Ethics in Research
Nursing research requires not only expertise and diligence but also honesty and integrity. Conducting research ethically starts with the identification of the study topic and continues through the publication of the study. Over the years, ethical codes and regulations have been developed to provide guidelines for (1) the selection of the study purpose, design, and subjects; (2) the collection and analysis of data; (3) the interpretation of results; and (4) the presentation and publication of the study. In 2003, an important regulation titled the Health Insurance Portability and Accountability Act (HIPAA) was enacted to protect the privacy of an individual’s health information. HIPAA has had an important impact on researchers and institutional review boards (IRBs), who review health care research for conduct in a variety of settings. This chapter provides an overview of this act and the other U.S. and international regulations that have been developed to promote ethical conduct in research worldwide.
An ethical problem that has received increasing attention since the 1980s is research misconduct. Misconduct has occurred during the performance, reporting, and publication of research, and the Office of Research Integrity (ORI) has investigated a number of research institutions in the United States for fraud (ORI, 2007, June 20). Many disciplines, including nursing, have had episodes of scientific misconduct that have affected the quality of research evidence generated.
Ethical research is essential to generate a sound evidence-based practice for nursing, but what does the ethical conduct of research involve? This is a question that has been debated for many years by researchers, politicians, philosophers, lawyers, and even research subjects. The debate continues, probably because of the complexity of human rights issues; the focus of research in new, challenging arenas of technology and genetics; the complex ethical codes and regulations governing research; and the various interpretations of these codes and regulations. Even though the phenomenon of the ethical conduct in research defies precise delineation, the historical events, ethical codes, and regulations presented in this chapter provide guidance for nurse researchers. The chapter also discusses the actions essential for conducting research ethically: (1) protecting the rights of human subjects, (2) balancing benefits and risks in a study, (3) obtaining informed consent from subjects, and (4) submitting a research proposal for institutional review. Current ethical issues related to scientific misconduct and the use of animals in research conclude the chapter.
HISTORICAL EVENTS AFFECTING THE DEVELOPMENT OF ETHICAL CODES AND REGULATIONS
The ethical conduct of research has been a focus since the 1940s because of the mistreatment of human subjects in selected studies. Four experimental projects have been highly publicized for their unethical treatment of subjects: (1) Nazi medical experiments, (2) the Tuskegee Syphilis Study, (3) the Willowbrook study, and (4) the Jewish Chronic Disease Hospital study. Although these were biomedical studies and the primary investigators were physicians, there is evidence that nurses were aware of the research, identified potential research subjects, delivered treatments to the subjects, and served as data collectors in these projects. The four projects demonstrate the importance of ethical conduct for anyone reviewing, participating in, and conducting nursing or biomedical research. These four projects and other incidences of unethical treatment of subjects and scientific misconduct in the development, implementation, and reporting of research have influenced the formulation of ethical codes and regulations to direct research today. In addition, the concern for patient privacy with the electronic storage and exchange of health information has resulted in HIPAA privacy regulations (Olsen, 2003).
Nazi Medical Experiments
From 1933 to 1945, the Third Reich in Europe implemented atrocious, unethical activities (Steinfels & Levine, 1976). The programs of the Nazi regime consisted of sterilization, euthanasia, and numerous medical experiments to produce a population of racially pure Germans, or Aryans, who the Nazis maintained were destined to rule the world. The Nazis encouraged population growth among the Aryans (“good Nazis”) and sterilized people they regarded as racial enemies, such as the Jews. They also practiced what they called “euthanasia,” which involved killing various groups of people whom they considered racially impure, such as the insane, deformed, and senile. In addition, numerous medical experiments were conducted on prisoners of war, as well as racially “valueless” persons who had been confined to concentration camps.
The medical experiments involved exposing subjects to high altitudes, freezing temperatures, malaria, poisons, spotted fever (typhus), and untested drugs and operations, usually without any anesthesia (Steinfels & Levine, 1976). For example, in the hypothermia studies, subjects were immersed in bath temperatures ranging from 2° to 12° C. The researchers noted that “immersion in water 5° C is tolerated by clothed men for 40 to 60 minutes, whereas raising the water temperature to 15° C increases the period of tolerance to four to five hours” (Berger, 1990, p. 1436). These medical experiments were conducted to generate knowledge about human beings but the end result often was to destroy certain groups of people. Extensive examination of the records from some of these studies showed that they were poorly designed and conducted. Thus, they generated little if any useful scientific knowledge.
The Nazi experiments violated numerous rights of human research subjects. The selection of subjects was racially based, demonstrating an unfair selection process. The subjects also had no opportunity to refuse participation; they were prisoners who were coerced or forced to participate. Subjects were frequently killed during the experiments or sustained permanent physical, mental, and social damage as a result (Levine, 1986; Steinfels & Levine, 1976). These studies were not conducted by a few isolated scientists and physicians; they were “the product of coordinated policymaking and planning at high governmental, military, and Nazi Party levels, conducted as an integral part of the total war effort” (Nuremberg Code, 1986, p. 425).
Nuremberg Code
The people involved in the Nazi experiments were brought to trial before the Nuremberg Tribunals, which publicized these unethical activities. The mistreatment of human subjects in these studies led to the development of the Nuremberg Code in 1949. This code contains guidelines for (1) subjects’ voluntary consent to participate in research; (2) the right of subjects to withdraw from studies; (3) protection of subjects from physical and mental suffering, injury, disability, and death during studies; and (4) the balance of benefits and risks in a study (Table 9-1). The Nuremberg Code was formulated mainly to direct the conduct of biomedical research; however, the rules it contains are essential to research in other sciences, such as nursing, psychology, and sociology. This code was developed to promote the ethical conduct of research in all countries and can be reviewed online at http://ohsr.od.nih.gov/guidelines/nuremberg.html.
TABLE 9-1
1. The voluntary consent of the human subject is absolutely essential.
2. The experiment should be such as to yield fruitful results for the good of society, unprocurable by other methods or means of study, and not random and unnecessary in nature.
3. The experiment should be so designed and based on the results of animal experimentation and knowledge of the natural history of the disease or other problem under study that the anticipated results will justify the performance of the experiment.
4. The experiment should be so conducted as to avoid all unnecessary physical and mental suffering and injury.
5. No experiment should be conducted where there is a priori reason to believe that death or disabling injury will occur, except, perhaps, in those experiments where the experimental physicians also serve as subjects.
6. The degree of risk to be taken should never exceed that determined by the humanitarian importance of the problem to be solved by the experiment.
7. Proper preparations should be made and adequate facilities provided to protect the experimental subject against even remote possibilities of injury, disability, or death.
8. The experiment should be conducted only by scientifically qualified persons. The highest degree of skill and care should be required through all stages of the experiment of those who conduct or engage in the experiment.
9. During the course of the experiment the human subject should be at liberty to bring the experiment to an end if he has reached the physical or mental state where continuation of the experiment seems to him to be impossible.
10. During the course of the experiment the scientist in charge must be prepared to terminate the experiment at any stage, if he has probable cause to believe, in the exercise of the good faith, superior skill, and careful judgment required of him, that a continuation of the experiment is likely to result in injury, disability, or death to the experimental subject.
From “The Nuremberg Code,” in Ethics and Regulation of Clinical Research (2nd ed., pp. 425–426), edited by R.J. Levine, 1986, Baltimore–Munich: Urban & Schwarzenberg.
Declaration of Helsinki
The Nuremberg Code provided the basis for the development of the Declaration of Helsinki, which was adopted in 1964 and amended in 1975, 1983, 1989, 1996, 2000, and 2002 by the World Medical Association; it can be viewed online at www.rotrf.org/information/Helsinki_declaration.pdf. The Declaration of Helsinki differentiated therapeutic research from nontherapeutic research. Therapeutic research gives the patient an opportunity to receive an experimental treatment that might have beneficial results. Nontherapeutic research is conducted to generate knowledge for a discipline, and the results from the study might benefit future patients but will probably not benefit those acting as research subjects.
The Declaration of Helsinki requires that (1) greater care be exercised to protect subjects from harm in nontherapeutic research; (2) there be a strong, independent justification for exposing a healthy volunteer to risk of harm just to gain new scientific information; (3) investigators must protect the life, health, privacy, and dignity of research subjects; and (4) extreme care must be taken in making use of placebo-controlled trial and used only in the absence of existing proven therapy (World Medical Association General Assembly, 2002). Thus, this legal document provides an ethical basis for the clinical trials and other research conducted today in the United States and internationally. Clinical trials must focus on improving diagnostic, therapeutic, and prophylactic procedures for patients with selected diseases without exposing subjects to any additional risk of serious or irreversible harm. Most institutions worldwide in which clinical research is conducted have adopted the Declaration of Helsinki. However, neither this document nor the Nuremberg Code has prevented some investigators from conducting unethical research (Beecher, 1966; Nelson-Marten & Rich, 1999; ORI, 2004).
Tuskegee Syphilis Study
In 1932, the U.S. Public Health Service (U.S. PHS) initiated a study of syphilis in black men in the small, rural town of Tuskegee, Alabama (Brandt, 1978; Rothman, 1982). The study, which continued for 40 years, was conducted to determine the natural course of syphilis in the adult black male. The research subjects were organized into two groups: one group consisted of 400 men who had untreated syphilis and the other consisted of a control group of 200 men without syphilis. Many of the subjects who consented to participate in the study were not informed about the purpose and procedures of the research. Some individuals were unaware that they were subjects in a study.
By 1936, it was apparent that the men with syphilis developed more complications than the control group. Ten years later, the death rate of the group with syphilis was twice as high as that for the control group. The subjects were examined periodically but were not treated for syphilis, even after penicillin was determined to be an effective treatment for the disease in the 1940s (Levine, 1986). Information about an effective treatment for syphilis was withheld from the subjects, and deliberate steps were taken to keep them from receiving treatment (Brandt, 1978).
Published reports of the Tuskegee Syphilis Study first started appearing in 1936, and additional papers were published every 4 to 6 years. No effort was made to stop the study; in fact, in 1969, the U.S. Centers for Disease Control (CDC) decided that it should continue. Numerous individuals were involved in conducting this study, including
three generations of doctors serving in the venereal disease division of the U.S. PHS, numerous officials at the Tuskegee Institute and its affiliated hospital, hundreds of doctors in the Macon County and Alabama medical societies, and numerous foundation officials at the Rosenwald Fund and the Milbank Memorial Fund. (Rothman, 1982, p. 5)
In 1972, an account of the study published in the Washington Star sparked public outrage. Only then did the U.S. Department of Health, Education, and Welfare (DHEW) stop the study. The Tuskegee Syphilis Study was investigated and found to be ethically unjustified, but its racial implications were never addressed (Brandt, 1978). There are still many unanswered questions about this study, such as where were the checks and balances in the government and health care systems that should have prevented this unethical study from continuing for 40 years and why was public outrage the only effective means of halting the study?
Willowbrook Study
From the mid-1950s to the early 1970s, Dr. Krugman at Willowbrook, an institution for the mentally retarded, conducted research on hepatitis (Rothman, 1982). The subjects, all children, were “deliberately infected with the hepatitis virus; early subjects were fed extracts of stool from infected individuals and later subjects received injections of more purified virus preparations” (Levine, 1986, p. 70). During the 20-year study, Willowbrook closed its doors to new inmates because of overcrowded conditions. However, the research ward continued to admit new inmates. To gain their child’s admission to the institution, the parents were forced to give permission for the child to be a subject in the study.
From the late 1950s to early 1970s, Krugman’s research team published several articles describing the study protocol and findings. Beecher (1966) cited the Willowbrook study as an example of unethical research. The investigators defended injecting the children with the virus by citing their own belief that most of the children would have acquired the infection after admission to the institution. The investigators also stressed the benefits the subjects received, which were a cleaner environment, better supervision, and a higher nurse-patient ratio on the research ward (Rothman, 1982). Despite the controversy, this unethical study continued until the early 1970s.
Jewish Chronic Disease Hospital Study
Another highly publicized example of unethical research was a study conducted at the Jewish Chronic Disease Hospital in the 1960s. Its purpose was to determine the patients’ rejection responses to live cancer cells. Twenty-two patients were injected with a suspension containing live cancer cells that had been generated from human cancer tissue (Hershey & Miller, 1976; Levine, 1986).
The patients were not informed that they were taking part in research or that the injections they received were live cancer cells. In addition, the Jewish Chronic Disease Hospital Institutional Review Board never reviewed the study; even the physicians caring for the patients were unaware that the study was being conducted. The physician directing the research was an employee of the Sloan-Kettering Institute for Cancer Research, and there was no indication that this institution had reviewed the research project (Hershey & Miller, 1976). The research project had the potential to cause the human subjects serious or irreversible harm and possibly death.
Other Unethical Studies
In 1966, Henry Beecher published a now classic article describing 22 of the 50 examples of unethical or questionably ethical studies that he had identified in the published literature. The examples, which included the Willowbrook and Jewish Chronic Disease Hospital studies, indicated a variety of ethical problems that were relatively widespread. Consent was mentioned in only 2 of the 50 studies, and many of the investigators had unnecessarily risked the health and lives of their subjects. Beecher (1966, p. 1356) believed that many of the abuses in the research were due to “thoughtlessness and carelessness, not a willful disregard of patients’ rights.” These studies reinforce the importance of conscientious institutional review and ethical researcher conduct.
U.S. Department of Health, Education, and Welfare Regulations
The continued conduct of harmful, unethical research made additional controls necessary. In 1973, the DHEW published its first set of regulations intended to protect human subjects. By May 1974, clinical researchers were presented with strict regulations for research involving humans, with additional regulations to protect persons having limited capacities to consent, such as the ill, mentally impaired, and dying (Levine, 1986).
In the 1970s, researchers went from a few vague regulations to almost overwhelming guidelines. All research involving human subjects had to undergo full institutional review, even nursing studies that involved minimal or no risks to the human subjects. Institutional review improved the protection of human subjects; however, reviewing all studies, without regard for the degree of risk involved, overwhelmed the review process and greatly prolonged the time required for a study to be approved (Levine, 1986).
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research
Because the DHEW regulations far from resolved the issue of protecting human subjects in research, the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (1978) was formed. This commission was established by the National Research Act (Public Law 93-348) passed in 1974. The goals of the commission were (1) to identify the basic ethical principles that should underlie the conduct of biomedical and behavioral research involving human subjects and (2) to develop guidelines based on these principles.
The commission developed The Belmont Report, which identified three ethical principles as relevant to research involving human subjects: the principles of respect for persons, beneficence, and justice. The principle of respect for persons holds that persons have the right to self-determination and the freedom to participate or not participate in research. The principle of beneficence requires the researcher to do good and “above all, do no harm.” The principle of justice holds that human subjects should be treated fairly. The ethical principles in this report continue to influence the conduct of ethical research in the United States and internationally. This commission developed ethical research guidelines based on these three principles, made recommendations to the U.S. Department of Health and Human Services (U.S. DHHS), and was dissolved in 1978 (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, 1978).
In response to the commission’s recommendations, in 1981 the U.S. DHHS developed a set of federal regulations to protect human research subjects, and these regulations were revised over the years (U.S. DHHS, 1981, 1983, 1991, 2001, 2005). The most current 2005 regulations are part of the Code of Federal Regulations (CFR), Title 45, Part 46, Protection of Human Subjects. These regulations are interpreted by the Office for Human Research Protection (OHRP), an agency within U.S. DHHS, whose functions are described online at www.hhs.gov/ohrp. These regulations provide direction for (1) the protection of human subjects in research, with additional protection for pregnant women, human fetuses, neonates, children, and prisoners; (2) the documentation of informed consent; and (3) the implementation of the institutional review board process (U.S. DHHS, 2005). These regulations apply to all research involving human subjects in the following areas: (1) studies conducted, supported, or otherwise subject to regulations by any federal department or agency; (2) research conducted in educational settings; (3) research involving the use of educational tests, survey procedures, interview procedures, or observation; and (4) research involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens.
Most of the biomedical and behavioral research conducted in the United States is governed by the U.S. DHHS Protection of Human Subjects Regulations (U.S. DHHS, 2005) or the U.S. Food and Drug Administration (FDA) (see www.fda.gov/oc/gcp). The FDA, within the U.S. DHHS, manages the CFR Title 21, Food and Drugs, Part 50, Protection of Human Subjects, and Part 56, Institutional Review Boards. The FDA has additional human subject protection regulations that apply to clinical investigations involving products regulated by the FDA under the Federal Food, Drug, and Cosmetic Act and research that supports applications for research or marketing permits for these products. Thus, these regulations apply to studies of drugs for humans, medical devices for human use, biological products for human use, human dietary supplements, and electronic products and can be viewed online at www.fda.gov/oc/gcp/regulations.html. The physician and nurse researchers conducting clinical trials to generate new drugs and refine existing drug treatments must comply with these FDA regulations. These regulations focus on the protection of human subjects’ rights, informed consent (FDA, 2006b), and institutional review boards (FDA, 2006b), with content that is consistent with Title 45, Public Welfare, Part 46, Protection of Human Subjects (U.S. DHHS, 2005).
The U.S. DHHS and FDA regulations provide guidelines to protect subjects in federally and privately funded research by ensuring that their privacy and the confidentiality of the information obtained through research. However, with the advent of electronic access and transfer, the public has become concerned about the potential abuses of the health information of individuals in all circumstances, including research projects. Thus, Public Law 104-191, the Health Insurance Portability and Accountability Act (HIPAA), was enacted in 1996 and implemented in 2003 to protect an individual’s health information. The U.S. DHHS developed regulations titled the Standards for Privacy of Individually Identifiable Health Information, and compliance with these regulations is known as the Privacy Rule (U.S. DHHS, 2002, 45 CFR Parts 160 and 164). The HIPAA Privacy Rule established the category of protected health information (PHI), which allows covered entities, such as health plans, health care clearinghouses, and health care providers that transmit health information, to use or disclose PHI to others only in certain situations. These situations are discussed later in this chapter.
The HIPAA Privacy Rule affects not only the health care environment but also the research conducted in this environment. An individual must provide his or her signed permission, or authorization, before his or her PHI can be used or disclosed for research purposes. Researchers must modify their current studies and develop their new research projects to comply with the HIPAA Privacy Rule. The U.S. DHHS developed a website, “HIPAA Privacy Rule: Information for Researchers,” to address the impact of this rule on the informed consent and institutional review board processes in research and to answer common questions about the HIPAA (see http://privacyruleandresearch.nih.gov). The full impact of the privacy rule on the protection of individuals’ privacy and on the conduct of research is yet to be determined (Conner, Smaldone, Bratts, & Stone, 2003; Olsen, 2003); however, you can view the privacy rule enforcement highlights online at www.hhs.gov/ocr/privacy/enforcement.
In summary, Table 9-2 was developed to clarify the overall objectives and applicability of the HIPAA Privacy Rule, U.S. DHHS Protection of Human Subjects Regulations Title 45 CFR Part 46, and FDA Protection of Human Subjects Regulations Title 21 CFR Parts 50 and 56 (U.S. DHHS, 2007, February 2, b). The specifics of these regulations are discussed later in this chapter in the sections on protecting human rights, obtaining informed consent, and institutional review.
TABLE 9-2
Clarification of the Focus of Federal Regulations and Impact on Research

From U.S. Department of Health and Human Services. (2007, February 2, b). How do other privacy protections interact with the privacy rule? HIPAA Privacy Rule: Information for researchers. Retrieved October 1, 2007, from http://privacyruleandresearch.nih.gov/pr_05.asp.
PROTECTION OF HUMAN RIGHTS
Human rights are claims and demands that have been justified in the eyes of an individual or by the consensus of a group of individuals. Having rights is necessary for the self-respect, dignity, and health of an individual (Sasson & Nelson, 1971). Researchers and reviewers of research have an ethical responsibility to recognize and protect the rights of human research subjects. The human rights that require protection in research are (1) the right to self-determination, (2) the right to privacy, (3) the right to anonymity and confidentiality, (4) the right to fair treatment, and (5) the right to protection from discomfort and harm (American Nurses Association [ANA], 2001; American Psychological Association [APA], 2002). Nurses in other countries have developed similar codes of ethics to protect the rights of their patients and to promote ethical conduct in research (Lin et al., 2007).
Right to Self-Determination
The right to self-determination is based on the ethical principle of respect for persons. This principle holds that because humans are capable of self-determination, or controlling their own destiny, they should be treated as autonomous agents who have the freedom to conduct their lives as they choose without external controls. As a researcher, you treat prospective subjects as autonomous agents by informing them about a proposed study and allowing them to voluntarily choose to participate or not. In addition, subjects have the right to withdraw from a study at any time without a penalty (Levine, 1986). Conducting research ethically requires that research subjects’ right to self-determination not be violated and that persons with diminished autonomy have additional protection during the conduct of studies.
Preventing Violation of Research Subjects’ Right to Self-Determination
A subject’s right to self-determination can be violated through the use of (1) coercion, (2) covert data collection, and (3) deception. Coercion occurs when, one person intentionally presents another with an overt threat of harm or the lure of excessive reward to obtain his or her compliance (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, 1978). Some subjects are coerced to participate in research because they fear that they will suffer harm or discomfort if they do not participate. For example, some patients believe that their medical or nursing care will be negatively affected if they do not agree to be research subjects. Sometimes students feel forced to participate in research to protect their grades or prevent negative relationships with the faculty conducting the research. Other subjects are coerced to participate in studies because they believe that they cannot refuse the excessive rewards offered, such as large sums of money, specialized health care, special privileges, and jobs. Most nursing studies do not offer excessive rewards to subjects for participating. A few nursing studies have included a small financial reward of $10 to $20 or support for transportation to increase participation, but this usually would not be considered coercive.
An individual’s right to self-determination can also be violated if he or she becomes a research subject without realizing it. Some researchers have exposed persons to experimental treatments without their knowledge, a prime example being the Jewish Chronic Disease Hospital study. Most of the patients and their physicians were unaware of the study. The subjects were informed that they were receiving an injection of cells, but the word cancer was omitted (Beecher, 1966). With covert data collection, subjects are unaware that research data are being collected because the investigator develops “descriptions of natural phenomena using information that is provided as a matter of normal activity” (Reynolds, 1979, p. 76). This type of data collection has more commonly been used by psychologists to describe human behavior in a variety of situations, but it has also been used by nursing and other disciplines (APA, 2002). Qualitative researchers have debated this issue, and some believe that certain group and individual behaviors are unobservable within the normal ethical range of research activities, such as the actions of cults or the aggressive or violent behaviors of individuals (Grbich, 1999). However, covert data collection is considered unethical when research deals with sensitive aspects of an individual’s behavior, such as illegal conduct, sexual behavior, or drug use (U.S. DHHS, 2005). With the HIPAA Privacy Rule (U.S. DHHS, 2002), the use of any type of covert data collection would be questionable and illegal if PHI data were being used or disclosed.
The use of deception in research can also violate a subject’s right to self-determination. Deception is the actual misinforming of subjects for research purposes (Kelman, 1967). A classic example of deception is the Milgram (1963) study, in which the subjects thought they were administering electric shocks to another person. The subjects were unaware that the person was really a professional actor who pretended to feel the shocks. Some subjects experienced severe mental tension, almost to the point of collapse, because of their participation in this study. The use of deception still occurs in some health care, social, and psychological investigations, but it is a controversial research activity (Grbich, 1999). If deception is to be used in a study, you must determine that there is no other way to get the essential research data needed and that the subjects will not be harmed. In addition, the subjects must be informed of the deception once the study is completed, and you must provide them with full disclosure of the study that was conducted (U.S. DHHS, 2005).
Protecting Persons with Diminished Autonomy
Some persons have diminished autonomy or are vulnerable and less advantaged because of legal or mental incompetence, terminal illness, or confinement to an institution (Levine, 1986). These persons require additional protection of their right to self-determination, because they have a decreased ability, or an inability, to give informed consent. In addition, these persons are vulnerable to coercion and deception. The U.S. DHHS (2005) has identified certain vulnerable groups of individuals, including pregnant women, human fetuses, neonates, children, mentally incompetent persons, and prisoners, who require additional protection in the conduct of research. Researchers need to justify their use of subjects with diminished autonomy in a study, and the need for justification increases as the subjects’ risk and vulnerability increase. However, in many situations, the knowledge needed to provide evidence-based care to these vulnerable populations can be gained only by studying them.
Legally and Mentally Incompetent Subjects: Neonates and children (minors), the mentally impaired, and unconscious patients are legally or mentally incompetent to give informed consent. These individuals lack the ability to comprehend information about a study and to make decisions regarding participation in or withdrawal from the study. Their vulnerability ranges from minimal to absolute. The use of persons with diminished autonomy as research subjects is more acceptable if (1) the research is therapeutic so that the subjects have the potential to benefit directly from the experimental process, (2) the researcher is willing to use both vulnerable and nonvulnerable individuals as subjects, (3) preclinical and clinical studies have been conducted and provide data for assessing potential risks to subjects, and (4) the risk is minimized and the consent process is strictly followed to secure the rights of the prospective subjects (Levine, 1986; U.S. DHHS, 2005).
Neonates: A neonate is defined as a newborn and is identified as either viable or nonviable on delivery. Viable neonates are able to survive after delivery, if given the benefit of available medical therapy, and can independently maintain a heartbeat and respiration. “A nonviable neonate means that a newborn after delivery, although living, is not viable” (U.S. DHHS, 2005, 45 CFR Section 46.202). Neonates are extremely vulnerable and require extra protection to determine their involvement in research. However, your research may involve viable neonates, neonates of uncertain viability, and nonviable neonates if the following conditions are met: (1) your study is scientifically appropriate and the preclinical and clinical studies have been conducted and provide data for assessing the potential risks to the neonates; (2) your study provides important biomedical knowledge, which cannot be obtained by other means and will not add risk to the neonate; (3) your research has the potential to enhance the probability of survival of the neonate; (4) both parents are fully informed about your research during the consent process; and (5) your research team will have no part in determining the viability of the neonate. In addition, for the “nonviable neonates, the vital functions of the neonate should not be artificially maintained because of the research and the research should not terminate the heartbeat or respiration of the neonate” (U.S. DHHS, 2005, 45 CFR Section 46.205).
Children: The unique vulnerability of children makes the decision to include them as research subjects particularly important. To safeguard their interests and protect them from harm, special ethical and regulatory considerations have been put in place for research involving children (Title 45, CFR 46 Subpart D). However, the laws defining the minor status of a child are statutory and vary from state to state. Often a child’s competency to consent is governed by age, with incompetence being nonrefutable up to age 7 years (Broome, 1999; Thompson, 1987). Thus, a child younger than 7 years is not believed to be mature enough to assent or consent to research. Developmentally by age 7, a child is capable of concrete operations of thought and can give meaningful assent to participate as a subject in studies (Thompson, 1987). With advancing age and maturity, a child should have a stronger role in the consent process.
To obtain informed consent, federal regulations require both “soliciting the assent of the children (when capable) and the permission of their parents or guardians” (U.S. DHHS, 2005, 45 CFR Section 46.408). “Assent means a child’s affirmative agreement to participate in research. Permission to participate in a study means the agreement of parents or guardian to the participation of their child or ward in research” (U.S. DHHS, 2005, 45 CFR Section 46.402). If a child does not assent to participate in the study, he or she should not be included as a subject even if parental permission is obtained.
Using children as research subjects is also influenced by the therapeutic nature of the research and the risks versus the benefits. Thompson (1987) developed a guide for obtaining informed consent that is based on the child’s level of competence, the therapeutic nature of the research, and the risks versus the benefits (Table 9-3). Children who are experiencing a developmental delay, cognitive deficit, emotional disorder, or physical illness must be considered individually (Broome, 1999; Broome & Stieglitz, 1992).
TABLE 9-3
Guide to Obtaining Informed Consent Based on the Relationship between a Child’s Level of Competence, the Therapeutic Nature of the Research, and Risk versus Benefit
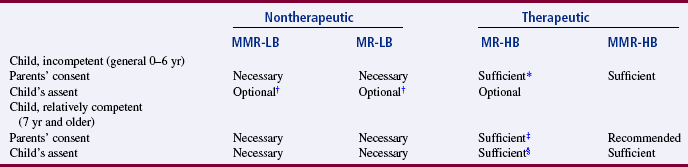
MMR, More than minimal risk; MR, minimal risk; LB, low benefit; HB, high benefit.
*A parent’s refusal can be superseded by the principle that a parent has no power to forbid the saving of a child’s life.
†Children making “deliberate objection” would be precluded from participation by most researchers.
‡In cases not involving the privacy rights of a “mature minor.”
§In cases involving the privacy rights of a “mature minor.”
From Thompson, P. J. (1987). Protection of the rights of children as subjects for research. Journal of Pediatric Nursing, 2(6): 397.
A child 7 years or older with normal cognitive development can provide assent or dissent to participation in a study, and the process for obtaining the assent should be included in the research proposal. In the assenting process, the child must be given developmentally appropriate information on the study purpose, expectations, and the benefit-risk ratio (discussed later). Videotapes, written materials, demonstrations, diagrams, role-modeling, and peer discussions are methods for communicating study information. The child also needs an opportunity to sign an assent form and to have a copy of this form. An example assent form is presented in Table 9-4. During the study, you must give the child the opportunity to ask questions and to withdraw from the study if he or she desires (Broome, 1999). Assent becomes more complex if the child is bilingual, because the researchers must determine the most appropriate language to use for the consent process for the child and the parents. Holaday, Gonzales, and Mills (2007) included a list of seven questions in their article to assist researchers in determining the language for communication during a study.
TABLE 9-4
Sample Assent Form for Children Ages 6 To 12 Years: Pain Interventions for Children with Cancer
Oral Explanation
I am a nurse who would like to know if relaxation, special ways of breathing, and using your mind to think pleasant things help children like you to feel less afraid and feel less hurt when the doctor has to do a bone marrow aspiration or spinal tap. Today, and the next five times you and your parent come to the clinic, I would like for you to answer some questions about the things in the clinic that scare you. I would also like you to tell me about how much pain you felt during the bone marrow or spinal tap. In addition, I would like to videotape (take pictures of) you and your mom and/or dad during the tests. The second time you visit the clinic I would like to meet with you and teach you special ways to relax, breathe, and use your mind to imagine pleasant things. You can use the special imagining and breathing then during your visits to the clinic. I would ask you and your parent to practice the things I teach you at home between your visits to the clinic. At any time you could change your mind and not be in the study anymore.
To Child
1. I want to learn special ways to relax, breathe, and imagine.
2. I want to answer questions about things children may be afraid of when they come to the clinic.
3. I want to tell you how much pain I feel during the tests I have.
4. I will let you videotape me while the doctor does the tests (bone marrow and spinal taps).
If the child says YES, have him/her put an “X” here. ___
From Broome, M. E. (1999). Consent (assent) for research with pediatric patients. Seminars in Oncology Nursing, 15(2): 101.
In addition to the assent of the child, you must obtain permission from the child’s parent or guardian for the child’s participation in a study. In studies of minimal risk, permission from one parent is usually sufficient for the research to be conducted. For research that involves more than minimal risk and has no prospect of direct benefit to the individual subject, “both parents must give their permission unless one parent is deceased, unknown, incompetent, or not reasonably available, or when only one parent has legal responsibility for the care and custody of the child” (U.S. DHHS, 2005, 45 CFR Section 46.408b).
Pregnant Women: Pregnant women require additional protection in research because of the fetus. Federal regulations define pregnancy as encompassing the period of time from implantation until delivery. “A woman is assumed to be pregnant if she exhibits any of the pertinent presumptive signs of pregnancy, such as missed menses, until the results of a pregnancy test are negative or until delivery” (U.S. DHHS, 2005, 45 CFR Section 46.202). Research conducted with pregnant women should have the potential to directly benefit the woman or the fetus. If your investigation is thought to provide a direct benefit only to the fetus, you must obtain the consent of the pregnant woman and father. Studies with “pregnant women should include no inducements to terminate the pregnancy and the researcher should have no part in any decision to terminate a pregnancy” (U.S. DHHS, 2005, 45 CFR Section 46.204).
Adults: Because of mental illness, cognitive impairment, or a comatose state, certain adults are incompetent and incapable of giving informed consent. Persons are said to be incompetent if, in the judgment of a qualified clinician, they have attributes that ordinarily provide the grounds for designating incompetence (Levine, 1986). Incompetence can be temporary (e.g., inebriation), permanent (e.g., advanced senile dementia), or subjective or transitory (e.g., behavior or symptoms of psychosis).
If an individual is judged incompetent and incapable of consent, you must seek approval from the prospective subject and his or her legally authorized representative. A legally authorized representative means an individual or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s participation in the procedure(s) involved in the research (Levine, 1986). However, individuals can be judged incompetent and can still assent to participate in certain minimal-risk research if they have the ability to understand what they are being asked to do, to make reasonably free choices, and to communicate their choices clearly and unambiguously.
Happ et al. (2007) conducted an ethnographic study to describe family members’ and clinicians’ interactions with intensive care unit (ICU) patients during their weaning from long-term mechanical ventilation (LTMV). Because some of the ICU patients were incompetent to give informed consent, the researchers obtained their assent if possible and their proxies’ or guardians’ permission for participation in the study. The following excerpt from this study will guide you in protecting the rights of persons with diminished autonomy by (1) obtaining institutional review board (IRB) approval for your study, (2) ensuring the privacy of patients during data collection, and (3) obtaining informed consent for all study participants based on their physical and mental capabilities.
A growing number of people have become permanently incompetent from the advanced stages of senile dementia of the Alzheimer type (SDAT). A minimum of 60% of nursing home residents have this condition (Floyd, 1988). Most long-term care settings have no IRB for research, and the families or guardians of patients in such settings are reluctant to give consent for the patients’ participation in research. Nursing research is needed to establish evidence-based interventions for comforting and caring for individuals with SDAT. Thus, there is a need to expand the development of IRBs in long-term care settings and the use of client advocates to assist in the consent process. Levine (1986) identified two approaches that families, guardians, researchers, or institutional review board might use when making decisions on behalf of these incompetent individuals: (1) best interest standard and (2) substituted judgment standard. The best interest standard involves doing what is best for the individual on the basis of balancing risks and benefits. The substituted judgment standard is concerned with determining the course of action that incompetent individuals would take if they were capable of making a choice.
Terminally Ill Subjects: When conducting research on terminally ill subjects, you should determine (1) who will benefit from the research and (2) whether it is ethical to conduct research on individuals who might not benefit from the study (U.S. DHHS, 2005). Participating in research could have greater risks and minimal or no benefits for these subjects. In addition, the dying subject’s condition could affect the study results and lead you to misinterpret the results (Watson, 1982). However, Hinds, Burghen, and Pritchard (2007) stressed the importance of conducting end-of-life studies in pediatric oncology to generate evidence that will improve the care for terminally ill children and adolescents and their families.
Some terminally ill individuals are willing subjects because they believe that participating in research is a way to contribute to society before they die. Others want to take part in research because they believe that the experimental process will benefit them. People with acquired immunodeficiency syndrome (AIDS) often want to participate in AIDS research to gain access to experimental drugs and hospitalized care. However, researchers are concerned because some of these individuals do not comply with the research protocol (Arras, 1990). This is a serious dilemma for researchers studying a population with any type of serious or terminal illness, because they must consider the rights of the subjects and be responsible for conducting high-quality research.
Subjects Confined to Institutions: Hospitalized patients have diminished autonomy because they are ill and are confined in settings that are controlled by health care personnel (Levine, 1986). Some hospitalized patients feel obliged to be research subjects because they want to assist a particular practitioner (nurse or physician) with his or her research. Others feel coerced to participate because they fear that their care will be adversely affected if they refuse. Some of these hospitalized patients are survivors of trauma (such as auto accidents, gunshot wounds, or physical and sexual abuse) who are very vulnerable and often have decreased decision-making capacities (McClain, Laughon, Steeves, & Parker, 2007). When conducting research with these types of patients, you must pay careful attention to the informed consent process and make every effort to protect these subjects from feelings of coercion and harm.
In the past, prisoners have experienced diminished autonomy in research projects because of their confinement. They might feel coerced to participate in research because they fear harm if they refuse or because they desire the benefits of early release, special treatment, or monetary gain. Prisoners have been used for drug studies in which there were no health-related benefits and there was possible harm for the prisoners (Levine, 1986). Current regulations regarding research involving prisoners require that “the risks involved in the research are commensurate with risks that would be accepted by nonprisoner volunteers and procedures for the selection of subjects within the prison are fair to all prisoners and immune from arbitrary intervention by prison authorities or prisoners” (U.S. DHHS, 2005, 45 CFR Section 46.305a).
Protecting subjects’ rights with diminished autonomy in research is regulated in many other countries as well. In Canada, the ethical conduct of research is governed by the Tri-Council Policy Statement on “Ethical Conduct for Research Involving Human Subjects” (Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and Social Sciences and Humanities Research Council of Canada, 2005). The Council for International Organizations of Medical Sciences (CIOMS) (2002) developed international ethical guidelines for biomedical research involving human subjects, and the guidelines require protection of vulnerable individuals, groups, communities, and populations during research. In summary, researchers must evaluate each prospective subject’s capacity for self-determination and protect subjects with diminished autonomy during the research process.
Right to Privacy
Privacy is an individual’s right to determine the time, extent, and general circumstances under which personal information will be shared with or withheld from others. This information consists of one’s attitudes, beliefs, behaviors, opinions, and records. An individual’s privacy was initially protected by the Privacy Act of 1974. As a result of this act, data collection methods were to be scrutinized to protect subjects’ privacy, and data cannot be gathered from subjects without their knowledge. Individuals also have the right to access their records and to prevent access by others (U.S. DHHS, 2002, 2005). The intent of this act was to prevent the invasion of privacy that occurs when private information is shared without an individual’s knowledge or against his or her will. Invading an individual’s privacy might cause loss of dignity, friendships, or employment or create feelings of anxiety, guilt, embarrassment, or shame.
The HIPAA Privacy Rule expanded the protection of an individual’s privacy, specifically his or her protected individually identifiable health information, and described the ways in which covered entities can use or disclose this information. Covered entities are the individual’s health care provider, health plan, employer, and health care clearinghouse (public or private entity that processes or facilitates the processing of health information). Individually identifiable health information (IIHI)
is information that is a subset of health information, including demographic information collected from an individual, and: (1) Is created or received by healthcare provider, health plan, or healthcare clearinghouse; and (2) Related to past, present, or future physical or mental health or condition of an individual, the provision of health care to an individual, or the past, present, or future payment for the provision of health care to an individual, and that identifies the individual; or with respect to which there is a reasonable basis to believe that the information can be used to identify the individual. (U.S. DHHS, 2002, 45 CFR, Section 160.103)
According to the HIPAA Privacy Rule, the IIHI is protected health information (PHI) that is transmitted by electronic media, maintained in electronic media, or transmitted or maintained in any other form or medium. Thus, the HIPAA privacy regulations affect nursing research in the following ways: (1) accessing data from a covered entity, such as reviewing a patient’s medical record in clinics or hospitals; (2) developing health information, such as the data developed when an intervention is implemented in a study to improve a subject’s health; and (3) disclosing data from a study to a colleague in another institution, such as sharing data from a study to facilitate development of an instrument or scale (Olsen, 2003).
The U.S. DHHS developed guidelines to help researchers, health care organizations, and health care providers determine when they can use and disclose IIHI. IIHI can be used or disclosed to a researcher in the following situations:
• The protected health information has been “de-identified” under the HIPAA Privacy Rule. (De-identifying PHI is defined in the following section.)
• The data are part of a limited data set, and a data use agreement with the researcher(s) is in place.
• The individual who is a potential subject for a study authorizes the researcher to use and disclose his or her PHI.
• A waiver or alteration of the authorization requirement is obtained from an IRB or a privacy board (U.S. DHHS, 2007, February 2, a) (see http://privacyruleandresearch.nih.gov/pr_08.asp).
The first two items are discussed in this section of the text. The authorization process is discussed in the section on obtaining informed consent, and the waiver or alteration of authorization requirement is covered in the section on understanding institutional review.
De-identifying Protected Health Information under the Privacy Rule
Covered entities, such as health care providers and agencies, can allow researchers access to health information if the information has been de-identified. De-identifying health data involves removing the 18 elements that could be used to identify an individual or his or her relatives, employer, or household members. The 18 identifying elements are as follows (U.S. DHHS, 2007 February 2, a):
2. All geographic subdivisions smaller than a state, including street address, city, county, precinct, zip code, and their equivalent geographical codes, except for the initial three digits of a zip code if, according to the current publicly available data from the Bureau of the Census:
a. The geographical unit formed by combining all zip codes with the same three initial digits contains more than 20,000 people
b. The initial three digits of a zip code for all such geographical units containing 20,000 or fewer people are changed to 000
3. All elements of dates (except year) for dates directly related to an individual, including birth date, admission date, discharge date, date of death; and all ages over 89 and all elements of dates (including year) indicative of such age, except that such ages and elements may be aggregated into a single category of age 90 or older
6. Electronic mail (e-mail) addresses
9. Health plan beneficiary numbers
11. Certificate/license numbers
12. Vehicle identifiers and serial numbers, including license plate numbers
13. Device identifiers and serial numbers
14. Web universal resource locators (URLs)
15. Internet protocol (IP) address numbers
16. Biometric identifiers, including fingerprints and voiceprints
17. Full-face photographic images and any comparable images
18. Any other unique identifying number, characteristic, or code, unless otherwise permitted by the Privacy Rule for re-identification
An individual’s health information can also be de-identified using statistical methods. However, the covered entity and you as the researcher must ensure that the individual subject cannot be identified or that there is a very small risk that the subject could be identified from the information used. The statistical method used for de-identification of the health data must be documented, and you must certify that the 18 elements for identification have been removed or revised to ensure that individual is not identified. You must retain this certification information for 6 years.
Limited Data Set and Data Use Agreement
Covered entities (health care provider, health plan, and health care clearinghouse) may use and disclose a limited data set to a researcher for a study without an individual subject’s authorization or an IRB waiver. However, a limited data set is considered protected health information, and the covered entity and the researcher must have a data use agreement. The data use agreement limits how the data set may be used and how it will be protected. The HIPAA Privacy Rule requires the data use agreement to do the following (U.S. DHHS, 2002):
1. Specifies the permitted uses and disclosures of the limited data set.
2. Identifies the researcher who is permitted to use or receive the limited data set.
3. Stipulates that the recipient (researcher) will
a. Not use or disclose the information other than permitted by the agreement.
b. Use appropriate safeguards to prevent the use or disclosure of the information, except as provided for in the agreement.
c. Hold any other person (co-researchers, statisticians, or data collectors) to the standards, restrictions, and conditions stated in the data use agreement with respect to the health information.
d. Not identify the information or contact the individuals whose data are in the limited data set.
Right to Autonomy and Confidentiality
On the basis of the right to privacy, the research subject has the right to anonymity and the right to assume that the data collected will be kept confidential. Anonymity exists if the subject’s identity cannot be linked, even by the researcher, with his or her individual responses (ANA, 2001; Sasson & Nelson, 1971). For studies that use de-identified health information or data from a limited data set, the subjects will be anonymous to the researcher. You will be unable to contact these subjects for additional information.
In most studies, researchers desire to know the identity of their subjects and promise that their identity will be kept anonymous from others. However, in these situations, you must receive authorization from the potential subject to use his or her health information, or you must have a waiver or an alteration of the authorization requirement from the IRB (see section on institutional review later in this chapter). Confidentiality is the researcher’s management of private information shared by a subject that must not be shared with others without the authorization of the subject. Confidentiality is grounded in the following premises:
• Individuals can share personal information to the extent they wish and are entitled to have secrets.
• One can choose with whom to share personal information.
• People who accept information in confidence have an obligation to maintain confidentiality.
• Professionals, such as researchers, have a “duty to maintain confidentiality that goes beyond ordinary loyalty” (Levine, 1986, p. 164; U.S. DHHS, 2005).
Breach of Confidentiality
A breach of confidentiality can occur when a researcher, by accident or direct action, allows an unauthorized person to gain access to the study raw data. Confidentiality can also be breached in the reporting or publication of a study when a subject’s identity is accidentally revealed, violating the subject’s right to anonymity (Ramos, 1989). Breaches of confidentiality can harm subjects psychologically and socially, as well as destroy the trust they had in you. Breaches of confidentiality can be especially harmful to a research subject if they involve (1) religious preferences; (2) sexual practices; (3) employment; (4) racial prejudices; (5) drug use; (6) child abuse; and (7) personal attributes, such as intelligence, honesty, and courage.
Some nurse researchers have encountered health care professionals who believe that they should have access to information about the patients in the hospital and will request to see the data the researchers have collected. Sometimes, family members or close friends would like to see the data collected on specific subjects. Sharing research data in these circumstances is a breach of confidentiality. When requesting permission to conduct a study, you should tell health care professionals, family members, and others in the setting that you will not share the raw data. However, you may elect to share the research report, including a summary of the data and findings from the study, with health care providers, family members, and other interested parties.
Maintaining Confidentiality
Researchers have a responsibility to protect the anonymity of subjects and to maintain the confidentiality of data collected during a study. You can protect anonymity by giving each subject a code number. Keep a master list of the subjects’ names and their code numbers in a locked place; for example, subject Mary Jones might be assigned the code number “001.” All of the instruments and forms that Mary completes and the data you collect about her during the study will be identified with the “001” code number, not her name. The master list of subjects’ names and code numbers is best kept separate from the data collected to protect subjects’ anonymity. You should not staple signed consent forms and authorization documents to instruments or other data collection tools, as this would make it easy for unauthorized persons to readily identify the subjects and their responses. Consent forms are often stored with the master list of subjects’ names and code numbers. When entering the data collected into the computer, use the code numbers for identification. Then lock the original data collection tools in a secure place.
Another way to protect your subjects’ anonymity is to have them generate their own identification codes (Damrosch, 1986). With this approach, each subject generates an individual code from personal information, such as the first letter of a mother’s name, the first letter of a father’s name, the number of brothers, the number of sisters, and middle initial. Thus, the code would be composed of three letters and two numbers, such as “BD21M.” This code would be used on each form that the subject completes. Even you as the researcher would not know the subject’s identity, only the subject’s code. If the data collected are highly sensitive, you might want to use this type of coding system.
The data collected should undergo group analysis so that an individual cannot be identified by his or her responses. If the subjects are divided into groups for data analysis and there is only one subject in a group, combine that subject’s data with that of another group or delete the data. In writing the research report, you should describe the findings in such a way that an individual or a group of individuals cannot be identified by their responses.
Maintaining confidentiality is often more difficult in qualitative research than in quantitative research. The nature of qualitative research requires that the “investigator must be close enough to understand the depth of the question under study, and must present enough direct quotes and detailed description to answer the question” (Ramos, 1989, p. 60). The small number of participants used in a qualitative study and the depth of detail gathered on each participant make it difficult to disguise the participant’s identity. Ford and Reutter (1990) have recommended that to maintain confidentiality, the researcher should (1) use pseudonyms instead of the participants’ names and (2) distort certain details in the participants’ stories while leaving the contents unchanged. You must respect participants’ privacy as they decide how much detail and editing of private information are necessary to publish a study (Munhall, 2001a; Orb, Eisenhauer, & Wynaden, 2001).
Researchers should also take precautions during data collection to maintain confidentiality. The interviews conducted with participants are frequently taped and later transcribed, so the participant’s name should not be mentioned on the tape. They have the right to know whether anyone other than you will be transcribing information from the interview. In addition, participants should be informed on an ongoing basis that they have the right to withhold information. To critique the rigor and credibility of qualitative research, produce an audit trail so that another researcher could examine the data to confirm the study findings. This process might create a dilemma regarding the confidentiality of participants’ data, so you must inform them if other researchers will be examining their data to ensure the credibility of the study findings (Munhall, 2001a; Orb et al., 2001).
Right to Fair Treatment
The right to fair treatment is based on the ethical principle of justice. This principle holds that each person should be treated fairly and should receive what he or she is due or owed. In research, the selection of subjects and their treatment during the course of a study should be fair.
Fair Selection of Subjects
In the past, injustice in subject selection has resulted from social, cultural, racial, and sexual biases in society. For many years, research was conducted on categories of individuals who were thought to be especially suitable as research subjects, such as the poor, charity patients, prisoners, slaves, peasants, dying persons, and others who were considered undesirable (Reynolds, 1979). Researchers often treated these subjects carelessly and had little regard for the harm and discomfort they experienced. The Nazi medical experiments, Tuskegee Syphilis Study, and Willowbrook study all exemplify unfair subject selection and treatment.
The selection of a population and the specific subjects to study should be fair, and the risks and benefits of a study should be fairly distributed on the basis of the subject’s efforts, needs, and rights. Subjects should be selected for reasons directly related to the problem being studied and not for “their easy availability, their compromised position, or their manipulability” (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, 1978, p. 10).
Another concern with subject selection is that some researchers select certain people as subjects because they like them and want them to receive the specific benefits of a study. Other researchers have been swayed by power or money to make certain individuals subjects so that they can receive potentially beneficial treatments. Random selection of subjects can eliminate some of the researcher bias that might influence subject selection.
A current concern in the conduct of research is finding an adequate number of appropriate subjects to take part in certain studies. As a solution to this problem in the past, some biomedical researchers have offered physicians a finder’s fee for identifying research subjects. For example, investigators studying patients with lung cancer would give a physician a fee for every patient with lung cancer the physician referred to them. However, the HIPAA Privacy Rule requires that individuals give their authorization before PHI can be shared with others. Thus, health care providers cannot recommend individuals for studies without their permission. Researchers can obtain a partial waiver from the IRB or privacy board so that they can obtain PHI necessary to recruit potential subjects (U.S. DHHS, 2002). This makes it more difficult for researchers to find subjects for their studies; however, researchers are encouraged to work closely with their IRBs to facilitate the conduct of quality studies essential for evidence-based practice.
Fair Treatment of Subjects
Researchers and subjects should have a specific agreement about what the subject’s participation involves and what the role of the researcher will be (APA, 2002). While conducting a study, you should treat the subjects fairly and respect that agreement. If the data collection requires appointments with the subjects, be on time for each appointment and terminate the data collection process at the agreed-on time. You should not change the activities or procedures that the subject is to perform unless you obtain the subject’s consent.
The benefits promised the subjects should be provided. For example, if you promise a subject a copy of the study findings, you should deliver your promise when the study is completed. In addition, subjects who participate in studies should receive equal benefits, regardless of age, race, and socioeconomic level. When possible, the sample should be representative of the study population and should include subjects of various ages, ethnic backgrounds, and socioeconomic status (Williams, 2002). Treating subjects fairly often facilitates the data collection process and decreases subjects’ withdrawal from a study (Orb et al., 2001).
Right to Protection from Discomfort and Harm
The right to protection from discomfort and harm is based on the ethical principle of beneficence, which holds that one should do good and, above all, do no harm. According to this principle, members of society should take an active role in preventing discomfort and harm and promoting good in the world around them (Frankena, 1973). Therefore, researchers should conduct their studies to protect subjects from discomfort and harm and try to bring about the greatest possible balance of benefits in comparison with harm.
Discomfort and harm can be physiological, emotional, social, and economic in nature. Reynolds (1979) identified the following five categories of studies, which are based on levels of discomfort and harm: (1) no anticipated effects, (2) temporary discomfort, (3) unusual levels of temporary discomfort, (4) risk of permanent damage, and (5) certainty of permanent damage. Each level is defined in the following discussion.
No Anticipated Effects
In some studies, the subjects expect neither positive nor negative effects. For example, studies that involve reviewing patients’ records, students’ files, pathology reports, or other documents have no anticipated effects on the subjects. In these types of studies, the researcher does not interact directly with the research subjects. Even in these situations, however, there is a potential risk of invading a subject’s privacy. The HIPAA Privacy Rule requires that the agency providing the health information de-identify the 18 essential elements, which could be used to identify an individual, to promote subjects’ privacy during a study.
Temporary Discomfort
Studies that cause temporary discomfort are described as minimal-risk studies, in which the discomfort encountered is similar to what the subject would experience in his or her daily life and ceases with the termination of the study. Many nursing studies require the subjects to complete questionnaires or participate in interviews, which usually involve minimal risk. The physical discomforts might be fatigue, headache, or muscle tension. The emotional and social risks might entail the anxiety or embarrassment associated with responding to certain questions. The economic risks might consist of the time spent participating in the study or travel costs to the study site. Participation in many nursing studies is considered a mere inconvenience for the subject, with no foreseeable risks of harm.
Most clinical nursing studies examining the impact of a treatment involve minimal risk. For example, your study might involve examining the effects of exercise on the blood glucose levels of diabetics. During the study, you ask the subjects to test their blood glucose level one extra time per day. There is discomfort when the blood is drawn and a risk of physical changes that might occur with exercise. The subjects might also experience anxiety and fear in association with the additional blood testing, and the testing is an added expense. The diabetic subjects in this study would experience similar discomforts in their daily lives, and the discomforts would cease with the termination of the study.
Unusual Levels of Temporary Discomfort
In studies that involve unusual levels of temporary discomfort, the subjects commonly experience discomfort both during the study and after its termination. For example, subjects might experience a deep vein thrombosis (DVT), prolonged muscle weakness, joint pain, and dizziness after participating in a study that required them to be confined to bed for 10 days to determine the effects of immobility. Studies that require subjects to experience failure, extreme fear, or threats to their identity or to act in unnatural ways involve unusual levels of temporary discomfort. In some qualitative studies, participants are asked questions that reopen old emotional wounds or involve reliving traumatic events (Ford & Reutter, 1990; Munhall, 2001a). For example, asking participants to describe their rape experience could precipitate feelings of extreme fear, anger, and sadness. In these types of studies, you should be vigilant about assessing the participants’ discomfort and should refer them for appropriate professional intervention as necessary.
Risk of Permanent Damage
In some studies, subjects have the potential to suffer permanent damage; this potential is more common in biomedical research than in nursing research. For example, medical studies of new drugs and surgical procedures have the potential to cause subjects permanent physical damage. However, nurses have investigated topics that have the potential to damage subjects permanently, both emotionally and socially. Studies examining sensitive information, such as sexual behavior, child abuse, or drug use, can be risky for subjects. These types of studies have the potential to cause permanent damage to a subject’s personality or reputation. There are also potential economic risks, such as reduced job performance or loss of employment.
Certainty of Permanent Damage
In some research, such as the Nazi medical experiments and the Tuskegee Syphilis Study, the subjects experience permanent damage. Conducting research that will permanently damage subjects is highly questionable, regardless of the benefits gained. Frequently, the benefits are for other people but not for the subjects. Studies causing permanent damage to subjects violate the fifth principle of the Nuremberg Code (1986, p. 426), which states, “No experiment should be conducted where there is an a priori reason to believe that death or disabling injury will occur except, perhaps, in those experiments where the experimental physicians (or other health professionals) also serve as subjects.”
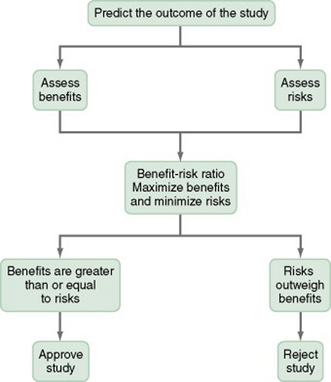
BALANCING BENEFITS AND RISKS FOR A STUDY
Researchers and reviewers of research must examine the balance of benefits and risks in a study. To determine this balance or benefit-risk ratio, you must (1) predict the outcome of your study, (2) assess the actual and potential benefits and risks on the basis of this outcome, and then (3) maximize the benefits and minimize the risks (see Figure 9-1). The outcome of a study is predicted on the basis of previous research, clinical experience, and theory.
Assessment of Benefits
The probability and magnitude of a study’s potential benefits must be assessed.
A research benefit is defined as something of health-related, psychosocial, or other value to a subject, or something that will contribute to the acquisition of knowledge for evidence-based practice. Money and other compensations for participation in research are not benefits but, rather, are remuneration for research-related inconveniences. (U.S. DHHS, 2005)
In most proposals, the research benefits are described for the individual subjects, subjects’ families, and society. Some optimistic researchers overestimate these benefits.
The type of research conducted, whether therapeutic or nontherapeutic, affects the potential benefits for the subjects. In therapeutic nursing research, the individual subject has the potential to benefit from the procedures, such as skin care, range of motion, touch, and other nursing interventions, that are implemented in the study. The benefits might include improvement in the subject’s physical condition, which could facilitate emotional and social benefits. In addition, the knowledge generated from the research might expand the subjects’ and their families’ understanding of health. The conduct of nontherapeutic nursing research does not benefit the subject directly but is important to generate and refine nursing knowledge for practice. By participating in research, subjects have the potential to increase their understanding of the research process and an opportunity to know the findings from a particular study.
Assessment of Risks
You must assess the type, severity, and number of risks that subjects will experience or might experience by participating in your study. The risks involved depend on the purpose of the study and the procedures used to conduct it. Research risks can be physical, emotional, social, and economic in nature and can range from no risk or mere inconvenience to the risk of permanent damage (Levine, 1986; Reynolds, 1979). Studies can have actual (known) risks and potential risks for subjects. In a study of the effects of prolonged bed rest, for example, an actual risk would be muscle weakness and the potential risk would be a DVT. Some studies have actual or potential risks for the subjects’ families and society. You must determine the likelihood of the risks and take precautions to protect the rights of subjects when implementing your study.
Benefit-Risk Ratio
The benefit-risk ratio is determined on the basis of the maximized benefits and the minimized risks. The researcher attempts to maximize the benefits and minimize the risks by making changes in the study purpose or procedures or both. If the risks entailed by your study cannot be eliminated or further minimized, you need to justify their existence. If the risks outweigh the benefits, you should revise the study or develop a new study. If the benefits equal or outweigh the risks, you can justify conducting the study, and an institutional review board (IRB) will probably approve it (see Figure 9-1).
Say, for example, that you want to balance the benefits and risks of a study that would examine the effect of an exercise and diet program on the participants’ serum lipid values (serum cholesterol, low-density lipoprotein [LDL], and high-density lipoprotein [HDL]) and cardiovascular (CV) risk level. The benefits to the participants are instruction about exercise and diet and information about their serum lipid values and CV risk level at the start of the program and 1 year later. The potential benefits are improved serum lipid values, lowered CV risk level, and better exercise and dietary habits. The risks consist of the discomfort of having blood specimens drawn twice for serum lipid measurements and the time spent participating in the study (Bruce & Grove, 1994). These discomforts are temporary, are no more than what the subject would experience in his or her daily life, and would cease with the termination of the study. The subjects’ time participating in the study can be minimized through organization and precise scheduling of research activities. When you examine the ratio of benefits to risks, you find that (1) the benefits are greater in number and importance than the risks and (2) the risks are temporary and can be minimized. Thus, you could justify conducting this study, and it would probably receive approval from the IRB.
The obligation to balance the benefits and risks of studies is the responsibility of the researcher, health professionals, and society. The researcher must balance the benefits and risks of a particular study and protect the subjects from harm during it. Health professionals participate on IRBs to ensure the conduct of ethical research. Society must be concerned with the benefits and risks of the entire enterprise of research and with the protection of all human research subjects from harm.
OBTAINING INFORMED CONSENT
Obtaining informed consent from human subjects is essential for the conduct of ethical research in the United States (FDA, 2006b; U.S. DHHS, 2005) and internationally (CIOMS, 2002; Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, Social Sciences and Humanities Research Council of Canada, 2005). Informing is the transmission of essential ideas and content from the investigator to the prospective subject. Consent is the prospective subject’s agreement to participate in a study as a subject, which the subject reaches after assimilating essential information. Prospective subjects, to the degree that they are capable, should have the opportunity to choose whether or not to participate in research. The phenomenon of informed consent was formally defined in the first principle of the Nuremberg Code as follows:
The voluntary consent of the human subject is absolutely essential…. This means that the person involved should have legal capacity to give consent; should be so situated as to be able to exercise free power of choice, without the intervention of any element of force, fraud, deceit, duress, over-reaching or other ulterior form of constraint or coercion; and should have sufficient knowledge and comprehension of the elements of the subject matter involved as to enable him to make an understanding and enlightened decision. (Nuremberg Code,1986, p. 425)
This definition of informed consent provided a basis for the discussion of consent in all subsequent codes and regulations and has general acceptance in the research community. As the definition indicates, informed consent consists of four elements: (1) disclosure of essential information, (2) comprehension, (3) competency, and (4) voluntarism. This section describes the elements of informed consent and the methods of documenting consent.
Information Essential for Consent
Informed consent requires the researcher to disclose specific information to each prospective subject. The following information has been identified as essential content for informed consent in research by CFR 45 Section 46.116 (U.S. DHHS, 2005) and CFR 21 Part 50 (FDA, 2006b).
Introduction of Research Activities
The introduction of the research must indicate that a study is being conducted and provide key information about the study. Each prospective subject is given “a statement that the study involves research, an explanation of the purposes of the research, and the expected duration of the subject’s participation” (U.S. DHHS, 2005, CFR 45 Section 46.116). In clinical nursing research, the patient, serving as a subject, must know which nursing activities are research activities and which are routine nursing interventions. If at any point the prospective subjects disagree with your goals or the intent of the study, they can decline participation.
Prospective subjects also need to receive a complete description of the procedures to be followed and identification of any procedures in the study that are experimental (FDA, 2006b; U.S. DHHS, 2005). Thus, you will describe the research variables and the procedures or mechanisms that will be used to observe, examine, manipulate, or measure these variables. In addition, you must inform prospective subjects about when the study procedures will be implemented, how many times, and in what setting.
Description of Risks and Discomforts
Inform prospective subjects about any reasonably foreseeable risks or discomforts (physical, emotional, social, or economic) that might result from the study (FDA, 2006b; U.S. DHHS, 2005). Indicate how the risks of the study have been or will be minimized. If the study involves greater than minimal risk, it is usually a good idea to encourage the prospective subjects to consult another person regarding their participation. A trusted advisor, such as a friend, family member, or another nurse, could serve as a consultant.
Description of Benefits
You should also describe any benefits to the subject or to others that may reasonably be expected from the research (FDA, 2006b; U.S. DHHS, 2005). The study might benefit the current subjects or might generate knowledge that will provide evidence-based care to patients in the future.
Disclosure of Alternatives
You must disclose the “appropriate, alternative procedures or courses of treatment, if any, that might be advantageous to the subject” (U.S. DHHS, 2002, 45 CFR Section 46.116a). For example, the researchers of the Tuskegee Syphilis Study should have informed the subjects with syphilis that penicillin was an effective treatment for their disease.
Assurance of Anonymity and Confidentiality
Prospective subjects must be given a statement describing the extent to which confidentiality of their records will be maintained (FDA, 2006b; U.S. DHHS, 2002, 2005). Thus, your subjects must know that their responses and the information obtained from their records during a study will be kept confidential. You also must assure them that their identity will remain anonymous in presentations, reports, and publications of the study.
Compensation for Participation in Research
For research involving more than minimal risks, the prospective subjects must be given an explanation as to whether any compensation or medical treatments or both are available if injury occurs. If medical treatments are available, describe the type and extent of the treatments. When appropriate, inform the prospective subject as to whether a treatment or procedure may involve potential risks to the subject or her fetus if she is or may become pregnant (FDA, 2006b; U.S. DHHS, 2005).
Offer to Answer Questions
The researcher offers to answer any questions that the prospective subjects may raise. Prospective subjects are provided an “explanation of whom to contact for answers to pertinent questions about the research and research subjects’ rights, and whom to contact in the event of a research-related injury” (FDA, 2006b, 21 CFR Section 50.25; U.S. DHHS, 2005, 45 CFR Section 46.116a) as well as a mechanism for contacting that person.
Noncoercive Disclaimer
A noncoercive disclaimer is a statement that participation is voluntary and refusal to participate will involve no penalty or loss of benefits to which the subject is entitled (FDA, 2006b; U.S. DHHS, 2005). This statement can facilitate a relationship between you and your prospective subjects, especially if the relationship has a potential for coercion.
Option to Withdraw
Subjects may discontinue participation or withdraw from a study at any time without penalty or loss of benefits. However, researchers do have the right to ask subjects if they think that they will be able to complete the study, to decrease the number of subjects withdrawing early. There may be circumstances “under which the subject’s participation may be terminated by the investigator without regard to the subject’s consent” (U.S. DHHS, 2005, 45 CFR Section 46.116b). For example, if a particular treatment becomes potentially dangerous to a subject, you have an obligation to discontinue the subject’s participation in the study. Thus, describe for prospective subjects the circumstances under which they might be withdrawn from the study, and make a general statement about the circumstances that could lead to the termination of the entire project.
Consent to Incomplete Disclosure
In some studies, subjects are not completely informed of the study purpose, because that knowledge would alter the subjects’ actions. However, prospective subjects must know that certain information is being withheld deliberately. You must ensure that there are no undisclosed risks to the subjects that are more than minimal and truthfully answer the subjects’ questions regarding the study. Subjects who are exposed to nondisclosure of information must know when and how they will be debriefed about the study. Debrief the subjects by informing them of the actual purpose of the study and the results that were obtained. If the subjects experience adverse effects related to the study, make every attempt to reconcile the effects (U.S. DHHS, 2005).
Comprehension of Consent Information
Informed consent implies not only the imparting of information by the researcher but also the comprehension of that information by the subject. Studies performed to determine subjects’ levels of comprehension after they received the essential information for consent have found the comprehension to be low (Levine, 1986). The amount of information to be taught depends on the subjects’ knowledge of research and the specific research project proposed. However, the purpose, benefits, and risks of the study must be presented clearly in the consent form. Subjects need to have a clear understanding of the therapeutic potential of participating in a study. They need to know if the research treatment is going to be nontherapeutic for them but has a potential to benefit future patients.
Federal regulations require that the information given to subjects or their representatives must be in a language they can understand (FDA, 2006b; U.S. DHHS, 2005). Thus, the consent information must be written and verbalized in lay terminology, not professional jargon, and must be presented without the use of loaded or biased terms that might coerce a subject into participating in a study. Meade (1999) identified the following tips for promoting the comprehension of a consent document by potential research subjects:
• Introduce the purpose of the study early in the consent form.
• Outline the study treatment with specificity and conciseness.
• Convey the elements of informed consent in an organized fashion.
• Define technical terms, and be consistent in the use of terminology.
• Use clear terminology, and avoid professional jargon.
• Develop the document using headings, uppercase and lowercase letters, and spacing to make it easy to read.
• Use headings for major elements in the consent form, such as “Purpose,” “Benefits,” and “Risks.”
• Use a readable font, that is, a minimum of 12- to 14-point font for text and a 16- to 18-point font for the headers.
• Address the subject directly, using phrases such as, “You are being asked to take part in this study … ”
• Estimate the reading level of the document with the use of a computerized readability formula, and revise it to achieve no higher than an eighth grade reading level.
Meade (1999) used these tips to simplify a paragraph from an example consent form, as shown in Table 9-5. Once you have developed the consent document, pilot-test it with patients who are comparable to the proposed subjects for the study. These patients can give feedback on the ease of reading, clarity, and understandability of the consent document. You can then revise the document as needed on the basis of the feedback. These guidelines will help you to develop a high-quality consent document that your study subjects can comprehend.
TABLE 9-5
Simplification of Consent Document
Origin Consent Document
Example A*:
Side effects of the marrow infusion are uncommon and consist primarily of an unusual taste from the preservative, occasional nausea and vomiting, and, rarely, fever and chills. In addition, your chest may feel tight for a while, but that will pass.
Example B†:
The standard approach to treating breast cancer is to give several “cycles” (repeated doses at regularly specified intervals) of a combination of two or more chemotherapy drugs (drugs that kill cancer cells). Recent information suggests that it may be more beneficial to give several cycles of one drug followed by several cycles of another drug. Some researchers think that the second approach may kill more cells that are resistant to chemotherapy. The approach being tested in this study is to administer four cycles of standard chemotherapy (doxorubicin/cyclophosphamide) followed by four cycles of the drug paclitaxel. Researchers hope to show that cancer cells resistant to the doxorubicin/cyclophosphamide chemotherapy may be sensitive to paclitaxel. This may then result in prolonged patient survival and result in a decrease in the number of patients experiencing a recurrence.
Revised Simplified Consent Document
Example A*:
Side effects of getting stem cells:
Example B†:
Why is this study being done?
The purpose of this research study is to find out whether adding the drug Taxol (paclitaxel) to a commonly used chemotherapy is better than the commonly used chemotherapy by itself at preventing your cancer from coming back. The study also will see what side effects there are from adding Taxol to the commonly used chemotherapy. Taxol has been found to be effective in treating patients with advanced breast cancer. In this study, we want to see whether Taxol will help to treat patients with early stage breast cancer and whether the side effects seem to be worth the possible benefit.
NCI Model Document Sub-Group: Comprehensive Working Group on Informed Consent, 1998.
*Standard doses versus myeloablative therapy for previously untreated symptomatic multiple myeloma. Phase III. SWOG 9321.
†A randomized trial evaluating the worth of paclitaxel (Taxol) following doxorubicin (Adriamycin)/cyclophosphamide (Cytoxan) in breast cancer.
From “Consent (Assent) for Research with Pediatric Patients,” by M.E. Broome, 1999, Seminars in Oncology Nursing, 15(2), p. 130.
In qualitative research, the participants might comprehend their participation in a study at the beginning, but unexpected events or consequences might occur during the study to obscure that understanding. These events might precipitate a change in the focus of the research and the type of participation by the participants. For example, the topics of an interview might change with an increased need for information from the participants to address these topics. Thus, informed consent is an ongoing, evolving process in qualitative research. The researcher must renegotiate the participants’ consent and determine their comprehension of that consent as changes occur in the study. By continually informing and determining the comprehension, of participants, you will establish trust with them and promote ethical, high-quality study outcomes (Munhall, 2001a).
Assessment of Subjects’ Comprehension
The researcher can take steps to determine the prospective subjects’ level of comprehension. Silva (1985) studied the comprehension of information for informed consent by spouses of surgical patients and found that 72 of the 75 spouses adequately comprehended the consent information. Silva assessed the subjects’ comprehension of consent information by asking the following questions:
(1) What is the purpose of this study? (2) What risks are involved in the study procedures? (3) What does your participation in this study involve? (4) Approximately how long will your participation in this study take? (5) When can you withdraw from this study? (6) How will your name be associated with the study data? (7) With whom will the study information be shared? and (8) What direct personal benefit will come to you as a result of participating in this study? (Silva, 1985, p. 121)
In complex, high-risk studies, it is more difficult for subjects to comprehend consent information. In some high-risk studies, the prospective subjects are tested on consent information, and they do not become subjects unless they pass the tests.
Competency to Give Consent
Autonomous individuals, who are capable of understanding and weighing the benefits and risks of a proposed study, are competent to give consent. The competence of the subject is often determined by the researcher (Douglas & Larson, 1986). Persons with diminished autonomy resulting from legal or mental incompetence, terminal illness, or confinement to an institution might not be legally competent to consent to participate in research (see the earlier discussion of the right to self-determination). However, the researcher makes every effort to present the consent information at a level potential subjects can understand, so that they can assent to the research. In addition, the researcher presents the essential information for consent to the legally authorized representative, such as the parents or guardian, of the prospective subject (U.S. DHHS, 2005).
Voluntary Consent
Voluntary consent means that the prospective subject has decided to take part in a study of his or her own volition without coercion or any undue influence. Voluntary consent is obtained after the prospective subject has been given essential information about the study and has shown comprehension of this information (FDA, 2006b; U.S. DHHS, 2005). Some researchers, because of their authority, expertise, or power, have the potential to coerce subjects into participating in research. Researchers must make sure that their persuasion of prospective subjects does not become coercion. Thus, the rewards offered in a study ought to be congruent with the risks taken by the subjects.
Documentation of Informed Consent
The documentation of informed consent depends on (1) the level of risk involved in the study and (2) the discretion of the researcher and those reviewing the study for institutional approval. Most studies require a written consent form, although in some studies, the consent form is waived.
Written Consent Waived
The requirements for written consent may be waived in research that “presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside of the research context” (U.S. DHHS, 2005, 45 CFR Section 46.117c). For example, if you were using questionnaires to collect relatively harmless data, you would not need to obtain a signed consent form from the subjects. The subject’s completion of the questionnaire may serve as consent. The top of the questionnaire might contain a statement such as “Your completion of this questionnaire indicates your consent to participate in this study.”
Written consent is also waived when “the only record linking the subject and the research would be the consent document and the principal risk would be potential harm resulting from a breach of confidentiality. Each subject will be asked whether the subject wants documentation linking the subject with the research, and the subject’s wishes will govern” (U.S. DHHS, 2005, 45 CFR Section 46.117c). Thus, in this situation, subjects are given the option to sign or not sign a consent form that links them to the research. The four elements of consent—disclosure, comprehension, competency, and voluntariness—are essential in all studies whether written consent is waived or required.
Written Consent Documents
Short Form Written Consent Document: The short form consent document includes the following statement: “The elements of informed consent required by Section 46.116 [see the section on information essential for consent] have been presented orally to the subject or the subject’s legally authorized representative” (U.S. DHHS, 2005, 45 CFR Section 46.117a). The researcher must develop a written summary of what is to be said to the subject in the oral presentation, and the summary must be approved by an IRB. When the oral presentation is made to the subject or to the subject’s representative, a witness is required. The subject or representative must sign the short form consent document. “The witness shall sign both the short form and a copy of the summary, and the person actually obtaining consent shall sign a copy of the summary” (U.S. DHHS, 2005, 45 CFR Section 46.117a). Copies of the summary and short form are given to the subject and the witness; the researcher retains the original documents and must keep these documents for 3 years after the end of the study. The short form written consent documents might be used in studies that present minimal or moderate risk to the subjects.
Formal Written Consent Document: The written consent document or consent form includes the elements of informed consent required by the U.S. DHHS (2005) and FDA (2006b) regulations (see the previous section on information essential for consent). In addition, a consent form might include other information required by the institution where the study is to be conducted or by the agency funding the study. Most universities provide consent form guidelines for researchers to use. A sample consent form is presented in Figure 9-2 with the common essential consent information. The subject can read the consent form, or the researcher can read it to the subject; however, it is wise also to explain the study to the subject. The subject signs the form, and the investigator or research assistant collecting the data witnesses it. This type of consent can be used for minimal to moderate risk studies. All persons signing the consent form must receive a copy of it. The researcher keeps the original consent form for 3 years.

Studies that involve subjects with diminished autonomy require a written consent form. If these prospective subjects have some comprehension of the study and agree to participate as subjects, they must sign the consent form. However, the subject’s legally authorized representative also must sign the form. The representative indicates his or her relationship with the subject under the signature (see Figure 9-2).
The written consent form used in a high-risk study often contains the signatures of two witnesses, the researcher and an additional person. The additional person signing as a witness must observe the informed consent process and must not be otherwise connected with the study (Hershey & Miller, 1976). The best witnesses are research subject advocates or patient advocates who are employed in the institution.
Sometimes nurses are asked to sign a consent form as a witness for a biomedical study. They must know the study purpose and procedures and the subject’s comprehension of the study before signing the form (Carico & Harrison, 1990). The role of the witness is more important in the consent process if the prospective subject is in awe of the investigator and does not feel free to question the procedures of the study.
Recording of the Consent Process
A researcher might elect to tape-record or obtain a DVD of the consent process. These methods document what was said to the prospective subject and record the subject’s questions and the investigator’s answers. Tape-recording and DVD are time- consuming and costly, however, and are not appropriate for studies of minimal or moderate risk. If your study is considered high risk, it might be wise to completely document the consent process on tape, because doing so might protect you and your subjects. Both of you would retain a copy of the tape recording or DVD.
Authorization for Research Uses and Disclosure
The HIPAA Privacy Rule provides individuals the right, as research subjects, to authorize covered entities (health care provider, health plan, and health care clearinghouse) to use or disclose their private health information (PHI) for research purposes. This authorization is regulated by the HIPAA and is in addition to the informed consent that is regulated by the U.S. DHHS (2005, CFR 45 Part 46) and the FDA (2006b, CFR 21 Part 50). The authorization focuses on the privacy risks and states how, why, and to whom the PHI will be shared. The authorization form must include the following information:
• Description of PHI to be used or disclosed (identifying the information in a specific and meaningful manner).
• The name(s) or other specific identification of person(s) or class of persons authorized to make the requested use or disclosure.
• The name(s) or other specific identification of the person(s) or class of persons who may use the PHI or to whom the covered entity may make the requested disclosure.
• Description of each purpose of the requested use or disclosure. Researchers should note that this element must be study specific, not for future unspecified research.
• Authorization expiration date or event that relates to the individual or to the purpose of the use or disclosure (the terms “end of the research study” or “none” may be used for research, including for the creation and maintenance of a research database or repository).
• Signature of the individual and date. If the Authorization is signed by an individual’s personal representative, a description of the representative’s authority to act for the individual. (U.S. DHHS, 2004)
The authorization information can be included as part of the consent form, but it is probably best to have two separate forms (Olsen, 2003). U.S. DHHS (2004) developed a sample authorization form that is presented in Figure 9-3.

INSTITUTIONAL REVIEW
In institutional review, a committee of the researcher’s peers examines his or her study for ethical concerns. The first federal policy statement on protection of human subjects by institutional review was issued by the U.S. PHS in 1966. The statement required that research involving human subjects must be reviewed by a committee of peers or associates to confirm that (1) the rights and welfare of the individuals involved were protected, (2) the appropriate methods were used to secure informed consent, and (3) the potential benefits of the investigation were greater than the risks (Levine, 1986). Internationally, CIOMS (2002) and the Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and Social Sciences and Humanities Research Council of Canada (Canadian Tri-Council) have developed regulations to guide the ethical review of research by peers.
In 1974, DHEW passed the National Research Act, which required that all research involving human subjects undergo institutional review. Currently, both the U.S. DHHS (2005, 45 CFR Sections 46.107–46.115) and the FDA (2006a, 21 CFR Sections 56.101–56.124) have similar regulations for institutional review of research. These regulations describe the membership, functions, and operations of an institutional review board. An institutional review board (IRB) is a committee that reviews research to ensure that the investigator is conducting the research ethically. Universities, hospital corporations, and many managed care centers have IRBs to promote the conduct of ethical research and protect the rights of prospective subjects at their institutions.
Each IRB has at least five members of various backgrounds (cultural, economic, educational, gender, racial) to promote a complete, scholarly, and fair review of research that is commonly conducted in an institution. If an institution regularly reviews studies with vulnerable subjects, such as children, neonates, pregnant women, prisoners, and mentally disabled persons, the IRB should include one or more members with knowledge about and experience in working with these subjects. The members must have sufficient experience and expertise to review a variety of studies, including quantitative, qualitative, outcomes, and intervention research (Munhall, 2001b). The IRB members must not have a conflicting interest related to a study conducted in an institution. Any member having a conflict of interest with a research project being reviewed must excuse himself or herself from the review process, except to provide information requested by the IRB. The IRB also must include other members whose primary concern is nonscientific, such as an ethicist, a lawyer, or a minister. At least one of the IRB members must be someone who is not affiliated with the institution (FDA, 2006b; U.S. DHHS, 2005). The IRBs in hospitals are often composed of physicians, lawyers, scientists, clergy, community laypersons, and, more recently, nurses.
Levels of Reviews Conducted by Institutional Review Boards
The functions and operations of an IRB involve the review of research at three different levels: (1) exempt from review, (2) expedited review, and (3) complete review. The level of the review required for each study is decided by the IRB chairperson or committee, but not by the researcher. Studies are usually exempt from review if they pose no apparent risks for the research subjects. The studies that are usually considered exempt from IRB review by the federal regulations are identified in Table 9-6. For example, studies by nurses and other health professionals that have no foreseeable risks or are a mere inconvenience for subjects might be identified as exempt from review by the chairperson of the IRB committee.
TABLE 9-6
Research Qualifying for Exemption from Review
Unless otherwise required by department or agency heads, research activities in which the only involvement of human subjects will be in one or more of the following categories are exempt from review:
(1) Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as (i) research on regular and special education instructional strategies, or (ii) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods.
(2) Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior, unless: (i) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects’ responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, or reputation.
(3) Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior that is not exempt under paragraph (b)(2) of this section, if: (i) the human subjects are elected or appointed public officials or candidates for public office; or (ii) Federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter.
(4) Research involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects.
(5) Research and demonstration projects which are conducted by or subject to the approval of Department or Agency heads, and which are designed to study, evaluate, or otherwise examine: (i) Public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives to those programs or procedures; or (iv) possible changes in methods or levels of payment for benefits or services under those programs.
(6) Taste and food quality evaluation and consumer acceptance studies, (i) if wholesome foods without additives are consumed or (ii) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the U.S. Department of Agriculture.
From U.S. Department of Health and Human Services. (2005, June 23). Protection of human subjects. Code of Federal Regulations, Title 45, Part 46. Retrieved October 1, 2007, from www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.htm.
Studies that have some risks, which are viewed as minimal, are expedited in the review process. Minimal risk means “that the risks of harm anticipated in the proposed research are not greater, considering probability and magnitude, than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests” (U.S. DHHS, 2005, 45 CFR Section 46.102). Expedited review procedures can also be used to review minor changes in previously approved research. Under expedited IRB review procedures, the review may be carried out by the IRB chairperson or by one or more experienced reviewers designated by the chairperson from among members of the IRB. In reviewing the research, the reviewers may exercise all of the authorities of the IRB except disapproval of the research. A research activity may be disapproved only after a complete review of the IRB (FDA, 2006b; U.S. DHHS, 2005). Table 9-7 identifies research that usually qualifies for expedited review.
TABLE 9-7
Research Qualifying for Expedited Institutional Review Board Review
Expedited review (by committee chairpersons or designated members) for the following research involving no more than minimal risk is authorized:
1. Collection of hair and nail clippings, in a nondisfiguring manner; deciduous teeth and permanent teeth if patient care indicates a need for extraction.
2. Collection of excreta and external secretions including sweat, uncannulated saliva, placenta removed at delivery, and amniotic fluid at the time of rupture of the membrane before or during labor.
3. Recording of data from subjects 18 years of age or older using noninvasive procedures routinely employed in clinical practice. This includes the use of physical sensors that are applied either to the surface of the body or at a distance and do not involve input of matter or significant amounts of energy into the subject or an invasion of the subject’s privacy. It also includes such procedures as weighing, testing sensory acuity, electrocardiography, electroencephalography, thermography, detection of naturally occurring radioactivity, diagnostic echography, and electroretinography. It does not include exposure to electromagnetic radiation outside the visible range (for example, x-rays, microwaves).
4. Collection of blood samples by venipuncture, in amounts not exceeding 450 ml in an 8-week period and no more than two times per week, from subjects 18 years of age or older and who are in good health and not pregnant.
5. Collection of both supragingival and subgingival dental plaque and calculus, provided the procedure is not more invasive than routine prophylactic scaling of the teeth and the process is accomplished in accordance with accepted prophylactic techniques.
6. Voice recordings made for research purposes such as investigations of speech defects.
7. Moderate exercise by healthy volunteers.
8. The study of existing data, documents, records, pathological specimens, or diagnostic specimens.
9. Research on individual or group behavior or characteristics of individuals, such as studies of perception, cognition, game theory, or test development, where the investigator does not manipulate subjects’ behavior and research will not involve stress to subjects.
Research on drugs or devices for which an investigational new drug exemption or an investigational device exemption is not required.
From U.S. Department of Health and Human Services. (2005, June 23). Protection of human subjects. Code of Federal Regulations, Title 45, Part 46. Retrieved October 1, 2007, from www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.htm.
A study that has greater than minimal risks must receive a complete IRB review. To obtain IRB approval, researchers must ensure that
(1) risks to subjects are minimized, (2) risks to subjects are reasonable in relation to anticipated benefits, (3) selection of subjects is equitable, (4) informed consent will be sought from each prospective subject or the subject’s legally authorized representative, (5) informed consent will be appropriately documented, (6) the research plan makes adequate provision for monitoring data collection for subjects’ safety, and (7) adequate provisions are made to protect the privacy of subjects and to maintain the confidentiality of data. (FDA, 2006b, 21 CFR 56.111; U.S. DHHS, 2005, 45 CFR Section 46.111)
Amdur (2003) provided a detailed discussion of the role of the IRB in health care research, and this information is a good resource for health care agencies and researchers. The process of seeking approval from a research review committee to conduct a study is described in Chapter 28.
Published studies often indicate that an IRB has approved the research project. For example, Bindler, Massey, Shultz, Mills, and Short (2007) conducted their study to examine metabolic syndrome in a multiethnic sample of schoolchildren. They described the following consent and IRB approval process for their study.
The subjects in this study were parents and school-age children from different ethnic backgrounds (English and Spanish) who were invited to participate in the study by a flyer sent to all affiliated with the selected school. The parents and children received information about the study in their appropriate language. Then the parents signed consent forms and the children signed assent forms documenting the informed consent process. The two versions of the consent forms (Spanish and English) probably increased the subjects’ ability to comprehend the consent form and the purpose of the study. IRB approvals were obtained from both the university and the school district where the study was conducted. Thus, the consent process and IRB approvals were clearly documented in this study
Influence of HIPAA Privacy Rule on Institutional Review Boards
Under the 2003 HIPAA Privacy Rule, IRBs or an institutionally established privacy board can act on requests for a waiver or an alteration of the authorization requirement for a research project. If an IRB and a privacy board both exist in an agency, the approval of only one board is required, and it will probably be the IRB for research projects. Researchers can choose to obtain a signed authorization form from potential subjects or can ask for a waiver or an alteration of the authorization requirement. An altered authorization requirement occurs when an IRB approves a request that some but not all of the required 18 elements be removed from health information that is to be used in research. The researcher can also request a partial or complete waiver of the authorization requirement from the IRB. For a partial waiver, discussed earlier, the researcher obtains PHI to contact and recruit potential subjects for a study. An IRB can give a researcher a complete waiver of authorization in studies where the informed consent requirements might also be waived. Thus, a waiver or alteration of the authorization requirement might occur when the following criteria have been met:
• The PHI use or disclosure involves no more than minimal risk to the privacy for research subjects based on (1) an adequate plan presented to the IRB to protect the PHI identifiers from improper use of disclosure; (2) an adequate plan exists to destroy the identifiers at the earliest opportunity; and (3) written assurance the PHI will not be reused or disclosed to any other person.
• The research could not reasonably be conducted without the waiver or alteration of the Authorization requirement.
• The research cannot be done without access to and use of the PHI. (U.S. DHHS, 2003)
The covered entity (health care provider, health plan, or health care clearinghouse) cannot release the PHI to the researcher until the following documentation has been received: (1) the identity of the approving IRB, (2) the date the waiver or alteration was approved, (3) IRB documentation that the criteria for waiver or alteration have been met, (4) a brief description of the PHI to which the researcher has been granted access or use, (5) a statement as to whether the waiver was approved under normal or expedited review procedures, and (6) the signature of the IRB chair or the chair’s designee.
The HIPAA Privacy Rule does not change the IRB membership and functions that are designated under the U.S. DHHS and FDA Regulations. For clarification, the responsibilities of the IRB/privacy board for HIPAA (U.S. DHHS, 2007, February 2, a) and the responsibilities of the IRB under the U.S. DHHS (2005) and FDA (2006a) are outlined in Table 9-8.
TABLE 9-8
Comparison of IRB/Privacy Board Responsibilities for HIPAA, U.S. DHHS, and FDA

From U.S. Department of Health and Human Services. (2007, February 2, a). How can covered entities use and disclose protected health information for research and comply with the Privacy Rule? HIPAA Privacy Rule: Information for researchers. Retrieved October 1, 2007, from http://privacyruleandresearch.nih.gov/pr_08.asp.
RESEARCH MISCONDUCT
The goal of research is to generate sound scientific knowledge, which is possible only through the honest conduct, reporting, and publication of studies. However, since the 1980s, a number of fraudulent studies have been conducted and published in prestigious scientific journals. An example of scientific misconduct was evident in the publications of Dr. Robert Slutsky, a heart specialist at the University of California, San Diego, School of Medicine. He resigned in 1986 when confronted with inconsistencies in his research publications. His publications contained “statistical anomalies that raised the question of data fabrication” (Friedman, 1990, p. 1416). In 6 years, Slutsky published 161 articles, and at one time, he was completing an article every 10 days. Eighteen of the articles were found to be fraudulent and have retraction notations, and 60 articles were judged to be questionable (Friedman, 1990; Henderson, 1990).
Stephen Breuning, a psychologist at the University of Pittsburgh, engaged in deceptive and misleading practices in reporting his research on retarded children. He used his fraudulent research to obtain more than $300,000 in federal grants. In 1988, he was criminally charged with research fraud, pleaded guilty, was fined $20,000, and was sentenced to up to 10 years in prison (Garfield & Welljams-Dorof, 1990).
In response to the increasing incidences of scientific misconduct, the federal government developed the Office of Research Integrity (ORI) in 1989 within the U.S. DHHS. ORI was to supervise the implementation of the rules and regulations related to scientific misconduct and to manage any investigations of scientific misconduct. In 1996, a review and revision of the existing scientific misconduct policy were initiated to (1) develop a uniform research misconduct policy across the agencies of the federal government, (2) establish a policy that addresses behavior that has the potential to affect the integrity of the research record, and (3) develop a procedure to safeguard the handling of allegations of research misconduct. Currently, ORI is responsible for the implementation of CFR 42, Parts 50 and 93, Policies of General Applicability (ORI, 2005).
Role of the Office of Research Integrity in Promoting the Conduct of Ethical Research
Currently, ORI promotes the integrity of biomedical and behavioral research in approximately 4000 institutions worldwide (ORI, 2007, September 10). ORI applies federal policies and regulations to protect the integrity of the U.S. PHS’s extramural and intramural research programs. The extramural program provides funding to research institutions, and the intramural program provides funding for research conducted within the federal government. The ORI carries out its responsibilities by doing the following:
• Developing policies, procedures, and regulations related to the detection, investigation, and prevention of research misconduct and the responsible conduct of research
• Reviewing and monitoring research misconduct investigations conducted by applicant and awardee institutions, intramural research programs, and the Office of Inspector General in the Department of Health and Human Services (HHS)
• Recommending research misconduct findings and administrative actions to the assistant secretary for health for decision, subject to appeal
• Assisting the Office of the General Counsel (OGC) to present cases before the HHS departmental appeals board
• Providing technical assistance to institutions that respond to allegations of research misconduct
• Implementing activities and programs to teach responsible conduct of research, promote research integrity, prevent research misconduct, and improve the handling of allegations of research misconduct
• Conducting policy analyses, evaluations, and research to build the knowledge base in research misconduct, research integrity, and prevention and to improve the HHS research integrity policies and procedures
• Administering programs for maintaining institutional assurances, responding to allegations of retaliation against whistleblowers, approving intramural and extramural policies and procedures, and responding to Freedom of Information Act and Privacy Act requests (ORI, 2005)
Since the mid-1990s, ORI has had a major role in the investigation of allegations of misconduct in research within several institutions. “Research misconduct means fabrication, falsification, or plagiarism in processing, performing, or reviewing research, or in reporting research results. It does not include honest error or differences in opinion” (ORI, 2005, 42 CFR Section 93.103). Fabrication in research is the making up of results and recording or reporting them. Falsification of research is manipulating research materials, equipment, or processes, or changing or omitting data or results such that the research is not accurately represented in the research record. Plagiarism is the appropriation of another person’s ideas, processes, results, or words without giving appropriate credit, including those obtained through confidential review of others’ research proposals and manuscripts.
The ORI classifies research misconduct as (1) an act that involves a significant departure from the acceptable practice of the scientific community for maintaining the integrity of the research record; (2) an act that was committed intentionally; and (3) an allegation that can be proved by a preponderance of evidence. ORI has a section on their website titled “Handling Misconduct” that includes a summary of the allegations and investigations managed by its office from 1992 to 2001 (ORI, 2004). The most common sites for the investigations were medical schools (68%), hospitals (11%), and research institutes (10%). The individuals charged with misconduct were primarily males holding a PhD or medical degree (MD) and were mostly associate professors, professors, and postdoctoral fellows. When research misconduct was documented, the actions taken against the researchers or agencies might have included debarment from receiving federal funding for periods ranging from 18 months to 8 years; prohibition from U.S. PHS advisory service; and other actions requiring supervised research, certification of data, certification of sources, and correction or retraction of articles. The regulations governing ORI and the current investigations and activities of ORI can be viewed online at http://ori.dhhs.gov.
Role of Journal Editors and Researchers in Preventing Scientific Misconduct
Editors of journals also have a major role in monitoring and preventing research misconduct in the published literature. Friedman (1990, p. 1416) identified criteria for classifying a publication as fraudulent, questionable, or valid and indicated research articles were “fraudulent if there was documentation or testimony from coauthors that the publication did not reflect what had actually been done.” Articles were considered questionable if no coauthor could produce the original data or if no coauthor had personally observed or performed each phase of the research or participated in the research publication. A research article was considered valid “if some coauthor had personally performed or participated in each aspect of the research and publication” (Friedman, 1990, p. 1416).
Preventing the publication of fraudulent research requires the efforts of authors, coauthors, reviewers, and editors (Hansen & Hansen, 1995; Hawley & Jeffers, 1992; Relman, 1990). Authors who are primary investigators for research projects must be responsible in their conduct, reporting, and publication of research. Coauthors and coworkers should question and, if necessary, challenge the integrity of a researcher’s claims. Sometimes, well-known scientists have been added to a research publication as coauthors to give it credibility. Individuals should not be listed as coauthors unless they were actively involved in the conduct and publication of the research.
Peer reviewers have a key role in determining the quality and publishability of a manuscript. They are considered experts in the field, and their role is to examine research for inconsistencies and inaccuracies. Editors must monitor the peer review process and be cautious about publishing manuscripts that are at all questionable. Editors also need procedures for responding to allegations of research misconduct. They must decide what actions to take if the journal contains an article that has proven to be fraudulent. Usually, fraudulent publications require retraction notations and are not to be cited by authors in future publications (ORI, 2005). However, Pfeifer and Snodgrass (1990) studied the continued citation of retracted, invalid scientific literature and found that articles commonly continued to cite retracted articles in U.S. publications and even more so in the literature of other countries.
The publication of fraudulent research is a major concern in medicine and a growing concern in nursing (Rankin & Esteves, 1997). The smaller pool of funds available for research and the greater emphasis on research publications could lead to a higher incidence of fraudulent publications. However, ORI (2007, June 20) has made major advances in addressing scientific misconduct since the regulations were enacted by doing the following:
• Providing a definition of research misconduct
• Designating responsibilities and actions related to research misconduct
• Identifying mechanisms to distribute the policy to scientists
• Designating the membership of investigating committees
• Identifying the administrative actions for acts of research misconduct
• Developing a process for notifying funding agencies and journals of acts of research misconduct
• Providing for public disclosure of the incidents of scientific misconduct
Each researcher is responsible for monitoring the integrity of his or her research protocols, results, and publications. In addition, nursing professionals must foster a spirit of intellectual inquiry, mentor prospective scientists regarding the norms for good science, and stress quality, not quantity, in publications (Wocial, 1995).
ANIMALS AS RESEARCH SUBJECTS
The use of animals as research subjects is a controversial issue of growing concern to nurse researchers. A small but increasing number of nurse scientists are conducting physiological studies that require the use of animals. Many scientists, especially physicians, believe that the current animal rights’ movement could threaten the future of health research. Animal rights groups are active in antiresearch campaigns and are backed by massive resources estimated in the millions of dollars (Pardes, West, & Pincus, 1991). Some of the animal rights groups are trying to raise the consciousness of researchers and society to ensure that animals are used wisely in the conduct of research and treated humanely.
Other animal rights groups have tried to frighten the public with sometimes distorted stories about inhumane treatment of animals in research. Some of the activist leaders have made broad comparisons between human life and animal life. For example, a major animal rights group called People for the Ethical Treatment of Animals (PETA) has stated, “There is no rational basis for separating out the human animal. A rat is a pig is a dog is a boy. They’re all mammals” (Pardes et al., 1991, p. 1641). Some of these activists have now progressed to violence, using “physical attacks, including real bombs, arson, and vandalism” (Pardes et al., 1991, p. 1642). Even more damage is being done to research through lawsuits that have blocked the conduct of research and the development of new research centers. Medical schools now spend millions of dollars annually for security, public education, and other efforts to defend research.
Two important questions must be addressed: Should animals be used as subjects in research, and if animals are used in research, what mechanisms ensure that they are treated humanely? In regard to the first question, the type of research project developed influences the selection of subjects. Animals are just one of a variety of types of subjects used in research; others are human beings, plants, and computer data sets. If possible, most researchers use nonanimal subjects, because they are generally less expensive. In studies that are low risk, which most nursing studies are, human beings are commonly used as subjects.
Some studies, however, require the use of animals to answer the research question. Approximately 17 to 22 million animals are used in research each year, and 90% of them are rodents, with the combined percentage of dogs and cats being only 1% to 2% (Goodwin & Morrison, 2000). Because animals are deemed valuable subjects for selected research projects, the second question, concerning their humane treatment, must also be answered. At least five separate types of regulations exist to protect research animals from mistreatment. The federal government, state governments, independent accreditation organization, professional societies, and individual institutions work to ensure that research animals are used only when necessary and only under humane conditions. At the federal level, animal research is conducted according to the guidelines of U.S. PHS Policy on Humane Care and Use of Laboratory Animals, which was adopted in 1986 and reprinted essentially unchanged in 1996 and is available on the Office of Laboratory Animal Welfare (OLAW) website at http://grants.nih.gov/grants/olaw/olaw.htm (OLAW, 2007).
The Humane Care and Use of Laboratory Animals Regulations define animal as any live, vertebrate animal used or intended for use in research, research training, experimentation, or biological testing or for related purposes. Any institution proposing research involving animals must have a written Animal Welfare Assurance statement acceptable to the U.S. PHS that documents compliance with the U.S. PHS policy. Every assurance statement is evaluated by the NIH’s Office for Protection from Research Risks (OPRR) to determine the adequacy of the institution’s proposed program for the care and use of animals in activities conducted or supported by the U.S. PHS (OLAW, 2007).
Institutions’ assurance statements about compliance with the U.S. PHS policy have promoted the humane care and treatment of animals in research. In addition, more than 700 institutions conducting health-related research have sought accreditation by the American Association for Accreditation of Laboratory Animal Care (AAALAC), which was developed to ensure the humane treatment of animals in research (Pardes et al., 1991). In conducting research, each investigator must carefully select the type of subject needed; if animals are used as subjects, they require humane treatment.
SUMMARY
• The ethical conduct of research starts with the identification of the study topic and continues through the publication of the study if quality research evidence is going to be developed for practice.
• The debate about ethics and research continues, probably because of (1) the complexity of human rights issues; (2) the focus of research in new, challenging arenas of technology and genetics; (3) the complex ethical codes and regulations governing research; and (4) the variety of interpretations of these codes and regulations.
• Two historical documents that have had a strong impact on the conduct of research are the Nuremberg Code and the Declaration of Helsinki. More recently, the U.S. Department of Health and Human Services (U.S. DHHS, 2005) and the Food and Drug Administration (FDA, 2006a, 2006b) have promulgated regulations that direct the ethical conduct of research. These regulations include (1) general requirements for informed consent, (2) documentation of informed consent, (3) institutional review board (IRB) review of research, (4) exempt and expedited review procedures for certain kinds of research, and (5) criteria for IRB approval of research.
• The Council for International Organizations of Medical Sciences revises and updates ethical guidelines for biomedical research conducted internationally. The Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and Social Sciences and Humanities Research Council of Canada (Tri-Council) (2005) developed a policy statement to promote the ethical conduct of research in this country.
• A relatively new federal regulation, Public Law 104-191, the Health Insurance Portability and Accountability Act (HIPAA), was enacted in 1996 and implemented in 2003. HIPAA includes privacy rules to protect an individual’s protected health information.
• Conducting research ethically requires protection of the human rights of subjects. Human rights are claims and demands that have been justified in the eyes of an individual or by the consensus of a group of individuals. The human rights that require protection in research are (1) self-determination, (2) privacy, (3) anonymity and confidentiality, (4) fair treatment, and (5) protection from discomfort and harm.
• The rights of research subjects can be protected by balancing benefits and risks of a study, securing informed consent, and submitting the research for institutional review.
• To balance the benefits and risks of a study, the type, level, and number of risks are examined, and the potential benefits are identified. If possible, the risks must be minimized and the benefits maximized to achieve the best possible benefit-risk ratio.
• Informed consent involves the transmission of essential information, comprehension of that information, competency to give consent, and voluntary consent of the prospective subject.
• In institutional review, a study is examined for ethical concerns by a committee of peers called an institutional review board (IRB). The IRB conducts three levels of review: exempt, expedited, and complete. The process for accessing protected health information according to the HIPAA Privacy Rule is also detailed.
• Research misconduct includes fabrication, falsification, and plagiarism during the conduct, reporting, or publication of research. The Office of Research Integrity (ORI) was developed to investigate and manage incidents of scientific misconduct so as to protect the integrity of research in all disciplines.
• Another current ethical concern in research is the use of animals as subjects. Two important questions are addressed: Should animals be used as research subjects, and if animals are used in research, what mechanisms ensure that they are treated humanely? The U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals provides direction for the conduct of research with animals as subjects.
REFERENCES
Amdur, R. Institutional review board: Member handbook. Sudbury, MA: Jones & Bartlett, 2003.
American Nurses Association (ANA). Code of ethics for nurses with interpretive statements. Washington, DC: American Nurses Association, 2001.
American Psychological Association (APA). Ethical principles of psychologists and code of conduct. Washington, DC: American Psychological Association, 2002.
Arras, J.D. Noncompliance in AIDS research. Hastings Center Report. 1990;20(5):24–32.
Beecher, H.K. Ethics and clinical research. New England Journal of Medicine. 1966;274(24):1354–1360.
Berger, R.L. Nazi science: The Dachau hypothermia experiments. New England Journal of Medicine. 1990;322(20):1435–1440.
Bindler, R.C.M., Massey, L.K., Shultz, J.A., Mills, P.E., Short, R. Metabolic syndrome in a multiethnic sample of school children: Implications for the pediatric nurse. Journal of Pediatric Nursing. 2007;22(1):43–58.
Brandt, A.M. Racism and research: The case of the Tuskegee Syphilis Study. Hastings Center Report. 1978;8(6):21–29.
Broome, M.E. Consent (assent) for research with pediatric patients. Seminars in Oncology Nursing. 1999;15(2):96–103.
Broome, M.E., Stieglitz, K.A. The consent process and children. Research in Nursing & Health. 1992;15(2):147–152.
Bruce, S.L., Grove, S.K. The effect of a coronary artery risk evaluation program on serum lipid values and cardiovascular risk levels. Applied Nursing Research. 1994;7(2):67–74.
Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and Social Sciences and Humanities Research Council of Canada Tri-Council Policy Statement: Ethical conduct of research involving humans, 2005. Retrieved October 2, 2002, from http://pre.ethics.gc.ca/english/policystatement/policystatement.cfm.
Carico, J.M., Harrison, E.R. Ethical considerations for nurses in biomedical research. Journal of Neuroscience Nursing. 1990;22(3):160–163.
Conner, J.A., Smaldone, A.M., Bratts, T., Stone, P.W. HIPAA in 2003 and its meaning for nurse researchers. Applied Nursing Research. 2003;16(4):291–293.
Council for International Organizations of Medical Sciences (CIOMS) International ethical guidelines for biomedical research involving human subjects, 2002. Retrieved October 2, 2007, from www.cioms.ch/guidelines_nov_2002_blurb.htm.
Damrosch, S.P. Ensuring anonymity by use of subject-generated identification codes. Research in Nursing & Health. 1986;9(1):61–63.
Douglas, S., Larson, E. There’s more to informed consent than information. Focus on Critical Care. 1986;13(2):43–47.
Floyd, J. Research and informed consent: The dilemma of the cognitively impaired client. Journal of Psychosocial Nursing and Mental Health Services. 1988;26(3):13–21.
Food and Drug Administration (FDA), Institutional review boards. Code of Federal Regulations, 2006, April 1. Title 21, Part 56. Retrieved October 1, 2007, from www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRsearch.cfm?CFRPart=56.
Food and Drug Administration (FDA), Protection of human subjects (informed consent). Code of Federal Regulations, 2006, April 1. Title 21, Part 50. Retrieved October 1, 2007, from www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRsearch.cfm?CFRPart=50.
Ford, J.S., Reutter, L.I. Ethical dilemmas associated with small samples. Journal of Advanced Nursing. 1990;15(2):187–191.
Frankena, W.K. Ethics, (2nd ed.). Englewood Cliffs, NJ: Prentice-Hall, 1973.
Friedman, P.J. Correcting the literature following fraudulent publication. JAMA. 1990;263(10):1416–1419.
Garfield, E., Welljams-Dorof, A. The impact of fraudulent research on the scientific literature: The Stephen E. Breuning case. JAMA. 1990;263(10):1424–1426.
Goodwin, F.K., Morrison, A.R. Science and self-doubt. Reason. 2000;32(5):22–28.
Grbich, C. Qualitative research in health: An introduction. London: Sage, 1999.
Hansen, B.C., Hansen, K.D. Academic and scientific misconduct: Issues for nursing educators. Journal of Professional Nursing. 1995;11(1):31–39.
Happ, M.B., Swigart, V.A., Tate, J.A., Arnold, R.M., Sereika, S.M., Hoffman, L.A. Family presence and surveillance during weaning from prolonged mechanical ventilation. Heart & Lung. 2007;36(1):47–57.
Hawley, D.J., Jeffers, J.M. Scientific misconduct as a dilemma for nursing. Image: Journal of Nursing Scholarship. 1992;24(1):51–55.
Henderson, J., When scientists fake it. American Way. 1990:56–101.
Hershey, N., Miller, R.D. Human experimentation and the law. Germantown, MD: Aspen, 1976.
Hinds, P.S., Burghen, E.A., Pritchard, M. Conducting end-of-life studies in pediatric oncology. Western Journal of Nursing Research. 2007;29(4):448–465.
Holaday, B., Gonzales, O., Mills, D. Assent of school-age bilingual children. Western Journal of Nursing Research. 2007;29(4):466–485.
Holditch-Davis, D., Brandon, D.H., Schwartz, T. Development of behaviors in preterm infants: Relation to sleeping and waking. Nursing Research. 2003;52(5):307–317.
Kelman, H.C. Human use of human subjects: The problem of deception in social psychological experiments. Psychological Bulletin. 1967;67(1):1–11.
Levine, R.J. Ethics and regulation of clinical research, (2nd ed.). Baltimore–Munich: Urban & Schwarzenberg, 1986.
Lin, C., Lu, M., Chiang, H, Chung, C., Lin, T., Yin, T.J., Yang, C. Using a citizen consensus conference to revise the code of ethics for nurses in Taiwan. Journal of Nursing Scholarship. 2007;39(1):95–101.
McClain, N., Laughon, K., Steeves, R., Parker, B. Balancing the needs of scientist and the subject in trauma research. Western Journal of Nursing Research. 2007;29(1):121–128.
Meade, C.D. Improving understanding of the informed consent process and document. Seminars in Oncology Nursing. 1999;15(2):124–137.
Milgram, S. Behavioral study of obedience. Journal of Abnormal and Social Psychology. 1963;67(4):371–378.
Munhall, P.L. Ethical considerations in qualitative research. In: Munhall P.L., ed. Nursing research: A qualitative perspective. 3rd ed. Sudbury, MA: Jones & Bartlett; 2001:537–549.
Munhall, P.L., Munhall, P.L. Institutional review of qualitative research proposals: A task of no small consequence. In Nursing research: A qualitative perspective, 3rd ed., Sudbury, MA: Jones & Bartlett; 2001:551–563.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. Belmont report: Ethical principles and guidelines for research involving human subjects. Washington, DC: U.S. Government Printing Office, 1978. [(DHEW Publication No. [05] 78-0012)].
Nelson-Marten, P., Rich, B.A. A historical perspective of informed consent in clinical practice and research. Seminars in Oncology Nursing. 1999;15(2):81–88.
Nuremberg Code. Levine R.J., ed. Ethics and regulation of clinical research, (2nd ed.). Baltimore–Munich: Urban & Schwarzenberg, 1986:425–426.
Office of Laboratory Animal Welfare (OLAW) Public Health Service policy on humane care and use of laboratory animals, 2007. Retrieved October 1, 2007, from http://grants.nih.gov/grants/olaw/olaw.htm.
Office of Research Integrity (ORI) Scientific misconduct investigations: 1992–2001, 2004. Retrieved October 1, 2007, from http://www.ori.dhhs.gov/misconduct/documents/NewInstitutionalResearchMisconductActivity.pdf.
Office of Research Integrity (ORI), Public Health Service Policies on Research Misconduct. Code of Federal Regulations, 2005, May 17. Title 42, Part 50 and 93, Policies of General Applicability. Retrieved October 1, 2007, from http://ori.dhhs.gov/documents/FR_Doc_05-9643.shtml.
Office of Research Integrity (ORI), Handling misconduct. Office of Research Integrity, 2007, June 20. Retrieved October 1, 2007, from http://ori.dhhs.gov/misconduct.
Office of Research Integrity (ORI), About ORI – History. Office of Research Integrity, 2007, September 10. Retrieved October 1, 2007, from http://ori.dhhs.gov/about/history.shtml.
Olsen, D.P. Methods: HIPAA privacy regulations and nursing research. Nursing Research. 2003;52(5):344–348.
Orb, A., Eisenhauer, L., Wynaden, D. Ethics in qualitative research. Journal of Nursing Scholarship. 2001;33(1):93–96.
Pardes, H., West, A., Pincus, H.A. Physicians and the animal-rights movement. New England Journal of Medicine. 1991;324(23):1640–1643.
Pfeifer, M.P., Snodgrass, G.L. The continued use of retracted, invalid scientific literature. JAMA. 1990;263(10):1420–1423.
Ramos, M.C. Some ethical implications of qualitative research. Research in Nursing & Health. 1989;12(1):57–63.
Rankin, M., Esteves, M.D. Perceptions of scientific misconduct in nursing. Nursing Research. 1997;46(5):270–275.
Relman, A.S. Publishing biomedical research: Roles and responsibilities. Hastings Center Report. 1990;20(5):23–27.
Reynolds, P.D. Ethical dilemmas and social science research. San Francisco: Jossey-Bass, 1979.
Rothman, D.J. Were Tuskegee and Willowbrook “studies in nature”? Hastings Center Report. 1982;12(2):5–7.
Sasson, R., Nelson, T.M. The human experimental subject in context. In: Jung J., ed. The experimenter’s dilemma. New York: Harper & Row; 1971:265–296.
Silva, M.C. Comprehension of information for informed consent by spouses of surgical patients. Research in Nursing & Health. 1985;8(2):117–124.
Steinfels, P., Levine, C. Biomedical ethics and the shadow of Naziism. Hastings Center Report. 1976;6(4):1–20.
Thompson, P.J. Protection of the rights of children as subjects for research. Journal of Pediatric Nursing. 1987;2(6):392–399.
U.S. Department of Health and Human Services (U.S. DHHS), Final regulations amending basic HHS policy for the protection of human research subjects. Code of Federal Regulations, 1981, January 26. [Title 45, Part 46].
U.S. Department of Health and Human Services (U.S. DHHS), Protection of human subjects. Code of Federal Regulations, 1983, March 8. [Title 45, Part 46].
U.S. Department of Health and Human Services (U.S. DHHS), Protection of human subjects. Code of Federal Regulations, 1991, June 18. [Title 45, Part 46].
U.S. Department of Health and Human Services (U.S. DHHS), Protection of human subjects. Code of Federal Regulations, 2001, November 23. [Title 45, Part 46].
U.S. Department of Health and Human Services (U.S. DHHS), Standards for privacy of individually identifiable health information: Final rule. Code of Federal Regulations, 2002, August 14. Title 45, Public Welfare, Parts 160 and 164. Retrieved October 1, 2007, from www.hhs.gov/ocr/hipaa/privrulepd.pdf.
U.S. Department of Health and Human Services (U.S. DHHS), Institutional review boards and the HIPAA Privacy Rule. HIPAA Privacy Rule: Information for researchers, 2003, September 25. Retrieved October 1, 2007, from http://privacyruleandresearch.nih.gov/irbandprivacyrule.asp.
U.S. Department of Health and Human Services (U.S. DHHS), HIPAA authorizations for research. HIPAA Privacy Rule: Information for researchers, 2004, April. Retrieved October 1, 2007, from http://privacyruleandresearch.nih.gov/authorization.asp.
U.S. Department of Health and Human Services (U.S. DHHS), Protection of human subjects. Code of Federal Regulations, 2005, June 23. Title 45, Part 46. Retrieved October 1, 2007, from www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.htm.
U.S. Department of Health and Human Services (U.S. DHHS). How can covered entities use and disclose protected health information for research and comply with the Privacy Rule? HIPAA Privacy Rule: Information for researchers. February 2, a Retrieved October 1, 2007, from http://privacyruleandresearch.nih.gov/pr_08.asp, 2007.
U.S. Department of Health and Human Services (U.S. DHHS), How do other privacy protections interact with the privacy rule?. HIPAA Privacy Rule: Information for researchers, 2007. February 2, b Retrieved October 1, 2007, from http://privacyruleandresearch.nih.gov/pr_05.asp.
Watson, A.B. Informed consent of special subjects. Nursing Research. 1982;31(1):43–47.
Williams, A.M. Issues of consent and data collection in vulnerable populations. Journal of Neuroscience Nursing. 2002;34(4):211–218.
Wocial, L.D. The role of mentors in promoting integrity and preventing scientific misconduct in nursing research. Journal of Professional Nursing. 1995;11(5):276–280.
World Medical Association (WMA) General Assembly World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. Washington, DC: WMA General Assembly, 2002. Retrieved October 1, 1007, from www.rotrf.org/information/Helsinki_declaration.pdf.