Cellular Homeostasis
Cell Growth and Cancer
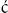 and
and Learning objectives
After reading this chapter you should be able to:
 Define the stages of the mammalian cell cycle.
Define the stages of the mammalian cell cycle.
Outline how the cell cycle is regulated by cyclins and cyclin-dependent kinases.
Describe the molecular events that enable growth factors to regulate cellular proliferation.
Discuss the different mechanisms that enable cells to cease proliferation or die.
Define the cellular and molecular events that define apoptosis and autophagy.
Explain how subversion of normal physiologic growth can lead to the development of tumors.
Distinguish between oncogenes and tumor suppressor genes and describe their role in tumor progression/suppression.
Introduction
The development and survival of multicellular organisms such as human beings is reliant on the appropriate regulation of growth, differentiation and death of individual cell types to maintain the integrity of the organism
While cells have evolved a complex set of control mechanisms to deter replication of damaged cells and to enable repair, if the growth control mechanisms become damaged, this can lead to the development of cancer. The majority of cells in an adult are not dividing. However, under certain conditions such as tissue repair and senescence, regulated growth and proliferation are promoted. Thus, as cells die, either by senescence or as a result of tissue damage, they must be replaced in a strictly regulated manner. Cellular homeostasis maintains organ integrity through controlled cell survival, proliferation and cell death, to ensure that healthy cells, unlike cancerous (transformed) cells, generally stop dividing when they contact neighboring cells.
Research in transformed cancerous cells has highlighted the important mechanisms that regulate cellular growth and cell division in normal cells
Through investigation of genetic alterations in cancer cells, it has been possible to identify a large number of genes that are critical for the regulation of normal cell proliferation. It is perhaps unsurprising to find that molecular mechanisms that favor cell survival and proliferation processes are commonly upregulated in human cancers. Mutated proliferative genes are called oncogenes (cancer-causing genes), the normal cellular counterparts of which are called proto-oncogenes. Proto-oncogenes are predominantly signal transducers that act to regulate normal cell growth and division; aberrant regulation of these processes leads to cellular transformation. Conversely, the proteins involved in suppressing proliferation or tumor suppressor genes are generally inhibited during oncogenesis, leading to uncontrolled proliferation. Exploring the role of these proteins under normal physiologic conditions can assist in the understanding of how they can be subverted when they become dysregulated during oncogenesis.
Cell cycle
Individual cells multiply by duplicating their contents and then dividing into two daughter cells
Cell division is tightly controlled by a complex mechanism called cell cycle. The duration of cell cycle varies between organisms, as well as between cell types within a single organism. In mammals, for example it can last from minutes to years. However, in immortalized cell lines, which are widely used as experimental model systems, one round of cell cycle is typically completed within 24 hours.
In recent years, extensive research of the cell cycle has defined a number of key control points
Traditionally, the cell cycle is divided into several phases (Fig. 42.1). Mitosis (M phase) is the stage of cell division, which is usually completed within an hour. The remainder of the cell cycle, during which cells prepare for division and duplicate deoxyribonucleic acid (DNA), is referred to as interphase. Nuclear DNA replication occurs in the synthesis (S) phase of interphase. The period between M and S phase is called G1 phase, while the interval between S and M phase is called G2 phase. During G1 and G2 phases, cells undergo several checkpoints to ensure appropriate cellular growth and accurate DNA synthesis occurs prior to cell division, thus preventing the incorporation of mutated DNA into daughter cells. As mentioned above, the duration of an individual cell cycle varies enormously and most of this variability can be attributed to the different length of the G1 phase. This is due to the fact that some cells, which are not stimulated to duplicate their DNA, can enter into the specialized form of G1 phase, called G0 phase.
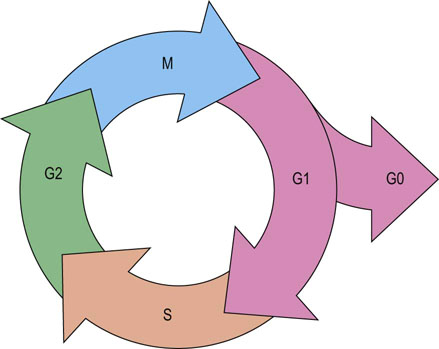
Fig. 42.1 The phases of the cell cycle.
Cell cycle is divided into interphase and mitotic phase. In interphase, composed of G1, S and G2 phases, cells grow, prepare for division and duplicate their DNA. Mitosis is the stage of cell division into two daughter cells. M, mitosis; S, synthetic phase of interphase; G1, interval between M and S phases; G2, interval between S and M phases; G0, resting, or quiescent, phase.
The G0 phase is a form of the resting state, or quiescence, in which cells reside until they receive appropriate signals: for example, from growth factors, urging them to re-enter and progress through the cell cycle
In mammals, the time required for a cell to transit from the beginning of S phase through mitosis is typically 12–24 hours, irrespective of the duration of G1 phase. Thus, the majority of variation in proliferation rates observed between different cell types is due to the amount of time spent in the G0/G1 phase. In conditions that favor cell growth, the total ribonucleic acid (RNA) and protein content of the cell increases continuously, except in M phase when the chromosomes are too condensed to enable transcription to occur.
Of note, the majority of cells in the human body have irreversibly withdrawn from the cell cycle into either a terminally differentiated state (neurons, myocytes or surface epithelial cells of skin and mucosa), or they are in the reversible quiescent G0 phase (stem cells, glial cells, hepatocytes or thyroid follicular cells). Only a minority of cells are normally actively cycling and these cells are located mainly in the stem/transit compartments of self-renewing tissues, which include the bone marrow and epithelia.
Regulation of cell proliferation and growth: growth factors
Cells of a multicellular organism have to receive positive signals in order to grow and divide
Many of these signals are in the form of polypeptide hormones (e.g. insulin), growth factors (e.g. epidermal growth factor, EGF) or cytokines (e.g. interleukins IL-1 to IL-36). These growth factors bind to specific cell surface receptors, initiating an intricate network of intracellular signaling cascades that counteract negative regulatory controls present in resting cells to block cell cycle progression and division.
In most cell types, proliferation is controlled by signals generated from a specific combination of growth factors rather than stimulation by a single growth factor
In this way, a relatively small number of growth factors can selectively regulate the proliferation of many cell types. In addition, some of the factors may induce cell growth without providing a signal for division. Indeed, neurons in the G0 phase of cell cycle grow very large without dividing. Moreover, although proliferating cells can stop growing when they are deprived of growth factors, they continue their progress through the cell cycle until they reach the point in the G1 phase at which they can enter G0 phase (the resting state) or undergo senescence.
Growth factors bind to their specific cell surface receptors. There are about 50 known growth factors
Growth factors bind to their specific cell surface receptors expressed on effector cells, which are generally transmembrane proteins with a growth factor (or ligand) binding domain and cytoplasmic protein tyrosine kinase (PTK) domain. There are about 50 known growth factors, of which platelet-derived growth factor (PDGF) was the first identified. The growth and proliferative responses to PDGF are prototypic for many growth factors, and these responses include:
Immediate increase in intracellular Ca2+ levels – indicating the initiation of transmembrane signaling.
Reorganization of actin stress fibers – to enable anchorage dependence of cell attachment, a requirement for cell cycle progression.
Activation and/or nuclear translocation of transcription factors that bind to regulatory regions of the DNA encoding genes responsive to a specific growth factor. These genes are referred to as immediate early genes, which usually code for transcription factors themselves that mediate expression of components of the cell cycle machinery, such as cyclins.
Growth factors initiate selective signaling cascades
Individual growth factors activate distinct cohorts of signaling molecules and transcription factors that in turn induce a unique repertoire of gene expression. In this way specific growth factors initiate characteristic differential responses that have a unique impact on cell behavior.
Growth factors initiate selective signaling cascades by binding to their receptors
The binding to a receptor causes receptor dimerization or oligomerization and activation of the intracellular tyrosine kinase domain, which in turn mediates receptor transphosphorylation at specific amino acids within the cytoplasmic domain. The phosphorylated region of the receptor is then able to act as a ‘docking site’ for the binding of specific proteins, thus enabling protein–protein interactions (Fig. 42.2). This in turn leads to the recruitment and activation of additional signaling molecules such as enzymes and ‘adapter’ molecules, which mediate the propagation of the intracellular signaling cascade from the surface of plasma membrane inside the cell. Transphosphorylation of receptor cytoplasmic domains creates a scaffold for binding of signal transduction elements such as phospholipase Cγ (PLC-γ), GTPase-activating proteins (GAPs), nonreceptor PTKs (Src, Fyn Abl), phosphotyrosine phosphatases (PTPases) and adapter molecules (Shc or Grb2), via their phosphotyrosine recognition domains.
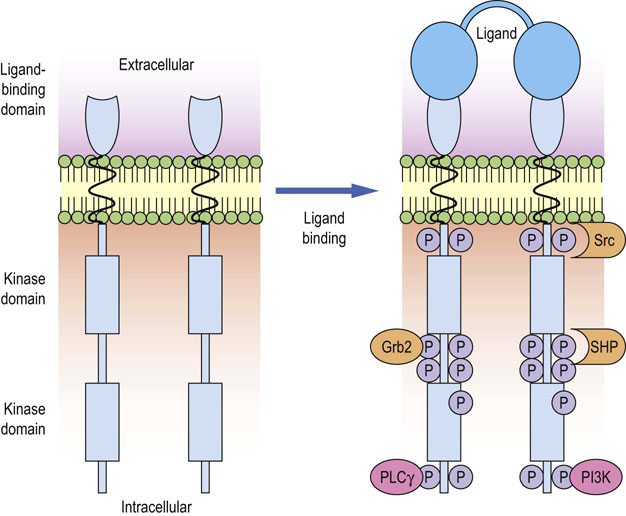
Fig. 42.2 Activation of growth factor receptor by ligand binding and recruitment of signaling molecules.
The binding of a growth factor (such as PDGF or EGF) to its receptor causes receptor dimerization and activation of the tyrosine kinase, intrinsic to the receptor cytoplasmic domains. This leads to the tyrosine phosphorylation of the dimerized receptors at specific sites within the cytoplasmic domains, by the process of transphosphorylation. The phosphorylation events create docking sites that enable protein–protein interactions between the receptor and downstream signaling components such as PLC-γ, protein tyrosine kinase Src, tyrosine phosphatase SHP, PI3K and adapter molecules like Grb2, which in turn recruits Ras/MAPK pathway. PLC-γ, phospholipase Cγ; PI3K, phosphatidylinositol 3-kinase; Grb2, growth factor receptor-bound protein 2; SHP, SH2 domain-containing phosphatase.
 Advanced concept box protein tyrosine kinases
Advanced concept box protein tyrosine kinases
Protein tyrosine kinases (PTKs) are enzymes that transfer the γ-phosphate group of ATP to tyrosine residues on target substrate proteins. The term ‘protein tyrosine kinase’ is a generic term for a large superfamily of enzymes, which includes both transmembrane-spanning receptors with an intrinsic tyrosine kinase activity in their cytoplasmic domains (e.g. some growth factor receptors), as well as a wide range of cytoplasmic tyrosine kinase subfamilies such as Src, Abl, Syk, Tec or Janus kinase (JAK) families.
Tyrosine phosphorylation is a covalent modification, which provides a rapid and reversible (by the action of protein tyrosine phosphatases) mechanism of changing the enzymatic activity of the target proteins, or modifying these proteins, so that they can act as adapters to recruit other signaling molecules.
For example, the tyrosine phosphorylation of receptors or signaling molecules creates ‘docking sites’ that enable protein–protein interactions via protein–protein interaction domains, such as those referred to as SH2 domains of other signaling transducers. SH2 stands for Src-homology region 2, from the cytoplasmic Src tyrosine kinase, in which this domain was first characterized. SH2 domains contain approximately 100 amino acids and specifically recognize a phosphotyrosine in the context of the three amino acids immediately C-terminal to that phosphotyrosine.
Role in the regulation of cellular proliferation, survival and differentiation.
PTKs play a critical role in the regulation of cellular proliferation, survival and differentiation, and the importance of these regulatory events is highlighted by the defects that are observed upon dysregulation of genes encoding PTKs. Indeed, dysregulated expression of growth factor receptors carrying intrinsic PTK activity can lead to the constitutive activation of the Ras/Raf/MEK/ERK and Ras/PI3K/Akt/mTor signaling pathways leading to enhanced cell growth, survival and proliferation and a subversion of molecular events that regulate apoptosis, events that are dysregulated in cancer. Mutations have been identified in PDGFR, EGFR, Kit and Flt3, in specific cancer types. Indeed, mutation in the EGFR family of receptors are responsible 30% of all epithelial cancers, including lung and brain cancers.
Nonreceptor tyrosine kinases also play critical roles in cellular responses, with mutations leading to a loss in kinase activity, resulting in serious abnormalities in B and T lymphocyte development. For example, loss of expression/activity of ZAP-70, a PTK that is essential for antigen-dependent T cell activation, can lead to severe combined immunodeficiency (SCID), due to a lack of T cell effector function during an immune challenge. Similarly, X-linked agammaglobulinemia, an immunodeficiency caused by a lack of IgG antibody production occurs as a result of loss of function mutations of Btk, a PTK that is important for B cell effector functions.
Epidermal growth factor (EGF) receptor signaling
Upon ligand binding, the EGF receptor (EGFR) leads to the activation of signals through phosphatidylintositol-, Ras/Raf/MAPK- and the PI3K/Akt/mTor-mediated signaling cascades
The EGF receptor (EGFR) can be bound and activated by a number of ligands including EGF and transforming growth factor-α (TGF-α). Ligation of the EGFR leads to the recruitment and activation of Src family of PTKs, which catalyze phosphorylation of PLC-γ, leading to the activation of this enzyme. Activated PLC-γ then catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the intracellular second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 stimulates release of Ca2+ from intracellular stores (mainly ER) and DAG activates an important signal transducer family of proteins, the protein kinase C (PKC) family. Ligation of the EGFR also induces activation of another lipid modifying enzyme, phosphatidylinositol 3-kinase (PI3K). This enzyme mediates phosphorylation of PIP2, generating the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3), which contributes to the activation of certain members of the PKC family (Fig. 42.3). Moreover, PIP3 can activate another kinase referred to as PIP3-dependent kinase (PDK1) or serve as docking site for proteins that contain so-called pleckstrin homology (PH) domains.
Fig. 42.3 Activation of ERK-MAPK kinase and PKC signaling cascades upon growth factor binding.
Growth factor signals activated at the plasma membrane as a result of ligand binding can regulate gene transcription, cell cycle progression, proliferation, differentiation or apoptosis. Growth factor receptor signals activate proximal signals to recruit the adapter molecules Grb2 and Shc, which lead to the activation of ERK-MAPK and PI3K/Akt and PKC pathways. Activation of these pathways leads to the activation of transcription factors, such as Jun and Fos (which dimerize to form AP1), NFAT, and Myc. These regulate the induction of components of the cell cycle machinery that controls cell cycle progression and identify the need for DNA repair. If DNA damage is detected, then cell cycle is arrested, and if the damage is too great, then apoptosis is induced. GF, growth factor; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; PI3K, phosphatidylinositol 3-kinase ; PIP2, phosphoinositol 4,5-bisphosphate; PIP3, phosphoinositol 3,4,5-trisphosphate; PKC, protein kinase C; PLC, phospholipase C; Shc, Src homology and collagen domain protein.
Signaling cascade involving Ras GTPase is important in regulating cell division
A quarter of all tumors have constitutively active mutations in the signaling component Ras, which is critical for the transmission of proliferation and differentiation signals from the extracellular receptors to the nucleus. Ras is constitutively bound to the plasma membrane via a post-translational modification involving the addition of a lipophilic farnesyl group. Ligation of the EGFR recruits Ras by its binding to the adaptor protein Grb2. Ras is a GTPase, which cycles between an active GTP-bound and inactive GDP-bound form (Fig. 42.4). Its intrinsic catalytic activity is low, and is enhanced by binding to a GTPase-activating protein (GAP). GDP/GTP exchange is promoted by binding to a guanine nucleotide exchange factor, called Sos, which returns Ras into an active state. One of the main functions of active Ras is to act as an allosteric regulator of the mitogen-activated protein kinase (MAPK) signaling cascade. Ras transduces signals from EGFR via activation of two intermediary kinases, Raf and MEK kinase. MEK is a MAPK-type kinase that activates MAPK by mediating dual-specificity phosphorylation of tyrosine and threonine residues within its TEY activation motif. MAPK exists in two isoforms, extracellular-signal regulated kinase (ERK) 1 and 2. Upon activation, they translocate into the nucleus and phosphorylate (on serine and threonine) key transcription factors involved in the regulation of transcription of genes that regulate DNA synthesis and cell division.
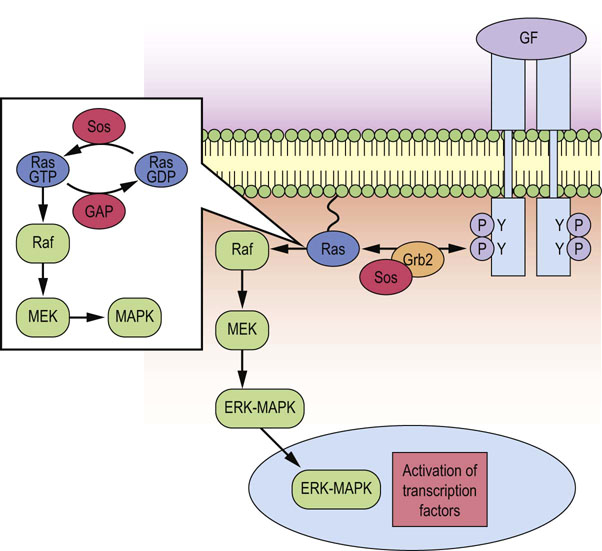
Fig. 42.4 Recruitment and activation of Ras by growth factor receptors.
Ras is anchored in the plasma membrane and recruited to the activated growth factor receptor via interaction with the Grb2-Sos complex. Receptor stimulated GTP/GDP exchange, and thus activation of Ras, is promoted by Sos, whereas GAP inactivates Ras by stimulating its intrinsic GTPase activity. Ras couples growth factor receptors to the MAPK signaling cascade, via stimulation of the intermediary kinases Raf and MEK. MAPK translocates into the nucleus and phosphorylates key transcription factors involved in the regulation of DNA synthesis and cell division. GF, growth factor; GAP, GTPase-activating protein; MAPK, mitogen-activated protein kinase; Sos, son of sevenless.
mTORC-1 and mTORC-2 complexes integrate mitogen and nutrient signals
In addition to acting on selective members of the PKC family, PI3K is also responsible for activating PDK1, which in turn activates Akt. This enzyme mediates the activity of mammalian target of rapamycin (mTor), a serine/threonine protein kinase. mTor can participate in two distinct signaling complexes mTORC-1 and mTORC-2, integrating mitogen and nutrient signals to promote cell survival, growth and proliferation. PI3K/Akt activation by receptor ligation leads to phosphorylation of the tuberous sclerosis protein 1/2 (TSC1/2), resulting in mTORC-1 activation. The best-characterized downstream effectors of mTORC-1 are translational regulators within the protein synthesis pathway, initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and ribosomal S6 kinase 1 (S6K1), which are stimulated by mTORC1-mediated phosphorylation (Fig. 42.5).
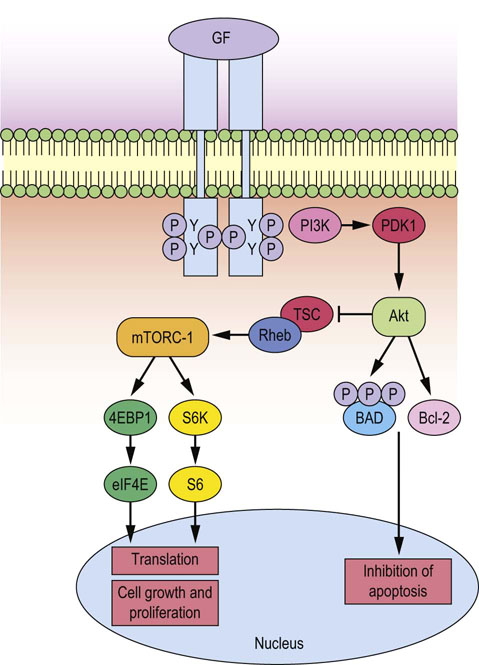
Fig. 42.5 Growth factor stimulation of mTor signaling.
Growth factor signals activated at the plasma membrane as a result of ligand binding can result in the recruitment of PI3K, thus activating PDK1/AKT, which enhances protein synthesis via mTOR (which is the catalytic subunit of the mTORC-1 protein complex shown), thereby contributing to cell cycle progression and inhibition of apoptosis by increasing the expression of cyclins and anti-apoptotic Bcl-2 family members. In addition, Akt inhibits apoptosis by mediating hyperphosphorylation of pro-apoptotic BAD, leading to the stabilization of mitochondria. GF, growth factor, PI3K, phosphatidylinositol 3-kinase; mTOR, mammalian target of rapamycin; BAD, Bcl-2-associated death promoter; TSC, tuberous sclerosis factor; Rheb, Ras homologue enriched in brain.
While the signaling pathways described above are linear, a significant amount of cross-talk occurs amongst these cascade elements
For example the ERK-MAPK signaling cascade is capable of regulating mTORC-1, by activating the kinase RSK1, which in turn phosphorylates and inhibits TSC1/2, thus resulting in activation of mTORC-1. Examples such as this illustrate the complexity that exists in intracellular signaling pathways downstream of receptors. Moreover, cross-talk goes some way to highlighting how a mutation in a single signaling component can impact on a wide array of biological responses within a single cell.
Cytokine receptor signaling
Cytokines are growth factors that mainly coordinate the development of hematopoietic cells and the immune response, although they also have multiple effects on nonhematopoietic cell types
As described above for growth factors, cytokines also exert their effects on cells by binding to cell surface receptors. There are many classes of cytokine receptors, many of which belong to a superfamily called the hematopoietic receptors. These are transmembrane glycoprotein receptors, characterized by the conserved extracellular ligand-binding domains, which contain characteristic cysteine pairs and a pentapeptide motif WSXWS (tryptophan-serine-X-tryptophan-serine, where X is any amino acid). Many consist of multisubunit receptors that contain a unique ligand-binding subunit that gives specificity, and a common signal transducer subunit that is often shared by several related cytokines. The sharing of the signal transducing subunit gives the basis for classification of cytokines into distinct subfamilies and helps to explain the severe immunodeficiencies that result from naturally occurring defects in these receptors.
Janus kinases (JAKs) link the hematopoietic receptors with the downstream signaling and gene transcription
In contrast to growth factor receptors, cytokine receptors do not possess an intrinsic catalytic activity. However, a family of cytosolic PTKs, called Janus kinases (JAKs), are essential for linking the hematopoietic receptors with the downstream signaling and gene transcription. Thus, after ligand engagement with the receptor, tyrosine kinase activity is induced, and most cytokines, in common with classic growth factors, can signal through PLC, PI3K and Ras-MAPK signaling cascades, JAKs associate with the receptors by binding to the conserved regions near the transmembrane domain. Upon cytokine binding, which causes receptor oligomerization, JAKs become phosphorylated and activated to mediate phosphorylation of their downstream targets, which are transcription factors called signal transducer and activators (STATs) (Fig. 42.6). In unstimulated cells, STATs are found in the cytoplasm in monomeric forms. Cytokine stimulation leads to the JAK-mediated STAT phosphorylation and dimerization. STAT dimers then translocate to the nucleus, where they mediate transcription of the target genes by binding to their specific DNA sequences. Recently, growth factors such as EGF and PDGF have also been shown to induce activation of JAK/STAT pathways; therefore, JAK/STAT signaling may be a universal mechanism utilized by growth factors to regulate gene induction and cellular responses.
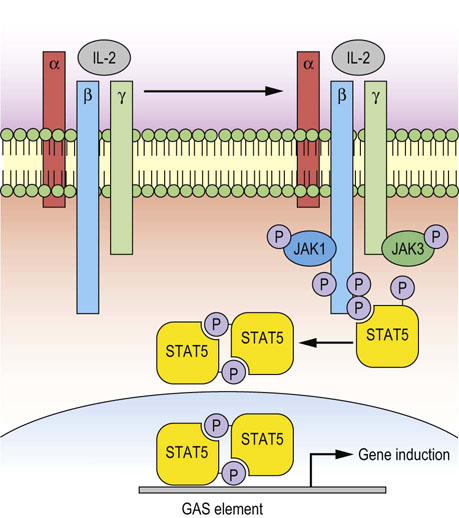
Fig. 42.6 Cytokine receptor signaling: IL-2 receptor.
The receptor is composed of the IL-2 receptor α-chain (IL-2Rα), IL-2/15Rß and the common cytokine-receptor γ-chain (γc). Binding of IL-2 to the α subunit causes association of this subunit with ß- and γ-subunits, thus forming a stable heterotrimer. JAK molecules associate with β-subunit (JAK1) and γc-subunit (JAK3), phosphorylate themselves and the key tyrosine residues within β- and γc-intracellular domains, thus enabling STAT5 recruitment and phosphorylation. -Phosphorylated STAT5 molecules dissociate from the receptors and form dimers, which rapidly translocate into the nucleus and act as transcription factors, binding to GAS elements. Different JAKs and STATs can be utilized to achieve the specific response of individual cytokines. These pathways control transcription, cell growth, proliferation and survival. JAK, Janus kinase; STAT, signal transducer and activator of transcription; GAS, gamma interferon activation site.
 Clinical box X-SCID: interleukin common gamma-chain deficiency
Clinical box X-SCID: interleukin common gamma-chain deficiency
Severe combined immunodeficiency (SCID) is a group of rare, sometimes fatal, inherited congenital disorders characterized by little or no immune response. SCID patients share the common characteristic of a block in T cell development, associated with a variable impairment in B/NK immunity depending on the mutation that has taken place (Chapter 38). Without a functional immune system, SCID patients are susceptible to recurrent infections such as pneumonia, meningitis and chickenpox, and can die before the first year of life. Though invasive, new treatments such as bone marrow and stem-cell transplantation save as many as 80% of SCID patients.
Around 60% of SCID patients have defective cytokine signaling. The gene responsible for SCID was initially reported to code γ-chain of the IL-2 receptor. It is now clear that this γ-chain is a shared common signaling subunit of receptors for IL-4, IL-7, IL-9, IL-13, and IL-15, highlighting the reason for such severe immunodeficiency, as the patient is unable to respond to any of these cytokines. The mutation is recessive, so heterozygous females are normal carriers, whereas the males who inherit the defective X chromosome develop the disease, hence being referred to as X-SCID. The intracellular domain of these proteins normally associates with the PTK, JAK3, whose activation leads to gene induction, cell growth and proliferation. In agreement with this, JAK3 mutations that lead to the absence or defective function of JAK3, are the cause of an autosomal recessive form of SCID, and result in a similar phenotype to X-SCID.
While treatment of this severe disorder can be through bone marrow transplantation, gene therapy has been trialled as an option to restore the immune system with the introduction of the patient's own bone marrow-derived B and T cell precursors. In these cells the γ-chain defect was repaired by the viral-mediated gene therapy, rendering the cells responsive to the cytokines, thus enabling restoration of the adaptive immune response. The finding that JAK3 mutations also resulted in SCID suggested that JAK3 may be good therapeutic target for the development of novel immunosuppressants. Indeed, the JAK3 inhibitor, tofacinib, is used as an immunosuppressant to prevent organ transplant rejection as well as autoimmune diseases such as psoriasis, psoriatic arthritis, inflammatory bowel syndrome and rheumatoid arthritis. Moreover, JAK3 inhibitors are potential antitumor therapeutics for cancers in which increased JAK3 activity has been shown, such as acute myeloid leukemia (AML), and colorectal and lung cancers.
Regulation of cell cycle
Cyclin-dependent kinase (CDK) family of serine/threonine kinases, and cyclins, regulate cell cycle transition points
As cells progress through the cell cycle in response to growth factor stimulation, they must pass through three switch-like transition/restriction points, positioned at the G1/S phase boundary and at the entry and exit of M phase. Master regulators of these transition points include members of the cyclin-dependent kinase (CDK) family of serine/threonine kinases and a family of proteins known as cyclins.
CDKs are expressed as heterodimers comprising a protein kinase subunit and a regulatory cyclin subunit. Their activity is tightly regulated by different mechanisms, including the phosphorylation status of the kinase subunit, levels of cyclins and/or interaction with inhibitory proteins (CDKIs) that block their catalytic activity. While CDK expression levels are relatively constant throughout the cell cycle, the expression levels of cyclins are strictly controlled at both the mRNA and the protein level (transcriptional and translational control). Indeed, cyclins were originally defined as proteins that were specifically degraded during every mitosis.
The traditional model of cell cycle states that a specific cyclin-CDK partner drives distinct parts of the cell cycle: D-type cyclins and CDK4/6 regulate the events in early G1 phase, cyclin E-CDK2 triggers S phase, cyclin A-CDK2 and cyclin A-CDK1 regulate the completion of the S phase, while cyclin B-CDK1 controls mitosis (Fig. 42.7).
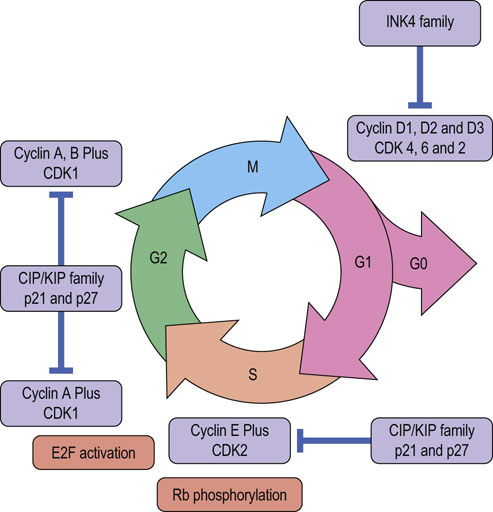
Fig. 42.7 Regulation of the cell cycle.
Progression through the cell cycle is regulated by interplay amongst specific cyclin-CDK partners and their inhibitors. Cyclin expression, particularly of those controlling G1 and S phase is regulated by growth factor signaling. INK4 family members inhibit cyclin D specific CDKs (CDK4 and 6), while CIP/KIP family inhibits all CDKs. CDK, cyclin-dependent kinase; CDKI, CDK inhibitor; E2F, transcription factor; Rb, retinoblastoma protein; INK4, inhibitors of CDK4 family.
Advanced concept box Changing the classical cell cycle paradigm
Pioneering studies assessing cells at different stages of the cell cycle, using classical biochemical approaches such as overexpression of kinase-dead CDK mutants and selective pharmacologic CDK inhibitors, revealed the preferred cyclin-CDK partners, leading to the formulation of the traditional model of cell cycle. However, results from the recent genetic studies, employing specific deletions of cyclins and CDKs in mice and yeast, demonstrate that this model should be revised.
Interestingly, mouse embryos are viable even in the absence of CDK2, CDK4 or CDK6, indicating that these CDKs have redundant functions and are dispensable for controlling cell proliferation. The absence of these CDKs, however, affects the proliferation and/or differentiation of some specific cell types. CDK2 knockout mice survive for up to two years of age, but they are sterile, indicating that CDK2 is absolutely required for meiosis.
Moreover, CDK4 controls proliferation/differentiation of pancreatic β-cells, or pituitary hormone producing cells, whereas CDK6 regulates proliferation/differentiation of some hematopoietic cells. In contrast, CDK1 is essential for driving the cell cycle in most cell types, at least until halfway through gestation.
From these studies, a new model has emerged, referred to as the minimal threshold model. In this model, either CDK1 or CDK2 coupled with cyclin A or E is sufficient to control interphase, while CDK1 coupled with cyclin B drives cells into mitosis. The differences in activity of the same cyclin-CDK complexes in interphase and mitosis may be attributed not only to the substrate specificity, but also to the different localization within the cell, and higher activity threshold for mitosis than for interphase. In addition to being regulated by binding of cyclins, CDK activation is also modulated by phosphorylation. CDK activating complex (CAK), composed of CDK7, cyclin H and MAT1 (ménage a trois), mediates phosphorylation/activation of CDK1, CDK2, CDK4 and CDK6, when bound to their cyclin partners. Moreover, CAK plays a role in gene transcription as a part of TFIIH, which is a general transcription factor. In this context, CAK phosphorylates the RNA polymerase II large subunit C-terminal domain (CTD). This event is part of the process of promoter clearance and progression from the pre-initiation to the initiation phase of transcription.
Mitogenesis
Mitogenic signals activated by growth factors exert their effects between the onset of G1 phase and a point late in the G1 phase, called the restriction point
The key event for the initiation of the cell cycle in G1 phase is the phosphorylation of retinoblastoma protein, Rb, at various residues. Rb controls the expression of genes that commit cells that have reached restriction point late in G1 phase to enter the S phase (DNA synthesis) of the cell cycle. In the early G1 phase, Rb molecules are in the hypophosphorylated state. This allows them to bind and repress the DNA-binding activity of the main regulators of the G1/S phase transition, members of E2F family of transcription factors, thus inhibiting cell cycle progression (Fig. 42.7). Additional molecules that play an important role at this stage are histone deacetylases and chromatin remodeling complexes that epigenetically regulate gene transcription. Stimulation with growth factors and/or mitogens affects the entry into the cell cycle by triggering the expression and/or activation of proto-oncogenes, such as Ras and Myc, which results in the induction of expression of cyclins from the D-type family (D1, D2 and/or D3), followed by the cyclins from E-type family (E1 and E2). The D-type cyclins partner with CDK4/6 and stimulate their activity, whereas E-type cyclins increase the kinase activity of CDK2. These cyclin-CDK partners modulate the phosphorylation state of Rb by converting it from hypophosphorylated into hyperphosphorylated state, thus inactivating Rb, and promoting the release of E2F from Rb inhibitory complex. Free E2F proteins mediate gene transcription, the products of which are important for entry into S phase, and beyond, such as A- and B-type cyclins (Fig. 42.7). From this point onward, cell cycle progression is independent of growth factors.
When E-type cyclins are degraded, CDK2 binds to the A-type cyclins and these complexes phosphorylate many protein targets necessary for the proper completion and exit from the S phase. At the end of S phase, A-type cyclins also associate with CDK1, and these complexes share substrates with cyclin A-CDK2. The importance of the existence of both cyclin A-CDK2 and cyclin A-CDK1 complexes is still not clear.
Nevertheless, during G2 phase A-type cyclins are degraded by ubiquitin-mediated proteolysis and B-type cyclins are synthesized and interact with CDK1. It is estimated that cyclin B-CDK1 complexes phosphorylate more than 70 target proteins, which are important mediators of both regulatory and structural processes (chromosomal condensation, fragmentation of the Golgi network and breakdown of the nuclear envelope) during G2/M transition. Finally, the inactivation of cyclin B-CDK1 complexes is necessary for the exit from mitosis. This is achieved by the ubiquitin-labeling and subsequent proteasomal degradation of B-type cyclins regulated by the anaphase-promoting complex (APC). The main phosphatase that mediates return to interphase after mitosis is a form of protein phosphatase-2A (PP2A), whose activity is increased after degradation of the mitotic cyclins.
Monitoring for DNA damage
Molecular checkpoints that mediate appropriate progression through the cell cycle sense problems that may occur during DNA synthesis and chromosome segregation
The ultimate role of these checkpoints is to inhibit cyclin-CDK activity and thus delay or arrest cell division. During replication, DNA is less condensed and therefore protected to a lesser degree from the attack of exogenous and endogenous genotoxic agents, which may cause DNA damage.
In the case of damage occurring, the DNA damage checkpoints sense alterations and activate signaling pathways that mediate DNA repair
If the damage is beyond repair, they induce apoptosis. The core molecules of the DNA damage checkpoint are sensor kinases, ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia Rad3-related (ATR), which detect double-strand breaks and replication stress, respectively, and checkpoint kinases, CHK1 and CHK2, which relay the signals from the sensor kinases. These molecules prevent G1/S and G2/M phase transitions by inhibiting the CDK activity through increasing the expression of CDK inhibitor (CDKI) protein, p21 via p53 stabilization and/or inhibiting the CDK activator Cdc25 phosphatases.
The tumor suppressor protein p53 is predominantly a DNA damage-sensing protein which monitors DNA damage throughout the cell cycle
If DNA damage is detected, ATM and subsequently CHK2 kinases are activated, action that contributes to the stabilization of p53 and thus enables the induction of DNA repair mechanisms. One of the roles of p53 is to act as a transcription factor, increasing the expression of the CDKI p21 (WAF1). In turn, p21 inhibits cyclinD-CDK4 complexes, preventing Rb phosphorylation and thus promoting Rb/E2F binding and suppression of E2F-mediated gene transcription. Moreover, type-E cyclin-CDK2 complexes are inhibited, enabling cell cycle arrest at the G1/S transition. This allows the cell time to repair the DNA damage, and hence prevents the incorporation of mutated genetic material into daughter cells. However, if the DNA damage is beyond repair, p53-dependent programmed cell death by apoptosis is triggered.
A p53-independent pathway involving the INK4 family of proteins can also induce cell cycle arrest in the G1 phase in response to DNA damage
These proteins, including p16 (INK4A), p15 (INK4B), p18 (INK4C) and p19 (INK4D), mediate cell cycle arrest by binding to CDK4/6 or their binding partner, cyclin D, thereby causing the inactivation of the cyclin cyclin D-CDK4/6 complexes. Another important checkpoint is the spindle assembly checkpoint (SAC), which ensures proper alignment and segregation of chromosomes in the metaphase of mitosis. The SAC signal is generated by the presence of unattached or improperly attached kinetochores (protein complexes on chromatids which mediate attachment to the mitotic spindle), ultimately leading to the inhibition of the anaphase-promoting complex (APC) and thus preventing the onset of anaphase.
Defects in the DNA damage checkpoints allow accumulation of the DNA alterations, thus contributing to the genomic instability, whereas defective SAC may lead to the unequal segregation of the genetic material among the daughter cells, thus creating chromosomal aberrations. Both genomic instability and chromosomal aberrations are major culprits in cell transformation and oncogenesis.
Clinical box Ataxia telangiectasia
Ataxia telangiectasia is a rare autosomal recessive disease caused by the mutations in the ataxia telangiectasia-mutated (ATM) gene. Patients suffering from this disease have the following symptoms and signs: an unsteady gut, telangiectasia, skin pigmentation, infertility, immune deficiencies, and increased incidence of cancer, especially lymphoreticular tumors.
ATM is a serine/threonine protein kinase involved in the induction of DNA damage checkpoint in response to ionizing radiation, anticancer treatment, or programmed DNA breaks during meiosis. Ataxia telangiectasia cells exhibit chromosomal instability, hypersensitivity to reagents that induce DNA strand breaks and defects in the G1/G2 phases and altered regulation of p53 and p21 expression. ATM is constitutively expressed during the cell cycle and is involved in induction of p53-mediated cell cycle arrest and/or apoptosis. In ataxia telangiectasia, cells with mutated ATM cannot properly activate p53, and hence induce cell cycle arrest or apoptosis in response to ionizing radiation. Moreover, when p53 is also mutated, the cell cycle is not adequately regulated, which increases the risk of tumor formation as a result of accumulation of mutations.
Cell death
Cell death is a fundamentally important part of a cell's life cycle, and appropriate regulation of this process is critical to maintain the homeostatic regulation of a multicellular organism
Cell death may be accidental or programmed, initiated and executed through distinct biochemical pathways. Programmed cell death (PCD) is genetically regulated and its role is to remove superfluous, damaged or mutated cells. For many years, apoptosis was a synonym for PCD; however, this concept is changing due to the recent findings identifying different modes of controlled cell death.
Both initiation and execution of cell death are complex processes, with scientists classifying the various forms of cell death on the basis of morphological and/or biochemical features as the most common criteria. According to morphological criteria, cell death can be classified as:
Apoptosis – the rounding up of the cell, retraction of pseudopods, reduction of cellular and nuclear volume (pycnosis), chromatin condensation and fragmentation (karyorrhexis), plasma membrane blebbing, formation of apoptotic bodies and engulfment by resident phagocytes in vivo.
Autophagy (often referred to as autophagic cell death) – massive vacuolization of the cytoplasm, accumulation of the double membrane autophagic vacuoles, without chromatin condensation and little or no uptake by phagocytes in vivo.
Necrosis or necrotic cell death – an enlargement of the cell volume, swelling of the organelles and plasma membrane bursting with concomitant loss of the intracellular content. Necrosis is often considered to be an accidental and uncontrollable form of cell death that occurs after severe insult to the cell. However, recent research has shown that necrosis can be controlled and initiated through specific signaling pathways involving mainly RIP1 serine/threonine kinase. This type of necrosis is called necrosis-like PCD or necroptosis and it has been observed in cancer cells, proliferating cells that have suffered DNA damage as well as cells infected with certain viruses (such as Vaccinia).
Cornification (also known as ‘cornified envelope’ formation or keratinization): an epidermis-specific form of PCD whose role is to create a barrier between the body and environment. It is characterized by elimination of cytosolic organelles, modifications of plasma membrane, accumulation of specific lipids and proteins both inside and outside of the cells to create skin elasticity, mechanical resistance and water resistance.
Apoptosis
Apoptosis is initiated and executed through either perturbation of intracellular homeostasis by the intrinsic (mitochondrial) or extrinsic pathways, the latter of which involves ligation of death receptors such as FAS, TNFR, TRAIL and TWEAK
For both of these pathways, two families of proteins are considered quintessential regulators: cysteine proteases called caspases, and the B cell lymphoma protein 2 (Bcl-2)-related family members, which interact to serve as modulators of life versus death decisions (Fig. 42.8). However, there is a growing body of evidence for other PCD pathways that sense stress and damage in other cellular organelles (such as endoplasmic reticulum and lysosomes), and result in the initiation of death programs. These programs can occur in association with, or independently of, the intrinsic mitochondrial pathway.
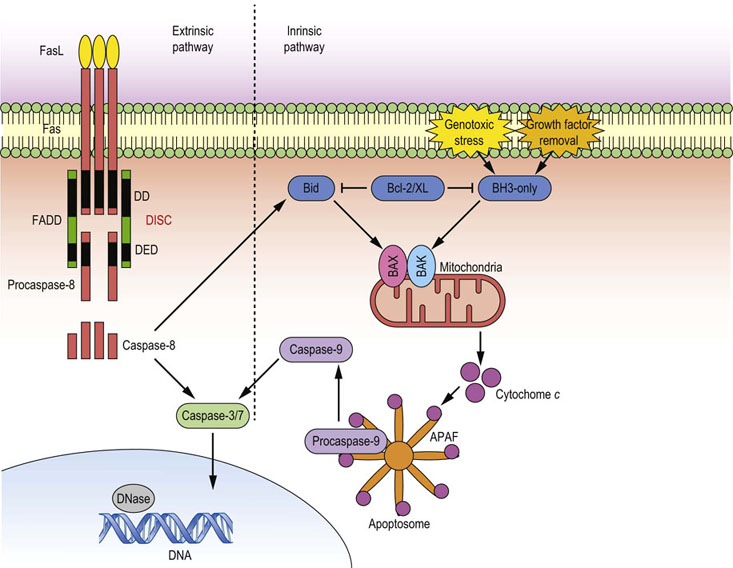
Fig. 42.8 Regulation of apoptosis.
Two main modes of apoptosis are induced through the ligation of death receptors, such as Fas, by growth factor deprivation, or genotoxic stress. Fas receptor ligation induces the extrinsic death pathway: the main initiator caspase is caspase-8. FasL binding causes receptor trimerization and formation of a macromolecular structure called DISC, which acts as a platform for caspase-8 activation. When activated, caspase-8 may directly cleave and activate effector caspase-3, or it may cleave BH3-only protein Bid, which acts directly on mitochondria, thus feeding into the second death pathway referred to as an intrinsic pathway. This pathway is initiated by the upregulation or activation of BH3-only family members, which relieve the inhibition mediated by Bcl-2/xL, thus allowing formation of the Bax/Bak pore on the mitochondrial outer membrane. This causes leakage of cytochrome c into cytoplasm, where it forms the apoptosome with APAF-1 and procaspase-9. Apoptosome is a platform for caspase-9 activation which, when cleaved, adopts active conformation, and can cleave effector caspase-3. Active caspase-3 then mediates bulk proteolysis either by activating other hydrolases, or by directly cleaving structural components. DISC, death-inducing signaling complex; Bid, BH3-interacting-domain death agonist; Bax, Bcl2-associated X protein; Bak, Bcl-2 homologous antagonist/killer; APAF-1, apoptotic protease activating factor 1; FADD, FAS-associated death domain protein; DD, death domain; DED death edffector domain.
Caspases
Caspases are cysteine proteases with aspartate substrate specificity
They are synthesized as inactive proenzymes (zymogens) referred to as procaspases. According to their role in the death pathways, caspases can be categorized as initiator or effector caspases.
Initiator caspases (caspase-2, -8, -9, -10) are synthesized as monomers and upon cell receiving a death signal they undergo activation, resulting from proximity-induced conformational changes and dimerization within multimeric complexes, as well as auto-proteolytic cleavage, which induces full enzymatic activity. Once cleaved/activated, caspases mediate proteolytic cleavage of other caspases in the death pathway cascade.
Effector caspase proenzymes are expressed as pre-formed dimers, which are activated by the direct proteolytic attack of initiator caspases. These effectors then execute the cell demise program by cleaving many vital cellular proteins (such as lamins, gelsolin), inducing cell cycle arrest, and disabling the initiation of homeostatic and repair mechanisms. All these events lead to the detachment of the cell from its surrounding tissue, dismantling structural components and ultimately flagging the phosphatidylserine (PS) ‘eat-me’ signal for phagocytosis. Overexpression of active caspases is sufficient to induce cellular apoptosis.
IAP gene family: its main function is to inhibit apoptosis
The inhibitor of apoptosis (IAP) gene family, composed of nine family members (X-linked IAP, cIAP1, cIAP2, melanoma IAP, IAP-like protein, neuronal apoptosis inhibitory protein, survivin, livin and apollon), is evolutionarily conserved from Drosophila through to humans. The main function of this gene family is, as their name indicates, to inhibit apoptosis by either directly blocking caspases and/or activating survival pathways via NFκB function. For example, X-linked IAP inhibits caspase-3, -7 and -9 by direct binding to the active pocket of caspase-3 and -7, while in the case of caspase-9 it prevents the dimerization necessary for full activation. On the other hand, cIAP1 and cIAP2 are positive regulators of both canonical and non-canonical pathways of NFκB activation. Survivin, in addition to inhibiting apoptosis, plays a role in cell cycle progression by blocking the activity of caspase-3, thus preserving the integrity of p21 within the survivin–caspase-3–p21 complex, and by mediating proper chromosome segregation as a part of the complex that binds chromosomes to kinetochores. It is not surprising, therefore, that IAP family members are found to contribute to tumor cell survival, cell invasion and metastasis in many human cancers. Interestingly, loss of cIAP1/cIAP2 is implicated in the development of multiple myeloma.
Bcl-2 gene family is composed of structurally related proteins that form homo- or heterodimers and act as positive or negative regulators of apoptosis
The Bcl-2 gene was initially discovered in a follicular B cell lymphoma as a protein that is constitutively expressed due to a t(14;18) chromosome translocation, which placed the Bcl-2 gene under the control of the immunoglobulin heavy chain promoter. Bcl-2 family members have traditionally been classified into three groups: pro-survival family members (Bcl-2, Bcl-xL, Bcl-W, Mcl-1); the pro-apoptotic BAX/BAK family; and the pro-apoptotic BH3-only proteins (BIM, BID, PUMA, NOXA, BAD, BIK). While the function of Bcl-2 appears to be to preserve the integrity of the outer mitochondrial membrane, pro-apoptotic family members BAX and BAK are responsible for inducing mitochondrial outer membrane permeabilization and subsequent release of apoptotic mediators (such as cytochrome c), leading to the caspase activation. Bcl-2 and Bcl-xL prevent apoptosis induction by inhibiting BAX and BAK, while BH-3-only family members prevent this inhibition by a direct binding to the Bcl-2 and other anti-apoptotic family members (Fig. 42.8).
There are alternative routes to apoptosis
The rupture of lysosomal membranes causes release and activation of lysosomal proteases (i.e. cathepsins), which either mediate direct proteolytic cleavage of cellular components or activation of the intrinsic pathway. The endoplasmic reticulum (ER) stress is generally caused by the accumulation of unfolded proteins in the lumen, giving rise to the unfolded protein response or irregular intracellular Ca2+ flux. These perturbations usually induce apoptosis through the intrinsic pathway. Accumulation of unfolded or misfolded protein aggregates is associated with many neurodegenerative disorders, including Alzheimer's, Huntington's and Parkinson's disease. For example, amyloid-β-protein aggregates and mutations in the ER-associated presenilin 1 have been associated with the development of the familial Alzheimer's disease. Interestingly, all these pathways may cross-talk and perform rather complex death programs in both the morphologic and biochemical sense.
Advanced concept box Intrinsic and extrinsic death pathway
The extrinsic apoptotic pathway is activated by the engagement of death receptors such as TNF family members (Fas, TNFR, TRAIL or TWEAK). For example, binding of homotrimeric FasL to Fas causes receptor oligomerization and assembly of an intracellular ‘death-inducing signaling complex’ (DISC). DISC contains procaspase-8, its adaptor/activator Fas-associated death domain (FADD) and its modulator cFLIP. Activation of caspase-8 occurs firstly by conformational change, which enables full enzymatic activity, and then by auto-proteolytic cleavage of the procaspase form. Cleaved caspase-8 molecules then leave the DISC and gain access to downstream targets, which include either effector caspases, such as caspase-3 and -7, or pro-apoptotic Bcl-2 family member BH3-interacting-domain death agonist (Bid). Cleaved Bid then feeds into the intrinsic pathway to amplify the death signal.
The intrinsic apoptotic pathway is also referred to as a Bcl-2 regulated pathway, due to the complex interplay between the pro- and anti-apoptotic Bcl-2 family members, which determines cell fate. It is usually activated either by the developmental cues, viral infections, DNA damage, and growth factor deprivation or other cytotoxic insults. These stress conditions increase the expression of BH3-only family members or, alternatively, their post-translational activation, depending on the context of death induction. Activated pro-apoptotic Bcl-2 family members relieve the inhibition of Bcl2-associated X protein (BAX) and Bcl-2 homologous antagonist/killer (BAK) at the pores of the outer mitochondrial membrane, enabling cytochrome c release from the mitochondrial intermembrane space into the cytoplasm. Cytoplasmic cytochrome c then binds to the apoptotic protease activating factor 1 (APAF1), allowing the formation of an apoptosome, which serves as a platform for caspase-9 activation (Fig. 42.8). Active caspase-9 then cleaves caspase-3 and/or -7, which, in turn, mediate a bulk proteolysis of vital cellular proteins, activate DNases, and orchestrate the demolition of the cell.
Autophagy
Autophagy is a degradation process of cellular components in which a part of cytoplasm is engulfed by a specific membrane, and the contents are subsequently degraded by lysosomal enzymes
Autophagy is a highly regulated homeostatic process, which plays a role in the turnover of long-lived proteins or in the elimination of damaged organelles such as mitochondria (mitophagy) or ER (reticulophagy). In addition to being associated with cell death, autophagy can also enable cells to survive starvation conditions in the circumstances of decreased availability of extra- and intracellular nutrients. In this case, autophagy induces catabolic processes, which generate metabolic substrates from ‘self’-components, thus allowing cells to meet their bioenergetic needs and initiate so-called adaptive protein synthesis in the time of scarcity.
Clinical box Deregulated apoptosis and autophagy can cause distinct pathologic conditions
Excessive apoptosis is linked to neurodegenerative diseases and immunodeficiency, while evasion of apoptosis is an important contributor to oncogenesis and the development of autoimmune diseases. Mutations (deletions/additions of one or a few nucleotides in the coding exons or splice sites) in the death receptor Fas lead to the defect in Fas-mediated apoptosis, which leads to the increased survival of activated lymphocytes causing autoimmune lymphoproliferative syndrome (ALPS). This rare inherited disorder usually presents in early childhood. Patients with ALPS have lymphoadenopathy, splenomegaly, and autoimmune cytopenias, and have increased risk for development of lymphomas. Aberrant expression and function of Bcl-2 family members is also implicated in the development of autoimmune diseases and cancer. Deletion of the Bcl-2 family member Bim in mice causes a systemic lupus erythematosus-like (SLE-like) disease, while chromosomal translocation of the Bcl-2 to the immunoglobulin heavy chain locus (t(14;18)) results in the constitutive expression of Bcl-2, leading to the development of follicular lymphoma.
Perturbations in autophagy induction and execution may also lead to a series of disorders and diseases. Monoallelic deletions in BECN1/ATG gene are tumorigenic in mice, and a decrease in expression of Beclin 1 is observed in human breast carcinoma. Loss of function mutations in the Pink1 and Parkin genes, which are regulators of mitophagy, are linked to familial Parkinson's disease in humans. Similar to the common neurodegenerative diseases are lysosomal storage disorders, which include over 40 genetic conditions, most of which are linked to the deficiency of lysosomal hydrolases. Reduced or impaired function of these enzymes leads to the accumulation of otherwise degraded macromolecules, while Huntington's and Parkinson's diseases are associated with accumulation of a mutant form of protein α-synuclein.
Autophagy is induced by a variety of stress stimuli, including nutrient and energy stress as well as hypoxia, redox stress, infections, ER stress, and mitochondrial damage
All of these stresses induce distinct signaling pathways that regulate autophagy. A characteristic signaling event that induces autophagy as a result of nutrient deprivation is the inhibition of mTORC-1 signaling and/or AMPK activation. The process of autophagy involves the formation of a membrane structure, termed a phagophore (most likely by the de novo synthesis), which encases part of the cytoplasm or a whole organelle, forming a double-membrane structure called the autophagosome. The autophagosome then may fuse with an endosome, creating an amphisome. Subsequently, these structures fuse with a lysosome, generating an autolysosome where acid hydrolases break down the inner membrane and the cargo. Digested macromolecule building blocks are then recycled back into the cytoplasm through protein channels referred to as permeases (Figs 42.9 and 42.10).
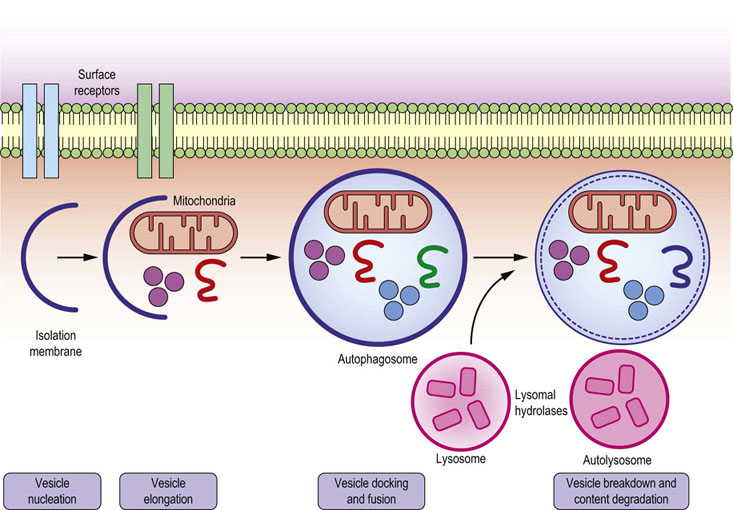
Fig. 42.9 Autophagy.
The process of autophagy starts by formation of isolation membrane (phagophore), which engulfs damaged mitochondria and/or misfolded proteins and forms the double-membraned vesicle autophagosome (autophagic vacuole). Autophagosomes then mature and fuse with lysosomes, thus creating autolysosomes, in which the inner membrane of autophagosome and its luminal content are degraded by the action of lysosomal acid hydrolases, such as cathepsins.
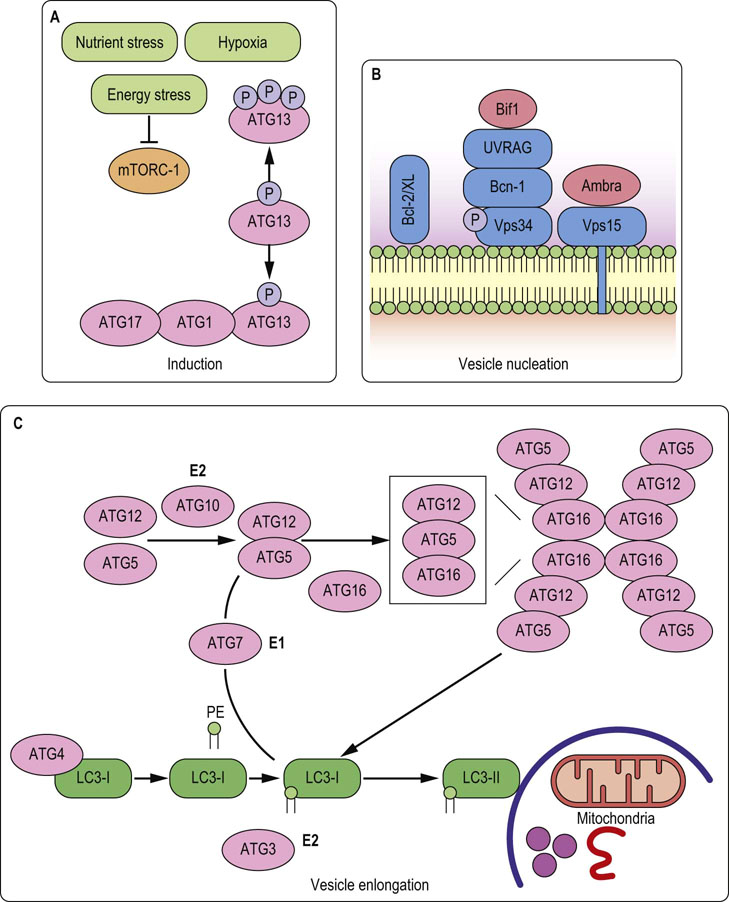
Fig. 42.10 Autophagy: important molecules.
A. Autophagy is usually induced by the inhibition of mTOR pathway, which relieves ATG13 protein from inhibitory phosphorylation and allows its association with ATG1. Within the ATG1–ATG13–ATG17 complex, ATG1 catalytic activity is increased and contributes to autophagy induction by an as yet unknown mechanism. B. Vesicle nucleation is initiated by the activation of Vps34, a mammalian class III PI3K, within a complex with Bcn-1, UVRAG and myristylated kinase Vps15. Active Vps34 catalyzes generation of PIP3 necessary for the autophagosomal membrane formation. C. Vesicle elongation and completion of autophagosome is regulated by the two ubiquitin-like conjugation systems: LC3 (ATG8) and ATG12. Phosphatidylethanolamine (PE) is conjugated to LC3 in a stepwise process controlled by the ATG4 protease, the E1-like enzyme ATG7 and E2-like enzyme ATG3. Binding of the PE to LC3 converts LC3 (LC3-I) from a soluble form into autophagosomal membrane-associated form (LC3-II). This conversion process is the foundation for many methods of autophagy detection. In the second pathway, ATG12 is conjugated to the ATG5 in a process mediated by the E1-like enzyme ATG7 and E2-like enzyme ATG10. ATG12-ATG5 then forms a multimeric complex with ATG16, which helps in targeting LC3 to the autophagosomal membrane, and accelerates lipid conjugation. ATG, autophagy-related gene; PI3K, phosphatidylinositol 3-kinase; UVRAG, UV radiation resistance-associated gene protein; LC3, microtubule-associated protein light chain 3. For more details see Advanced Concepts Box on the next page.
Apart from its roles in maintenance of cell homeostasis, autophagy plays a role in both innate and adaptive immunity. For example, autophagy is used for the elimination of intracellular bacteria such as Streptococcus pyogenes and Mycobacterium tuberculosis. Moreover, the Epstein–Barr virus nuclear antigen 1 (EBNA1) is processed through an autophagic pathway, and loaded on MHC class II molecules for presentation to the CD4+ T lymphocytes.
Advanced concept box Regulation of autophagy by atg gene family
Autophagy is a stepwise process whose main regulators are members of the autophagy-related gene (ATG) family, composed of 31 genes to date. Since their initial discovery in Saccharomyces cerevisiae, orthologues (genes in different species that evolved from a common ancestral gene) have been identified in mammals, indicating this gene family is conserved from yeast to humans.
The inhibition of mTOR, more precisely mTORC-1, relieves the inhibition of ATG13, which can associate with ATG1 kinase, thereby increasing its catalytic activity and inducing autophagy by a mechanism that is not well defined. Initial steps of autophagosome formation are mediated by the PI3K complex, which contains mammalian Vps34 (class III PI3K, which generates phosphatidylinositol-3-phosphate PIP3), BECN-1 and UV irradiation resistance-associated tumor suppressor gene (UVRAG; Fig. 42.10). The next step, vesicle elongation, is mediated by the two ubiquitin-like conjugation systems. Firstly, E1-like enzyme ATG7 and E2-like enzyme ATG10 conjugate ATG12 to ATG5. Secondly, protease ATG4, E1-like enzyme ATG7 and E2-like enzyme ATG3 conjugate phosphatidylethanolamine (PE) to microtubule-associated protein light chain 3 (LC3/ATG8). Binding of PE to LC3 is crucial for its translocation to the autophagosomal membrane (Fig. 42.10). The conversion of soluble (LC3-I) to the membrane-bound form (LC3-II) is widely used as an experimental marker for autophagy. After their formation, autophagosomes mature into the autolysosomes by fusion with lysosomes, which allows the process of degradation to be completed.
Cancer
Cells that develop mutations affecting normal regulation of the cell cycle are able to undergo unchecked proliferation, resulting in a loss of homeostatic regulation and the development of a tumor or neoplasm
As long as the neoplastic cells remain as an intact tumor, the tumor is considered benign and can be removed surgically. However, if further mutations allow such tumor cells to invade and colonize other tissues, creating widespread secondary tumors or metastases, the tumor is described as malignant and classified as a cancer. Each cancer is derived from a single cell that has undergone some germline mutation that allows it to outgrow its surrounding cells; by the time they are first detected, tumors typically contain a billion cells. Cancers are classified according to the tissue and cell type that they are derived from: those from epithelial cells are carcinomas, those from connective tissue or muscle cells are termed sarcomas, and those from the hematopoietic system are called leukemias. About 90% of human cancers are carcinomas, the five most common being those of the lung, stomach, breast, colon/rectum, and uterine cervix.
In the majority of cases, a single mutation is not sufficient to convert a healthy cell to a cancer cell; several rare mutations have to occur together
Mutations in DNA occur spontaneously at a rate of 10−6 mutations per gene, per cell division (even more in the presence of mutagens). Thus, since approximately 1016 cell divisions occur in the human body over an average lifetime, every human gene is likely to undergo mutation on about 1010 occasions. Clearly then, a single mutation is not normally sufficient to convert a healthy cell to a cancer cell; several rare mutations have to occur together, as demonstrated by epidemiologic studies showing that, for any given cancer, the incidence increases exponentially with age. It has been estimated that 3–7 independent mutations are usually required, leukemias apparently needing the fewest mutations and carcinomas the most.
Mutations need to occur in the appropriate cells in order to enable the neoplasm to develop, indicating that cell context has an important bearing on the type of cancer that subsequently develops
In addition to developing cancer-promoting mutations, the cell in which a mutation occurs must then be permissive to being a cancer-initiating cell. This relates to the cellular context in which an oncogene is expressed, and the properties bestowed upon a cancer cell by expression of a particular oncogene. In order to become oncogenic and promote cancer cell growth, the cell must possess self-renewal capacity. If oncogene expression occurs in a stem cell, then oncogene expression could inhibit the negative regulatory networks in place that would ordinarily stop cell growth and proliferation, thus generating a cancer stem cell. Cancers such as chronic myeloid leukemia (CML) and acute myeloid leukemia (AML) can arise from mutations in stem cells. However, it is not essential for the cell of origin of individual cancers to arise from stem cells. Indeed, cancers can arise from mutations arising in committed progenitor cells that enable those cells to acquire the potential to self-renew, therefore providing a cellular source for cancer.
Mutations that lead to the expression of established oncogenes do not necessarily lead to the development of cancer if it occurs in nonsusceptible cells
For example, the Philadelphia chromosome (t(9;22)) that generates a fusion protein BCR-Abl, encoding a constitutively active from of the protein tyrosine kinase (PTK) c-Abl (which is the causative mutation of the development of >95% CML cases), is detected at a very low level in circulating peripheral blood cells isolated from around 30% of healthy individuals. Studies have demonstrated that expression of BCR-Abl alone does not confer self-renewal properties on committed progenitor cells, indicating that secondary mutations are required to make committed progenitor cells cancerous. This finding suggests that the expression of oncogenes such as BCR-Abl within a stem cell environment can enable it to hardwire into the initiation of the neoplastic program.
Advanced concept box Oncogenic events are cell-context dependent
While genetic mutations are common, and expected, with loss of function in p53, PTEN and Rb, and gain of function with Ras, genome sequencing has also identified mutations in ‘unexpected’ genes in individual cancers. For example the Notch gene is mutated to form a constitutively active Notch protein, and is linked to the generation of around 50% of T lymphocyte- acute lymphoblastic leukemia (T-ALL) cases. However, activating mutations in Notch1 have also been identified in B lineage diseases including B lymphocyte chronic lymphocytic leukemia (B-CLL) and mantle cell lymphoma (MCL). Interestingly, an inactivating mutation of Notch has recently been identified in head and neck squamous cell carcinoma, indicating that while in hematopoietic lineages Notch acts as a tumor promoter, it can behave as a tumor suppressor in squamous cell carcinogenesis.
Alterations in gene expression have also been identified as a mechanism for tumor development/promotion. Indeed, expression patterns of specific protein kinase C (PKC) isoforms are dysregulated, possibly through alterations in epigenetic regulation of gene expression, in a number of cancers. In particular, PKCα is upregulated in breast, gastric, prostate and brain cancers, suggesting that it contributes to tumorigenesis. Moreover, PKCα expression levels have also been linked with the aggressiveness and invasive capacity of breast cancer cells. However, PKCα expression is downregulated in epidermal, pancreatic, colon cancers and B-CLL, suggesting that PKCα can also function as a tumor suppressor. Taken together, these findings indicate that the cell in which the mutation or modulation of expression of a protein occurs has a direct bearing on whether cancer formation is a likely outcome.
Tumor promoters: oncogenes
Mutations that lead to the uncontrolled proliferation of cancer cells can result either from disruption of the control of normal cell division or, alternatively, from a reduction in the normal processes of terminal differentiation or apoptosis. This distinction is reflected by the two major groups of genes targeted for mutation in cancer: oncogenes and tumor suppressor genes.
Oncogenes were first identified as viral genes that infect normal cells and transform them into tumor cells
The Rous sarcoma virus, which is a retrovirus that causes connective tissue tumors in chickens, will infect and transform fibroblast cells grown in cell culture. The transformed cells outgrow the normal cells and exhibit a number of growth abnormalities, such as a loss of cell contact-mediated inhibition of growth and loss of anchorage dependence of growth. In addition, the cells have a rounded appearance and can proliferate in the absence of growth factors. Moreover, the cells are immortal, do not senesce, and can induce tumor formation when injected into a suitable animal host, confirming their ability to self-renew.
The key to understanding cell transformation lies in the mutation of a normal cellular gene that controls cell growth
The use of mutant Rous sarcoma viruses that, despite multiplying normally, have lost the ability to transform host cells, showed that it was the Src gene that was responsible for such cell transformation. The breakthrough in our understanding of how this single gene could transform cells in culture came when it became apparent that the viral oncogene was a mutated homologue of a normal cellular gene. This gene is now called the c-Src proto-oncogene, and has been identified as a PTK signal transducer involved in the normal control of cell growth. As expression of this gene is not essential to the survival of the retrovirus, it is likely that Src was accidentally incorporated by the virus from a previous host genome and was somehow mutated in the process. In the case of the Rous sarcoma virus, the introns normally present in c-Src are spliced out and, in addition, there are a number of mutations causing amino acid substitutions, resulting in a constitutively active PTK.
Cell transformation can, however, also result from oncogenes that are not constitutively activated but, rather, are overexpressed in an abnormally high number of copies, as a consequence of the gene being under the control of powerful promoters or enhancers in the viral genome. Alternatively, for retroviruses, DNA copies of the viral RNA can be inserted into the host genome at or near sites of proto-oncogenes (insertional mutation), causing abnormal activation of these proto-oncogenes. In this situation, the altered genome is inherited by all progeny of the original host cell.
Clinical box mTOR in cancer and metabolic disorders
Owing to its vital role in regulating cell proliferation/survival and its intricate relationship with the phosphatidylinositol-3-kinase(PI3K) pathway, components of the mTOR pathway are often dysfunctional in many types of cancers and metabolic disorders. Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder that occurs as a result of inactivating mutations within the TSC1 or TSC2 genes. TSC is characterized by multiple benign tumors such as angiofibroma of the skin, lymphangioleiomyoma of the lungs, renal angiomyolipoma and astrocytoma of the brain.
In the context of the mTORC-1 axis, Tsc1/2 serve as a relay center for tumor microenvironmental cues. Under normal circumstances, hypoxia (via Hif1α), DNA damage (via p53) and nutrient deprivation (via LKB1 transcription factor) activate Tsc1/2 to regulate mTORC-1, thus controlling biosynthetic processes. These pathways are inactivated during tumorigenesis, usually by the cooperative action of oncogenic PI3K/PDK1 and Ras/MAPK pathways to reduce Tsc1/2 activity. Upregulation of mTORC-1 signaling leads to overactivated protein and lipid biosynthesis, which supports the bioenergetic needs of energy-demanding proliferating tumor cells. Enhanced protein synthesis often promotes increased expression of cell cycle regulators such as cyclin D1 and cyclin E, while constitutively active Akt contributes to the inactivation of cell cycle inhibitors p27 and p21. The role of mTORC-2 in tumorigenesis is still not very well defined; however, part of the mTORC-2 complex called Rictor is overexpressed in many gliomas. Higher expression promotes mTORC-2 complex assembly and activation, allowing cells to have higher proliferation and increased invasion. These events suggest that cancer could be considered a metabolic disorder.
Indeed, mTOR pathway deregulation has also been shown to contribute towards the development of various metabolic disorders such as obesity, nonalcoholic fatty liver and diabetes type 2. For example, in the hypothalamus, leptin relays signals via mTORC-1 for the reduction in food intake. Overactivation of mTORC-1 due to the high-fat diet may promote obesity by favoring resistance to the leptin-induced anorexic signals, thus promoting hyperphagia. Moreover, increased activation of mTORC-1 promotes adipogenesis and adipose tissue expansion, inhibits insulin signaling in skeletal muscles, liver and pancreas by promoting insulin resistance, and contributes to the induction of apoptosis in pancreatic β-cells by exhausting the homeostatic process of β-cell compensation.
Most human tumors are nonviral in origin and arise from spontaneous or induced mutations
Approximately 85% of human tumors arise as a result of point or deletion mutations. These mutations may be spontaneous or induced by carcinogens or radiation, resulting in overexpression or hyperactivity of the proto-oncogenes. Indeed, Ras, which has been found to be mutated to a constitutively activated form in approximately 25% of all tumors, appears to exert many, if not all, of its effects ultimately by upregulating concentrations of cyclin D and, hence, by stimulating cell cycle progression. This upregulation of cyclin D expression results from stimulation of the MAPK cascade by Ras, and the induction of the transcription factor AP-1, which regulates induction of cyclin D expression.
Whole-exome/genome sequencing of individual patients, utilized to determine the specific mutational landscape within cancer subtypes, has enabled links to be established between seemingly diverse cancers that result from similar genetic mutations
Of note, B-RafV600E mutation was recently found to be present in all in hairy cell leukemia (HCL) patients assessed. B-RafV600E is oncogenic in a number of tumors, including melanoma, encoding active B-Raf kinase leading to the constitutive activation of the MEK/ERK signaling pathway. This mutation has a major impact on cell cycle. Interestingly, now that the B-RafV600E mutation has been linked as a causal/driver event of HCL, it presents the opportunity for targeted therapies, such as PLX-4720, a specific inhibitor of active B-Raf, which would otherwise have not been considered for the treatment of HCL. Identification of specific driver mutations in a particular cancer cell opens up the possibility of treating patients with a targeted therapy towards that mutation.
Karyotyping of tumor cells has also shown that chromosomal translocation can bring the oncogene under the control of an inappropriate promoter. For example, in Burkitt's lymphoma, overexpression of the Myc gene occurs through its translocation into the vicinity of one of the Ig loci. Because Myc normally acts as a nuclear proliferative signal, overexpression of Myc induces the cell to divide, even under conditions that would normally dictate growth arrest.
Tumor suppressor genes: subversion of the cell cycle
Mutations in tumor suppressor genes are recessive and thus, mutations in both copies of the gene are usually required for transformation. As it is very difficult to identify the loss of function of a single gene in a cell, much of the initial information relating to tumor suppressor genes was obtained by studying a range of inherited cancer syndromes (Table 42.1).
Table 42.1
Selected inherited cancer syndromes
| Syndrome | Cancer | Gene product |
| Li–Fraumeni | Sarcomas, adrenocortical, carcinomas of breast, lung, larynx. Colon and brain tumors and leukemias | p53: transcription factor, DNA damage and stress |
| Familial retinoblastoma | Retinoblastoma, osteosarcoma | Rb1: cell cycle and transcriptional regulation |
| Familial adenomatous polyposis (FAP) | Colorectal cancer; colorectal adenomas, duodenal and gastric tumors, jaw osteomas and desmoid tumors (Gardner syndrome), medulloblastoma (Turcot syndrome) | APC: regulation of β-catenin, microtubule binding |
| Wiedmann–Beckwith syndrome | Wilms' tumor, organomegaly, hemihypertrophy, hepatoblastoma, adrenocortical cancer | P57/KIP2: cell cycle regulator |
| PTEN hamartoma tumor syndrome | Benign tumors: breast, thyroid, colorectal, endometrial and kidney cancers | PTEN: protein and lipid phosphatase, regulating Akt kinase and cell cycle |
| Neurofibromatosis type 1 (NF1) | Neurofibrosarcoma, AML, brain tumors | GTP-ase activating protein (GAP) for Ras |
| Hereditary papillary renal cancer | Renal cancer | MET receptor for HGF |
| Familial melanoma | Melanoma, pancreatic cancer, dysplastic neviatypical moles | P16 (CDK): inhibitor of cyclin-dependent kinase (CDK4/6) |
AML, acute myeloid leukemia; HGF, hepatocyte growth factor; KIP2, 57 kDa inhibitor of cyclin–CDK complexes; HGF, hepatocyte growth factor.
p53: guardian of the genome
The protein p53 plays a critical role in regulating the G1/S phase transition of the cell cycle and monitoring for DNA damage, and becomes activated upon sensing damage, stress and oncogenic signals to induce cell cycle arrest and/or death. Therefore it is perhaps unsurprising that p53 function is commonly disrupted in cancer cells. This can either be through inactivating mutations of p53 function directly, abrogating its transcriptional activity, or by dysregulating pathways responsible for p53 activation. The importance of p53 is highlighted in individuals who only have one functional copy of the p53 gene. People with this syndrome, called Li–Fraumeni syndrome, are predisposed to develop a wide range of tumors, including sarcomas, carcinomas of the lung, breast, larynx and colon, brain tumors and leukemias. This syndrome is rare, and tumor cells in affected patients exhibit defects in both copies of p53. Deletion of p53, in addition to allowing uncontrolled cell cycle progression, also permits replication of damaged DNA, leading to further carcinogenic mutations or gene amplification.
Phosphatase and TENsin homologue (PTEN)
The tumor suppressor PTEN is one of the most commonly inactivated proteins in sporadic cancer
As described above, PI3K-mediated signaling pathways are activated in response to a multitude of growth factor stimuli, resulting in the promotion of cell growth, survival and proliferation. The main protein responsible for attenuating PI3K activity and downstream pathways is the dual-specificity protein and lipid phosphatase PTEN. It reverses the activity of PI3K by dephosphorylating PIP3. The tumor suppressor PTEN is one of the most commonly inactivated proteins in sporadic cancer, resulting in sustained PI3K/Akt signaling and uncontrolled cell survival and proliferation. Mutations in PTEN have been identified in a wide range of cancers, including breast, thyroid, prostate and brain cancers. Interestingly, individuals with inherited germline mutations in PTEN, known as PTEN hamartoma tumor syndrome, develop benign tumors associated with breast, thyroid, colorectal, endometrial and kidney. However, these patients also carry an elevated lifetime risk of developing malignant cancers in these tissues. The increased susceptibility to cancer in cells carrying germline PTEN mutations underlines the importance of this protein as a tumor suppressor.
Clinical box Specific mutations that define a cancer therapy
In the majority of cases a number of mutations are required in to order to change a healthy cell into a cancer cell. One possible exception to this rule is chronic myeloid leukemia (CML), which in 95% of cases develops as a result of a translocation between chromosomes 9 and 22 (t(9;22)), to develop the Philadelphia chromosome, in the hematopoietic stem cell compartment. This type of translocation can also be found in 25–30% of acute lymphoblastic leukemia (ALL) cases and a minority of acute myeloid leukemia (AML) cases. The translocation forms a fusion product between the breakpoint cluster regions (BCR) and the Abl protein tyrosine kinase gene ABL Abl, resulting in the constitutive production of fusion protein BCR-Abl, which exhibits enhanced protein tyrosine kinase (PTK) activity. As Abl regulates a number of proteins involved with cell cycle, the result is that BCR-Abl expression increases cell division, leading to an overproduction of myeloid lineage cells. Moreover, BCR-Abl inhibits DNA repair mechanisms, leading to genomic instability, which enables cells to accumulate several additional mutations that precipitate disease progression. This transforms the disease from the chronic phase that is stable for several years, towards an acute/blast crisis phase.
BCR-Abl was one of the first proteins for which drugs were designed specifically to antagonize the signal transducer of interest, a so-called targeted therapy. Imatinib (Glivec) is a tyrosine kinase inhibitor (TKI) developed by Novartis to specifically inhibit the Abl kinase activity. While unable to completely eradicate the CML cells, it reduced their proliferation rate and delayed the onset of blast crisis. While imatinib is an important drug for the treatment of the majority of CML patients, a small percentage of patients are resistant, or become resistant to imatinib treatment, possibly due to acquired mutations within BCR-Abl, which has lead to the development of second- (e.g. dasatinib and nilotinib) and third-line therapies for CML. Of note, TKI therapies to date, have been unsuccessful at eradicating the CML stem cells, the root of the disease; therefore there is a risk of relapse in TKI-treated patients if the therapy is withdrawn or the patients develop chemoresistance.
Summary
Most proto-oncogenes and tumor suppressor genes have a function associated with signal transduction, mimicking the effects of persistent mitogenic stimulation, and thereby uncoupling cells from normal external controls.
These signaling pathways converge on the machinery that controls the transition of the cell through the G1 phase and prevent cell cycle exit.
Additional genes, many of which are targeted by cancer-specific chromosomal translocations and epigenetic regulation, lead to aberrant cell fate decisions that would normally induce apoptosis. The two tumor suppressor proteins, PTEN and p53, which have key roles in determining cell cycle progression and apoptosis, and the genes coding for these proteins, are most frequently disrupted in cancer cells.
Chiara Maiuri, M, Zalckvar, E, Kimchi, A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007; 8:741–752.
Dick, JE. Stem cell concepts renew cancer research. Blood. 2008; 112:4793–4807.
Hollander, MC, Blumenthal, GM, Dennis, PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011; 11:289–301.
Hotchkiss, RS, Strasser, A, McDunn, JE, et al. Cell Death. 2009; 361:157–183.
Laplante, M, Sabatini, DM. mTOR signaling in growth control and disease. Cell. 2012; 149:274–293.
Levine, AJ, Oren, M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009; 9:749–758.
Malumbres, M, Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009; 9:153–166.
Taylor, RC, Cullen, SP, Martin, SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008; 9:231–241.
Nature's Encyclopedia of Life Science. www.els.net/
Kimball's Biology Page. http://users.rcn.com/jkimball.ma.ultranet/BiologyPages/A/Apoptosis.html
KEGG – Human Cell Cycl. www.genome.jp/kegg/pathway/hsa/hsa04110.html