Chapter 9 Diseases of the hemolymphatic and immune systems
ABNORMALITIES OF PLASMA PROTEIN CONCENTRATION 439
HEMORRHAGIC DISEASE 441
PURPURA HEMORRHAGICA 443
DISORDERS OF RED CELL NUMBER OR FUNCTION 450
DISORDERS OF WHITE CELLS 460
LYMPHADENOPATHY (LYMPHADENITIS) 464
DISEASES OF THE SPLEEN AND THYMUS 464
IMMUNE DEFICIENCY DISORDERS (LOWERED RESISTANCE TO INFECTION) 466
AMYLOIDOSES 467
PORPHYRIAS 469
Abnormalities of plasma protein concentration
Plasma contains hundreds of proteins, including albumin, immunoglobulins, clotting factors, acute phase proteins, hormones and cytokines. The proteins in plasma are produced by the liver (albumin, acute phase proteins (fibrinogen, serum amyloid A), clotting factors) and lymphoid organs (the gamma-globulins and many cytokines). The plasma proteins serve as sources of amino acids for tissues, as carrier molecules, maintain plasma oncotic pressure, regulate immune function, and effect hemostasis and fibrinolysis. Defects or deficits of individual proteins can result in specific diseases, including immune deficiency, defective hemostasis and endocrinopathy. The individual diseases resulting from loss of activity of specific proteins are dealt with under the headings of those diseases. Provided here is an overview of hypoproteinemic and hyperproteinemic states.
HYPOPROTEINEMIA
ETIOLOGY
Hypoproteinemia is a plasma or serum total protein concentration that is below that expected in animals of the same age, sex, physiological state and species. Hypoproteinemia can be a result of a reduction in concentration of albumin and globulin, or a reduction in either albumin or globulin concentrations. Abnormalities in plasma protein concentration include:
• Panhypoproteinemia with hypoalbuminemia and hypoglobulinemia
• Hypoproteinemia with hypoalbuminemia and normal globulin concentration
• Hypoproteinemia with hypoglobulinemia and normal albumin
• Normal total protein concentration with hypoalbuminemia and hyperglobulinemia, and less commonly, hyperalbuminemia and hypoglobulinemia.
The specific deficiency has important diagnostic significance.
Panhyproteinemia
Hypoproteinemia with hypoalbuminemia and hypoglobulinemia can be either relative or absolute.
Relative hypoproteinemia occurs when plasma protein concentrations are lower than normal but the absolute content of protein in the vascular space is normal. This is a dilutional hypoproteinemia and is attributable to either excessive fluid therapy or excessive water intake. These causes are readily determined from a review of the history and treatment of the animal and resolve within hours of discontinuation of fluid therapy or restriction of fluid intake.
Absolute hypoproteinemia occurs when there is a reduction in the amount of plasma proteins in the vascular space in the presence of normal or almost normal plasma volume. The reduced protein concentration can be the result of impaired production or accelerated loss. Reduced production of all plasma proteins occurs only as part of malnutrition and starvation. Liver disease can cause a reduction in the concentration in plasma of those proteins produced by the liver (see below) but in large animals is an unusual cause of hypoproteinemia. Loss of protein is a more common cause of hypoproteinemia.
The loss of proteins can be either from the vascular space into the extravascular compartment (e.g. endotoxemia, vasculitis) or from the body (compensated hemorrhage, glomerulonephritis, protein-losing enteropathy). This situation is evident as a reduction in concentrations of both albumin and globulins, and in hemorrhagic disease by a reduction in hematocrit. Loss because of vascular leakage is usually evident as hypoproteinemia with normal or elevated hematocrit. Diseases causing panhypoproteinemia include:
• Hemorrhage – hypoproteinemia occurs when plasma volume is restored after severe hemorrhage, or in normovolemic anemia when there is persisting loss of blood (see page 451 for a list of diseases causing chronic hemorrhagic anemia). All causes of chronic blood loss can cause hypoproteinemia
• Endotoxemia – protein loss is secondary to leakage of protein from the vascular space into interstitial spaces because of increased capillary permeability
• Vasculitis – causing increased capillary permeability and leakage of protein. Evident in many systemic diseases, including African horse sickness, purpura hemorrhagica, swine fever, and malignant catarrhal fever
• Protein-losing enteropathy – the initial change is in plasma albumin concentration, but panhypoproteinemia ensues as the disease progresses. Diseases causing protein-losing enteropathy include:
• Urinary tract disease including cystic calculi, pylonephritis, glomerulonephritis
• Acute, severe inflammation of the peritoneal or pleural membranes (peritonitis, pleuritis). The hypoproteinemia occurs early in the disease but if the disease becomes chronic hypergammaglobulinemia ensues
Hypoalbuminemia
Hypoalbuminemia with normal or elevated plasma globulin concentration occurs in diseases in which there is insufficient production of albumin by the liver or excessive or selective loss of albumin compared to loss of globulin. Insufficient production of albumin occurs in diseases of the liver, although these animals might not necessarily be hypoproteinemic,1 and malnutrition or starvation. Diseases of the liver that cause hypoalbuminemia are usually diffuse, severe and chronic. The prolonged half-life of albumin in cattle and horses (approximately 18 d) renders them less liable to hypoalbuminemia than smaller animals.
Albumin has a lower molecular weight than most globulins, especially the immunoglobulins, and can be lost selectively in renal or gastrointestinal disease. Diseases associated with hypoalbuminemia and normal to elevated globulin concentrations include:
• Amyloidosis – loss of albumin into urine or the gastrointestinal tract is sometimes offset, in terms of plasma protein concentration, by increases in plasma globulin concentration
• Chronic peritonitis or pleuritis – loss of albumin into the inflammatory exudate is offset, in terms of plasma total protein concentration, by increases in plasma globulin concentration
Hypogammaglobulinemia
Hypoglobulinemia with normal plasma albumin concentration occurs in few diseases. Notably, it is a feature of failure of transfer of passive immunity in neonates (Ch. 3). Hypoglobulinemia is an unusual isolated defect in other diseases. It can be detectable in immunodeficiencies causing decreased production of gammaglobulins, such as combined variable immunodeficiency in horses.2
PATHOPHYSIOLOGY
Panhypoproteinemia and hypoalbuminemia cause a reduction in concentration in plasma of proteins essential for a variety of functions. Overall, the reduction in plasma albumin concentration results in a low plasma oncotic pressure. Low plasma oncotic pressure allows movement of fluid from the vascular space, causing a reduction in plasma volume and increases in extravascular volume. The reduction in plasma volume lowers blood flow to tissues and can result in organ dysfunction. The increased extravascular volume is evident as edema.
Low plasma albumin concentration, in addition to the reduction in plasma oncotic pressure, reduces opportunities for transport of substances in the plasma, including hormones and electrolytes (calcium).
Hypogammaglobulinemia increases the risk of infectious disease (see Ch. 3).
CLINICAL SIGNS
The clinical signs of hypoproteinemia are lethargy, ill-thrift and edema. The edema is usually distributed symmetrically, with some species predilection for site of accumulation – ventral edema in horses, submandibular edema in cattle and sheep. Affected animals are often tachycardic because of the reduced plasma volume.
Signs of the inciting disease will also be present (weight loss, diarrhea, melena, polyuria).
CLINICAL PATHOLOGY
Detection of hypoproteinemia is readily achieved by routine hematologic or serum biochemical testing. The albumin to globulin (A:G) ratio can be useful in assessment of the hypoproteinemia. Hypoalbuminemia with normal globulin concentration results in a low A:G ratio, whereas panhypoproteinemia results in a normal A:G ratio. Selective deficiencies can be detected by protein electrophoresis or measurement of concentrations of specific proteins, such as the immunoglobulins by enzyme-linked immunosorbent assay (ELISA), radial immunodiffusion (RID), or immunoturbidimetric analysis (see Failure of transfer of passive immunity in Ch. 3).
Measurement of plasma oncotic pressure is useful in detecting low plasma oncotic pressure, which contributes to a reduction in plasma volume and increases in extravascular fluid which can lead to formation of edema. Plasma oncotic pressure is proportional to the plasma protein concentration, with the greatest correlation being with plasma albumin concentration in animals that have not received dextran solutions. Intravenous administration of dextran or hydroxyethyl starch increases plasma oncotic pressure.3
NECROPSY
The changes are those of the inciting disease, or secondary infection in animals with hypogammaglobulinemia. Edema can be present in subcutaneous and internal connective tissues.
TREATMENT
The principles of therapy are treatment of the inciting disease, and correction of hypoproteinemia or low plasma oncotic pressure. Correction of hypoproteinemia (hypoalbuminemia, hypogammaglobulinemia) is achieved by administration of plasma by transfusion. Unless anemia is also present, plasma transfusion is preferred over blood transfusion. The amount of plasma transfused to neonates is discussed in Chapter 3. Plasma transfusion to adult horses and cattle is often limited by the cost of the plasma. Ideally, plasma should be transfused to increase plasma albumin concentrations to more than 2.0 g/dL (20 g/L). This can be calculated as (where 0.05 is the proportion of body weight that is plasma):
Current plasma albumin content = body weight (kg) × 0.05 × (plasma albumin concentration in g/L)
Desired plasma albumin content = body weight (kg) × 0.05 × (desired albumin concentration in g/L)
Amount of albumin required (g) = Desired plasma albumin content – current albumin content
Volume of plasma required (L) = Amount of albumin required (g)/albumin concentration in transfused plasma (g/L).
A numerical example is of a 500 kg horse with a plasma albumin concentration of 1.5 g/dL (15 g/L) and a target plasma albumin concentration of 2.5 g/dL (25 g/L):
Current plasma albumin content = 500 (kg) × 0.05 × 15 (g/L) = 375 g
Desired plasma albumin content = 500 (kg) × 0.05 × 25 (g/L) = 525 g
Amount of albumin required (g) = 525 – 375 = 150 g
Volume of plasma required (L) = 150 (g)/30 (g/L) = 5 L.
It is a frequent observation that transfusion of the calculated volume of plasma, while improving clinical signs, does not result in the expected increase in plasma albumin concentration. This is probably because transfusion of albumin results in an increase in plasma oncotic pressure and a net movement of fluid from the extravascular space into the vascular space with subsequent expansion of the plasma volume. The expansion of plasma volume dilutes the administered albumin and attenuates the increase in plasma protein concentration.
Plasma oncotic pressure can be increased by intravenous administration of hydroxyethyl starch or high-molecular-weight dextrans. The dose is 8–10 mL/kg of 6% solution delivered intravenously over 6–12 hours.3,4
HYPERPROTEINEMIA
ETIOLOGY
Panhyperproteinemia
An increase in concentration of all plasma proteins occurs only in situations in which there is a reduction in plasma water content. This occurs in animals that are severely dehydrated through lack of access to water, inability to drink, loss of protein-poor body fluids (diarrhea, vomitus) or excessive polyuria with inadequate water intake.
Hyperglobulinemia
Hyperglobulinemia occurs as a consequence of chronic inflammation or abnormal production of globulins. Chronic inflammation causes a polyclonal gammopathy whereas plasma cell neoplasia (plasmacytoma, myeloid leukemia, see ‘Leukoproliferative disease’) causes a monoclonal gammopathy. Any chronic inflammatory disease, including those of infectious, toxic or neoplastic origin, can cause hyperglobulinemia.
PATHOPHYSIOLOGY
Chronic inflammation results in chronic stimulation of the immune system with subsequent increased production of immune globulins and acute-phase proteins. Monoclonal gammaglobulinemia occurs as a result of unrestrained production of gammaglobulins by neoplastic plasma cells.
CLINICAL PATHOLOGY
Measurement of plasma protein concentration reveals hyperglobulinemia and/or hyperfibrinogenemia. Serum protein electrophoresis demonstrates whether the abnormality is a polyclonal or monoclonal gammaglobulinopathy. Measurement of specific immunoglobulins (IgG, IgA, etc.) can be useful. Fibrinogen concentration must be measured in plasma as it is consumed during the clotting process when blood is allowed to clot.
Hemorrhagic disease
Hemorrhagic disease is manifest as the presence of hemorrhage of unusual duration or severity, either externally from apparently minor wounds, or into body cavities, or as the presence of petechial and ecchymotic hemorrhages in mucous and conjunctival membranes and the skin. Petechial and ecchymotic hemorrhage, spontaneous hemorrhage or excessive bleeding after minor injury may result from increased capillary fragility, disorders in platelet function or defects in the coagulation mechanism of the blood.
Diagnosis
Diagnosis of hemorrhagic disease is based on the demonstration of abnormalities in the activity, concentration or function of components of blood coagulation and fibrinolysis. The exception is diagnosis of vasculitis, which is achieved by biopsy, usually of skin, and histologic examination and demonstration of inflammatory lesions in the walls of blood vessels.
Demonstration of prolonged bleeding time is achieved using devices that inflict a controlled wound on either the skin or a mucous membrane (template bleeding time). A wound is inflicted in the skin and blood is periodically collected on to absorbent filter paper until bleeding ceases. The time from discharge of the device until bleeding stops is the ‘bleeding time’. The mean template bleeding time is less than 5 minutes in most healthy animals.1
Care must be taken when collecting specimens of blood for measurement of factors involved in coagulation or fibrinolysis. Blood samples collected into containers that do not contain an anticoagulant will rapidly clot and the resulting serum sample will be minimally useful for any tests of clotting or fibrinolysis. The ideal anticoagulant for assays of clotting and fibrinolysis is trisodium citrate (1 part of 3.8% trisodium citrate to 9 parts of blood). Sodium citrate decreases the concentration of ionized calcium in blood and thereby inhibits platelet activity.2 Heparin, both unfractionated (conventional) and low-molecular-weight inhibits thrombin activity and activates platelets and is not a suitable anticoagulant for measurement of clotting times or platelet activity.2 Potassium ethylenediamine tetraacetic acid (EDTA) interferes with platelet function.
An integrated measure of the capacity of blood to clot is the activated clotting time. In this test, blood is collected into plastic syringes that do not contain an anticoagulant and then immediately injected into glass tubes containing diatomaceous earth. The tubes are gently agitated and then incubated for 1 minute in a water bath at 37°C. The tubes are then removed from the water bath and examined for clotting of blood by gently rolling the tube. The tube is then returned to the water bath and reexamined every 30–60 minutes.
The rate of clot retraction of blood collected into a glass tube that does not contain anticoagulants is a measure of platelet activity. The time until maximum clot retraction is 1–2 hours in most species when the blood is held at 37°C.
Measurement of prothrombin time (an indicator of activity of the extrinsic clotting system), activated partial thromboplastin time (an indicator of functionality of the intrinsic clotting system) and thrombin time (common pathway) are routinely performed for animals. The tests are reliable when performed properly; however, values for normal animals can vary and the recommendation is that, when submitting a sample from an animal with suspected coagulopathy, a sample from a similar healthy animal should also be examined. If prothrombin or activated partial thromboplastin time are prolonged, other tests to determine the specific factor(s) involved might be warranted.
Measurement of the activity or concentration of blood clotting factors is routine in human medicine and many of these tests have been adapted for use in animals. Chromogenic assays of factors VII, VIII:C, IX and X developed for testing of human plasma are reliable when used for testing of horse plasma.3 An ELISA for von Willebrand factor is available that is suitable for use in a number of species, including horses, pigs, and cattle.4 While most functional assays, including chromogenic assays, are suitable for use among species, most immunologically based assays developed for use in humans are not suitable for use in animals.5 It is important that assays should be validated in the species of interest before clinical use in animals.
Fibrinogen is an essential substrate for clot formation, and low plasma concentrations of fibrinogen, such as can be encountered in animals with disseminated intravascular coagulation, can impair blood clotting. Measurement of fibrin (fibrinogen) degradation products (FDP) has been used to detect disseminated intravascular coagulation in horses but the test has poor sensitivity and specificity. Measurement of D-dimer concentration has the potential to be much more useful than FDP in assessment of fibrinolysis and detection of thromboembolic disease, including disseminated intravascular coagulation and coagulopathies.6-9 Performance characteristics vary among assays and kit suppliers. The FDP assays had low sensitivity (< 40%), whereas the most accurate d-dimer kit had 50% sensitivity and 97% specificity for diagnosis of disseminated intravascular coagulation in horses with colic.9 The activity of antithrombin (previously antithrombin III), a cofactor of heparin, is measured in horses as a means of assessing the anticoagulant activity of plasma. Activity of antithrombin is reduced in animals with coagulopathies secondary to gastrointestinal disease. This factor is best measured in concert with thrombin–antithrombin complex concentration, and protein C and plasminogen activity to detect hypercoagulable states.10
Platelet count in blood should be evaluated in any animal with a hemorrhagic diathesis. Caution should be exercised in interpreting low platelet counts determined by automated analyzers, as clumping of platelets can cause artificially low values. This pseudothrombocytopenia can be a result of anticoagulant induced ex-vivo aggregation of platelets,11 which can be readily detected by microscopic examination of the blood smear. Platelets counts of less than 100 000 cells/μL are considered abnormal, although excessive hemorrhage is usually not apparent until platelet counts are below 40 000 cells/μL. Determination of the proportion of platelets that stain with thiazole orange dye (reticulated platelets) can be useful in determining the bone marrow regenerative response in horses, and probably other species, with thrombocytopenia.12,13 Reticulated platelets are those platelets that have been recently released from bone marrow. Healthy ponies have 1.3–2.8%, and horses have 1–3.4% of platelets in circulation that stain with thiazole orange. Thrombocytopenic, equine-infectious-anemia-positive ponies have 11–48% and thrombocytopenic, equine-infectious-anemia-negative horses have 2–9% thiazole-orange-staining platelets in the circulation.13 Platelet function of horses can be evaluated using platelet function analyzers designed for use with human blood, ultrastructure and flow cytometry. Impaired platelet aggregation can be detected as a prolongation in closure time using cartridges with collagen– adenosine diphosphate (CT-ADP) and collagen– epinephrine (CT-Epi) as platelet agonists. In normal horses calculated reference ranges are 60.5–115.9 seconds and 158.5–>300 seconds for CT-ADP and CT-Epi, respectively.14
Values of the above variables in normal animals have been reported inconsistently and are available in textbooks dealing with hematology. Values in foals and calves of increasing age are reported.15,16
Treatment of coagulopathies
Plasma is often administered to animals with hemorrhagic diatheses to replace clotting factors that are deficient because of failure of production (e.g. warfarin intoxication), increased consumption (e.g. in disseminated intravascular coagulation) or dilution (e.g. in animals with severe hemorrhage treated by administration of large quantities of fluids). The actual concentration or activity of factors involved in clotting or fibrinolysis depends upon the methods used to collect and store the plasma. Fresh frozen plasma kept at −80°C retains much of the activity of clotting factors (VII, VIII, etc.) and inhibitors of coagulation, including antithrombin, protein C, protein S and antitrypsin, for up to 1 year, whereas plasma stored at higher temperatures might not retain as much activity. The dosage varies from 2–10 mL/kg body weight (BW) intravenously, but this has not been critically evaluated. Platelet-rich plasma, which requires more sophisticated collection techniques, is useful for treatment of severe thrombocytopenic purpura. Platelet rich plasma can be prepared by centrifugation of blood at 150 × g for 20–30 minutes. Plasma is preferred over whole blood for treatment of nonanemic hemorrhagic diatheses.
Aminocaproic acid (30–100 mg/kg intravenously) reduces plasma fibrinogen concentration and decreases partial thromboplastin time of horses for up to 5 hours after administration. At the higher dose, alpha-2-antiplasmin activity is increased and fibrinogen concentration is decreased, consistent with an action of the drug to inhibit fibrinolysis.17 The utility of aminocaproic acid to inhibit bleeding in clinical situations has not been determined.
Tranexamic acid inhibits fibrin degradation and is used as adjunctive treatment in animals with hemorrhagic diathesis. Its efficacy in farm animals has not been reported. Carbazochrome is a compound that stabilizes capillary membranes and is used for treatment of exercise-induced pulmonary hemorrhage in horses, although with undetermined efficacy.
Formalin has been suggested as an effective treatment of excessive hemorrhage in horses, although it does not appreciably alter bleeding time or indices of coagulation.17 A common dose for an adult horse is 1 L isotonic electrolyte solution (saline or lactated Ringer’s solution) with a final concentration of formalin of 0.37–0.74%. Adult goats administered a 5.5% solution of formalin in lactated Ringer’s solution intravenously had a marked decrease in clotting time. However, this dose in horses is expected to be toxic.1
Administration of aspirin to horses inhibits platelet function in a dose-dependent fashion for 48 hours after a single dose of 12 mg/kg.18 This is not the case in cattle, in which aspirin does not inhibit platelet aggregation even at doses of 100 mg/kg orally.19 Aspirin irreversibly inhibits activity of thromboxane synthetase in both cattle and horses for a prolonged period (days) despite having a short plasma elimination half-life (hours). The bleeding time in horses is not restored until the affected platelets have been replaced by unaffected platelets.18 Dosages of aspirin in horses range from 15–100 mg/kg orally every 8–12 hours to 10 mg/kg orally every 48 hours.
Warfarin reduces the concentration of vitamin-K-dependent clotting factors by inhibiting hepatic production of these compounds. Therapeutic use of warfarin was limited to treatment of navicular disease in horses, although its use for this purpose is now archaic.
Heparin, and the newer heparin-related compounds (low-molecular-weight heparins) dalteparin and enoxaparin, have been used in horses with, or at risk of developing, coagulopathies.20,21 The low-molecular-weight heparins appear to be effective in reducing the frequency of coagulopathy in horses with colic20 without the adverse effect of heparin on hematocrit and clotting time.22 Calcium heparin causes in-vivo red cell aggregation in horses, with a resultant reduction in hematocrit and hemoglobin concentration and a reduction in platelet count.21-23 The low-molecular-weight heparins are dosed on the basis of anti-factor-Xa activity. At doses of these compounds that prolong factor Xa activity and thrombin time, they have a minimal effect on bleeding time or activated partial thromboplastin time in horses.20,22,24 A range of doses of heparin calcium have been employed, ranging from 40 IU/kg BW intravenously or subcutaneously every 12–24 hours to 150 IU/kg BW initially followed by 125 IU/kg BW every 12 hours for 3 days and then 100 IU/kg BW every 12 hours.20,21 Dalteparin (50 and 100 anti-Xa U/kg) and enoxaparin (40 and 80 anti-Xa U/kg) can be administered once daily to horses.24
Sodium pentosan polysulfate, a compound with heparin-like activity used for treatment of arthritis in horses, at doses of 3, 6 or 10 mg/kg, causes dose-dependent increases in partial prothrombin time that persist for 24–48 hours.25 This drug is not used for treatment of coagulopathies.
Hirudin, an anticoagulant originally derived from leaches but now available as a recombinant compound, is a specific inhibitor of thrombin that is independent of antithrombin activity. The compound could be useful in treatment of hypercoagulable states in which there is diminished thrombin activity. Recombinant hirudin has been investigated in horses in which the maximum plasma concentration occurred at approximately 130 minutes and declined with a terminal half-life of approximately 600 minutes. A doubling of activated partial thromboplastin time occurred 1.5 hours after subcutaneous administration of 0.4 mg/kg.26 The clinical efficacy of recombinant hirudin has not been determined.
Tissue plasminogen activator increases the activity of plasmin, thereby facilitating dissolution of clots. Its use in farm animals has not been reported, with the exception of its injection into the anterior segment of the eye to dissolve fibrin associated with uveitis in horses.
Streptokinase and urokinase have been used to facilitate dissolution of fibrin clots in farm animals, but there has been no critical analysis of their effectiveness.
DISEASES CAUSING HEMORRHAGE
Vasculitis
Septicemic and viremic diseases
The vasculitis is associated with endothelial damage occurring as a direct result of infection of the endothelium (e.g. equine herpesvirus-1 myeloencephalopathy, African horse sickness) or from immune-mediated events centered on the endothelium (e.g. purpura hemorrhagica). It may be complicated by defects in blood coagulation and platelet disorders depending upon the infection. In many instances coagulation defects are a manifestation of early disseminated intravascular coagulation. Clinically, petechial and ecchymotic hemorrhages associated with septicemia are most obvious in the mucous membrane of the mouth, vulva and conjunctiva or in the sclera but they are widely distributed throughout the body on postmortem examination. Diseases causing vasculitis include:
• Systemic viral diseases: equine viral arteritis, equine infectious anemia, African horse sickness, malignant catarrhal fever, bovine ephemeral fever, bovine virus diarrhea, bluetongue, hog cholera, swine fever, equine herpesvirus-1 myeloencephalopathy
• Chlamydial and rickettsial diseases: Anaplasma phagocytophila
• Bacterial diseases: salmonellosis, Histophilus somni infection, Actinobacillus sp. pleuropneumonia infection, pasteurellosis, erysipelas in pigs
• Miscellaneous: aspergillosis, Strongylus vulgaris infection.
Purpura hemorrhagica
This is a hemorrhagic disease of horses associated with a leukocytoclastic vasculitis. The majority of cases occur as a sequel to strangles. Cases also occur following immunization against Streptococcus equi and as a sequel to infection with other streptococci. The disease appears to be an immune complex-mediated disease with deposition of IgA-containing immune complexes on vessel walls.27,28 Hemorrhagic tendencies in the disease include petechial and ecchymotic hemorrhages but also may result in large extravasations of blood and serum into tissues. The hemorrhage and exudation of serum may result in anemia and a depression in the circulating blood volume. Hemorrhage associated with purpura is usually treated with blood transfusions and corticosteroids. A fuller description of the syndrome is given elsewhere.
Necrotizing vasculitis
Of unknown etiology but possibly immune mediated, this syndrome occurs in all species.29 It is similar to purpura and may be local or generalized with petechial hemorrhage and serosanguineous exudation subcutaneously and into tissue spaces. Hemorrhagic tendencies associated with vasculitis may be confused with those associated with a defect in the clotting mechanisms as the primary cause. Differentiation depends on accurate laboratory examination.
Treatment
Treatment of vasculitis centers on removal of the inciting cause and minimizing or eliminating inflammation in the vessel. Disease-specific treatments are discussed under each of those topics. Inflammation can be reduced by administration of glucocorticoids, the selection and dose of which vary with species (see Formulary in the Appendix). General supportive treatment can include the administration of blood or plasma if severe anemia or hypoproteinemia occur.
Coagulation defects
Coagulation defects can be either acquired or inherited. Acquired defects are usually related to exposure to compounds that interfere with production of clotting factors, or that cause depletion of these factors. Inherited defects usually present in young animals, but defects that only marginally increase clotting time might not be detected until the animal undergoes surgery or suffers trauma. Demonstration of defects in blood clotting is based on observation of signs of excessive hemorrhage, with confirmation achieved by measurement of bleeding time and laboratory examination of the activity or concentration of soluble blood clotting factors.
Acquired hemostatic defects
Acquired clotting defects include those associated with intoxications that impair production or function of clotting factors, and those related to depletion of clotting factors. Disseminated intravascular coagulation is a common cause of hemorrhagic diathesis in animals that is discussed in detail under that heading. Protein losing nephropathy is associated with loss of antithrombin in urine and increased risk of thrombosis in cattle. Similarly, horses with protein-losing enteropathy have low plasma concentrations of antithrombin, which could contribute to the thrombotic tendency noted in these animals.30
Reduction of vitamin K1-dependent clotting factors II, VII, IX and X may result from coumarol poisoning following ingestion of coumarol containing plants such as Melilotus alba, Anthoxanthum odoratum, Apium nodiflorum, Ferula communis (giant fennel) or warfarin, brodifacoum and related compounds.31-33 This syndrome is discussed in detail under each of those headings. Vitamin K deficiency, other that induced by intoxication with the compounds listed above and the disorder in postweaned pigs discussed below, has not been reported in farm animals, probably because forage contains high concentrations of this compound.
A hemorrhagic syndrome in postweaned pigs is recorded from the USA, New Zealand, France, Japan, Germany, Brazil and South Africa.34-36 The syndrome occurs as an outbreak with anemia, hemarthrosis, spontaneous hemorrhage under the skin of the legs and body and hemorrhage following management procedures such as castration. There is a high case fatality and the syndrome is particularly common in pigs a few weeks after weaning. There is prolongation of the prothrombin time and activated partial thromboplastin time. The outbreaks resolve promptly following the injection of vitamin K or its inclusion in the diet. An association has been made with housing on mesh floors.36 The disease is believed to be due to a deficiency of vitamin K in the diet coupled with housing that precludes intake of vitamin K2 from the feces or bedding and decreased synthesis in the gut as a result of antibiotics in the feed, especially sulfonamides.
Snake venom may have procoagulant or anticoagulant action.37 In both cases coagulation defects may occur as procoagulant toxins result in the activation, consumption and depletion of prothrombin and fibrinogen, leading to a coagulopathy, prolonged clotting times and epistaxis.38,39
Carcass hemorrhage or blood splash in slaughter lambs has been associated with extended prothrombin times because of prior grazing of coumarin-producing plants. The method of electrical stunning at slaughter can also result in carcass hemorrhage.40
Parafilaria bovicola produces large extravasations of blood under the skin of cattle and to some extent in tissue spaces.41 Bleeding from the skin may be the presenting sign of infestation.
A number of fungal toxins can cause hemorrhagic disease when ingested:
• Aflatoxins produced by Aspergillus spp. do so in association with increased prothrombin time in cattle, swine and horses
• Trichothecene toxins produced by fungal infestations of feed by Fusarium spp., Myrothecium spp., Cephalosporium spp.
• Trichothecium spp. also produce hemorrhagic disease
Hydroxyethyl starch solution (Hetastarch), used to increase plasma oncotic pressure in animals with hypoproteinemia, prolongs cutaneous bleeding, prothrombin and activated partial thromboplastin times and decreases fibrinogen concentration and von Willebrand antigen and factor VIII:C activities in ponies, apparently by dilution of clotting factors.42
Navel (umbilical) bleeding in newborn piglets is a syndrome of unknown etiology. Following birth and for periods up to 2 days afterwards, blood drips or oozes from the umbilicus of affected pigs to produce severe anemia, with death frequently occurring from crushing. The navel cords are abnormally large and fleshy and fail to shrink after birth. The defect appears to be one of immaturity of collagen so that a proper platelet clot does not form. Ear-notching for identification is also followed by excessive bleeding. A variable number of piglets within the litter may be affected and the syndrome may have a high incidence on certain problem farms. The addition of vitamin K and folic acid to the sows’ ration may be followed by a drop in incidence but controlled trials with menadione have shown no effect. Vitamin C given to pregnant sows for at least 6 days before farrowing appears to prevent the syndrome.
Jejunal hemorrhage syndrome in cattle is discussed in Chapter 5.43
Infestation of sheep by Fasciola hepatica shortens activated partial thromboplastin time and prolongs prothrombin and thrombin times.44
Inherited or congenital defects in hemostasis
Hemophilia A
Factor VIII:coagulant (VIII:C) deficiency, hemophilia A, is recorded in Thoroughbreds,45 Standardbreds, Arabian and Quarter horse colt foals46 and sheep47 and is associated with a deficiency in factor VIII. The disease in the Quarter horse colt also involved deficiency of factors IX and XI. It is inherited as a sex-linked recessive trait with the defective gene located on the X chromosome. Clinically affected foals show signs of a hemorrhagic tendency within a few weeks of birth, with the development of hematomas, persistent nasal bleeding, bleeding from injection sites and sudden death from massive internal hemorrhage. Affected foals are anemic. The diagnosis is made by the finding of very low plasma factor VIII:C activity (usually < 10% of values in unaffected animals). Treatment requires fresh frozen plasma or plasma concentrates but is not recommended because of the unavailability of sufficient plasma concentrates, the recurrent nature of the problem and the poor prognosis for long-term soundness.46
Von Willebrand disease (factor VIII:vWF deficiency)
Von Willebrand factor is a large adhesive glycoprotein that mediates adhesion of platelets to exposed subendothelium and that also is a carrier for coagulation factor VIII, protecting it from degradation in the circulation. There are three variations of von Willebrand’s disease – types I, II and III. Two variations of von Willebrand’s disease are recorded in pigs47,48 – both inherited as simple autosomal recessive traits. The disease in pigs is used as an experimental model for the disease in humans and the pig gene for von Willebrand factor is similar in size and complexity to its human counterpart, with affected pigs having a point mutation within the vWF gene. Type 1 von Willebrand’s disease occurred in an 8-day-old Quarter horse colt that was examined because of extensive purpura.49 The colt had a concentration of von Willebrand factor that was 9% of that of normal horses. The dam of the colt also had prolonged bleeding time and a concentration of von Willebrand factor 30% of that of normal animals,49 suggesting a familial and possibly heritable trait. Type II von Willebrand disease is recorded in a Quarter horse with hemorrhage associated with trauma, prolonged bleeding, lasting several hours, at an injection site, and spontaneous conjunctival hemorrhage,50 and in a Thoroughbred mare and her colt.51 The hemorrhage in the Thoroughbred mare and foal was not life-threatening. Type III von Willebrand’s disease is usually associated with low concentrations of factor VIII. Suspect factor VIII deficiency has been reported in Hereford calves.52 The prime manifestation was death shortly following castration with bleeding from the surgical site, intra-abdominal hemorrhage and severe anemia. The disease also occurs in sheep, and is linked to the X chromosome.53
Diagnosis is based on the observation of prolonged bleeding after minor trauma or surgery, prolonged activated partial thromboplastin time, although this can be minimal, normal prothrombin time, and decreased factor VIII:C activity and von Willebrand antigen concentration.49,51 The ristocetin cofactor activity of von Willebrand factor is reduced in animals with type II von Willebrand’s disease.49,51 An ELISA is available that is suitable for use in a number of species, including horses, pigs and cattle.4 Chromogenic assays of factors VII, VIII:C, IX and X developed for testing of human plasma are reliable when used for testing of horse plasma.3 Desmopressin increases the release of von Willebrand factor from vascular endothelium and is used for treatment of the disease in humans. There are no reports to date of its use in farm animals or horses. The disease can be managed by housing to minimize trauma and administration of plasma before elective surgery, although this did not completely prevent excessive bleeding.
Factor XI deficiency
Factor XI deficiency is recorded in Holstein–Friesian cattle in Canada54 and in Great Britain.55 It is transmitted as an autosomal recessive gene and occurrence in Britain has been traced to the importation of Canadian semen, with genetic links between carriers in the two countries.56 Heterozygous cattle have decreased levels of factor XI but are usually asymptomatic. The severity of clinical disease varies in homozygous cattle but they usually show prolonged or repeated bleeding episodes after trauma such as dehorning and hemorrhage following venepuncture. There are occasional deaths associated with multiple hemorrhages. Heterozygote carriers have decreased factor XI coagulant activity but measurement of factor XI activity is not a sensitive test for the carrier status, and a DNA-based tested is available that accurately identifies heterozygotes.54,56-58
Other clotting factor disorders
Prekallikrein deficiency is recorded in a family of miniature horses.59 The condition was not associated with clinical disease in these horses but blood samples failed to clot. It has also been recorded in three Belgian horses and was detected in this group because one hemorrhaged following castration.60 The mode of transmission is not known, but the familial nature of the disease suggests that it is heritable.
Heritable fibrinogen deficiency was found in a Border Leicester lamb manifest with inflammation and bleeding at the umbilicus and ear tag wound at 7 weeks of age61 and in Saanen goats.
Platelet disorders
Disorders of platelets include alterations in the number of platelets in blood (thrombocytopenia or thrombocytosis) and their function (with reduced function referred to as thrombasthenia). The physiology of platelets varies in important ways among the farm animal species. For instance, aggregation of platelets from horses, but not cattle, is inhibited by aspirin (acetylsalicylic acid), and horse platelets adhere to immobilized autologous fibrinogen while those of sheep do not.62 Platelets of different species respond differently to some agonists of platelet aggregation, such as ristocetin.63
Thrombocytopenia
Clinical signs associated with thrombocytopenia or thrombasthenia are petechial and ecchymotic hemorrhages, prolonged bleeding after venipuncture or from injection sites, epistaxis, hyphema and melena. A combination of some or all of these clinical signs is referred to as purpura. Thrombocytopenia can result from decreased production of platelets in the bone marrow or by increased consumption, increased peripheral destruction or a combination of these factors.64
Decreased platelet production is commonly associated with disorders that impair bone marrow function and there is usually simultaneous suppression of granulocyte and erythrocyte production. Thrombocytopenia and neutropenia develop before anemia because of the short life span of these cells relative to erythrocytes. Increased destruction, that is, the abnormal consumption of platelets, is most commonly immune-mediated. Increased consumption also occurs with severe trauma and disseminated intravascular coagulation, both of which increase the rate at which platelets are incorporated into clots.
Pseudothrombocytopenia occurs as a result of aggregation of platelets after collection into a glass tube containing EDTA.11 Ex-vivo aggregation can occur with other anticoagulants, including heparin. This situation can be recognized by the presence of abnormally low platelet counts in animals without evidence of excessive hemorrhage, or by the presence of clumps of platelets on microscopic examination of blood smears. Collection of blood into an anticoagulant other than EDTA or heparin, such as citrate, and measurement of a normal platelet count confirms the diagnosis of pseudothrombocytopenia.
Differentiation of the causes of thrombocytopenia is by clinical examination to detect underlying disease, hematology, examination for anti-platelet antibody and examination of bone marrow aspirates.
Decreased production
Thrombocytopenia due to decreased production, as opposed to increased destruction within the marrow, usually occurs with granulocytopenia because of the short intravascular half-life of both granulocytes and platelets.
This occurs with poisonings by Pteridium spp. (bracken fern) or Cheilanthes seiberi in cattle; the fungus Stachybotrys spp. (which produces a trichothecene) in cattle, pigs, sheep and horses; chronic furazolidone poisoning in calves; poisoning caused by trichloroethylene-extracted soybean meal; drugs that cause bone-marrow suppression, and radiation injury.65,66 Severe myelophthisis, such as is associated with myeloid dysplasia or myelofibrosis, causes thrombocytopenia in association with anemia and leukopenia. A familial myelofibrosis is reported in pygmy goats with anemia, granulocytopenia and thrombocytopenia.67
The syndrome is predominantly one of spontaneous hemorrhage but is complicated by bacteremia and fulminant infections facilitated by severe leukopenia. A granulocytopenic syndrome of unknown origin, occurring in all ages of cattle and manifest with a severe hemorrhagic diathesis, high morbidity and high case fatality, has been reported on various occasions in Australia.68
Familial diseases resulting in thrombocytopenia secondary to decreased production are recorded in Standardbred horses in which there is generalized bone marrow hypoplasia.69 Pancytopenia secondary to bone marrow aplasia is reported in a Holstein heifer.70
Increased destruction
The most common causes of thrombocytopenia are severe gastrointestinal disease (strangulating intestinal obstruction, anterior enteritis, colitis) and infectious and inflammatory disease, where a combination of increased destruction and increased consumption is the cause.64,71
Infection by a variety of viral, bacterial or rickettsial agents causes mild to severe thrombocytopenia. African horse sickness, equine infectious anemia, Anaplasma phagocytophila (equine granulocytic ehrlichiosis) and infection by Neorickettsia risticii (Potomac horse fever) cause mild to moderate thrombocytopenia.
Infection such as occurs in hog cholera and African swine fever can result in thrombocytopenia and contributes to the hemorrhagic tendency seen in these diseases.72 Outbreaks of hemorrhagic disease due to thrombocytopenia in veal calves have been attributed to infection with bovine virus diarrhea (BVD) virus type 2 and the disease has been reproduced experimentally with noncytopathic BVD virus.73 The appearance of hemorrhage was directly related to the number of circulating platelets and bleeding was seen when platelet numbers fell below 500/mL. Calves that developed thrombocytopenia had low (+ 1:32) BVD neutralizing titers. The virus infects megakaryocytes.74 Thrombocytopenia with a bleeding tendency (bloody diarrhea, petechial and ecchymotic hemorrhage) is also recorded in approximately 10% of adult cattle with acute BVD infection.75 Infection by Theileria annulata causes thrombocytopenia and prolonged prothrombin time in cattle.76 Other infectious causes in cattle include bovine leukemia virus, sarcocytosis and salmonellosis.
Immune-mediated (idiopathic) thrombocytopenia
Most instances of thrombocytopenic purpura in the past have been described as idiopathic. However, the development of newer diagnostic tests, including flow cytometry, that can reveal the presence of antibodies on the surface of platelets has permitted the classification of many of these cases as immune-mediated.12,77 Among the immune-mediated thrombocytopenias there are autoimmune and isoimmune diseases.
Isoimmune thrombocytopenia can be a complication in neonatal isoerythrolysis in foals and mules as a result of absorption of colostrum containing antiplatelet antibodies.78,79 The disease also occurs as only thrombocytopenia in mule foals and is attributable to antiplatelet IgG antibodies in the mule foal’s serum.80 In addition to thrombocytopenia, there is also depression of platelet aggregation in these foals, probably because of binding of IgG to collagen-binding sites on the platelet surface. Thrombocytopenia and neutropenia, assumed to be isoimmune-mediated, were found in foals with ulcerative dermatitis and mucosal ulceration.81 The foals had purpura and responded to supportive treatment and administration of corticosteroids.
The disease is observed in newborn pigs as a result of maternal isoimmunization, and has been reproduced experimentally. Piglets are normal at birth but become thrombocytopenic after suckling colostrum containing antiplatelet antibody. Clinical signs do not develop until after the fourth day of life. There is a heavy mortality rate, death being preceded by a generalized development of submucosal and subcutaneous hemorrhages, drowsiness, weakness and pallor. There is no practicable treatment. The sow should be culled. Thrombocytopenic purpura has occurred in a group of lambs given a single cow’s colostrum and was manifest with multiple hemorrhages and death by two days of age.82
Thrombocytopenia in adult animals with normal prothrombin times and partial thromboplastin times and with no evidence of disseminated intravascular coagulation is considered most likely to develop by autoimmune-mediated mechanisms.83-85 Immune-mediated thrombocytopenia can be induced by drugs or can be secondary to infectious or neoplastic disease64 but most cases are idiopathic.83,84 The disease is reported most commonly in horses but does occur in cattle.55
Horses of any age can be affected. The cause is usually not identified but the disease can be associated with administration of drugs, especially penicillin. The disease is caused by binding of IgG to platelets or to megakaryocytes, with subsequent impaired maturation, enhanced clearance or both, resulting in low platelet counts in blood.86 Petechiation and hemorrhage may be confined to single systems, such as the respiratory system with epistaxis and hematomas in the nasal sinuses, or the genital tract producing a bloody vulval discharge, with no detectable abnormality at other mucous membranes. More generalized involvement with widespread petechiation of mucous membranes, epistaxis and melena can also occur. Diagnosis is by measurement of blood platelet concentration and elimination of disseminated intravascular coagulation or primary diseases. Demonstration of platelet-surface-bound IgG on a significant proportion of platelets is diagnostic. Fewer than 0.15% of platelets from normal horses have IgG bound to the surface, whereas more than 4% of platelets of thrombocytopenic horses have IgG on the surface.77 Treatment includes the immediate removal from any medication and the administration of dexamethasone (0.040 mg/kg intramuscularly, intravenously or orally, once daily) or prednisolone (1 mg/kg orally), but not prednisone. This is usually effective in restoring platelet count and controlling hemorrhage. Resolution of hemorrhage can occur even before there are marked changes in platelet count. Treatment might need to be continued for days to weeks. Most horses do not require long-term treatment. For horses with life-threatening hemorrhage, administration of platelet-rich plasma or blood is needed. A transfusion volume of 10 mL blood per kg BW can be effective. Successful treatment with azathioprine in horses that do not respond to glucocorticoids is reported (0.5–1.5 mg/kg orally every 24 h).77,83 Splenectomy has been used to treat horses with chronic, idiopathic thrombocytopenia that is refractory to medical therapy. However, surgical treatment should be undertaken only in extreme cases, and with attention to the effect of thrombocytopenia on hemostasis during surgery.
Idiopathic thrombocytopenia purpura is also recorded in a 10-month-old bull,87 and immune-mediated thrombocytopenia and anemia occurred in a cow after vaccination with a polyvalent botulism vaccine.88 Corticosteroid-responsive thrombocytopenia occurred in two beef-breed cows with subcutaneous hematomas and epistaxis.89
Increased consumption
Increased consumption of platelets occurs in animals with severe trauma or disseminated intravascular coagulation.
Other causes
Thrombocytopenia occurs in horses with lymphosarcoma or myeloproliferative disease.64 Thrombocytopenia is usually associated with myelophthisic disease and is therefore a result of reduced platelet production. Heparin causes thrombocytopenia in horses, but the mechanism has not been determined.21,22
Thrombasthenia
Disorders of platelet function can result in purpura even in the presence of normal platelet counts. Disorders of platelet function can be congenital or acquired.
Acquired defects of platelet function are usually secondary to severe metabolic abnormalities such as uremia, liver failure or septicemia, or to administration of drugs. Among the compounds commonly administered to animals, aspirin is most notable in that it inhibits platelet aggregation in horses but not in cattle, despite inhibition of platelet generation of thromboxane-2 in both species.19 Other NSAIDs have minimal, if any, effect on platelet function. Dextran inhibits platelet function when administered to horses.90
A bleeding tendency is present in the Chédiak–Higashi syndrome in cattle. A prolonged bleeding time is demonstrable despite the presence of normal soluble coagulation factors and platelet numbers, and is due to a defect in platelet aggregation.91 Thrombasthenia,92,93 possibly also associated with variant von Willebrand factor,94 is also recorded in bleeding disorders in Simmental and Simmental crossbred cattle in Canada and the USA, and manifests with epistaxis in cold weather, subcutaneous hematomas and prolonged bleeding following minor procedures such as vaccination and ear-tagging.92,93 Platelet dysfunction and purpura were diagnosed in a 5-day-old Simmental heifer.95 Umbilical bleeding in calves has also been reported as an inherited condition in Japanese black cattle96,97 with low ADP-induced platelet aggregation. Affected cattle die by 1 year of age from repeated umbilical cord hemorrhage.
Thrombasthenias are also reported in horses. Glanzmann’s disease is reported in horses with a prolonged history of epistaxis not associated with exercise.98 The horses had prolonged bleeding time, markedly delayed clot retraction and a decrease in concentration of fibrinogen receptors on the platelet surface. Treatment with glucocorticoids was not effective in preventing epistaxis. Another form of platelet defect was diagnosed in a Thoroughbred filly with excessive hemorrhage after pin firing. The filly had prolonged bleeding time and normal clot retraction. The filly’s platelets did not bind to collagen and the defect was deduced to be in calcium signaling within the platelets.99
Thrombocytosis
Thrombocytosis is not usually associated with purpura or a tendency to hemorrhage unless the platelets have abnormal function. Thrombocytosis is considered to be either primary or secondary.100,101 Primary thrombocytosis is a result of excessive production of megakaryocytes in the absence of any inciting disease or increased release into the circulation. While exercise, epinephrine and vincristine can increase platelet counts, the most important cause of primary thrombocytosis is myeloproliferative disorder resulting in abnormal rate of platelet production. Primary thrombocytosis is rare in farm animal species.
Secondary thrombocytosis occurs in animals with severe systemic inflammatory or infectious diseases of more than several days duration, and usually of several weeks duration. Young animals appear to be more susceptible but the condition can occur in animals of any age. Detection of thrombocytosis should prompt a thorough clinical examination for a cause of chronic inflammation in the animal. Common causes in horses include pneumonia, Rhodococcus equi infection, septic arthritis and colitis.97 Thrombocytosis with Heinz body anemia is reported in cattle fed cabbage.
DISSEMINATED INTRAVASCULAR COAGULOPATHY AND HYPERCOAGULABLE STATES
There is increasing recognition that abnormalities in blood clotting and fibrinolysis exist in both subclinical and clinical forms in many diseases of farm animals, and that the presence and severity of these disorders is related to prognosis for survival. There is a spectrum of abnormalities ranging from mild changes in concentration or activity of clotting factors and indicators of fibrinolysis, through evidence of excessive coagulation or impaired fibrinolysis, to a hemorrhagic diathesis. Previously, the most extreme form of this disorder was recognized as a hemorrhagic diathesis and termed disseminated intravascular coagulation (DIC). Increasing sophistication and availability of measures of coagulation and fibrinolysis have revealed that abnormalities of hemostasis exist even in animals without clinical evidence of excessive hemorrhage. These milder changes in hemostasis and fibrinolysis are, not surprisingly, much more common than is DIC, but are still associated with am increased case fatality rate.
ETIOLOGY AND EPIDEMIOLOGY
DIC and hypercoagulable states are acquired disorders of hemostasis in animals that occur as a consequence of severe disease that induces systemic inflammation (systemic inflammatory syndrome). DIC is now regarded as a component and consequence of systemic inflammation, rather than being an isolated disorder of hemostasis.100 DIC and hypercoagulable states are therefore associated with any severe disease that initiates a systemic inflammatory response. A partial listing is: colitis, enteritis, infarctive lesions of the intestines, septicemia, abomasal torsion, metritis, severe trauma, immune-mediated inflammation (e.g. purpura hemorrhagica), hyperthermia and neoplasia. A common, but not universal feature, of diseases that induce DIC or a hypercoagulable state is the presence of presumed or documented endotoxemia, although DIC can be induced by most infectious organisms. It is important to recognize that any severe disease that causes a systemic inflammatory response can incite changes in hemostatic function.
The presence of a hypercoagulable state or DIC is most well recognized in horses with gastrointestinal disease.10,102-104 It also occurs in cattle with abomasal displacement and in endotoxemic calves.105,106 Low plasma antithrombin concentrations occur in cows with hepatopathy, peritonitis or acute enteritis.107 The disease has been reproduced experimentally in pigs, and probably occurs naturally in that species in many diseases, including African swine fever.108,109 The prevalence of DIC (clinically evident hemorrhage) is uncommon, while the prevalence of a hypercoagulable state detectable only by clinicopathologic testing is much more common.30,104
The prevalence of the syndrome is not well defined, partly because of problems in achieving a confirmatory diagnosis by laboratory assessment of factors involved in coagulation or fibrinolysis because of the lack of laboratories providing the necessary assays, and partly because of lack of recognition of the disease. Five of 20 cattle with either left or right displacement of the abomasum had a hypercoagulable state.106 Of horses examined at a referral institution for colic, 3.5% had clinical signs consistent with DIC and supportive laboratory evidence.110 All these horses had severe inciting disease, with most requiring surgical intervention for infarctive intestinal disease. This study probably under-represents the proportion of horses with severe gastrointestinal disease that have hemostatic abnormalities, given that many horses with severe gastrointestinal disease have subclinical abnormalities in hemostasis and fibrinolysis.30 The survival rate among horses with colic and DIC or a hypercoagulable state was 19%, whereas that in horses with colic but no clinicopathologic evidence of a hemostatic disorder was 80%.102 Twelve of 37 horses with colitis had clinicopathological evidence of a hypercoagulable state, although none had clinical signs of DIC at the time of sample collection.30 Of the 12 horses with a hypercoagulable state, five died, compared to two of 25 horses with colitis that did not have evidence of a coagulopathy.
Clinically relevant alternations in hemostatic and fibrinolytic indices occur in neonatal foals with septicemia.111 Derangements in hemostatic or fibrinolytic indices were helpful in identification of septic foals with increased risk of coagulopathy, but were not helpful in predicting hemorrhage as compared to thrombus formation. Twenty-three of the 34 septic foals did not survive. Survival of septicemic foals was correlated with Gram-negative bacteremia but not with the presence of endotoxin or coagulopathy.111
PROGNOSIS
The prognosis for animals with clinical signs of disseminated coagulation is very poor. Horses without physical signs of hemorrhage or defective fibrinolysis but with clinicopathological evidence of a hypercoagulable state have a worse prognosis than horses without evidence of a hypercoagulable state.10,30,102-104 When evaluating the prognosis of an animal with evidence of a coagulopathy as part of the systemic inflammatory syndrome it must be borne in mind that the coagulopathy is secondary to the initiating disease; the more severe the initiating disease the greater the likelihood that the animal will have a coagulopathy, and the more severe the initiating disease the poorer the prognosis. DIC and lesser abnormalities of hemostasis can therefore be regarded as markers of disease severity and considered accordingly when determining a prognosis. This is not to minimize the importance of DIC and hemostatic defects of lesser severity in the pathogenesis of severe disease, and the need to institute effective preventive measures and treatment.
PATHOPHYSIOLOGY
DIC, or consumption coagulopathy, can develop in a number of diseases which, in themselves, are not diseases that primarily affect hemostatic mechanisms. The pathogenesis involves systemic activation of coagulation with intravascular deposition of fibrin leading to thrombosis of small and medium-sized blood vessels with subsequent organ failure. Depletion of platelets as a result of platelet activation and binding to fibrin to form clots, and of coagulation factors, results in excessive bleeding. The systemic formation of fibrin results from increased generation of thrombin and the simultaneous suppression of anticoagulation mechanisms (which are detectable in animals as reduced concentration of antithrombin) and impaired fibrinolysis.112 Products of fibrinogen activation, including fibrinopeptides A and B, contribute to systemic vasoconstriction and the hypoperfusion of some organs. The disorder, in its most extreme form, involves both excessive coagulation and, seemingly paradoxically, bleeding.
Systemic activation of coagulation is part of the systemic inflammatory response syndrome, which is dominated by interleukins 1 and 6 and tumor necrosis factor-alpha.100,112 There might be a contribution of complement activation to the hypercoagulability. Activation of clotting occurs through either damage to endothelium or activation and release of tissue factor. Tissue factor expression is increased by one or more of the proinflammatory cytokines (interleukin-1, -6, -8 and tumor necrosis factor), which are almost universally increased in diseases that feature systemic inflammation. Generation of tissue factor results in activation of the extrinsic clotting cascade with resultant increases in thrombin. The increased activity of the coagulation cascade is temporally associated with impaired activity of anticoagulant mechanisms, demonstrable as decreases in plasma concentration of antithrombin and protein C. Further exacerbating the effect of increased rate of fibrin synthesis is impaired fibrinolysis, indicated by diminished activity of plasminogen and increased activity of plasminogen-activator inhibitor.112
In summary DIC is a hemorrhagic diathesis, characterized by an augmentation of normal clotting mechanisms that results in depletion of coagulation factors, deposition of fibrin clots in the microvasculature and the secondary activation of fibrinolytic mechanisms. The augmentation of clotting mechanisms can result in a depletion of platelets and factors V, VIII, IX, XI and XIIa, and the depletion of fibrinogen in association with the formation of fibrin clots in the microvasculature. These fibrin clots decrease tissue perfusion, which can then lead to further activation and depletion of clotting factors by the release of tissue thromboplastin as a result of tissue hypoxia. The bleeding tendency occasioned by the depletion of these clotting factors is further accentuated by the secondary activation of the thrombolytic system with the production of fibrin degradation products that have anticoagulant properties.
Impaired capacity of the monocyte phagocytic system contributes to the disease. Macrophages in the reticuloendothelial system remove fibrin degradation products and activated clotting factors from the circulation. Loss or diminution of the capacity to remove hemostatic and fibrinolytic compounds causes increases in plasma concentration of these products and exacerbation of the disease. Damage to the reticuloendothelial system, notably in the liver and spleen, resulting from damage as a consequence of the underlying disease (endotoxemia) or lack of perfusion of liver and spleen as part of DIC, decreases removal of these compounds and induces a viscous cycle of disease.
DIC can be initiated by a variety of different mechanisms.
• Extensive tissue necrosis, such as occurs in trauma, rapidly growing neoplasm, acute intravascular hemolysis and infective diseases such as blackleg, can cause extensive release of tissue thromboplastin and initiate exuberant coagulation via the extrinsic coagulation pathway
• Exuberant activation of the intrinsic pathway can occur when there is activation of the Hageman factor by extensive contact with vascular collagen, as occurs in disease with vasculitis, or those associated with poor tissue perfusion and tissue hypoxia with resultant endothelial damage
• Factors that initiate platelet aggregation, such as endotoxin, that cause reticuloendothelial blockage, such as excessive iron administration to piglets, or that cause hepatic damage to interfere with clearance of activated clotting factors, can contribute to the occurrence of disseminated intravascular coagulation.
CLINICAL SIGNS
As discussed above, defects in hemostasis and fibrinolysis range from those that are detectable by clinicopathologic examination but are not associated with clinical signs of excessive bleeding or coagulation, through fulminant hemorrhagic diathesis.
The presence of a hypercoagulable state that is not associated with signs of excessive bleeding or thrombosis nonetheless worsens the prognosis of severe diseases. This is probably due to DIC-induced injury to organs that is not detectable against the background of damage caused by the primary disease but that has an important or pivotal effect on the animal’s well being. The next progression of the disease is evidence of enhanced thrombosis, most evident as thrombosis of large vessels after minor damage such as that associated with intravascular catheterization or simple venipuncture. In some cases vessels can thrombose without obvious inciting cause. An example of a common manifestation of this stage of the disease is jugular vein thrombosis in horses or cattle with severe disease and low plasma concentration of antithrombin.30,107
An unusual, but severe, manifestation of DIC in horses is thrombosis of the distal limbs, resulting in ischemic necrosis of the limb and death of the animal. This clinical manifestation of DIC occurs in foals and, to a lesser extent, in adults with evidence of septicemia or severe gastrointestinal disease.113
The most severe acquired hemostatic defect in animals with systemic disease is DIC. This extreme of the clotting disorder is manifested by local or generalized bleeding tendencies that vary in severity from occurrence of petechial hemorrhages in mucous membranes to life-threatening hemorrhage or infarction of organs. Ischemic damage to a wide variety of organs is possible, with the gastrointestinal tract and kidneys being commonly affected.49
CLINICAL PATHOLOGY
There are a large number of hemostatic and fibrinolytic factors that can be measured in research laboratories, only a few of which are routinely available in clinical laboratories. The following measures are commonly used to detect hypercoagulable states or DIC in clinical situations:
• Platelet count. The abnormality consistent with DIC is thrombocytopenia
• Prothrombin time. This is usually prolonged in animals with DIC but can occasionally be shortened in animals with a hypercoagulable state
• Activated partial thromboplastin time. This measure of hemostasis is usually prolonged in animals with coagulopathies
• Serum markers of fibrinogen activation/fibrin degradation. FDPs have poor sensitivity and specificity for detection of DIC. Plasma d-dimer concentration is more sensitive for detection of abnormalities in hemostatis/fibrinolysis9
• Fibrinogen concentration. Classical descriptions of DIC include hypofibrinogenemia as a common finding. However, this is uncommonly the case in horses and cattle,103,106 probably because fibrinogen is an acute-phase protein the concentration of which increases in inflammatory diseases in these species. Declines in plasma fibrinogen concentration, with values remaining above the lower bound of the reference range, are often noted in horses with coagulopathy and impending death30
• Antithrombin activity is often reduced in animals with a hypercoagulable state or DIC.
A number of studies provide detailed description of the occurrence, and time course, of abnormalities in hemostatic and fibrinolytic function in horses with gastrointestinal disease.26,102-104,114 The general pattern is that of prolonged clotting times (prothrombin (PT), activated partial thromboplastin (APTT)) with diminished activity of antithrombin and protein C, and increased plasma concentrations of fibrinogen and fibrin degradation products. d-dimer concentration has been reported to increase in horses undergoing surgery for colic115 or to be lower in horses with colic than in healthy horses.102 Platelet concentration is reduced in horses with colic and evidence of coagulopathy. Abnormalities in hemostatic factors are more common in peritoneal fluid than in blood of horses with colic.114 Tissue plasminogen activator, plasminogen, protein C, antithrombin III, and alpha-2-antiplasmin activities and concentrations of fibrinogen and fibrin degradation products are greater in peritoneal fluid from horses with colic than in peritoneal fluid of healthy horses.
Compared to healthy foals, the PT, APTT and whole blood recalcification times are significantly longer in septic foals. The fibrinogen and fibrin degradation products concentrations, percentage plasminogen, alpha-2-antiplasmin and plasminogen activator inhibitor activities, and tumor necrosis factor and interleukin-6 activities are greater, and protein C antigen and antithrombin III activity are lower in septic foals.111
Cattle with displaced abomasum often have abnormalities in one or more of PT, APTT, thrombin time, platelet count and plasma concentration of fibrin degradation products.106 Pigs with induced endotoxemia have increases in activity of tissue factor, plasminogen activator and plasminogen activator-inhibitor, and concentrations of thrombin–antithrombin complexes and fibrin monomer, and a decline in fibrinogen and factor VII concentrations.108
NECROPSY EXAMINATION
It is important to differentiate the abnormalities at necropsy caused by DIC from those of the underlying disease. This can be challenging. The presence of DIC is suspected by the presence of hemorrhage in the carcass. Hemorrhage can vary from occasional petechiation to frank hemorrhage into body cavities. Horses dying of DIC usually have widespread lesions, including petechiation of mucosal and serosal surfaces, including the mesentery and pleura. There is often hemorrhage into parenchymatous organs (kidneys, adrenals), lungs and the myocardium, and infarcts in the adrenals and kidney. Microthrombi are detectable in the intestine and kidney of some horses with DIC.
DIAGNOSTIC CONFIRMATION
The presence of a hypercoagulable state is determined by clinicopathological testing. DIC is diagnosed by the presence of clinical signs of a hemorrhagic diathesis and laboratory confirmation of abnormalities in hemostasis and fibrinolysis. A conventional definition of DIC requires the presence of clinical evidence of coagulopathy and the presence of at least three abnormal measures of coagulation or fibrinolysis. It is likely that this definition will change as our understanding of the spectrum of abnormalities and manifestations of the disorder matures.
Differential diagnoses include all of the acquired or inherited coagulopathies. However, the cardinal differentiating attribute of DIC or the lesser hypercoagulable states is the presence of severe inciting disease.
TREATMENT
Most recommended therapies for DIC have been extrapolated from the human literature and may not be applicable to farm animals. However, generally stated, the principles of therapy are:
• Treatment of the underlying disease and correction of acid–base, inflammatory, electrolyte and perfusion abnormalities
• Restoration of normal activity or concentration of clotting factors in blood
• Halting or attenuating the increased coagulopathy
• Minimizing effect of microthrombi and thrombi on organ function.
Disseminated intravascular coagulation is invariably secondary to an initiating primary disease. Vigorous therapy should consequently be directed toward correction of the primary initiating disease. Aggressive intravenous fluid administration to maintain tissue perfusion and to correct any acid–base and electrolyte imbalance is also very important. There should be aggressive treatment of endotoxemia and of diseases likely to induce endotoxemia. Treatment of endotoxemia is discussed elsewhere in this text, and current reviews are available.116
The plasma concentration of clotting factors should be restored, or supplemented, in horses with clinical or clinicopathological evidence of a coagulopathy. The practice of blood component therapy is well accepted in human medicine but because of technological limitations is not generally available in farm animals. However, stored plasma, preferably fresh frozen, can be administered to increase the concentration of clotting factors that are depleted during hypercoagulable states or DIC. Antithrombin is often readily measured and horses with low plasma antithrombin activity should be administered plasma. The dose of plasma necessary to increase blood antithrombin activity to appropriate levels has not been determined. However, many clinicians use a plasma antithrombin activity 60% of that of healthy horses as a minimal acceptable activity. This choice has not been verified empirically. Dosages of plasma vary from 2–10 mL/kg, intravenously. Platelet-rich plasma, or whole blood, can be used to treat thrombocytopenia.
Heparin and low-molecular-weight heparin is used to treat horses with hypercoagulable states2,24 and its use is discussed above. The aim is to prevent formation of thrombi and microthrombi. Heparin requires antithrombin as a cofactor, and it might not exert its full therapeutic activity in horses with abnormally low blood antithrombin concentrations.
Aspirin is used to inhibit platelet activity in horses with prothrombotic states. Its efficacy in reducing morbidity or mortality has not been determined.
Darien BJ. Hemostasis — a clinical review. Equine Vet Educ. 1993;5:33-36.
Sellon DC, Grindem CB. Quantitative platelet abnormalities in horses. Compend Contin Educ Pract Vet. 1994;16:1335-1346.
Davis EG, et al. Flow cytometry: clinical applications in Equine medicine. J Vet Intern Med. 2002;16:404-410.
Dallap BL. Coagulopathy in the Equine critical care patient. Vet Clin North Am. 2004;20:231-251.
1 Taylor EL, et al. Am J Vet Res. 2000;61:1191.
2 Kingston JK, et al. Am J Vet Res. 2001;62:547.
3 Topper MJ, Prasse KW. Am J Vet Res. 1998;59:538.
4 Benson RE, et al. J Lab Clin Med. 1992;119:420.
5 Munster AB, et al. Comp Med. 2002;52:39.
6 Heidmann P, et al. Am J Vet Res. 2005;66:313.
7 Feige K, et al. J Vet Med A. 2003;50:30.
8 Monreal L. J Vet Intern Med. 2003;17:757.
9 Stokol T, et al. Vet Clin Pathol. 2005;34:375.
10 Topper MJ, Prasse KW. Am J Vet Res. 1996;57:456.
11 Hinchcliff KW, et al. J Am Vet Med Assoc. 1993;203:1715.
12 Davis EG, et al. J Vet Intern Med. 2002;16:404.
13 Russell KE, et al. Am J Vet Res. 1997;58:1092.
13a Segura D, et al. J Vet Int Med. 2006;20:581.
14 Segura D, et al. Vet J. 2005;170:108.
15 Barton MH, et al. J Vet Diagn Invest. 1995;7:380.
16 Gentry PA, et al. Res Vet Sci. 1994;57:336.
17 Heidmann P, et al. Am J Vet Res. 2005;66:313.
18 Cambridge H, et al. Equine Vet J. 1991;23:123.
19 Gentry PA, et al. Am J Vet Res. 1989;50:919.
20 Feige K, et al. Equine Vet J. 2002;35:506.
21 Moore BR, Hinchcliff KW. J Vet Intern Med. 1991;8:26.
22 Monreal L, et al. Am J Vet Res. 1995;56:1281.
23 Moore JN, et al. Am J Vet Res. 1987;48:68.
24 Schwarzwald CC, et al. Am J Vet Res. 2002;63:868.
25 Dart A, et al. Aust Vet J. 2001;79:624.
26 Fiege K, et al. Equine Vet J. 2004;36:135.
27 Roberts MC, Kelley WR. Vet Rec. 1982;110:144.
28 Galan JE, Timoney JF. J Immunol. 1985;13:3134.
29 Werner LL, et al. J Am Vet Med Assoc. 1984;185:87.
30 Dolente BA, et al. J Am Vet Med Assoc. 2002;220:1034.
31 McConnico RS, et al. J Am Vet Med Assoc. 1997;211:882.
32 Tligui N, Ruth GR. Am J Vet Res. 1994;55:1558.
33 Runciman DJ, et al. Aust Vet J. 2002;80:28.
34 Sasaki Y, et al. Jpn J Vet Sci. 1985;47:435.
35 Nuwsholme SJ, et al. J S Afr Vet Assoc. 1985;56:101.
36 Hoppe PP. Prakt Tierarzt. 1987;68:32.
37 Crawford AM, Mills JN. Aust Vet J. 1985;62:185.
38 Meier J, Stocker K. Crit Rev Toxicol. 1991;21:171.
39 Dickinson CE, et al. J Am Vet Med Assoc. 1996;208:1866.
40 Devine CE, et al. Meat Sci. 1983;9:247.
41 Lundquist H. Nord Vet Med. 1983;35:57.
42 Jones PA, et al. Am J Vet Res. 1997;58:541.
43 Abutarbusch SM, Radostits OM. Aust Vet J. 2005;46:711.
44 Joachim A, et al. Parasitol Res. 2003;89:53.
45 Littlewood JD, et al. Equine Vet J. 1991;23:70.
46 Henninger RW. J Am Vet Med Assoc. 1988;193:91.
47 Neuenschwander S, et al. Thromb Haemost. 1992;68:618.
48 Thiele GL, et al. J Hered. 1986;77:179.
49 Laan MTTJ, et al. Vet Rec. 2005;157:322.
50 Brooks M, et al. J Am Vet Med Assoc. 1991;198:114.
51 Rathgeber RA, et al. J Vet Intern Med. 2001;15:63.
52 Healy PJ, et al. Aust Vet J. 1984;61:132.
53 Backfisch W, et al. J Hered. 1994;85:474.
54 Brush PJ, Gentry PA. Vet Rec. 1988;122:134.
55 Yasuda J, et al. J Vet Med Sci. 2002;64:87.
56 Gentry PA, Brush PJ. Aust Vet J. 1987;28:110.
57 Gentry PA, Ross ML. Am J Vet Res. 1994;58:242.
58 Marron BM, et al. Anim Genet. 2004;35:454.
59 Turrentine MA, et al. Am J Vet Res. 1986;47:2464.
60 Geor RJ, et al. J Am Vet Med Assoc. 1990;197:741.
61 Fecteau G, et al. Can Vet J. 1997;38:443.
62 Pelagalli A, et al. J Comp Pathol. 2003;128:127.
63 Pelagalli A, et al. J Comp Pathol. 2002;127:126.
64 Sellon DC. J Vet Educ. 1998;10:133.
65 Handagama PJ, Feldman BF. Vet Res Communn. 1986;10:1.
66 Jeffers M, Lenghaus C. Aust Vet J. 1986;63:262.
67 Cain GR, et al. Comp Haematol Int. 1994;4:167.
68 Nicholls TJ, et al. Aust Vet J. 1985;62:67.
69 Kohn CW, et al. J Vet Intern Med. 1995;9:315.
70 Ammann VJ, et al. Can Vet J. 1996;37:493.
71 Sellon DC, et al. J Vet Intern Med. 1996;10:127.
72 Gomez-Villamandos JC, et al. J Comp Pathol. 1998;118:1.
73 Corapi WV, et al. J Am Vet Med Assoc. 1990;196:590.
74 Woods RD, et al. Can J Vet Res. 2004;68:42.
75 Rebhun WC, et al. J Vet Intern Med. 1989;3:42.
76 Singh A, et al. Vet Res Commun. 2001;25:289.
77 McGurrin MK, et al. J Am Vet Med Assoc. 2004;224:83.
78 Buechner-Maxwell V, et al. J Vet Intern Med. 1997;11:304.
79 Traub-Dargatz JL, et al. J Am Vet Med Assoc. 1995;296:67.
80 Ramirez S, et al. J Vet Intern Med. 1999;13:534.
81 Perkins GA, et al. J Vet Intern Med. 2005;19:211.
82 Simpson VR, Done SH. Vet Rec. 1994;135:327.
83 Humber KA, et al. J Am Vet Med Assoc. 1991;199:591.
84 Cohen ND, et al. J Am Vet Med Assoc. 1991;199:790.
85 Sockett DC, et al. J Am Vet Med Assoc. 1987;190:308.
86 Nunez R, et al. BMC Blood Disorders. 2001;1:1. (available on line at: http://www.biomedcentral.com/1471-2326/1/1).
87 Lunn DP, Butler DG. Aust Vet J. 1991;32:559.
88 Yeruham I, et al. Vet Rec. 2003;153:502.
89 Hoyt PG, et al. J Am Vet Med Assoc. 2000;217:717.
90 Heath MF, et al. Equine Vet J. 1998;30:408.
91 Shiraishi M, et al. J Vet Med Sci. 2002;64:751.
92 Searcy GP, et al. Can J Vet Res. 1990;54:394.
93 Gentry PA, et al. Can J Vet Res. 1997;61:128.
94 Sullivan PS, et al. J Am Vet Med Assoc. 1994;205:1763.
95 Navarre CB, et al. J Vet Intern Med. 1996;9:283.
96 Akuzawa M, et al. J Vet Med Sci. 1991;53:107.
97 Tu CH, et al. J Jpn Vet Med Assoc. 1996;49:237.
98 Livesey L, et al. J Vet Intern Med. 2005;19:917.
99 Fry MM, et al. J Vet Intern Med. 2005;19:359.
100 Dallap BL. Vet Clin North Am. 2004;20:231.
101 Sellon DC, et al. J Vet Intern Med. 1997;11:24.
102 Monreal L, et al. Equine Vet J Suppl. 2000;32:19.
103 Topper MJ, Prasse KW. Am J Vet Res. 1998;59:542.
104 Prasse KW, et al. J Am Vet Med Assoc. 1993;203:685.
105 Thomson GW, et al. Can J Comp Med. 1974;38:457.
106 Irmak K, Turgut K. Vet Res Communn. 2005;29:61.
107 Pusterla N, et al. Vet Rec. 1997;140:17.
108 Kutzsche S, et al. Thromb Res. 2000;98:517.
109 Villeda CJ, et al. Arch Virol. 1993;133:467.
110 Welch RD, et al. J Vet Intern Med. 1992;6:29.
111 Barton MH, et al. J Vet Intern Med. 1998;12:26.
112 Levi M, Cate HT. N Engl J Med. 1999;341:586.
113 Brianceau P, Divers TJ. Equine Vet J. 2001;33:105.
114 Collatos C, et al. J Am Vet Med Assoc. 1995;15:465.
THROMBOSIS (HYPERCOAGULABILITY)
Abnormal formation of thrombi is often a consequence of diminished concentrations or activity of anticoagulant factors, such as antithrombin, protein C and antiplasmin, increased concentrations of plasminogen activator–inhibitor or abnormalities of vessel walls. Thrombotic disease is usually a consequence of a primary disease that depletes anticoagulant factors and involves mechanisms discussed above under Disseminated intravascular coagulopathy. Thrombosis of the jugular vein is discussed elsewhere. Primary diseases involving thrombosis are thromboembolic colic in horses and aortoiliac thrombosis in horses.
An apparently primary defect in protein C activity in a Thoroughbred colt with a hypercoagulable state has been described.1 The colt had repeated episodes of venous thrombosis and developed renal failure. Plasma concentrations of protein C were within the reference range for healthy horses, but the activity of protein C in plasma was 32% of that of healthy horses, suggesting a defect in the protein.
Disorders of red cell number or function
ANEMIA
Etiology
Deficiency of circulating erythrocytes associated with hemorrhage, increased destruction or the inefficient production of erythrocytes. There are a large number of specific etiologies
Clinical findings
Pallor of mucosae, tachycardia, lethargy, exercise intolerance, arrhythmia, ileus, decreased ruminations and colic. Petechial and ecchymotic hemorrhages, icterus, hemoglobinuria, bleeding tendencies can be seen if the underlying cause is excessive hemorrhage
ETIOLOGY
Anemia can be classified as hemorrhagic anemia, hemolytic anemia, or anemia due to decreased production of erythrocytes. Another classification system is based on evidence of regeneration of anemia, with anemia classified as either regenerative or nonregenerative. Both classifications are useful in determining the cause, treatment and prognosis. Diseases causing anemia in horses are listed in Table 25.2 and those causing hemolytic anemia in cattle in Table 20.4.
Hemorrhagic anemia
Acute hemorrhage and hemorrhagic shock are discussed in Chapter 2. The diseases discussed here are those that cause normovolemic anemia. While anemia occurs after restitution of plasma volume in animals with severe hemorrhage, most diseases that cause normovolemic anemia do so because of the chronic loss of blood either from the body or into a body cavity. The most common route of loss is through the gastrointestinal tract. Diseases include:
• Parasitism – intestinal nematodiasis: Teladorsagia circumcincta or Haemonchus contortus in lambs and sheep, Ostertagia ostertagi in cattle, Strongylus sp. and cyathostomes in horses; trematodiasis including Fasciola hepatic in sheep and cattle; hematophagous lice and ticks, including Linognathus vituli in calves
• Gastrointestinal disease, including:
• Genitourinary tract disease, including:
Hemolytic anemia
Cattle and sheep
• Babesiosis, anaplasmosis, Mycoplasma ovis comb. nov. (formerly Eperythrozoon ovis) eperythrozoonosis, trypanosomiasis, nagana, theileriosis, alone or in various combinations1
• Leptospirosis (L. interrogans serovar pomona)
• Bovine virus diarrhea and mucosal disease2
• Postparturient hemoglobinuria
• Associated with grazing Brassica spp. rape, kale, chou moellier, turnips, cabbages
• Associated with the excessive feeding of culled onions3 or cannery offal, especially tomatoes and onions
• Poisoning by Mercurialis, Ditaxis, Pimelia and Allium spp.
• Poisoning by miscellaneous agents, including phenothiazine, guaifenesin
• Poisoning – chronic copper poisoning. In sheep secondary to pyrrolizidine alkaloids as in toxemic jaundice or primary from the feeding of diets too high in copper. Cattle are much less susceptible than sheep, although preruminant calves are very susceptible
• Treatment with long-acting oxytetracycline
• Water intoxication and drinking cold water in calves,4 and in goat kids fed water from a nipple bottle5
• Inadvertent administration of hypotonic fluids intravenously
• Part of a transfusion reaction
• Rare cases of alloimmune hemolytic anemia (isoerythrolysis) in calves from vaccination of the dam with blood-derived vaccines such as anaplasma vaccine
• Autoimmune hemolytic anemia is recorded in calves but is rare.6 All reported cases have occurred in calves under 6 months of age
• Immune-mediated anemia can occur in lambs that are fed cow colostrum as a source of immunoglobulin. This is not a common sequel to the feeding of cows’ colostrum and occurs only with the colostrum from certain cows. Anemia is evident at 7–20 days of age but jaundice and hemoglobinuria are not usually present.7,8 The syndrome must be differentiated from immune-mediated thrombocytopenia, which occurs at a younger age in some lambs fed bovine colostrum
• Rarely, in adults after vaccination9
• Congenital anemia associated with dyserythropoiesis and accompanied by dyskeratosis and progressive alopecia is recorded in Polled Hereford calves. The anemia is present at birth and the disease is probably inherited10,11
• A congenital anemia with jaundice is recorded in Murray Grey calves and it is postulated that a defect in the red cell membrane leads to intravascular hemolysis.12
Horses
• Equine infectious anemia, although the pathogenesis of the anemia is probably multifactorial, including hemolysis and decreased red cell production
• Phenothiazine poisoning. This anthelmintic is now used rarely in horses
• Red maple leaf (Acer rubrum) toxicity13
• Ingestion of dried garlic (> 0.2 g/kg BW) results in development of Heinz bodies and hemolytic anemia14
• Intravenous administration of hypotonic or hypertonic fluids (water, 20% dimethylsulfoxide)
• As a sequela to severe cutaneous burns.15 The severity of the hemolysis correlates with the amount of skin area burned. Hemolysis is due to oxidative damage of red cell membranes that occurs within minutes of the burn. Prevention and treatment include immediate administration of polyionic fluids to prevent hemoconcentration and to prevent hemolytic uremia
• As a sequel to clostridial abscessation. The anemia occurs more than 10 days after development of the abscess and is associated with the presence of IgG or IgM on the surface of red cells16
• Alloimmune hemolytic anemia (isoerythrolysis) of foals
• Autoimmune hemolytic anemia. Not common but several series have been recorded17-20
• Immune-mediated hemolytic anemia and thrombocytopenia (Evans syndrome)21
• Penicillin-induced hemolytic anemia. This is a rare event but can occur when horses develop IgG antipenicillin antibodies. These antibodies bind to penicillin on erythrocytes with resultant red cell destruction. Penicillin-coated erythrocytes agglutinate with patient serum.22,23 It is probable that other immune-mediated hemolytic anemias in the horse are also associated with the development of antibody to therapeutic agents24
• Some snake envenomations cause intravascular hemolysis in dogs and cats25 and hemolytic anemia can occur in snakebite in horses26 and calves
• Lead intoxication in horses causes mild anemia, but signs of peripheral neuropathy are the more obvious manifestation
• Abnormalities in red cell function can lead to increased removal of red cells from blood (extravascular hemolysis) and are discussed under ‘Abnormalities of red cell function’.
Anemia due to decreased production of erythrocytes or hemoglobin (nonregenerative anemia)
The diseases in this group tend to affect all species so that they are divided up according to cause rather than according to animal species.
Nutritional deficiency
Nutritional deficiencies impair production of hemoglobin or red cells. A number of specific deficiencies that result in anemia are described:
• Cobalt and copper. These elements are necessary for all animals, but clinically occurring anemia is observed in only ruminants. Copper deficiency induced by zinc toxicity causes anemia in pigs27
• Iron, but as a clinical occurrence this is limited to rapidly growing animals, including baby pigs, young calves designated for the white veal market, housed lambs and foals. This predilection of young animals for iron deficiency is attributable to their rapid growth and hence requirement for relatively large intakes of iron (which, in addition to production of hemoglobin, is used in production of myoglobin and other iron-containing compounds), the low concentration of iron in milk, and management practices that deny access of the animals to pasture or soil from which they can obtain iron
• Potassium deficiency is implicated in causing anemia in calves
• Pyridoxine deficiency, produced experimentally, can contribute to the development of anemia in calves
• Folic acid deficiency is rare in horses, has not been reported as a spontaneous disease in pigs, and is unlikely to occur in ruminants because of the constant production of folic acid by rumen bacteria.37 Plasma folic acid concentrations vary in pregnant mares kept at pasture, and in their foals, but there is no evidence of folate deficiency in either mares or foals.38 Administration of antifolate drugs (trimethoprim, sulfonamides, pyrimethamine, methotrexate) could, theoretically, cause folate deficiency in horses. Folate deficiency causing anemia and leukopenia is reported in a horse treated for equine protozoal myelitis with antifolate drugs concurrent with oral supplementation with folic acid.39 Intravenous administration of folic acid (0.055–0.11 mg/kg BW) resulted in rapid resolution of leukopenia and anemia. Paradoxically, oral administration of folic acid in monogastric animals receiving antifolate drugs impairs absorption of folic acid in the small intestine and causes folate deficiency.39 Administration of folic acid, sulfonamides and pyrimethamine orally to pregnant mares results in congenital signs of folate deficiency in foals, including anemia and leukopenia.40
Chronic disease
Chronic inflammatory disease causes mild to moderate anemia in all species of large animals. The anemia can be difficult to differentiate from that of mild iron-deficiency anemia. The genesis of anemia of chronic inflammation is multifactorial and includes sequestration of iron stores such that iron availability for hematopoiesis is reduced despite adequate body stores of iron, reduced erythrocyte life span and impaired bone marrow response to anemia. The result is normocytic, normochromic anemia in animals with normal to increased serum ferritin concentrations. The clinicopathologic features of both iron deficiency anemia and anemia of chronic disease are detailed in Table 9.1.
• Chronic suppurative processes can cause severe anemia by depression of erythropoiesis
• Poisoning by bracken, trichloroethylene-extracted soybean meal, arsenic, furazolidone41 and phenylbutazone, cause depression of bone marrow activity
• A sequel to inclusion body rhinitis infection in pigs
• Porcine dermatitis and nephropathy syndrome42
• Intestinal parasitism, e.g. ostertagiasis, trichostrongylosis in calves and sheep, have this effect.7
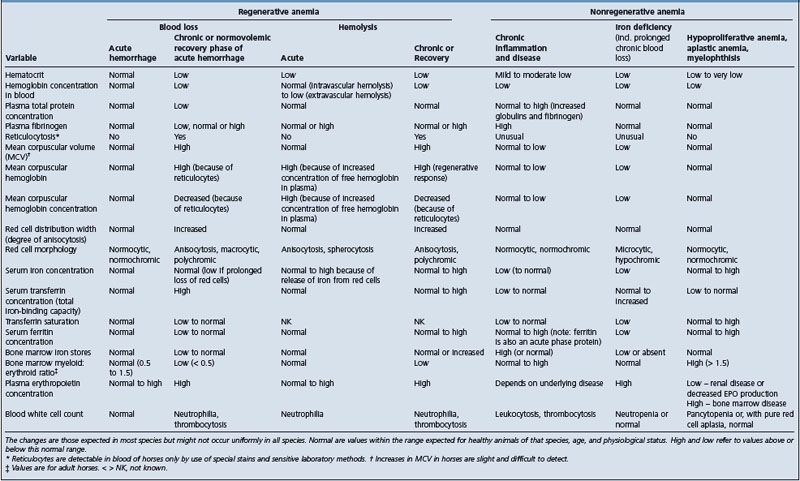
Red cell aplasia
• Red cell hypoplasia is a fatal syndrome of anemia, immunodeficiency and peripheral gangliopathy that develops at 4–8 weeks of age in some Fell pony foals
• Anemia in some horses follows the administration of recombinant human erythropoietin.43-45 The anemia is due to pure red cell aplasia and is manifest as normocytic, normochromic anemia. The disease is attributable to injection of horses with recombinant human erythropoietin with subsequent development of substances in blood, presumably antibodies to rhEPO, that cross-react with and neutralize endogenous erythropoietin in affected horses.43,45 Not all horses administered rhEPO develop anemia, but the disease is reported as an outbreak in a stable of Thoroughbred race horses administered the compound.45 Severely affected horses die. Treatment of severely affected horses is futile, but mildly affected horses can recover.43 Whether the recovery was spontaneous or because of administered glucocorticoids is unknown. Administration of cyclophosphamide and glucocorticoids was not effective in treatment of several severely affected horses. Blood transfusion provides temporary relief
• Pure red cell aplasia not associated with administration of rhEPO occurs but rarely in horses. The disease can be transient.
Myelophthisic anemia
Myelophthisic anemia, in which the bone marrow cavities are occupied by other, usually neoplastic tissues, is rare in farm animals. Clinical signs, other than of the anemia, which is macrocytic and normochromic, include skeletal pain, pathological fractures and paresis due to the osteolytic lesions produced by the invading neoplasm. Cavitation of the bone may be detected on radiographic examination.
• Lymphosarcoma with bone marrow infiltration occurs in most species
• Plasma cell myelomatosis has been observed as a cause of such anemia in pigs, calves and horses
• Infiltration of neoplastic cells, other than lymphoma or myeloma, such as melanoma in horses46
• Myelophthisic anemia due to myelofibrosis is reported in a pony47 and as a familial disease in pygmy goats.48
PATHOGENESIS
Anemic hypoxia
The most important abnormality in anemia is the hypoxemia and subsequent tissue hypoxia that results from the reduced hemoglobin concentration and oxygen-carrying capacity of blood. The anemia becomes critical when insufficient oxygen is delivered to tissue to maintain normal function.
Oxygen delivery is described mathematically by the Fick equation:
Oxygen delivery = cardiac output × arteriovenous oxygen content difference.
Oxygen delivery is therefore the rate at which oxygen is delivered to the tissue – it is a combination of the rate at which oxygen arrives at the tissue in arterial blood and the proportion of that oxygen extracted from the capillary blood.
Cardiac output is determined by heart rate and stroke volume, whereas the arteriovenous difference in blood oxygen content is determined by the hemoglobin concentration, hemoglobin saturation with oxygen in both arterial and venous blood, and the extraction ratio. The extraction ratio is the proportion of oxygen that is removed from the blood during its passage through tissues. In animals with a normal hematocrit and cardiac output, oxygen delivery to tissues exceeds the oxygen requirements of the tissue by a large margin, with the result that the oxygen extraction ratio is small (< 40%). However, as the oxygen-carrying capacity per unit of blood declines (usually expressed as mL of oxygen per 100 mL of blood) then either blood flow to the tissue or the extraction ratio must increase to maintain oxygen delivery.49,50 In reality, both of these compensatory mechanisms occur during the acute and chronic responses to anemia. Heart rate increases to increase cardiac output and therefore the delivery of oxygen to tissue, and blood flow is preferentially directed to those tissue beds that are most essential for life or are most sensitive to deprivation of oxygen (heart, brain, gut, kidney). Extraction ratio increases and is evident as a decrease in venous blood hemoglobin saturation. Hemoglobin in arterial blood is usually thoroughly saturated with oxygen and the limitation to oxygen delivery to tissue is the low hemoglobin concentration and consequent low arterial oxygen content. Assessment of arterial blood oxygen tension and content is discussed in Chapter 10.
Reductions in hemoglobin concentration are compensated for by increases in cardiac output and extraction ratio so that oxygen delivery to tissue is maintained in mild to moderate anemia.49 As the severity of anemia increases, these compensatory mechanisms are inadequate and oxygen delivery to tissues declines. At some point the delivery of oxygen fails to meet the oxygen needs of the tissue and organ function is impaired. It is important to realize that this is not an all-or-none phenomenon and that there is not a particular point at which decompensation occurs. In fact, with progressive anemia there are progressive increases in cardiac output and oxygen extraction ratio (evident as a progressive decline in venous hemoglobin saturation) until these compensatory mechanisms are maximal.49 Arterial pH and lactate concentration are maintained until the degree of anemia cannot be compensated for by increases in cardiac output and extraction ratio, at which point blood lactate concentration rises and blood pH and base excess decline. This is the degree of anemia at which oxygen use by tissue is entirely dependent on blood flow – decreases in blood flow decrease oxygen utilization and increases in blood flow increase oxygen utilization until the point where oxygen delivery exceeds oxygen consumption.
Compensation for slowly developing anemia is more complete than for rapidly evolving anemia such that animals with chronic anemia can tolerate a degree of anemia that would be intolerable for animals with acute anemia of a similar severity. Part of this chronic compensation includes changes in the affinity of hemoglobin for oxygen, which is due in part to increases in 2,3-diphosphoglycerate concentration in red cells.
When anemia is sufficiently severe that it reduces oxygen delivery to tissue to rates that are less than the oxygen needs of tissue, tissue hypoxia develops and the proportion of energy generated by anaerobic metabolism increases. Anaerobic metabolism cannot be sustained for more than a short period of time (minutes) before tissue function is impaired. Impaired organ function is evident as decreased myocardial contractility, decreased cerebral function, decreased gastrointestinal motility and abnormal renal function, to list just a few of many important abnormalities. The severity of these abnormalities depends on the metabolic activity of the tissue with more metabolically active tissues (e.g. the heart) being more sensitive to hypoxia. Death usually results from acute heart failure due to arrhythmia.
The effect of anemia is also dependent on the metabolic state of the animal. Exertion, even mild exertion such as grazing or following a herd or flock, can increase oxygen demands above that which can be sustained by the degree of anemia. Similarly, increases in body temperature, such as with fever, increase oxygen demand noticeably – an increase in body temperature of 1°C increases oxygen need by 12%.
Anemia induces increases in plasma erythropoietin concentration, which stimulates erythropoiesis in bone marrow and, in young animals or those with extreme anemia, in extramedullary sites. The increase in plasma erythropoietin concentration is prompt, occurring within hours of the development of anemia. The compensatory erythropoietic response is slower, with new red cells being detectable in 1–2 days in most species and bone marrow reticulocytosis detectable in less than 1 week.50,51
Autoimmune hemolytic anemia
The disease is believed to result from an aberrant production of antibodies targeted against surface antigens of the erythrocyte as a result of an alteration in the erythrocyte membrane from systemic bacterial, viral or neoplastic disease. An alternate hypothesis is the development of immunocompetent clones that direct antibody at the red cell membrane.17,24 Red cells are lost by intravascular hemolysis or removal by macrophages of the reticuloendothelial system and anemia occurs when the capacity of the bone marrow to compensate for increased red cell destruction is exceeded. Autoimmune hemolytic anemia is considered to be idiopathic if it cannot be associated with an underlying disease and is considered to be secondary if associated with another condition. Often this is neoplastic disease. The antibodies are of the IgG or IgM class, may be agglutinating or nonagglutinating and can also be temperature-dependent. The antiglobulin test has been used to confirm the diagnosis in cases of nonagglutinating autoimmune hemolytic anemia, but demonstration of immunoglobulin on the surface of red cells by immunofluorescent cell staining and flow cytometry is much more sensitive and specific.52,53
Hemolysis
Hemolysis results from rupture of red cell membranes as a consequence of injury to the membrane or osmotic lysis when serum tonicity is lower than normal. Hemolytic disease of any cause has the potential to overwhelm the normal clearance mechanisms for hemoglobin, with the result that hemoglobin concentrations in plasma are abnormally high. This can result in hemoglobinuric nephrosis (see Ch. 11).
Methemoglobinemia and oxidative damage
Methemoglobinemia results from oxidative damage of hemoglobin and occurs in disease such as red maple leaf toxicosis in horses and nitrate poisoning in ruminants. Methemoglobinemia is reversible but important as an indicator of oxidative damage and because methemoglobin cannot transport oxygen. Oxidative damage to red cells results in denaturation of hemoglobin with subsequent formation of Heinz bodies. Red cells damaged in this way are sensitive to osmotic lysis and fragmentation. Intravascular hemolysis and removal of damaged red cells by the reticuloendothelial system contributes to anemia.
CLINICAL FINDINGS
The clinical signs and their severity depend on the degree of anemia. Mild anemia in animals that are not required to be physically active, such as veal calves or housed lambs, might be apparent only as failure to achieve optimal weight gain. More severe degrees of anemia, or mild anemia in animals required to be physically active, such as foals at pasture or race horses, can be evident as exercise intolerance, failure to perform athletically, or lethargy. Behavioral signs of anemia include prolonged recumbency, depressed mentation, reduced nursing, foraging or grazing and, in extreme anemia, belligerence.
Physical findings include pallor of the mucosae but appreciable degrees of anemia can occur without clinically visible change in mucosal or skin color. The mucous membranes, and skin in pale-skinned, sparsely haired animals such as pigs, can be almost white in animals with severe anemia. Hemolytic anemia causes jaundice in most cases.
A chart for examination of conjunctival color in sheep and goats has been validated as a means of assessing severity of anemia in these species. The chart (FAMACHA®) was developed to aid in parasite control programs.54 Conjunctival color is assessed on a scale of 1–5 in which 1 = red and 5 = white.54 The correlation between FAMACHA score and hematocrit was very good (R = −0.52 in sheep and –0.30 in goats). The sensitivity and specificity for detection of a hematocrit below 15% for FAMACHA scores of 4 and 5 were 83% and 89% for sheep, and 83% and 71% for goats. This methodology appears to be very useful for detection of anemia in small ruminants.
The heart rate is increased, the pulse has a large amplitude and the absolute intensity of the heart sounds is markedly increased in anemic animals. Terminally the moderate tachycardia of the compensatory phase is replaced by a severe tachycardia, a decrease in the intensity of the heart sounds and a weak pulse. A hemic murmur might be heard and is likely a result of the low viscosity of blood in anemic animals combined with increased ejection velocity of blood from the heart as a consequence of increased heart rate and cardiac output.
Dyspnea is not pronounced in anemia, the most severe degree of respiratory distress appearing as an increase in depth of respiration without much increase in rate. Labored breathing occurs only in the terminal stages and at those times the animals can be severely distressed.
Other signs of decompensated anemia include anxious expression, absent rumination, ileus, colic, anuria and cardiac arrhythmia. Animals can appear quiet and comfortable unless they are forced to move or an event occurs that increases oxygen consumption and causes decompensation. An example is an animal that has compensated for its severe anemia but then develops a fever. Fever can increase whole body oxygen requirement by 12% for each 1°C increase in temperature and this can cause a finely balanced animal to decompensate.
There can be signs of the inciting disease and these can include edema, jaundice, petechial and ecchymotic hemorrhages in the mucosa and hemoglobinuria.
Adjunctive examination can include gastrointestinal, urinary or upper respiratory endoscopy; radiography of the chest or abdomen; and ultrasonographic examination of affected regions.
CLINICAL PATHOLOGY
The clinicopathological characteristics of the common forms of anemia are provided in Table 9.1.
Hematology
Anemia is definitively diagnosed by measurement of red cell indices and demonstration of low hematocrit, red cell count and hemoglobin concentration. Examination of various red cell indices can yield important information about the cause of anemia and evidence of regeneration. In addition to providing the diagnosis, serial monitoring of the hemogram is useful in detecting evidence of a regenerative response. At a minimum, repeated measurement of hematocrit will reveal a gradual increase when there is a regenerative response. Hematocrit of horses with induced anemia increases by approximately 1% (0.01 L/L) every 3 days.55
Red cell morphological abnormalities include variations in size, shape, and content:
Agglutination of red cells is apparent as irregularly shaped agglomerations of red cells. The clumps of red cells do not dissociate when blood is diluted 1:4 with 0.9% saline, as happens with rouleaux. Rouleaux are normal findings in blood of horses and are apparent as rows of erythrocytes.
Coombs testing or use of direct immunofluorescent flow cytometry can provide evidence of immune-mediated hemolytic anemia.52,53
Other hematologic changes in severe anemia include leukocytosis and thrombocytosis.
Bone marrow
Examination of bone marrow is useful for demonstrating a regenerative response, especially in horses in which a regenerative response can be difficult to detect in peripheral blood, and for determining the cause of nonregenerative anemia.
Collection of bone marrow
Samples of bone marrow can be obtained by aspiration, with samples submitted for cytological examination, or biopsy, with core samples submitted for histological examination. Bone marrow aspirates are useful in that they provide samples in which the relative proportions of myeloid and erythroid cell lines can be determined. However, samples obtained by aspiration do not allow examination of the over all cellularity of the marrow or its architecture.
Samples of bone marrow can be obtained from the sternebrae, proximal aspects of the ribs or tuber coxae. The preferred site in adult animals, and in calves, is the cranial sternum. The procedure is performed on standing adult animals or laterally recumbent calves. Animals should be adequately restrained, which could include administration of sedatives and analgesics. A site on the ventral midline over the second or third sternebra is clipped and aseptically prepared. Local analgesia is induced by injection of lidocaine or similar local anesthetic (5–10 mL). The local anesthetic is injected subcutaneously and to the surface of the sternebra. A small skin incision is made and the aspiration needle or biopsy instrument is introduced. Bone marrow aspirates can be collected using a 13–15-gauge, 5–7 cm needle and stylet. Bone marrow core biopsies are performed using an 8-, 11- or 13-gauge 100–150 mm bone marrow biopsy needle (TrapSystem®).
Bone marrow aspirates are collected from adult horses by advancing the needle approximately 2–3 cm into the sternebra. The stylet is then removed, a 5–10 mL syringe is attached and bone marrow is aspirated. The samples should be placed on a clean glass slide and air-dried, or put in a Petri dish containing 0.5–1.0 mL of 2% EDTA.
Core samples of bone marrow are obtained by inserting the biopsy needle 2 cm into the cortex of the sternebra. The stylet is then removed and the needle is advanced with a rotating motion. This can require considerable effort in adult animals. The needle is advanced approximately 2–3 cm and then rapidly withdrawn. A sample of bone marrow will be evident as pink to red bone. The sample should be rolled on a clean, dry glass slide (for cytological examination) and then placed in 10% neutral buffered formalin for histological examination.
Interpretation of bone marrow
Bone marrow is examined for overall architecture, cellularity, the ratio of myeloid to erythroid cells (M:E ratio) and the presence of inflammation, necrosis or abnormal cells. A subjective assessment of iron stores can be made by staining sections of marrow with Prussian blue stain.
A regenerative response is evident as a low M:E ratio due to erythrocyte hyperplasia, and the presence of erythroid series cells in all stages of maturity. There are increased counts of reticulocytes in bone marrow and the number of nucleated cells relative to the hematocrit increases. The MCV and reticulocyte hemoglobin content are high in regenerative bone marrow. These responses are evident as soon as 3 days after acute anemia and peak at approximately 9 days.51,57
Abnormal white cells, such as seen in myelophthisic disease caused by myeloma or lymphosarcoma, cause displacement of erythroid series cells and a high M:E ratio. Similarly, a high M:E ratio is obtained when there is primary red cell aplasia. A normal M:E ratio is obtained when there is aplasia of both myeloid and erythroid series of cells, highlighting the need to evaluated overall cellularity of the marrow. Normal marrow is approximately 50% fat and 50% combined myeloid and erythroid series cells.
Blood gas analysis, oximetry and lactate
Arterial blood gas analysis
Arterial blood oxygen tension (mmHg, kPa) in animals with anemia is almost always within the reference range for healthy animals unless there is coexisting lung disease. Anemia does not interfere with diffusion of oxygen from the alveolus into capillary blood. However, the arterial oxygen content (mL O2 per 100 mL blood) is reduced because of the reduced arterial blood hemoglobin concentration (see Ch. 10). Arterial carbon dioxide tension is often reduced in severe anemia as a result of alveolar hyperventilation that is a response to arterial hypoxemia. Arterial pH and base excess decline as the severity of anemia increases and compensatory mechanisms are no longer able to ensure delivery of sufficient oxygen to tissue the, indicative of metabolic acidosis resulting from tissue anaerobiosis.
Venous blood gas analysis
The ideal sample is mixed venous blood collected from the pulmonary artery or right atrium. However, these sites are only infrequently available for collection so samples should be collected from a a major vein (jugular vein, cranial vena cava). Samples collected from small leg veins are less than ideal. Measurement of venous blood gas tensions, pH, base excess, hemoglobin saturation and oxygen content are useful in evaluating the physiological effect of anemia. As discussed under pathophysiology, reductions in oxygen content of arterial blood cause an increase in oxygen extraction ratio in an attempt to maintain oxygen delivery to tissue. The increased extraction ratio is evident as a reduction in venous oxygen tension, hemoglobin saturation and oxygen content.49 When oxygen delivery to tissue is less than that needed to maintain aerobic metabolism, venous pH, bicarbonate concentration and base excess decline.
Methemoglobinemia
Measurement of methemoglobin concentration is useful in documenting the severity of diseases such red maple leaf toxicosis and nitrate poisoning. Methemoglobinemia is reversible but is a sign of oxidative damage to red cells. Oxidative damage to red cells causes Heinz body formation and eventual lysis of the cell. Methemoglobin is measured using a co-oximeter and is combined with measurement of oxygen saturation. Methemoglobin concentration in blood of healthy animals is usually less than 3%.
Lactate
Concentrations of lactate can be measured in blood (‘whole blood lactate’) or plasma. Whole blood lactate concentrations are lower than lactate concentrations in plasma because red blood cells have lower lactate concentration than does plasma. Lactate concentrations can be measured using point-of-care analyzers, some of which have been validated for use in animals. Lactate concentration in blood or plasma increases when compensatory mechanisms are no longer effective and aerobic metabolism is impaired.
Serum biochemistry
Serum biochemical abnormalities are those of the inciting disease or reflect damage to organs as a result of the anemia. Severe anemia can damage many organs, resulting in increases in serum concentration or activity of urea nitrogen, creatinine, sorbitol dehydrogenase, gamma-glutamyl transpeptidase, bile acids, bilirubin, aspartate aminotransferase, creatine kinase and troponin, among others. Hemolytic anemia causes increases in plasma hemoglobin concentration (evident grossly as pink-tinged plasma or serum) and hyperbilirubinemia (unconjugated).
Iron metabolism in anemic animals is defined by measurement of serum iron concentration, serum transferrin concentration (total iron-binding capacity), transferrin saturation and serum ferritin concentration. Serum ferritin concentration correlates closely with whole body iron stores. Values of these variables in anemia of differing cause are provided in Table 9.1.
Other evaluations
Feces should be examined for the presence of parasites (ova, larvae or adult parasites), frank blood (hematochezia or melena) and occult blood. Detection of occult blood can be difficult and samples should be collected on multiple occasions. Samples should not be collected soon after rectal examination, as false-positive results can be found because of trauma to the rectal mucosa.
Urine should be evaluated for the presence of pigmenturia, red cells and casts. Pigmenturia should be differentiated into hemoglobinuria or myoglobinuria. Microscopic examination will reveal red cells, or ghost red cells, in animals with hematuria. Casts and isosthenuria can be present in urine of animals with hemoglobinuric nephrosis.
Serum erythropoietin concentration should be evaluated in animals with nonregenerative anemia. It is not a readily available assay. Concentrations of erythropoietin in adult horses are usually less than 37 mU/mL, but values are probably dependent on the assay used.
Tests for specific diseases should be performed as appropriate:
NECROPSY FINDINGS
Findings indicative of anemia include pallor of tissues, thin, watery blood and contraction of the spleen. Icterus may be evident where there has been severe hemolytic anemia and petechial and ecchymotic hemorrhages with thrombocytopenia. Necropsy findings specific to individual diseases are given under those disease headings.
TREATMENT
The principles of treatment of anemia are ensuring adequate oxygen transport to tissues, prevention of the deleterious effects of anemia or hemolysis, and treatment of the inciting disease. The individual inciting diseases are discussed elsewhere in this text.
Correction of anemia
The discussion here deals with normovolemic anemia. Acute anemia with hypovolemia (hemorrhagic shock) is dealt with in Chapter 2.
Transfusion
The oxygen-carrying capacity of blood should be restored in the short term to at least the level at which oxygen use by tissue is not flow-dependent, and to normal levels in the longer term. Short-term restoration of the oxygen-carrying capacity of blood is achieved by transfusion of whole blood or packed red cells, or administration of a commercial stromal free hemoglobin solution.
The decision to transfuse an anemic animal should not be undertaken lightly for a number of reasons. Transfusion of blood or packed red cells is not without risk to the recipient, there is usually considerable cost in identifying a suitable donor and collecting blood, and the process can be time-consuming. An important concern in performing a blood transfusion is the risk to the recipient. Acute reactions, include anaphylaxis and acute host versus graft reaction (hemolysis of transfused red cells), and graft versus host disease (hemolysis of recipient red cells). Development of alloantibodies in the recipient with consequent problems with repeat transfusion or development of neonatal alloimmune hemolytic anemia in progeny of female recipients is a concern. The incidence of these adverse events has not been recorded for large animals but can be minimized by crossmatching donor and recipient.
Crossmatching and the mechanics of blood transfusion are discussed in Chapter 2 and elsewhere.58 Briefly, both major (donor red cells and recipient plasma) and minor (donor plasma and recipient red cells) crossmatching should be performed. Ideally, blood typing and examination of plasma for alloantibodies of both donor and recipient would be performed before transfusion, but these are rarely available in an appropriate time frame.
Indications for transfusion are not straightforward. Because of the risk to the recipient and cost, blood transfusion should be performed only when indicated. Conversely, the severe adverse effects of anemia mean that animals should not be denied a transfusion if it is needed. There is no one variable for which a single value is a ‘transfusion trigger’, and the decision to provide a transfusion should not be based on hematocrit, hemoglobin concentration or red cell count alone. Rather, the decision to provide a transfusion should be based on a holistic evaluation of the animal, including the history, physical abnormalities and clinicopathological data. This information should be considered in total before a decision is made to provide a transfusion. Considerations regarding transfusion include:
• History – animals with acutely developing anemia are more likely to require transfusion at a given hematocrit than are animals with slowly developing anemia. Similarly, young animals with higher intrinsic metabolic rates might require transfusion at hematocrit values that would be tolerated by adults
• Physical findings – these are some of the most important indicators of the need for transfusion and include:
Transfusion to correct anemia in normovolemic animals should be done cautiously to minimize the risk of excessive expansion of the intravascular volume. Ideally, packed red cells can be administered to reduced the extent of blood volume expansion. However, preparation of packed red cells can be difficult and time-consuming. An alternative with horses is simply to allow the collected blood to sit undisturbed for 1–2 hours, during which time the cells will settle to the bottom. The red cells can then be siphoned off and administered to the recipient.
Details of donor selection, blood collection and blood administration are provided in Chapter 2.
An alternative to transfusion of whole blood or packed red cells is the administration of a commercial preparation of stromal free hemoglobin. This product is effective in increasing oxygen-carrying capacity of blood in anemic horses and has been used for support of a foal with alloimmune hemolytic anemia until a blood transfusion was available.59,60 The compound is stable at room temperature and can therefore be stored for long periods of time and be readily available for use. However, it is expensive and its effect is short-lived (< 48 h and probably less). The recommended dose is 15 mL/kg BW intravenously, but lower doses have been used. The compound increases oncotic pressure of plasma and causes expansion of the plasma volume.
The efficacy of transfusion can be assessed by examination of the animal and measurement of venous blood oxygen tension and saturation, and blood lactate concentration. Venous blood oxygen tension and saturation improve promptly with transfusion of an adequate red cell mass.
Hematinics
Hematinic preparations are used in less severe cases and in animals with anemia due to iron deficiency or severe external blood loss (see Table of Drug Doses in Appendix). Iron is administered to prevent iron deficiency in young animals denied access to pasture or soil. The use of recombinant human erythropoietin in horses has a risk of inducing anemia.43,45 Given that there are no known causes of low erythropoietin concentrations causing anemia in horses, with the exception of those horses with anemia subsequent to rhEPO administration, the use of this compound in horses is specifically contraindicated.
Supportive care
The oxygen requirements of anemic animals should be minimized. This can be achieved by housing them individually in quiet stalls the temperature of which is maintained in the animal’s thermoneutral zone, minimizing the need for exercise (such as grazing or following the mare to nurse), and maintaining a normal body temperature.
Animals with hemolytic anemia and hemoglobinuria should be administered polyionic isotonic fluids intravenously to reduce the risk of hemoglobinuric nephrosis.
Treatment of autoimmune hemolytic anemia
Some animals with autoimmune hemolytic anemia respond well to administration of corticosteroids.6,21,24 Compounds used include prednisolone and dexamethasone. Horses with refractory aplastic anemia or hemolytic anemia have been administered cyclophosphamide (2 mg/kg intravenously every 14–21 d) in addition to glucocorticoids.
Lassen ED, Swardson CJ. Hematology and hemostasis in the horse: Normal functions and common abnormalities. Vet Clin North Am Equine Pract. 1995;11:351.
Durham AE. Blood and plasma transfusion in the horse. Equine Vet Educ. 1996;8:8-12.
Knight R, et al. Diagnosing anemia in horses. Compend Contin Educ Pract Vet Equine Ed. October 2005:23-33.
1 Hofmann-Lehmann R, et al. J Clin Microbiol. 2004;42:3775.
2 Braun U, et al. Vet Rec. 2005;157:452.
3 Lincoln SD, et al. J Am Vet Med Assoc. 1992;200:1090.
4 Gilchrist F. Can Vet J. 1996;37:490.
5 Middleton JR, et al. J Vet Intern Med. 1997;11:382.
6 Fenger CK, et al. J Am Vet Med Assoc. 1992;201:97.
7 Winter A, Clarkson M. In Pract. 1992;14:283.
8 Nappert GN, et al. Can Vet J. 1995;36:104.
9 Yeruham I, et al. Vet Rec. 2003;153:502.
10 Steffen DJ, et al. Vet Pathol. 1992;29:203.
11 Steffan DJ, et al. J Hered. 1993;84:263.
12 Nicholls TJ, et al. Aust Vet J. 1992;69:39.
13 Semrad SD. Compend Contin Educ Pract Vet. 1993;15:261.
14 Pearson W, et al. Am J Vet Res. 2005;66:457.
15 Norman TE, et al. J Am Vet Med Assoc. 2005;226:2039.
16 Weiss DJ, Moritz A. Vet Clin Pathol. 2003;32:22.
17 Beck DJ. Equine Vet J. 1990;22:292.
18 Messer NT, Arnold K. J Am Vet Med Assoc. 1991;198:1415.
19 Sockett D, et al. J Am Vet Med Assoc. 1987;190:308.
20 Mair TS, et al. Vet Rec. 1990;126:51.
21 Lubas G, et al. Equine Pract. 1997;19:27.
22 Step DL, et al. Cornell Vet. 1990;81:13.
23 McConnico RS, et al. J Am Vet Med Assoc. 1992;201:1402.
24 Morris DD. Equine Pract. 1990;11:34.
25 Meier J, Stocker K. Crit Rev Toxicol. 1991;21:171.
26 Dickinson CE, et al. J Am Vet Med Assoc. 1996;208:1866.
27 Pritchard GC, et al. Vet Rec. 1985;117:545.
28 Szabo P, Bilkei G. J Vet Med A. 2002;49:390.
29 Lindt F, Blum JW. Zentralbl Veterinarmed A. 1994;41:333.
30 Moser M, et al. Zentralbl Veterinarmed A. 1994;41:343.
31 Vatn S, Framstad T. Acta Vet Scand. 2000;41:273.
32 Kohn CW, et al. Am J Vet Res. 1990;51:1198.
33 Brommer H, van Oldruitenborgh-Oosterbaan MM. J Vet Intern Med. 2001;15:482.
34 Grace ND, et al. Aust Vet J. 1999;77:177.
35 Mullaney TP, Brown CM. Equine Vet J. 1988;20:119.
36 Pearson EG, Andreasen CB. J Am Vet Med Assoc. 2001;218:400.
37 Girard CL, Matte JJ. J Dairy Sci. 1998;81:1412.
38 Ordakowski-Burk AL, et al. Am J Vet Res. 2005;66:1214.
39 Piercy RJ, et al. Equine Vet J. 2002;34:311.
40 Toribio RE, et al. J Am Vet Med Assoc. 1998;212:697.
41 Finnie JW. Aust Vet J. 1992;69:21.
42 Sipos W, et al. Vet Immunol Immunopathol. 2005;107:303.
43 Piercy RJ, et al. J Am Vet Med Assoc. 1998;212:244.
44 Woods PR, et al. Equine Vet J. 1997;29:326.
45 Schwarzwald CR, Hinchcliff KW. Proc Am Assoc Equine Pract. 2004;50:270.
46 MacGillivray KC, et al. J Vet Intern Med. 2002;16:452.
47 Angel KL, et al. J Am Vet Med Assoc. 1991;198:1039.
48 Cain GR, et al. Comp Haematol Int. 1994;4:167.
49 Widness JA, et al. J Appl Physiol. 2000;88:1397.
50 Malikides N, et al. Vet J. 2001;162:44.
51 Cooper C, et al. J Appl Physiol. 2005;99:915.
52 Wilkerson MJ, et al. J Vet Intern Med. 2000;14:190.
53 Davis EG, et al. J Vet Intern Med. 2002;16:404.
54 Kaplan RM, et al. Vet Parasitol. 2004;123:105.
55 Radin MJ, et al. Vet Pathol. 1986;23:656.
56 Harper SB, et al. ASAIO J. 1994;40:M816.
57 Malikides N, et al. Res Vet Sci. 1999;67:285.
58 Durham AE. Equine Vet Educ. 1996;8:8.
59 Perkins GA, Divers TJ. J Vet Emerg Crit Care. 2001;11:141.
ERYTHROCYTOSIS
Erythrocytosis is an increase in erythrocyte count, hemoglobin concentration and hematocrit in blood. Polycythemia vera, a disease of humans and rarely small animals, and scarcely reported in cattle,1 is due to an increase in concentration of all blood cellular elements (erythrocytes, granulocytes and platelets). Erythrocytosis, which is due solely to an increase in red cell count, is either relative or absolute.
Relative erythrocytosis occurs when the total body red cell mass (i.e. the total amount of red cells in the body) is not elevated above normal, but the red cell count in peripheral blood is higher than expected. This is the most common form of erythrocytosis. Relative erythrocytosis occurs both as an abnormality and as a physiological response to physical or psychological stress in animals with a capacious and capricious spleen. Abnormal relative erythrocytosis results from hemoconcentration and is evident as an increase in concentration of red cells and serum total protein. The cause is a reduction in plasma volume, which is usually associated with dehydration due either to lack of water intake or to excessive losses (diarrhea, vomition). The diagnosis is usually obvious, based on the presence of hemoconcentration and other signs of the underlying disease. Physiological relative erythrocytosis occurs most noticeably in horse as a result of either excitement or exercise. The blood in the spleen of horses has a hematocrit much higher than that of blood (70–80%) and when relaxed the spleen contains many liters of blood. Excitement or exercise cause splenic contraction through an alpha-1-mediated event and ejection of the red-cell-rich blood into the peripheral circulation, with subsequent marked increases in hematocrit.2,3 The spleen of an adult horse can eject 5–10 L of blood into the circulation, which, together with a decline in plasma volume during exercise, increases hematocrit to 55–60% (0.55–0.60 L/L).2
Absolute erythrocytosis occurs because of an increase in the number of red blood cells in the body. It is classified as primary or secondary, and within secondary erythrocytosis there is a further classification of appropriate or inappropriate. Primary erythrocytosis is attributable to proliferation of erythroid progenitors with maturation of the red cell series in the absence of arterial hypoxemia or increases in plasma erythropoietin concentration. It is a myeloproliferative disorder. Disorders resembling primary erythrocytosis are described in horses.4,5 These horses had marked increases in red cell count without evidence of diseases causing arterial hypoxemia or tissue hypoxia and without increases in serum erythropoietin concentration. A familial erythrocytosis is documented in cattle, but the disease resolved as animals matured, which is not consistent with primary erythrocytosis due to a myeloproliferative disorder.6
Secondary erythrocytosis is classified as either appropriate or inappropriate. Appropriate secondary erythrocytosis occurs as a consequence of diseases that cause tissue hypoxia with subsequent increases in plasma erythropoietin concentration. Tissue hypoxia is often inferred from the low arterial blood oxygen tension or content in these diseases. Tissue hypoxia can occur in the face of normal arterial blood oxygen tension when there is an abnormality in hemoglobin (such as chronic methemoglobinemia or carboxyhemoglobinemia), although this has not been reported as a cause of erythrocytosis in large animals. Diseases causing appropriate secondary erythrocytosis include chronic lung or respiratory disease, and congenital cardiac anomalies in which there is right-to-left shunting (such as Eisenmenger’s complex in cattle). Physiological appropriate secondary erythrocytosis occurs in animals living at high altitude.
Inappropriate secondary erythrocytosis occurs in animals that do not have arterial hypoxemia or diseases causing tissue hypoxia. Plasma erythropoietin concentrations are elevated despite there being normal arterial oxygen tension and content, hence the term ‘inappropriate’. The disease is usually associated with hepatic or renal neoplasia. The disease in horses is described in foals or young animals with hepatoblastoma7,8 and adults with hepatic carcinoma.9,10 Erythrocytosis is recorded in a mare with a lymphoma that expressed the gene for equine erythropoietin, suggesting that anomalous production was the cause of the secondary inappropriate erythrocytosis.11 Erythrocytosis also occurs in horses with liver disease.12 The cause is not known, but could involve increased production of erythropoietin or decreased clearance because of reduced hepatic function. Inappropriate secondary erythrocytosis in ruminants or pigs is not reported, but probably occurs.
The clinical signs of secondary erythrocytosis are those of the underlying disease (dyspnea, congestive heart failure, cyanosis). In addition, the erythrocytosis can be evident as dark red or slightly purplish mucous membranes, lethargy and an increased propensity for thrombosis. These signs occur because of the increase in blood viscosity that results from marked increases in red cell concentration. Treatment is directed toward the inciting disease. For animals with primary erythrocytosis, repeated phlebotomy and restriction of iron intake has been used to reduce the red cell count.5
A syndrome is described in Standardbred trotters in Sweden that have normal red cell count at rest but counts during maximal exercise that are higher than expected.13 The syndrome is referred to as ‘red cell hypervolemia’ and is associated with poor race performance. Diagnosis is based on a history of poor performance and hematocrit or red cell counts during maximal exercise or after administration of epinephrine that are higher than expected. Treatment is prolonged rest, although some horses have had phlebotomy and therapeutic bleeding.
1 Fowler ME, et al. Cornell Vet. 1964;54:153.
2 McKeever KH, et al. Am J Physiol. 1993;265:R404.
3 Hardy J, et al. Am J Vet Res. 1994;55:1570.
4 Beech J, et al. J Am Vet Med Assoc. 1984;184:986.
5 McFarlane D, et al. J Vet Intern Med. 1998;12:384.
6 Tennant B, et al. J Am Vet Med Assoc. 1967;150:1493.
7 Cantile C, et al. Equine Vet J. 2001;33:214.
8 Lennox TJ, et al. J Am Vet Med Assoc. 2000;216:718.
9 Cook G, et al. Equine Vet J. 1995;27:316.
10 Roby KA, et al. J Am Vet Med Assoc. 1990;196:465.
11 Koch TG, et al. J Vet Intern Med. 2006;20:000.