ALTERATIONS OF PULMONARY FUNCTION



Pulmonary disease is often classified as acute or chronic, obstructive or restrictive, and infectious or noninfectious. Because skillful and knowledgeable clinical care plays a major role in decreasing respiratory morbidity and mortality, the clinician with a clear understanding of the pathophysiology of common respiratory problems can greatly affect the outcome for each individual.
CLINICAL MANIFESTATIONS OF PULMONARY ALTERATIONS
Signs and Symptoms of Pulmonary Disease
Pulmonary disease is associated with many signs and symptoms. The most common of these are cough and dyspnea. Other manifestations include chest pain, abnormal sputum, hemoptysis, altered breathing patterns, cyanosis, clubbing of the digits, and fever. The signs and symptoms and their specific characteristics often help in identifying the underlying disorder.
Cough
Cough is an important reflex that helps clear the airways of large amounts of inhaled material, excessive secretions, or abnormal substances, such as edema or pus. Individuals with an inability to cough normally are at greater risk for pneumonia. The cough reflex results from a complex interaction of sensory receptors in the upper and lower airways, including stimulation of mechanical and chemical “irritant” receptors.1,2 There are few such receptors in the most distal bronchi and the alveoli; thus it is possible for significant amounts of secretions to accumulate in the distal respiratory tree without cough being initiated. Other cough receptors are located in the external auditory canal, diaphragm, pericardium, pleura, and stomach. Stimulation of cough receptors is transmitted centrally through the vagus nerve, and central modulation of the cough reflex can be influenced by opiates and serotonergic agents.3
Acute cough is cough that resolves within 2 to 3 weeks of the onset of illness or resolves with treatment of the underlying condition. It is most commonly the result of upper respiratory infections, allergic rhinitis, acute bronchitis, pneumonia, congestive heart failure, pulmonary embolus, or aspiration.4 Chronic cough is defined as cough that has persisted for more than 3 weeks, although some researchers have suggested that 7 or 8 weeks is a more appropriate timeframe because acute cough and bronchial hyperreactivity can be prolonged in some cases of viral infection. In nonsmokers, chronic cough is almost always caused by postnasal drainage syndrome, nonasthmatic eosinophilic bronchitis, asthma, or gastroesophageal reflux disease.4,5 In smokers, chronic bronchitis is the most common cause of chronic cough, although lung cancer must always be considered. Up to 33% of individuals taking angiotensin-converting enzyme inhibitors for cardiovascular disease develop chronic cough that resolves with discontinuation of the drug.
Dyspnea
Dyspnea is the subjective sensation of being unable to get enough air. It is often described as breathlessness, air hunger, shortness of breath, labored breathing, and preoccupation with breathing. Dyspnea is a common symptom of respiratory disease.
The severity of the sensation of dyspnea may not directly correlate with the severity of underlying pulmonary disease.6,7 Either diffuse or focal disturbances of ventilation, gas exchange, or ventilation-perfusion relationships can cause dyspnea, as can increased work of breathing or diseases that damage lung tissue (lung parenchyma). Many mechanisms have been proposed to explain the complex sensation of dyspnea, but no single mechanism has been found to be responsible in all situations. One commonly accepted mechanism involves an impaired sense of effort. This is a situation in which the perceived work of breathing is greater than the actual motor response generated. Mechanoreceptors in the chest wall respond to the length and tension in muscles and can contribute to the sensation of dyspnea as can upper airway receptors that signal the brain through trigeminal nerve fibers.7 A second explanation involves the stimulation of central and peripheral chemoreceptors. It has long been known that decreased pH, hypercapnia, and hypoxemia can cause dyspnea.6,7 (The neurochemical control of ventilation is described in Chapter 32.) Stimulation of chemoreceptors causes dyspnea in many lung diseases in which oxygenation and gas exchange are impaired.
A third explanation is stimulation of the afferent receptors in the lung (the stretch receptors, irritant receptors, and J-receptors), which send impulses to the central nervous system through the vagus nerve.6,7 Stretch receptors are stimulated in asthma and may be the primary cause of the sensation of dyspnea and chest tightness in that disorder. J-receptors also trigger dyspnea in individuals with pulmonary edema and pulmonary microemboli. Finally, the sensation of dyspnea also can be caused by increased work of breathing, respiratory muscle fatigue, decreased breathing reserve, and strong emotions, particularly anxiety and anger. There is general agreement that neurologic control and function of respiratory muscles are the common elements in most clinical experiences of dyspnea and that the sensation perceived is that of increased respiratory effort.
The signs of dyspnea include flaring of the nostrils, use of accessory muscles of respiration, and retraction (pulling back) of the intercostal spaces. In dyspnea caused by parenchymal disease (e.g., pneumonia), retractions of tissue between the ribs (subcostal and intercostal retractions) are observed more often than supercostal retractions (retractions of tissues above the ribs), which predominate in upper airway obstruction. Retractions of any type are more commonly seen in children or in adults who are thin and have poorly developed thoracic musculature. Dyspnea can be quantified by the use of ordinal rating scales or visual analog scales.7
Dyspnea can occur transiently or can become chronic. The first episode commonly occurs with exercise and is called dyspnea on exertion. Dyspnea also can be associated with body positioning. Orthopnea is dyspnea that occurs when an individual lies flat and is common in individuals with heart failure. The recumbent position redistributes body water, causes the abdominal contents to exert pressure on the diaphragm, and decreases the efficiency of the respiratory muscles. Orthopnea is generally relieved by sitting up in a forward-leaning posture or supporting the upper body on several pillows. Another type of positional dyspnea is termed paroxysmal nocturnal dyspnea (PND) in which individuals with heart failure or lung disease wake up at night gasping for air and must sit up or stand to relieve the dyspnea.
Pain
Pain caused by pulmonary disorders originates in the pleurae, airways, or chest wall. Pleural pain is the most common pain caused by pulmonary disease and is usually sharp or stabbing in character. Infection and inflammation of the parietal pleura (pleuritis or pleurisy) cause pain when the pleura stretch during inspiration. The pain is usually localized to a portion of the chest wall, where a unique breath sound called a pleural friction rub may be heard over the painful area. Laughing or coughing makes pleural pain worse. Pleural pain is also common with pulmonary infarction (tissue death) caused by pulmonary embolism and emanates from the area around the infarction.
Pulmonary pain is central chest pain that is pronounced after coughing and occurs in individuals with infection and inflammation of the trachea or bronchi (tracheitis or tracheobronchitis). Central chest pain can be difficult to differentiate from cardiac pain (see Chapter 30). High blood pressure in the pulmonary circulation (pulmonary hypertension) can cause pain during exercise that is often mistaken for cardiac pain (angina pectoris).
Pain in the chest wall is muscle pain or rib pain. The common causes of chest wall pain are excessive coughing, which makes the muscles sore, and rib fractures. Inflammation of the costochondral junction (costochondritis) also can cause chest wall pain. Chest wall pain can often be reproduced by pressing on the sternum or ribs.
Abnormal Sputum
The color, consistency, odor, and amount of sputum vary with different pulmonary disorders. A distinctive color or odor may suggest infection by a specific microorganism. Changes in the amount and consistency of sputum provide information about progression of disease and effectiveness of therapy. The gross and microscopic appearances of sputum enable the clinician to identify cellular debris or microorganisms that aid in diagnosis and choice of therapy.
Hemoptysis
Hemoptysis is the coughing up of blood or bloody secretions. Hemoptysis is sometimes confused with hematemesis, which is the vomiting of blood. Blood that is coughed up is usually bright red, has an alkaline pH, and is mixed with frothy sputum, whereas blood that is vomited is dark, has an acidic pH, and is mixed with food particles.
The most common causes of hemoptysis are bronchiectasis, lung cancer, bronchitis, and pneumonia. Tuberculosis remains an important cause of hemoptysis but is less common in the United States than in many other parts of the world. Hemoptysis results from damage to the lung parenchyma with rupture of pulmonary vessels or from inflammation, injury, or cancer of the bronchial tree. The amount and duration of bleeding (i.e., a sudden large amount versus a persistent slight amount) provide important clues about the source of the bleeding.8 Bronchoscopy, combined with chest computed tomography (CT), can identify the cause in the majority of cases of hemoptysis.
Abnormal Breathing Patterns
Normal breathing (eupnea) is rhythmic and effortless. Ventilatory rate is 8 to 16 breaths per minute, and tidal volume ranges from 400 to 800 ml. A short expiratory pause occurs with each breath, and the individual takes an occasional deeper breath or sigh. Sigh breaths, which help maintain normal lung function, are usually 1.5 to 2 times the normal tidal volume and occur approximately 10 to 12 times per hour.
The rate, depth, regularity, and effort of breathing undergo characteristic alterations in response to physiologic and pathophysiologic conditions. Patterns of breathing automatically adjust to minimize the work of respiratory muscles. Strenuous exercise or metabolic acidosis induces Kussmaul respiration (hyperpnea). Kussmaul respiration is characterized by a slightly increased ventilatory rate, very large tidal volume, and no expiratory pause.
Labored breathing occurs whenever there is an increased work of breathing, especially if the airways are obstructed, as in chronic obstructive pulmonary disease (COPD). If the large airways are obstructed, a slow ventilatory rate, increased effort, prolonged inspiration or expiration, and stridor (high-pitched sounds made during inspiration) or audible wheezing (whistling sounds on expiration) are typical. In small airway obstruction, like that seen in asthma and chronic obstructive pulmonary disease, a rapid ventilatory rate, small tidal volume, increased effort, prolonged expiration, and wheezing are often present.
Restricted breathing is commonly caused by disorders such as pulmonary fibrosis that stiffen the lungs or chest wall and decrease compliance. Restricted breathing is characterized by small tidal volumes and rapid ventilatory rate (tachypnea).
Shock and severe cerebral hypoxia (insufficient oxygen in the brain) contribute to gasping respirations that consist of irregular, quick inspirations with an expiratory pause. Anxiety can cause sighing respirations that consist of irregular breathing characterized by frequent, deep sighing inspirations.
Cheyne-Stokes respirations are characterized by alternating periods of deep and shallow breathing. Apnea lasting 15 to 60 seconds is followed by ventilations that increase in volume until a peak is reached, after which ventilation (tidal volume) decreases again to apnea. Cheyne-Stokes respirations result from any condition that slows the blood flow to the brainstem, which in turn slows impulses sending information to the respiratory centers of the brainstem. Neurologic impairment above the brainstem is also a contributing factor (see Table 16-5).
Hypoventilation and Hyperventilation
Hypoventilation is inadequate alveolar ventilation in relation to metabolic demands. It is caused by alterations in pulmonary mechanics or in the neurologic control of breathing such that minute volume (tidal volume times respiratory rate) is reduced. When alveolar ventilation is normal, carbon dioxide (CO2) is removed from the lungs at the same rate at which it is produced by cellular metabolism. This maintains arterial CO2 (PaO2) at normal levels (40 mmHg). With hypoventilation, CO2 removal does not keep up with CO2 production and PaCO2 increases, causing hypercapnia (PaCO2 greater than 44 mmHg). (Table 32-2 contains the definition of gas partial pressure and other pulmonary abbreviations.) This results in an increase in hydrogen ion in the blood, termed respiratory acidosis, which can affect the function of many tissues throughout the body.
Hypoventilation is often overlooked until it is severe because breathing pattern and ventilatory rate may appear normal. Blood gas analysis (i.e., measurement of the PaCO2 of arterial blood) reveals the hypercapnia. Pronounced hypoventilation can cause somnolence or disorientation. In addition, hypoventilation with hypercapnia results in secondary hypoxemia.
Hyperventilation is alveolar ventilation that exceeds metabolic demands. The lungs remove CO2 at a faster rate than it is produced by cellular metabolism, resulting in decreased PaCO2 or hypocapnia (PaCO2 less than 36 mmHg). Hypocapnia results in a respiratory alkalosis that also can interfere with tissue function. Like hypoventilation, hyperventilation can be determined only by arterial blood gas analysis. Hyperventilation commonly occurs with severe anxiety, acute head injury, and conditions that cause insufficient oxygenation of the blood.
Cyanosis
Cyanosis is a bluish discoloration of the skin and mucous membranes caused by increasing amounts of desaturated or reduced hemoglobin (which is bluish) in the blood. Cyanosis generally develops when 5 g of hemoglobin is desaturated, regardless of hemoglobin concentration. For example, if total hemoglobin concentration is 15 g/dl of blood, 5 g/dl must be desaturated to cause cyanosis. If total hemoglobin is 11 g/dl, 5 g/dl must still be desaturated for cyanosis to occur.
Peripheral cyanosis is most often caused by poor circulation resulting from intense peripheral vasoconstriction, like that seen in Raynaud disease, cold environments, or severe stress. Central cyanosis is caused by decreased arterial oxygenation (low PaO2) from pulmonary diseases or pulmonary or cardiac right-to-left shunts. In adults, cyanosis is not evident until severe hypoxemia is present and therefore is an insensitive indicator of respiratory distress. Lack of cyanosis does not necessarily indicate that oxygenation is normal. For example, severe anemia (inadequate hemoglobin concentration) and carbon monoxide poisoning (in which hemoglobin binds to carbon monoxide instead of binding to oxygen) can cause inadequate oxygenation of tissues without causing cyanosis. Individuals with polycythemia (an abnormal increase in numbers of red blood cells), however, may have cyanosis when tissue oxygenation is adequate. Because polycythemia causes hemoglobin concentration to be greater than normal, 5 g/dl can be desaturated, causing cyanosis, without having much effect on oxygenation. Therefore, the significance of cyanosis as a clinical finding must be interpreted in relation to the underlying pathophysiology. Central cyanosis is best seen in buccal mucous membranes and lips. Peripheral cyanosis is best seen in nail beds. If cyanosis is suggested, the PaO2 should be measured.
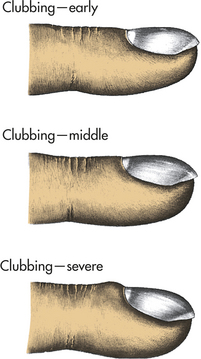
Clubbing
Clubbing is the selective bulbous enlargement of the end (distal segment) of a digit (finger or toe) (Figure 33-1) whose severity can be graded from 1 to 5 based on the extent of nail bed hypertrophy and the amount of changes in the nails themselves. Usually it is painless. Clubbing is commonly associated with diseases that interfere with oxygenation, such as bronchiectasis, cystic fibrosis, pulmonary fibrosis, lung abscess, and congenital heart disease. It is usually reversible with treatment of the underlying pulmonary condition. Lung cancer is sometimes associated with clubbing even in the absence of significant hypoxemia. This syndrome is called hypertrophic osteoarthropathy (HOA) and its pathogenesis is unknown, although tumor-associated production of inflammatory cytokines and growth factors have been implicated.
Conditions Caused by Pulmonary Disease or Injury
Hypercapnia, or increased CO2 in the arterial blood (increased PaCO2), is caused by hypoventilation of the alveoli. As discussed in Chapter 32, CO2 is easily diffused from the blood into the alveolar space; thus minute volume (respiratory rate × tidal volume) determines not only alveolar ventilation but also PaCO2. Hypoventilation is often overlooked because breathing pattern and ventilatory rate may appear normal; it is important to obtain blood gas analysis to determine the severity of hypercapnia and resultant respiratory acidosis (acid-base balance is described in Chapter 3).
There are many causes of hypercapnia. Most are a result of decreased drive to breathe or an inadequate ability to respond to ventilatory stimulation. Causes include (1) depression of the respiratory center by drugs; (2) diseases of the medulla, including infections of the central nervous system or trauma; (3) abnormalities of the spinal conducting pathways, as in spinal cord disruption or poliomyelitis; (4) diseases of the neuromuscular junction or of the respiratory muscles themselves, as in myasthenia gravis or muscular dystrophy; (5) thoracic cage abnormalities, as in chest injury or congenital deformity; (6) large airway obstruction, as in tumors or sleep apnea; and (7) increased work of breathing or physiologic dead space, as in emphysema.
Hypercapnia and the associated respiratory acidosis can result in several important clinical manifestations. Of greatest concern are electrolyte abnormalities that occur in response to the low pH that may cause dysrhythmias. Individuals also may have somnolence and even be in a coma because of changes in intracranial pressure associated with high levels of arterial carbon dioxide, which causes cerebral vasodilation. Alveolar hypoventilation with increased alveolar carbon dioxide limits the amount of oxygen available for diffusion into the blood, leading to secondary hypoxemia.
Hypoxemia
Hypoxemia, or reduced oxygenation of arterial blood (reduced PaO2), is caused by respiratory alterations, whereas hypoxia, or reduced oxygenation of cells in tissues, may be caused by alterations of other systems as well. Although hypoxemia can lead to tissue hypoxia, tissue hypoxia can result from other abnormalities, such as low cardiac output or cyanide poisoning, that have no relation to alterations of pulmonary function.
Hypoxemia results from problems with one or more of the major mechanisms of oxygenation:
1. Oxygen delivery to the alveoli
Table 33-1 lists some of the common clinical causes of these problems.
Table 33-1
| Mechanism | Common Clinical Causes |
| Decrease in inspired oxygen (decreased FiO2) | High altitudeLow oxygen content of gas mixtureEnclosed breathing spaces (suffocation) |
| Hypoventilation of the alveoli | Lack of neurologic stimulation of the respiratory center (oversedation, drug overdose, neurologic damage) |
| Defects in chest wall mechanics (neuromuscular disease, trauma, chest deformity, air trapping), | |
| Large airway obstruction (laryngospasm, foreign body aspiration, neoplasm) | |
| Increased work of breathing (emphysema, severe asthma) | |
| Ventilation-perfusion mismatch | AsthmaChronic bronchitisPneumoniaAcute respiratory distress syndromeAtelectasisPulmonary embolism |
| Alveolocapillary diffusion abnormality | EdemaFibrosisEmphysema |
| Decreased pulmonary capillary perfusion | Intracardiac defectsIntrapulmonary arteriovenous malformations |
The amount of oxygen in the alveoli is called the PAO2 and is dependent on two factors. The first factor is the presence of adequate oxygen content of the inspired air. The amount of oxygen in inspired air is expressed as the percentage or fraction of air that is composed of oxygen called the FiO2. The FiO2 of air at sea level is approximately 21% or 0.21. Anything that decreases the FiO2 (such as high altitude) decreases the PAO2. The second factor is the amount of alveolar minute ventilation (tidal volume × respiratory rate). Hypoventilation results in an increase in PACO2 and a decrease in PAO2 such that there is less oxygen available in the alveoli for diffusion into the blood. This type of hypoxemia can be completely corrected if alveolar ventilation is improved by increases in the rate and depth of breathing. Hypoventilation causes hypoxemia in unconscious persons; in people with neurologic, muscular, or bone diseases that restrict chest expansion; and in individuals who have COPD.
Diffusion of oxygen from the alveoli into the blood is also dependent on two factors. The first is the balance between the amount of air getting into alveoli (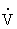 ) and the amount of blood perfusing the capillaries around the alveoli (
) and the amount of blood perfusing the capillaries around the alveoli (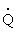 ). An abnormal ventilation-perfusion ratio (
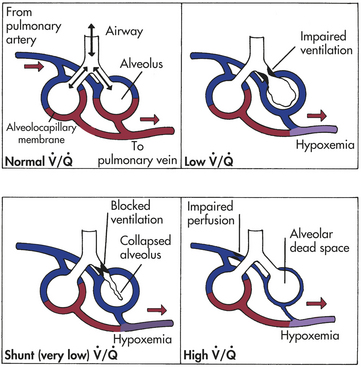
). An abnormal ventilation-perfusion ratio (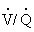 ) is the most common cause of hypoxemia (Figure 33-2). Normally, alveolocapillary lung units receive almost equal amounts of ventilation and perfusion. The normal is 0.8 to 0.9 because perfusion is somewhat greater than ventilation in the lung bases and because some blood is normally shunted to the bronchial circulation. mismatch refers to an abnormal distribution of ventilation and perfusion. Hypoxemia can be caused by inadequate ventilation of well-perfused areas of the lung (low ). Mismatching of this type, called shunting, occurs in atelectasis, in asthma as a result of bronchoconstriction, and in pulmonary edema and pneumonia when alveoli are filled with fluid. When blood passes through portions of the pulmonary capillary bed that receive no ventilation, right-to-left shunt occurs, resulting in decreased systemic PaO2 and hypoxemia. Hypoxemia also can be caused by poor perfusion of well-ventilated portions of the lung (high ), resulting in wasted ventilation. The most common cause of high is a pulmonary embolus that impairs blood flow to a segment of the lung. An area where alveoli are ventilated but not perfused is termed alveolar dead space.
) is the most common cause of hypoxemia (Figure 33-2). Normally, alveolocapillary lung units receive almost equal amounts of ventilation and perfusion. The normal is 0.8 to 0.9 because perfusion is somewhat greater than ventilation in the lung bases and because some blood is normally shunted to the bronchial circulation. mismatch refers to an abnormal distribution of ventilation and perfusion. Hypoxemia can be caused by inadequate ventilation of well-perfused areas of the lung (low ). Mismatching of this type, called shunting, occurs in atelectasis, in asthma as a result of bronchoconstriction, and in pulmonary edema and pneumonia when alveoli are filled with fluid. When blood passes through portions of the pulmonary capillary bed that receive no ventilation, right-to-left shunt occurs, resulting in decreased systemic PaO2 and hypoxemia. Hypoxemia also can be caused by poor perfusion of well-ventilated portions of the lung (high ), resulting in wasted ventilation. The most common cause of high is a pulmonary embolus that impairs blood flow to a segment of the lung. An area where alveoli are ventilated but not perfused is termed alveolar dead space.
The second factor affecting diffusion of oxygen from the alveoli into the blood is the alveolocapillary barrier. Diffusion of oxygen through the alveolocapillary membrane is impaired if the alveolocapillary membrane is thickened or the surface area available for diffusion is decreased. Abnormal thickness, as occurs with edema (tissue swelling) and fibrosis (formation of fibrous lesions), increases the time required for diffusion across the alveolocapillary membrane. If diffusion is slowed enough, the oxygen in the alveolar gas (PAO2) and capillary blood does not have time to equilibrate during the fraction of a second that blood remains in the capillary. Destruction of alveoli, such as that which occurs in emphysema, decreases the surface area available for diffusion. Hypercapnia is rarely produced by impaired diffusion because carbon dioxide diffuses so easily from capillary to alveolus that the individual with impaired diffusion would die from hypoxemia before hypercapnia could occur.
Hypoxemia most often is associated with a compensatory hyperventilation and resultant respiratory alkalosis (i.e., decreased PaCO2 and increased pH). However, in individuals with associated ventilatory difficulties, hypoxemia may be complicated by hypercapnia and respiratory acidosis. Hypoxemia results in widespread tissue dysfunction and, when severe, can lead to organ infarction. In addition, hypoxic pulmonary vasoconstriction can contribute to increased pressures in the pulmonary artery and lead to right-sided heart failure and cor pulmonale (see p. 1298). Clinical manifestations of acute hypoxemia may include cyanosis, confusion, tachycardia, edema, and decreased renal output.
Acute Respiratory Failure
Respiratory failure is defined as inadequate gas exchange, that is, hypoxemia, in which PaO3 is ≤50 mmHg, or hypercapnia, in which PaCO2 is ≥50 mmHg with a pH of ≤7.25. Respiratory failure can result from direct injury to the lungs, airways, or chest wall or indirectly because of injury to another body system such as the brain. It can occur in individuals who have an otherwise normal respiratory system or in those with underlying chronic pulmonary disease. Most pulmonary diseases can cause episodes of acute respiratory failure. If the respiratory failure is primarily hypercapnic, it is the result of inadequate alveolar ventilation (see Hypercapnia, p. 1269) and the individual must receive ventilatory support, such as with a bag-valve mask, noninvasive positive pressure ventilation, or intubation and placement on mechanical ventilation. If the respiratory failure is primarily hypoxemic, it is the result of inadequate exchange of oxygen between the alveoli and the capillaries (see Hypoxemia, p. 1269) and the individual must receive supplemental oxygen therapy. Many individuals have a combined hypercapnic and hypoxemic respiratory failure and require both kinds of support.
Respiratory failure is an important potential complication of any major surgical procedure, especially those that involve the central nervous system, thorax, or upper abdomen. Smokers are at risk, particularly if they have preexisting lung disease. Limited cardiac reserve, chronic renal failure, chronic hepatic disease, and infection also increase the tendency to develop postoperative respiratory failure. The most common postoperative pulmonary problems are atelectasis, pneumonia, pulmonary edema, and pulmonary emboli (these conditions are discussed later in this chapter).
Prevention of postoperative respiratory failure includes frequent turning, deep breathing, and early ambulation to prevent atelectasis and accumulation of secretions. Humidification of inspired air can help loosen secretions. Incentive spirometry gives individuals immediate feedback about tidal volumes, which encourages them to breathe deeply. Supplemental oxygen is given for hypoxemia, and antibiotics are given as appropriate to treat infection. If respiratory failure develops, the individual may require mechanical ventilation for a time.
DISORDERS OF THE CHEST WALL AND PLEURA
There are many conditions that can affect the chest wall and/ or pleura that affect the function of the respiratory system. Chest wall disorders primarily affect tidal volume and therefore result in hypercapnia. Pleural diseases affect ventilation and oxygenation.
Disorders of the Chest Wall
If the chest wall is deformed, traumatized, immobilized, or made heavy by fat, the work of breathing is increased and ventilation may be compromised because of a decrease in tidal volume. The degree of ventilatory impairment depends on the severity of the chest wall abnormality. Grossly obese individuals are often dyspneic on exertion or when recumbent. Individuals with severe kyphoscoliosis (lateral bending and rotation of the spinal column with distortion of the thoracic cage) often have dyspnea on exertion that can progress to respiratory failure. Such individuals are also susceptible to lower respiratory tract infections. Obesity and kyphoscoliosis are risk factors for respiratory disease in individuals admitted to a hospital for other problems, particularly those who require surgery. Other musculoskeletal abnormalities that can impair ventilation are ankylosing spondylitis and pectus excavatum (a deformity characterized by depression of the sternum) (see Chapters 42 and 43, respectively). Pain from chest wall injury, surgery, or disease is also an important cause of restriction and decreased tidal volume. This can cause significant hypoventilation, especially in those with underlying lung disease.
Impairment of respiratory muscle function caused by neuromuscular disease also can restrict the chest wall or impair pulmonary function. Muscle weakness can result in hypoventilation and hypercapnia, inability to remove secretions, and hypoxemia. The most common cause of hospital admission for individuals with neuromuscular diseases, such as poliomyelitis, muscular dystrophy, myasthenia gravis, and Guillain-Barré syndrome, is respiratory difficulty. (See Unit V for a more complete discussion of these disorders.)
Because chest wall restriction results in a decrease in tidal volume, an increase in respiratory rate can temporarily compensate and restore minute ventilation. However, many individuals will eventually progress to hypercapnic respiratory failure. Diagnosis of chest restriction is made by pulmonary function testing (reduction in forced vital capacity [FVC]), arterial blood gas measurement (hypercapnia), and radiographs. Treatment is aimed at any reversible underlying cause but is otherwise supportive. In severe cases, mechanical ventilation may be indicated.
Flail Chest
Flail chest results from the fracture of several consecutive ribs in more than one place, or the fracture of the sternum plus several consecutive ribs. These multiple fractures result in instability of a portion of the chest wall, causing paradoxical movement of the chest with breathing. During inspiration the unstable portion of the chest wall moves inward and during expiration it moves outward, impairing movement of gas in and out of the lungs (Figure 33-3). Flail chest is usually associated with significant underlying lung contusion.
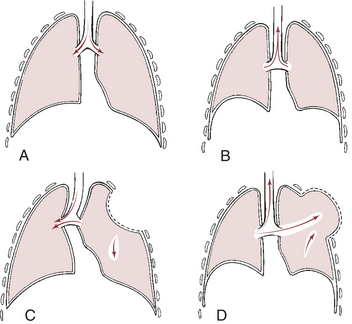
Figure 33-3 Flail chest. Normal respiration: A, inspiration; B, expiration. Paradoxical motion: C, inspiration, area of lung underlying unstable chest wall sucks in on inspiration; D, expiration, unstable area balloons out. Note movement of mediastinum toward opposite lung during inspiration.
The clinical manifestations of flail chest are pain, dyspnea, unequal chest expansion, hypoventilation, and hypoxemia. Treatment is internal fixation by controlled mechanical ventilation until the chest wall has stabilized.
Pleural Abnormalities
Pneumothorax is the presence of air or gas in the pleural space caused by a rupture in the visceral pleura (which surrounds the lungs) or the parietal pleura and chest wall (see Chapter 32). As air separates the visceral and parietal pleurae, it destroys the negative pressure of the pleural space. This disrupts the state of equilibrium that normally exists between elastic recoil forces of the lung and chest wall. No longer held in check by the recoil forces of the chest wall, the lung fulfills its tendency to recoil by collapsing toward the hilum (Figure 33-4).
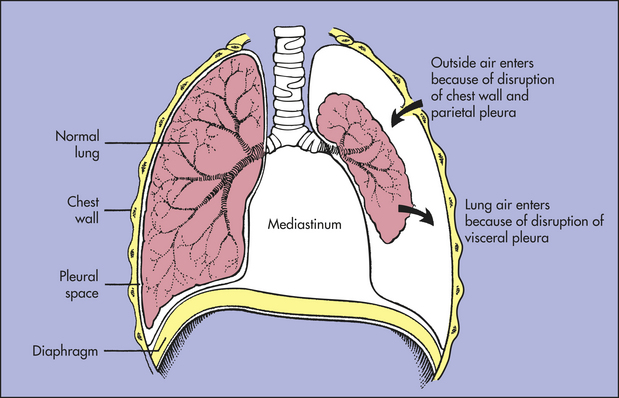
Figure 33-4 Pneumothorax. Air in the pleural space causes the lung to collapse around the hilus and may push mediastinal contents (heart and great vessels) toward the other lung.
Primary (spontaneous) pneumothorax, which occurs unexpectedly in healthy individuals (usually men) between ages 20 and 40 years, is most often caused by the spontaneous rupture of blebs (blister-like formations) on the visceral pleura.9,10 The cause of bleb formation is not known, although more than 80% of these individuals have been found to have emphysema-like changes in their lungs even if they have never smoked or have no known genetic disorder. Approximately 10% of affected individuals have a significant family history of primary pneumothorax that has been linked to mutations in the folliculin gene.11 Bleb rupture can occur during sleep, rest, or exercise. The ruptured bleb or blebs are usually located in the apexes of the lungs. A secondary pneumothorax can be caused by chest trauma, such as a rib fracture, stab or bullet wounds, or surgical procedure that tears the pleura; rupture of a bleb or bulla (larger vesicle) as occurs in COPD; or mechanical ventilation, particularly if it includes positive end-expiratory pressure (PEEP).10
Spontaneous and secondary pneumothorax can present as either open or tension. In open pneumothorax (communicating pneumothorax), air pressure in the pleural space equals barometric pressure because air that is drawn into the pleural space during inspiration (through the damaged chest wall and parietal pleura or through the lungs and damaged visceral pleura) is forced back out during expiration. In tension pneumothorax, however, the site of pleural rupture acts as a one-way valve, permitting air to enter on inspiration but preventing its escape by closing up during expiration. As more and more air enters the pleural space, air pressure in the pneumothorax begins to exceed barometric pressure. The pathophysiologic effects of tension pneumothorax are life threatening. Air pressure in the pleural space pushes against the already recoiled lung, causing compression atelectasis, and against the mediastinum, compressing and displacing the heart and great vessels.
Clinical manifestations of spontaneous or secondary pneumothorax begin with sudden pleural pain, tachypnea, and possibly mild dyspnea. The manifestations depend on the size of the pneumothorax. Physical examination may reveal absent or decreased breath sounds and hyperresonance to percussion on the affected side. Clinical manifestations of tension pneumothorax may also include severe hypoxemia, dyspnea, tracheal deviation away from the affected lung, and hypotension (low blood pressure).
Diagnosis of open pneumothorax is made with chest radiographs and CT. A thoracostomy (chest) tube is placed, and its efficacy in relieving the pneumothorax is documented on repeat chest radiograph. The diagnosis of tension pneumothorax is made on physical examination alone. It requires immediate treatment and a chest tube must be placed quickly. If a chest tube is not readily available, a large-bore needle is inserted into the pleural space to decompress it until a chest tube can be placed. An outward gush of air as the needle or chest tube is inserted confirms the presence of tension pneumothorax. For both open and tension pneumothorax, the chest tube is connected to a water-seal drainage and suction until the damaged pleura is healed.
In some situations, the pleural tear does not heal spontaneously and it is necessary to prevent recurrence of the pneumothorax by a process called pleurodesis. This procedure uses the chest tube to instill a caustic substance, such as talc, into the pleural space. The resultant inflammation and scarring as the pleura heals result in closure of the pleural tear. Some individuals require thoracotomy with pleurectomy.
Pleural Effusion
Pleural effusion is the presence of fluid in the pleural space. The source of the fluid is usually blood vessels or lymphatic vessels lying beneath either pleura, but occasionally an abscess or other lesion may drain into the pleural space. Because the pleura is a relatively permeable membrane, fluids that accumulate in the lung can cross into the pleural space.
The most common mechanism of pleural effusion is migration of fluids and other blood components through the walls of intact capillaries bordering the pleura. Pleural effusions that enter the pleural space from the intact blood vessels can be transudative or exudative. In transudative effusion, the fluid, or transudate, is watery and diffuses out of the capillaries as a result of disorders that increase intravascular hydrostatic pressure or decrease capillary oncotic pressure. Examples are congestive heart failure, in which venous and left atrial pressures are increased, and liver or kidney disorders that cause hypoproteinemia. Hypoproteinemia decreases capillary oncotic pressure, which promotes diffusion of water out of the capillaries. (This mechanism is discussed in Chapter 3).
Exudative effusion is less watery and contains high concentrations of white blood cells and plasma proteins. Exudative effusion occurs in response to inflammation, infection, or malignancy and involves inflammatory processes that increase capillary permeability (see Chapter 6). When stimulated by biochemical mediators of inflammation, junctions in the capillary endothelium separate slightly, enabling leukocytes and plasma proteins to migrate out into affected tissues. Other types of pleural effusion are characterized by the presence of pus (empyema), blood (hemothorax), or chyle (chylothorax). Mechanisms of pleural effusion are summarized in Table 33-2.
Table 33-2
Mechanisms of Pleural Effusion
| Type of Fluid/Effusion | Source of Accumulation | Primary or Associated Disorder |
| Transudate (hydrothorax) | Watery fluid that diffuses out of capillaries beneath the pleurae (i.e., capillaries in lung or chest wall) | Cardiovascular disease that causes high blood pressure; liver or kidney disease that disrupts plasma protein production, causing hypoproteinemia (decreased oncotic pressure in the blood vessels) |
| Exudate | Fluid rich in proteins (leukocytes, plasma proteins of all kinds; see Chapter 8) that migrates out of the capillaries | Infection, inflammation, or malignancy of the pleurae that stimulates mast cells to release biochemical mediators that increase capillary permeability |
| Empyema (pus) | Detritus of infection (microorganisms, leukocytes, cellular debris) dumped into the pleural space by blocked lymphatic vessels | Pulmonary infections, such as pneumonia; lung abscesses; infected wounds |
| Hemothorax (blood) | Hemorrhage into the pleural space | Traumatic injury, surgery, rupture, or malignancy that damages blood vessels |
| Chylothorax (chyle) | Chyle (milky fluid containing lymph and fat droplets) that is dumped by lymphatic vessels into the pleural space instead of passing from the gastrointestinal tract to the thoracic duct | Traumatic injury, infection, or disorder that disrupts lymphatic transport |
NOTE: The principles of diffusion are discussed in Chapter 1; mechanisms that increase capillary permeability and cause exudation of cells and proteins are discussed in Chapter 8.
Small pleural effusions may not affect lung function and go undetected. Most will be removed by the lymphatic system once the underlying condition is resolved. Like pneumothorax, larger pleural effusions can cause compression atelectasis and displace mediastinal contents. Unlike pneumothorax, however, pleural effusion does not cause the lung to collapse. Because there is no communication between the pleural space and environmental air, pressure in the pleural space remains negative and atelectasis is caused solely by pressure exerted by the effusion.
The most common symptom associated with pleural effusion is dyspnea. Pleuritic chest pain may be present if the pleura is inflamed. Physical examination usually reveals decreased breath sounds and dullness to percussion on the affected side, and a pleural friction rub may be heard. In large, rapidly developing effusions, compression atelectasis may cause hypoxemia and mediastinal shift. Inability to expand the lungs may impair ventilation, leading to hypercapnia. Diagnosis is confirmed by chest x-ray and thoracentesis (needle aspiration) with pleural fluid analysis, which can determine the type of effusion and provide symptomatic relief.12 Small effusions will usually resolve with treatment of the underlying disorder. Large pleural effusions can contain several liters of fluid and require the placement of a thoracostomy (chest) tube.
Empyema
Empyema (infected pleural effusion) is the presence of pus in the pleural space. It is thought to develop when the pulmonary lymphatics become blocked, leading to an outpouring of contaminated lymphatic fluid into the pleural space. Empyema occurs most commonly in older adults and children and usually develops as a complication of pneumonia, surgery, trauma, or bronchial obstruction from a tumor.13 Commonly documented infectious microorganisms include Staphylococcus aureus, Escherichia coli, anaerobic bacteria, and Klebsiella pneumoniae.
Individuals with empyema are usually quite ill and may have cyanosis, fever, tachycardia (rapid heart rate), cough, and pleural pain. Breath sounds are decreased directly over the empyema. Diagnosis is made by chest radiographs and thoracentesis. Identification of the causative microorganism by positive cultures from the pleural fluid is obtained only about 50% of the time, and empiric antibiotics may be needed. The treatment for empyema includes the administration of appropriate antimicrobials, and thoracentesis is performed to drain the pleural space. Continuous drainage with a chest tube may be required. In severe cases, instillation of fibrinolytic agents or deoxyribonuclease (DNase) into the pleural space may be required to mobilize the fluid and facilitate drainage. Surgical débridement of the pleural space also may be performed to prevent reaccumulation and achieve adequate drainage.
PULMONARY DISORDERS
Restrictive lung disorders are characterized by decreased compliance of lung tissue. This means that it takes more effort to expand the lungs during inspiration, which increases the work of breathing. Individuals with lung restriction complain of dyspnea and have an increased respiratory rate and decreased tidal volume. Pulmonary function testing reveals a decrease in FVC. Restrictive lung diseases can cause ventilation and perfusion mismatch or can affect the alveolocapillary membrane. In both cases there is decreased diffusion of oxygen from the alveoli into the blood, resulting in hypoxemia. Some of the most common restrictive lung diseases in adults are aspiration, atelectasis, bronchiectasis, bronchiolitis, pulmonary fibrosis, inhalational disorders, pneumoconiosis, allergic alveolitis, pulmonary edema, and acute respiratory distress syndrome.
Aspiration
Aspiration is the passage of fluid and solid particles into the lung. It tends to occur in individuals whose normal swallowing mechanism and cough reflex are impaired by a decreased level of consciousness or central nervous system abnormalities. It has been estimated that more than 10% of all hospital admissions for community-acquired pneumonias and up to 30% of admissions for pneumonia in residents of long-term facilities are the result of aspiration.13 Predisposing factors include altered level of consciousness caused by substance abuse, sedation, or anesthesia; seizure disorders; cerebrovascular accident; and neuromuscular disorders that cause dysphagia. In individuals who require enteral feeding (through a nasogastric feeding tube), aspiration is common and frequently leads to bacterial pneumonia.14 The right lung, particularly the right lower lobe, is more susceptible to aspiration than the left lung because the branching angle of the right mainstem bronchus is straighter than the branching angle of the left mainstem bronchus.
The effects of aspiration depend on the material aspirated. The aspiration of large food particles or gastric fluid with pH of less than 2.5 has serious consequences. Solid food particles can obstruct a bronchus, resulting in bronchial inflammation and collapse of airways distal to the obstruction. If the aspirated solid is not identified and removed by bronchoscopy, a chronic, local inflammation develops that may lead to recurrent infection and bronchiectasis (permanent dilation of the bronchus). Once the pathologic process has progressed to bronchiectasis, surgical resection of the affected area is usually required.
Aspiration of oral or pharyngeal secretions can lead to aspiration pneumonia, especially if the oral cavity is colonized with bacteria (e.g., individuals with poor dentition). Intubation of the trachea also can cause aspiration and bacterial pneumonia. Aspiration of acidic gastric fluid may cause severe pneumonitis. Bronchial damage includes inflammation, loss of ciliary function, and bronchospasm. In the alveoli, acidic fluid damages the alveolocapillary membrane. This allows plasma and blood cells to move from capillaries into the alveoli, resulting in hemorrhagic pneumonitis. The lung becomes stiff and noncompliant as surfactant production is disrupted, leading to further edema and collapse. Hypoventilation may develop as this progresses, and systematic complications, such as hypotension, may occur.
The clinical manifestations of aspiration include the sudden onset of choking and intractable cough with or without vomiting, fever, dyspnea, and wheezing. Some individuals have no symptoms acutely; instead they have recurrent lung infections, chronic cough, or persistent wheezing over months and even years.
Preventive measures for individuals at risk are more effective than treatment of known aspiration. The most important preventive measures include a semirecumbent position, surveillance of enteral feeding, use of promotility agents, and avoidance of excessive sedation. Individuals undergoing general anesthesia should not receive food or fluid for several hours before or after surgery. Antacids are sometimes given to individuals at risk for aspiration to keep gastric pH greater than 2.5. Individuals who have difficulty swallowing are fed with extreme caution and positioned to minimize the likelihood of aspiration. Nasogastric tubes, which often are used to remove stomach contents and reduce the risk for aspiration, also can cause aspiration if fluid and particulate matter are regurgitated as the tube is being placed. For those who suffer from swallowing difficulties, speech-language pathologists can often improve swallowing abilities and prevent recurrence.
The rate of deaths resulting from aspiration-caused pneumonitis has been estimated to be as high as 50%. Treatment includes supplemental oxygen and may require mechanical ventilation with PEEP. Fluids are restricted to decrease blood volume and minimize pulmonary edema. Steroids often are administered during the first 72 hours after aspiration, although their effectiveness is not well documented. Bacterial pneumonia may develop as a complication of aspiration pneumonitis and must be treated with broad-spectrum antibiotics.
Atelectasis
Atelectasis is the collapse of lung tissue. There are three types of atelectasis: compression, absorption, and surfactant impairment15:
1. Compression atelectasis is caused by the external pressure exerted on lung tissue, such as occurs with tumors, or by fluid or air in the pleural space. Atelectasis at the base of the lungs can be caused by abdominal distention pressing on a portion of the lung, causing the alveoli to collapse.
2. Absorption atelectasis results from gradual absorption of air from obstructed or hypoventilated alveoli or from inhalation of concentrated oxygen or anesthetic agents.
3. Surfactant impairment results from decreased production or inactivation of surfactant that is necessary to reduce surface tension in the alveoli and thus prevent lung collapse during expiration. Surfactant impairment can occur because of prematurity, acute respiratory distress syndrome, anesthesia, or mechanical ventilation.
Atelectasis tends to occur after surgery. Intraoperative high-dose supplemental oxygen in combination with general anesthesia increases the likelihood of postoperative atelectasis.15 In addition, individuals are often in pain, breathe shallowly, are reluctant to change position, and produce viscous secretions that tend to pool in dependent portions of the lung after surgical procedures, especially those involving the thorax or upper abdomen.
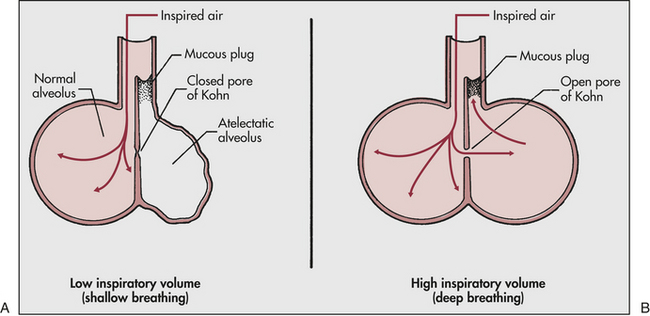
Clinical manifestations of atelectasis are similar to those of pulmonary infection: dyspnea, cough, fever, and leukocytosis. Prevention and treatment of postoperative atelectasis usually include deep breathing (often with the aid of an incentive spirometer), frequent position changes, and early ambulation. Deep breathing is beneficial because it (1) promotes the ciliary clearance of secretions, (2) stabilizes the alveoli by redistributing surfactant, and (3) permits collateral ventilation of the alveoli through pores of Kohn in the alveolar septa. The pores of Kohn, which open only during deep breathing, allow air to pass from well-ventilated alveoli to obstructed alveoli, minimizing their tendency to collapse and facilitating expectoration of the bronchial obstruction (Figure 33-5).
Bronchiectasis
Bronchiectasis is persistent abnormal dilation of the bronchi. It usually occurs in conjunction with other respiratory conditions that are associated with chronic bronchial inflammation. Causes include obstruction of an airway with mucous plugs, atelectasis, aspiration of a foreign body, infection, cystic fibrosis, tuberculosis, congenital weakness of the bronchial wall, or impaired defense mechanisms. Bronchiectasis is also associated with a number of systemic disorders such as rheumatologic disease, inflammatory bowel disease, and immunodeficiency syndromes (e.g., acquired immunodeficiency syndrome [AIDS]).16
Chronic inflammation of the bronchi leads to destruction of elastic and muscular components of their walls and permanent dilation.16 Bronchial dilation (Figure 33-6) may be cylindrical (cylindrical bronchiectasis), with symmetrically dilated airways as can be seen after pneumonia and is reversible; saccular (saccular bronchiectasis), in which the bronchi become large and balloon-like; or varicose (varicose bronchiectasis), in which constrictions and dilations deform the bronchi. In both varicose and saccular bronchiectasis, the smaller bronchial divisions are plugged with secretions or obliterated by fibrosis. Large anastomoses (connections) develop between the bronchial and pulmonary blood vessels, increasing blood flow through the bronchial circulation. These anastomoses are thought to cause the hemoptysis experienced by individuals with bronchiectasis. Airway damage leads to bronchospasm and copious production of purulent mucus. Ventilation-perfusion abnormalities develop and result in hypoxemia. In severe cases, minute ventilation is also compromised and PaCO2 may become elevated.
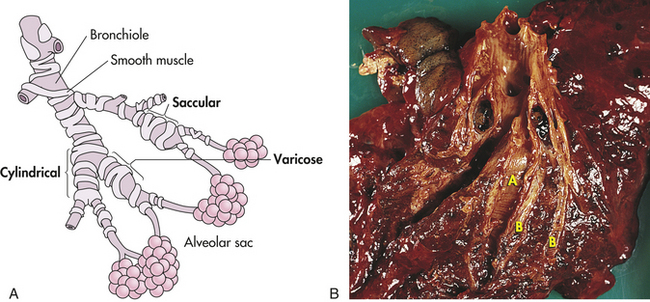
Figure 33-6 Bronchiectasis. A, Types of bronchiectasis. B, Cylindrical bronchiectasis. The dilated bronchi (A) and bronchioles (B) can be dissected almost to the pleural surface. (B, From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
The primary symptom of bronchiectasis is chronic productive cough. The symptoms of bronchiectasis may date back to a childhood illness or infection. The disease is commonly associated with recurrent lower respiratory tract infections and expectoration of voluminous amounts of purulent sputum (measured in cupfuls). If the individual is not receiving antibiotics, the sputum has a foul odor. Hemoptysis and clubbing of the fingers are common. Pulmonary function studies show decreases in FVC and expiratory flow rates. The diagnosis is usually confirmed by the use of high-resolution CT. Bronchiectasis is treated with antibiotics, bronchodilators, chest physiology, and supplemental oxygen.16 In selected individuals with localized areas of involvement, surgery may be indicated to remove the affected portion of the lung.
Bronchiolitis
Bronchiolitis is inflammation of the small airways or bronchioles. It is most common in children (see Chapter 34). In adults it usually occurs with chronic bronchitis but can occur in otherwise healthy individuals in association with an upper or lower airway viral infection (e.g., respiratory syncytial virus [RSV]), or with inhalation of toxic gases. Atelectasis or emphysematous destruction of the alveoli may develop distal to the inflammatory lesion. Bronchiolitis is usually diffuse. The resulting decrease in the ventilation-perfusion ratio results in hypoxemia. A decrease in minute ventilation with resulting carbon dioxide retention also may occur as lung restriction worsens.
Clinical manifestations include a rapid ventilatory rate; marked use of accessory muscles; low-grade fever; dry, nonproductive cough; and hyperinflated chest. If bronchiolitis is caused by an inhalation injury, pulmonary edema occurs rapidly and then quickly clears. One to 2 weeks later, respiratory distress develops, and infiltrates are seen on chest radiographs. Bronchiolitis is treated with appropriate antibiotics, steroids, and chest physical therapy (humidified air, coughing and deep breathing, postural drainage).
Bronchiolitis obliterans is a late-stage fibrotic process that occludes the airways and causes permanent scarring of the lungs. This process can occur in all causes of bronchiolitis but is most common after lung transplantation. Bronchiolitis obliterans can be further complicated by the development of pneumonia (called bronciolitis obliterans organizing pneumonia [BOOP]) in which the alveoli and bronchioles become filled with plugs of connective tissue.16 This complication of lung transplant has a high morbidity. Diagnosis is made by spirometry and bronchoscopy with biopsy. Treatment includes corticosteroids and other immunosuppressive agents.17
Pulmonary Fibrosis
Pulmonary fibrosis is an excessive amount of fibrous or connective tissue in the lung. When no specific cause for the development of fibrosis is known, it is called idiopathic pulmonary fibrosis. Although fibrosis can complicate healing after active pulmonary diseases, such as ARDS or tuberculosis, specific causes most often include inhalation of harmful substances, such as toxic gases, inorganic dusts, or organic dusts, and underlying autoimmune systemic disorders, such as rheumatologic disease. The fibrotic process results from chronic inflammation, alveolar epithelialization, and myofibroblast proliferation. Fibrosis causes a marked loss of lung compliance. The lung becomes stiff and difficult to ventilate, and the diffusing capacity of the alveolocapillary membrane may decrease, causing hypoxemia.
Idiopathic Pulmonary Fibrosis: Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial lung disorder. It is more common in men than in women and most cases occur after age 60. The median survival is only 2 to 4 years after diagnosis. IPF is characterized by chronic inflammation and fibroproliferation of the interstitial lung tissue around the alveoli. This causes decreased oxygen diffusion across the alveolocapillary membrane and hypoxemia. As the disease progresses decreased lung compliance leads to increased work of breathing, decreased tidal volume, and resultant hypoventilation with hypercapnia. Acute exacerbations of IPF can occur with rapid decompensation and a mortality as high as 40%.18 The primary symptom of IPF is increasing dyspnea on exertion; examination reveals diffuse inspiratory crackles and diagnosis is confirmed by pulmonary function testing (decreased FVC), high-resolution CT, and lung biopsy. Treatment with corticosteroids alone causes remission in approximately 50% of individuals. Combined treatment with cytotoxic drugs has a higher success rate but also higher toxicity. Newer therapies include antifibrotic drugs (such as N-acetylcysteine and pirfenidone), interferon, and anticoagulation.19 Selected individuals may benefit from lung transplantation.
Exposure to Toxic Gases: Inhalation of gaseous irritants can cause significant respiratory dysfunction. Commonly encountered toxic gases include ammonia, hydrogen chloride, sulfur dioxide, chlorine, phosgene, and nitrogen dioxide. Inhalation injuries in burns can include toxic gases from household or industrial combustants, heat, and smoke particles. Inhaled toxic particles cause damage to the airway epithelium, mucus secretion, inflammation, mucosal edema, ciliary damage, pulmonary edema, and surfactant inactivation. The cellular effects of toxic gases are described in Chapter 2. Acute toxic inhalation is frequently complicated by the ARDS and pneumonia.20 Initial symptoms include burning of the eyes, nose, and throat; coughing; chest tightness; and dyspnea. Hypoxemia is common. Treatment includes supplemental oxygen, mechanical ventilation with PEEP, and support of the cardiovascular system. Steroids sometimes are used, although their effectiveness has not been well documented. Most individuals respond quickly to therapy. Some, however, may improve initially and then deteriorate as a result of bronchiectasis or bronchiolitis.
Prolonged exposure to high concentrations of supplemental oxygen can result in a relatively rare iatrogenic condition known as oxygen toxicity. The basic underlying mechanism of injury is a severe inflammatory response mediated primarily by oxygen radicals. The result is damage to alveolocapillary membranes, disruption of surfactant production, interstitial and alveolar edema, and decrease in compliance. Treatment involves ventilatory support and reduction of inspired oxygen concentration to less than 60% as soon as tolerated.
Pneumoconiosis: Pneumoconiosis represents any change in the lung caused by inhalation of inorganic dust particles, which usually occurs in the workplace. As in all cases of environmentally acquired lung disease, the individual’s history of exposure is important in determining the diagnosis. Pneumoconiosis often occurs after years of exposure to the offending dust, and manifestations are often difficult to differentiate from those resulting from smoking.
The dusts of silica, asbestos, and coal are the most common causes of pneumoconiosis. Others include talc, fiberglass, clays, mica, slate, cement, cadmium, beryllium, tungsten, cobalt, aluminum, and iron. Deposition of these materials in the lungs leads to chronic inflammation with scarring of the alveolar capillary membrane, leading to pulmonary fibrosis and progressive pulmonary deterioration (see p. 1277). Clinical manifestations with advancement of disease may include cough, sputum production, dyspnea, decreased lung volumes, and hypoxemia. In most cases, diagnosis is made by chest x-ray, CT, and careful occupational history.21 Treatment is usually palliative and focuses on preventing further exposure and improving working conditions, along with pulmonary rehabilitation and management of associated hypoxemia and bronchospasm.
Silicosis is a type of pneumoconiosis resulting from the inhalation of free silica (silicon dioxide) and silica-containing compounds. Silica exposure occurs in mining and other industries involved with the extraction and processing of ores; preparation and use of sand; and manufacture of pipe, building, and roofing materials. Silica exposure activates innate and adaptive immune mechanisms and causes tissue injury and cellular apoptosis.22 Acute inflammation contributes to bronchospasm and wheezing. Persistent alveolitis progresses to diffuse fibrosis and nodules within the lung. Release of proteolytic enzymes and toxic oxygen radicals increases the risk for lung cancer.22 Exposed individuals may remain asymptomatic long after the nodules are visible on chest radiography. When clinical manifestations do appear, they include cough and dyspnea. There is no specific treatment for the disease, although corticosteroids may produce some improvement in the early, more acute stages.
Coal worker pneumoconiosis (coal miner lung, black lung) is caused by coal dust deposits in the lung. Although coal dust itself is relatively well tolerated by the lung, it is frequently inhaled as a mixture of coal, silica, and quartz, which is strongly inflammatory.23 Its mild form is asymptomatic, except for possible chronic bronchitis. Its advanced form consists of severe pulmonary fibrosis. Individuals usually are seen with a productive cough and wheezing. Symptoms are more severe with advanced disease and mimic those of chronic bronchitis (see p. 1287). Diagnosis is made by history of exposure and characteristic chest radiographs. There is no specific treatment for coal worker pneumoconiosis. Individuals with the mild form of the disease usually do well. Those with more complicated forms often develop marked cardiopulmonary dysfunction.
Asbestos exposure affects not only factory workers but also individuals who live in areas of asbestos emission. Asbestos exposure can result in a type of pulmonary fibrosis called asbestosis, but can also cause lung cancer; mesothelioma (cancer of the pleura); or pleural plaques, especially in those also exposed to cigarette smoke.24 Asbestosis is caused by inhalation of hydrous silicates of various metals in fibrous form. Asbestos fibers cause inflammation, release of toxic oxygen radicals, and cellular apoptosis leading to both fibrosis and malignancy. The most prominent clinical manifestations of asbestosis with fibrosis are dyspnea on exertion, a nonproductive cough, diffuse inspiratory crackles on examination, hypoxemia, and decreased lung volume. Progressive disease may lead to respiratory failure and cardiac complications. Diagnosis is made by chest x-ray, pulmonary function testing, and CT. Therapy is supportive.
Allergic Alveolitis: Inhalation of organic dusts can result in an allergic inflammatory response called allergic alveolitis (hypersensitivity pneumonitis). Many allergens can cause this disorder, including grains, silage, bird droppings or feathers, wood dust (particularly redwood and maple), cork dust, animal pelts, coffee beans, fish meal, mushroom compost, and molds that grow on sugar cane, barley, and straw. The immune response to these allergens results in immunoglobulin G (IgG) antibody production and cellular immune activation with initiation of the inflammatory response.25 Granuloma formation is common.
Allergic alveolitis can be acute, subacute, or chronic. The acute form causes a fever, cough, dyspnea, and chills a few hours after exposure that resolve without treatment in 1 to 3 days. With continued exposure, the disease becomes chronic and pulmonary fibrosis develops. (The mechanisms of hypersensitivity reactions are discussed in Chapter 8.) Chronic allergic alveolitis causes weight loss, fever, fatigue, and gradually progressive respiratory failure. Diagnosis is made by obtaining a history of allergen exposure and by serum antibody testing, chest x-ray, bronchoscopy, and CT.26 Treatment consists of avoidance of the offending agent and corticosteroid administration.
Systemic Disorders
Several systemic diseases affect the airways, pleurae, or lung parenchyma, causing fibrosis, vasculitis, pulmonary hemorrhage, or granuloma formation. Clinical manifestations of lung involvement are usually nonspecific, and the diagnosis is based on involvement of other organs. There is usually no specific treatment, although corticosteroids often are used. Some of the systemic diseases affecting the lung are granulomatous disorders such as sarcoidosis, Wegener granulomatosis, lymphomatoid granulomatosis, and eosinophilic granuloma; connective tissue diseases such as rheumatoid arthritis, systemic lupus erythematosus, scleroderma, polymyositis or dermatomyositis, Sjögren syndrome, and polyarteritis nodosa; angioimmunoblastic or immunoblastic lymphadenopathy (a disease of the lymph nodes); cystic fibrosis (see Chapter 34); and Goodpasture syndrome (a pulmonary and renal disorder).
Pulmonary Edema
Pulmonary edema is excess water in the lung. The normal lung contains very little fluid. It is kept dry by lymphatic drainage and a balance among capillary hydrostatic pressure, capillary oncotic pressure, and capillary permeability. In addition, surfactant lining the alveoli repels water, keeping fluid from entering the alveoli. Predisposing factors for pulmonary edema include heart disease, ARDS, and inhalation of toxic gases. The pathogenesis of pulmonary edema is illustrated in Figure 33-7.
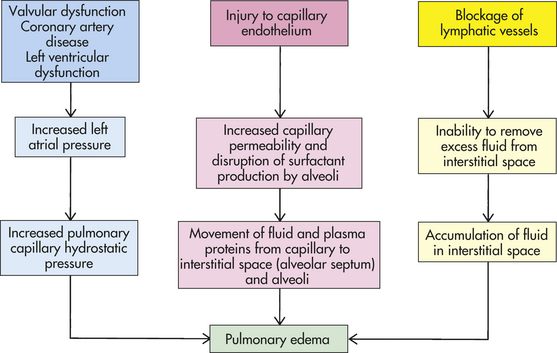
The most common cause of pulmonary edema is heart disease (see Chapter 30). When the left ventricle fails, filling pressures on the left side of the heart increase and cause a concomitant increase in pulmonary capillary hydrostatic pressure. When the hydrostatic pressure exceeds oncotic pressure, fluid moves out into the interstitium, or interstitial space (the space within the alveolar septum between alveolus and capillary). Fluid is initially picked up by lymphatic vessels and removed from the lung. When the flow of fluid out of the capillaries exceeds the lymphatic system’s ability to remove it, pulmonary edema develops. Pulmonary edema usually begins to develop at a pulmonary capillary wedge pressure or left atrial pressure of 20 mmHg. If the capillary oncotic pressure is decreased for any reason (e.g., anemia or decreased plasma proteins), pulmonary edema develops at a lower hydrostatic pressure.
Another cause of pulmonary edema is capillary injury that increases capillary permeability. Capillary injury causes edema in cases of ARDS or inhalation of toxic gases, such as ammonia. Capillary injury causes water and plasma proteins to leak out of the capillary and move into the interstitium. When plasma proteins move into the lung interstitium, they increase the interstitial oncotic pressure, which is usually very low. As the interstitial oncotic pressure begins to equal capillary oncotic pressure, water moves out of the capillary and into the lung. (Mechanisms of edema are discussed in Chapter 3.)
Pulmonary edema also can result from obstruction of the lymphatic system. Drainage can be blocked by compression of lymphatic vessels caused by edema, tumors, and fibrotic tissue or by increased systemic venous pressure that elevates hydrostatic pressure of the large pulmonary veins into which the pulmonary lymphatic system drains. This can happen in left-sided heart failure.
Clinical manifestations of pulmonary edema include dyspnea, orthopnea, hypoxemia, and increased work of breathing. Physical examination may reveal inspiratory crackles (rales), dullness to percussion over the lung bases, and evidence of ventricular dilation (S3 gallop and cardiomegaly). In severe edema, pink, frothy sputum is expectorated, hypoxemia worsens, and hypoventilation with hypercapnia may develop.
The treatment of pulmonary edema depends on its cause. If the edema is caused by increased hydrostatic pressure caused by heart failure, therapy is geared toward improving cardiac output and volume status with diuretics, vasodilators, and drugs that improve the contraction of the heart muscle. If edema is the result of increased capillary permeability resulting from injury, the treatment is focused on removing the offending agent and supportive therapy to maintain adequate oxygenation, ventilation, and circulation. Individuals with either type of pulmonary edema require supplemental oxygen. Mechanical ventilation may be needed if edema significantly impairs ventilation and oxygenation.
Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) is a fulminant form of respiratory failure characterized by acute lung inflammation and diffuse alveolocapillary injury. In the United States, acute lung injury (a slightly milder form of lung injury) and ARDS complicate more than 30% of all intensive care unit (ICU) admissions.27 Advances in therapy have decreased the overall mortality rate to approximately 40%, although older people and those with severe infections or are immunocompromised continue to have a much higher mortality rate.27 Most survivors, however, have almost normal lung function 1 year after the acute illness. ARDS is the result of injury to the lung by numerous unrelated causes. The most common predisposing factors are sepsis and multiple trauma (especially when multiple transfusions are received); however, there are many other causes, including pneumonia, burns, aspiration, cardiopulmonary bypass surgery, pancreatitis, drug overdose, smoke or noxious gas inhalation, oxygen toxicity, radiation therapy, and disseminated intravascular coagulation.27
PATHOPHYSIOLOGY All disorders that result in ARDS cause massive pulmonary inflammation that acutely injures the alveolocapillary membrane and produces severe pulmonary edema (Figure 33-8). The alveolocapillary damage can occur directly, as with the aspiration of highly acidic gastric contents or inhalation of toxic gases, or indirectly from chemical mediators released in response to systemic disorders, as with sepsis and trauma. Because the form of pulmonary edema is not secondary to heart failure, ARDS is often referred to as noncardiogenic pulmonary edema.
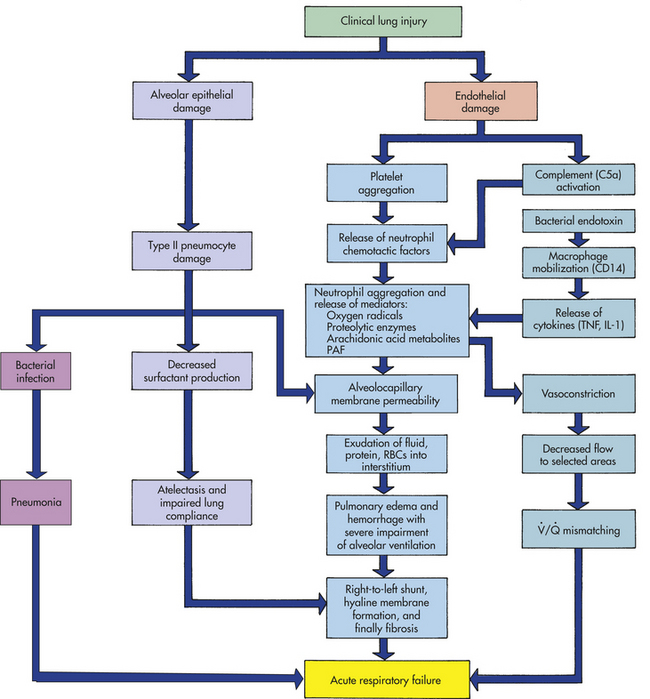
Figure 33-8 Pathogenesis of acute respiratory distress syndrome (ARDS). IL-1, Interleukin-1; PAF, platelet-activating factor; RBCs, red blood cells; TNF, tumor necrosis factor.
The initial injury to the lungs damages the pulmonary capillary endothelium, activating complement and stimulating platelet aggregation and intravascular thrombus formation. Platelets release substances that attract and activate neutrophils. In ARDS caused by sepsis, bacterial toxins are recognized by the CD14 receptors on macrophages and result in chemotaxis of large numbers of neutrophils to the lungs. A cascade of inflammatory mediators is released by the macrophages, including tumor necrosis factor (TNF), interleukin-1 (IL-1), alpha and beta chemokines, and other interleukins.28,29 Complement is also activated and contributes to lung capillary damage.
The role of neutrophils is central to the development of ARDS. Activated neutrophils release a battery of inflammatory mediators, among them proteolytic enzymes, oxygen-free radicals (superoxide radicals, hydrogen peroxide, hydroxyl radicals), arachidonic acid metabolites (prostaglandins, thromboxanes, leukotrienes), and platelet-activating factor. These mediators cause extensive damage of the alveolocapillary membrane and greatly increase capillary membrane permeability.
Increased capillary permeability, a hallmark of ARDS, allows fluids, proteins, and blood cells to leak from the capillary bed into the pulmonary interstitium and alveoli. The resulting pulmonary edema and hemorrhage severely reduce lung compliance and impair alveolar ventilation (Figure 33-9).
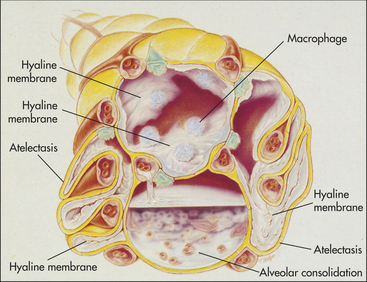
Figure 33-9 Acute respiratory distress syndrome (ARDS). Cross-sectional view of alveoli in ARDS. (Modified from Des Jardins T, Burton GG: Clinical manifestations and assessment of respiratory disease, ed 3, St Louis, 1995, Mosby.)
Mediators released by neutrophils, and to a certain extent by macrophages, also cause pulmonary vasoconstriction. Pulmonary hypertension occurs early in the course of the disease secondary to vasoconstriction and to vascular occlusion by aggregated neutrophils, macrophages, and platelets. Because vasoconstriction occurs more in some vascular beds than others, blood flow to selected areas of the lungs is decreased, resulting in mismatching.
Lung inflammation and injury damages the alveolar epithelium and the vascular endothelium. Surfactant is inactivated, and its production by type II alveolar cells is impaired as alveoli and respiratory bronchioles fill with fluid or collapse. The lungs become less compliant, resulting in increased work of breathing and decreased minute ventilation and hypercapnia.
Within 24 to 48 hours after the acute hemorrhagic phase of ARDS, hyaline membranes form, and after approximately 7 days, fibrosis progressively obliterates the alveoli, respiratory bronchioles, and interstitium. This leads to a decrease in functional residual capacity (FRC) and even more mismatching with severe right-to-left shunting. The result of this overwhelming inflammatory response by the lungs is acute respiratory failure.
The same chemical mediators responsible for the alveolocapillary damage of ARDS often cause widespread inflammation, endothelial damage, and capillary permeability throughout the body, resulting in the systemic inflammatory response syndrome (SIRS), which then leads to multiple organ dysfunction syndrome (MODS). In fact, death may not be caused by respiratory failure alone but by MODS associated with ARDS. (MODS is discussed in Chapter 46.)
CLINICAL MANIFESTATIONS The primary symptom of ARDS is progressive dyspnea. The initial physical examination may only reveal tachypnea, followed by gradually increasing inspiratory crackles heard throughout the lungs. Over the first 24 to 48 hours after injury, interstitial and alveolar infiltrates appear on chest radiographs. Hypoxemia and respiratory alkalosis are common at this stage. As pulmonary edema worsens, hypoxemia becomes refractory to oxygen therapy, and hypoventilation develops with increasing PaCO2. Worsening hypoxemia and hypercapnia lead to respiratory failure. Decreased oxygen delivery to tissues results in metabolic acidosis and organ dysfunction (e.g., decreased urine output and a decline in cognitive functioning). Decreased cardiac output and hypotension eventually lead to death. The clinical course of progressive ARDS can be summarized as follows: dyspnea and hypoxemia → hyperventilation and respiratory alkalosis → decreased tissue perfusion, organ dysfunction, and metabolic acidosis → decreased tidal volume and hypoventilation → respiratory acidosis and further hypoxemia → decreased cardiac output and hypotension → death.
EVALUATION AND TREATMENT Diagnosis is made on the basis of a history of systemic insult, physical examination, analysis of arterial blood gases, and chest x-ray. Initial physical examination may show fine inspiratory crackles, and the chest film may be clear or show a few scattered infiltrates. With progressive respiratory involvement, crackles are heard throughout the lungs and radiographs show extensive bilateral infiltrates. The criteria for diagnosis of ARDS include refractory hypoxemia, a chest x-ray with bilateral infiltrates, and the exclusion of cardiogenic pulmonary edema. Further diagnostic testing may include CT of the chest and bronchoscopy.
Treatment is based on early detection, supportive therapy, and prevention of complications such as pneumonia. Traditional therapy involves mechanical ventilation with PEEP and high oxygen concentrations. Numerous alternative modalities of ventilation are being tested, including low volume ventilation, noninvasive positive pressure ventilation, permissive hypercapnia, prone positioning, extracorporeal gas exchange, and partial liquid ventilation; some of these methods have shown apparent reductions in mortality rates.30
Many studies are investigating new ways to prevent or treat ARDS. Anticoagulant therapy with recombinant human-activated protein C improves outcomes in sepsis associated with ARDS and continues to be evaluated. Prophylactic immunotherapy, antibodies against endotoxins, antioxidants, surfactant replacement, nitric oxide inhalation, and inhibition of various inflammatory mediators are among other possibilities being tested.31 Steroid administration remains controversial but may improve overall outcomes when given in physiologic doses.32
Obstructive Pulmonary Disease
Obstructive pulmonary disease is characterized by airway obstruction that is worse with expiration. Either more force (i.e., use of accessory muscles of expiration) or more time is required to expire a given volume of air, or both. The unifying symptom of obstructive pulmonary disease is dyspnea; the unifying sign is wheezing. Individuals have an increased work of breathing, ventilation-perfusion mismatching, and a decreased forced expiratory volume in one second (FEV1). The most common obstructive diseases are asthma, chronic bronchitis, and emphysema. Because many individuals have chronic bronchitis and emphysema, these diseases together are often called COPD (Figure 33-10).
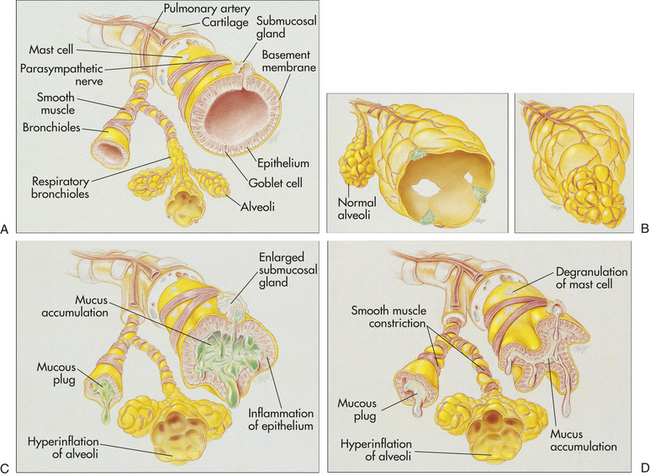
Figure 33-10 Airway obstruction caused by emphysema, chronic bronchitis, and asthma. A, The normal lung. B, Emphysema: enlargement and destruction of alveolar walls with loss of elasticity and trapping of air; (left) panlobular emphysema showing abnormal weakening and enlargement of all air spaces distal to the terminal bronchioles (normal alveoli shown for comparison only); (right) centrilobular emphysema showing abnormal weakening and enlargement of the respiratory bronchioles in the proximal portion of the acinus. C, Chronic bronchitis: inflammation and thickening of mucous membrane with accumulation of mucus and pus leading to obstruction; characterized by cough. D, Bronchial asthma: thick mucus, mucosal edema, and smooth muscle spasm causing obstruction of small airways; breathing becomes labored and expiration is difficult. (Modified from Des Jardins T, Burton GG: Clinical manifestations and assessment of respiratory disease, ed 3, St Louis, 1995, Mosby.)
Asthma
Asthma is defined as “a chronic disorder of the airways that involves a complex interaction of airway obstruction, bronchial hyperresponsiveness and an underlying inflammation.”33 Many cells and cellular elements contribute to the inflammatory response including mast cells, eosinophils, neutrophils, T lymphocytes, macrophages, and damaged epithelial cells (especially in sudden onset, fatal exacerbations, occupational asthma, and individuals who smoke). In susceptible individuals, this inflammation causes recurrent episodes of coughing (particularly at night or early in the morning), wheezing, breathlessness, and chest tightness. These episodes are usually associated with widespread but variable airflow obstruction that is often reversible either spontaneously or with treatment.33
Asthma occurs at all ages, with approximately half of all cases developing during childhood and another third before age 40. In the United States, asthma affects more than 22 million persons and is a major global health problem affecting more than 300 million people worldwide.33,34 With the projected increase in the proportion of the world’s population that lives in cities, it is estimated that there may be an additional 100 million people with asthma by 2025.34 Mortality rates have declined since 1995 in the United States, but the incidence of asthma has increased over the past two decades, especially in urban areas.33
Asthma is a familial disorder and more than 100 genes have been identified that may play a role in the susceptibility and pathogenesis of asthma, including those that influence the production of IL-4, IL-5, and IL-13; IgE; eosinophils; mast cells; adrenergic receptors; leukotrienes; and bronchial hyperresponsiveness.35–36 The ADAM 33 (a disintegrin and metalloprotease)33 gene is particularly associated with asthma and bronchial hyperresponsiveness.37 Furthermore, there is increasing understanding of the specific interactions of susceptibility genes with the environment that can guide prevention and treatment interventions.38–40 Risk factors for asthma, in addition to family history, include allergen exposure, urban residence, exposure to air pollution and cigarette smoke, recurrent respiratory viral infections, and obesity.33,41
There is significant evidence that allergy and inflammation involving the upper airway (allergic rhinitis) can contribute to lower airway inflammation and bronchospasm (“one airway hypothesis”) (see What’s New? The Link Between the Upper and Lower Airways in Asthma and COPD).42 Based on this understanding, it has been shown that prevention and treatment of allergic rhinitis may prevent the development of asthma.43
Of great interest in recent years has been the effect of recurrent allergen exposure during childhood on the subsequent development of asthma. A significant amount of evidence indicates that exposure to high levels of most allergens (e.g., dust mites) is correlated with an increased risk of asthma, although the age at the time of exposure may influence the risk.33 It also has been noted that children who live on farms or have certain childhood infections may have a decreased
risk for asthma, theoretically because they become less immunologically “primed” to be allergic (see What’s New? New Understandings of Gene and Environmental Interactions in Asthma).33,41,44 These relationships have been described as the hygiene hypothesis, and many studies are being conducted to further elucidate the relationship between allergen exposure and infection and asthma risk and to see if asthma can be prevented through allergen reduction and exposure to probiotics.33,41,44,45
Inflammation resulting in hyperresponsiveness of the airways is the major pathologic feature of asthma. Airway epithelial cell irritation combined with exposure to antigens initiates both an innate and an adaptive immune response (see Chapter 8). In sensitized individuals, allergen exposure leads to activation of T-helper cells. These cells release what are called T-helper 2 cytokines, especially IL-4, IL-5, IL-8, and IL-13. IL-4 stimulates B-cell activation, proliferation, and production of antigen-specific IgE. IgE causes mast cell degranulation with the release of a large number of inflammatory
mediators, such as histamine, prostaglandins, and leukotrienes33,41,46 (Figure 33-11; see Figure 34-14, p. 1331, for additional detail). IL-5 stimulates the activation, migration, and proliferation of eosinophils, which cause direct tissue injury and release toxic neuropeptides that contribute to increased bronchial hyperresponsiveness, fibroblast proliferation, and airway scarring.33,47 IL-8 activates polymorphonucleocytes that contribute to a more exaggerated inflammatory response. IL-13 impairs mucociliary clearance, enhances fibroblast secretion, and contributes to bronchoconstriction. The resulting inflammatory process produces bronchial smooth muscle spasm, vascular congestion, increased vascular permeability, edema formation, production of thick tenacious mucus, impaired mucociliary function (see Figure 33-10), thickening of airway walls, and increased contractile response of bronchial smooth muscle.48 Other inflammatory cytokines, such as TNF and IL-1, have been found to alter muscarinic receptor function, leading to increased levels of acetylcholine, which cause bronchial smooth muscle contraction and mucus secretion.33,49,50 These changes, combined with the epithelial cell damage caused by eosinophil infiltration, produce acute airway hyperreponsiveness and obstruction. Recent studies have identified important roles for nitric oxide and reduced airway pH in the pathogenesis of asthma and airway inflammation.51,52 Untreated inflammation can lead to long-term airway damage that is irreversible (airway remodeling).33,49
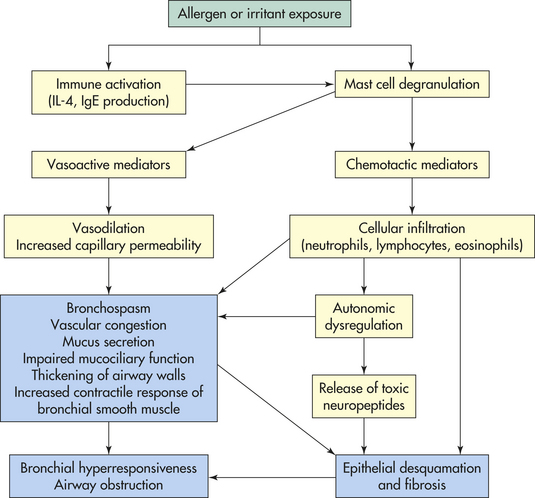
Figure 33-11 Pathophysiology of asthma. Allergen or irritant exposure results in a cascade of inflammatory events leading to acute and chronic airway dysfunction. IgE, Immunoglobulin E; IL-4, interleukin-4.
Airway obstruction increases resistance to airflow and decreases flow rates, especially expiratory flow. Impaired expiration causes air trapping, hyperinflation distal to obstructions, altered pulmonary mechanics, and increased work of breathing. Changes in resistance to airflow are not uniform throughout the lungs and the distribution of inspired air is uneven, with more air flowing to the less resistant portions. Continued air trapping increases intrapleural and alveolar gas pressures and causes decreased perfusion of the alveoli. Increased alveolar gas pressure, decreased ventilation, and decreased perfusion lead to variable and uneven ventilation-perfusion relationships within different lung segments. Hyperventilation is triggered by lung receptors responding to increased lung volume and obstruction. The result is early hypoxemia without CO2 retention. Hypoxemia further increases hyperventilation through stimulation of the respiratory center, causing PaCO2 to decrease and pH to increase (respiratory alkalosis). As the obstruction becomes more severe, the number of alveoli being inadequately ventilated and perfused increases. As air trapping in the lungs because of obstruction of expiratory airflow progresses, the lungs and thorax become hyperexpanded putting the respiratory muscles at a mechanical disadvantage. This leads to CO2 retention and respiratory acidosis. Respiratory acidosis signals respiratory failure.
CLINICAL MANIFESTATIONS Individuals are asymptomatic between attacks and pulmonary function tests are normal. No clinical symptoms are present during partial remission but pulmonary function tests are abnormal. At the beginning of an attack, the individual experiences chest constriction, expiratory wheezing, dyspnea, nonproductive coughing, prolonged expiration, tachycardia, and tachypnea. Severe attacks involve the use of accessory muscles of respiration, and wheezing is heard during both inspiration and expiration. A pulsus paradoxus (decrease in systolic blood pressure during inspiration of more than 10 mmHg) may be noted. Because the severity of blood gas alterations is difficult to evaluate by clinical signs alone, arterial blood gas tensions should be measured. In cases of significant allergen exposure, asthma symptoms can recur 4 to 12 hours after the initial attack because of persistent eosinophil and lymphocyte activation. This is called the late asthma response and it can be even more severe than the initial attack.
If bronchospasm is not reversed by usual measures, the individual is considered to have severe bronchospasm or status asthmaticus. If status asthmaticus continues, hypoxemia worsens, expiratory flows decrease further, and effective ventilation decreases. Acidosis develops as arterial PaCO2 begins to rise. Asthma becomes life threatening at this point. A silent chest (no audible air movement) and a PaCO2 greater than 70 mmHg are ominous signs of impending death.
EVALUATION AND TREATMENT The evaluation of an acute asthma attack requires the rapid assessment of arterial blood gases, expiratory flow rates (using a peak flowmeter), and a search for underlying triggers, such as infection. The degree of decrease in peak expiratory flow is a useful indicator of the severity of the attack. Hypoxemia and respiratory alkalosis are expected early in the course of an acute attack. The development of hypercapnia with respiratory acidosis signals the need for mechanical ventilation. Repeated peak flow rates help to determine whether the individual is responding to treatment. Management of the acute asthma attack requires immediate administration of oxygen and inhaled beta-agonist bronchodilators. In addition, oral corticosteroids should be administered early in the course of management.33,51,53 Careful monitoring of gas exchange and airway obstruction in response to therapy provides information necessary to determine whether hospitalization is necessary. Antibiotics are not indicated for acute asthma unless there is a documented bacterial infection.33
Further evaluation is indicated once the individual is stable. A careful family and personal history of allergies and other illness should be obtained. Pulmonary function testing (spirometry) will reveal decreases in expiratory flow rate as measured by the FEV1 (see Chapter 32). FVC will also be reduced, but much less so relative to the FEV1. FRC and total lung capacity (TLC) are increased. Responses to bronchodilator treatment also can be documented.
In 2007 the National Asthma Education and Prevention Program (NAEPP) classified asthma on clinical severity (intermittent, mild persistent, moderate persistent, and severe persistent). The NAEPP then offered six sequential guidelines for the management of chronic asthma based on this classification scheme. Chronic management of asthma begins with avoidance of allergens and other triggers. In the mildest form of asthma (intermittent), short acting beta-agonist inhalers are prescribed. However, individuals tend to underestimate the severity of their asthma and should receive extensive patient education, including the use of a peak flowmeter and the adherence to an action plan should symptoms worsen.33,34 For all categories of persistent asthma, anti-inflammatory medications are essential and inhaled corticosteroids are the mainstay of therapy.33,51 In individuals who are not adequately controlled on inhaled corticosteroids, leukotriene antagonists can be considered.46 In more severe asthma, long-acting beta agonists can be used to control persistent bronchospasm; however, they can actually worsen asthma in some individuals with certain genetic polymorphisms (see What’s New? Pharmacogenetics and Beta Agonists in the Treatment of Asthma).33,49,54,55 For those persons who do not achieve adequate asthma control on inhaled corticosteroids and a beta agonist, a monoclonal antibody that
blocks IgE (omalizumab) is recommended.33 Long-term therapy with oral corticosteroids should be avoided if possible. Immunotherapy has been shown to be highly effective in selected allergic children in preventing and treating asthma.56 Treatments, such as phosphodiesterase-4 inhibitors and specific cytokine blockers, are continually being developed and tested.51
Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease (COPD) has been defined as pathologic lung changes consistent with emphysema or chronic bronchitis. A recent consensus report defines COPD as a “preventable and treatable disease with some significant extrapulmonary effects that may contribute to the severity in individual patients. Its pulmonary component is characterized by airflow limitation that is not fully reversible. The airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lung to noxious particles or gases.”57 It is the fourth leading cause of death in the United States and is the sixth leading cause of death worldwide.57 Overall mortality from COPD has increased 103% in the United States over the past 30 years; however, mortality in women has increased more than twice that much.58 COPD is primarily caused by cigarette smoke, and active as well as passive smoking have been implicated. Other risks include occupational exposures, indoor and outdoor air pollution, and history of severe childhood respiratory infections. Genetic susceptibilities have been identified, including polymorphisms of genes that code
for TNF, surfactant, proteases, and antiproteases.59,60 Gender differences in the genes that code for the breakdown and removal of cigarette smoke metabolites may explain the increased susceptibility of women smokers to COPD and lung cancer.58 An inherited mutation in the α1-antitrypsin gene results in the development of COPD at an early age, even in nonsmokers.
Chronic Bronchitis: Chronic bronchitis is defined as hypersecretion of mucus and chronic productive cough that continues for at least 3 months of the year (usually the winter months) for at least 2 consecutive years. Incidence is increased in smokers (up to 20-fold) and even more so in workers exposed to air pollution. It is a major health problem for older adults. Repeated infections are common.
PATHOPHYSIOLOGY Inspired irritants increase not only mucus production but also the size and number of mucous glands and goblet cells in airway epithelium. The mucus produced is thicker and more tenacious than normal. This sticky mucus coating makes it much more likely that bacteria, such as Haemophilus influenzae and Streptococcus pneumoniae, will become embedded in the airway secretions, where they reproduce rapidly. Ciliary function is impaired, reducing mucus clearance further. The lung’s defense mechanisms are therefore compromised, which increases susceptibility to pulmonary infection and injury. As infection and injury increase mucus production further, the bronchial walls become inflamed and thickened from edema and accumulation of inflammatory cells. Persistent inflammation and recurrent infection lead to bronchospasm and eventual permanent narrowing of the airways.61 (The pathogenesis of chronic bronchitis is shown in Figure 33-12.)
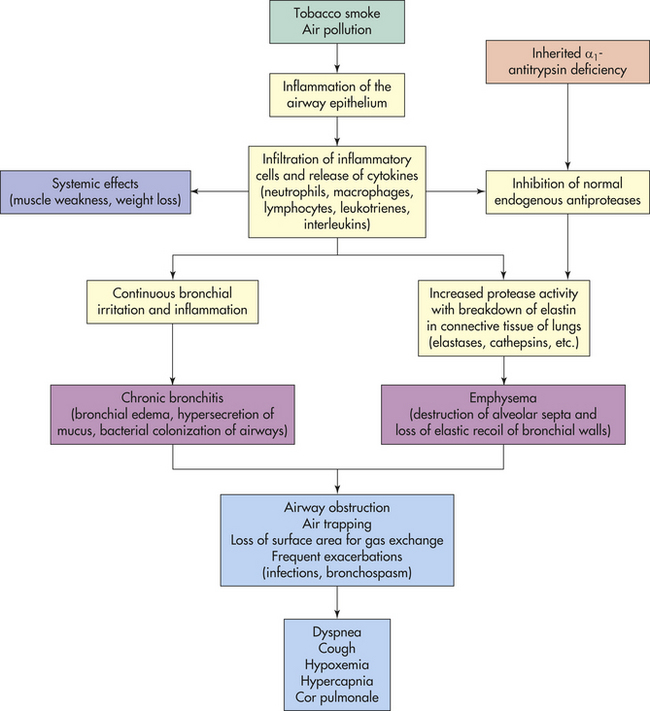
Figure 33-12 Pathogenesis of chronic bronchitis and emphysema (chronic obstructive pulmonary disease [COPD]).
Initially chronic bronchitis affects only the larger bronchi, but eventually all airways are involved. The thick mucus and hypertrophied bronchial smooth muscle obstruct the airways and lead to obstruction, particularly during expiration when the airways are narrowed (Figure 33-13). Obstruction eventually leads to ventilation-perfusion mismatch with hypoxemia. The airways collapse early in expiration, trapping gas in the distal portions of the lung. Air trapping expands the thorax, putting the respiratory muscles at a mechanical disadvantage. This leads to decreased tidal volume, hypoventilation, and hypercapnia.
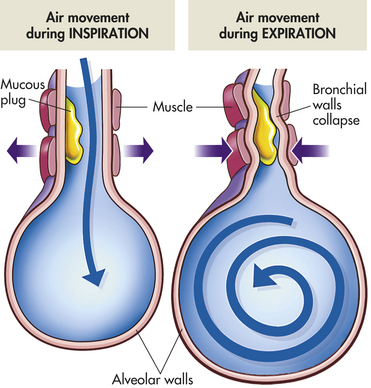
Figure 33-13 Mechanisms of air trapping in chronic obstructive pulmonary disease (COPD). Mucous plugs and narrowed airways cause air trapping and hyperinflation on expiration. During inspiration the airways are pulled open, allowing gas to flow past the obstruction. During expiration decreased elastic recoil of the bronchial walls results in collapse of the airways and prevents normal expiratory airflow.
CLINICAL MANIFESTATIONS The symptoms that lead individuals with chronic bronchitis to seek medical care include decreased exercise tolerance, wheezing, and shortness of breath. Individuals usually have a productive cough (“smoker’s cough”), and evidence of airway obstruction (decreased FEV1) is shown by spirometry. Hypoxemia may occur with exercise. As the disease progresses, copious amounts of sputum are produced, accompanied by frequent pulmonary infections.62 FVC and FEV1 become markedly reduced, and FRC and residual volume (RV) are increased as airway obstruction and air trapping become more pronounced.
Airway obstruction results in decreased alveolar ventilation and increased PaCO2. Marked hypoxemia leads to polycythemia (overproduction of erythrocytes) and cyanosis. If not reversed, hypoxemia leads to pulmonary hypertension and eventually results in cor pulmonale (see p. 1298) and can lead to severe disability or death. (Table 33-3 lists the common clinical manifestations of chronic bronchitis.)
Table 33-3
Clinical Manifestations of Chronic Pulmonary Disease
| Clinical Manifestations | Chronic Bronchitis | Emphysema |
| Productive cough | Classic sign | Late in course with infection |
| Dyspnea | Late in course | Common |
| Wheezing | Intermittent | Minimal |
| History of smoking | Common | Common |
| Barrel chest | Occasionally | Classic |
| Prolonged expiration | Always present | Always present |
| Cyanosis | Common | Uncommon |
| Chronic hypoventilation | Common | Late in course |
| Polycythemia | Common | Late in course |
| Cor pulmonale | Common | Late in course |
EVALUATION AND TREATMENT Diagnosis is made on the basis of history of symptoms, physical examination, chest radiograph, pulmonary function tests, and blood gas analyses; these tests reflect the progressive nature of the disease. The best “treatment” for chronic bronchitis is prevention because pathologic changes are not reversible. By the time an individual seeks medical care for symptoms, considerable airway damage is present. If the individual stops smoking, disease progression can be halted. If smoking is stopped before symptoms occur, the risk of chronic bronchitis decreases considerably and eventually reaches that of nonsmokers.
Bronchodilators and expectorants are prescribed to control cough and reduce dyspnea. Chest physical therapy may be helpful and includes deep breathing and postural drainage. Teaching of individuals includes nutritional counseling, respiratory hygiene, recognition of the early signs of infection, and techniques that relieve dyspnea, such as pursed-lip breathing.57 The role of antibiotics in the management of acute exacerbations of chronic bronchitis has been controversial. Good evidence now indicates that antibiotics should be used for all acute exacerbations of chronic bronchitis (change in sputum amount or color, increased dyspnea and wheezing).63,64 Oral steroids are used for acute exacerbations but should be avoided for chronic use. Inhaled corticosteroids may be useful in the treatment of chronic bronchitis in selected individuals.57,65
Individuals with severe hypoxemia require oxygen therapy to prevent cor pulmonale.66 Oxygen is administered with care to individuals with severe hypoxemia and CO2 retention. Because of the chronic elevation of PaCO2, the central chemoreceptors no longer act as the primary stimulus for breathing. (Chemoreceptors are described in Chapter 32.) This role is taken over by the peripheral chemoreceptors, which are sensitive to changes in PaO2. Peripheral chemoreceptors do not stimulate breathing if the PaO2 is much more than 60 mmHg. Therefore, if oxygen therapy causes PaO2 to exceed 60 mmHg, the stimulus to breathe decreases, PaCO2 increases, and apnea results. However, severe hypoxemia must be reversed, especially if there are comorbidities (e.g., heart disease, tissue injury) that require adequate tissue oxygenation. If adequate oxygenation cannot be achieved without resulting in respiratory depression, the individual must be mechanically ventilated.
Emphysema: Emphysema is abnormal permanent enlargement of gas-exchange airways (acini) accompanied by destruction of alveolar walls without obvious fibrosis. The major mechanism of airflow limitation in emphysema is loss of elastic recoil. Some degree of emphysema is considered normal in older adults but results in a slow and predictable decline in lung function with aging. When it occurs earlier in life, however, it is usually secondary to cigarette smoking or indoor and outdoor air pollution, although it may be primary emphysema in rare cases.
Primary emphysema, which accounts for 1% to 3% of all cases of emphysema, is commonly linked to an inherited deficiency of the enzyme α1-antitrypsin.67 Normally α1-antitrypsin inhibits the action of many proteolytic enzymes (enzymes that break down proteins). Individuals who have α1-antitrypsin deficiency (an autosomal recessive trait) have an increased likelihood of developing emphysema because proteolysis in lung tissues is not inhibited. Homozygous individuals have a 70% to 80% likelihood of developing lung disease. (Mechanisms of genetic inheritance are described in Chapter 4.) Persons with α1-antitrypsin deficiency who smoke are even more susceptible to emphysema than those with the deficiency alone. α1-Antitrypsin deficiency is suggested in individuals who develop emphysema before age 40 years (or in their early 40s) and in nonsmokers who develop emphysema. (The principles of risk factor analysis are discussed in Chapter 5.)
Secondary emphysema also is caused by an inability of the body to inhibit proteolytic enzymes in the lung. It results from an insult to the lungs from inhaled toxins, such as cigarette smoke and air pollution. Not all smokers develop emphysema, but approximately 20% are especially susceptible and develop significant lung damage if they continue to smoke.57
PATHOPHYSIOLOGY Emphysema is characterized by destruction of alveoli through the breakdown of elastin within the septa by proteases.60,68 In most individuals this process is initiated through the inhalation of inflammatory oxidants such as cigarette smoke. Toxins in smoke lead to airway epithelial inflammation with infiltration of numerous cells such as neutrophils, macrophages, and lymphocytes (see Figure 33-12). Inflammatory cytokines are released that increase protease activity and inhibit the normal endogenous antiproteases in the lung.60,68 Some of the most important proteases activated in emphysema are elastases, cathepsins, and matrix metalloproteases. The imbalance between proteases and antiproteases leads to breakdown of elastin in the alveolar septa.60,68 Septal destruction eliminates portions of the pulmonary capillary bed and results in ventilation-perfusion mismatching and hypoxemia. In addition, destruction of elastin in the bronchial walls reduces elastic recoil of the airways. Expiration becomes difficult because loss of elastic recoil reduces the volume of air that can be expired passively. Hyperinflation of alveoli causes large air spaces within the lung parenchyma (bullae) and air spaces adjacent to pleura (blebs) to develop. Septal destruction also affects airway caliber because the force that normal alveoli exert on bronchiolar walls is diminished. The combination of increased RV in the alveoli and diminished caliber of the bronchioles causes part of each inspiration to be trapped in the acinus. Air trapping causes hyperexpansion of the chest, which puts the muscles of respiration at a mechanical disadvantage. This results in increased work of breathing so that many individuals will develop hypoventilation and hypercapnia late in the course of the disease. Persistent inflammation in the airways can result in hyperreactivity of the bronchi with bronchoconstriction, which may be partially reversible with bronchodilators. Chronic inflammation also can have significant systemic effects including weight loss, muscle weakness, and increased susceptibility to comorbidities, such as infection.57
Emphysema can be centriacinar (centrilobular) or panacinar (panlobular), depending on the site of involvement (Figure 33-14). In centriacinar emphysema septal destruction occurs in the respiratory bronchioles and alveolar ducts, usually in the upper lobes of the lung. The alveolar sac (alveoli distal to the respiratory bronchiole) remains intact. It tends to occur in smokers with chronic bronchitis. Panacinar emphysema involves the entire acinus, with damage more randomly distributed and involving the lower lobes of the lung. It tends to occur in older adults and in those with α1-antitrypsin deficiency.
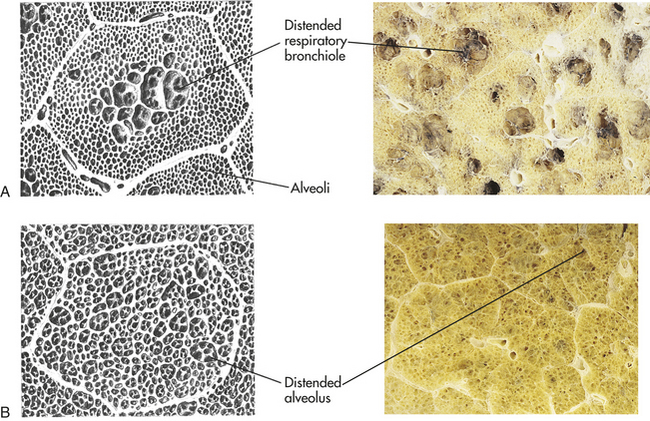
Figure 33-14 Types of emphysema. A, Centriacinar emphysema. B, Panacinar emphysema. (Micrographs from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
CLINICAL MANIFESTATIONS Individuals with emphysema usually have dyspnea on exertion that later progresses to marked dyspnea, even at rest (see Table 33-3). Little coughing and very little sputum are produced. The individual often is thin, has tachypnea with prolonged expiration, and must use accessory muscles for ventilation. The anteroposterior diameter of the chest is increased (barrel chest), and the chest has a hyperresonant sound with percussion. To increase lung capacity, the individual often leans forward with arms extended and braced on knees when sitting. In addition, people with emphysema often exhale through pursed lips, which helps prevent expiratory airway collapse.
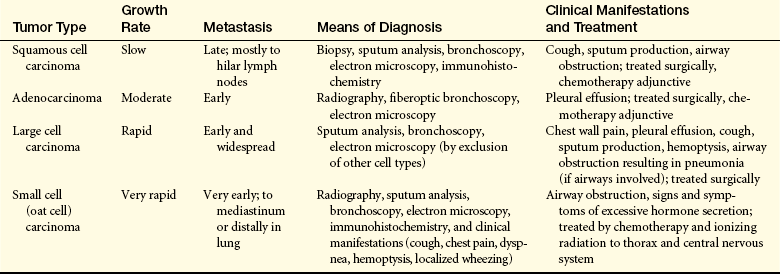
EVALUATION AND TREATMENT Emphysema is usually diagnosed and staged by pulmonary function measures. In COPD, pulmonary function tests indicate obstruction to gas flow during expiration with a marked decrease in FEV1. Airway collapse and air trapping in distal portions of the lung lead to a decrease in FVC (but less so than FEV1) and an increase in FRC, RV, and TLC.57 Diffusing capacity is decreased because of destruction of the alveolocapillary membranes. On radiographs the diaphragm appears flattened and the lung fields appear overdistended. In individuals for whom pulmonary function testing is not definitive for the diagnosis, high-resolution CT scanning may be indicated.57,69 Arterial blood gas measurements reveal varying degrees of hypoxemia and/or hypercapnia. The disease course is usually prolonged, with increasing dyspnea and intermittent bouts of infection that culminate in failure of the right side of the heart (cor pulmonale) and death.
Management of acute exacerbations of emphysema is similar to that for chronic bronchitis and requires obtaining a chest radiograph, serum white blood cell count, arterial blood gas, and sputum sample.57,63 Individuals should receive oxygen and may require noninvasive positive pressure ventilation or mechanical ventilation. Inhaled bronchodilators should be administered by either inhaler or nebulizer. Oral corticosteroids and antibiotics should begin immediately.57,62,63 Chronic management of emphysema begins with smoking cessation. Pharmacologic management is based on clinical severity as defined by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) as mild, moderate, severe, or very severe.57 Inhaled anticholinergic agents and beta agonists should be prescribed.57 A trial of inhaled corticosteroids should be used for severe COPD, although long-term therapy with oral steroids should be avoided if possible.57,65 Pulmonary rehabilitation, improved nutrition, and breathing techniques all can improve symptoms.70 Oxygen therapy is indicated in chronic hypoxemia but must be administered with care. Progressive pulmonary dysfunction with hypoxemia and hypercapnia may require long-term oxygen therapy and ventilation if indicated.66 In selected patients, lung reduction surgery or transplantation can be considered.57 A new class of drugs called phosphodiesterase E4 (PDE4) inhibitors is proving to be effective in selected patients with severe COPD.71 α1-Antitrypsin augmentation may be indicated for primary emphysema.72
Respiratory Tract Infections
Respiratory tract infections are the most common cause of short-term disability in the United States. Most of these infections—the common cold, pharyngitis (sore throat), and laryngitis—involve only the upper airways. Although the lungs have direct contact with the atmosphere, they remain sterile under most circumstances. Infections of the lower respiratory tract occur most often in the very young, the very old, or individuals with impaired immunity or underlying disease. In all cases the body’s normal defense mechanisms are impaired.
Pneumonia
Pneumonia is infection of the lower respiratory tract caused by bacteria, viruses, fungi, protozoa, or parasites. It is the sixth leading cause of death in the United States and is responsible for more disease and death than any other infection.73 More than half of those hospitalized for pneumonia are older than age 65; mortality from pneumonia is highest in older adults.74,75 Risk factors for pneumonia include advanced age, immunocompromise, underlying lung disease (especially COPD), alcoholism, altered consciousness, impaired swallowing, smoking, endotracheal intubation, malnutrition, immobilization, underlying cardiac or liver disease, and residence in a nursing home. The causative microorganism influences the symptoms and signs with which the patient presents, how the pneumonia should be treated, and the prognosis.73 Community-acquired pneumonia tends to be caused by different microorganisms than those infections acquired in the hospital (nosocomial).76 In addition, the characteristics of the individual are important in determining which etiologic microorganism is likely; for example, immunocompromised persons tend to be susceptible to opportunistic infections that are uncommon in normal adults. In general, nosocomial infections and those affecting immunocompromised individuals have a higher mortality rate than community-acquired pneumonias.77 Some of the most common causal microorganisms include the following74–77:

The most common community-acquired pneumonia is caused by S. pneumoniae (also known as pneumococcus), which has a relatively high mortality in older adults.74,75,78 M. pneumoniae and C. pneumoniae are common causes of pneumonia in young people, especially those living in group housing, such as dormitories and army barracks.79 Influenza is the most common viral community-acquired pneumonia in adults and causes more than 200,000 hospitalizations and more than 30,000 deaths annually in the United States.80 Legionella species can contaminate cooling systems and are an important cause of community-acquired pneumonia. Legionnaire disease has increased in incidence since it was first described in water supplies leading to outbreaks of disease, such as the 1976 incident at the American Legion convention in Philadelphia.81 Nosocomial pneumonia is a frequent complication in the ICU, most often in individuals placed on mechanical ventilation (ventilator-associated pneumonia [VAP]). P. aeruginosa, other gram-negative microorganisms, and S. aureus (including methicillin-resistant Staphylococcus aureus [MRSA]) are the most common etiologic agents in nosocomial pneumonia.82 Immunocompromised individuals are especially susceptible to P. jiroveci, other fungal infections, viruses, and mycobacterial infections of the respiratory tract.83–85
PATHOPHYSIOLOGY Aspiration of oropharyngeal secretions is the most common route of lower respiratory tract infection; thus the nasopharynx and oropharynx constitute the first line of defense for most infectious agents. Another route of infection is through the inhalation of microorganisms that have been released into the air when an infected individual coughs, sneezes, or talks, or from aerosolized water, such as that from contaminated respiratory therapy equipment. This route of infection is most important in viral and mycobacterial pneumonias and in Legionella outbreaks. Pneumonia also can occur when bacteria are spread to the lungs in the blood from bacteremia that can result from infection elsewhere in the body or from intravenous drug use.
In healthy individuals, pathogens that reach the lungs are expelled or held in check by mechanisms of self-defense (see Chapters 6, 7, and 32). If a microorganism gets past the upper airway defense mechanisms, such as the cough reflex and mucociliary clearance, the next line of defense is the airway epithelial cell. Airway epithelial cells can recognize some pathogens directly (e.g., P. aeruginosa and S. aureus).73 However, the most important guardian cell of the lower respiratory tract is the alveolar macrophage. This phagocyte can recognize pathogens through its pattern-recognition receptors (e.g., Toll-like receptors) which then activates both innate and adaptive immune responses.86 Release of TNF-α and IL-1 from macrophages contributes to widespread inflammation in the lung with recruitment of polymorphonuclear neutrophils (PMNs). PMNs migrate from the capillaries of the lungs into the alveoli. PMNs are critical phagocytes that kill microbes through the formation of phagolysosomes filled with degredative enzymes, antimicrobial proteins, and toxic oxygen radicals.73 PMNs have also been found to extrude a meshwork of proteins called a neutrophil extracellular trap (NET) that can capture and kill bacteria that have not yet been phagocytosed. Unfortunately many pathogens, such as the pneumococcus, can release a DNase that cleaves the NET and thus escape PMN defense. In addition to activating PMNs, macrophages also present infectious antigens to the adaptive immune system activating T cells and B cells with the induction of cellular and humoral immunity. The release of inflammatory mediators and immune complexes can damage bronchial mucous membranes and alveolocapillary membranes, causing the acini and terminal bronchioles to fill with infectious debris and exudate. In addition, some microorganisms release toxins from their cell walls that can cause further lung damage. The accumulation of exudate in the acinus leads to dyspnea and to mismatching and hypoxemia.
Pneumococcal Pneumonia: The pathogenesis of pneumococcal pneumonia (S. pneumoniae) has been well documented and serves as a model for understanding other forms of bacterial pneumonia (Figure 33-15). S. pneumoniae microorganisms initiate innate and adaptive immune responses (see Chapters 6 and 7). The immune response includes complement activation and the production of antibodies, which are crucial for opsonizing the encapsulated bacterium. Rapid lysis of pneumococcal bacteria (as occurs with antibiotic treatment) results in the release of intracellular bacterial proteins that can be toxic. The best known of these proteins is pneumolysin, which is cytotoxic to virtually every cell in the lung and is partially responsible for the worsening in clinical symptoms sometimes seen in individuals immediately after they begin antibiotic treatment.87 Inflammatory cytokines and cells are released that cause alveolar edema.73 Edema creates a medium for the multiplication of bacteria and aids in the spread of infection into adjacent portions of the lung. The involved lobe undergoes consolidation (solidification of the tissue caused by filling with exudate). A stage of red hepatization follows in which alveoli fill with blood cells, fibrin, edematous fluid, and pneumococci, giving lung tissue a red appearance. This passes into the stage of gray hepatization, in which affected tissues become gray because of fibrin deposition over the pleural surfaces and the presence of fibrin and leukocytes (neutrophils) in the consolidated alveoli, where phagocytosis is rapidly taking place. With resolution, increasing numbers of macrophages appear in the alveolar spaces, the neutrophils degenerate, and the fibrin threads and remaining bacteria are digested by macrophages and removed by lymphatic vessels. Usually infection is limited to one or two lobes.
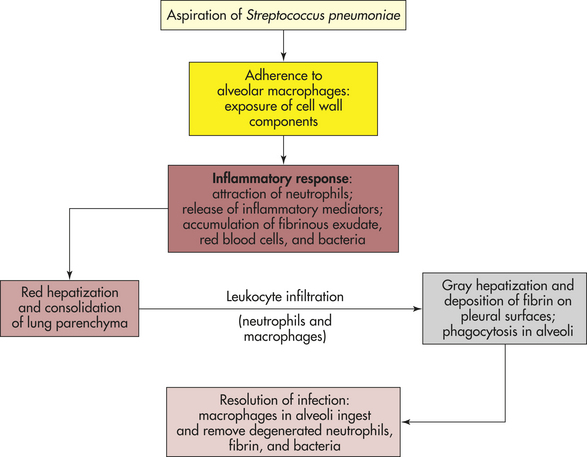
Viral Pneumonia: Viral pneumonia is usually mild and self-limiting, but it can set the stage for a secondary bacterial infection (especially by S. aureus microorganisms) by providing an ideal environment for bacterial growth and by damaging ciliated epithelial cells, which normally prevent pathogens from reaching the lower airways. Viral pneumonia can be a primary infection (e.g., influenza pneumonia) or a complication of another viral illness (e.g., chickenpox, measles). The virus not only destroys the ciliated epithelial cells but also invades the goblet cells and bronchial mucous glands. Sloughing of destroyed bronchial epithelium occurs throughout the respiratory tract, preventing mucociliary clearance. Bronchial walls become edematous and infiltrated with leukocytes.
Some forms of viral pneumonia can progress to severe systemic illness with many complications and a high morbidity and mortality. Severe viral pneumonia can include common types of influenza which can be fatal, especially in older adults. Other severe viral infections are considered opportunistic infections, such as cytomegalovirus pneumonia in immunocompromised individuals. New or atypical forms of viral infection, such as influenza A (H1N1) virus, avian influenza and the virus that causes the severe acute respiratory syndrome (SARS), are affecting previously healthy populations and pose a considerable threat for pandemics.88–90
CLINICAL MANIFESTATIONS Most cases of pneumonia are preceded by an upper respiratory infection, which is usually viral. This is then followed by the onset of cough, dyspnea, and fever. The cough is often productive but may be nonproductive, especially in viral pneumonia. Other symptoms include chills, malaise, and pleuritic chest pain. Physical examination may reveal signs of pulmonary consolidation, such as inspiratory crackles, increased tactile fremitus, egophony, and whispered pectoriloquy. Individuals also may demonstrate symptoms and signs of underlying systemic disease or sepsis.
EVALUATION AND TREATMENT Diagnosis is made on the basis of physical examination, white blood cell count, chest x-ray, stains and cultures of blood, cultures of respiratory secretions, and blood cultures.91 The white blood cell count is usually elevated, although it may be low if the individual is debilitated or immunocompromised. Chest radiographs show infiltrates that may involve a single lobe of the lung or may be more diffuse. Once the diagnosis of pneumonia has been made, the pathogen is identified by means of sputum characteristics (Gram stain, color, odor) and cultures or, if sputum is absent, blood cultures.91 Because many pathogens exist in the normal oropharyngeal flora, the specimen may be contaminated with pathogens from oral secretions. If sputum studies fail to identify the pathogen, the individual is immunocompromised, or the individual’s condition worsens, further diagnostic studies may include molecular testing of blood or urine, bronchoscopy, or lung biopsy.92–94
Prevention of pneumonia includes prevention of aspiration, respiratory isolation of immunocompromised individuals, vaccination for appropriate populations, and reduction of ventilator-associated pulmonary infections through a variety of dental and endotracheal tube interventions.95–97 The first step in the management of pneumonia is establishing adequate ventilation and oxygenation. Most individuals have hypoxemia and a respiratory alkalosis, although persons with underlying lung disease may require ventilation. Adequate hydration and good pulmonary hygiene (e.g., deep breathing, coughing, chest physical therapy) are also important.
Antibiotics are used to treat bacterial pneumonia; however, resistant strains of Pneumococcus are on the rise.91,97 In individuals for whom a specific microorganism is not identified, empiric antibiotics are chosen based on the likely causative microorganism.91,97,98 Viral pneumonia is usually treated with supportive therapy alone (unless secondary bacterial infection is present); however, antivirals may be needed in severe cases. Infections with opportunistic microorganisms may be polymicrobial and require multiple drugs, including antifungals.
Tuberculosis
Tuberculosis (TB) is an infection caused by M. tuberculosis, an acid-fast bacillus that usually affects the lungs but may invade other body systems. TB is the leading cause of death from a curable infectious disease throughout the world. TB cases increased greatly during the mid-1990s because of AIDS.99 Many ambitious programs for prevention and treatment have been initiated worldwide and the World Health Organization (WHO) 2008 Global Tuberculosis Control Report indicates that global TB incidence and prevalence have declined in most regions of the world in recent years, although southern Africa continues to carry a huge portion of the burden of TB infection and mortality.100 In the United States, the incidence of TB has reached its lowest level since 1953, but the rate of decline has begun to slow with more than half of new cases of TB occurring in foreign-born individuals, especially those from Mexico, the Philippines, India, and Vietnam.101 Individuals with AIDS are highly susceptible to respiratory infections, including multidrug-resistant TB. Emigration of infected individuals from high-prevalence countries, transmission in crowded institutional settings, homelessness, substance abuse, and lack of access to medical care have contributed to the spread of TB.100,101
PATHOPHYSIOLOGY TB is highly contagious and is transmitted from person to person in airborne droplets. Host susceptibility to infection is influenced by genetic polymorphisms, including those that affect macrophages, tumor necrosis factor, and interleukins.100 In immunocompetent individuals, the microorganism is usually contained by the inflammatory and immune response systems, and latent TB infection (LTBI) develops with no clinical evidence of disease.102 Microorganisms lodge in the lung periphery, usually in the upper lobe. Once the bacilli are inspired into the lung, they multiply and cause nonspecific pneumonitis (lung inflammation). Some bacilli migrate through the lymphatics and become lodged in the lymph nodes, where they encounter lymphocytes and initiate the immune response.
Inflammation in the lung causes neutrophils and macrophages to migrate to the area. These cells are phagocytes that engulf the bacilli and begin the process by which the body’s defense mechanisms isolate the bacilli, preventing their spread. However, the bacterium is successful as a pathogen because it can survive within macrophages, resist lysosomal killing, and multiply within the cell. In defense, macrophages and lymphocytes release interferon, which inhibits the replication of the microorganism and stimulates more macrophages to attack the bacterium.103 Apoptotic infected macrophages also can activate cytotoxic T cells (CD8). Neutrophils, lymphocytes, and macrophages seal off the colonies of bacilli, forming a granulomatous lesion called a tubercle.104 Infected tissues within the tubercle die, forming cheeselike material called caseation necrosis. (Necrosis is described in Chapter 2.) Collagenous scar tissue then grows around the tubercle, completing isolation of the bacilli. The immune response is complete after 10 days or so, preventing further multiplication of the bacilli.
Once the bacilli are isolated in tubercles and immunity develops, TB may remain dormant for life.102 If the immune system is impaired, however, or if live bacilli escape into the bronchi, active disease occurs and may spread through the blood and lymphatics to other organs. Infection with human immunodeficiency virus (HIV) is the single greatest risk factor for reactivation of tuberculosis infection. Other medical conditions that can cause reactivation include cancer, immunosuppressive medications, antirejection medications, and renal failure. Endogenous reactivation of dormant bacilli in older adults may be caused by poor nutritional status, insulin-dependent diabetes, long-term corticosteroid therapy, and other debilitating diseases.
CLINICAL MANIFESTATIONS Latent TB infection is asymptomatic. In some individuals, symptoms develop so gradually that they are not noticed until the disease is advanced. However, symptoms can appear in immunosuppressed individuals within weeks of exposure to the bacillus. Common clinical manifestations include fatigue, weight loss, lethargy, anorexia (loss of appetite), and a low-grade fever that usually occurs in the afternoon. (These are common signs and symptoms of all chronic infections.) A cough that produces purulent sputum develops slowly and becomes more frequent over several weeks or months. Night sweats and general anxiety are often present. Dyspnea, chest pain, and hemoptysis also may occur as the disease progresses. Extrapulmonary TB disease is common in HIV-infected individuals and may cause neurologic deficits, meningitis symptoms, bone pain, and urinary symptoms.
EVALUATION AND TREATMENT TB is diagnosed by a positive tuberculin skin test (TST; purified protein derivative [PPD]), sputum culture, immunoassays, and chest radiographs.85,105 A positive tuberculin skin test indicates that an individual has been infected and has mounted an immune response against the bacillus; however, the skin test does not differentiate between past, latent, or active disease. In addition, those individuals who have received the TB vaccine with bacille Calmette-Guérin (BCG) will have a positive TST even if they have never had TB. Two immunoassays (enzyme-linked immunospot and quantitative blood interferon-gamma assay) are available.85 These new tests are more sensitive and specific for the diagnosis of latent and active TB and are not confounded by previous BCG vaccination.106
When active pulmonary disease is present, the tubercle bacillus can be cultured from the sputum and may be seen with an acid-fast stain. However, sputum culture can take up to 6 weeks to become positive. Chest radiographs of individuals with current or previous active disease demonstrate characteristic changes. Nodules, calcifications, cavities, and hilar enlargement (enlarged mediastinal lymph nodes) commonly are seen in the upper lobes. A positive skin test indicates the need for yearly chest radiographs to detect active disease.
Prevention of tuberculosis infection is a complex challenge. Isolating individuals with active tuberculosis, limiting use of immunosuppressive medications, and treating underlying immunocompromising diseases, such as AIDs, are all critical steps. Development of an effective TB vaccine has been elusive. A recent report states that although eight new vaccines are in clinical trials, none are yet approved.107
Treatment consists of antibiotic therapy to control active disease or prevent reactivation of latent TB infection. The choice of drugs and the duration of treatment depend on the individual’s health history, the likelihood of bacterial resistance to certain drugs, and the presence of active disease. The waxy coat of M. tuberculosis renders it impermeable to many common drugs. Today, with the increased numbers of immunosuppressed and susceptible individuals and drug-resistant bacilli, the recommended treatment for those with active infection is a combination of drugs to which the microorganism is susceptible, including isoniazid, rifampin, pyrazinamide, ethambutol, rifapentine, and streptomycin. Treatment must be continued for a minimum of 6 months.85,108 Infection in immunocompromised individuals and multidrug-resistant strains of mycobacteria require the use of newer drugs for longer periods.108,109 Newer drugs being tested include immune amplifiers.110
In the past, individuals with active TB were isolated from the community and their families in sanitariums. Today individuals remain at home or, rarely, in the hospital, until sputum cultures show that the active bacilli have been eliminated. This usually takes a few weeks to 2 months if the antibiotics are taken conscientiously. If the individual’s cooperation is in question, it is advisable for the administration of the drugs to be supervised by healthcare workers.111
Abscess Formation and Cavitation
An abscess is a circumscribed area of suppuration and destruction of lung parenchyma. Abscess formation follows consolidation of lung tissue, in which inflammation causes alveoli to fill with fluid, pus, and microorganisms. Necrosis (death and decay) of consolidated tissue may progress proximally until it communicates with a bronchus. If this occurs, the abscess empties into the bronchus, leaving a cavity that has a radiographic appearance similar to that of a lesion of tuberculosis. Cavitation is the process of abscess emptying and cavity formation. Diagnosis is made by radiography.
Pneumonia caused by aspiration, Klebsiella, or Staphylococcus is the most common cause of abscess formation. Aspiration abscess is usually associated with alcohol abuse, seizure disorders, general anesthesia, and swallowing disorders. Immunocompromised individuals also are at greater risk for lung abscesses and may be infected with opportunistic microorganisms, such as fungi and mycobacteria. The clinical manifestations of abscess formation are similar to those of pneumonitis: fever, cough, chills, sputum production, and pleural pain. Abscess communication with a bronchus causes a severe cough, copious amounts of often foul-smelling sputum, and occasionally hemoptysis.
Treatment includes the administration of appropriate antibiotics and chest physical therapy, including chest percussion and postural drainage. Bronchoscopy is sometimes performed to drain the abscess. Mortality rates are influenced by the severity of the primary disease that initially caused consolidation and by the virulence of the causative microorganism.
Acute Bronchitis
Acute bronchitis is acute infection or inflammation of the airways or bronchi. The vast majority of acute bronchitis is caused by viruses.112 Many of the clinical manifestations are similar to those of pneumonia (i.e., fever, cough, chills, malaise), but physical examination does not reveal signs of pulmonary consolidation and chest radiographs do not show infiltrates. Individuals with viral bronchitis usually have a nonproductive cough that occurs in paroxysms and is aggravated by cold, dry, or dusty air. However, purulent sputum may be produced with some viral infections. Chest pain often develops from the effort of coughing. Treatment consists of rest, aspirin, humidity, and a cough suppressant, such as codeine.
Individuals with bacterial bronchitis have a productive cough, fever, and pain behind the sternum (breast bone) that is aggravated by coughing. It is rare in previously healthy adults except after viral infection but is common in those with COPD. Bacterial bronchitis is treated with rest, aspirin, humidity, and antibiotics.112
Pulmonary Vascular Disease
Blood flow through the lungs can be disrupted by a number of disorders that result in occlusion of the vessels, an increase in pulmonary vascular resistance, or destruction of the vascular bed. The consequences of altered pulmonary blood flow may be of no functional significance or can result in severe and life-threatening changes in ventilation-perfusion ratios. Major disorders include pulmonary embolism, pulmonary hypertension, and cor pulmonale.
Pulmonary Embolism
Pulmonary embolism (PE) is occlusion of a portion of the pulmonary vascular bed by an embolus that can be a thrombus (blood clot), a tissue fragment, lipids (fats), or an air bubble (Figure 33-16). The most common emboli are thrombi dislodged from deep veins in the thigh and pelvis, termed venous thromboembolism. Symptomatic pulmonary embolism occurs in approximately 50% of cases of untreated deep venous thrombosis (DVT).113 PE has an incidence of nearly 300,000 cases per year in the United States, although it is estimated that if undiagnosed cases also were counted, the number would be closer to 900,000. Many with PE die before reaching the hospital, and mortality at 3 months remains at 15% to 18% despite adequate anticoagulation therapy.114
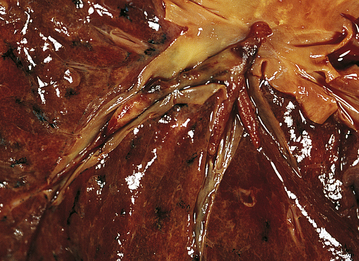
Figure 33-16 Pulmonary embolus. The embolus extends into major branches of the pulmonary artery. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Risk factors for pulmonary thromboembolism include many conditions and disorders that promote blood clotting (see Chapter 25). The three categories of pathologic risks are called the Virchow triad and include (1) venous stasis (slowing or stagnation of blood flow through the veins), (2) hypercoagulability (increased tendency of the blood to form clots), and (3) injuries to the endothelial cells that line the vessels. Venous stasis is usually caused by immobility with prolonged bed rest or sitting, for example, with air travel, neurologic disorders, advanced age, or immobilzer and cast use. Other causes of venous stasis include obesity, pregnancy, congestive heart failure, and sickle cell disease. Hypercoagulability can result from inherited or acquired conditions. Some of the most important acquired hypercoagulable states include malignancy, antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, polycythemia vera, hyperhomocysteinemia, and hormone use (oral contraceptives or hormone replacement therapy).113–115 Some of the most common inherited clotting disorders include factor V Leiden or prothrombin mutations and deficiencies of antithrombin III, protein C, protein S, or plasminogen.113–115 Endothelial injury and clot formation also proceed if vessel damage occurs, as in traumatic injury, surgery (especially obstetric or orthopedic procedures), indwelling venous catheters, or caustic intravenous medication use. No matter what its source, a blood clot becomes an embolus when all or part of it breaks away from the site of formation and begins to travel in the bloodstream. (Thromboembolism is described further in Chapter 30.)
PATHOPHYSIOLOGY The effect of the embolus depends on the extent of pulmonary blood flow obstruction, the size of the affected vessels, the nature of the embolus, and the secondary effects. Pulmonary emboli can occur as any of the following:
1. Embolus with infarction: an embolus that causes infarction (death) of a portion of lung tissue
2. Embolus without infarction: an embolus that does not cause permanent lung injury (perfusion of the affected lung segment is maintained by the bronchial circulation)
3. Massive occlusion: an embolus that occludes a major portion of the pulmonary circulation (i.e., main pulmonary artery embolus)
4. Multiple pulmonary emboli: multiple emboli may be chronic or recurrent
As a result of the thrombus lodging in the pulmonary circulation, there is a release of neurohumoral substances, such as serotonin, histamine, catecholamines and angiotensin II, and inflammatory mediators, such as endothelin, leukotrienes, thromboxanes, and toxic oxygen radicals. This causes widespread vasoconstriction that further impedes blood flow to the lung. Hemodynamically, this results in increased pulmonary artery pressures and can lead to right heart failure.114 Absent blood flow to a lung segment causes a ventilation-perfusion mismatch (increased dead space) and a decrease in surfactant production. The resulting atelectasis of the affected lung segments further contributes to hypoxemia. If the thrombus is large enough, infarction of lung tissue, dysrhythmias, decreased cardiac output, shock, and death are possible.114 The pathogenesis of venous thromboembolism is summarized in Figure 33-17.
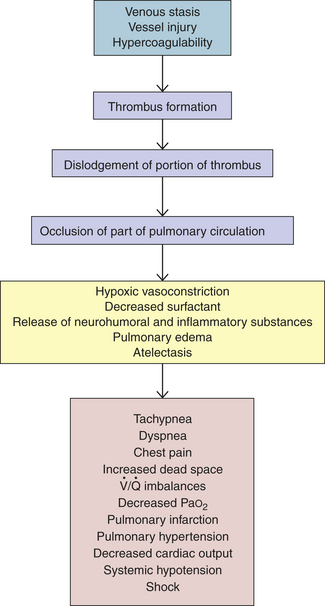
Figure 33-17 Pathogenesis of massive pulmonary embolism caused by a thrombus (pulmonary thromboembolism).
If the embolus does not cause infarction, the clot is dissolved by the fibrinolytic system (see Chapter 25) and pulmonary function returns to normal. If pulmonary infarction occurs, shrinking and scarring develop in the affected area of the lung. The risk of recurrent venous thromboembolism is 30% over the next 10 years, and is much higher in those individuals who have irreversible risk factors for the disease.113
CLINICAL MANIFESTATIONS In most cases the clinical manifestations of PE are nonspecific; therefore, evaluation of risk factors and predisposing factors is an important aspect of diagnosis. Consequently, the recognition of individuals at high risk for PE is crucial to assessing the clinical presentation.113–117 A list of an individual’s predisposing factors for venous thromboembolism can be inserted into one of several clinical prediction models (e.g., Wells Prediction Rule model) to obtain a prediction score that helps determine risk probability.114,116,117
In suspected PE, assessment for DVT may indicate the presence of a lower extremity source for the thromboembolism. Calf pain and tenderness, along with calf asymmetry when documented with a tape measure, are some of the most important findings in DVT. Unfortunately, DVT is often asymptomatic and clinical examination has low sensitivity for the presence of a clot, especially in the thigh and pelvis. Therefore, the lack of clinical indicators for DVT does not rule out the possibility for PE.
An individual with PE usually presents with the sudden onset of pleuritic chest pain, dyspnea, tachypnea, tachycardia, and unexplained anxiety. Occasionally syncope (fainting) or hemoptysis occurs. With large emboli, a pleural friction rub, pleural effusion, fever, and leukocytosis may be noted. Recurrent pulmonary emboli occur in individuals with a history of previous emboli. Recurrent small emboli may not be detected until progressive incapacitation, precordial pain, anxiety, dyspnea, and right ventricular enlargement are exhibited. Massive occlusion causes profound shock, hypotension, tachypnea, tachycardia, severe pulmonary hypertension, and chest pain.
EVALUATION AND TREATMENT When an individual is suspected of having a PE based on the presence of risk factors, symptoms, and physical findings, a chest x-ray, arterial blood gas, and ECG are obtained immediately.114–117 Chest x-ray findings are nonspecific in PE and often can be normal for the first 24 hours until atelectasis occurs in the lung. The arterial blood gas commonly reveals hypoxemia with a respiratory alkalosis (most individuals will hyperventilate in response to PE). The ECG may show evidence of strain on the right side of the heart. A serum D-dimer measures a product of thrombus degradation by the fibrinolytic system and, if normal, makes the presence of a PE highly unlikely.118 If the D-dimer is elevated, further evaluation is conducted using single or multidetector spiral CT arteriography.114,115,117 This highly sensitive and specific test has replaced the radionucleotide ventilation-perfusion scan in most hospitals. In rare cases, a pulmonary angiogram is necessary to confirm the diagnosis of PE. Recently, the measurement of elevated serum troponin levels has been useful in stratifying the risk and severity of PE.114,115,117
The ideal treatment of PE is prevention through risk factor recognition and elimination of predisposing factors. Venous stasis in hospitalized individuals is minimized by bed exercises, frequent position changes, early ambulation, and pneumatic calf compression.119 Most at-risk individuals also will receive prophylactic anticoagulation with unfractionated heparin, low-molecular-weight heparin, warfarin, or fondaparinux.119,120 In individuals who have contraindications to anticoagulation, the placement of a filter in the inferior vena cava can prevent emboli from reaching the lungs.119,121
Management of PE begins with administration of oxygen and hemodynamic stabilization with fluids, if needed, followed by rapid administration of anticoagulation, usually unfractionated or low-molecular-weight heparin.114,115,117,122 This is usually followed by weeks or months of outpatient warfarin. Newer anticoagulants that target factor Xa or thrombin are showing considerable clinical promise and include fondaparinux, idraparinux, rivaroxaban, and apixaban.123 If a massive life-threatening embolism occurs, a fibrinolytic agent, such as streptokinase, can be used and may be infused through a pulmonary artery catheter.124 Some individuals require emergent percutaneous or surgical embolectomy.124,125 Reversal of the underlying cause of the thrombus is important in preventing recurrent venous thromboembolism.
Pulmonary Artery Hypertension
Pulmonary artery hypertension (PAH) is defined as a mean pulmonary artery pressure above 25 mmHg at rest or 30 mmHg with exercise.126 Pulmonary artery pressure is lower than systemic arterial pressure and is normally 15 to 18 mmHg. Box 33-1 contains the WHO127 categories for PAH. Idiopathic PAH (IPAH) is rare with only one or two cases per million people. It is more common in women than in men and presents in women in the third decade of life and in men in the fourth decade.126,127 Familial PAH (FPAH) describes those individuals with PAH who have a family history of the disorder, most often due to mutations in the gene encoding the bone morphogenetic protein receptor type II (BMPR2).127,128 Associated PAH (APAH) is a leading cause of mortality in many connective tissue disorders and affects up to 1 in 200 individuals infected with HIV.127 Diet drugs, amphetamines, and cocaine also have been linked to an increased risk for PAH. Pulmonary artery hypertension associated with left heart failure or valvular disease is caused by increased pulmonary venous pressure and is discussed in Chapter 30. COPD is the most common lung disease associated with PAH, but any condition that causes chronic hypoxemia can result in pulmonary hypertension. Recurrent pulmonary embolism may be subclinical in its presentation and unrecognized until the signs and symptoms of PAH are detected.
PATHOPHYSIOLOGY PAH is characterized by endothelial dysfunction with overproduction of vasoconstrictors (e.g., thromboxane and endothelin) and decreased production of vasodilators (e.g., nitric oxide and prostacyclin).126,128 In individuals with BMPR2 gene mutations, intracellular signaling abnormalities result in vascular proliferation. This, along with release of vascular growth factors such as vascular endothelial growth factor, cause changes in the vascular smooth wall called remodeling.128 Endothelial-derived nitric oxide is an important vasodilator that also reduces smooth muscle cell proliferation and vascular thrombosis, and dysfunction of nitric oxide pathways is considered an important component of PAH pathophysiology.128,129 Other important mediators, including phosphodiesterases, serotonin, and adrenomedullin, also play a role in the pathogenesis of this disorder.128,129 Together, this results in pathologic changes in the pulmonary vasculature characterized by fibrosis and thickening of the vessel wall with luminal narrowing and abnormal vasoconstriction. These changes cause resistance to pulmonary artery blood flow, thus increasing the pressure in the pulmonary arteries. As resistance and pressure increase, the workload of the right ventricle increases and subsequent right ventricular hypertrophy, followed by failure, may occur (cor pulmonale). This eventually results in the death of most individuals with PAH.
Pulmonary artery hypertension associated with lung respiratory diseases and hypoxia is usually mild to moderate; however, resultant cor pulmonale is a significant cause of morbidity and mortality in late-stage chronic lung disease. Chronic hypoxemia, especially in association with respiratory acidosis, results in vasoconstriction and in vascular remodeling with significant smooth muscle hypertrophy, fibrosis, and luminal narrowing.130 The pathogenesis of pulmonary artery hypertension and cor pulmonale, resulting from disease of the respiratory system or hypoxia, is shown in Figure 33-18.
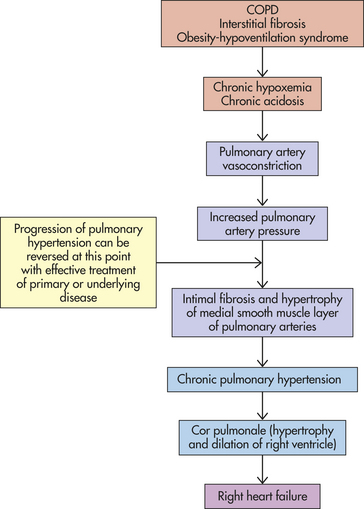
Figure 33-18 Pathogenesis of pulmonary hypertension and cor pulmonale caused by disease of the respiratory system or hypoxia. COPD, Chronic obstructive pulmonary disease.
CLINICAL MANIFESTATIONS Pulmonary artery hypertension may not be detected until it is quite severe. The symptoms are often masked by primary pulmonary or cardiovascular disease. The first indication of pulmonary hypertension is often an abnormality seen on a chest radiograph (enlarged pulmonary arteries and right heart border) or an electrocardiogram that shows right ventricular hypertrophy. Symptoms of fatigue, chest discomfort, tachypnea, and dyspnea on exertion, palpitations, and cough are common.126,127 Examination may reveal peripheral edema, jugular venous distention, a precordial heave, and accentuation of the pulmonary compartment of the second heart sound.126,127
EVALUATION AND TREATMENT Definitive diagnosis and accurate assessment of pulmonary artery pressure can be made only with right-sided heart catheterization. Laboratory studies, including arterial blood gas testing, liver function testing, HIV serology, electrocardiography, chest x-ray and CT scanning, pulmonary function testing, polysomnography, ventilation-perfusion scanning, and echocardiography, are used to detect underlying causes of PAH.126,127 Disease severity is quantified using the New York Heart Association/WHO classification of functional status of patients with pulmonary artery hypertension.127
General therapies for PAH include oxygen, diuretics, anticoagulants, and avoidance of contributing factors such as air travel, decongestant medications, nonsteroidal anti-inflammatory medications, pregnancy, and tobacco use.126, 130 Medications used in the treatment of PAH include prostacyclin analogs (epoprostenol, beraprost, iloprost), nitric oxide agonists (arginine, inhaled nitric oxide, sildenafil, dipyridamole), endothelin blockers (bosentan, ambrisentan), and phosphodiesterase-5 inhibitors.126,131 Those who fail medical therapy require lung transplantation to survive.
The most effective treatment for secondary pulmonary artery hypertension is treatment of the primary disorder. However, once pulmonary hypertension has persisted long enough for hypertrophy of the medial smooth muscle layer to develop (as it does with chronic hypoxemia), it is no longer reversible. Treatment relies on the use of supplemental oxygen to reverse hypoxic vasoconstriction.
PATHOPHYSIOLOGY Cor pulmonale develops as pulmonary artery hypertension creates chronic pressure overload in the right ventricle similar to that created in the left ventricle by systemic hypertension. (Systemic hypertension is discussed in Chapter 30.) Pressure overload increases the work of the right ventricle and causes hypertrophy of the normally thin-walled heart muscle. Acute hypoxemia, such as might occur with pneumonia, can exaggerate pulmonary hypertension and dilate the ventricle as well. Right ventricular filling pressures are normal until failure occurs. The right ventricle usually fails when pulmonary artery pressure equals systemic blood pressure.
CLINICAL MANIFESTATIONS The clinical manifestations of cor pulmonale may be obscured by primary respiratory disease and appear only during exercise testing. The heart appears normal at rest, but with exercise, cardiac output falls. The electrocardiogram shows right ventricular hypertrophy. Chest pain is common. The pulmonary component of the second heart sound, which represents closure of the pulmonic valve, may be accentuated, and a pulmonic valve murmur also may be present. Tricuspid valve murmur may accompany the development of right ventricular failure. Peripheral edema, hepatic congestion, and jugular venous distention often may be detected.
EVALUATION AND TREATMENT Diagnosis is made on the basis of physical examination, radiologic examination, and electrocardiogram or echocardiogram, or both. The goal of treatment for cor pulmonale is to decrease the workload of the right ventricle by lowering pulmonary artery pressure. Treatment is the same as for pulmonary artery hypertension, and its success depends on reversal of the underlying lung disease.
Malignancies of the Respiratory Tract
Cancer of the lip is more prevalent in men, with 3100 new cases per year accounting for about 1% of all cancers in men.132 Long-term exposure to sun, wind, and cold over a period of years results in dryness, chapping, hyperkeratosis, and predisposition to malignancy. The lower lip is the most common site.
PATHOPHYSIOLOGY The most common form of lower lip cancer is termed exophytic. The lesion usually develops in the outer part of the lip along the vermilion border. The lesion becomes thickened and evolves to an ulcerated center with a raised border (Figure 33-19). Verrucous-type lesions are less common. They have an irregular surface, follow cracks in the lip, and tend to extend toward the inner surface. Squamous cell carcinoma is the most common cell type. Basal cell carcinoma does not develop unless there is extension beyond the mucous membrane or vermilion border of the lip.
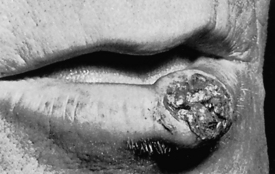
Figure 33-19 Lip cancer. Carcinoma of the lower lip with central ulceration and raised, rolled borders. (From del Regato JA, Spjut HJ, Cox JD: Ackerman and del Regato’s cancer, ed 2, St Louis, 1985, Mosby.)
CLINICAL MANIFESTATIONS Malignant lesions often are preceded by the development of a blister that evolves into a superficial ulceration. In some cases there is a history of recurrent scales that precede development of a bleeding ulceration. Metastases to the cervical lymph nodes have a low rate of occurrence and are more likely when the primary lesion is thicker and exists for a longer period.
EVALUATION AND TREATMENT Diagnosis is commonly made by clinical history and presentation of the lesion. Biopsy confirms the presence of malignant cells. The staging for lip cancer is based on the size of the primary tumor, the extent of lymph node involvement, and the presence of metastases. Surgical excision, such as the Mohs micrographic surgery technique, is effective for smaller lesions. Larger lesions that require extensive resection may need subsequent cosmetic surgeries. Interstitial irradiation and radioactive implants have proved effective for control of primary lesions. The prognosis for recovery is excellent, and deaths are usually the result of delayed or inadequate treatment.
Laryngeal Cancer
Cancer of the larynx represents approximately 2% to 3% of all cancers in the United States. There were an estimated 12,290 new cases in 2009, 9920 of them in men.132 The risk of laryngeal cancer is increased by the amount of tobacco smoked; risk is further heightened with the combination of smoking and alcohol consumption. Gastroesophageal reflux disease is also a risk factor.133 The human papillomavirus (HPV) has been linked to both benign and malignant disease of the larynx.134 The highest incidence is in men between 50 and 75 years of age.
PATHOPHYSIOLOGY Carcinoma of the true vocal cords (glottis) is more common than that of the supraglottic structures (epiglottis, aryepiglottic folds, arytenoids, and false cords). Tumors of the subglottic area are rare. Squamous cell carcinoma is the most common cell type, although small cell carcinomas also occur (Figure 33-20). Metastasis develops by spreading to the draining lymph nodes, and distant metastasis, usually to the lung, is rare.
Figure 33-20 Laryngeal cancer. A, Mirror view of carcinoma of right false cord partially hiding true cord. B, Lateral view. (From del Regato JA, Spjut HJ, Cox JD: Ackerman and del Regato’s cancer, ed 2, St Louis, 1985, Mosby.)
CLINICAL MANIFESTATIONS The presenting symptoms of laryngeal cancer include hoarseness, dyspnea, and cough. Progressive hoarseness is the most significant symptom and can result in voice loss. Dyspnea is rare in the case of supraglottic tumors but can be severe in subglottic tumors. Cough occurs less commonly and may follow swallowing. Laryngeal pain or a sore throat is likely to be present with supraglottic lesions.
EVALUATION AND TREATMENT Evaluation of the larynx includes external inspection and palpation of the larynx and the lymph nodes in the neck. Indirect laryngoscopy provides a stereoscopic view of the structure and movement of the larynx. A biopsy also can be obtained during this procedure. Direct laryngoscopy provides specific visualization of the tumor. Plain films of the larynx and CT facilitate the identification of tumor boundaries and the degree of extension to surrounding tissue. Magnetic resonance imaging (MRI) and positron-emission tomography (PET) can be used for staging.
Combined chemotherapy and radiation can result in cure in selected cases; however, sequelae such as swallowing and speech difficulties may result.135 Photodynamic therapy improves outcomes while preserving function in many individuals with early stage disease.136 Partial laryngectomies are the preferred treatment for small supraglottic and subglottic malignancies.137 Total laryngectomy is required when lesions are extensive and involve the cartilage. Swallowing and speech therapy after treatment can significantly improve recovery.
Lung Cancer
Lung cancers (bronchogenic carcinomas) arise from the epithelium of the respiratory tract. As such, the term lung cancer excludes other pulmonary tumors, including sarcomas, lymphomas, blastomas, hematomas, and mesotheliomas. Lung cancer is the number one cancer killer in the United States and the world. In the United States, there were an estimated 219,440 new cases and 159,390 deaths in 2009. It accounts for 15% of all cancers in men and 14% in women but is responsible for 31% of all cancer deaths in men and 26% of all cancer deaths in women (Box 33-2). Lung cancer is more common in blacks, for whom survival rates are lower.132 Although the mortality rate for lung cancer has leveled off in men, it is still rising in women. Overall 5-year survival remains low at 20%.
The most common cause of lung cancer is cigarette smoking, and approximately 10% to 15% of active smokers will develop lung cancer.138 About 10% of lung cancers occur in never-smokers and it is estimated that one fourth of lung cancer cases among never-smokers could be attributed to exposure to passive cigarette smoke.138,139 Cigarette smoke contains several organ-specific carcinogens, and smoking has been causally related to carcinogenesis at several sites, including the larynx, oral cavity, esophagus, and urinary bladder. Underlying smoking-related COPD is also a risk factor.138,139 Many genes have been implicated in the risk for lung cancer including genes for glutathione S-transferase M1, growth factors, tumor suppressor, detoxification enzymes, proteases, addiction, and deoxyribonucleic acid (DNA) repair have been identified138–141 (see What’s New? The Genetics of Lung Cancer). Theories of carcinogenesis are discussed in Chapter 11.
Environmental or occupational risk factors associated with lung cancer include benzopyrene and radon particles associated with uranium mining, radiation, and nuclear bombs. Others are polycyclic aromatic hydrocarbons and arsenicals, asbestos fibers, diesel exhaust, nitrogen mustard gases, nickel, silica, vinyl chloride, and chloromethyl methyl ether. Air pollution, coal, and iron mining are also considered risk factors.139
Primary lung cancers arise from the bronchi within the lungs and are therefore called bronchogenic carcinomas. Although there are many types of lung cancer, they are divided into two major categories: non–small cell lung carcinoma (NSCLC, 75% to 85% of all lung cancers) and small cell lung carcinoma (SCLC, 15% to 20% of all lung cancers). The NSCLC can be subdivided into three common types of lung cancer: squamous cell carcinoma, adenocarcinoma, and large cell undifferentiated carcinoma.141 The clinical and pathologic features that most commonly characterize these cancer types are illustrated in Figure 33-21 and described in Table 33-4. Many cancers that arise in other organs of the body metastasize to the lungs; however, these are not considered lung cancers and are categorized by their primary site of origin.
Figure 33-21 Cancer of the lung. A, Squamous (epidermoid) cell carcinoma. B, Small cell (oat cell) carcinoma. C, Adenocarcinoma. D, Large cell carcinoma. (From Des Jardins T, Burton GG: Clinical manifestations and assessment of respiratory disease, ed 3, St Louis, 1995, Mosby.)
Non–Small Cell Lung Cancer (NSCLC):
Squamous Cell Carcinoma: Squamous cell carcinoma accounts for about 30% of bronchogenic carcinomas, representing a sharp decline in incidence in the past two decades. These tumors are typically located centrally near the hilus and project into bronchi.
Because of the location in the central bronchi, obstructive manifestations are nonspecific and include nonproductive cough or hemoptysis. Pneumonia and atelectasis are often associated with squamous cell carcinoma (Figure 33-22, A). Chest pain is a late symptom associated with large tumors. These tumors can remain fairly well localized and tend not to metastasize until late in the course of the disease. The preferred treatment is surgical resection, although once metastasis has taken place, total surgical resection is difficult and survival rates dramatically decrease. Although chemotherapy has limited effectiveness, adjuvant treatment with newer agents has been shown to improve survival and quality of life.142,143
Figure 33-22 Lung cancer. A, Squamous cell carcinoma. This hilar tumor originates from the main bronchus. B, Peripheral adenocarcinoma. The tumor shows prominent black pigmentation, suggestive of having evolved in an anthracotic scar. C, Small cell carcinoma. The tumor forms confluent nodules. On cross sectioning, the nodules have an encephalid appearance. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Adenocarcinoma: Adenocarcinoma (tumor arising from glands) of the lung constitutes 35% to 40% of all bronchogenic carcinomas (Figure 33-22, B). The recent increase in incidence of adenocarcinoma has been ascribed to the increasing frequency of lung cancer in women, environmental and occupational carcinogens, and changes in the histologic criteria for diagnosis. These tumors, which are usually smaller than 4 cm, more commonly arise in the peripheral regions of the pulmonary parenchyma. They may be asymptomatic and discovered by routine chest roentgenogram in the early stages, or the individual may seek treatment for pleuritic chest pain and shortness of breath from pleural involvement by the tumor.
Included in the category of adenocarcinoma is bronchoalveolar cell carcinoma. These tumors tend to arise from the terminal bronchioles and alveoli. They are slow-growing tumors with an unpredictable pattern of metastasis. Metastasis occurs through the pulmonary arterial system and mediastinal lymph nodes. This cell type has the weakest association with smoking.144
Surgical resection is possible in a high proportion of adenocarcinoma cases, but because metastasis occurs early, the 5-year survival rate remains below 15%. Newer chemotherapeutic agents are resulting in increased survival rates.142,143
Large Cell Carcinoma (Undifferentiated): Large cell carcinomas constitute 10% to 15% of bronchogenic carcinomas. This cell type has lost all evidence of differentiation and is therefore commonly referred to as undifferentiated large cell anaplastic cancer. Because large cell carcinomas show none of the histologic findings of squamous cell carcinoma or adenocarcinoma, they are diagnosed by a process of exclusion. The cells are generally larger than leukocytes and contain large, darkly stained nuclei. These tumors commonly arise peripherally but are found centrally and can grow to distort the trachea and cause widening of the carina.
Once metastasis has occurred, surgical therapy is limited to palliative procedures (comfort measures) designed to relieve obstructive pneumonitis or prevent recurrence of pleural effusion. Neither radiation therapy nor chemotherapy has been successful in increasing survival.
Small Cell Carcinoma: Small cell lung carcinomas (SCLC) constitute about 15% of bronchogenic carcinomas but cause 25% of lung cancer deaths.145 Most tumors arise from the central part of the lung (see Figures 33-21 and 33-22, C). Cell sizes range from 6 to 8 μm. This cell type has the strongest correlation with cigarette smoking. Because these tumors show a rapid rate of growth and tend to metastasize early and widely, this type of carcinoma has the worst prognosis of all lung cancers. Staging for small cell carcinoma is divided into only two categories: limited disease (20% to 30%) and extensive disease (70% to 80%).145 Survival time for untreated small cell carcinoma is usually 1 to 3 months with treatment. With chemotherapy, radiation, or both, approximately 90% of individuals respond to treatment, but virtually all relapse within 2 years.145
Small cell carcinoma arises from neuroendocrine cells that contain neurosecretory granules and exist throughout the tracheobronchial tree. Thus small cell carcinoma is often associated with ectopic hormone production. Ectopic hormone production is important to the clinician because resulting signs and symptoms (called paraneoplastic syndromes) may be the first manifestation of the underlying cancer. The most common paraneoplastic syndrome associated with SCLC is the syndrome of inappropriate antidiuretic hormone secretion (see Chapter 21). Small cell carcinomas also commonly produce gastrin-releasing peptide, calcitonin, arginine vasopressin, and adrenocorticotropic hormone (ACTH). As a result of ACTH secretion, individuals with lung cancer secrete large quantities of 17-hydroxysteroids and 17-ketosteroids, leading to the development of Cushing syndrome. Signs and symptoms related to this condition include muscular weakness, facial edema, hypokalemia, alkalosis, hyperglycemia, hypertension, and increased pigmentation.
PATHOGENESIS Tobacco smoke contains as many as 20 documented lung carcinogens and is responsible for the vast majority of lung cancers. These carcinogens, along with probable inherited genetic predisposition to cancers, result in multiple genetic abnormalities in bronchial cells, including deletions of chromosomes, activation of oncogenes, and inactivation of tumor-suppressor genes.138 The most common genetic abnormality associated with lung cancer is a mutation of the tumor-suppressor gene TP53. Mutations in this gene have been found in 45% to 55% of NSCLCs and 75% to 100% of small cell cancers.146 Once lung cancer is initiated by these carcinogen-induced mutations, further tumor development is promoted by growth factors, such as epidermal growth factor, and by production of inflammatory mediators, such as toxic oxygen radicals.146–148
The bronchial mucosa suffers multiple carcinogenic “hits” because of repetitive exposure to cigarette smoke, and eventually epithelial cell changes begin to be visible on biopsy. These changes progress from metaplasia to carcinoma in situ and finally to invasive carcinoma.141 tumor progression includes invasion of surrounding tissues and, finally, metastasis to distant sites, including the brain, bone marrow, and liver.
CLINICAL MANIFESTATIONS Symptoms of early stage, localized disease are nonspecific and are likely to be attributed by the individual to the effects of smoking. The clinical manifestations are ambiguous and insidious; they include coughing, chest pain, sputum production, hemoptysis, pneumonia, airway obstruction, and pleural effusions. By the time manifestations are severe enough to motivate the individual to seek medical advice, the disease is usually advanced, and symptoms and signs of metastatic disease (e.g., neurologic deficits, bone pain) or paraneoplastic syndromes may be evident.
EVALUATION AND TREATMENT Although it is clear that diagnosing and treating lung cancer early in its development are crucial for long-term survival, screening for the presence of asymptomatic tumors in high-risk individuals remains controversial. The latest guidelines state that the evidence remains insufficient to recommend for or against screening asymptomatic individuals with sputum cytology, chest x-ray, or CT.149,150 However, many clinicians and researchers continue to examine these and other modalities in an effort to find more effective ways of catching this deadly disease when it is still curable. The diagnosis of lung cancer relies on the history of risk factors and symptoms, a careful physical examination, and a constellation of diagnostic tests including sputum cytology, chest x-ray, CT scanning, PET scanning, bronchoscopy, biopsy, and search for potential metastatic disease.151 The goal of these evaluations is to (1) establish the presence of a primary lung cancer, (2) determine its cell type, and (3) stage the tumor. As stated, SCLC is staged as either limited or extensive. The staging of NSCLC uses the TNM classification system in which T denotes the extent of the primary tumor, N indicates nodal involvement, and M describes the extent of metastasis and is illustrated in Figure 33-23. The use of biomarkers has been explored as a way of early detection and staging of lung cancer.152,153
Figure 33-23 Staging of lung cancer by the TNM classification system. A, B, Stage I disease includes tumors classified as T1, with or without metastasis to the lymph nodes in the ipsilateral hilar region. C, Also included in stage I are tumors classified as T2 but having no nodal or distant metastases. D, Stage II disease includes those tumors classified as T2, with metastasis only to the ipsilateral hilar lymph nodes. E, Stage III includes all tumors more extensive than T2 or any tumor with metastasis to the lymph nodes in the mediastinum or with distant metastasis.
The choice of treatment for lung cancer relies on an accurate description of the type of cancer cell and the stage of the tumor. In general, surgical removal of the entire tumor is the only certain cure. NSCLC is less responsive to chemotherapy than is small cell carcinoma, but chemotherapy and radiation are commonly used as adjuvant or palliative care.138,142,143,154 Small cell carcinoma is usually widely metastasized by the time of diagnosis, and treatment, although palliative, can markedly extend survival. Small cell carcinoma is most often treated with chemotherapy or radiation.145 Other therapies for lung cancer that can be used in selected individuals are laser phototherapy, photodynamic therapy, cryotherapy, and brachytherapy. New and exciting treatments for lung cancer are under investigation, including antiangiogenic therapy, targeting growth factor receptors, tumor sensitizing agents, gene therapy, and immunotherapy.138,146,155–160
Prevention of lung cancer relies primarily on reduction of exposure to carcinogens. For most individuals this means smoking cessation, and numerous governmental and private organizations are working toward the complete end of cigarette smoking. Other forms of prevention also are being explored.161
Other Lung Cancers: Bronchial carcinoid tumors represent about 1% of all lung tumors. The tumor cells have dense granules containing neuroendocrine-like hormones, but they rarely produce endocrine symptoms (carcinoid syndrome).162
Carcinoid tumors tend to occur earlier in life than bronchogenic carcinoma, although they can occur through the seventh decade of life. The average age at diagnosis is about 45 years. Carcinoid tumors are not related to smoking. They arise more commonly in the main or segmental bronchi, are easily visualized bronchoscopically, and are found on routine chest radiographs. Cells are not recovered from bronchial washings because the tumor is covered with normal mucosa. These tumors are slow-growing cancers, and 50% of individuals with bronchial carcinoid tumors are asymptomatic. Local surgical resection is curative if metastasis has not occurred; this can often be done by bronchoscopic laser electrocautery. Adenocystic tumors (cylindromas) and mucoepidermoid carcinomas are rare bronchial gland tumors. They arise predominantly in the trachea or large airways and cause obstruction. They can be malignant and metastasize early, although distal pulmonary metastases are usually slow growing. Thus it is not unusual for an individual to survive 10 to 15 years after diagnosis.
Mesotheliomas can be benign but most often are aggressive malignant tumors arising from the epithelium covering the serous membranes.163 Most arise from the pleural surface (80%). Benign pleural mesotheliomas have a slow clinical onset and are usually asymptomatic, but over a period of years they can cause dyspnea and mild pleuritic pain. These tumors can grow to be very large and fill the entire pleural cavity.
There is a clear association between asbestos exposure and malignant mesothelioma, especially in asbestos workers, although the minimum amount of exposure that constitutes risk has not been determined. A long latent interval between exposure to asbestos and appearance of mesothelioma usually occurs, and onset of symptoms may take 20 to 40 years. Clinical manifestations include dyspnea and chest pain that result from tumor-derived pleural fluid and invasion of the chest wall. Diagnosis is made by chest x-ray, CT scan, and thoracentesis with cytologic examination of the pleural fluid. Thoracoscopy also may be used for biopsy. Osteopontin and mesothelin are being explored as potential tumor markers for early diagnosis.163 Current management of malignant mesothelioma includes a combination of pleuropneumonectomy, chemotherapy, radiation, and hyperthermia.163,164
Abscess 1294
Absorption atelectasis 1275
Acute cough 1266
Acute respiratory distress syndrome (ARDS) 1279
Adenocarcinoma 1301
Adenocystic tumor (cylindroma) 1304
Allergic alveolitis (hypersensitivity pneumonitis) 1278
Alveolar dead space 1270
Asbestosis 1278
Aspiration 1274
Asthma 1283
Atelectasis 1275
Bronchial carcinoid tumor 1303
Bronchiectasis 1275
Bronchiolitis 1277
Bronchiolitis obliterans 1277
Bronchiolitis obliterans organizing pneumonia (BOOP) 1277
Cavitation 1294
Centriacinar emphysema 1289
Cheyne-Stokes respiration 1268
Chronic bronchitis 1286
Chronic cough 1266
Chronic obstructive pulmonary disease (COPD) 1286
Chylothorax 1273
Clubbing 1269
Coal worker pneumoconiosis (coal miner lung, black lung) 1278
Compression atelectasis 1275
Consolidation 1294
Cor pulmonale 1298
Cough 1266
Cyanosis 1268
Cylindrical bronchiectasis 1276
Dyspnea 1267
Dyspnea on exertion 1267
Emphysema 1288
Empyema (infected pleural effusion) 1273
Flail chest 1272
Hemoptysis 1268
Hemothorax 1273
Hypercapnia 1268
Hyperventilation 1268
Hypocapnia 1269
Hypoventilation 1268
Hypoxemia 1269
Hypoxia 1269
Idiopathic pulmonary fibrosis (IPF) 1277
Kussmaul respiration (hyperpnea) 1268
Large cell carcinoma 1301
Laryngeal cancer 1299
Lip cancer 1298
Lung cancer 1299
Mesothelioma 1304
Mucoepidermoid carcinoma 1304
Obstructive pulmonary disease 1282
Open pneumothorax (communicating pneumothorax) 1273
Orthopnea 1267
Oxygen toxicity 1277
Pain 1267
Panacinar emphysema 1289
Paroxysmal nocturnal dyspnea (PND) 1267
Pleural effusion 1273
Pneumoconiosis 1278
Pneumonia 1290
Pneumothorax 1272
Primary (spontaneous) pneumothorax 1272
Pulmonary artery hypertension (PAH) 1296
Pulmonary edema 1279
Pulmonary embolism (PE) 1294
Pulmonary fibrosis 1277
Pulmonary thromboembolism 1295
Pulsus paradoxus 1284
Respiratory failure 1271
Saccular bronchiectasis 1276
Secondary pneumothorax 1273
Shunting 1270
Silicosis 1278
Small-cell lung carcinoma 1302
Sputum 1267
Status asthmaticus 1284
Surfactant impairment 1275
Tension pneumothorax 1273
TNM classification system 1303
Transudative effusion 1273
Tuberculosis (TB) 1293
Undifferentiated large cell anaplastic cancer 1301
Varicose bronchiectasis 1276
REFERENCES
1. Millqvist, E., Bende, M. Role of the upper airways in patients with chronic cough. Curr Opin Allergy Clin Immunol. 2006;6(1):7–11.
2. Canning, B.J. Anatomy and neurophysiology of the cough reflex: ACCP evidence-based clinical practice guidelines. Chest. 2006;129:33S–47S.
3. Boulet, L.P. Future directions in the clinical management of cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129:287S–292S.
4. Brashers, V.L., Haden, K. Differential diagnosis of cough: focus on lung malignancy. Lippincott Prim Care Pract. 2000;4(4):374–389.
5. Pratter, M.R. Overview of common causes of chronic cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129:59S–62S.
6. Lumb, A.B. Nunn’s applied respiratory physiology, ed 6. St. Louis: Butterworth-Heinemann; 2005.
7. Spector, N., et al. Dyspnea: applying research to bedside practice. AACN Adv Crit Care. 2007;18(1):45–58. [quiz 59-60].
8. Corder, R. Hemoptysis. Emerg Med Clin North Am. 2003;21(2):421–435.
9. Noppen, M., Baumann, M.H. Pathogenesis and treatment of primary spontaneous pneumothorax: an overview. Respiration. 2003;70(4):431–438.
10. Currie, G.P., et al. Pneumothorax: an update. Postgrad Med J. 2007;83(981):461–465.
11. Chiu, H.T., Garcia, C.K. Familial spontaneous pneumothorax. Curr Opin Pulm Med. 2006;12(4):268–272.
12. Sahn, S.A. The value of pleural fluid analysis. Am J Med Sci. 2008;335(1):7–15.
13. Shigemitsu, H., Afshar, K. Aspiration pneumonias: under-diagnosed and under-treated. Curr Opin Pulm Med. 2007;13(3):192–198.
14. Metheny, N.A., Meert, K.L., Clouse, R.E. Complications related to feeding tube placement. Curr Opin Gastroenterol. 2007;23(2):178–182.
15. Duggan, M., Kavanagh, B.P. Atelectasis in the perioperative patient. Curr Opin Anaesthesiol. 2007;20(1):37–42.
16. Drakopanagiotakis, F., Polychronopoulos, V., Judson, M.A. Organizing pneumonia. Am J Med Sci. 2008;335(1):34–39.
17. Gottlieb, J., et al. Long-term azithromycin for bronchiolitis obliterans syndrome after lung transplantation. Transplantation. 2008;85(1):36–41.
18. Hyzy, R., et al. Acute exacerbation of idiopathic pulmonary fibrosis. Chest. 2007;132(5):1652–1658.
19. Noth, I., Martinez, F.J. Recent advances in idiopathic pulmonary fibrosis. Chest. 2007;132(2):637–650.
20. Palmieri, T.L. Inhalation injury: research progress and needs. J Burn Care Res. 2007;28(4):549–554.
21. Chong, S., et al. Pneumoconiosis: comparison of imaging and pathologic findings. Radiograph. 2006;26(1):59–77.
22. Huaux, F. New developments in the understanding of immunology in silicosis. Curr Opin Allergy Clin Immunol. 2007;7(2):168–173.
23. Cohen, R., Velho, V. Update on respiratory disease from coal mine and silica dust. Clin Chest Med. 2002;23(4):811–826.
24. O’Reilly, K.M., et al. Asbestos-related lung disease. Am Fam Physician. 2007;75(5):683–688.
25. Greenberger, P.A. 7. Immunologic lung disease. J Allergy Clin Immunol. 2008;121(2 Suppl):S393–S397. [quiz S418].
26. Madison, J.M. Hypersensitivity pneumonitis: clinical perspectives. Arch Pathol Lab Med. 2008;132(2):195–198.
27. Rubenfeld, G.D., Herridge, M.S. Epidemiology and outcomes of acute lung injury. Chest. 2007;131(2):554–562.
28. Ware, L.B., Matthay, M.A. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349.
29. Bhatia, M., Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202(2):145–156.
30. Girard, T.D., Bernard, G.R. Mechanical ventilation in ARDS: a state-of-the-art review. Chest. 2007;131(3):921–929.
31. Calfee, C.S., Matthay, M.A. Nonventilatory treatments for acute lung injury and ARDS. Chest. 2007;131(3):913–920.
32. Peter, J.V., et al. Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: meta-analysis. BMJ. 2008;336:1006–1009.
33. National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program Expert Panel Report 3: Guidelines for the diagnosis and management of asthma, 2007, p.12. Available at http://www.nhlbi.nih.gov/guidelines/asthma/asthgdin.pdf.
34. Global Initiative for Asthma (GINA): Global strategy for asthma management and prevention, 2006. Available from www.ginasthma.org.
35. Zhang, J., Pare, P.D., Sandford, A.J. Recent advances in asthma genetics. Resp Res. 2008;9:4.
36. Holloway, J.W., Koppelman, G.H. Identifying novel genes contributing to asthma pathogenesis. Curr Opin Allergy Clin Immunol. 2007;7(1):69–74.
37. Yang, Y., et al. Epigenetic mechanisms silence a disintegrin and metalloprotease 33 expression in bronchial epithelial cells. J Allergy Clin Immunol. 2008;121(6):1393–1399.
38. McLeish, S., Turner, S.W. Gene-environment interactions in asthma. Arch Dis. 2007;92(11):1032–1035.
39. Miller, R.L., Ho, S.M. Environmental epigenetics and asthma: current concepts and call for studies. Am J Respir Crit Care Med. 2008;177(6):567–573.
40. Yang, I.A., et al. Gene-environmental interaction in asthma. Curr Opin Allergy Clin Immunol. 2007;7(1):75–82.
41. Effros, R.M., Nagaraj, H. Asthma: New developments concerning immune mechanisms, diagnosis and treatment. Curr Opin Pulm Med. 2007;13(1):37–43.
42. Corren, J. The connection between allergic rhinitis and bronchial asthma. Curr Opinin Pulm Med. 2007;13(1):13–18.
43. Passalacqua, G., Durham, S.R. Global Allergy and Asthma European Network: allergic rhinitis and its impact on asthma update: allergen immunotherapy. J Allergy Clin Immunol. 2007;119(4):881–891.
44. Kiechl-Kohlendorfer, U., et al. Neonatal characteristics and risk of atopic asthma in schoolchildren: results from a large prospective birth-cohort study. Acta Paediatr. 2007;96(11):1606–1610.
45. Cabana, M.D., et al. Examining the hygiene hypothesis: the Trial of Infant Probiotic Supplementation. Paediatr Perinat Epidemiol. 2007;21(Suppl 3):23–28.
46. Peters-Golden, M., Henderson, W.R., Jr. Leukotrienes. N Engl J Med. 2007;357(18):1841–1854.
47. Trivedi, S.G., Lloyd, C.M. Eosinophils in the pathogenesis of allergic airways disease. Cell Mol Life Sci. 2007;64(10):1269–1289.
48. Tliba, O., Amrani, Y., Panettieri, R.A., Jr. Is airway smooth muscle the “missing link” modulating airway inflammation in asthma? Chest. 2008;133(1):236–242.
49. Moore, W.C., Peters, S.P. Update in asthma 2006. Am J Respir Crit Care Med. 2007;175(7):649–654.
50. Frieri, M. Advances in the understanding of allergic asthma. Allergy Asthma Proc. 2007;28(6):614–619.
51. Holgate, S.T., Polosa, R. Treatment strategies for allergy and asthma. Nat Rev Immunol. 2008;8(3):218–230.
52. Stewart, L., Katial, R. Exhaled nitric oxide. Immunol Allergy Clin North Am. 2007;27(4):571–586. [v].
53. Kaza, V., Bandi, V., Guntupalli, K.K. Acute severe asthma: recent advances. Curr Opin Pulm Med. 2007;13(1):1–7.
54. Oppenheimer, J., Nelson, H.S. Safety of long-acting beta-agonists in asthma: a review. Curr Opin Pulm Med. 2008;14(1):64–69.
55. Taylor, D.R. beta-adrenergic receptor polymorphisms: relationship to the beta-agonist controversy and clinical implications. Exp Opin Pharmacother. 2007;8(18):3195–3203.
56. Jacobsen, L., Valovirta, E. How strong is the evidence that immunotherapy in children prevents the progression of allergy and asthma? Curr Opin Allergy Clin Immunol. 2007;7(6):556–560.
57. Rabe, K.F., et al, Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176(6):532–555. (Available at http://ajrccm.atsjournals.org/cgi/content/full/176/6/532)
58. Ben-Zaken Cohen, S., et al. The growing burden of chronic obstructive pulmonary disease and lung cancer in women: examining sex differences in cigarette smoke metabolism. Am J Respir Crit Care Med. 2007;176(2):113–120.
59. Molfino, N.A. Current thinking on genetics of chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2007;13(2):107–113.
60. Hallberg, J., et al. Interaction between smoking and genetic factors in the development of chronic bronchitis. Am J Resp Critical Care Med. 2008;177:486–490.
61. Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–1892.
62. Braman, S.S. Chronic cough due to chronic bronchitis: ACCP evidence-based clinical practice guidelines. Chest. 2006;129(1 Suppl):104S–115S.
63. Quon, B.S., Gan, W.Q., Sin, D.D. Contemporary management of acute exacerbations of COPD: a systematic review and metaanalysis. Chest. 2008;133(3):756–766.
64. Martinez, F.J., Anzueto, A. Appropriate outpatient treatment of acute bacterial exacerbations of chronic bronchitis. Am J Med. 2005;118(Suppl 7A):39S–44S.
65. Sin, D.D., Man, S.F. Do chronic inhaled steroids alone or in combination with a bronchodilator prolong life in chronic obstructive pulmonary disease patients. Curr Opin Pulm Med. 2007;13(2):90–97.
66. O’Reilly, P., Bailey, W. Long-term continuous oxygen treatment in chronic obstructive pulmonary disease: proper use, benefits and unresolved issues. Curr Opin Pulm Med. 2007;13(2):120–124.
67. Stoller, J.K. Aboussouan LS:.Alpha 1-antitrypsin deficiency. Lancet. 2005;365(9478):2225–2236.
68. Taraseviciene-Stewart, L., Voelkel, N.F. Molecular pathogenesis of emphysema. J Clin Invest. 2008;118(2):394–402.
69. Friedman, P.J. Imaging studies in emphysema. Proc Am Thorac Soc. 2008;5(4):494–500.
70. Ries, A.L., et al. Pulmonary rehabilitation: joint ACCP/AACVPR evidence-based clinical practice guidelines. Chest. 2007;131(5 Suppl):4S–42S.
71. Boswell-Smith, V., Spina, D. PDE4 inhibitors as potential therapeutic agents in the treatment of COPD-focus on roflumilast. Int J COPD. 2007;2(2):121–129.
72. Heresi, G.A., Stoller, J.K. Augmentation therapy in alpha-1 antitrypsin deficiency. Exp Opin Biol Ther. 2008;8(4):515–526.
73. Mizgerd, J.P. Acute lower respiratory tract infection. N Engl J Med. 2008;358(7):716–727.
74. Niederman, M.S., Brito, V. Pneumonia in the older patient. Clin Chest Med. 2007;28(4):751–771. [vi].
75. Donowitz, G.R., Cox, H.L. Bacterial community-acquired pneumonia in older patients. Clin Geriatr Med. 2007;23:515–534.
76. File, T.M. Community-acquired pneumonia. Lancet. 2003;362(9400):1991–2001.
77. Kollef, M.H., et al. Epidemiology and outcomes of health-care-associated pneumonia: results from a large US database of culture positive patients. Chest. 2005;128:3854–3862.
78. Ortqvist, A., Hedlund, J., Kalin, M. Streptococcus pneumoniae: epidemiology, risk factors, and clinical features. Semin Respir Crit Care Med. 2005;26(6):563–574.
79. Blasi, F., et al. Chlamydia pneumoniaeand Mycoplasma pneumoniae. Semin Respir Crit Care Med. 2005;26(6):617–624.
80. Lynch, J.P., 3rd., Walsh, E.E. Influenza: evolving strategies in treatment and prevention. Semin Respir Crit Care Med. 2007;28(2):144–158.
81. Diederen, B.M. Legionella spp. and legionnaires’ disease. J Infect. 2008;56(1):1–12.
82. Mueller, E.W., et al. Repeat bronchoalveolar lavage to guide antibiotic duration for ventilator-associated pneumonia. J Trauma. 2007;63(6):1329–1337. [discussion 1337].
83. Davaro, R.E., Thirumalai, A. Life-threatening complications of HIV infection. J Intern Care Med. 2007;22(2):73–81.
84. Morris, A., et al. Epidemiology and clinical significance of pneumocystis colonization. J Infect Dis. 2008;197(1):10–17.
85. Yew, W.W., Leung, C.C. Update in tuberculosis 2007. Am J Respir Crit Care Med. 2008;177(5):479–485.
86. Gerold, G., Zychlinsky, A., de Diego, J.L. What is the role of toll-like receptors in bacterial infections? Semin Immunol. 2007;19(1):41–47.
87. Shoma, S., et al. Critical involvement of pneumolysin in production of interleukin-1 alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infect Immun. 2008;76(4):1547–1557.
88. Rothberg, M.B., Haessler, S.D., Brown, R.B. Complications of viral influenza. Am J Med. 2008;121(4):258–264.
89. Writing Committee of the Second World Health Organization Consultation on Clinical Aspects of Human Infection with Avian Influenza(H5N1) virus, A., et al. update on avian influenza A (H5N1) virus infection in humans. N Engl J Med. 2008;358(3):261–273.
90. Centers for Disease Control and Prevention (CDC). Update: novel influenza A (H1N1) virus infections—worldwide, May 6, 2009. MMWR Morb Mortal Wkly Rep. 2009;58(17):453–458.
91. Mandell, L., et al. Infectious Diseases Society of America/American Thoracic Society Guidelines on the management for community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(Suppl 2):S27–S72.
92. Chan, Y.R., Morris, A. Molecular diagnostic methods in pneumonia. Curr Opin Infect Dis. 2007;20(2):157–164.
93. Soto, G. J: Diagnostic strategies for nosocomial pneumonia. Curr Opin Infect Dis. 2007;20(2):157–164.
94. Ramirez, P., Valencia, M., Torres, A. Bronchoalveolar lavage to diagnose respiratory infections. Semin Respir Crit Care Med. 2007;28(5):525–533.
95. Oosterhuis-Kafeja, F., Beutels, P., Van Damme, P. Immunogenicity, efficacy, safety and effectiveness of pneumococcal conjugate vaccines (1998-2006). Vaccine. 2007;25(12):2194–2212.
96. Ramirez, P., Ferrer, M., Torres, A. Prevention measures for ventilator-associated pneumonia: a new focus on the endotracheal tube. Curr Opin Infect Dis. 2007;20(2):190–197.
97. Armitage, K., Woodhead, M. New guidelines for the management of adult community-acquired pneumonia. Curr Opin Infect Dis. 2007;20(2):170–176.
98. Aarts, M.A., et al. Empiric antibiotic therapy for suspected ventilator-associated pneumonia: a systematic review and meta-analysis of randomized trials. Crit Care Med. 2008;36(1):108–117.
99. Dye, C. Global epidemiology of tuberculosis. Lancet. 2006;367(9514):938–940.
100. WHO 2008 Report: Global tuberculosis control—surveillance, planning, financing. Available at www.who.int/tb/publications/global_report/2008/download_centre/en/index.html.
101. Centers for Disease Control and Prevention: Trends in tuberculosis—United States, 2007, JAMA 299(18):2142–2144, 2008.
102. Cardona, P.J. New insights on the nature of latent tuberculosis infection and its treatment. Inflamm Allergy Drug Targets. 2007;6(1):27–39.
103. Bottasso, O., et al. The immuno-endocrine component in the pathogenesis of tuberculosis. Scand J Immunol. 2007;66(2-3):166–175.
104. Russell, D.G. Who puts the tubercle in tuberculosis? Nat Rev Microbiol. 2007;5(1):39–47.
105. Lalvani, A. Diagnosing tuberculosis infection in the 21st century: new tools to tackle an old enemy. Chest. 2007;131(6):1898–1906.
106. Campbell, I.A., Bah-Sow, O. Pulmonary tuberculosis: diagnosis and treatment. BMJ. 2006;332(7551):1194–1197.
107. Gupta, U.D., Katoch, V.M., McMurray, D.N. Current status of TB vaccines. Vaccine. 2007;25(19):3742–3751.
108. Zhang, Y. Advances in the treatment of tuberculosis. Clin Pharmacol Ther. 2007;82(5):595–600.
109. Furin, J. The clinical management of drug-resistant tuberculosis. Curr Opin Pulm Med. 2007;13(3):212–217.
110. Roy, E., Lowrie, D.B., Jolles, S.R. Current strategies in TB immunotherapy. Curr Mol Med. 2007;7(4):373–386.
111. Volmink, J., Garner, P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst Rev. (4):2007. [CD003343].
112. Braman, S.S. Chronic cough due to acute bronchitis: ACCP evidence-based clinical practice guidelines. Chest. 2006;129(1 Suppl):95S–103S.
113. Tapson, V.F. Acute pulmonary embolism. N Engl J Med. 2008;358(10):1037–1052.
114. Heit, J.A. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol. 2008;28(3):370–372.
115. Dimarsico, L., Cymet, T. Pulmonary embolism—a state of the clot review. Comprehen Ther. 2007;33(4):184–191.
116. Hunt, D. Determining the clinical probability of deep venous thrombosis and pulmonary embolism. South Med J. 2007;100(10):1015–1021.
117. Minichiello, T., Fogarty, P.F. Diagnosis and management of venous thromboembolism. Med Clin North Am. 2008;92(2):443–465.
118. Di Nisio, M., et al. Diagnostic accuracy of D-dimer test for exclusion of venous thromboembolism: a systematic review. J Thromb Haemost. 2007;5(2):296–304.
119. Francis, C.W. Clinical practice: prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med. 2007;356(14):1438–1444.
120. Sjalander, A., et al. Efficacy and safety of anticoagulant prophylaxis to prevent venous thromboembolism in acutely ill medical inpatients: a meta-analysis. J Intern Med. 2008;263(1):52–60.
121. Barral, F.G. Vena cava filters: why, when, what and how? J Cardiovasc Surg. 2008;49(1):35–49.
122. Segal, J.B., et al. Management of venous thromboembolism: a systematic review for a practice guideline. Ann Internal Med. 2007;146(3):211–222.
123. Gross, P.L., Weitz, J.I. New anticoagulants for treatment of venous thromboembolism. Arterioscler Thromb Vasc Biol. 2008;28(3):380–386.
124. Carlbom, D.J., Davidson, B.L. Pulmonary embolism in the critically ill. Chest. 2007;132(1):313–324.
125. Uflacker, R., Schonholz, C. Percutaneous interventions for pulmonary embolism. J Cardiovasc Surg. 2008;49(1):3–18.
126. Highland, K.B. Pulmonary arterial hypertension. Am J Med Sci. 2008;335(1):40–45.
127. Heresi, G.A., Dweik, R.A. Pulmonary hypertension: evaluation and management. Comprehen Ther. 2007;33(3):150–161.
128. Chan, S.Y., Loscalzo, J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44(1):14–30.
129. Jain, S., Ventura, H., deBoisblanc, B. Pathophysiology of pulmonary arterial hypertension. Sem Cardiothorac Vasc Anesth. 2007;11(2):104–109.
130. Tuder, R., et al. Hypoxia and chronic lung disease. J Molec Med. 2007;85(12):1317–1324.
131. Driscoll, J.A., Chakinala, M.M. Medical therapy for pulmonary arterial hypertension. Exp Opin Pharmacother. 2008;9(1):65–81.
132. American Cancer Society, Inc., Surveillance and Health Policy Research: Estimated New Cancer Cases and Deaths by Sex, U.S. 2009 available at: http://www.cancer.org/downloads/stt/CFF2009_EstCD_3.pdf
133. Karamanolis, G., Sifrim, D. Developments in pathogenesis and diagnosis of gastroesophageal reflux disease. Curr Opin Gastroenterol. 2007;23(4):428–433.
134. Torrente, M.C., Ojeda, J.M. Exploring the relation between human papillomavirus and larynx cancer. Acta Otolaryngol. 2007;127(9):900–906.
135. Genden, E.M., et al. Recent changes in the treatment of patients with advanced laryngeal cancer. Head Neck. 2008;30(1):103–110.
136. Biel, M.A. Photodynamic therapy treatment of early oral and laryngeal cancers. Photochem Photobiol. 2007;83(5):1063–1068.
137. Marioni, G., et al. Organ-preservation surgery following failed radiotherapy for laryngeal cancer. Evaluation, patient selection, functional outcome and survival. Curr Opin Otolaryngol Head Neck Surg. 2008;16(2):141–146.
138. Dubey, S., Powell, C.A. Update in lung cancer 2007. Am J Respir Crit Care Med. 2008;177(9):941–946.
139. Alberg, A.J., et al. Epidemiology of lung cancer: ACCP evidence-based clinical practice guidelines (ed 2). Chest. 2007;132(3 Suppl):29S–55S.
140. Kiyohara, C., et al. Lung cancer susceptibility: are we on our way to identifying a high-risk group? Future Oncol. 2007;3(6):617–627.
141. Maitra, A., Kumar, V. The lung. In Kumar V., et al, eds.: Robbins basic pathology, ed 8, Philadelphia: Saunders, 2007.
142. Malingam, S., Belani, C. Systemic chemotherapy for advanced non-small cell lung cancer: recent advances and future directions. Oncologist. 2008;13(Suppl 1):5–13.
143. Alberts, W.M. American College of Chest Physicians: Introduction: diagnosis and management of lung cancer: ACCP evidence-based clinical practice guidelines (2nd ed). Chest. 2007;132(3 Suppl):20S–22S.
144. Garfield, D.H., Cadranel, J., West, H.L. Bronchioloalveolar carcinoma: the case for two diseases. Clin Lung Can. 2008;9(1):24–29.
145. Sher, T., Dy, G.K., Adjei, A.A. Small cell lung cancer. Mayo Clin Proc. 2008;83(3):355–367.
146. Hudkinson, P.S., MacKinnin, A., Sethi, T. Targeting growth factors in lung cancer. Chest. 2008;133:1209–1216.
147. Krysan, K., et al. Inflammation, epithelial to mesenchymal transition, and epidermal growth factor receptor tyrosine kinase inhibitor resistance. J Thorac Oncol. 2008;3(2):107–110.
148. Azad, N., Rojanasakul, Y., Vallyathan, V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev. 2008;11(1):1–15.
149. Bach, P.B., et al. Screening for lung cancer: ACCP evidence-based clinical practice guidelines (2nd ed). Chest. 2007;132(3 Suppl):69S–77S.
150. Aberle, D.R., Brown, K. Lung cancer screening with CT. Clin Chest Med. 2008;29(1):1–14.
151. Rivera, P.M., Mehta, A.C. Initial diagnosis of lung cancer: ACCP evidence-based clinical practice guidelines (2nd ed). Chest. 2007;132(3 Suppl):1315–1485.
152. Patz, E.F., Jr., et al. Panel of serum biomarkers for the diagnosis of lung cancer. J Clin Oncol. 2007;25:5578–5583.
153. D’Amico, T.A. Molecular biologic staging of lung cancer. Ann Thorac Surg. 2008;85(2):S737–S742.
154. de Marinis, F., Grossi, F. Clinical evidence for second- and third-line treatment options in advanced non-small cell lung cancer. Oncologist. 2008;13(Suppl 1):14–20.
155. Chada, S., et al. Cancer targeting using tumor suppressor genes. Front Biosci. 2008;13:1959–1967.
156. Deng, W.G., et al. Enhancement of antitumor activity of cisplatin in human lung cancer cells by tumor suppressor FUS1. Cancer Gene Ther. 2008;15(1):29–39.
157. Gutierrez, M., Giaccone, G. Antiangiogenic therapy in nonsmall cell lung cancer. Curr Opin Oncol. 2008;20(2):176–182.
158. Ruttinger, D., et al. Current immunotherapeutic strategies in lung cancer. Surg Oncol Clin North Am. 2007;16(4):901–918. [x].
159. Sequist, L.V., Lynch, T.J. EGFR tyrosine kinase inhibitors in lung cancer: an evolving story. Annu Rev Med. 2008;59:429–442.
160. Wheatley-Price, P., Shepherd, F.A. Epidermal growth factor receptor inhibitors in the treatment of lung cancer: reality and hopes. Curr Opin Oncol. 2008;20(2):162–175.
161. Omenn, G.S. Chemoprevention of lung cancers: lessons from CARET, the beta-carotene and retinol efficacy trial, and prospects for the future. Eur J Cancer Prev. 2007;16(3):184–191.
162. Garcia-Yuste, M., Matilla, J.M., Gonzalez-Aragoneses, F. Neuroendocrine tumors of the lung. Curr Opin Oncol. 2008;20(2):148–154.
163. Zervos, M.D., Bizekis, C., Pass, H.I. Malignant mesothelioma 2008. Curr Opin Pulm Med. 2008;14(4):303–309.
164. Fennell, D.A., et al. Advances in the systemic therapy of malignant pleural mesothelioma. Nat Clin Pract Oncol. 2008;5(3):136–147.