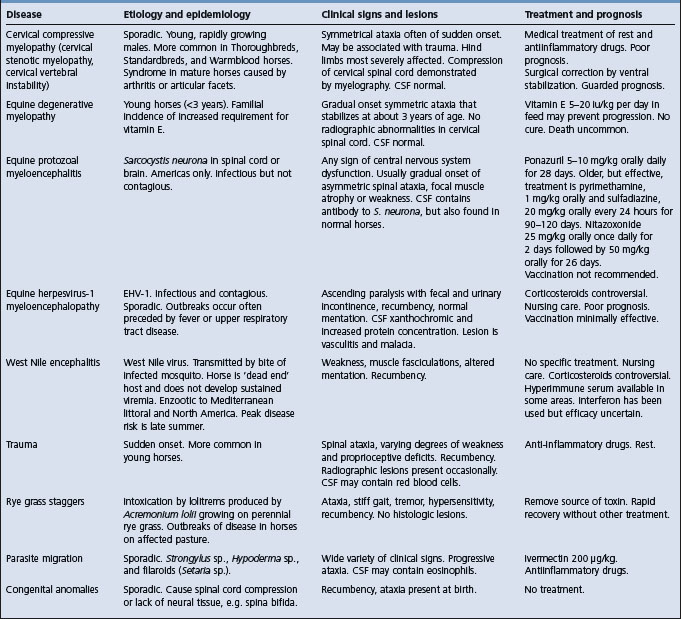
EQUINE NEONATAL ENCEPHALOPATHY (NEONATAL MALADJUSTMENT SYNDROME OF FOALS, DUMMY FOAL, BARKERS, AND WANDERERS)
This is a syndrome of foals less than 36 hours of age characterized by a spectrum of changes in mentation ranging from failure to suckle, abnormal behavior, seizures, through coma in otherwise apparently healthy foals.
ETIOLOGY
A number of diseases cause the clinical signs consistent with this syndrome. These include antenatal, natal or postnatal hypoxia, congential and metabolic anomalies, placental abnormalities, intracranial hemorrhage, prematurity, and thoracic trauma.1 Of these causes, hypoxia before, during or soon after birth is considered the most common cause of neonatal encephalopathy, although hypoxia is rarely documented. It is important to realize the neonatal encecphalopathy is one of many manifestations of hypoxia of the fetus or neonate, the other manifestations including gastrointestinal and renal damage.2,3
EPIDEMIOLOGY
The disease is sporadic and occurs worldwide with an annual incidence in foals of approximately 1%.4 Foals of either sex and of any breed born to mares of any age or reproductive history can be affected. The case fatality rate is very low for appropriately treated foals without other systemic illness.
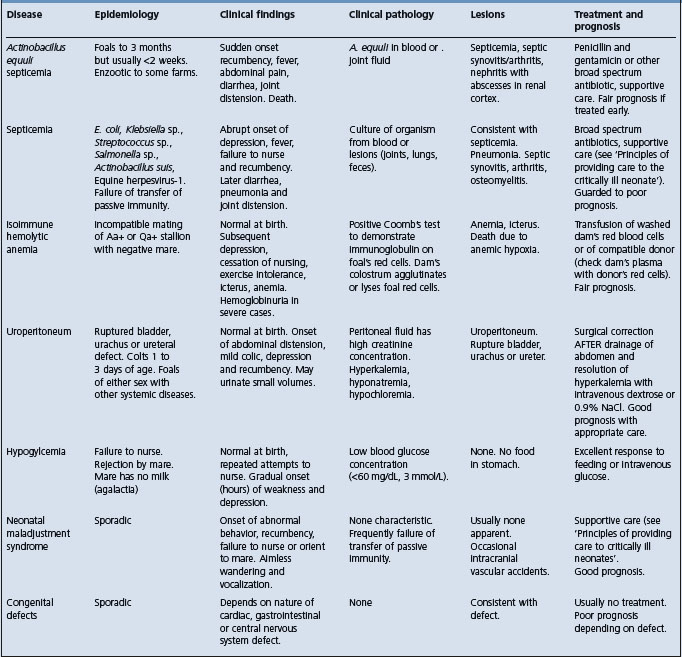
PATHOGENESIS
It is speculated that hypoxia resulting from intracranial vascular accidents,5 asphyxia at birth or placental insufficiency before birth damages the central nervous system. Neurological abnormalities and a failure to nurse, result in a failure of the transfer of maternal immunoglobulins, which predisposes the foal to septicemia and hypoglycemia.
CLINICAL SIGNS
Foals that are abnormal at birth can display a range of behavioral abnormalities, from lack of suckle reflex to convulsions with extensor rigidity. The placenta of affected foals is often abnormal or there is a history of prolonged parturition. Affected foals either do not develop or lose the suck reflex, and have no affinity for the mare and are unable to locate the udder or teat. Aimless wandering and a characteristic ‘barking’ vocalization are sometimes present. Recumbent foals struggle wildly and in an uncoordinated fashion to stand. Convulsing foals usually display opisthotonos with extensor rigidity. Other signs of convulsive activity include facial twitching and grimacing, nystagmus, rapid blinking, sucking, chewing, and drooling.1 Between episodes foals are usually depressed or somnolent. Affected foals display little or no interest in the mare. Convulsing foals are tachypneic, tachycardic (>180 bpm), and hyperthermic (>39°C, 102°F) during and immediately after convulsions. It is important to recognize that the severity of clinical signs varies from very mild (foals are often described by owners as being a bit slow or dimwitted) through to grand mal seizures.
Foals that are normal at birth may develop signs by 24 hours of age. The signs are similar to those described above, with the exception that the foals are initially able to ambulate. It is important to realize that healthy newborn foals lack a mence reflex, have a hypermetric gait and intention tremor, and become flaccid when restrained.
Affected foals can take days to weeks to recover completely. Blind foals that do not have ocular lesions can take as long as 4–6 weeks to regain vision.
Ancillary testing is not usually indicated unless the foal fails to respond after approximately 7 days. At that time, CT or MRI examination of the brain might be indicated to detect congenital anomalies such as hydrocephalus. Examination of cerebrospinal fluid should be performed in any foal with signs of CNS dysfunction in the presence of fever or other signs of sepsis.
CLINICAL PATHOLOGY
There are no hematological or serum biochemical abnormalities characteristic of the disease:
DIAGNOSTIC CONFIRMATION
Definitive diagnosis of the disease is difficult and is based on exclusion of other diseases that can cause similar signs and, at necropsy, demonstration of intracranial lesions consistent with the disease.
NECROPSY FINDINGS
Gross changes are typically limited to diffuse pulmonary congestion with a variable degree of atelectasis. In cases in which dystocia has been a contributing factor, fractured ribs, and foci of subcutaneous edema and hemorrhage are sometimes noted. Occasionally, macroscopic cerebral hemorrhages are visible. Histologicially, the key findings are hemorrhagic foci within the brain and areas of ischemic necrosis in the cerebral cortex.6 Meconium and other components of aspirated amniotic fluid accompanied by atelectasis and a mild inflammatory response may be present within the lung. In less affected foals the brain lesions are restricted to hemorrhage, cerebral swelling, and edema. Many affected foals have evidence of intracranial vascular accidents.7 Affected foals that are euthanased often have no detectable lesions in the brain.
Samples for post-mortem diagnostic confirmation
Formalin-fixed brain, including cerebral cortex, cerebellum and brain stem, and lung for light microscopic examination.
The disease must be differentiated from other diseases that cause neurological or behavioral abnormalities in foals including: sepsis; renal, hepatic, or gastrointestinal disease, which can occur secondary to fetal hypoxia; hydrocephalus; hypoglycemia; meningitis; neonatal isoimmune hemolytic anemia; and prematurity, dysmaturity, or immaturity.
TREATMENT
The principles of treatment are:
• Treatment of cerebral edema and hemorrhage
• Correction of failure of transfer of passive immunity
• Nutritional support and general nursing care. The management of affected foals is mainly supportive and is time consuming and labor intensive.
Provision of nutritional support, treatment of failure of passive transfer of maternal immunoglobulins, and nursing care is dealt with in detail in the section ‘Principles of care of the critically ill neonate’.
For other than emergency treatment of seizures, in which diazepam (0.1–0.4 mg/kg, intravenously, as required) or midazolam (0.05–0.1 mg/kg IV, as required) are useful, phenobarbital (phenobarbitone), phenytoin and primidone are the drugs of choice for long-term control of seizure activity. Phenobarbital is administered initially at a dose of 1–3 mg/kg intravenously in 30 mL of isotonic saline infused over 15–30 minutes. Maintenance therapy is a similar dose intravenously or orally, every 8 hours, and the dose adjusted to provide control of seizures while minimizing the degree of sedation. Because of the long elimination half life of phenobarbitol in foals (∼200 hours) and the transient nature of the disease, once seizure control is achieved administration of phenobarbitol can be discontinued. Drug concentrations will be at or above the target concentration (5–30 μg/mL) for several days after the final dose. Phenytoin (5–10 mg/kg intravenously or orally initially, then 1–5 mg/kg every 4 hours) or primidone (20–40 mg/kg orally every 12–24 hours, to effect) are also used to control convulsions.
Definitive demonstration of the presence of cerebral edema or intracranial hemorrhage is impossible without sophisticated imaging devices, such as magnetic resonance imaging or computed tomography (CT).8 However, treatment is often initiated on the basis of clinical signs. None of the treatments have demonstrated efficacy, and some are controversial. Dimethyl sulfoxide (DMSO) is given intravenously at 0.5–1 mg/kg once or twice daily for 3 days as a 10% solution. Mannitol (0.25 g/kg, intravenously as a 20% solution) may be effective in treating cerebral edema but is contraindicated if intracranial hemorrhage is present. Glucocorticoids (dexamethasone, 0.2–1 mg/kg or prednisone, 1–2 mg/kg) might reduce intracranial inflammation and swelling. They might be contraindicated in foals with sepsis.
Magnesium sulfate (0.05 mg/kg per hour for one hour, then 0.025 mg/kg/h IV for up to 48 hours) is often administered to foals with suspected hypoxic encephalopathy in an attempt to minimize neuronal damage. There is no objective evidence of its efficacy in foals.
Foals with respiratory depression can be administered caffeine (10 mg/kg orally once and then 3.0 mg/kg orally q 24 hours).3 Adverse effects include agitation, hyperesthesia, tachycardia, and convulsions.
Good nursing care is critical in affected foals, and a concerted and persistent effort should be made to encourage the foal to nurse the mare. Encouraging the foal to nurse can be frustrating for the handler and mare, but should be done regularly, about every 4 hours, and preferably when the foal is hungry. Affected foals often begin to nurse quite suddenly.
Affected foals can require up to 4–6 weeks to recover completely, although most do so within 1 week of birth, and hasty decisions regarding euthanasia should not be made without recognition of the sometimes long time required for complete recovery.
1 Hess-Dudan F, Rossdale PD. Equine Vet Educ. 1996;8:24.
2 Rossdale PD. Proc Am Assoc Equine Pract. 2004;50:75.
3 Vaala WE. Proc Am Assoc Equine Pract. 1999;45:247.
4 Rossdale PD. Equine Vet J. 1972;4:117.
5 Rossdale PD, et al. Vet Rec. 1976;99:111.
6 Palmer AC, Rossdale PD. Equine Vet Sci. 1976;20:267.