e.g., IL-4, IgE, and leukotrienes), nitric oxide, and transmembrane proteins in the endoplasmic reticulum.111,112 In addition, genes may impart associated phenotypes, such as bronchial hyperresponsiveness (BHR), sensitization to allergens, and responsiveness to asthma therapies. For example, a specific polymorphism in the gene for the β-adrenergic receptor that is prevalent in black children is associated with poor response to β-adrenergic inhaled medication.108
Environmental exposures such as allergens, viruses, smoke, and air pollutants interact with an individual’s genetic vulnerabilities to induce the onset of clinical asthma.106,111,113,114 One hypothesis for the high prevalence of asthma in westernized cultures is called the “hygiene hypothesis.” This theory suggests that certain early childhood respiratory viral infections could favor the Th2-predominant phenotype and contribute to inducing asthma in susceptible individuals. It also suggests that lack of sufficient early exposure to other types of infections (e.g., hepatitis) also favors the Th2 phenotype in the airways and permits induction of asthma.115 Asthma develops because the Th2 response (in which CD4 T-helper [Th] cells produce specific cytokines, such as IL-4, IL-5, and IL-13) promotes an atopic/allergic response in the airways as opposed to a Th1 response characteristic of delayed-type hypersensitivity and phagocyte-mediated host defense. IL-4 and IL-13 are particularly important for B-cell switching to favor IgE production, and IL-5 is crucial for local differentiation and enhanced survival of eosinophils within the airways. This theory about the origins of asthma is consistent with many of the epidemiologic and pathophysiologic features of childhood asthma, but is not yet proven.
PATHOPHYSIOLOGY The pathophysiology of asthma in children is similar to that for adults (see Chapter 33, Figure 33-11, p. 1285). However, good criteria for predicting adult asthma in persons with childhood asthma remains unclear.116,117 For acute allergen-induced asthma, the paradigm of the early asthmatic response remains useful (Figure 34-14, A and 34-15). This begins immediately after exposure and lasts up to 2 hours. The allergen binds to preformed IgE on the surface of mucosal mast cells, and crosslinking of these IgE molecules triggers degranulation of the mast cell, releasing mediators such as histamine, leukotrienes, prostaglandins, platelet-activating factor, chemotactic chemokines, and certain cytokines (e.g., IL-1).106 These mediators cause airway smooth muscle constriction (bronchospasm), increased vascular permeability (mucosal edema), and mucus secretion. The late asthmatic response starts 4 to 8 hours after exposure and may persist up to 24 hours (Figure 34-14, B). The response is characterized by inflammatory cell recruitment (neutrophils, eosinophils, basophils, and T lymphocytes) that was triggered earlier by chemotactic factors and up-regulation of endothelial adhesion molecules. Another wave of mediator release occurs, again inciting bronchospasm, edema, and mucus secretion. Epithelial damage and impaired mucociliary function may be seen because of direct toxic effects of cellular products such as major basic protein from eosinophils. This local injury stimulates local nerve endings, which may aggravate bronchoconstriction and mucus secretion through autonomic pathways (Figure 34-15).
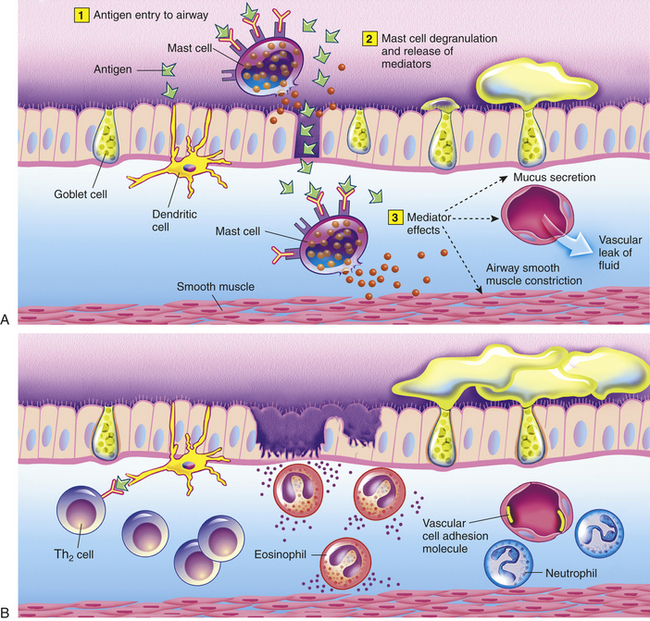
Figure 34-14 Asthmatic responses. A, In the early asthmatic response, inhaled antigen (1) binds to preformed IgE on mast cells. Mast cells degranulate (2) and release mediators such as histamine, leukotrienes, prostaglandin D2, platelet-activating factor, and others. Acute inflammation opens intercellular tight junctions, allowing antigen to penetrate and activate submucosal mast cells. Secreted mediators (3) induce active bronchospasm, edema, and mucus secretion. Inflammatory responses are set in motion by chemotactic factors and upregulation of adhesion molecules (not shown). At the same time, as shown on the left, antigen may be received by dendritic cells that process and later present it, either in regional lymph nodes to naive (Tho) T lymphocytes or locally to memory Th2 cells in the airway mucosa (see B). B, In the late asthmatic response are areas of epithelial damage caused at least in part by toxicity of eosinophil products (major basic protein, eosinophilic cationic protein, eosinophil-derived neurotoxin, and eosinophil peroxidase). Many inflammatory cells have been recruited by chemokines and upregulation of vascular cell adhesion molecules. Local T lymphocytes display a predominant Th2 cytokine profile. They produce interleukin-4 (IL-4) and IL-13, which promote switching of B cells to favor immunoglobulin E (IgE) production, and IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor, which encourage eosinophil differentiation and survival.
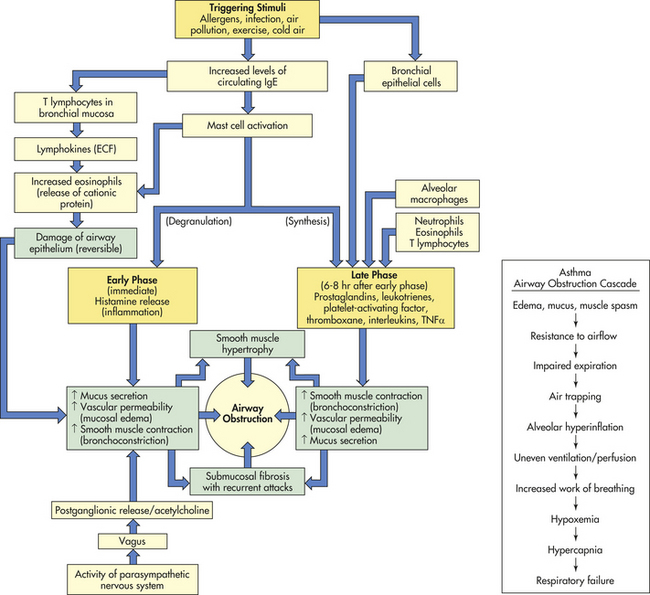
Figure 34-15 Pathophysiology of asthma. ECF, Eosinophil chemotactic factor; IgE, immunoglobulin E; TNF, tumor necrosis factor.
Although allergen-associated immune and inflammatory processes are considered the primary etiologic processes in asthma, neutrophilic airway inflammation likely accounts for a portion of asthma cases. Those individuals with a predominance of inflammatory neutrophils (rather than eosinophils) have allergies less often and are less responsive to steroids.106,111 As the inflammatory process continues, bronchospasm develops which narrows the airways especially during expiration. Mucous plugging, edema, and cellular infiltration lead to further airway narrowing. A partial obstruction is present that creates a “ball-valve” effect, leading to segmental hyperinflation, which may become extreme and compromise effective tidal volume. Measures of expiratory flow rates, such as forced expiratory volume in 1 second (FEV1) and peak flow, are markedly reduced.
Examination of postmortem lung specimens of individuals who died from asthma reveals abnormalities consistent with acute and chronic changes in the airways. These include extensive mucous plugging, mucosal edema, and denudation of bronchial and bronchiolar epithelium. Eosinophilia is present in the submucosa, and a multicellular inflammatory infiltrate accumulates in the airways. Thickening of the basement membrane, airway smooth muscle hypertrophy, and mucous gland hypertrophy are often noted. In chronic asthma, chronically increased numbers of inflammatory cells may lead to long-term changes, such as goblet cell hyperplasia and airway wall remodeling (subepithelial fibrosis, smooth muscle hypertrophy).
The typical arterial blood gas abnormalities in acute asthma are hypoxemia, hypocarbia, and respiratory alkalosis. Because bronchial obstruction is nonuniform, ventilation is likewise uneven, causing ventilation mismatch and hypoxemia. The degree of hypoxemia is usually mild, however, and arterial saturations of less than 90% indicate severe airway obstruction. Pulmonary circulation may be altered by regional hypoxic vasoconstriction, as well as the effect of increased intra-alveolar pressure (caused by expiratory airway obstruction and alveolar hyperinflation) to decrease perfusion of alveolar capillaries. Typically, respiratory rate is elevated to compensate for hypoxemia and reduces arterial PCO2 (respiratory alkalosis). If airway resistance and hyperinflation become severe, increased respiratory rate can no longer compensate for decreasing VT causing a reduction in minute ventilation and a gradual rise in arterial PCO2, thus even a normal PaCO2 value should be of concern if respiratory distress is significant. Retention of CO2 is a late finding, usually occurring only if FEV1 falls to around 15% to 20% of predicted values, and reflects inadequate alveolar ventilation and increased functional dead space. With severe and prolonged airway obstruction (status asthmaticus), the end result of the pathophysiologic processes may be respiratory failure with acute CO2 retention and respiratory acidosis. Metabolic acidosis may accompany life-threatening asthma, especially when left ventricular filling and thus cardiac output become compromised because of severe hyperinflation.
CLINICAL MANIFESTATIONS In a typical acute asthma attack, the major complaints are cough, wheeze, and shortness of breath. Signs of a preceding upper respiratory infection, such as rhinorrhea or low-grade fever, may be present. In children, 70% to 80% of acute wheezing episodes are associated with viral respiratory infections. In infants and toddlers younger than 2 years old, the most common of these is RSV. In older children and adults, the major viral trigger is rhinovirus. In fact, rhinovirus is reportedly associated with 60% to 80% of asthma exacerbations in school-age children.118
On physical examination, expiratory wheezing that is often described as high pitched and musical is found, along with prolongation of the expiratory phase of the respiratory cycle. Sometimes hyperinflation is visible. Respiratory rate is elevated, as is heart rate. Nasal flaring and accessory muscle use are evident, with retractions in the substernal, subcostal, intercostal, suprasternal, or sternocleidomastoid areas. Infants may appear to be “head bobbing” because of sternocleidomastoid muscle use. Pulsus paradoxus (decrease in systolic blood pressure of more than 10mmHg during inspiration) may be present. The child may appear anxious or diaphoretic, important signs of respiratory compromise.
Findings in chronic asthma may include hyperinflation of the thorax (barrel chest) or pectus excavatum. Clubbing should not be seen in those with asthma and, if present, should trigger evaluation for other conditions, such as cystic fibrosis.
EVALUATION AND TREATMENT Asthma is often underdiagnosed and untreated, especially in preschool-age children. The fact that many of the symptoms overlap with other respiratory illness, like bronchitis or upper respiratory infections, may confuse the diagnosis. Because of the changing appearance of asthma in some children, tools to help confirm diagnosis are recommended. The modified asthma predictive index (API) is recommended by the National Institutes of Health (NIH) guidelines. Diagnosis is based on episodes of wheezing noted on an annual basis as well as concurrent risk factors. The major historical and physical factors that contribute to the diagnosis of asthma are parental history of asthma, physician-diagnosed atopic dermatitis, and evidence of sensitization to aeroallergens. Minor factors include evidence of sensitization to foods, 4% or greater peripheral blood eosinophilia, or wheezing not associated with upper respiratory illnesses.106 To firmly establish the diagnosis, objective information is gathered by medical history, physical examination, and, when the child is 5 or older, pulmonary function testing (spirometry).
Characteristic abnormalities of spirometry would be reduced expiratory flow rates, namely FEV1 and to an even greater extent, the midexpiratory flow rate (or forced expiratory flow rate between 25% and 75% [FEF25%-75%] of total exhaled volume); also the ratio of FEV1 to forced vital capacity (FVC) would be typically decreased. Unlike asthmatic adults, however, asthmatic children often have normal or near-normal spirometry between attacks. Other potentially useful supportive diagnostic findings would include evidence of air trapping on lung volume measurement (by plethysmography), documentation of bronchial hyperreactivity (in response to challenge such as exercise or inhaling methacholine) or increased expiratory flow rates in response to an inhaled bronchodilator. Often it is not feasible to obtain the above tests on children, so in practice an empiric trial of asthma-directed medications is commonly initiated, using clinical symptoms (wheeze, cough, exercise tolerance, handling of respiratory infections, etc.) as a guideline. During a significant asthma attack, individuals may be too dyspneic to perform spirometry, and it may precipitate excessive coughing.
For home management of asthma, peak flowmeters are often used. Peak flow measures are less reliable and less reproducible than those obtained by spirometry but can be helpful. For serial tracking, peak flow measurements should be obtained at consistent times of day because of the natural diurnal variation in peak flow, which is usually lowest at approximately 4 AM and highest at approximately 4 PM. Once a baseline value has been established on the basis of repeated measurements over a period of time, decreases in peak flow can be interpreted meaningfully to help assess the child and modify treatment in the face of increased symptoms or intercurrent illness.
The goal of asthma therapy is to achieve long-term control by reduction in impairment and risk.106 Management of asthma medications in children is often difficult because remission is common. Care providers must periodically assess asthma control in children to decide if a “step up” (increase) or “step down” (decrease) is indicated in their asthma therapy. Algorithms can help assess asthma control and offer information about how to adjust therapy accordingly. Key features to assess include nighttime awakenings, interference with normal activities, use of short-acting beta2 agonist, lung function, and exacerbations requiring oral steroids. Before therapy is augmented, reassessment of inhaler technique, adherence, environmental controls, and comorbidities should occur. For a reduction in therapy, the child’s asthma should be well controlled for a minimum of 3 months.106
For management of intermittent (formerly called mild) asthma, rapid-acting bronchodilators, such as albuterol (a β2-adrenergic agonist) or levalbuterol, may be sufficient with addition of oral systemic steroids for more significant attacks to decrease inflammatory responses in the lung.106 Inhaled ipratropium bromide is an anticholinergic agent that contributes to bronchodilation by inhibiting vagal tone; it is sometimes used together with albuterol for acute treatment or sometimes as an alternative, though generally considered less potent for those who cannot tolerate β2-adrenergic agonists because of side effects. Environmental controls and monitoring would also be included in the treatment plan for children as noted below.
There are a growing number of options for management of persistent asthma depending on chronicity and severity of symptoms, as well as on individual compliance issues. Guidelines have been outlined and widely distributed by an NIH expert panel.106 In addition to medications, environmental controls are instituted. There also is a new focus on child-parent education and monitoring control of symptoms more closely and by a variety of methods. For individuals with persistent symptoms, daily “controller” medication is recommended. The most widely preferred controller therapy remains inhaled corticosteroids (ICS), although there is some debate about their safety in children younger than 2 years of age.119 Montelukast (an oral leukotriene receptor antagonist) is frequently used as supplemental therapy or, for milder allergic or exercise-induced asthma, as monotherapy.120 Inhaled cromolyn and nedocromil remain available anti-inflammatory therapies, but their use has declined in the United States in favor of other therapies. Long-acting β2-adrenergic agonists, such as salmeterol, also may be applied to pediatric asthma except in the youngest children. All classifications of asthma also require intermittent use of a short-acting bronchodilator during times of exacerbation or illness. Depending on the severity of symptoms, short courses of oral systemic corticosteroids also are prescribed. For allergic asthma, immunotherapy has been shown to be an important tool in reducing asthma symptoms and exacerbations, and can now be given sublingually.121 Anti-IgE therapy is indicated for select individuals with severe asthma.122 There also may be utility in using inhaled nitric oxide as an objective measure to determine asthma relapse after cessation of ICS.123 Research in the area of pediatric asthma is targeted at evaluating the effects of vitamin D supplementation on wheezing; the associations between asthma and other underlying illnesses, such as sickle cell disease; the role of intermittent inhaled steroid use; and the effect of functional polymorphisms of antioxidant genes in regard to air pollution (in vitro).111
Regardless of the type of asthma, written asthma action plans and extensive child and family education is necessary for everyone. Asthma management education is necessary for children and their parents to ensure disease control and compliance. Follow-up care is needed every 3 to 6 months to evaluate control and need for change in the treatment plan.
Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) is a condition that can result from either a direct or indirect pulmonary insult. It is defined as respiratory failure of acute onset characterized by severe hypoxemia that is refractory to treatment with supplemental oxygen, bilateral infiltrates on chest x-ray, and no evidence of heart failure.124 Individuals with conditions such as pneumonia, aspiration, near-drowning, and smoke inhalation or a systemic insult such as sepsis, multiple trauma, or burns are susceptible to triggering this process. All of these clinical scenarios activate an inflammatory response that causes alveolocapillary injury. ARDS accounts for approximately 10% of total patient days and one third of all deaths in pediatric intensive care units. The overall mortality in children is less than that experienced by the adult population but can be as high as 40%, depending on age and associated conditions and complications.125 Children younger than 5 years appear to have diminished mortality compared with those older than 5 years of age. Children with underlying disease processes, such as Down syndrome,126 systemic lupus erythematosus, and those who have undergone lung transplantation have decreased survival. Genetic predisposition to developing ARDS has been documented in adults; however, this is less well characterized in the pediatric population.127
PATHOPHYSIOLOGY The hallmark of ARDS is lung inflammation. Destruction of the capillary-alveolar unit leads to activation of a number of systems and mediators (Figure 34-16), including complement, cytokines, arachidonic acid metabolites, platelet-activating factor, reactive oxygen species, and others (specifically TNF, interferon-alpha, and lipopolysaccharide).128 Sources of these mediators include neutrophils, activated platelets, macrophages, and injured endothelium. Early, during the inflammatory or exudative phase of ARDS, there is pulmonary neutrophil influx along with intraluminal fibrin and platelet aggregation. Injury to the endothelial barriers results in capillary leak and noncardiogenic pulmonary edema. The presence of inflammatory mediators and edema fluid in the alveoli inactivates surfactant, contributing further to alveolar collapse.125 This fluid also has procoagulant activity, leading to fibrin clotting within air spaces. Similarly, the pulmonary microcirculation is compromised by the formation of thrombi composed of fibrin, platelets, and leukocytes.
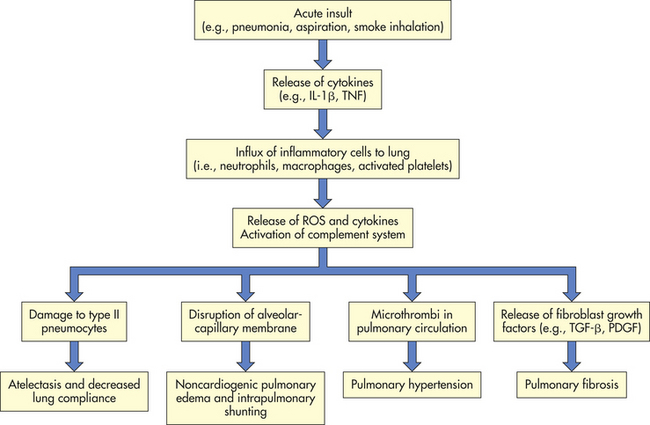
Figure 34-16 Proposed mechanisms for the pathogenesis of acute respiratory distress syndrome (ARDS). IL-1β, Interleukin-1β; PDGF, platelet-derived growth factor; ROS, reactive oxygen species; TGF-β, transforming growth factor-beta; TNF, tumor necrosis factor. (From Soubani AO, Pieroni R: South Med J 92[5]:452, 1999.)
The early accumulation of edema fluid in the air spaces results in decreased lung compliance, decreased functional residual volume, and increased dead space. Ventilation-perfusion mismatching, intrapulmonary shunting, and hypoxemia occur. Diffuse pulmonary thrombosis contributes further to the formation of pulmonary edema by increasing capillary hydrostatic pressure and may lead to pulmonary hypertension.
In the fibroproliferative phase, type II alveolar cells proliferate, and there is alveolar septal thickening and collagen deposition. Interstitial fibrosis can be evident as early as 10 days after the initial insult. Similarly, vascular changes may occur, including obliteration of the microcirculation and thickening of the walls of pulmonary arterioles and arteries, which can lead to chronic pulmonary hypertension in survivors.
CLINICAL MANIFESTATIONS ARDS develops acutely after the initial insult, usually within 24 hours (although occasionally it is delayed up to a few days). There is progressive respiratory distress and severe hypoxemia with poor response to oxygen supplementation. Initially, hyperventilation occurs, but CO2 retention may ultimately develop because of inadequate functional air space, atelectasis, decreased pulmonary compliance, and respiratory muscle fatigue. Severity of the clinical course is modified by comorbid factors, such as the presence of sepsis or multisystem organ failure, and whether complications develop, such as nosocomial pneumonia.
EVALUATION AND TREATMENT Treatment of ARDS remains supportive in nature. Any underlying condition, such as sepsis, must be treated. The goals of therapy are to preserve and restore oxygen delivery, minimize acute lung injury, and decrease mortality by avoiding iatrogenic pulmonary complications. Most individuals with ARDS require mechanical ventilation and often high levels of positive end-expiratory pressure to promote alveolar recruitment, stabilization, and redistribution of alveolar edema fluid into the interstitium. Various ventilation strategies may be used for ARDS, such as low tidal volumes, prone positioning, inverse inspiratory/expiratory ratio, high-frequency oscillatory ventilation, and airway pressure release ventilation.129–131 Liquid ventilation through perfluorocarbons instilled in the lungs is being investigated. These fluids have low surface tension and are efficient carriers of oxygen and CO2. Other therapies, such as surfactant and nitric oxide, offer promise, but their efficacy is still being evaluated in the treatment of pediatric ARDS.130 Surfactant appears to improve oxygenation and increase survival time in some investigations, whereas in others its benefits remain inconsequential.125,132 Systemic corticosteroids have proven effective for decreasing postextubation stridor and may reduce reintubation rates.130,131
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive inherited disorder that is associated with defective epithelial ion transport. CF affects primarily whites (approximately 1 in 3200) and is one of the most common lethal genetic diseases in this ethnic group. Approximately 30,000 individuals in the United States and 70,000 worldwide manifest the disease. The incidence in blacks is 1:15,000 and in Asian Americans is 1:31,000.133 Estimated symptomless carrier frequency is higher affecting 10 million or 1 in 29 whites in the United States. Approximately 1000 new cases of CF are diagnosed each year, and the median age at diagnosis is 6 months; 75% of cases are diagnosed by 1 year of age. Approximately 10% of cases are not diagnosed until after age 10; however, these cases usually have milder symptoms. The median age of survival in the United States is 36.5 years of age (2005 data).133
PATHOPHYSIOLOGY On a simplistic level, CF is characterized by abnormal secretions that cause obstructive problems within the respiratory, digestive, and reproductive tracts. However, research suggests that there may be additional CF-associated primary defects, such as an intrinsic proinflammatory state and abnormal local immune defenses in the lungs. The CF gene (CFTR) has been located on chromosome 7. Its mutation results in the abnormal expression of cystic fibrosis transmembrane conductance regulator (CFTCR) protein, which is a cyclic adenosine monophosphate (cAMP)–activated chloride channel present on the surface of many types of epithelial cells, including those lining airways, bile ducts, pancreas, sweat ducts, and vas deferens. Despite knowing that chloride transport is a fundamental abnormality, the exact disease mechanisms in CF have still not been clearly defined at the cellular and end-organ levels.
Although CF is a multiorgan disease, the lungs are the most critical site of involvement, and respiratory failure is almost always the cause of death. The typical features of CF lung disease are mucous plugging, chronic inflammation, and infection. The abnormalities primarily involve the airways, with progressive bronchiectasis that becomes widespread. Parenchymal involvement occurs much later and includes microabscess formation, patchy consolidation and pneumonia, peribronchial fibrosis, and cyst formation (Figure 34-17). The pathophysiology for these changes is outlined in a simplified form in Figure 34-18. Peripheral bullae may develop because of obstruction and airway wall weakening, and pneumothorax can occur. Hemoptysis, sometimes life threatening, may occur because of erosion of enlarged bronchial arteries that develop in response to the inflammation associated with bronchiectasis. Over a long period of time, pulmonary vascular remodeling occurs because of localized hypoxia and arteriolar vasoconstriction; pulmonary hypertension and cor pulmonale may develop with end-stage disease.
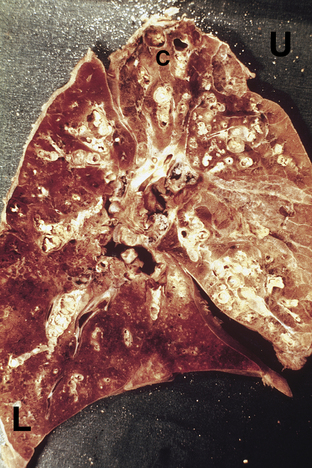
Figure 34-17 Pathology of the lung in end-stage cystic fibrosis. Key features are widespread mucus impaction of airways and bronchiectasis (especially in upper lobe, U), with hemorrhagic pneumonia in the lower lobe (L). Small cysts (C) are present at the apex of the lung. (From Kleinerman J, Vauthy P: Pathology of the lung in cystic fibrosis, Atlanta, 1976, Cystic Fibrosis Foundation.)
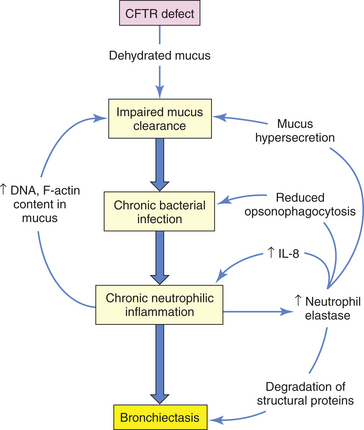
Figure 34-18 Pathogenesis of cystic fibrosis lung disease. CFTR, Cystic fibrosis transmembrane conductance regulator; IL-8, interleukin-8.
The mucus plugging seen in CF probably results from the combination of increased production of mucus, altered physicochemical properties of the mucus, and reduced mucociliary clearance.134 Mucus-secreting airway cells (goblet cells and submucosal glands) are increased in number and size. Dysregulation of the airway epithelial sodium channel (ENaC) causes abnormal chloride secretion and exaggerated sodium absorption, resulting in depletion of the airway surface liquid volume and therefore dehydration of airway mucus.135 This appears to facilitate mucus adherence to the epithelium, subsequent impairment of ciliary mobility, and retention of bacteria that can then form biofilms. Finally, after secretion, CF mucus becomes even more viscous because of deoxyribonucleic acid (DNA) and filamentous (F) actin released from degraded neutrophils, which are present in very high numbers in CF airways.
Chronic, intense neutrophil-dominated inflammation occurs in CF airways, and plays a critical role in long-term damage.136 Abnormal cytokine profiles have been documented in CF airway fluids, including deficient IL-10 and excessive IL-1, IL-8, and TNF-α, all changes conducive to promoting inflammation. Neutrophils are present in great excess in CF airways and release damaging oxidants, such as myeloperoxidase136 and proteases in massive amounts that overwhelm local antiprotease defenses. One protease in particular, neutrophil elastase, has the following detrimental effects: (1) direct damage to lung structural proteins, such as elastin; (2) induction of airway cells to produce IL-8, a strong attractant for neutrophils and thus a means for augmenting a local “vicious cycle” of inflammation; (3) destruction of IgG and complement components important for opsonization and phagocytosis of pathogens; and (4) direct stimulation of mucus secretion by mucus-producing cells. Other contributors to CF pathogenesis are being explored including errant epithelial oxidant defense with exposure to airborne bacteria, major vault protein migration with pseudomonas infection, and antiprotease activity relating to the inflammatory process.137
Children with CF have a propensity for chronic endobronchial infection that remains poorly understood. It is likely that local factors in the CF airway microenvironment favor bacterial colonization because no systemic immune defect has been found. Staphylococcus aureus is common, and Pseudomonas aeruginosa ultimately colonizes airways in 75% of children with CF. Infecting colonies of Pseudomonas appear to adopt a mucoid phenotype and organize themselves into adherent biofilms, making it difficult for antibiotics and local defenses to reach them. Pseudomonas acquisition has been linked with more rapid decline in pulmonary function.138 Persistence of infection incites chronic local inflammation, airway damage, bronchiectasis, microabscess formation, and foci of hemorrhagic pneumonia. New evidence suggests that anaerobes present in the CF lung may contribute to the cycle of infection and ongoing inflammation. The amount of smooth muscle present in the airway in this group of children, increased from normal, is also a contributor to the chronic inflammatory process.139,140
CLINICAL MANIFESTATIONS The most common manifestations are respiratory and gastrointestinal, including the pancreas and biliary tract. Respiratory symptoms at presentation may include persistent (but not necessarily severe) cough or wheeze, sputum production, and recurrent or severe pneumonia. More subtle respiratory tract presentations of CF include chronic sinusitis and nasal polyps. With appropriate treatment, cough, sputum production, and exercise limitation do not usually reach debilitating levels during childhood. Digital clubbing may appear quite early and in the absence of significant pulmonary impairment. Development of barrel chest or persistent crackles occurs much later in the course of the disease.
Classic gastrointestinal manifestations include meconium ileus at birth, which is almost pathognomonic for CF. Approximately 15% to 20% of individuals with CF present with this. Another classic presentation is failure to thrive and malabsorptive symptoms, such as frequent loose and oily stools. Metabolic abnormalities, trace element deficiencies, fat-soluble vitamin alterations, and electrolyte imbalances may occur early in the course of disease.137 Rectal prolapse is an occasional presenting sign that should always prompt testing for CF. About 10% of CF patients do not experience gastrointestinal problems and are termed “pancreatic sufficient.” Several specific CFTR mutations are predictive of this milder phenotype. Males with CF are typically infertile (98%). Other complications of CF may include liver disease (approximately 5%), diabetes mellitus (10% to 25%), and decreased bone mineral density as early as preschool age.137
Overall severity of CF lung disease is highly variable. Even affected siblings may have disparate courses despite identical CFTR mutations, environment, and treatment strategy. We now know that some genetic variants significantly influence survival among children with the same mutations.133 Mutations in the CFTR gene have been classified based on the type of defect. Classes 1 through 3 are associated with more severe disease and 4 and 5 with milder pulmonary disease (generally pancreatic sufficient). Mortality rates correlate respectively with the classes noted above. Researchers continue to explore identification of “gene modifiers,” other than CFTR, that may serve to lessen or aggravate the degree of CF lung disease and increase or decrease the risk of developing other CF complications, such as severe liver disease.141 Other variables that have long been associated with decline in pulmonary status for those with CF and, thus earlier demise, include colonization with P. aeruginosa, female sex, and poor nutritional status.142
EVALUATION AND TREATMENT The standard method of diagnosis has been the sweat test, which will reveal sweat chloride concentration in excess of 60 mEq/L. Genotyping for CFTR mutations is also available as an alternative or supplemental method but may fail to confirm up to 10% of cases because of a lack of ability to screen for every described CF-associated mutation. There are more than 1500 specific mutations, but most standard laboratory panels include fewer than 100 mutations.133 Newborn screening for CF is currently mandated in 32 of the 50 states in the United States and will continue to expand, on a voluntary, state-by-state basis subsequent to recent recommendations developed by the Centers for Disease Control and Prevention (CDC) and an advisory panel of CF experts143 (see What’s New? Newborn Screening for Cystic Fibrosis).
Treatment is primarily focused on pulmonary health and nutrition. Because the pulmonary decline in CF is slow and insidious, and because of the early onset of chronic inflammation and infection, treatment strategies begin immediately at diagnosis and are modified over time as disease progresses. Pulmonary therapies include techniques to promote mucus clearance, such as chest physical therapy and related equipment (such as the high-frequency chest wall oscillation vest) and an assortment of handheld positive expiratory pressure (PEP) devices. Aerosol therapy includes bronchodilators and nebulized deoxyribonuclease (DNase), which acts to liquefy mucus and may even have anti-inflammatory effects.144 Antibiotic practices vary, with prophylactic and treatment strategies being used. Increasing emphasis has been placed on delaying and controlling Pseudomonas colonization, such as the maintenance use of inhaled antibiotics.145 Macrolide antibiotics, such as azithromycin, have been reported to improve pulmonary function through improvement in the airway inflammation process.136 Hypertonic saline also has proven beneficial to improve airway surface water, thus improving mucus viscosity.146 Ibuprofen has been shown to improve lung function in several clinical trials.137,147 Intravenous antibiotics are used to treat major exacerbations of pulmonary infection, which may be either subacute or acute. Individuals with end-stage lung disease may consider double lung transplant as a life-lengthening measure.148
Nutritional problems are extremely common in CF, and poor nutrition is correlated with worse outcomes including progression of lung disease and onset of additional complications such as decreased bone mineral density.149 Elements of aggressive nutritional support include meticulous monitoring of growth parameters, controlling fat malabsorption, ensuring adequate caloric intake, and keeping overall health stable. Approximately 90% of children with CF have pancreatic insufficiency. This is the result of abnormal ion transport causing decreased fluid and bicarbonate secretion from the pancreatic acinar cells, which leads to thickened secretions plugging the smaller pancreatic ducts, and eventual autodigestion or
atrophy of the acinar cells. Therefore, patients must take exogenous pancreatic enzymes with meals and snacks in order to absorb nutrients and control malabsorptive symptoms. Fat-soluble vitamins (A, D, E, and K) must be supplemented. Caloric needs are high, especially with advancing lung disease, and high-calorie supplements or even gastrostomy feeding may be warranted.
Future treatments for CF in trial phases are aimed at improving inhaled antibiotic therapy (duramycin, denufosol and dry powder tobramycin), a vaccine against Pseudomonas, and the use of growth hormone. Gene transfer and stem cell therapies also are being investigated.150
There is a growing contingent of adults with CF living into their 40s and 50s. Care for these individuals shifts away from a pediatric focus because their care needs are unique and often extremely complex. Challenges noted in this specialty area are pregnancy and details about balancing disease management and adult life issues.
SUDDEN INFANT DEATH SYNDROME
Sudden infant death syndrome (SIDS) remains a disease of unknown cause and is the most common cause of unexplained infant death in Western countries.151,152 It is defined as “sudden death of an infant under 1 year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of the clinical history.”152
The incidence of SIDS is low during the first month of life but sharply increases in the second month of life, peaks at 2 to 4 months old, and is unusual after 6 months of age. The incidence of SIDS in the United States is 0.57 per 1000 live births.153 It is more common in male (60%) than female (40%) infants. It almost always occurs during nighttime sleep, when infants are least likely to be observed. A seasonal variation has been noted, with higher frequencies during the winter months. This has been related to a higher rate of respiratory tract infection during those months, and such infections are often reported to have preceded the death, leading to speculation regarding etiology.
Clinical risk groups include babies who were preterm or low birth weight, multiple births, and siblings of prior SIDS victims (fourfold to sixfold increased risk). SIDS is more prevalent among infants of low or adverse socioeconomic status and occurs more frequently when family size is larger.154 Nevertheless, about three quarters of all SIDS victims have no known predisposing clinical risk factor. Maternal factors that predict increased SIDS risk are maternal smoking, young maternal age (younger than 20 years), unmarried mother, less prenatal care, poverty, and illicit drug use or binge-drinking. Risk factors that relate to the baby’s sleeping situation are prone positioning (and to a lesser extent, side sleeping), sleeping on soft bedding, and overheating. Prone sleeping was concluded to be a major and modifiable risk factor. Epidemiologic studies have shown that SIDS rates decreased by 50% to 90% in countries, including the United States, where massive public campaigns warned against prone sleeping for infants.151–153 Other avoidable risk factors include loose bedding materials and sleeping on top of any soft surface (such as sheepskins, quilts, comforters, pillows, adult-type mattresses, or waterbeds). Bed sharing with parents increases risk in some situations.155 Overwrapping the infant or overheating the room also appear to increase risk, particularly if the infant is sleeping prone.
The etiology of SIDS remains unknown, but probably involves a combination of predisposing factors along with external stressors.151,154 There has been long-standing interest in hypotheses involving impaired autonomic regulation and failure of cardiovascular, ventilatory, and arousal responses to hypoxemia or hypercarbia, or to airway obstruction events.156 This blunted responsiveness could be related to developmental immaturity, or may be inducible by external factors such as exposure to maternal smoking or recent infection.157 Infection may be linked to SIDS on the basis of exaggerated inflammation, eosinophil degranulation, and massive cytokine release, causing pulmonary or airway edema in response to either bacterial pathogens from the nasopharynx or viral respiratory tract infections. Finally, there is growing evidence that genetic factors may predispose certain individuals to SIDS.158 A number of candidate gene polymorphisms (16) have been proposed based on epidemiologic evidence, including defects in sodium and potassium cardiac ion channels; 5-HTT, the serotonin transporter gene; and genes linked with the autonomic nervous system.159 The complement components make up the many other gene polymorphisms: C4A and B, IL-6, IL-10, and vascular endothelial growth factor.154,159 One study has also revealed a reduction in surfactant protein A (SP-A) expression in the first months after birth, which correlates with the peak age of SIDS.160 Although still being elucidated, the responsiveness of the immune and inflammatory systems appears to be a causal mechanism to increased rates of SIDS.159
Currently, the best strategy to reduce SIDS is avoidance of all the controllable risk factors, particularly unsafe sleeping practices and maternal smoking. Parents of infants with clinical risk should be taught cardiopulmonary resuscitation as a precaution. Although home monitoring has not been proven to decrease the incidence of SIDS, some at-risk infants may warrant cardiorespiratory monitoring after careful consideration of the individual situation.152 A number of challenges continue to face healthcare providers in continuing to lower the SIDS rate. There is a lack of testing available to identify and stratify the risk of SIDS and no universally accepted or proven intervention. Recent studies have shown that the epidemiology of this health issue is changing and that perhaps combined with the newer knowledge linking genetics to sudden infant death, identified risk factors can be used more effectively.161
Acute epiglottitis 1317
Acute respiratory distress syndrome (ARDS) 1334
Angioedema 1318
Aspiration pneumonitis 1329
Asthma 1330
Atypical pneumonia 1329
Bacterial pneumonia 1327
Bacterial tracheitis 1315
Bronchiolitis 1326
Bronchiolitis obliterans (BO) 1330
Bronchopulmonary dysplasia (BPD) 1323
Community-acquired pneumonia (CAP) 1327
Croup 1316
Cystic fibrosis (CF) 1336
Cystic fibrosis transmembrane conductance regulator (CFTCR) protein 1336
Diphtheria 1316
Foreign body aspiration 1317
Functional residual capacity (FRC) 1312
Hyaline membrane disease (HMD) 1321
Laryngomalacia 1319
Laryngotracheobronchitis 1316
Obstructive sleep apnea syndrome (OSAS) 1320
Peritonsillar abscess 1316
Pneumonia 1327
Respiratory distress syndrome (RDS) of the newborn 1321
Retropharyngeal abscess 1316
Spasmodic croup 1316
Stridor 1314
Subglottic stenosis 1318
Sudden infant death syndrome (SIDS) 1339
Surfactant 1311
Tracheomalacia (tracheobronchomalacia) 1319
Upper airway obstruction (UAO) 1313
Viral pneumonia 1328
REFERENCES
1. Graf, J., Stein, F. Tracheitis in pediatric patients. Sem Pediatric Infect Dis. 2006;17:11–13.
2. Hopkins, A., et al. Changing epidemiology of life-threatening upper airway infections: the reemergence of bacterial tracheitis. Pediatrics. 2006;118(4):1418–1421.
3. Abdel-Haq, N., et al. Retropharyngeal abscess in children: the emerging role of group A beta hemolytic streptococcus. South Med J. 2006;99(9):927–931.
4. Ochoa, T., et al. Community-associated methicillin-resistant Staphylococcus aureus in pediatric patients. Emerg Infect Dis. 2005;11(6):966–968.
5. Ossowski, K., et al. Increased isolation of methicillin-resistant Staphylococcus aureus in pediatric head and neck abscesses. Arch Otolaryngol Head Neck Surg. 2006;132:1176–1181.
6. Hammer, J. Acquired upper airway obstruction. Paediatr Respir Rev. 2004;5(1):25–33.
7. Cabrera, C., et al. Increased incidence of head and neck abscesses in children. Otolaryngol Head Neck Surg. 2007;136:176–181.
8. Brooke, I. Microbiology and management of peritonsillar, retropharyngeal, and parapharyngeal abscesses. J Oral Maxillofac Surg. 2004;62(12):1545–1550.
9. Sobol, S.E., Zapata, S. Epiglottitis and croup. Otolaryngol Clin North Am. 2008;41(3):551–566. [ix].
10. Fitzgerald, D. The assessment and management of croup. Paediatr Respir Rev. 2006;7:73–81.
11. Bjornson, B., et al. A randomized trial of a single dose of oral dexamethasone for mild croup. N Engl J Med. 2004;351:1306–1313.
12. Glynn, F., Fenton, J. Diagnosis and management of supraglottitis (epiglottitis). Curr Infect Dis Rep. 2008;10:200–204.
13. Alcaide, M., Bisno, A. Pharyngitis and epiglottitis. Infect Dis Clin North Am. 2007;21:449–469.
14. Kay, M., Wyllie, R. Pediatric foreign bodies and their management. Curr Gastroenterol Rep. 2005;7(3):212–218.
15. Lea, E., et al. Diagnostic evaluation of foreign body aspiration in children: a prospective study. J Pediatr Surg. 2005;40(7):1122–1127.
16. Chiu, C., et al. Factors predicting early diagnosis of foreign body aspiration in children. Pediatr Emerg Care. 2005;21(3):161–164.
17. Farkas, H. Management of hereditary angioedema in pediatric patients. Pediatrics. 2007;120(3):e713–e722.
18. Zuran, B.L. Current and future therapy for hereditary angioedema. Clin Immunol. 2005;114(1):10–16.
19. Bracho, F.A. Hereditary angioedema. Curr Opin Hematol. 2005;12(6):493–498.
20. Frank, M.M. Hereditary angioedema. J Allergy Clin Immunol. 2008;121(2 Suppl):S398–S401.
21. Daniel, S. The upper airway: congenital malformations. Paediatr Respir Rev. 2006;7S:S260–S263.
22. Alvarez-Neri, H., et al. Primary cricotracheal resection with thyrotracheal anastomosis for the treatment of severe subglottic stenosis in children and adolescents. Ann Otol Rhinol Laryngol. 2005;114(1 pt 1):2–6.
23. Dauer, E.H., et al. Airway manifestations of pediatric eosinophilic esophagitis: a clinical and histopathologic report of an emerging association. Ann Otol Rhinol Laryngol. 2006;155(7):507–517.
24. Erickson, V.R., Hwang, P.H. Wegener’s granulomatosis: current trends in diagnosis and management. Curr Opin Otolaryngol Head Neck Surg. 2007;15(3):170–176.
25. Khariwala, S., Lee, W., Koltai, P. Laryngotracheal consequences of pediatric cardiac surgery. Arch Otolaryngol Head Neck Surg. 2005;131(4):336–339.
26. Sidman, J., Jaguan, A., Couser, R. Tracheotomy and decannulation rates in a level 3 neonatal intensive care unit: a 12 year study. Laryngoscope. 2006;116:136–139.
27. Bluestone, C. Humans are born too soon: impact on pediatric otolaryngology. Int J Pediatr Otorhinolaryngol. 2005;69:1–8.
28. Hueman, E., Simpson, B. Airway complications from topical mitomycin C. Otolaryngol Head Neck. 2005;133(6):831–835.
29. Smith, J.L., et al. State-dependent laryngomalacia in sleeping children. Ann Otol Rhinol Laryngol. 2005;114(2):111–114.
30. Thompson, D.M. Abnormal sensorimotor integrative function of the larynx in congenital laryngomalacia: a new theory of etiology. Laryngoscope. 2007;117(6 pt 2 Suppl 114):1–33.
31. Richter, G., et al. Late-onset laryngomalacia. Arch Otolaryngol Head Neck Surg. 2008;134(1):75–80.
32. Weinberger, M., Abu-Hasan, M. Pseudo-asthma: when cough, wheezing and dyspnea are not asthma. Pediatrics. 2007;120(4):855–864.
33. Masters, I., et al. Quantified tracheobronchomalacia disorders and their clinical profiles in children. Chest. 2008;133(2):461–467.
34. Ishman, S., et al. Management of vocal paralysis: a comparison of adult and pediatric practices. Otolaryngol Head Neck Surg. 2006;135:590–594.
35. Doshi, D., Weinberger, M. Long-term outcome of vocal chord dysfunction. Ann Allergy Asthma Immunol. 2006;96:794–799.
36. McLaren, C., Elliott, M., Roebuck, D. Vascular compression of the airway in children. Paediatr Respir Rev. 2008;9:85–94.
37. Tarasuik, A., et al. Elevated morbidity and health care use in children with obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 2007;175:55–61.
38. Ievers-Landis, C., Redline, S. Pediatric sleep apnea. Am J Respir Crit Care Med. 2007;175:436–441.
39. Arens, R., Marcus, C.L. Pathophysiology of upper airway obstruction: a developmental perspective. Sleep. 2004;27(5):997–1019.
40. Goldbart, A., et al. Inflammatory mediators in exhaled breath condensate of children with obstructive sleep apnea syndrome. Chest. 2006;130:143–148.
41. Fauroux, B., Aubertin, G., Clement, A. What’s new in paediatric sleep in 2007? Paediatr Respir Rev. 2008;9:139–143.
42. Leung, L., et al. Twenty-four-hour ambulatory BP in snoring children with obstructive sleep apnea syndrome. Chest. 2006;130:1009–1017.
43. American Academy of Pediatrics. Clinical practice guidelines: diagnosis and management of childhood obstructive sleep apnea. Pediatrics. 2002;109(4):704–712.
44. Gozal, D., O’Brien, L.M. Snoring and obstructive sleep apnoea in children: why should we treat? Paediatr Respir Rev. 2004;5(Suppl A):S371–S376.
45. Rosen, C.L. Obstructive sleep apnea syndrome in children: controversies in diagnosis and treatment. Pediatr Clin North Am. 2004;51(1):153–167. [vii].
46. Ray, M., Bower, C. Pediatric obstructive sleep apnea: the year in review. Curr Opin Otolaryngol Head Neck Surg. 2005;13(6):360–365.
47. Tran, K., et al. Child behavior and quality of life in pediatric obstructive sleep apnea. Arch Otolaryngol Head Neck Surg. 2005;131(1):52–57.
48. Fraser, J., Walls, M., McGuire, W. Respiratory complications of preterm birth. Br Med J. 2004;329:962–965.
49. Dalziel, S., et al. Long term effects of antenatal betamethasone on lung function: 30 year follow up of a randomised controlled trial. Thorax. 2006;61:678–683.
50. American Heart Association (AHA). Guidelines for cardiopulmonary resuscitation (CPR) and emergency cardiovascular care (ECC) of pediatric and neonatal patients: neonatal resuscitation guidelines (2005). Pediatrics,. 2005;117(5):e1029–e1038.
51. Sweet, D., et al. European consensus guidelines on the management of neonatal respiratory distress syndrome. J Perinatal Med. 2007;35:175–186.
52. NIH Consensus Development Conference Statement: Effect of corticosteroids for fetal maturation on perinatal outcomes. Am J Obstet Gynecol. 1995;173:246.
53. Mazela, J., Merritt, T., Finer, N. Aerolsolized surfactants. Curr Opin Pediatr. 2007;19:155–162.
54. Engle, W. Committee on Fetus and Newborn: Surfactant-replacement therapy for respiratory distress in the preterm and term neonate. Pediatrics,. 2008;121(2):419–432.
55. Howlett, A., Ohlsson, A. Inositol for respiratory distress syndrome in preterm infants. Cochrane Database Syst Rev. (4):2004. [CD000366].
56. Shah, S. Is elective high frequency oscillatory ventilation better than conventional mechanical ventilation in very low birth weight infants? Arch Dis Child. 2003;88(9):833–834.
57. Firer, N.N., Barrington, K.J. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev. (1):2005. [CD000399].
58. Kinsella, J., et al. Early inhaled nitric oxide therapy in premature newborns with respiratory failure. N Engl J Med. 2006;355(4):354–364.
59. Ballard, P., et al. Plasma biomarkers of oxidative stress: relationship to lung disease and inhaled nitric oxide therapy in premature infants. Pediatrics. 2008;121(3):555–561.
60. Doyle, L., et al. Outcome at 2 years of age of infants from the DART study: a multicenter, international, randomized, controlled trial of low-dose dexamethasone. Pediatrics. 2007;119(4):716–721.
61. Asikainen, T.M., White, C.W. Pulmonary antioxidant defenses in the preterm newborn with respiratory distress and bronchopulmonary dysplasia in evolution: implications for antioxidant therapy. Antioxid Redox Signal. 2004;6(1):155–167.
62. Baier, R.J., et al. CC chemokine concentrations increase in respiratory distress syndrome and correlate with development of bronchopulmonary dysplasia. Pediatr Pulmonol. 2004;37(2):137–148.
63. Jobe, A.H. Antenatal factors and the development of bronchopulmonary dysplasia. Semin Neonatol. 2003;8(1):9–17.
64. Morley, C., et al. Nasal CPAP or intubation at birth for very preterm infants. N Engl J Med. 2008;358(14):700–708.
65. Northway, W.H., Jr., Rosan, R.C., Porter, D.Y. Pulmonary disease following respiratory therapy of hyaline-membrane disease: bronchopulmonary dysplasia. N Engl J Med. 1967;276(7):357–368.
66. Baraldi, E., Filippone, M. Chronic lung disease after premature birth. N Engl J Med. 2007;357(19):1946–1955.
67. Bancalari, E., Claure, N., Sosenko, I.R.S. Bronchopulmonary dysplasia: changes in pathogenesis, epidemiology, and definition. Semin Neonatol. 2003;8(1):63–71.
68. Eichenwald, E., Stark, A. Management and outcomes of very low birth weight. N Engl J Med. 2008;358(16):1700–1711.
69. Mahut, B., et al. Chest computed tomography findings in bronchopulmonary dysplasia and correlation with lung function. Arch Dis Child Fetal Neonatal Ed. 2007;92(6):F459–F464.
70. Vento, G., et al. Serum levels of seven cytokines in premature ventilated newborns: correlations with old and new forms of bronchopulmonary dysplasia. Intensive Care Med. 2006;32(5):723–730.
71. Geary, C., et al. Decreased incidence of bronchopulmonary dysplasia after early management changes, including surfactant and nasal continuous positive airway pressure treatment at delivery, lowered oxygen saturation coals, and early amino acid administration: a historical cohort study. Pediatrics. 2008;121:89–96.
72. Morley, C., et al. Nasal CPAP or intubation at birth for very preterm infants. N Engl J Med. 2008;358(7):700–708.
73. Ballard, P.L., et al. Plasma biomarkers of oxidative stress: relationship to lung disease and inhaled nitric oxide therapy in premature infants. Pediatrics. 2008;121(3):555–561.
74. Barrington, K., Finer, N. Inhaled nitric oxide for preterm infants: a systematic review. Pediatrics. 2007;120:1088–1099.
75. Bhandari, A., et al. Effect of a short course of prednisolone in infants with oxygen-dependent bronchopulmonary dysplasia. Pediatrics. 2008;121:e344–e349.
76. Kobaly, K., et al. Outcomes of extremely low birth weight (<1 kg) and extremely low gestational age (<28 weeks) infants with bronchopulmonary dysplasia: effects of practice changes in 2000 to 2003. Pediatrics. 2008;121:73–81.
77. Aghai, Z., et al. Azithromycin suppresses activation of nuclear factor-kappa B and synthesis of pro-inflammatory cytokines in tracheal aspirate cells from premature infants. Pediatric Res. 2007;62(4):483–488.
78. Mentro, A.M. Vitamin A and bronchopulmonary dysplasia: research, issues and related clinical practice. Neonatal Netw. 2004;23(4):19–21.
79. Kesson, A. Respiratory virus infections. Paediatr Respir Rev. 2007;8:240–248.
80. Smyth, R., Openshaw, P. Bronchiolitis. Lancet. 2006;368:312–322.
81. Piippo-Savolainen, E., Korppi, M. Wheezy babies—wheezy adults? Review on long-term outcome until adulthood after early childhood wheezing. Acta Paediatr. 2008;97(1):5–11.
82. Christakis, D., et al. Variation in inpatient diagnostic testing and management of bronchiolitis. Pediatrics. 2005;115(4):878–884.
83. Martinon-Torres, F., Rodriguez-Nunez, A., Martinon-Sanchez, J. Nasal continuous positive airway pressure with heliox versus air oxygen in infants with acute bronchiolitis: a crossover study. Pediatrics. 2008;121:E1190–E1195.
84. Patel, H., et al. Glucocorticoids for acute viral bronchiolitis in infants and young children. Cochrane Database Syst Rev. (3):2004. [CD004878].
85. Steiner, R.W. Treating acute bronchiolitis associated with RSV. Am Fam Physician. 2004;15(69):325–330.
86. Flood, R., Badik, J., Aronoff, S. The utility of serum C-reactive protein in differentiating bacterial from nonbacterial pneumonia in children,. Pediatr Infect Dis J. 2008;27(2):95–99.
87. Atkinson, M., et al. Comparison of oral amoxicillin and intravenous benzyl penicillin for community acquired pneumonia in children (PIVOT trial): a multicentre pragmatic randomized controlled equivalence trial. Thorax. 2007;62:1102–1106.
88. Wolf, J., Daly, A. Microbiological aspects of bacterial lower respiratory tract illness in children: typical pathogens. Paediatr Respir Rev. 2007;8:204–211.
89. Sandora, T., Harper, M. Pneumonia in hospitalized children. Pediatr Clin North Am. 2005;52:1059–1081.
90. Ozcelik, C., et al. Management of postpneumonic empyemas in children. Eur J Cardiothorac Surg. 2004;25(6):1072–1078.
91. Schultz, K.D., et al. The changing face of pleural empyemas in children: epidemiology and management. Pediatrics. 2004;113(6):1735–1740.
92. Sinaniotis, C.A. Viral pneumoniae in children: incidence and aetiology. Paediatr Respir Rev. 2004;5(Suppl A):S197–S200.
93. Moreno, L., et al. Development and validation of a clinical predication rule to distinguish bacterial from viral pneumonia in children. Pediatr Pulmonol. 2006;41:331–337.
94. Hammerschlag, M.R. Pneumonia due to Chlamydia pneumoniae in children: epidemiology, diagnosis, and treatment. Pediatr Pulmonol. 2003;36(5):384–390.
95. Waites, K.B. New concepts of Mycoplasma pneumoniae infections in children. Pediatr Pulmonol. 2003;36(4):267–278.
96. Bradley, J., et al. Comparative study of levofloxacin in the treatment of children with community-acquired pneumonia. Pediatr Infect Dis J. 2007;26(10):868–878.
97. Klig, J., Shah, N. Office pediatrics: current issues in lower respiratory infections in children. Curr Opin Pediatr. 2005;17(1):111–118.
98. Moussaoui, E., deBorgie, C., van den Broek, P. Short course antibiotics in community acquired pneumonia. BMJ. 2006;332:1355–1358.
99. Clark, J., et al. Children with pneumonia: how do they present and how are they managed? Arch Dis Child. 2007;92:394–398.
100. Posfay-Barbe, K.M., Wald, E.R. Pneumococcal vaccines: do they prevent infection and how? Curr Opin Infect Dis. 2004;17(3):177–184.
101. Boesch, R.P., et al. Advances in the diagnosis and management of chronic pulmonary aspiration in children. Eur Respir J. 2006;28:847–861.
102. Kurland, G., Michelson, P. Bronchiolitis obliterans in children. Pediatr Pulmonol. 2005;39:193–208.
103. Smith, K., Fan, L. Insights into post-infectious bronchiolitis obliterans in children. Thorax. 2006;61:462–463.
104. Colom, A., et al. Risk factors for the development of bronchiolitis obliterans in children with bronchiolitis. Thorax. 2006;61:503–506.
105. Verleden, G., et al. Azithromycin reduces airway neutrophilia and interleukin-8 in patients with bronchiolitis obliterans syndrome. Am J Respir Crit Care Med. 2006;174:566–570.
106. National Heart, Lung, and Blood Institute: National Asthma Education and Prevention Program Expert Panel Report 3: guidelines for the diagnosis and management of asthma. 2007. Available at www.nhlbi.nih.gov/guidelines
107. Pearce, N., Douwes, J. The global epidemiology of asthma in children. Int J Tuberc Lung Dis. 2006;10(2):125–132.
108. Clement, L., Jones, C., Cole, J. Health disparities in the United States: childhood asthma. Am J Med Sci. 2008;335(4):260–265.
109. Chen, A., Escarce, J. Family structure and the treatment of childhood asthma. Med Care. 2008;46(2):174–184.
110. Van Schayck, C., Smit, H. The prevalence of asthma in children: a reversing trend. Eur Respir J. 2005;26(4):647–650.
111. Grigg, J. Asthma year in review 2006-7. Paediatr Respir Rev. 2008;9:134–138.
112. Zhang, J., Pare, P.D., Sandford, A.J. Recent advances in asthma genetics. Respir Res. 2008;9:4.
113. McLeish, S., Turner, S.W. Gene-environment interactions in asthma. Arch Dis Child. 2007;92(11):1032–1035.
114. Morgenstern, V., et al. Atopic diseases, allergic sensitization, and exposure to traffic-related air pollution in children. Am J Respir Crit Care Med. 2008;177:1331–1337.
115. Kiechl-Kohlendorfer, U., et al. Neonatal characteristics and risk of atopic asthma in schoolchildren: results from a large prospective birth-cohort study. Acta Paediatrica. 2007;96(11):1606–1610.
116. Vonk, J.M., Boezen, H.M. Predicting adult asthma in childhood. Curr Opin Pulm Med. 2006;12(1):42–47.
117. Panettierri, R.A., Jr. Natural history of asthma: persistence versus progression—does the beginning predict the end? J Allergy Clin Immunol. 2008;121(3):607–613.
118. Brownlee, J., Turner, R. New developments in the epidemiology and clinical spectrum of rhinovirus infections. Curr Opin Pediatr. 2008;20:67–71.
119. Lenney, W. Pro-con debate: inhaled corticosteroids should not be prescribed in primary care to children under two years of age—the case against. Prim Care Respir J. 2008;17(3):181–184.
120. Bisgaard, H., et al. Montelukast reduces asthma exacerbations in 2 to 5 year old children with intermittent asthma. Am J Respir Crit Care Med. 2005;171:315–322.
121. Penagos, M., et al. Metaanalysis of the efficacy of sublingual immunotherapy in the treatment of allergic asthma in pediatric patients, 3 to 18 years of age. Chest. 2008;133(3):599–609.
122. Niven, R., et al. Effectiveness of omalizumab in patients with inadequately controlled severe persistent allergic asthma: an open-label study. Respir Med. 2008;102(10):1371–1378.
123. Pijnenburg, M., et al. Exhaled nitric oxide predicts asthma relapse in children with clinical asthma remission. Thorax. 2005;60:215–218.
124. Bernard, G.R., et al. The American-European Consensus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824.
125. Willson, D.F., Chess, P.R., Notter, R.H. Surfactant for pediatric acute lung injury. Pediatr Clin North Am. 2008;55(3):545–575. [ix].
126. Bruijn, M., et al. High incidence of acute lung injury in children with Down syndrome. Intensive Care Med. 2007;33(12):2179–2182.
127. Cincotta, D., et al. Fatal acute fibrinous and organizing pneumonia in an infant: the histopathologic variability of acute respiratory distress syndrome. Pediatr Crit Care Med. 2007;8(4):378–382.
128. Bhatia, M., Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202(2):145–156.
129. Ferguson, N.D., Slutsky, A.S. Point: high-frequency ventilation is the optimal physiological approach to ventilate ARDS patients. J Appl Physiol. 2008;104(4):1230–1231.
130. Turner, D., Arnold, J. Insights in pediatric ventilation: timing of intubation, ventilatory strategies, and weaning. Curr Opin Crit Care. 2007;13:57–63.
131. Wunsch, H., Mapstone, J. High-frequency ventilation versus conventional ventilation for the treatment of acute lung injury and acute respiratory distress syndrome: a systematic review and Cochrane analysis. Crit Care Trauma. 2005;100:1765–1772.
132. Been, J.V., Zimmerman, L.J. What’s new in surfactant? A clinical view on recent developments in neonatology and paediatrics. Eur J Pediatr. 2007;166(9):889–899.
133. Strausbaugh, S., Davis, P. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med. 2007;28:279–288.
134. Rogers, D.F. Physiology of airway mucus secretion and pathophysiology of hypersecretion. Respir Care. 2007;52(9):1134–1146. [discussion 1146-1149].
135. Donaldson, S.H., Boucher, R.C. Sodium channels and cystic fibrosis. Chest. 2007;132(5):1631–1636.
136. Elizur, A., Cannon, C.L., Ferkol, T.W. Airway inflammation in cystic fibrosis. Chest. 2008;133(2):489–495.
137. Accurso, F. Update in cystic fibrosis 2007. Am J Respir Crit Care Med. 2008;177:1058–1061.
138. Elkin, S., Geddes, D. Pseudomonal infection in cystic fibrosis: the battle continues. Expert Rev Anti Infect Ther. 2003;1(4):609–618.
139. Tunney, M., et al. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med. 2008;177:995–1001.
140. Regamey, N., et al. Increased airway smooth muscle mass in children with asthma, cystic fibrosis and non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2008;177:837–843.
141. Sontag, M.K., Accurso, F.J. Gene modifiers in pediatrics: application to cystic fibrosis. Adv Pediatr. 2004;51:5–36.
142. Konstan, M., et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr. 2007;151:134–139.
143. Grosse, S.D., et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Morb Mortal Wkly Rep. 2004;53(RR-13):1–36.
144. Paul, K., et al. Effect of treatment with dornase alpha on airway inflammation in patients with cystic fibrosis. Am J Respir Crit Care Med. 2004;169(6):719–725.
145. Rosenfeld, M., Ramsey, B.W., Gibson, R.L. Pseudomonas acquisition in young patients with cystic fibrosis: pathophysiology, diagnosis, and management. Curr Opin Pulmon Med. 2003;9(6):492–497.
146. Boucher, R.C. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med. 2007;13(6):231–240.
147. Konstan, M. Ibuprofen theory for cystic fibrosis lung disease: revisited. Curr Opin Pulmon Med. 2008;14(6):567–573.
148. Aurora, P. Lung transplantation for cystic fibrosis. J Royal Soc Med. 2007;100(Suppl 47):46–52.
149. Milla, C.E. Nutrition and lung disease in cystic fibrosis. Clin Chest Med. 2007;28(2):319–330.
150. Sueblinvong, V., Suratt, B.T., Weiss, D.J. Novel therapies for the treatment of cystic fibrosis: new developments in gene and stem cell therapy. Clin Chest Med. 2007;28(2):361–379.
151. Daley, K.C. Update on sudden infant death syndrome. Curr Opin Pediatr. 2004;16(2):227–232.
152. American Academy of Pediatrics Task Force on Sudden Infant Death Syndrome. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics. 2005;116(5):1245.
153. Moon, R., Horne, S., Hauck, F. Sudden infant death syndrome. Lancet. 2007;370:1578–1587.
154. Blair, P., Fleming, P. Recurrence risk of sudden infant death syndrome. Arc Dis Child. 2008;93(4):269–270.
155. Berkowitz, C.D. Cosleeping: benefits, risks, and cautions. Adv Pediatr. 2004;51:329–349.
156. Horne, R.S., Parslow, P.M., Harding, R. Respiratory control and arousal in sleeping infants. Paediatr Respir Rev. 2004;5(3):190–198.
157. Horne, R.S., et al. Influences of maternal cigarette smoking on infant arousability. Early Hum Dev. 2004;79(1):49–58.
158. Hunt, C.E. Gene-environment interactions: implications for sudden unexpected deaths in infancy. Arch Dis Child. 2005;90(1):48–53.
159. Hunt, C. Small for gestational age infants and sudden infant death syndrome: a confluence of complex conditions. Arch Dis Child. 2007;92:428–430.
160. Stray-Pedersen, A., et al. Post-neonatal drop in alveolar SP-A expression: biological significance for increase vulnerability to SIDS? Pediatr Pulmonol. 2008;43:160–168.
161. Leiter, J., Bohm, I. Mechanisms of pathogenesis in the sudden infant death syndrome. Resp Physiol Neurobiol. 2007;159(2):127–138.