Chapter 35 Diseases associated with the inheritance of undesirable characters
INTRODUCTION 1938
DISEASES CHARACTERIZED BY CHROMOSOMAL ANOMALIES 1939
INHERITED DEFECTS OF THE BODY AS A WHOLE 1940
INHERITED DEFECTS OF THE ALIMENTARY TRACT 1943
INHERITED DEFECTS OF THE CIRCULATORY SYSTEM 1944
INHERITED DEFECTS OF THE URINARY TRACT 1948
INHERITED DEFECTS OF THE NERVOUS SYSTEM 1948
INHERITED DEFECTS OF THE MUSCULOSKELETAL SYSTEM 1957
INHERITED DISEASES OF JOINTS 1961
INHERITED DISEASES OF MUSCLES 1962
INHERITED DEFECTS OF THE SKIN 1973
MISCELLANEOUS INHERITED DEFECTS 1976
Introduction
GENERAL
Genetic disorders are a small but important cause of wastage in farmed animals. Most occur in pure-bred animals and are inherited as autosomal recessive traits because dominant disorders tend to be self limiting, or affected animals are excluded from the breeding pool. Incomplete dominants occasionally occur in which there are three potential phenotypes, i.e. normal, affected, and more severely affected. The classic example is in the original Dexter cattle where the slightly dwarfed Dexter phenotype is dominant to the normal and is selected for. Animals homozygous for the gene abort with a non-viable ‘bulldog’ type fetus.1,2 Sex-linked disorders may occur but are uncommon. Some monogenic disorders may arise de novo due to new mutations of germ plasm. Those with a dominant mode of inheritance are present in offspring of the animal in question, usually affecting genes for structural proteins such as collagen. A new mutation should be considered with disorders such as osteogenesis imperfecta or skin fragility.3 The proportion of offspring with the defect may vary depending on what stage of gametogenesis the mutation occurred.
In-breeding, knowingly or unknowingly practiced, is an important feature in the manifestation of most outbreaks of a recessive disorder. Founder effect is an aspect of this that has been important when new breeds have been introduced to a country by importation of genetic material from a small number of individuals. Artificial breeding on a large and international scale has sometimes exacerbated this, particularly in cattle.
Genetic disorders may be manifested as disease or bodily malformation. When diagnosed, an entity may reflect the tip of an iceberg only and it can be expected that many other cases go undiagnosed. Spread across an industry their economic importance may be limited, but as particular disorders tend to be concentrated in certain herds/flocks they may have considerable importance to an individual breeder, particularly those involved with pedigree breeding. Animal welfare is also a concern to be addressed, being driven by a greater awareness of ethical standards in livestock production and by potential market access requirements.
The two main problems for the clinician investigating a suspected inherited disorder are to confirm a primary genetic cause and then to institute control in a cost-effective manner.
DIAGNOSIS
For a number of inherited diseases or malformations known to occur in a breed, morphology or histopathology may be so characteristic as to be essentially pathognomonic. However, for some disorders environmental agents (teratogens) may cause similar morphological anomalies, e.g. arthrogryposis, so care should be taken. Pedigree analysis may help if there are sufficient animals of known breeding to show that the incidence of the disorder follows a Mendelian pattern. However, in many herds/flocks animals may be closely related and pedigree analysis can sometimes be misleading and produce a fictitious relationship between inheritance and disease. As the biochemical anomaly is now known for many diseases, or perhaps can be deduced from histopathological lesions, laboratory tests may confirm a presumptive diagnosis. Test mating of a sire to daughters, related females, or females that have given birth to an affected animal is the ultimate confirmation of the genetic nature of a disorder, provided the appropriate numbers of progeny are generated. Disproving a genetic cause of a disorder may be as important as proving it. Matings of a sire to produce a minimum of 24 progeny from his daughters or 12 from putative heterozygotes (females that have given birth to affected individuals) are usually considered satisfactory numbers to exclude a likely inherited cause if no affected individuals are born (P<0.5). The birth of a proportion of affected offspring is strong evidence of inheritance. The use of super-ovulation and embryo transfer techniques may facilitate this, particularly if insufficient daughters or putative heterozygotes are available. The time to accomplish this may be decreased by caesarian section of the surrogate dams if the defect can be detected in the fetuses.
The degree of in-breeding is an important indicator of whether a congenital disorder is inherited or not. Consistency of the defect is a characteristic of inherited disorders but there may be some variation in age of onset or expressivity of lesions. Other epidemiological factors include the occurrence of the defect over more than 1 year and occurrence or repetition of it in the same mating group, but not in others on the property.
CONTROL OF INHERITED DISEASE
Appropriate control measures may vary, depending on the importance of the disorder and may be aimed at the herd/flock level or at the breed as a whole. It may be prospective but, at the farm level, it is mainly reactive with the purpose of preventing further losses by immediate action. This should include not breeding from putative heterozygous sires or females which should preferably be culled. Replacement sires are best acquired from another breeder but, if the defect is common in the breed, then the risk may remain and cross breeding with a sire from another breed may be considered if the type of farm operation permits it. If a test is available for detecting heterozygous animals then this can be used in new sire selection.
Control of genetic disorders in pedigree herds is more complex and to be effective depends on ability to detect heterozygotes or prove animals do not carry the recessive gene in question. Test mating is time consuming, expensive, and of limited application. The explosion of knowledge concerning the biochemical and molecular genetic basis of inherited diseases across species has opened up effective means of diagnosing genotype for many of them. Control may be at an individual herd/flock level or applied to all at risk. It is best instigated with the help of breed societies who may exert control over the fate of animals diagnosed as heterozygous through control of registrations. Apart from the accuracy of genetic tests in genotype diagnosis, there is the added advantage that particularly valuable animals may be kept within the herd/flock for breeding as their offspring can in turn be tested as normal or heterozygous.
The first generation of tests for heterozygotes was biochemical being based on knowledge of the enzyme deficiency and the gene dosage phenomenon. Heterozygous animals having one normal and one mutant gene have enzyme values midway between normal and diseased values, although there may be some overlap. Supplementary tests or knowledge of the parents’ genotype may assist with clarification of equivocal results. Such tests were used to control the economically important lysosomal storage diseases α-mannosidosis in Angus and Murray Grey cattle in New Zealand and Australia4 and Glycogen storage disease type II in Shorthorn and Brahman cattle in Australia.5 These have now given way to more accurate second-generation technology based on DNA for these diseases as well as a number of others.6-8 Such tests may be performed on blood samples but are increasingly being done on hair roots.
The genome for the major farm species is essentially known and, given the will and enough affected families, the technology exists to develop tests for most disorders. If the disorder in question is poorly defined biochemically, then tests are still possible by finding a closely linked polymorphic gene marker. This is usually the first step in investigating the molecular genetics of an unknown disease but it is expensive. In contrast, if a candidate gene can be deduced from the pathology of the disease, then it is much simpler to define the mutation and through polymerase chain reaction (PCR) technology develop a DNA based test.
Artificial breeding techniques have the capacity to spread undesirable genotypes widely before they are recognized. Many artificial breeding organizations involved in the dairy industry prospectively screen for genetic disorders by mating prospective sires over a proportion of their daughters. This is possible because of the time taken to prove a sire before he enters the industry on a large scale.
ONLINE MENDELIAN INHERITANCE IN ANIMALS (OMIA)
Online Mendelian Inheritance in Animals (OMIA) is a database of genes, inherited disorders and traits in animal species (other than human and mouse) authored by Professor Frank Nicholas of the University of Sydney, Australia, with help from many people over the years. The database contains textual information and references, as well as links to relevant records from OMIM, PubMed, Gene, and soon to be NCBI’s phenotype database.
Agerholm JS, et al. Investigations on the occurrence of hereditary diseases in the Danish cattle population. Acta Vet Scand. 1993;34:245.
Healy PJ. Testing for undesirable traits in cattle. J Anim Sci. 1996;74:917.
Huston K. Hereditability and diagnosis of congenital abnormalities in food animals. Vet Clin North Am: Food Anim Pract 9.1. 1993:1.
Jolly RD, Healy PJ. Screening for carriers of genetic diseases by biochemical means. Vet Rec. 1986;119:264.
Jolly RD, Blair HT, Johnstone AC. Genetic disorders of sheep in New Zealand: A review and perspective. NZ Vet J. 2004;52:52-64.
Nicholas FW, Harper PAW. Inherited disorders; the comparative approach. Aust Vet J. 1995;73:64.
1 Harper PAW, et al. Aust Vet J. 1998;76:199.
2 Harper PAW. Proc. First World Congress on Dexter Cattle. (Ed., A. Sheppy), The Dexter cattle Society, Dulverton, U.K. 1998; 92.
3 Arthur DG, et al. New Z Vet J. 1992;40:112.
4 Jolly RD. New Z Vet J. 2002;50;Suppl).. 90:2002.
5 Healy PJ. Diagnosis of genotypes for generalized glycogenosis in cattle. Biochem Med. 1982;28:224.
6 Berg T, et al. Res Vet Sci. 1997;63:279.
Diseases characterized by chromosomal anomalies
The principal advance in cytogenetics in agricultural animals has been in the field of fertility in cattle but freemartinism and structural translocations are the only groups to have been well documented. Other chromosomal abnormalities in other species are recorded but not in sufficient numbers to make a coherent account possible; details of them should be sought in the theriogenology literature. Besides the many identifications of individual infertile males and females by this means there is the probability that chromosomal aberrations characterized by aneuploidy are the cause of many stillbirths and multisystemic anomalies that make neonates non-viable.1,2 A true hermaphrodite in a horned goat with 60 XX/60 XY chimerism has been described.3
Chromosomal analysis techniques are based on tissue culture using leukocytes collected in a highly aseptic manner. The blood is placed in tissue culture medium containing a stimulant to cell division. After brief incubation, cell division is arrested by the addition of a cytotoxic agent and the cells are then swollen osmotically by treatment with a hypotonic solution, then fixed and dried onto slides and stained. Microscopic examination of dividing cells allows the total number of chromosomes in each cell to be counted, and the sex chromosomes and abnormal chromosomes identified. The individual chromosomes are cut out from photographic prints, paired, and stuck onto cards to create the document used for identification – the karyotype. The number of cells subjected to chromosomal analysis in each case is usually 10–20, but 50 are recommended for satisfactory accuracy.
FREEMARTINISM IN CALVES
A freemartin is defined as a sterile female partner of a pair of heterosexual twins. In cattle, 92% of females born co-twins to males are freemartins.
In normal calves the chromosomal identification of females is 60 XX (60 chromosomes, both X chromosomes) and of males is 60 XY (the Y being smaller and not readily paired with its opposite X chromosome).
The freemartin is the classical example of the chimera in cytogenetics. They are the individuals which contain two or more cell types which originated in separate individuals. The only way in which chimera can develop is via the fusion of circulations or zygotes in utero. Sex chromosome chimerism is also reported in goats, sheep, and pigs, and, although the male partners of female twins are usually anatomically normal, they often have reduced fertility. Bulls born co-twin with freemartin females may also be chimeric and have low reproductive efficiency.
The diagnosis of freemartinism has been based on physical examination, karyotyping, or blood typing and each has its limitations. There is variation in the degree of reproductive tract abnormalities in freemartins. The external genitalia may appear normal, the vulval hair may be coarser than usual or the clitoris may be enlarged. The vagina is generally expected to be shorter than normal. The cervix, uterus, uterine tubes, and ovaries may be absent, present in underdeveloped form, or may appear normal on rectal palpation.
Special cytogenetic techniques are also available which facilitate the diagnosis of freemartinism in a female calf of a male–female twinning. In freemartins (phenotypically female, but carrying also male cells) there is a mixture of mostly 60 XX chromosomes to a cell, and a small proportion of 60 XY cells. A large number of cells need to be analyzed if only the freemartin calf is available because the proportion of abnormal cells present may be as low as 2%. It is, however, possible to make a diagnosis on the examination of 10–20 cells, provided the male twin is also analyzed; the female may have very few XY chromosomes but the male will have a very high proportion of XX chromosomes. The technique is much more accurate than blood group analysis, or clinical observations of a short vagina, enlarged clitoris and the presence of a vulval tuft of hair. Karyotyping is a definitive method of freemartin diagnosis but it is tedious, time consuming, and expensive. Blood typing analysis may be performed on both the male and female co-twins in order to demonstrate two blood group populations, but it is expensive and requires blood samples from both co-twins.
The polymerase chain reaction (PCR) method of freemartin diagnosis using sex-specific DNA sequences is rapid, accurate, relatively simple, and inexpensive to perform and a blood sample is required only from the female co-twins.4 It allows for the accurate decision of freemartinism down to a level of 0.05% of male chimeric cells present.
CHROMOSOMAL TRANSLOCATIONS IN CATTLE
When two chromosomes which have previously been broken have fused to form a morphologically distinct chromosome this is known as a translocation. It is further identified by the chromosomal series involved. Thus a 1/29 translocation represents a fusion between a chromosome of each of the pairs numbered 1 and 29.
Translocation 1/29 has been identified in many breeds of cattle and has been associated with significant reductions in the fertility of cows bred by artificial insemination (AI) services. Early embryonic death occurs in embryos produced by fertilization of affected gametes or fertilization of normal gametes by spermatozoa carrying the 1/29 translocation. There is no abnormality of serving behavior or semen quality. The translocation has been shown to be inherited in most European beef breeds including the Blonde d’Aquitaine, Swedish Red and White, Charolais,5 Danish Limousin,6 British Friesian7 and Red Poll breeds and in the wild British White cattle. In Bolivian Creole cattle breeds, the Creole-like cattle, the average frequency was 10.42% with a variation from 0 to 28.2%.8 In contrast, Yacumeno and Creole-type cattle did not show the centric fusion. The highly significant differences between Creole cattle breeds in relation to the 1/29 translocation could be the consequence of factors such as founder group, genetic drift, and selection. The low frequency observed in the Saavendreho Creole dairy cattle might be due to breeding under a more intensive system, and selection according to milk yield and fertility traits. The frequency of affected animals in a breed may vary between 1 and 20%. Karyotyping and culling of abnormal bulls in most artificial breeding centers has reduced the impact of the defect.
Translocations 1/21, 2/4, 14/20, and 13/2 have also been identified in bulls, the 1/21 in Holstein Friesian cattle,9 and the latter two seeming to be widespread in Simmental cattle.10 None of them has been linked with a disease but it is becoming accepted practice not to use such animals for artificial insemination, and in some countries to refuse their importation.
A cytogenetic survey of Holstein bulls at a commercial AI unit to determine the prevalence of bulls with centric fusion and chimeric anomalies found that chimeric fusion is extremely rare in Holstein blood lines available by AI in the United States.11 However, chimeric bulls are more common and reportedly have decreased reproductive performance. Because of the possibility of de novo onset of chimeric fusion at any time, early cytogenetic screening should be encouraged for prospective bulls intended for AI programs.
Translocation 27/29 is suspected of being associated with reduced fertility in Guernsey cattle. These and other abnormalities of chromosomal structure were detected in an examination of a large number of infertile dairy heifers.
CHROMOSOMAL TRANSLOCATIONS IN SHEEP
The literature on chromosomal aberrations in sheep has been reviewed.12 Centric-fusion (Robertsonian) translocations have been described in sheep in New Zealand. There is no evidence to suggest that these centric-fusions, in a variety of combinations, affect the overall total reproductive performance of domestic sheep, as unbalanced spermatids failed to mature and take part in fertilization.
Jolly RD, Blair HT, Johnstone AC. Genetic disorders of sheep in New Zealand: A review and perspective. New Z Vet J. 2004:52-64.
McFeely RA. Chromosomal abnormalities. Vet. Clin. North Am. 1993;9.1:11.
Weber AF, et al. Low fertility related to the 1/29 centric fusion anomaly in cattle. J Am Vet Med Assoc. 1989;195:643-646.
1 Coates JW, et al. Can J Vet Res. 1988;52:258.
2 Schmutz SM, et al. J Vet Diagn Investig. 1996;8:91.
3 Batista M, et al. Can Vet J. 2000;41:662.
4 Ennis S, et al. Res Vet Sci. 1999;67:111.
5 Buoen LC. Can Vet J. 1988;29:455.
6 Nielsen JS. Christensen K Dansk. Veterinaertidssk. 1990;73:1036.
7 Wilson TD. Vet Rec. 1990;126:37.
8 De Luca JC, et al. Therigenol. 2002;58:1273.
9 Miyake Y, et al. J Vet Med Sci. 1991;53:113.
10 Weber AF, et al. J Am Vet Med Assoc. 1992;200:1216.
Inherited defects of the body as a whole
Many inherited defects, other than those listed in this section, probably have a metabolic defect as their basic cause, for example the abiotrophies, but they are listed elsewhere because of lack of certainty about their exact cause. Three known inherited enzyme deficiencies – bovine citrullinemia, bovine protoporphyria – are described in system groupings where their clinical signs are most prominent. Inherited vitamin C, D, and, E deficiencies are dealt with in the chapter on nutritional deficiency diseases. Inherited immune deficiencies are listed in this section.
DEFICIENCY OF UMP SYNTHASE (DUMPS)
This is a partial deficiency of an enzyme which is involved in the conversion of orotate to uridine 5′-monophosphate (UMP) as a step in the synthesis of pyrimidine nucleotides. It is recorded at a high prevalence in Holstein Friesian cattle in the US and is characterized by an autosomal recessive form of inheritance and the secretion of high levels of orotate in the milk. Heterozygous animals have a partial deficiency of UMP synthase1; they have no individual or herd clinical abnormalities but can be detected biochemically by their half-normal levels of erythrocyte UMP synthase. Bovine homozygotes die at about the 40th day of pregnancy.2 Embryonic mortality is the only form of loss.
INHERITED GOITER
This disease is recorded in Merino sheep, Afrikaner cattle, crossbred Saanen dwarf goats, Boer goats, possibly Poll Dorset sheep, and pigs,1 and appears to be inherited as a recessive character. The essential defect is in the synthesis of abnormal thyroid hormone leading to increased production of thyrotropic factor in the pituitary gland, causing in turn a hyperplasia of the thyroid gland. In Afrikaner cattle the defect stems from an abnormality of the basic RNA, and heterozygotes can be identified by blot hybridization analysis.2
Clinically in sheep there is a high level of mortality, enlargement of the thyroid above the normal 2.8 g, but varying greatly up to 222 g, and the appearance of ‘lustrous’ or ‘silky’ wool in the fleeces of some lambs. Other defects which occur concurrently are edema and floppiness of ears, enlargement of, and outward or inward bowing of, the front legs at the knees, and dorsoventral flattening of the nasal area. The thyroglobulin deficiency in the neonatal lamb may result in defective fetal lung development and the appearance of a neonatal respiratory distress syndrome; there is dyspnea at birth.
The clinical picture in goats is the same as for lambs. It includes retardation of growth, sluggish behavior, rough, sparse hair coat, which worsens as the goats get older, and a thick scaly skin.
In Afrikaner cattle most of the losses are from stillbirths or from early neonatal deaths. Some are caused by tracheal compression from the enlarged gland. It is the calves with the largest glands that have the greatest mortality. In these cattle there may be a concurrent inherited gray coat color, a defect in a red breed.
In pigs hairless and swollen piglets with enlarged thyroid glands occur, in the proportions with normal piglets consistent with an autosomal recessive mode of inheritance.
INHERITED IMMUNODEFICIENCIES
Many of these diseases have been previously characterized as diseases of other body systems but are now correctly classified by the wider acceptance of clinicopathological examinations, especially those involving immunological studies. The group is probably still in a period of transition (see Chapter 9).
BOVINE LEUKOCYTE ADHESION DEFICIENCY (BLAD)
This granulocytopathy is inherited as an autosomal recessive trait in Holstein-Friesian cattle. Homozygotes are not viable because of their low resistance to infection. Heterozygotes are unaffected. Signs are observed first between 2 weeks and 8 months, and are characterized in most cases by bouts of infectious disease, e.g. persistent fever, diarrhea, cough, dyspnea, delayed wound healing, and stunted growth.1 Some cases exhibit a striking periodontal gingivitis with marked retraction of the gingiva, and severe resorption of mandibular bone causing premature teeth loss.2 In a small proportion of cases the signs are limited to unthriftiness.3 Severe ulcers on oral mucosa, severe periodontitis, loss of teeth, chronic pneumonia, and recurrent or chronic diarrhea are common.1
The clinical pathology is characterized by a severe and persistent neutrophilia, without a left shift, and a significantly increased cellularity of bone marrow.4 At necropsy there are very large numbers of intravascular neutrophils in all tissues, especially the spleen, but not in infected tissues, which may include bronchopneumonia, pseudomembranous or necrotizing enteritis, and granulomatous gingivitis.5 Intestinal ulcers are an essential part of the pathogenesis of the disease in chronically affected animals which receive intensive medical care.6 Affected animals are stunted and unlikely to live as long as 2 years.
The gene frequency is widespread in the Holstein-Friesian breed. The genetic basis for the disease is a single point mutation in the gene coding for CD18, a subunit of the beta3,1 integrins, surface glycoproteins which are important to cell adhesion processes, causing a deficiency of adhesion on the surface of leukocytes.7 Neutrophils from BLAD cattle have impaired expression of β2 integrin (CD11a, b, c/CD 18) of the leukocyte adhesion molecule. The biochemical basis for the disease is a deficiency of interaction between receptors on the leukocytes with adhesion glycoproteins in the mediation of immunological functions.8 A polymerase chain reaction test is available for the detection of heterozygotes9 and an eradication program is operating in Japan10 and the US.11 Heterozygotes have poorer feed utilization and growth rates than non-carriers of the inheritance.12 Control of BLAD in Holstein cattle requires publishing the genotypes and avoiding the mating between BLAD carriers which is successful.1 Heterozygote calves are not affected.13
1 Nagahata H. J Vet Med Sci. 2004;66:1475-1482.
2 Nagahata H, et al. Can J Vet Res. 1995;59:316.
3 Agerholm JS, et al. Acta Vet Scand. 1993;34:237.
4 Nagahata H, et al. Am J Vet Res. 1995;56:167.
5 Stadler P, et al. J South Afr Vet Assoc. 1993;64:172.
6 Ackerman MR, et al. Vet Pathol. 1996;33:273.
7 Mirck T, et al. Cell Mol Biol. 1995;41:695.
8 Worku M, et al. Am J Vet Res. 1995;56:435.
9 Tammen I, et al. Res Vet Sci. 1996;60:218.
10 Nagahata H, et al. World Assoc. Buiatrics, 19th Congress, 1996; 1–3, 489.
11 Powell RL, et al. J Dairy Sci. 1996;79:895.
12 Jorgensen JM. Madsen P Acta Agric Scand Section A. Animal Sci. 1997;47:1.
INHERITED DEFICIENCY OF LYMPHOCYTE MATURATION (LETHAL TRAIT A46, PARAKERATOSIS, ADEMA DISEASE)
This defect is recorded in Black Pied Danish cattle but probably occurs in a number of European breeds of cattle, including Friesian-type cattle, and in beef Shorthorn calves in the US.1 It is a defect of lymphocyte maturation and is inherited as an autosomal recessive character.
Calves are normal at birth and signs appear at 4–8 weeks of age; untreated animals die at about 4 months of age. There is exanthema and loss of hair, especially on the legs, parakeratosis in the form of scales or thick crusts around the mouth and eyes, under the jaw, and on the neck and legs, and a very poor growth rate. Lymphocyte numbers and function are reduced when the patient is in a zinc-deficient state,2 and antibody responses are suppressed.
At necropsy the characteristic skin lesion is acanthosis and hyperkeratosis and there is atrophy of the thymus, spleen, lymph nodes, and gut-associated lymphoid tissue.
There is a significant response to oral treatment with zinc (0.5 g zinc oxide/day) and an apparently complete recovery can be achieved in a few weeks if treatment is continued. The disease reappears if treatment is stopped. The dose rate needs to be increased as body weight increases. It is thought that the disease is an inherited excessive requirement for zinc and that the thymic hypoplasia is due to the dietary deficiency. Absorption studies with radioactive zinc have shown that there is impaired absorption of the element.
INHERITED DEFICIENCY OF IMMUNOGLOBULIN SYNTHESIS
An inherited complete deficiency of IgG2 occurs in Red Danish cattle at a low level of incidence. Affected animals are unusually susceptible to severe infections including gangrenous mastitis and pneumonia.
CHEDIAK–HIGASHI SYNDROME
This inherited disease occurs in humans, in mink, and in Hereford, Japanese Black, Brangus cattle,1,2 and possibly other breeds of cattle. Clinically affected animals grow poorly; they are incomplete albinos with generalized oculocutaneous hypopigmentation, e.g. pale gray hair, ocular iridal and fundic hypopigmentation, photophobia and lacrimation, and have anemia, enlarged, edematous lymph nodes, and a defect in immune defense mechanisms so that they often die of septicemia. Their average life span is about 1 year. The immunological defect has been identified as one of insufficient bactericidal activity within abnormal leukocytes. The clinical, morphologic, and biochemical characteristics of Chediak–Higashi syndrome in Japanese cattle has been described.2
A mutation in the Chediak–Higashi 1/LST gene is likely responsible for the disease in Japanese Black cattle.3 The LYST gene responsible for the mutation has been cloned.4
The disease is readily diagnosed by the detection of anomalous enlarged cytoplasmic granules in neutrophils, lymphocytes, monocytes, and eosinophils. The granules are swollen lysosomes, and the disease is a lysosomal storage disease. There is also a defect in blood clotting, and this has been identified as a metabolic defect within structurally abnormal platelets. The platelets have a storage pool deficiency of dense granules and produce much less serotonin, ATP, and ADP than normal platelets. The platelets also fail to aggregate normally in response to the presence of collagen.5 The disease is conditioned by a factor inherited as a single autosomal recessive.6
A DNA diagnostic system using allele-specific PCR for detection of the nucleotide substitution has been developed as an effective DNA diagnostic aid.7
PSEUDOALBINISM AND LETHAL WHITES
There are a number of forms of pseudoalbinism in domestic animals. There is a non-lethal form in cattle and a lethal dominant in horses in which 25% of conceptions produced by mating dominant white horses die in utero in early gestation. The only pigment in the affected foals is in the eyes.
The disease in cattle occurs in Angus, Brown Swiss, Holstein, and Hereford cattle. The Angus cattle have a brown coat and two-tone irises with an outer pale brown ring and an inner blue one. There appears to be no defect in digestion or metabolism. Hereford incomplete albinos have the Chediak–Higashi syndrome. The other breeds do not appear to have defects other than in pigmentation and the defect in Angus is probably more accurately called ‘oculocutaneous hypopigmentation’. They do have one problem; they are photophobic and prefer to be out of the sun.
A complete albinism in Icelandic sheep is manifested by white skin color, pink eyes, and impaired vision in bright light. It is an autosomal recessive. Albinism is also a problem in Karakul sheep.
True albino horses rarely if ever occur in nature, but white horses with pigmented eyes do. They are more accurately called pseudoalbinos. A recessive lethal white can also be produced by mating two Overo paint horses (an Overo is a horse with a coat color pattern where white is continuous over the body, but there is pigmented hair in a patch stretching from the ears to the tail). Affected foals develop colic soon after birth, fail to pass meconium, and die at 2–4 days of age. At necropsy there is an irreparable atresia or contraction of the colon associated with a congenital absence of myoenteric ganglia in the terminal portion of the ileum and the cecum and colon. The colon is patent but unable to dilate. Another variety of paint horses, the Tobiano (white markings extend from the dorsal midline ventrally with limbs usually white, and white covering a large part of the horse or very little), are expected to produce lethal whites also but there is no record of it.1 Both of these varieties of paints have blue or heterochromic irises.
FELL PONY SYNDROME
This is a familial disease of Fell ponies characterized by immunodeficiency, anemia, opportunistic infection, and death at 2–3 months of age.
ETIOLOGY
The disease is a putative autosomal recessive genetic defect of the immune system, although the exact nature of the defect has yet to be determined.
EPIDEMIOLOGY
The disease is restricted to Fell pony foals less than 3 months of age. The disease is reported in this breed in the United Kingdom, one case is reported in the United States (which has a total population of Fell ponies of <200) and a case is reported in the Czech Republic.1-37 The frequency of the disease is not reported. The case fatality rate is 100%.
PATHOGENESIS
There is immunodeficiency associated with low concentrations of immunoglobulins in serum and B lymphocytes in blood. T lymphocytes are present in normal concentration and respond appropriately to in vitro proliferation tests. Death is coincident with declines in concentrations of antibodies derived from colostrum. Immunodeficiency results in development of opportunistic infections including glossitis, adenoviral pneumonia, and cryptosporidial diarrhea. Aplastic anemia develops and can contribute to death. Anemia is associated with aplasia of red cell series in bone marrow and is not due to hemolysis or blood loss.
CLINICAL FINDINGS
Affected foals are lethargic at birth, are unable to keep up with the herd, and do not establish a strong bond with the dam. Foals develop ill-thrift, exercise intolerance, and diarrhea beginning at approximately 3 weeks of age. Clinical signs are attributable to anemia and opportunistic infections. At the time that foals develop other clinical abnormalities they are pyrexic and tachypneic. Most foals have bilateral mucopurulent nasal discharge and abnormal lung sounds consistent with pneumonia. The tongue is covered by a pseudomembranous, hyperkeratotic membrane suggestive of Candida sp. infection. Foals develop diarrhea and progressive illness with death occurring by 3–4 months of age even in cases treated aggressively.
CLINICAL PATHOLOGY
The underlying hematology and serum biochemistry is influenced by the opportunistic infections that develop in all foals affected with Fell pony syndrome. Abnormalities consistently associated with the syndrome and likely as a result of the underlying disease, include anemia, B cell lymphopenia, and variable to low concentrations of immunoglobulins in serum.1,2,4
Normocytic, normochromic anemia (6–26%, 6–26 L/L) is present in almost all affected foals. There is no evidence of regeneration in the blood and examination of bone marrow reveals an elevated myeloid:erythroid ratio (21:1 to 62:1, reference values 0.5:1 to 1.5:1).1 There is no evidence of hemolysis.
White blood cell concentration in affected foals is usually below or in the lower range of the reference range of normal, age-matched foals, and is attributable to a B-cell lymphopenia.5,6 Concentrations of CD4+ and CD8+ cells in blood are normal in affected foals.5,6 The concentration of neutrophils is often elevated in affected foals.
Concentrations of immunoglobulins (IgGa, IgGb, IgG(T), and IgM) in serum are variable and depend on the amount of immunoglobulin ingested in colostrum and the age of the foal. Affected foals are unable to produce immunoglobulins and therefore have declining concentrations of immunoglobulins with age. Serum concentrations of IgM and IgA become undetectable before does IgG – a consequence of the shorter half-life of the former immunoglobulins in foals. Measurement of low to undetectable concentrations of IgM at >4 weeks of age provides a reasonable means of diagnosing the disease.6
NECROPSY FINDINGS
Gross lesions include pale bone marrow, small thymus and lymph nodes, pneumonia, and pseudomembranous glossitis. The underlying disease is characterized by lesions in bone marrow and lymphoid tissue. Bone marrow has evidence of abnormal hemopoiesis with an elevated myeloid:erythroid ratio. Lymph nodes have sparse to moderate numbers of lymphocytes in cortices and paracortices. The thymus has no clear demarcation of cortex and medulla and the thymic lobules are small.1,2 Germinal centers are not present in the spleen and the red pulp is markedly contracted and contains siderophages. Ganglionopathy reported in foals in the original report of the disease has not been found in subsequent cases.
The differential diagnosis of immunodeficiency in foals is provided in Table 35.1.
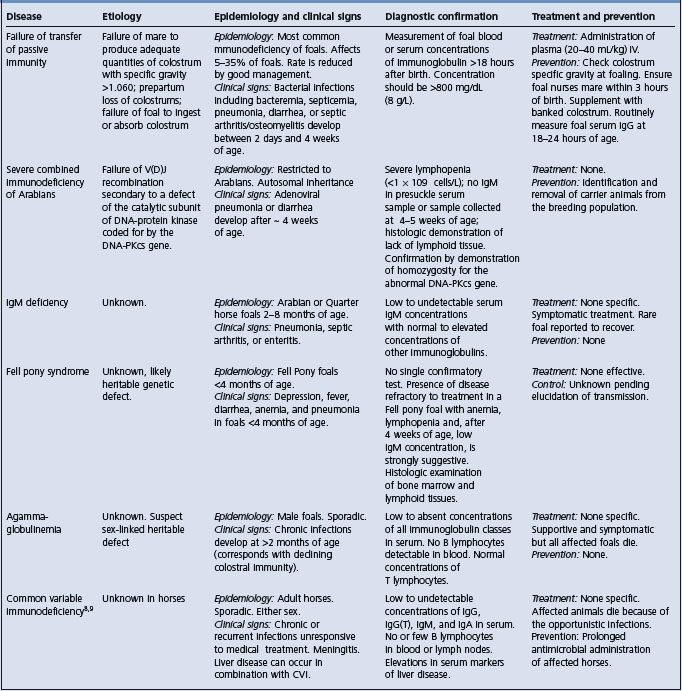
CONFIRMATION OF DIAGNOSIS
The disease is confirmed by presence of characteristic lesions at necropsy. Ante-mortem diagnosis is confounded by the presence of opportunistic infections, but should be suspected in any Fell pony foal with anemia, ill-thrift, and declining serum concentrations of immunoglobulin.
TREATMENT
There is no effective treatment. Supportive treatment of transfusions of blood or plasma, and administration of antibiotics does not affect the eventual outcome of the disease.
CONTROL
There are no control measures for the disease pending determination of the nature of its genetic transmission. Should the presumed autosomal recessive inheritance be demonstrated, then identification of carriers and elimination of these animals from the breeding population will permit control of the disease.
1 Richards AJM, et al. Equine Vet J. 2000;32:386.
2 Scholes SFE, et al. Vet Rec. 1998;142:128.
3 Flaminio MJ, et al. J Vet Int Med. 2005:19.
4 Thomas GW, et al. Vet Rec. 2003;152:618.
5 Bell SC, et al. Equine Vet. 2001;33:687.
6 Thomas GW, et al. Equine Vet J. 2005;37:48.
7 Jelinek F, et al. Vet Med A. 2006;53:69.
8 Flaminio MJ, et al. J Am Vet Med Assoc. 2002;221:1296.
9 Pellegrini-Masini A, et al. J Am Vet Med Assoc. 2005;227:114.
Inherited defects of the alimentary tract
INHERITED DEFECTS OF THE MOUTH AND JAW
Harelip in cattle often has a distinct familial tendency but little work appears to have been done on the mode of inheritance. An apparently inherited harelip combined with poor growth and accompanying cryptorchidism is recorded in Holstein-Friesian cattle. Bilateral cleavage of the lip which also involves the maxilla is recorded in Texel sheep as being conditioned by a single recessive autosomal gene.
Cleft palate is inherited as a simple recessive character in Hereford and Charolais cattle, concurrent with arthrogryposis in the latter, and is commonly thought to be inherited in sheep and pigs. The progeny of a commercial swine herd (Landrace x Duroc) and Large White Boar contained a number of piglets with cleft palates.1 Chromosomal analysis of affected piglets found all had identical unbalanced karyotype with partial monosomy of chromosomes 16 and partial trisomy of chromosome 3, compared to normal piglets in the litters with balanced karyotypes.
Jaw deformity. Shortness of the maxilla is thought to be inherited in Jersey cattle and Large White pigs, sometimes in association with chondrodysplasia. Shortness of the mandible is also inherited in cattle, in Angus in combination with cerebellar hypoplasia and osteopetrosis.
Smooth tongue (epitheliogenesis imperfecta linguae bovis). A defect of Holstein-Friesian and Brown Swiss cattle, this condition is inherited as an autosomal recessive factor. The filiform papillae on the tongue are small, there is hypersalivation and poor hair coat, and the calves do not fare well. The heterozygote is normal.
Tongue aplasia. Congenital absence of the median part of the tip of the tongue occurs rarely in piglets, often in association with cleft palate and/or harelip.
Rectal prolapse may be inherited in piglets as a result of agenesis of the anal sphincter (see below).
INHERITED RECTOVAGINAL CONSTRICTION
The defect is inherited in Jersey cattle and is manifested as stenosis of the rectum in either sex and stenosis of the vaginal vestibule in females. The tone of both rectal and vaginal sphincters is increased but attempts to detect heterozygotes by electromyographic measurement of these tones have been unsuccessful.2 The defect is regulated by an autosomal recessive gene. Affected cows are difficult to inseminate and have difficulty in calving. Their udders are small and hard and productivity is low. The condition is due to the presence of bands of non-elastic fibrous tissue. Edema of the udder is also a common complication. Some assistance in the identification of affected animals is available by the detection of collagen type II in muscle biopsies. 50% of heterozygotes also test positively, and so do a small percentage of normals.3
HEPATIC LIPODYSTROPHY IN GALLOWAY CALVES
Hepatic lipodystrophy has been reported occurring in Galloway calves on 5 farms in the UK over a 10-year period.4 Calves appear normal after birth but die by 5 months of age. Clinically there is tremor, opisthotonus, and dyspnea before affected calves become recumbent and die. At necropsy the liver is enlarged, pale and mottled. Histologically there is evidence of hepatic encephalopathy. The cause is unknown, but limited evidence suggests a storage disease is possible.
INHERITED ATRESIA OF ALIMENTARY TRACT SEGMENTS
Anal sphincter atresia occurs rarely in piglets, causing rectal prolapse.
Atresia ani occurs quite commonly in pigs, sheep, and, to a less extent, in cattle. Affected animals may survive for up to 10 days and are identified by their depression, anorexia, colic, marked abdominal distension and lack of feces, feces being replaced by thick white mucus.1 Abdominal distension in utero occasionally causes dystocia.2 Surgical repair is possible in some cases but in others a large segment of rectum is missing and creation of a colonic fistula in the inguinal region is necessary. The condition is thought to be inherited in pigs and calves but supporting evidence is slim: the evidence is less clear still in sheep. A suggestion that the defect may be also associated with the manipulation of the fetus during pregnancy examination has not been supported.2 A calf with atresia ani and diphallus and separate scrota has been described.
Inherited atresia coli, with complete closure of the ascending colon at the pelvic flexure, has been recorded in Percheron horses. A clinically similar defect in Overo horses, described in the section on pseudoalbinism, is in fact an aganglionosis. Death occurs during the first few days of life. The defect appears to be inherited as a simple recessive character.
Inherited atresia ilei has been recorded in Swedish Highland cattle. Affected calves manifest marked abdominal distension causing fetal dystocia. The distension is caused by accumulation of intestinal contents. Inheritance of a single recessive gene conditions the occurrence of the defect in some species and breeds but the prevalence may be higher than would be expected with that form of inheritance, especially in Jersey cattle with atresia coli.
Inherited defects of the circulatory system
BOVINE HEREDITARY DILATED CARDIOMYOPATHY
Bovine hereditary dilated cardiomyopathy is a group of progressive degenerative diseases of the myocardium causing congestive heart failure.1 At least three different types have been reported: dilatated cardiomyopathy in cattle of Canadian Holstein origin; cardiomyopathy in Japanese Black cattle; and, cardiomyopathy in Hereford cattle.
Pedigree analysis of 75 animals in three age classes and five diagnostic classes based on clinical and pathological findings using the Pedigree Analysis Package provided strong evidence for autosomal recessive inheritance of a single major gene responsible for the disease.2 Pedigree analyses of affected animals in Canada, Japan, and Switzerland, revealed that the Holstein bull ‘ABC Reflection Sovereign’, the son of Canadian Holstein sire Montwick Red Apple Sovereign, as the common ancestor.2 The disease in cattle is being used as a research model of human dilated cardiomyopathy.3 Using proteomic analysis, the examination of tissue from younger that are genetically diseased but have not yet developed clinical disease, a number of proteins have been identified whose abundance is altered significantly to suggest a possible pathogenetic mechanism for the onset of the disease.3
The disease has now been reported in Denmark in the Red Danish Dairy breed, Holsteins, and Red Holsteins.1,4 Pedigree analysis of 12 cases found both maternal and paternal relationship to the Canadian sire Monwick Red Apple Sovereign, and several sires were identified as carriers of the disease. These sires originated from breeding lines used to upgrade Danish cattle populations and pose a potential animal health problem. The introduction of the defect into the Danish cattle population is an example of how widespread a genetic disease can become in a short period of time. During their active life, two sires used for artificial insemination obtained a total of approximately 62 000 living progeny.
There can be a high incidence in certain herds, possibly associated with some unrecognized environmental precipitating factor but probably caused by autosomal recessive trait concentrated by a high coefficient of inbreeding.
There are three types recorded in cattle:
Type 1 calf – acute heart failure
Sudden death of Poll Hereford and horned Hereford5 calves up to 3 months of age may be due to inherited cardiomyopathy. The calves are often identifiable before death by their very rapid growth rate, short curly coat, and moderate bilateral exophthalmos. Death is usually precipitated by stress or exercise and is characterized by dyspnea, the passage of bloody froth from the nose, and a course of a few minutes to a few hours. Less acute cases have a syndrome of congestive heart failure for several days before death. Life expectancy is less than 6 months. At necropsy there is an obvious patchiness of the myocardium, reminiscent of a bad case of white muscle disease. The disease appears to be conditioned by a single autosomal recessive gene.
Type 2 calf – pulmonary edema
A second form of inherited cardiomyopathy is recorded in Japanese Black cattle. Death is preceded by a brief period (a few minutes to a few hours) of agonizing dyspnea in calves aged 30 up to 120 days. At necropsy there is edema, ascites, hydrothorax, and marked dilatation of the left ventricle. This is matched by acute myocardial necrosis. A new autosomal recessive gene is credited with initiating the disease.
Type 3 – young adult congestive heart failure
This occurs in young adult cattle and has been reported in Holstein-Friesian cattle in Japan, Canada, the UK,6,7 and Australia,8 and in Simmental-Red Holstein crossbred9 cattle and Black Spotted Friesian cattle in Switzerland. Similar family lines in Holstein breed have been identified in affected cattle in all three countries and it has been suggested that there is an inherited predisposition to cardiomyopathy in the Holstein breed.8 Pedigree analysis of hereditary dilatated cardiomyopathy in Holstein-Friesian cattle in Japan suggests an association with hereditary myopathy of the diaphragmatic muscles.10 The disease is endemic in Switzerland, occurring mainly in the Simmentaler x Red Holstein crossbreed of cattle.3
The disease occurs in cattle from 1.5 to 6 years of age with the peak prevalence in 3- and 4-year-old cattle. The stress of pregnancy and lactation may precipitate clinical disease and the majority of cases occur in late pregnancy or early lactation. The onset is sudden and the majority of cases have signs of congestive right heart failure. Edema of the submandibular area, brisket, ventral abdomen, and udder is prominent and there is venous engorgement, hepatomegaly, interstitial nephritis, pleural and pericardial effusion, and ascites. Muffling of the heart sounds, tachycardia, and a gallop rhythm are evident on ausculation of the heart. There is no characteristic biochemical or hematological change.7
Necropsy findings include congestive heart failure and histological findings compatible with congestive cardiomyopathy. There is dilation of the chambers of the heart, thickening or thinning of the ventricular wall, subcutaneous, mesenteric and pulmonary edema, hydrothorax, hepatomegaly, and ascites. Histologically there is fibrosis, myocardial degeneration, and vacuolation of cardiomyocytes and infiltration of mononuclear cells into the myocardium. In some cases, interstitial non-suppurative nephritis is present.7 Electron microscopically, the sarcoplasm of the hypertrophic fibers is filled with fine structures of low electron-density, together with thin filamentous material, suggesting myofibillar lysis.10
Following introduction of an eradication program based on culling of carriers in the sire population, the incidence in Switzerland has decreased.1
1 Leifsson PS, Agerholm JS. J Vet Med A. 2004;51:332.
2 Dolf G, et al. J Anim Sci. 1999;76:1824.
3 Weekes J, et al. Electrophoresis. 1999;20:898.
4 Agerholm J, Leifsson P. 23rd World Buiatrics Congress. Quebec City, Canada. July 11–16. 2004; Abstact No. 622(157).
5 Storie GJ, et al. Aust Vet J. 1991;68:119.
6 Bradley R, et al. J Comp Pathol. 1991;104:101.
7 Nart P, et al. Vet Rec. 2004;155:355.
8 McLennan MW, Kelly WR. Aust Vet J. 1990;67:75.
9 Graber HU, et al. Dtsch Tierärztl Wochenschr 97. 1990;447:451.
INHERITED LYMPHATIC OBSTRUCTION
This defect has been recorded in Ayrshire and Hereford1 calves. Males are more often affected than females; it has been suggested that some affected females may not be detected. The defect is inherited as a single, autosomal recessive character, with variable expressivity and incomplete penetrance.
The outstanding clinical feature is edema, the degree varying from slight to severe; severe cases causing dystocia to the point where embryotomy or cesarean sections is necessary. Some mortality occurs among the dams. Many calves are dead at birth and those born alive may be reared but the edema persists. Before parturition the cow may show evidence of hydrops amnii and have difficulty in rising. In calves the edema may be generalized or, more commonly, be localized to the head, neck, ears, legs, and tail. Drooping of the ears caused by increased weight is characteristic, and accessory lobes are commonly situated behind and at the base of the ears.
The edema is caused by a developmental abnormality of the lymphatic system. The lymph nodes are small and contain cystic dilatations and the lymphatic vessels are enlarged, tortuous, and dilated. Edema of the subcutaneous tissues and body cavities varies in degree; the skin is usually thickened and there is edema of the stomach wall.
INHERITED VENTRICULAR SEPTAL DEFECT
Ventricular septal defects are common in food animals; reports of their occurrence in lambs and Hereford cattle suggest that the condition can be inherited.
INHERITED AORTIC ANEURYSM
An inherited defect of the abdominal aorta, resulting in a high mortality from intra-abdominal hemorrhage, has been observed in an unidentified breed of cattle in Holland, and is an important feature in Marfan syndrome of humans.
BOVINE MARFAN SYNDROME
A model of human Marfan syndrome, this disease of cattle is an autosomal dominant disorder caused by mutations in the fibrillin-1-gene.1 It is manifested primarily by cardiovascular lesions and signs, but lacks the skeletal abnormalities of the human disease.2 Necropsy lesions include aortic and pulmonary artery aneurysm, with consequent cardiac tamponade in some cases. Fragmentation of elastic laminae in the vessels is also characteristic.3
INHERITED CONGENITAL PORPHYRIA
A congenital defect of porphyrin metabolism in cattle and swine characterized by excessive excretion of porphyrins in urine and feces and deposition of porphyrins in tissues, especially bones and teeth. Photosensitization occurs in affected cattle.
ETIOLOGY
Congenital porphyria is similar to erythropoietic or Gunther’s porphyria of humans. Most cases in cattle are due to the inheritance of a single recessive factor, heterozygotes being clinically normal. A deficiency of uroporphyrinogen III cosynthetase results in the accumulation of porphyrin type I isomers. Although there is no strict sex linkage in the mode of inheritance, the incidence is higher in females than in males. In pigs the pattern of inheritance is uncertain but may be due to one or more dominant genes.
EPIDEMIOLOGY
Porphyria is recorded only in cattle and pigs. Shorthorn, Holstein, Black and White Danish, Jamaica Red and Black cattle, and Ayrshires carry the defect.
There are no serious losses except that affected cattle suffer from incapacitating photosensitization when exposed to sunlight and must be kept indoors. In countries where sunlight hours are limited the disease may go unnoticed. Affected pigs appear to suffer little harm. Porphyria is of little economic importance because of its rarity.
PATHOGENESIS
The porphyrins are natural pigments but in these diseases they are present in larger than normal concentrations in the blood, urine, and feces. In porphyria the metabolic defect is one of abnormal synthesis of heme due to an enzymatic insufficiency at the stage of conversion of pyrrol groups to series 3 porphyrins. Excess series 1 porphyrins, physiologically inactive substances are produced as a result, and there is flooding of the tissues with these coloring and photosensitizing substances. The high tissue levels of porphyrins sensitize the skin to light and photosensitive dermatitis follows.
CLINICAL FINDINGS
In cattle the passage of amber to port wine colored urine, a pink to brown discoloration of the teeth and bones, and severe photosensitization are characteristic. Additional signs include pallor of the mucosae and retardation of growth.
Affected pigs are usually normal and photosensitivity does not occur, but the disease can be recognized by the red-brown discoloration of the bones and teeth, which is present even in the newborn.
CLINICAL PATHOLOGY
In porphyria the urine is amber to port wine in color when voided, due to the high content of porphyrins. The urine of affected cattle may contain 500–1000 μg/dL of uroporphyrins and 356–1530 μg/dL of coproporphyrins. The urine of normal cattle contains 1.84 μg/dL of coproporphyrins and no significant quantity of uroporphyrins. The color of the urine darkens to brown on exposure to light. Spectroscopic examination is necessary to identify the pigment as porphyrin. Erythrocyte survival time is reduced considerably. A macrocytic, normochromic anemia occurs and its severity appears to be related to the level of uroporphyrins in the erythrocytes, and there is evidence of a hemolytic anemia. Cattle with the highest erythrocyte uroporphyrin levels are also the most sensitive to sunlight.
NECROPSY FINDINGS
In porphyric animals the teeth and bones are stained brown or reddish purple, the pigment occurring chiefly in the dentine in teeth and often in concentric layers in the bones. Affected bones and teeth show a red fluorescence under illumination with ultraviolet light. The histological findings are unique to this disease.
Confirmation of the diagnosis depends on identification of greatly increased levels of porphyrins in the blood and urine. Presumed affected cattle and pigs can be detected at birth by the discoloration of the teeth. Breeding trials are necessary to detect heterozygous, normal carrier animals.
Differential diagnosis list. Other causes of photosensitive dermatitis.
INHERITED ERYTHROCYTIC PROTOPORPHYRIA
Inherited erythrocytic protoporphyria is an autosomal recessive disease which occurs in Limousin1 and Blonde d’Aquitaine2 cattle. It is similar to the same disease in humans and to porphyria but is milder. There is deficient activity of the enzyme ferrochelatase, resulting in excessive photosensitizing protoporphyrin accumulation with high levels appearing in the erythrocytes and feces. The total amount of the enzyme is normal but up to 96% of it is non-functional.3
Protoporphyria is clinically differentiated from porphyria by the absence of anemia and discoloration of the teeth and urine. The major clinical abnormality is photosensitive dermatitis affecting particularly the tips of the ears and the edges of the nostrils. There may be intense pruritus and exudative dermatitis involving the head and upper aspect of the thorax.4 The hematocrit values are within normal ranges, the teeth are normochromic, and there is no fluorescence of urine; however, whole blood fluoresces under ultraviolet light.4 Protoporphyrin binds to proteins that are not excreted by the kidney and thus protoporphyrin will not be detected in the urine. In a Limousin calf, the disease was characterized by ataxia, and intermittent seizures.5 At necropsy there is hepatic portal fibrosis, bile ductule hyperplasia, and swelling of parenchyma cells. Phagocytic cells in the dermis contain large heterogeneous lysosomes.6 Histologically, in some cases, the earliest lesions are moderate to severe acanthosis, hyperkeratosis, and parakeratosis with dermal angiofibroplasia. There may be intercellular edema, and intraepithelial vesicles and pustules.4 Elimination of affected carrier animals from the breeding program is the only control measure available.
HEMOCHROMATOSIS
Hemochromatosis is rare in domestic animals.1 This inherited defect of iron metabolism in humans has also been observed in yearling Salers cattle in circumstances in which an inherited etiology is suggested.1,2 The pattern of inheritance is uncertain because of the small number of pedigrees available from affected cattle.1 Hemochromatosis occurs when inappropriately large amounts of iron are absorbed from the intestine over an extended period. The excessive accumulation of iron causes iron-induced lysosomal injury and peroxidation by free radicals, which are the two major mechanisms responsible for hepatocellular necrosis and for sequelae such as fibrosis, bile duct hyperplasia, veno-occlusive disease, and hepatic neoplasia. Unlike hemosiderosis, hemochromatosis is associated with high transferrin saturation values in serum (>60%). Clinical disease develops between 9 and 22 months of age. Animals are normal until weaning but then lose weight, develop rough hair coats, and lose incisor teeth. The skeletal changes in hemochromatosis are due to abnormal bone development.3 Bone analysis reveals iron levels in affected animals may be 30 to 50 times greater than normal and decreased percent ash in the outer cortex. Periosteal dysplasia and osteopenia are responsible for the pathologic fractures and tooth loss.3
At necropsy, there is emaciation, firm dark brown livers and lymph nodes, soft bones, and brown-colored small intestine.1 The major histological changes are hepatocellular siderosis and periportal bridging, and perivenular fibrosis. Heavy deposits of iron in the liver, and deposits of hemosiderin are visible in liver tissue obtained by biopsy. Hepatic iron concentrations in clinically affected cattle range from 1500 to 10 500 wet weight (reference range for cattle = <300 g/g. Ultrastructurally, the heaviest intrahepatic deposition is in the hepatocyte. Iron in bone is associated with osteopenia.
FAMILIAL POLYCYTHEMIA
This inherited defect has been observed only in Jersey cattle. Attention is drawn to the presence of the disease by early calfhood deaths and a clinical syndrome including congestion of mucosae, dyspnea, and poor growth. Hematologically there is marked elevation of erythrocyte count, hemoglobin concentration, and packed cell volume. The disease appears to be a primary polycythemia inherited as a simple autosomal recessive.
INHERITED BLEEDING DISORDERS
Hemophilia
Hemophilia A (deficiency of factor VIII, or classic hemophilia) occurs in Thoroughbred, Standardbred, Arab, and Quarter horses,1,2 causing uncontrollable bleeding after injury or surgery, or spontaneously. This may be manifested by the sudden appearance of swellings over joints or the upper cervical region causing dyspnea, or into joints or body cavities causing acute hemorrhagic anemia, and blood can be aspirated from them. The platelet-dependent bleeding time is normal but the fibrin-dependent bleeding time is markedly prolonged. The defect is genetically transmitted as a sex-linked recessive trait, appearing clinically only in males.
von Willebrand’s disease has been identified in a Quarter horse which experienced bleeding episodes and a low blood level of vWF, after a normal platelet count and negative coagulation screening tests.3
Bovine factor XI deficiency
Factor XI (partial thromboplastin antecedent) is involved in coagulation but animals afflicted by a deficiency of it may be clinically normal even though their whole blood clotting time is very prolonged. Others may have a severe bleeding tendency but the frequency of hemorrhagic episodes with factor XI deficiency is very low.4 Heterozygotes experience minor episodes, homozygotes may have serious ones, especially neonates, which may die at birth and be classified as uncomplicated neonatal mortality. Affected cows also experience an increase in repeat breeder problems, apparently associated with a slower luteolysis and the development of small graafian follicles.5 Both males and females transmit the trait6 which is inherited as an autosomal recessive.
Prekallikrein deficiency
Prekallikrein is necessary to activate factor XII in the coagulation process. An inherited deficiency of it is recorded in a family of Belgian horses,7 as a cause of a bleeding tendency in the presence of the conventional coagulation factors. The deficiency is of the order of <1% of normal levels of 63–150%. The activated partial thromboplastin time is markedly prolonged.
Inherited thrombopathia
Thrombopathia causes uncontrolled bleeding in Simmental cattle.8,9 Collected blood undergoes good clot retraction but platelet aggregation in response to adenosine diphosphate and collagen in a whole blood aggregation system is badly impaired. Clinical findings include epistaxis, hematuria, the sudden development of subcutaneous swellings, hemorrhagic anemia due to internal bleeding, or bleeding after external lacerations or surgery. Restriction of the problem to the Simmental breed suggests inheritance of a recessive trait. A study of the inheritance of the abnormality using embryo transfer technology and superovulating a donor cow which previously had a calf with platelet aggregation disorder found a very low incidence of the abnormality.10 This suggests inheritance of the defect is not simple Mendelian recessive.
Afibrinogenemia (related diseases are hypofibrinogenemia, dysfibrinogenemia)
Afibrinogenemia is a rare cause of bleeding diathesis recorded in cattle, sheep, and goats, especially the newborn. Confirmation of the diagnosis and differentiation from the related diseases (above) requires sophisticated laboratory technology.11
1 Henninger RW. J Am Vet Med Assoc. 1988;193:91.
2 Littlewood JD, et al. Equine Vet J. 1991;23:70.
3 Brooks M, et al. J Am Vet Med Assoc. 1991;198:114.
4 Gentry PA, Ross ML. Am J Vet Res. 1994;58:242.
5 Liptrap RM, et al. Vet Res Commun. 1995;19:463.
6 Brush PJ, Gentry PA. Vet Rec. 1988;122:134.
7 Geor RJ, et al. J Am Vet Med Assoc. 1990;197:741.
8 Searcy GP, et al. Can J Vet Res. 1990;54:394.
9 Searcy GP, Petrie L. Aust Vet J. 1990;31:101.
INHERITED ANEMIAS
Inherited dyserythropoiesis and dyskeratosis (bovine congenital anemia, dyskeratosis, and progressive alopecia)
This disease occurs in 1–16-month old calves in some Poll Hereford families.1 It is thought to be inherited as a simple autosomal recessive character.2 Clinical signs commence at about 2 months of age and include skin lesions which commence on the face and neck, especially around the muzzle and along the edges of the ears, then extend in the midline down the back, then down the sides, and onto the limbs; the long hairs on the tail tip are shed.3 The hyperkeratotic muzzle accumulates dust. The skin lesions comprise alopecia, with surviving hairs wiry, kinked or tightly curled, accumulations of sebum, and hyperkeratosis and marked wrinkling. The alopecia and hyperkeratotic dermatitis extend and the calves do not thrive, becoming small in stature, intolerant of exercise, susceptible to heat stress, and eventually pining away until they die or are euthanized. Histologically the skin is affected by dyskeratosis, hyperkeratosis, and orthokeratosis, and there are morphological abnormalities of the nucleus in erythroid precursors; anemia results from ineffective erythropoiesis. There is a persistent, non-regenerative anemia due to a failure of maturation of erythrocytes; the blood contains many nucleated erythrocytes, and bone marrow aspirates contain increased numbers of erythroid precursors.1
Inherited glucose-6-phosphate dehydrogenase enzyme deficiency
A single case of this persistent hemolytic disease has been recorded in a yearling American Saddlebred colt.1 The disease in humans is well recognized as an inherited defect. A similar clinical disease is recorded in Murray Grey calves in Australia.2 Signs are observed first when affected calves are 3–8 weeks old. Both sexes are affected. Signs include poor growth, exercise intolerance, progressive weakness, severe jaundice, and death. There is a severe regenerative anemia, hemoglobin levels of 25–30 g/L, and an absolute nucleated erythrocyte count of 9–18 × 109 L.
Necropsy lesions include jaundice, a grossly enlarged, in some cases misshapen, greenish liver, and brown urine. Histological lesions are suggestive of a persistent intravascular hemolysis. A series of cases of hemolytic anemia of undetermined origin, recorded in a familial pattern in horses, was characterized by high blood levels of methemoglobin.3
Inherited defects of the urinary tract
INHERITED NEPHROSIS (MESANGIOCAPILLARY GLOMERULONEPHRITIS)
An apparently inherited mesangiocapillary glomerulonephritis occurs in Finnish Landrace lambs less than 4 months old,1 and in newborn Yorkshire pigs.2 The lambs absorb an agent from the colostrum which causes an immunological response in the lamb and the deposition of immune complexes within the glomerular capillary walls. Many affected lambs are asymptomatic before being found dead. Some have tachycardia, conjunctival edema, nystagmus, walking in circles, and convulsions. Enlarged, tender kidneys are palpable, and there is severe proteinuria, low plasma albumin, hyperphosphatemia, hypocalcemia, and BUN levels greater than 100 μg/100 mL. Necropsy findings include pale swollen kidneys and severe vascular lesions in the choroid plexuses and the lateral ventricles of the brain. Uremia supervenes in 75% of lambs by 27 days of age. Cases also occur in crossbred lambs.
CHRONIC INTERSTITIAL NEPHRITIS
Chronic interstitial nephritis with diffuse zonal fibrosis (CINF) occurs in Japanese Black cattle (Wagyu) as an autosomal recessive disorder leading to death prior to puberty.3 Clinically there is growth retardation between 3 and 5 months of age. A genome-wide scan using microsatellite markers in a Wagyu pedigree segregated for CINF, mapped the CINF locus to bovine chromosome 1.
INHERITED CYSTIC RENAL DYSPLASIA
Inherited, possibly conditioned by an autosomal dominant gene, this disease occurs in lambs sired by carrier Suffolk rams out of mixed ewes. Signs include recumbency and coma by days 2–3. Abortions and stillbirths occur in the same flocks at the same time. The kidneys are enlarged and cystic.4
INHERITED BILATERAL RENAL HYPOPLASIA
This usually lethal condition is inherited as an autosomal recessive character in Large White pigs.
INHERITED RENAL TUBULAR DYSPLASIA
This occurs in Japanese Black cattle.5 Affected calves have intermittent diarrhea at 2 months of age, are unthrifty, behave sluggishly, have a rough hair coat, and overgrown hooves from 2 to 5 months of age. There is progressive renal failure, and a high level blood urea nitrogen and serum creatinine. However, appetites are almost normal or only slightly depressed. All affected calves have been sired by the same bull and the defect is due to an autosomal recessive trait.
At necropsy there are no gross lesions of the kidneys. Histologically, there are streaky masses of renal tubules without lumens. In calves over 3 months of age, there is interstitial fibrosis surrounding the abnormal tubules. It is suggested that the genes associated with the adhesion of epithelial cells, such as adhesion molecules, extracellular matrix components, and growth factors are abnormal in animals with renal tubular dysplasia. The causative gene (rtd) has been mapped to chromosome 1 by linkage analysis. A part of the paracellin-1 gene, which encodes the renal epithelial tight junction protein is contained in the deletion and this deletion can be considered to be the cause of renal tubular dysplasia.6,7 A DNA specific test for this mutation has been developed.8
INHERITED RENAL LIPOFUSCINOSIS IN DANISH SLAUGHTER CATTLE
Dark brown or black discolored kidneys (‘black kidneys’) have been reported as incidental findings in slaughter cattle for more than 100 years.9 A pigment with characteristics similar to those lipofuscin is present in secondary liposomes in epithelial cells of the proximal tubules. Cases occurred only in Holstein cattle or the Red Danish Dairy Breed and mainly in animals aged 3 years or older. The prevalences of the abnormality were 0.44% and 2.51%, respectively. Epidemiological and genealogical analyses strongly indicate an autosomal recessive inheritance.
1 Frelier PF, et al. Vet Pathol. 1990.
2 Jansen JH. Acta Path Microbiol. Immunol Scand. 1993;101:281.
3 Kobayashi N, et al. Animal Genetics. 2000;31:91.
4 Jones TO, et al. Vet Rec. 1990;127:421.
5 Sasaki Y, et al. Vet Rec. 2002;150:628.
6 Ohba Y, et al. Vet Rec. 2001;149:153.
7 Ohba Y, et al. Vet Rec. 2001;149:115.
Inherited defects of the nervous system
INHERITED LYSOSOMAL STORAGE DISEASES
These are diseases in which there is a genetically determined deficiency of a specific lysosomal hydrolase enzyme causing a defective glycoprotein metabolism. As a result of the deficiency, metabolic substrates accumulate in the lysosomes. The lysosomes themselves are concerned with hydrolyzing polymeric material, which enters the vacuolar system, and converting it to monomeric units, such as amino acids, monosaccharides, and nucleotides, which can be dealt with by the better known metabolic processes.
There are other lysosomal storage diseases caused by poisonings and these are dealt with elsewhere. The best known ones are caused by poisoning with Swainsona, Astragalus, Oxytropis, and Phalaris spp. (the chronic form of that disease).
The diseases included in this section are not strictly diseases of the nervous system because the lysosomes in both neuronal and visceral sites are affected, but the effects of the disease are most obvious in terms of nervous system function.
MANNOSIDOSIS
Mannosidosis is the best known group of the inherited lysosomal storage diseases in agricultural animals.
α-Mannosidosis
This is a lysosomal storage disease in which a deficiency of the enzyme α-mannosidase results in the accumulation of a metabolite rich in mannose and glucosamine in secondary lysosomes in neurons, macrophages, and reticuloendothelial cells of lymph nodes, causing apparent vacuolations in them. Similar vacuoles are found in exocrine cells in pancreas, abomasum, and lacrimal and salivary glands. Storage appears to be cumulative in the fetus, but after birth stored material is lost from the kidney into the urine via desquamated tubular epithelium. On the other hand, postnatal storage continues in the brain, pancreas, and lymph nodes. The disease occurs in Angus, Murray Grey, and Galloway cattle, is inherited as a simple recessive, and is recorded as occurring in the United States, Australia, and New Zealand.
Clinically it is characterized by ataxia, fine lateral head tremor, slow vertical nodding of the head, intention tremor, an aggressive tendency, failure to thrive, and death or the necessity of euthanasia at about 6 months of age. These signs appear almost immediately after birth up to several months later and worsen over a period of up to 3–4 months. The signs are bad enough to require euthanasia during the first week of life in many cases.1 The first sign observed is a swaying of the hindquarters, especially after exercise or with excitement. The stance becomes wide based and the gait jerky, stilted and high stepping, with slight overflexion of the hindquarters so that the animal appears to be squatting as it moves.
The nervous signs are exacerbated by excitement, diarrhea is common, and the calves are usually stunted and unthrifty. They are also aggressive and attempt to charge but are usually impeded by their incoordination. Many calves die after having shown general ill thrift and with minimal nervous signs. Death may occur due to paralysis and starvation, or to misadventure, and some calves appear to die during a ‘fit’ following a period of excitement. Many others are euthanized because of persistent recumbency. The nervous syndrome of mannosidosis is well known; affected calves will die. An α-mannosidosis is recorded in Galloway cattle and is manifested by stillbirth, moderate hydrocephalus, enlargement of the liver and kidneys, and arthrogryposis.
CLINICAL PATHOLOGY
Normal heterozygotes carrying genes for mannosidosis are identifiable because of their reduced tissue or plasma levels of α-mannosidase. The mannosidase test for α-mannosidase in goats is specific and does not cross-react with α-mannosidase.
Advances in molecular biology have now led to the development of a more accurate test based on DNA technology.2 DNA tests based on the PCR have been developed for the detection of two breed-specific mutations responsible α-mannosidosis.3 One of the mutations is responsible for α-mannosidosis in Galloway cattle. The other mutation is uniquely associated with α-mannosidosis in Angus, Murray Grey, and Brangus cattle from Australia. The latter mutation was also detected in ion Red Angus cattle exported from Canada to Australia as embryos. The two breed-specific mutations may have arisen in Scotland and by the export of animals and germplasm disseminated to North America, New Zealand, and Australia.3
CONTROL
A control program can be based on the identification of heterozygotes using the PCR based assays for detection of breed-specific mutations.3 A program of screening cattle in herds which produce bulls for sale to commercial herds should stop the spread of the disease very quickly because the number of heterozygous females in the population will be irrelevant to the continuation of the disease in the absence of affected sires.
The α-mannosidosis gene prevalence is now insignificant and disease incidence has been reduced from an estimated 3000 cases/year to neglibile levels.2
CLINICAL FINDINGS
All cases are affected at birth with craniofacial deformity and inability to stand. The cranium is domed and there is mild prognathism, narrow palpebral fissures, and a tough, hidebound skin. When in sternal recumbency the head is moved in a combined motion of circling and bobbing, eventually converting the calf to lateral recumbency, in which it remains until passively returned to the sternal position, where nystagmus and tremor become evident. There is no suck reflex at any time. In lateral recumbency there is opisthotonos and paddling convulsions.
NECROPSY FINDINGS
Include a deficiency of cerebral cortical and cerebellar substance, distended lateral ventricles, and bilateral renomegaly. The biochemical defect is one of acidic β-mannosidase,6 and is conditioned by an autosomal recessive character. The carrier rate of the causative gene is very high in the Saler breed.7
By the biochemical examination of ultrasound-guided fetal fluid aspiration at between 59 and 65 days of pregnancy in ewes and goat does is accurate but not without the risk of causing abortion.8
In the goats the condition is present at birth and characterized clinically by tetraplegia, tremor, deafness, and nystagmus, and an inexorably fatal termination. Additional signs include bilateral Horner’s syndrome, carpal contractures, pastern joint hyperextension, thickened skin, and a dome-shaped skull. Although retinal ganglion cells are obviously badly affected, there appears to be no defect of vision.5 It is an autosomal recessive defect, very similar to α-mannosidosis.
1 Healy PJ, et al. Res Vet Sci. 1990;49:82.
2 Jolly RD. New Z Vet J. 2002;50(Suppl):90.
3 Berg T, et al. Res Vet Sci. 1997;63:279.
4 Abbitt B, et al. J Am Vet Med Assoc. 1991;198:109.
5 Render JA, et al. Vet Pathol. 1989;26:437-444.
6 Jolly RD, et al. NZ Vet J. 1990;38:102.
GANGLIOSIDOSES
At least five types of gangliosidosis are known to occur in humans and animals. Two have so far been identified in agricultural animals.
GM1 gangliosidosis
GM1 gangliosidosis occurs in cattle and sheep. In Friesian cattle it is inherited as an lysosomal storage disease in which the activity of an enzyme, β-galactosidase, in nervous tissue is greatly reduced. As a result there is an accumulation of the ganglioside (GM1) in the tissue. Clinical signs of progressive neuromotor dysfunction and a reduction in growth rate appear at about 3 months of age. The growth rate is reduced, and the animal is in poor condition, blind, and has a staring coat. The neuromotor signs include lack of response to external stimuli, sluggish mastication and swallowing, hindquarter sway while walking, a wide stance, a tendency to fall, reluctance to move, stiff high-stepping gait, aimless walking, head-pressing, and convulsions. Abnormal ECG tracings are common. The blindness results from lesions in the retina and the optic nerve. Ophthalmoscopic examination of the retina is recommended as an aid to diagnosis. A positive diagnosis is made on the grounds of intraneuronal lipid storage plus reduced β-galactosidase activity plus identification of the stored lipid. The stored ganglioside is visible under the electron microscope as stacks and concentric whorls of lamellae. In the live animal enzyme assays are carried out on leukocytes. The enzymatic defect is also detectable in liver, skin, and leukocytes.
The disease is also present in Suffolk and Suffolk-cross sheep. Visceral and neuronal lysosomal storage are both evident but the neuronal lesion is more severe. Deficiencies of β-galactosidase and α-neuraminidase are evident. Affected sheep become ataxic at 4–6 months old and worsen to recumbency and death in up to 2 months.
GM1 gangliosidosis has been reported from England in ‘Coopworth Romney’ lambs closely related to a ram imported from New Zealand.1
GM2 gangliosidosis
This has been identified in Yorkshire pigs and also causes decreased growth rate, incoordination appearing after 3 months of age, gray-white spots in the retina and dark blue granules in neutrophils, and azurophilic granules in lymphocytes. A serum enzyme assay is a suitable method of detecting ‘carrier’ heterozygous pigs. The test is based on the amount of N-acetyl-β-D-hexosaminidase in tissues.
CEROID LIPOFUSCINOSIS
The ceroid-lipofuscinoses are a group of inherited storage diseases, inherited as autosomal recessive traits. They are grouped together because of common clinical and pathological phenomena related to brain and retinal atrophy, premature death, and accumulation of a fluorescent lipopigment in neurons and many other cell types within the body.
The disease is recorded in South Hampshire sheep,1 Rambouillet sheep,2,3 Borderdale sheep,4 Merino sheep,5 Nubian goats, and Devon cattle.6 It resembles neuronal ceroid lipofuscinosis of humans and is not strictly a primary lysosomal disorder; it is classified as a proteolipid proteinosis, and provides a good animal model for discussing the similar disease (Batten’s disease) of humans. It is inherited as an autosomal recessive trait.7 Secondary lysosomes fill with subunit C of mitochondrial ATP synthase because of excessive peroxidation of polyunsaturated fatty acids.8 The mechanism of the accumulation is that protein is formed which is normal for mitochondria but is misdirected so that it accumulates in the lysosome.9 The brains of affected lambs grow till they are 4 months old, then they commence to atrophy.10 The disease in Merino sheep is a subunit c-storing abnormality, clinically and pathologically similar to ceroid-lipofuscinosis in South Hampshire sheep.5 The South Hampshire form of the disease maps to chromosome 7q13-15, which is synthetic with human chromosome 15q21-23, the site of human CLN6.1
The occurrence of a neuronal ceroid-lipofuscinosis in Borderdale sheep in New Zealand has been described.4 The severity of neurodegeneration and minor differences in the ultrastructure of storage material suggests this is a different disease from other forms of ovine ceroid-lipofuscinosis which accumulate subunit-c of mitochondrial ATP synthase.4 An autosomal recessive mode of inheritance is considered probable.
Clinical findings include slowly progressive ataxia of the hindlimbs, commencing usually at about 4 months but possibly as late as 18 months of age, and lasting for 6 months leading to euthanasia at up to 4 years. Inability to keep up with the flock is noticed first, followed by a sawhorse stance, obvious ataxia, severe depression, and an increasing failure of the menace and pupillary light reflexes. Terminal blindness is a constant sign. Positional nystagmus, circling, and head pressing occur in some. Eating, drinking, and defecation are normal, but there is slight weight loss.11
The lesion in lambs and calves12 is atrophy of the cerebrum, especially the optic cortex, with eosinophilic granulation of neurons and macrophages in the central nervous system followed by progressive retinal atrophy. There is a progressive storage of lipopigment in nervous tissue, especially retinal photoreceptors; its presence can be demonstrated by quantitative autofluorescence using a modified slit lamp microscope.11 Other clinicopathological aids include lysosomal enzyme assay, organ biopsy, and computed tomography, which reveals the enlargement of the lateral ventricles of the brain resulting from cerebral atrophy.12
Neuronal ceroid lipofuscinosis has been described in three horses.13 Clinically, there was developmental retardation, slow movements and loss of appetite at 6 months of age. Torticollis, ataxia, head tilt, and loss of eyesight were present at 1 year of age. There were abnormalities in posture and movements, decreased spinal reflexes, and some cranial nerve dysfunction, dorsal strabismus, and absence of the menace reflex. At necropsy, there was flattening of the gyri and discoloration of the brain. Histologically, eosinophilic, autofluorescent material in the perikarya of neurons were present throughout the brain, spinal cord, neurons of the retina, submucosa, and myenteric ganglia and in glial cells.
1 Jolly RD, et al. New Z Vet J. 2004;52:52.
2 Edwards JF, et al. Vet Pathol. 1994;31:48.
3 Woods PR, et al. J Vet Intern Med. 1994;8:37.
4 Jolly RD, et al. New Z Vet J. 2002;50:199.
5 Cook RW, et al. New Z Vet J. 2002;80:292.
6 Harper PAW, et al. Acta Neuropathol. 1988;75:632.
7 Prieur DJ, et al. J Hered. 1990;81:245.
8 Fiske RA, Shorts RW. Vet Pathol. 1988;25:171.
9 Fearnley IM, et al. Biochem J (Lond). 1990;268:751.
10 Jolly RD, et al. Can J Vet Res. 1990;54:15.
11 Armstrong D, et al. Vet Res Commun. 1988;12:453.
GLOBOID CELL LEUKODYSTROPHY (GALACTOCEREBROSIDOSIS)
Globoid cell leukodystrophy has been identified in Poll Dorset sheep in Australia1. Incoordination in the hindlimbs progresses until the animals are tetraplegic. Only histological changes are evident at necropsy. These include myelin destruction and the accumulation of characteristic globoid cells in nervous tissue. There is greatly decreased galactocerebrosidase activity in affected tissue.
INHERITED NERVOUS SYSTEM ABIOTROPHIES
These diseases are characterized by premature, progressive loss of nervous function due to spontaneous, late degeneration of nervous tissue. As a result most affected animals are born normal but develop signs of a progressive neurological disease that is either fatal or leads to such a serious neurological deficit that euthanasia is the only reasonable solution. In a few rare diseases the patient is abnormal at birth but worsens, and usually dies, during the neonatal period. Again there are exceptions and rare cases in calves recover completely. The genetic nature of some of the cases included may not be certain; they are included here if the evidence that they are inherited can be reasonably presumed. The lysosomal storage diseases, listed in the preceding section, are a specific group of abiotrophic diseases.
CEREBELLAR ABIOTROPHY
This disease occurs in Holstein and Poll Hereford cross1 calves, pigs, and Merino sheep.
Cattle
In the calves ataxia appears for the first time when they are 3–8 months old. The calves are not blind but they often fail to exhibit a menace reflex. The onset of clinical signs is sudden but progression is slow or inapparent. Some become recumbent. Those that remain standing have a spastic, dysmetric ataxia, and a broad-based stance; they fall easily and have a fine head tremor. All are strong and have good appetites. Abiotrophy, or premature aging, is evident only microscopically and consists of axonal swellings and segmental degeneration and loss of cerebellar Purkinje cells. The disease appears to be inherited, but recovery of some late cases is recorded.
Sheep
The disease in sheep does not appear until about 3 years of age. There is incoordination and dysmetria so that the gait is awkward and disorganized and there is frequent falling. There are also a reduced menace response, an apprehensive manner, and a wide-based stance in the hindlimbs. At necropsy there is diffuse cerebellar degeneration and severe loss of Purkinje cells.
Pigs
A congenital progressive cerebellar abiotrophy is also reported in piglets of the offspring of Saddleback sows and an unrelated Large White boar. The disorder behaves epidemiologically like an inherited disease conditioned by a simple autosomal recessive trait. Clinical signs include dysmetria, ataxia, and tremor at standing but not at rest. There is gradual adjustment so that the piglets can walk and stand at 5 weeks of age but by 15 weeks they are no longer able to do so. Affected pigs also have a coarse matted hair coat caused by a disproportionate number of coarse hairs to fine hairs. Histopathological lesions are confined to the cerebellum where there is a significant loss of Purkinje cells.
Horses
The disease is recorded principally in Arabs but occurs also in the Australian pony, which was developed from the Arab, and in the Gotland breed from Sweden. A similar clinical syndrome occurs in the Oldenberg breed but the pathological picture is quite different.
The disease may be present at birth but is often not observed until the foal is 6–9 months old. The characteristic signs are vertical head-nodding (some cases show horizontal head tremors), especially when excited, and ataxia which is most noticeable at a fast gait. It may not be evident while the foal is walking. Very badly affected foals are unable to stand or suckle at birth, less severe ones are normal until about 4 months of age when head-nodding becomes obvious. The degree of ataxia varies from slight incoordination to inability to stand. A ‘goose-stepping’ gait which slams the front feet into the ground occurs in some. All foals can see but there is an absence of the menace reflex in many. Nystagmus is not recorded as occurring in this disease.
Necropsy findings are limited to histopathological lesions in the cerebellum. These include widespread loss of Purkinje cells and the presence of a gliosis. There are no degenerative lesions in the spinal cord. In the similar disease in Oldenberg horses the cerebellum is often reduced in size. The disease is an abiotrophy, a premature aging of tissues.
There have been doubts about the heritability of the disease but analysis of European data confirms that it can be inherited as an autosomal recessive.2
Familial convulsions and ataxia, especially of Angus cattle, is an abiotrophy and on histopathological criteria belongs in this group, but because it is usually present at birth it is included, for ease of diagnosis, in the section on congenital cerebellar defects.
CAPRINE PROGRESSIVE SPASTICITY
A possibly inherited progressive paresis of Angora goats is recorded in Australia.1 Signs first appear at about 2 months of age, commencing with lethargy, followed by ataxia, then paresis progressing to sternal recumbency and eventual euthanasia. Tendon reflexes are normal but the kids have difficulty getting to their feet, especially in the hindlimbs. The gait is ataxic with frequent stumbles, and the kids are unwilling to run. At necropsy there are many large, clear vacuoles in many neurons of the spinal cord, posterior brainstem and midbrain, and degeneration of nerve fibers in the same areas and peripheral nerves.
INHERITED NEURODEGENERATION (SHAKER CALF SYNDROME)
This is an inherited, degenerative disorder of horned Hereford calves. Newborn calves show severe tremor, difficulty in rising, spastic gait, and aphonia. Terminally there is spastic paraplegia. Histologically there are accumulations of neurofilaments within neurons. A similar disease in Holstein-Friesians occurs only in males. There are severe degenerative changes in the spinal cord with spongiform lesions and some cavitation. It has the epidemiological distribution of a sex-linked recessive mutation.1
INHERITED PROGRESSIVE DEGENERATIVE MYELOENCEPHALOPATHY (WEAVER SYNDROME)
The defect is inherited in Brown Swiss cattle. It appears first in calves when they are 6 months to 2 years old with a small number more than 2 years, and is manifested by progressive bilateral hindlimb weakness and proprioceptive deficits causing difficulty in rising and a weaving, hypermetric gait, goose stepping with the forelimbs, and dragging the hindlimbs. The limb reflexes are normal. The calves are bright and alert throughout. There is a broad-based stance and finally recumbency1 and, after a course of 12–18 months, inevitable euthanasia. Necropsy lesions include axonal degeneration, including spheroid formation, and vacuolation of white matter in the cerebellum and at all levels of the spinal cord2 but especially in the thoracic segment.3 There is some neurogenic atrophy of muscles but there is no muscular dystrophy.4 The defect can be identified by examination of chromosomes.5 The defect appears to be linked chromosomally with high milk yield traits.6
INHERITED SPINAL DYSMYELINATION
Bovine spinal dymyelination is a congenital neurological disease occurring in several national cattle breeds upgraded with American Brown Swiss cattle.1 The disease was first described in Red Danish Dairy breed. In Denmark, all cases are genetically related to the ABS bull White Cloud Jason’s Elegant. It is inherited as an autosomal recessive trait. Genetic mapping of the gene in cross-bred American Brown Swiss cattle to the bovine Chromosome II has been done.1
Clinically, in calves there is lateral recumbency, opisthotonos, limb extension, normal to increased reflexes and mental alertness and by dysmyelination, including axonal degeneration and astrogliosis, in spinal tracts, especially the ascending gracile funiculus and dorsolateral spinocerebellar tracts and the descending sulcomarginal tract.2,3 This is probably the same defect as spinal muscular atrophy.
INHERITED PROGRESSIVE ATAXIA
This well-recognized disease occurs in Charolais cattle.1 The first onset of signs is at about 12 months of age when the gait is seen to be stiff and stumbling, especially in the hindlimbs, and the hind toes are dragged. The ataxia may be asymmetric, and the animal cannot back up. The ataxia progresses over a period of 1–2 years. Affected animals tend to be down a lot and have difficulty in rising and posturing for urination. Urination is abnormal, being a squirting but continuous flow which soils the tail. Some affected animals nod their heads from side to side when excited. Both males and females are affected. It has been described occurring in 2-year-old Charolais steer in New Zealand.1 Characteristic necropsy lesions are confined to the central nervous system and are histopathological. The white matter of the cerebellum and internal capsule contains multiple foci of oligodendroglial dysplasia. The somatic lymph nodes contain nodules of hyperplastic lymphoid follicles, some catarrh of the medullae of the nodes, and an accumulation of eosinophils.
INHERITED SPINAL DYSRAPHISM
This is found as a congenital defect in Charolais calves, and is associated with arthrogryposis and cleft palate.
INHERITED SPINAL MYELINOPATHY
There is a progressive spinal myelinopathy of Murray Grey cattle, similar to that seen in Charolais cattle. It is possibly genetic in origin. Some calves are affected at birth; others do not become affected until 1 year old. The syndrome is one of a progressing paresis, without significant ataxia leading to paresis and permanent recumbency. There are degenerative lesions in spinal cord, midbrain, and cerebellum. The disease is conditioned by an autosomal recessive gene.1,2
INHERITED MULTIFOCAL SYMMETRICAL ENCEPHALOPATHY
Two forms of the disease are recorded, in Simmentals1 and in Limousin and Limousin-cross cattle.2 The Limousin calves are normal at birth but from about 1 month of age develop a progressive forelimb hypermetria, hyperesthesia, blindness, nystagmus, weight loss, and behavioral abnormalities, especially aggression. The signs gradually worsen for up to 4 months when euthanasia is necessary. Necropsy lesions include brain swelling, optic chiasma necrosis, and multifocal, symmetrical areas of pallor, up to 0.5 cm diameter in the brain. These lesions show partial cavitation and multiple, pathological abnormalities especially myelin lysis and vacuolation and demyelination.3 The distribution of cases suggests an inherited defect.
The disease in Simmental and Simmental-cross cattle recorded in Australia and New Zealand4 also has a distribution suggesting an inherited defect. The disease is clinically similar to that in Limousin cattle except that affected animals are not blind and it develops later, 5–8 months; calves may survive longer, up to 12 months and, although the characteristic abnormality of gait is hypermetria, the hindlimbs are affected, not the forelimbs. Other signs observed are dullness, a swaying gait and, terminally, gradually developing opisthotonos and forelimb hypertonia in extension. Necropsy lesions are also similar to those in the Limousins but the distribution is in the midbrain and the entire brainstem.5
A multifocal symmetrical necrotizing encephalomyelopathy in Angus calves has been described.6 Clinically calves exhibited ataxia, nystagmus, strabismus, muscular tremors, opisthotonus, bruxism, hyperaesthesis, tetanic spasms, and episodic convulsions at 2–6 weeks of age. Death occurred 4–7 days after the onset of clinical signs. Lesions consisted of symmetrical degenerative foci affecting the dorsal vagal motor, lateral cuneate and olivary nuclei in the medulla oblongata and occasionally in the spinal cord, substantia nigra and cerebellar peduncles. While an inherited basis for the disease is suspected, the etiology is unknown.
INHERITED BOVINE DEGENERATIVE AXONOPATHY
Reported in Holstein-Friesian calves in Australia,1 most affected calves are affected at birth by recumbency, hyperesthesia or depression, rigidity of limbs, tremor, especially of the head, nystagmus, apparent blindness, and the development of opisthotonos and tetanic spasms when stimulated. At necropsy the consistent lesion is a severe, diffuse, axonal swelling and loss in the spinal cord and brainstem. The cause is unknown but the indicators point to heredity.
INHERITED OVINE DEGENERATIVE AXONOPATHY (NEURAXONAL DYSTROPHY)
A heterogeneous group of diseases of genetic or acquired etiology is characterized by spheroidal swellings of axons, the result of accumulation of axoplasmic organelles including neurofilaments.1
This is reported in Suffolk, Merino, and Coopworth2 sheep. An inherited defect is suspected in all three diseases.
In Coopworths the lambs are affected at birth but have a progressive syndrome in which cerebellar and proprioceptive signs predominate. Most die by 6 weeks of age. Large axonal spheroids are present in the spinal cord and midbrain, and there is a severe depletion of Purkinje cells in the cerebellum.
In Suffolks the disease does not appear until 1–6 months; signs are a gradual onset of ataxia, followed by recumbency, leading to death or euthanasia. Spheroids in central nervous system axons are characteristic, mostly in the spinal cord and cerebellum.
The disease in Merinos is in fine-wool sheep, is probably the same disease as that previously called Murrurrundi disease, and does not appear until 4–6 years of age. Most cases require to be euthanized after about 2 months but some mild cases survive for up to 3 years. The clinical signs include a wide-based stance, dysmetria of all limb movements with a pronounced hypermetria of the forelimbs resulting in frequent falling, a fine intention tremor of the head, and a diminished menace reflex. A similar disease of medium-wool Merinos, characterized by progressive posterior ataxia and degeneration of sensory tracts in thoracic segments of spinal cord, commencing after 5 months of age and terminating fatally before 2 years of age, is also recorded in Australia.3 The probability is that it is also an inherited defect.
INHERITED SPONTANEOUS LOWER MOTOR NEURON DISEASE
This progressive disease of Yorkshire piglets 5–10 weeks of age is presumed to be inherited.1 Clinical signs include hindlimb tremor, weakness, and ataxia appearing at 2–5 weeks of age. The gait includes fetlock knuckling, short choppy steps, and a tendency to collapse after a few steps. Segmental and postural reflexes are normal. By 10 weeks there is complete hindlimb paralysis, the pig is in sternal recumbency and front limb paralysis has begun. The appetite is good and the pig is bright and alert. On necropsy there is symmetrical degeneration and loss of motor neurons in the spinal cord, in some ventral spinal nerve roots.
A lower motor neuron disease in newborn Romney lambs has been described.2 Lambs are normal at birth but within 1 week they developed weakness and ataxia, which progressed until they were unable to stand. The principal histological lesions were degeneration and loss of neurons in the ventral horns of the spinal cord and brain stem, Wallerian degeneration of ventral rootlets and motor nerves, and associated denervation atrophy of skeletal muscle fibers. Large fibrillar spheroids were found in white and gray matter including nuclei in the brain stem. A similar, though not identical, disease of newborn lambs has been recorded in a Dorset Down flock affecting about 20% of lambs. They lay with hindlimbs tucked under the body and forelimbs splayed sideways.
INHERITED MAPLE SYRUP URINE DISEASE (Branched Chain KETOACID DECARBOXYLASE DEFICIENCY – BCKAD)
Calves affected by this disease may be stillborn. Live calves are normal at birth and develop signs only at 1–3 days of age. It is inherited as an autosomal recessive and occurs principally in Poll Hereford, Hereford, and Poll Shorthorn1 cattle but probably occurs also in other breeds.2 There is molecular heterogeneity between the breeds, and tests based on detection of the mutation could be prone to error.3 Hair roots are good sources of target DNA for genotyping cattle for the mutation. This avoids the errors created by hemopoietic chimerism when blood is used for the test.4 The disease is caused by an accumulation of branched chain amino acids – including valine, leucine, and isoleucine – due to an absence of branched chain ketoacid decarboxylase.5 The mutation responsible for maple syrup urine disease in Poll Shorthorns and genotyping Poll Shorthorns and Poll Herefords for the maple syrup urine disease alleles has been determined.6 The mutations responsible for maple syrup urine disease and inherited congenital myoclonus are present in the Australian Poll Hereford population.7
Clinical signs include dullness, recumbency, tremor, tetanic spasms and opisthotonos, a scruffy coat, blindness, and severe hyperthermia. When held in a standing position some calves have tetanic paralysis, others have flaccid paralysis. Terminal coma is followed by death after a course of 48–72 hours. The urine smells of burnt sugar.
At necropsy there is a characteristic severe spongiform encephalopathy similar to that found in comparable hereditary aminoacidurias in humans. Final identification can be made based on the elevated ratios of branched:straight chain amino acids in nervous tissue.8
1 Healy PJ. Aust Vet J. 1992;69:143.
2 Baird JD, et al. Can Vet J. 1987;28:505.
3 Healy PJ, Dennis JA. Anim Genet. 1994;25:329.
4 Healy PJ, Dennis JA. Aust Vet J. 1995;72:346.
5 Zang B, et al. J Biol Chem. 1990;265:2425.
6 Dennis JA, Healy PJ. Res Vet Sci. 1999;67:1.
INHERITED CITRULLINEMIA
This autosomal recessive disease is inherited in Australian Holstein-Friesians, USA Holsteins,1 and Red Holsteins in Europe.2 A previous report of a high incidence of the disease in a German Holstein herd has been refuted by a negative result in the breed in a wide-ranging survey.3
Affected calves are normal at birth but develop signs in the first week of life and die 6–12 hours after the onset of illness. The signs are depression, compulsive walking, blindness, head-pressing, tremor, hyperthermia, recumbency, opisthotonos, and convulsions. Arginosuccinate synthetase deficiency is the likely cause. Blood citrulline levels are of the order of 40–1200 times normal and the assay can be used to detect heterozygotes.4 The alternative method of detecting heterozygotes is to use a polymerase chain reaction, a restriction endonuclease test designed to identify the mutation that causes the disease.5 Prenatal diagnosis has been achieved by examination of cell cultures derived from amniotic fluid.6
EQUINE DEGENERATIVE MYELOENCEPHALOPATHY
Equine degenerative myeloencephalopathy is characterized by symmetrical, slowly progressive spasticity and ataxia in foals and horses less than 2 years of age. The disease occurs in most breeds in North America and Europe and is reported in captive zebra and Mongolian Wild Horses in North America.1,2 Neuronal dystrophy of the cuneate and gracilus nuclei is considered a form of equine degenerative myeloencephalopathy.3
The prevalence of the disease varies widely, with up to 40% of susceptible animals on a farm being affected, although the disease is usually sporadic. There is a familial predisposition to the disease apparently involving an increased requirement for vitamin E, although other factors, including housing, are contributory.4-7
The pathogenesis of the disease is unknown. Abnormal expression of integral synaptic vesicle, synaptic vesicle-associated presynaptic plasma membrane and cytosolic proteins was observed in two Arabian horses with equine degenerative myeloencephalopathy.8 These proteins have a role in trafficking, docking, and fusion of neuronal synaptic vesicles, and this finding suggests that there is disruption of axonal transport in equine degenerative myeloencephalopathy.8 Loss of axons leads to defects in neurological function and consequent gait abnormalities.
The clinical signs are those of a slowing progressive spinal ataxia that stabilizes when the animal is 2–3 years of age. Affected foals and yearlings have symmetrical signs that are most severe in the hind limbs, of ataxia characterized by pivoting, circumduction, truncal sway, and difficulty performing complex movements such as backing or walking with the head elevated. At rest, severely affected horses may have an abnormal posture. The cutaneous trunci reflex may be absent. Spontaneous recovery does not occur, but progression to death is unusual. Radiography and myelography of the cervical spine does not reveal evidence of compression of the spinal cord.
Serum vitamin E concentrations may be normal or low. The hemogram, serum biochemical profile, and cerebrospinal fluid analysis are normal. There are no gross lesions on necropsy. Histological lesions include neuronal atrophy, accumulation of lipofuscin-like pigment, and glial cell proliferation.
Differential diagnoses are listed in Table 36.1. Diagnosis is achieved by exclusion of other causes, of abnormal gait without fever or disease in other body systems in horses, such as compressive myelopathy and equine protozoal myeloencephalopathy.
No treatment is curative, but vitamin E (6000 IU orally once daily) may prevent progression of signs. Supplementation of at-risk foals and yearlings with vitamin E can prevent the disease.2
1 Montali RJ, et al. Vet Path. 1974;11:68.
2 Liu K, et al. J Am Vet Med Assoc. 1983;183:1266.
3 Beech J. Vet Pathol. 1984;21:384.
4 Blythe LL, et al. J Am Vet Med Assoc. 1991;198:1005.
5 Mayhew IG, et al. J Vet Intern Med. 1987;1:45.
6 Dill SG, et al. Am J Vet Res. 1990;51:1300.
INHERITED CONGENITAL HYDROCEPHALUS
Cattle
Congenital hydrocephalus without abnormality of the frontal bones occurs sporadically but is also known to be an inherited defect in Holstein and Hereford and possibly in Ayrshire and Charolais cattle. Two specific inherited entities have been described. In one there is obstruction to drainage of the cerebrospinal fluid from the lateral ventricles which become distended with fluid and may cause bulging of the forehead, often sufficient to cause fetal dystocia. Hereford calves with this defect have partial occlusion of the supraorbital foramen, a domed skull, and poorly developed teeth; at necropsy the cerebellum is found to be small and there may be microphthalmia and skeletal muscle myopathy. They are usually born a few days prematurely, are small in size and unable to stand or suck. In some cows the amniotic fluid is increased in volume.
Another form of inherited hydrocephalus due to malformation of the cranium and with no enlargement of the cranium has also been observed in Hereford cattle. The ventricular dilatation is not marked, and microphthalmia and cerebellar hypoplasia are not features. Affected calves may be alive at birth but are blind and unable to stand. Some bawl continuously and some are dumb. They do not usually survive for more than a few days. At necropsy there is internal hydrocephalus of the lateral ventricles with marked thinning of the overlying cerebrum. Other lesions include constriction of the optic nerve, detachment of the retina, cataract, coagulation of the vitreous humor, and a progressive muscular dystrophy. The condition is inherited as a recessive character.
Internal hydrocephalus inherited in combination with multiple eye defects in White Shorthorns is dealt with elsewhere, as are non-inherited forms of the disease.
Sheep
A defect comparable to the Dandy–Walker syndrome in humans and characterized by internal hydrocephalus caused by obstruction of the foramina of Magendie and Lushka occurs in several breeds of sheep, especially Suffolks,1 and in cattle. Affected lambs are stillborn or die within a few hours of birth; because of the grossly enlarged cranium many cause dystocia which can only be relieved by a fetotomy.
Pigs
Congenital hydrocephalus in Yorkshire and European pigs has been recorded. The abnormality varies from a small protrusion of dura (meningocele) to an extensive brain hernia in which the cerebral hemispheres protrude through the frontal suture, apparently forced there by increased fluid pressure in the lateral and third ventricles. The condition is thought to be inherited in a recessive manner, but exacerbated in its manifestation by a coexisting hypovitaminosis-A. An outbreak of congenital meningoencephalocele in Landrace pigs is recorded in circumstances suggesting that it was inherited.
Horses
A Standardbred stallion sired a number of hydrocephalic foals in a pattern that suggested the inheritance of a dominant mutation in the germ line and in the form of a single locus defect.2 Affected foals caused dystocia and were all stillborn.
INHERITED PROSENCEPHALY
Recorded in Border Leicester sheep, this defect takes the form of fusion of the cerebral hemispheres and a single lateral ventricle. It is widespread in the breed in Australia1 and is inherited as an autosomal recessive character. Most affected lambs are stillborn. Live ones have dyspnea due to gross shortening of the nasomaxillary region creating a severely overshot mandible and interference with sucking. Blindness, nystagmus, and recumbency are constant signs. The cerebrum and the cranial cavity are much smaller than normal.
INHERITED HYDRANENCEPHALY AND ARTHROGRYPOSIS
The defect is recorded in Corriedale sheep, and breeding trials indicate that it is inherited as an autosomal recessive character.1 Most affected lambs are found dead but facial deformity, including shortening of the mandible and distortion of the facial bones will be evident. At necropsy the predominant finding is the fixation and deformity of the joints of the limbs and vertebral column, and the almost complete absence of a cerebral cortex.
INHERITED CONGENITAL CEREBELLAR DEFECTS
Several inherited cerebellar defects occur congenitally in calves, lambs, and foals. Lesions of the cerebellum may or may not be obvious. They all need to be differentiated from similar defects known to be caused by intrauterine viral infections such as swine fever, bovine mucosal disease, and bluetongue.
Cerebellar hypoplasia
This occurs in Herefords, Guernseys, Holsteins, Shorthorns, and Ayrshires and appears to be conditioned by a factor inherited in a recessive manner. Most calves are obviously affected at birth. While lying down there is no marked abnormality, although a moderate lateral tremor of the neck occurs, causing a gentle side-to-side swaying of the head. Severely affected calves are blind; they have widely dilated pupils and their pupils do not react to light. Such calves are unable to stand, even when assisted, because of flaccidity of limb muscles. When less severely affected animals attempt to rise the head is thrown back excessively, the limb movements are exaggerated in force and range and are grossly incoordinated, and many calves are unable to rise without assistance. If they are placed on their feet the calves adopt a straddle-legged stance with the feet wide apart and the legs and neck extended excessively. On attempting to move, limb movements are incoordinated and the calf falls, sometimes backwards because of overextension of the forelimbs. Affected animals drink well but have great difficulty in getting to the teat or pail, attempts usually being wide of the mark. There are no defects of consciousness and no convulsions. Tremor may be evident while standing and there may be postrotational nystagmus after rapid lateral head movements. Sight and hearing are unimpaired and, although complete recovery does not occur, the calf may be able to compensate sufficiently to enable it to be reared to a vealing weight. Diagnosis can be confirmed by magnetic resonance imaging.1
At necropsy the most severe defect comprises complete absence of the cerebellum, hypoplasia of the olivary nuclei, the pons and optic nerves and partial or complete absence of the occipital cortex. Less severe defects include a reduction in size of the cerebellum and absence of some neuronal elements in a cerebellum of normal size.
Although the disease is dealt with generally as an inherited one there is no firm evidence to substantiate this view, and there are sporadic, non-inherited cases in other breeds.
Cerebellar atrophy of lambs (daft lamb disease 1)
This has been recorded in many sheep breeds in Britain, Corriedales in Canada and New Zealand, and in Drysdales.2 Affected lambs are normal at birth but are weak and unable to rise without assistance. At 3 days of age it is obvious that there is severe incoordination of limb movement, opisthotonos, tremor, and a straddle-legged stance. At necropsy the cerebellum may be of normal size but on histological examination there is gross atrophy of cerebellar neurons. The disease appears to be conditioned by a recessive gene but not as a simple homozygous recessive. A clinically similar disease has been observed in Border Leicester lambs. There is no histopathological lesion in the cerebellum, but there are significant lesions in the cervical muscles and the nerve supply to them. The disease is inherited, most likely as an autosomal recessive trait.
Star-gazing lambs (daft lamb disease 2)
An hereditary disease clinically similar to cerebral cortical atrophy has been described in newborn Leicester lambs in the UK but without histological evidence of Purkinje cell loss or reactive changes, considered the hallmark of ‘cerebellar cortical atrophy’. Affected lambs exhibit ‘dorsal arching of the neck with the head being pressed backwards’, which has is also described as ‘star-gazing’. Histological lesions are present in neck muscles and nerves but it is uncertain if these are primary or secondary.
Inherited ataxia of calves
This is a true cerebellar ataxia inherited as a recessive character in Jerseys, Shorthorns, and Holsteins. Clinically the condition resembles cerebellar hypoplasia except that signs may not occur until the calves are a few days to several weeks old. At necropsy the cerebellum is normal in size but histologically aplasia of neurons is evident in the cerebellum and also in the thalamus and cerebral cortex. An inherited condition, manifested by cerebellar ataxia which does not develop until calves are 6 weeks to 5 months old, has also been recorded but the cerebellum is small and macroscopically abnormal. Conspicuous degeneration of cerebellar Purkinje cells is evident on histological examination.
Cerebellar abiotrophy
This fourth disease in this group is described under the heading of abiotrophies.
Familial convulsions and ataxia in cattle
A neurological disease is recorded as being inherited in Aberdeen Angus cattle and their crossbreds and Charolais. In young calves there are intermittent attacks of convulsions, and in older animals these are replaced by a residual ataxia. The first signs appear within a few hours of birth; up to several months later there are single or multiple tetanic convulsions lasting for 3–12 hours. As these episodes disappear a spastic goose-stepping gait becomes apparent in the forelimbs and there is difficulty placing the hindlimbs. The characteristic necropsy lesion is a very selective cerebellar cortical degeneration. A proportion of cases make a complete recovery.3 The epidemiology of the disease is consistent with the operation of an autosomal dominant gene with incomplete penetrance.
Inherited congenital spasms of cattle
This condition has been recorded only in Jersey cattle and appears to be conditioned by a factor inherited in a recessive manner. Affected calves show intermittent, vertical tremor of the head and neck, and there is a similar tremor of all four limbs which prevents walking and interferes with standing. Although the calves are normal in all other respects, they usually die within the first few weeks of life. No histological examinations have been reported but a cerebellar lesion seems probable.
INHERITED SPASTIC PARESIS (ELSO HEEL)
This disease occurs in the Holstein, Aberdeen Angus, Red Danish, Ayrshire, Beef Shorthorn, Poll Hereford, Murray Grey, and many other breeds of cattle. It has been observed in crossbred Brahman cattle1 and in an Ayrshire x Beef Shorthorn crossbred steer. The disease occurs principally in calves, with signs appearing from several weeks to 6 months or more after birth. Occasional cases are reported as developing in adult European cattle and there is one report of the occurrence of the disease in adult Indian cattle.
It has been held for a long time that the disease is inherited, and the principal argument has centered on the mode of inheritance. Attempts to determine this have shown that the rate of occurrence in planned test matings is so low that, if inheritance is involved, it can only be the inheritance of a susceptibility to the disease. It is suggested that different time appearances represent a single disease entity with varying expressivity, the late forms being affected by cumulative environmental factors. A hypothesis proposed is of a gene with increased penetrance in the homozygote, with weak penetrance in the heterozygote, acting on a polygenic basis dependent on external factors.2,3 Infectious agents causing transmissible sub-acute spongiform encephalopathies interacting with trace elements such as lithium have been suggested as etiological agents but there is no evidence to support the hypothesis.3
In all forms of the disease there is excessive tone of the gastrocnemius muscle and straightness of the hock, usually more marked in one hindleg. If only one leg is affected it may be thrust out behind while the calf is walking and advanced with a restricted, swinging motion often without touching the ground. There is no resistance to passive flexion of the limb. The gastrocnemius and perforatus muscles are rigid and in a state of spastic contraction. There is a characteristic elevation of the tail. The lameness becomes progressively worse and affected animals spend much time lying down. Much body weight is lost and the animal is usually destroyed between 1 and 2 years of age.
Minor lesions described as regressive changes in the neurons of the red nucleus, in the reticular substance and the lateral vestibular nucleus are of doubtful significance, as are the observed reduction in inorganic phosphate and ascorbic acid levels in the blood and cerebrospinal fluid of affected calves. A lower CSF concentration than normal of a central neurotransmitter, dopamine, could also be an effect rather than a cause.
There are demonstrable lesions on radiologic examination of the tarsus but exhaustive examinations of muscles and tendons fail to reveal histological abnormalities. The absence of any structural lesion and the variation in intensity of the abnormality suggests that it is a functional one. The hypersensitivity of the myotic reflex which has been observed could be such a defect.
In Europe affected animals are kept for breeding purposes, especially if they are double-muscled. They are kept for this reason because of the efficacy of the curative surgical operation of partial tibial neurectomy and in view of the high incidence of double muscling in such calves. In the Holstein breed, and several German breeds, bulls which sire affected calves have been observed to have very straight hocks and to suffer from various forms of stifle and hock lameness early in life.
Several surgical techniques including tenectomy, partial tibial neurectomy, and triple tenectomy have been described.2 Partial tibial neurectomy under caudal epidural anaesthesia was done on 113 Belgian blue calves with spastic paresis.2 A telephone follow-up of the owners 3 months later revealed good results in 83%; a considerable improvement in 4.4%; and severe hyperflexion of the hock necessitated early culling for slaughter; and in 8% there was little or no improvement.2