DISORDERS OF THYROID FUNCTION (HYPOTHYROIDISM, HYPERTHYROIDISM, CONGENITAL HYPOTHYROIDISM, THYROID ADEMONA)
Disorders of thyroid function due to abnormalities in the thyroid gland, pituitary gland or hypothalamus are uncommon in the domestic species and are best documented for the horse. Thyroid disorders secondary to excessive or inadequate intake of iodine, or selenium deficiency are discussed under those headings.
ETIOLOGY
Disorders of thyroid function result in hypothyroidism or hyperthyroidism. Hypothyroidism can result from diseases of the thyroid gland (primary hypothyroidism), pituitary gland (secondary hypothyroidism due to reduced secretion of thyroid stimulating hormone), or hypothalamus (tertiary hypothyroidism, decreased thyrotropin [thyroid releasing hormone] secretion). Autoimmune thyroiditis has not been described in horses. Lymphocytic thyroiditis occurs in goats.1 Consumption of propylthiouracil (4 mg/kg body weight orally once daily for 4–6 weeks) induces hypothyroidism in adult horses.2,3 Administration of trimethoprim-sulfadiazine (30 mg/kg orally q.24 h for 8 weeks), which can induce hypothyroidism in humans and dogs, does not impair thyroid function of most horses.4
Hereditary congenital hypothyroidism secondary to defects in thyroglobulin production occurs in sheep, goats, and Afrikander cattle.5 The disease is inherited as an autosomal recessive trait.5 The cause of congenital hypothyroidism in foals is uncertain, although ingestion of nitrates by the pregnant dam is strongly suspected.6 Partial thyroidectomy of equine fetuses results in birth of foals with clinical and pathological characteristics similar to the spontaneous disease.7
Hyperthyroidism in horses is attributable to functional adenocarcinoma or adenoma of the thyroid gland8,9 but most thyroid tumors, are not functional.10,11
EPIDEMIOLOGY
The frequency with which hypothyroidism occurs in adult horses is unknown. It is relatively common practice to administer thyroid hormone or iodinated casein to fat horses, those with laminitis, rhabdomyolysis, or anhidrosis, or to enhance fertility, but documentation of abnormal thyroid function in these animals is rare. None of 79 clinically normal brood mares had an abnormal response to thyroid stimulating hormone administration,12 indicating that hypothyroidism is uncommon. Importantly, horses with non-thyroid related illness often have low concentrations of thyroid hormones in blood without evidence of thyroid dysfunction– this is referred to as the euthyroid sick or non-thyroidal illness syndrome and is not indicative of thyroid disease.13
Abnormalities of the thyroid gland were detected in 12% of 1972 goats examined in India.1 Of thyroid glands examined from 1000 goats in India, 2.4% had colloid goiter, 39% parenchymatous goiter, 1.8% lymphocytic thyroiditis, and 2.1% were fibrotic.1
Congenital hypothyroidism in foals occurs in western Canada and the western and northern USA. One survey of necropsy records of almost 3000 equine fetuses and neonatal foals in western Canada found that 2.7% had histologic evidence of thyroid and musculoskeletal abnormalities consistent with congenital hypothyroidism.14 Congenital hypothyroidism occurs in Dutch goats, Merino sheep, and Afrikander cattle.5,15 Hypothyroidism is reported in an East Friesian ram.16
Hyperthyroidism is a sporadic disease of older horses for which other risk factors are not identified.9,17
Thyroid tumors are common in older horses with ∼50% having adenomas evident on histologic examination of the thyroid gland.11 The clinical course of such tumors is benign, although their size can be quite impressive. Thyroid adenocarcinoma is much less common but has a malignant course.10,11
CLINICAL FINDINGS
Clinical characteristics of hypothyroidism in adult horses are poorly defined, largely because of the difficulty of confirming the diagnosis and the pharmacological effect of exogenous thyroid hormones. Clinical abnormalities anecdotally attributed to hypothyroidism include exercise intolerance, infertility, weight gain, maldistribution of body fat, agalactia, anhidrosis, and laminitis, among others. Definitive association of these clinical syndromes with abnormalities of thyroid function is lacking.
Thyroidectomy of horses causes a reduction in resting heart rate and body temperature, docility, decreased food intake, increased cold sensitivity, dull hair coat, and delayed shedding of hair.18,19 Blood and plasma volumes of horses increased after removal of the thyroid glands.19 Effects of thyroidectomy were reversed by administration of thyroxine, with the exception of blood and plasma volume which did not return to euthyroid values.19 Thyroidectomized horses did not become obese or develop laminitis.
Induced hypothyroidism in goats is evident as a loss of body weight, facial edema, weakness, profound depression, and loss of libido.20
Congenitally hypothyroid foals have a prolonged gestation but are born with a short silky hair coat, soft pliable ears, difficulty in standing, lax joints, and poorly ossified bones. The foals are referred to as dysmature. Characteristic musculoskeletal abnormalities include inferior (mandibular) prognathism, flexural deformities, ruptured common and lateral extensor tendons, and poorly ossified cuboidal bones.14
Horses with hyperthyroidism are tachycardic, cachexia, and have hyperactive behavior.9,17 There is usually detectable enlargement of the thyroid gland.
Thyroid adenomas are evident as unilateral, non-painful, enlargement of the thyroid gland of older (>15 years) horses. Thyroid adenocarcinoma presents as metastatic disease with both local and distant spread. Some affected horses have signs of hyperthyroidism, although this is unusual.
CLINICAL PATHOLOGY
Hematologic abnormalities in hypothyroid horses are not well documented. Induced-hypothyroidism in horses causes increases in serum concentrations of very low density lipoprotein, triglycerides, and cholesterol, and decreased concentrations of non-esterified fatty acids.21 Induced hypothyroidism in goats caused hypoglycemia, hypercholesterolemia, and anemia.20 Hypothyroidism in a ram caused hypercholesterolemia.16
Thyroid hormone assays
Assays are available for measurement of serum concentrations of T3, T4, free T4 (radioimmunoassay or equilibrium dialysis), and TSH.13 Values of each of these analytes varies depending on the method of analysis, physiologic status of the animal, and administration of other compounds (Table 29.8). Serum concentrations of thyroid hormones is high at birth and declines with age.22-25 There are statistically significant diurnal variations in serum concentrations of T3 and T4 in adult horses with lowest concentrations observed during the early morning hours, likely coincident with the time at which metabolic rate is lowest (Table 29.8).26 Feed restriction for 3–5 days lowers serum concentrations of T3, T4, and fT4 in horses by 24–42%.27 Administration of phenylbutazone decreases concentrations of fT4 (measured by equilibrium dialysis) and T4 by 4 days of treatment, and can persist for up to 10 days after discontinuation of phenylbutazone.28 The decrease in T4 is suggested to be attributable to displacement of T4 from protein binding sites by phenylbutazone, but this does not explain the decrease in fT4. The clinical significance of phenylbutazone-induced decreases in thyroid hormones is uncertain, but should be considered when assessing thyroid function in horses.
Table 29.8 Serum or plasma concentrations of thyroid hormones and thyroid stimulating hormone (TSH) in foals and horses
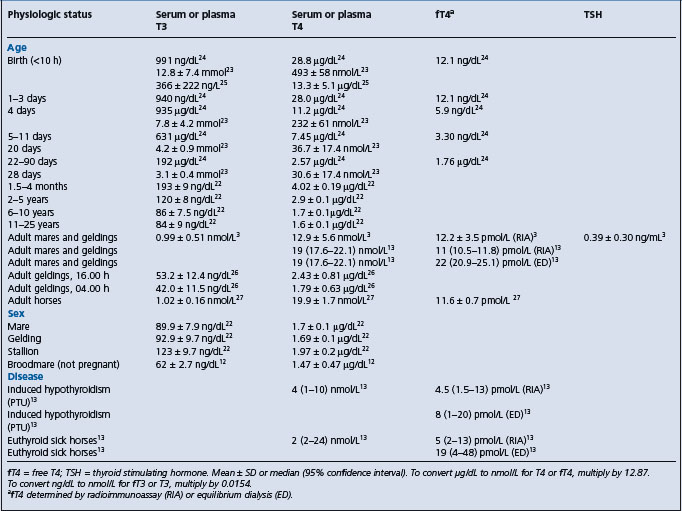
Because of the number of analytical and physiological factors that affect serum thyroid hormone concentrations, values considered normal vary considerably, as illustrated by the finding that 44 of 79 clinically normal non-pregnant broodmares had serum T4 concentrations below the reference range, although responses to TRH were normal.12 This example illustrates the need to determine reference ranges based on the methodology used and with well defined definition of the physiological state of the animals being tested.
Diagnosis of hypothyroidism is aided by demonstration of inappropriate responses of the thyroid gland to administration of TSH or TRH, although the use of these tests depends on determining the increase in serum T3 and/or T4 that is expected in normal horses and in horses with thyroid disease. Of 79 clinically normal mares, all had some increase in T3 and 77 had an increase in T4 2 hours after IV administration of 1 mg of TRH intravenously.12 The mean increase in serum T3 concentration was 4.5 times that of resting values (from 0.62 ng/mL to 2.44 ng/mL), whereas serum T4 concentration increased to a mean of 2.1 times that of resting value (from 14.7 ng/mL to 28.6 ng/mL).12 While responses to administration of TSH are reported, responses, other than complete lack of response, indicative of abnormal thyroid function have not been determined and the utility of the test has been questioned.27
The TSH response test involves administration of 5 IU of TSH intravenously. Blood samples are collected before, 30 min and 2 and 4 h after administration.29 Serum concentrations of T3 and T4 in healthy horses double after administration of TSH. An alternative involves administration of 5 IU intramuscularly and collection of blood before and 3 and 6 h after a TSH administration.30 TSH is currently unavailable.
The TRH response test requires administration of 0.5–1 mg of TRH intravenously. Serum concentrations of T3 and T4 at 2 and 4 h are double those before TRH administration in horses with normal thyroid function.31
Measurement of fT4 in serum is useful for assessment of thyroid function.3 fT4 concentrations can be normal in horses with low concentrations of T3 and T4, and in this situation are likely indicative of normal thyroid function.
Measurement of serum concentrations of TSH is useful in determining thyroid responsiveness to endogenous TSH. Elevated TSH concentrations in horses with low serum concentrations of T3, T4, or fT4 is indicative of thyroid dysfunction.13,32
Diagnosis of hypothyroidism in horses should be based on the presence of compatible clinical signs, low serum concentrations of thyroid hormones (T3, T4, fT4); elevated concentrations of thyroid stimulating hormone, lack of an increase in serum concentrations of thyroid hormones in response to administration of thyroid releasing hormone (TRH), and increased TSH concentration in serum in response to TRH administration. Diagnosis of hypothyroidism should not be based solely on clinical signs, or on the measurement of resting (unstimulated) serum T3 or T4 concentrations. At a minimum, appropriate clinical signs and documentation of an abnormal response to stimulation testing (TSH or TRH) are essential for diagnosis of hypothyroidism in horses. Measurements of fT4 concentrations determined by equilibrium dialysis are useful in determining thyroid function in sick horses in which T3 and T4 concentrations are low as fT4 concentrations will be normal in horses without thyroid disease.13
Foals with congenital hypothyroidism have abnormally low concentrations of T3 and T4, and less than expected increases in serum concentrations of these hormones in response to TSH administration.33
Horses with hyperthyroidism have markedly elevated concentrations of T3 and T4.9,17 Concentrations of T4 do not decline in response to administration of T3.17 T3 (2.5 mg) is administered intramuscularly twice daily for 3 days and serum concentrations of T3 and T4 measured. T4 concentrations in serum of healthy horses decline by approximately 80% whereas those of horses with hyperthyroidism do not decline.
NECROPSY FINDINGS
Findings on necropsy examination of hypothyroid horses have not been reported. Foals with congenital hypothyroidism have histologic evidence of thyroid hyperplasia, but no gross signs of goiter.
TREATMENT
Treatment of confirmed hypothyroidism in horses is achieved by administration of levo-thyroxine (20 μg/kg PO q24 h).34 Serum T3 concentrations peak in 1 h and then decline while concentrations of T4 peak in 2 h and persist for 24 h.34 The clinical status of the horse should be monitored during treatment and serum concentrations of T3 and T4 measured every several months. Iodinated casein, which is no longer readily available in the USA, is administered at 5 g/450 kg body weight orally once daily. Administration of thyroxine or iodinated casein for treatment of low serum thyroid hormone concentrations in horses with non-thyroidal illness syndrome (euthyroid sick syndrome) should be done judiciously.
A response to thyroxine administration is not necessarily confirmation of hypothyroidism as thyroxine can have marked effects in horses with normal thyroid function. Administration of thyroxine (up to 96 mg/470 kg horse, orally once daily) increases serum concentrations of T4 and, to a lesser extent, fT4, and decreases concentrations of TSH.35 The increases in T4 are associated with a loss of body weight, decreases in serum concentrations of triglycerides, cholesterol, and very low density lipoproteins, and an increase in whole body insulin sensitivity.35,36 Thyroxine should be administered with caution to horses with normal thyroid function.
CONTROL
There are no recognized control measures for hypothyroidism in adult horses. Minimizing intake of nitrates by pregnant mares appears warranted, but definitive proof of the efficacy of this practice is lacking. Pregnant mares should not be fed fodder or supplements that interfere with thyroid function.
The inherited disorder in sheep, cattle, and goats can be prevented by selective breeding.
1 Ramakrishna C, et al. Ind Vet Med J. 1992;16:37.
2 Johnson PJ, et al. Equine Vet J. 2003;35:296.
3 Breuhaus BA. J Vet Intern Med. 2002;16:109.
4 Rothschild CM, et al. J Vet Intern Med. 2004;18:370.
5 Veenboer GJM, et al. Endocrinol. 1993;132:377.
6 Allen AL, et al. Can Vet J. 1996;37:349.
7 Allen AL, et al. Equine Vet J. 1998;30:53.
8 Alberts MK, et al. J Am Vet Med Assoc. 2000;217:1051.
9 Ramirez S, et al. J Vet Intern Med. 1998;12:475.
10 Hovda LR, et al. J Am Vet Med Assoc. 1990;197:1187.
11 Dalefield RR, Palmer DN. J Comp Pathol. 1994;110:57.
12 Meredith TB, Dobrinski I. J Am Vet Med Assoc. 2004;224:892.
13 Breuhaus BA, et al. J Vet Intern Med. 2006;20:371.
14 Allen AL, et al. Can Vet J. 1994;35:31.
15 Dolling CE, Good BF. J Endocrinol. 1976;71:179.
16 Sipos W, et al. J Vet Med A. 2004;51:90.
17 Alberts MK, et al. J Am Vet Med Assoc. 2000;217:1051.
18 Lowe JE, et al. Cornell Vet. 1974;64:276.
19 Vischer CM, et al. Am J Vet Res. 1999;60:14.
20 Ramakrishna C, et al. Ind Vet J. 1995;71:1107.
21 Frank N, et al. Am J Vet Res. 1999;60:730.
22 Chen CL, et al. Am J Vet Res. 1981;42:1415.
23 Murray MJ, Luba NK. Equine Vet J. 1993;25:237.
24 Irvine CHG, Evans MJ. J Reprod Fertil Suppl. 1975;23:709.
25 Shaftoe S, et al. J Equine Vet Sci. 1988;8:310.
26 Duckett WM, et al. Equine Vet J. 1989;21:123.
27 Messer NT, et al. Am J Vet Res. 1995;56:116.
28 Ramirez S, et al. J Vet Intern Med. 1997;11:371.
29 Held JP, Oliver JW. J Am Vet Med Assoc. 1984;184:326.
30 Morris DD, Garcia M. Am J Vet Res. 1983;44:503.
31 Lothrop CD, Nolan HL. Am J Vet Res. 1986;47:942.
32 Rothschild CM, et al. J Vet Intern Med. 2004;18:370.
33 McLaughlin BG, et al. Can Vet J. 1986;27:264.
34 Chen CL, et al. J Equine Vet Sci. 1984;4:5.