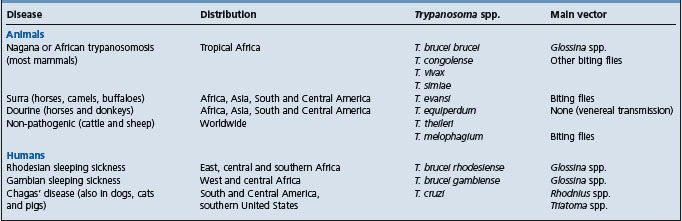
THEILERIOSES
Theilerioses are those tick-borne protozoan diseases associated with Theileria spp. in cattle, sheep, and goats as well as in wild and captive ungulates. The genus Theileria belongs to the Amplicomplexa group which includes Babesia, Toxoplasma, Neospora, Plasmodium, among others. The life cycle of Theileria spp. involves cyclical development in ticks to form sporozoites which, on being injected with tick saliva into the mammalian host, develop into schizonts in leukocytes and then piroplasms (merozoites) in erythrocytes. The diseases in ruminants are characterized by fever and lymphoproliferative disorders and are associated with varying degrees of leukopenia and/or anemia.
Theileria spp. are found throughout the world and their nomenclature and classification, though still controversial, are being gradually elucidated through molecular characterization. The important pathogens of cattle are restricted to certain geographical regions after which the diseases are named1-3 (Table 26.6). East coast fever (ECF) associated with T. parva and tropical theileriosis (or Mediterranean coast fever) associated with T. annulata are the most important and are dealt with separately below. Japanese bovine theileriosis associated with T. sergenti is probably next in importance.
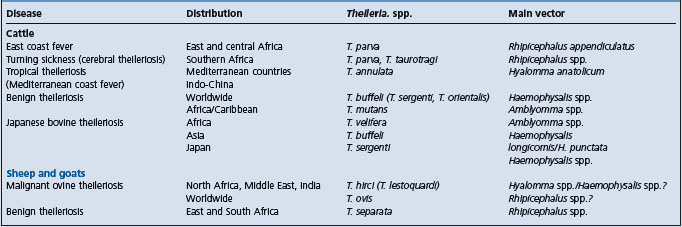
T. orientalis is responsible for oriental theileriosis, a milder disease than ECF or bovine tropical theileriosis, and hence also called benign theileriosis. It has been proposed that the species found worldwide and responsible for benign theileriosis be named T. buffeli (replacing T. orientalis in Asia and T. sergenti in Japan1) but genetic differences have been shown between T. buffeli and T. sergenti.4,5 Furthermore, T. sergenti causes a more severe disease in southeastern Asia, also called Japanese bovine theileriosis. Both T. buffeli and T. sergenti are transmitted by Haemophysalis ticks which occur in Europe, the Mediterranean basin, Asia, and Australia. Although this tick does not occur in the United States of America, T. buffeli was diagnosed in a herd of beef cattle in Missouri and was associated with severe clinical illness and death (by euthanasia) in a pregnant cow.6
In general, benign theileriosis is characterized by moderate to severe anemia in heavily parasitized cattle and moderate enlargement of lymph nodes. More severe clinical signs and economic losses have been reported from eastern Asia. The pathogenesis of the anemia is not clear but a hemolytic factor has been reported in the serum of acutely affected cattle.7 In addition, it has been shown that oxidative bursts of macrophages in experimentally infected cattle can damage red blood cells and that this may contribute to the anemia in Japanese bovine theileriosis.8 European breeds are more susceptible than zebu breeds. Even for the more pathogenic T. sergenti, native and crossbred cattle in Korea were found to be more resistant to infection than Holsteins.9 Furthermore, transplacental (vertical) transmission of T. sergenti from pregnant cows to calves has been reported.10 Transmission to the calves was confirmed by parasitological, serological, and polymerase chain reaction (PCR) assays. These are also the methods generally recommended for diagnosis.5 Calves used for the production of live vaccines against babesiosis and anaplasmosis should be free of benign theileriosis. In Australia, concurrent treatment with primaquine phosphate (six doses at 2 mg/kg) and halofuginone lactate (two doses at 1 mg/kg) was effective for this purpose.11
T. mutans, confined to Africa and the Caribbean Islands, causes a usually innocuous disease, but it may be manifested by fever, anorexia, and anemia. Another species, T. velifera, is associated with very mild theileriosis in tropical Africa. Both are transmitted by Amblyomma ticks. T. taurotragi of the eland antelope is generally non-pathogenic to cattle, but is one of the causes of cerebral theileriosis (turning sickness) in southern Africa (cerebral theileriosis can also be associated with T. parva). Parasitized lymphoblasts accumulate in cerebral, spinal, and meningeal arteries, with resultant thrombosis and infarction of affected organs. T. taurotragi is transmitted by Rhipicephalus spp.
The important pathogen of sheep and goats is T. hirci (synonym T. lestoquardi), the cause of malignant ovine theileriosis. The disease is enzootic from North Africa through the Middle East to India and China, approximately the same geographical region as bovine tropical theileriosis. Malignant theileriosis in sheep and goats is similar to bovine tropical theileriosis due to T. annulata. Like the latter, it is also transmitted by Hyalomma spp. but in China, the main vector is Haemophysalis spp. The disease can be acute, subacute, or chronic, depending on the resistance of the sheep or goats, and is seasonal, depending on availability of ticks. The acute disease is characterized by fever and very high mortality in 3–6 days.12,13 Anemia, jaundice, and enlargement of lymph nodes are characteristic, and both piroplasms and schizonts can be demonstrated in smears of blood and tissues, respectively. In subacute and chronic cases, signs are generally less marked except for anemia and emaciation. An indirect fluorescent antibody test is available. Parvaquone and buparvaquone may be used to treat early cases. Benign ovine theileriosis is caused either by T. ovis or by T. separata in Africa. Piroplasms are found in blood but there are no overt clinical signs.
In general, the pathogenesis of various forms of theileriosis is dependent on the production of schizonts in lymphocytes and piroplasms in erythrocytes. Thus, T. parva, T. annulata, and T. hirci produce numerous schizonts and piroplasms and are very pathogenic; T. mutans, T. buffeli, and T. ovis rarely produce schizonts but may cause varying degrees of anemia when piroplasms are many in red blood cells; and with T. velifera and T. separata, no schizonts have been described, the parasitemia is usually scanty and the infection is mild or subclinical.
1 Uilenberg NP, et al. Trop Anim Health Prod. 1996;28:81.
2 Coetzer JAW, Tustin RC, editors. Infectious diseases of livestock, 2nd edn., vol 1. Cape Town: Oxford University Press, 2004;448-501.
3 Brown CGD. Theileriosis. In: Sewell MMH, Brocklesby DW, editors. Handbook on animal diseases in the tropics. 4th edn. London: Bailliére Tindall; 1990:183-199.
4 Chansiri K, et al. Vet Parasitol. 1998;79:143.
5 OIE. Manual of diagnostic tests and vaccines for terrestrial animals. http://www.oie.int/eng/normes/mmanual/A_00062.htm, 2004. chapter 2.3.11. 5th edn.
6 Stockman SL, et al. Vet Pathol. 2000;37:11.
7 Hagiwara K, et al. Parasitol Res. 1995;81:470.
8 Shiono H, et al. Parasitol Res. 2003;89:228.
9 Kim GH, et al. J Protozool Res. 1999;9:103.
10 Baek BK, et al. Can J Vet Res. 2003;67:278.
11 Stewart NP, et al. Trop Anim Health Prod. 1990;22:109.