Chapter 32 Diseases associated with inorganic and farm chemicals
Principles of treatment in cases of poisoning
DISEASES ASSOCIATED WITH INORGANIC POISONS 1799
DISEASES ASSOCIATED WITH FARM CHEMICALS 1830
MISCELLANEOUS FARM CHEMICALS 1846
Suspicion of poisoning is aroused when illness occurs in a number of previously healthy animals; all affected at the same time and showing the same signs and necropsy findings, to the same degree of severity. These conditions, of course, may also apply to some infections, metabolic and nutritional deficiency diseases. It is only by acquaintance with the syndromes produced by the common poisons, particularly those likely to occur locally, that this primary differentiation can be made.
Poisonous plants often show a geographical limitation in distribution; particular industrial enterprises may create poison hazards in local areas; certain agricultural practices, including the spraying of orchards, the dipping or spraying of cattle for ectoparasites, and the use of prepared concentrate feed for pigs and cattle, may also lead to poisoning in groups of animals. So many chemical agents are used in agriculture today that a section of miscellaneous farm chemicals likely to be associated with the poisoning of animals has been included.
The appearance of clinical illness soon after feeding, after a change of ration, after medication or spraying, or after change to new pasture, is a common history in many outbreaks of disease associated with chemical agents. The report which accompanies material for toxicological analysis should include a full record of history, clinical signs and necropsy findings and particularly the results of a search of the environment for access to a poison. If the animal has been treated, the drugs that were used and the dates of administration should be given as they may create difficulties for the analyst. The poison or group of poisons suspected should be defined.
Specimens for analysis should include a sample of the suspected source material. Next most important is a specimen of alimentary tract contents, so that ingestion of the material can be proven, and a sample of tissue, usually liver, to prove that absorption of the poison has occurred. Kidney also provides a route for concentrating many toxicants for excretion and is an important specimen for chemical poisoning. Most toxic chemicals are ingested but percutaneous absorption and inhalation must be considered as possible portals of entry. One of the advantages of an examination of alimentary tract contents is that qualitative tests can be carried out and in many cases this determines whether or not further examination of tissues is necessary.
Additional specimens required other than liver and alimentary tract and contents, vary with the poison and the following list is suggested for the common chemicals:
• Arsenic – kidney, skin, and hair
• Lead – kidney, liver, bones, and whole blood
• Phosphorus – kidney and muscle
• Mercury – kidney, brain if organomercurials are suspected
• Copper – kidney, liver, and blood
• Sodium chloride – alimentary tract and contents, brain, and serum
• Fluorine – bones, teeth, and urine, contaminated forages
• Hydrocyanic acid – ingesta in a filled and airtight container, blood and muscle
• Nitrate and nitrite – ingesta (plus chloroform or formalin) in an airtight, filled container, blood, ocular vitreous humor
Careful packing of specimens is necessary to avoid loss of some poisons by escape as gas or conversion by bacterial fermentation, and to prevent contamination. No preservative should be added except in the case of suspected nitrite poisoning. If a preservative is necessary because of distance from the laboratory, packing in dry ice or ethyl alcohol (1 mL/g of tissue) is advisable; in the latter instance a specimen of the alcohol should also be sent. Ingesta and tissues must be kept separate as diffusion is likely to occur between the two. Specimens should be packed in glass or plastic to prevent contamination by lead in soldered joints of cans. Metal tops on jars should also be separated from the tissues by a layer of plastic or other impervious material. A suitable amount of material should be submitted for analysis: 1 kg of ingesta, 1 kg of liver, 0.5 kg of kidney, and proportionate amounts of other viscera are suggested to cover all contingencies. Urine (200 mL or whatever is available) may allow quick analysis of some toxicants. Both blood and serum are helpful for rapid testing of some toxicants and for characterizing a potential poisoning through complete blood count and clinical chemistry. Special action is needed when plant poisoning is suspected. First examine the premises for evidence that known toxic plants actually appear to have been eaten. It is possible to ascertain the identity of the plants eaten recently by a careful examination of the ruminal contents. The freshest, least macerated material is best and whole leaves preferred. Atlases of epidermal plant fragments are available to aid in identification of ingested plant species in agricultural animals. Laboratories with sophisticated equipment can now identify most plant toxins in ruminal contents by spectrometric analysis, leaving the veterinarian with the simpler task of deciding whether any of the plants which contain the toxin are present in the environment. At such times access to a computerized data bank of plants and their toxins1 is a great advantage.
Commonly, with plant poisonings, there are perplexing epidemiological features. For example, animals already grazing in the dangerous field are often unaffected and only those recently introduced may be poisoned. Some of the factors which affect susceptibility to plant poisonings are:
• Hungry, ravenous animals are more likely to be affected
• Animals thirsty or recently moved to a new location may sample toxic plants
• Curious, excited animals are likely to sample the plants they would not otherwise eat
• Young animals are less discerning and are less easily put off
• Plants that are different in texture, e.g. sprayed weeds, lopped foliage, often appear to be attractive
• Pica due to mineral deficiency or some other association may encourage toxic plant consumption
• Genetic selection of animals toward tolerance of a particular poison, e.g. fluoroacetate.
Poisoning is in most instances accidental, although it may occasionally be deliberate. Deliberate or criminal poisoning is often suspected but is rarely proved. If there is a strong suspicion of criminal poisoning, or if litigation appears possible in accidental poisoning, specimens should be collected in duplicate and placed in sealed containers in the presence of witnesses. A complete set of specimens should be available to both plaintiff and defending parties for independent analysis. Also, if litigation appears possible, the veterinarian should make detailed observations of the clinical, pathological, and epidemiological findings and record them in detail. The taking of photographs of affected animals and the environmental surroundings is also recommended for future reference and documentation if necessary.
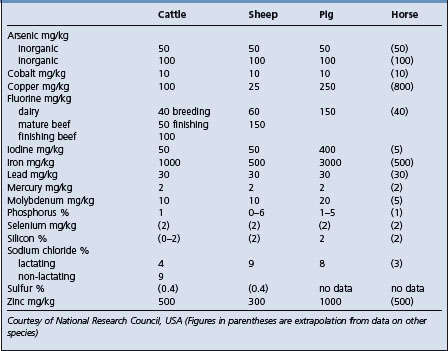
MINERAL TOLERANCE OF ANIMALS
One of the very important aspects of toxicology as it applies to agricultural animals is the determination of levels of dietary constituents which the animals will tolerate for a limited period without impairing their performance and without producing unsafe residues in products destined for the human food chain. There is a great deal of information on this subject and it has been collated and published.2 Table 32.1 is an adapted summary of the information.
PRINCIPLES OF TREATMENT IN CASES OF POISONING
There are certain principles which apply to all cases of poisoning and they are listed briefly below. The three main principles are:
• Removal of the residual poison from the alimentary tract or skin
• Provision of chemical and physiological antidotes to the poison that has been absorbed
• Effective supportive care, nursing, and convalescent care.
In farm animals, gastric lavage and emetics are of little or no practical value and the removal of residual poison from the alimentary tract depends largely upon the use of adsorbents and purgatives. The only effective adsorbent is activated charcoal. The dose rate is 1–3 g/kg BW repeated as necessary. It adsorbs chlorinated hydrocarbons, organophosphorus compounds, mycotoxins and plant alkaloids, the common feed additives, antibacterial agents and bacterial toxins. It does not adsorb cyanide, heavy metals, halogens, nitrite, alcohols, caustics, sodium chloride, or chlorate. A purgative is necessary to remove the combined adsorbent and poison; it can be administered simultaneously with the adsorbent. The use of irritant purgatives is not advisable when the poison is an irritant and has already been associated with gastroenteritis, and non-absorbable oily purgatives (e.g. mineral oil) are preferable in these cases. Saline purgatives (sodium sulfate) are of value in the treatment of non-irritant poisons such as cyanogenetic glucosides. Neutralization of residual poison in the alimentary tract includes use of oxidizing agents or tannic acid preparations for precipitating alkaloids; proteins, including milk and eggs, are effective chemical antidotes for poisons that coagulate proteins; lead is precipitated by the addition of sulfates to the alimentary tract contents.
Poison that has already been absorbed can in some instances be inactivated or its excretion facilitated by the provision of chemical antidotes. For instance, sodium nitrite and sodium thiosulfate are effective systemic antidotes to hydrocyanic acid, and calcium versenate is an effective antidote against lead.
Treatment of the effects of a poison includes provision of physiological antidotes, e.g. the injection of a calcium salt in cases of overdosing with magnesium salts. Ancillary or supportive treatment, including the provision of fluids in dehydration due to diarrhea, demulcents in gastroenteritis, sedatives in excitement, stimulants in cases of central nervous system depression, all treat the effects of poisoning.
It is essential when undertaking the treatment of animals for poisoning, especially those which are producing milk or which are destined to become meat in a short time, to take into account the possible unsuitability of the product for human consumption because of the presence of the poison or the antidote. Carefully planned sampling in concert with regulatory authorities can avoid unwanted contamination of the human food supply.
Diseases associated with inorganic poisons
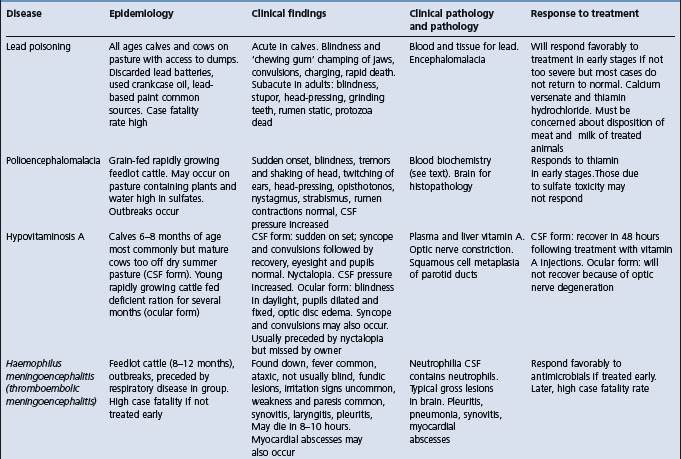
LEAD POISONING (PLUMBISM)
Etiology Accidental ingestion of lead or ingestion of feed or grazing pasture containing excessive lead.
Epidemiology Occurs in all age groups. One of the most common poisonings of farm livestock especially in young calves after turn out in spring. In cattle, usually sporadic and due to ingestion of single source of lead but outbreaks occur when feed is contaminated. High case fatality rate if untreated. Sources include discarded lead batteries, lead-based paints, industrial sources of lead, pastures near motor vehicle highways and smelters. Occurs in sheep and horses grazing contaminated pastures.
Signs
Cattle: Acute – convulsions, blindness, tremors, charging, rapid death unless treated. Subacute – blindness, stupor, head-pressing, rhythmic ear tics, blepharospasm, rumen stasis and eventual death.
Sheep: Lambs on pasture with posterior paresis.
Horses: On pasture. Signs highly variable. Inspiratory dysnea, roughened hair coat, weight loss most commonly. Occasionally convulsions.
Clinical pathology Lead levels in blood, feces, liver, kidney; elevated porphyrins in blood
Lesions Encephalopathy, degeneration of liver and kidney; pale musculature, brain laminar cortical necrosis, intranuclear renal inclusion bodies.
Diagnostic confirmation Toxic levels of lead in blood and tissues.
Cattle: See Table 32.3.
Table 32.3 Differential diagnosis of diseases of cattle with clinical findings referable to brain dysfunction


Horses: See Table 22.1.
• Viral encephalomyelitides/West Nile virus
• Hepatoencephalopathy due to hepatotoxic plants
• Equine degenerative myeloencephalopathy
ETIOLOGY
Lead poisoning is associated with the accidental ingestion of sources of lead metal or compounds or the ingestion of feed, usually forage, containing lead usually from pollution of the environment.
EPIDEMIOLOGY
Occurrence
Lead is one of the most common poisonings in farm animals, especially young cattle. Sheep and horses are also affected but not as commonly. Pigs are not often exposed to lead and appear to be more tolerant to it than other species.
Data from diagnostic toxicology laboratories illustrate that lead poisoning is the most common toxicosis in cattle.1,2 The disease occurred most commonly in younger cattle with 52% of the cases reported in animals 6 months of age or less. Approximately 60% of the cases occurred during the summer months from May to August, when the cattle have ready access to lead-containing materials such as crankcase oil and batteries that are being changed in agricultural machinery. In many countries the incidence of the disease is highest in cattle in the spring of the year a few days after the animals have been turned out onto pasture.3 Lead poisoning occurs most commonly in young cattle soon after spring turnout when animals gain access to discarded waste materials including batteries, dump oil, oil paint containers, and bonfire ash where painted lumber has been burned.4 In Alberta, Canada, over a period of 22 years, lead poisoning was the most frequently diagnosed toxicosis of cattle, representing 0.68% of all bovine submissions to the provincial diagnostic laboratories.5 Young cattle, particularly, are curious and amazingly seem to find sources of lead.
Lead poisoning in cattle is usually due to the accidental ingestion of a toxic quantity of lead over a short period of time. The natural curiosity, licking habits, and lack of oral discrimination of cattle makes any available lead-containing material a potential source of poisoning. Cattle will readily drink crankcase oil, lick machinery grease, and chew batteries. Present-day machinery grease does not commonly contain lead. Compared to earlier times, used crankcase oil may not contain lead if leaded gasoline is banned in a country or region, or if diesel engines are used where lead additive is not utilized in the fuel. In ruminants there is a tendency for metallic lead particles to settle in the reticulum and poisoning results from the gradual conversion of lead particles to soluble lead acetate. Horses, on the other hand, are much more selective than cattle in their eating habits. They usually do not lick old paint cans, lead storage batteries, and peeling paint, nor do they seem to find the taste of used motor oil attractive.6 Several epidemics of lead poisoning in domestic animals have been recorded throughout the world where the source of the metal was contamination of pasture or crops by nearby industrial lead operation.6 Animals eating vegetation in these areas may accumulate amounts of lead sufficient to produce clinical signs of lead poisoning.
Sheep are usually affected by eating forage contaminated by environmental sources of lead.
Lead poisoning in horses occurs most commonly when they graze lead-contaminated pastures rather than by the accidental ingestion of a toxic amount of lead. Young horses are particularly more susceptible than older horses and cattle grazing on the same pasture. Some cases in horses have been due to ingestion of paint chips from a fence on pasture.7
Lead poisoning in buffalos has been reported and provides interesting comparative data8; they may have a higher tolerance to lead than cattle.
Morbidity and case fatality
Where groups of animals have access to the same source of lead, outbreaks occur and the morbidity rate ranges from 10 to 30%. The case fatality rate may reach 100% but early intensive therapy can be successful and reduce the figure to less than 50%. In one recorded outbreak, in which a discarded 24-volt battery was accidentally mixed and ground up into the feed of 80 heifers, 55 of the animals died or were destroyed on humane grounds.9
Sources of lead
Lead poisoning occurs most commonly in cattle at pasture, particularly if the pasture is poor and the animals are allowed to forage in unusual places, such as rubbish dumps. Phosphorus deficiency may also be a predisposing factor, in that affected animals will chew solid objects as a manifestation of osteophagia. However, cattle on lush pasture may also seek out foreign material to chew. Confined housing of calves with or without overcrowding is often followed by the appearance of pica which may be associated with boredom or mineral deficiency.
Lead paint and lead batteries.
The common sources of lead are lead-bearing paints and metallic lead. Discarded lead batteries are one of the most common sources of lead poisoning in cattle. In Alberta, Canada, over a period of 22 years, discarded batteries or used crankcase oil accounted for more than 80% of cases for which the source of lead was determined: batteries, 39.5%; used crankcase oil, 31.6%.10 The batteries are commonly placed in garbage dumps on the farm and, in temperate climate countries, the batteries freeze during the winter months and break open, exposing the plates which are attractive and palatable for cattle to lick and chew.
The contamination of forage supplies with shotgun lead pellets used in hunting and shooting exercises, can serve as a source of lead for cattle grazing the pasture or consuming haylage or silage made from the contaminated field. Automobile batteries have been accidentally added to feed mixers where they are ground by powerful augers and mixed into the feed supply of cattle.9 Feed accidentally contaminated with lead affected some 15 000 cattle on 330 farms in the Netherlands within a 3-month period.11,12 Discarded lead-based paint cans are particularly dangerous but fences, boards, and the walls of pens, painted canvas and burlap are also common sources in calves. Painted silos may cause significant contamination of the ensilage. One outbreak of lead poisoning in cattle was associated with silage containing 1200 mg/kg DM lead which had become contaminated by ash and debris left after burning an old lead-containing electrical cable in the silo before it had been filled.13
Metallic lead in the form of lead shot, solder, or leaded windows has been associated with mortalities, although, experimentally, sheet lead is not toxic. Lead sheeting which has been exposed to the weather or subjected to acid corrosion appears to be more damaging, possibly because of the formation of a fine coating of a soluble lead salt. Lead poisoning can be a major hazard in the vicinity of oil fields, and engine sump oil may contain over 500 mg lead per 100 mL. Automotive and other mineral oils are very palatable to young beef calves. In one study, used crankcase oil was the most common source of lead poisoning in cattle, followed by paint, grease, and lead car batteries. As lead use becomes more restricted in many countries, grease and lead contaminated engine oil have become less common sources of lead. Less common but still potent sources of lead are linoleum, roofing felt, putty, automobile oil filters, and aluminum paint. Some of the latter paints contain large quantities of lead, others none at all. Only lead-free aluminum paint should be used on fixtures to which animals have access.
Lead parasiticide sprays, particularly those containing lead arsenate, was once associated with heavy losses in cattle grazing in recently sprayed orchards or vegetable crops. These are not commonly used now, except in some countries, but cattle may accidentally ingest old stores of the compound.14
Environmental pollution with lead
Environmental pollution with lead is a common occurrence in cities and on their edges. For farm animals, significant pollution is more likely to occur near smelters or other industrial enterprises, or near major highways where pasture is contaminated by exhaust fumes of automobiles, only if leaded gasoline is still used in the region. Much of the poisoning is subclinical because of the low level of absorption, and any program intending to use domestic animals as monitors of pollution would need to be based on tissue lead levels.
Lead in pastures near highways.
The lead levels in whole blood of sheep grazing near main highways in three areas of the Nile delta region of Egypt were 0.062, 0.067, and 0.083 mg/mL (ppm).15 Pasture adjacent to heavily used roads may carry as much as 390 mg/kg of lead, in contrast to 10 mg/kg on lightly used roads. The concentration of lead on pasture varies markedly with proximity to the traffic, falling rapidly the greater the distance, and with the time of the year. Pastures contaminated by smelters are recorded as carrying 325 mg/kg of lead (equivalent to a daily intake for an animal of 6.4 mg/kg BW). In some locations near lead smelters, lead poisoning is considered to be a predictable occurrence in horses which are allowed to graze on local pastures. As a result horses are either not raised in these areas or hay is imported from other areas. Although ingestion is the principal method of poisoning of animals, inhalation may also be a significant method of entry for cattle grazing close to smelters or highways.
Lead as an environmental contaminant is often combined with cadmium which has some effects similar to those of lead so that the effects may be somewhat additive. Experimental poisoning with both elements is associated with reduced weight gains in calves at dose levels up to 18 mg/kg BW of each, and clinical signs appear at levels above 18 mg/kg BW of each. Lead is also combined with chromate for industrial purposes. It is not toxic when combined with lead at lead intake levels of 100 mg/kg BW.
Environmental pollution in the vicinity of lead and zinc-ore processing factories can result in varying degrees of poisoning with lead, zinc, and cadmium. These can be monitored by the analysis of blood, hair, and tissues obtained at necropsy.16,17
There is some concern that toxic levels of lead may occur in the human food chain. Canadian studies of the lead and mercury residues in kidney and liver of slaughter animals have shown that all levels were below the official tolerance level of 2 mg/kg WW for lead and 0.5 mg/kg for mercury. Levels of lead in beef randomly selected from supermarkets in the US were for muscle 0.46, liver 0.50, and kidney 0.45 mg/kg (WW).18 The upper range of liver levels exceeded the 1 mg/kg WW guideline which may cause for concern about the source of the lead. Moderate exposure of people to meat, including liver and kidney, from animals exposed to lead poisoning is thought not to represent a human health hazard. The biological monitoring of cadmium and lead from contaminated sandy soil into sudan-sorghum hay consumed by pregnant dairy goats over a period of 98 days revealed that only miniscule amounts of soil cadmium and lead were retained in the selected animal tissues (liver and kidney) via the ingestion of the hay.19 It was concluded that if these animal tissues were used as food, no deleterious effect to human health would be induced.
Lead in blood, milk, kidneys, and liver.
The relationship between lead concentrations in blood of cattle with lead poisoning and those in the milk is exponential.20 The lead level in milk is relatively constant up to a blood level of 0.2–0.3 mg/L, and increases sharply at higher blood levels. The biological half-life of lead in blood is approximately 9 weeks. Recent studies in six affected dairy herds reported a variable half-life ranging from 48 to 2507 days.21 A probable reason for this great variance is the ability of the ruminant to retain variable amounts of metallic lead in the rumen which thus acts as a continuing reservoir. Since classical biological half-life studies do not account for variable intake and retention of a persistent reservoir of toxicant, the concept of half-life in dealing with lead-poisoned cattle is likely not accurate. Owners of such cattle should be advised of the potentially long withdrawal period. It may be advisable to test periodically and allow marketing based on actually measured levels, or to estimate the costs of such a plan and consider salvage. This recent work casts doubt on the economic utility of holding recovered animals. In acutely sick cows which were emergency slaughtered, the range of lead levels in edible muscle tissue was 0.23–0.50 mg/kg WW. The concentrations in the kidneys ranged from 70 to 330 mg/kg WW and in the livers 10–55 mg/kg WW.
Background blood lead is the concentration of lead in whole blood resulting from the daily exposure to lead which does not produce any clinical evidence of disease.21 The background levels of blood lead which have been reported ranged from 3 to 50 μg/dL,22 but most were less than 10 μg/dL. A recent analysis of 266 blood samples from dairy cattle submitted to the diagnostic laboratory at the New York State College of Veterinary Medicine, for reasons other than heavy metal toxicity, indicated that 259 (97%) had levels lower than minimum detectable level of 2.5 μg/dL (0.025 ppm).22 Six had concentrations between 2.6 and 5.8 and one had 10 μg/dL. Blood lead concentrations in cows with lead poisoning ranged from 56.4 to 1390.0 μg/dL.
Toxic levels of lead
The toxic level of lead varies between species and the chemical composition of the compound containing lead may influence its toxicity. Lead acetate is very soluble and more toxic than insoluble lead oxide, or solid lead sheeting.
In calves, acute single lethal doses range from 400 to 600 mg/kg body weight (BW), in adult cattle 600–800 mg/kg and for goats 400 mg/kg. The acute dose for horses is less than for ruminants, one horse having survived 1000 mg/kg BW on two occasions 6 months apart. Pigs are also less susceptible but single lethal dose levels are not recorded. Buffalo calves given a single oral dose of 600 mg/kg BW of lead acetate died within 120 hours.23
Young animals, e.g. milk-fed calves, are more susceptible. A daily intake of 2.7 mg/kg BW of lead can be associated with the death of calves fed a milk diet in 20 days or less, while 5 mg/kg BW of lead is consistently associated with signs of poisoning or death within 7 days. The absorption rate of lead is rapid and tissue depositions are high in calves on a milk replacer diet and given lead. In toxicity studies, calves on a milk diet absorb lead much more quickly than calves fed a grain diet. The addition of lactose to a grain diet will also increase the absorption of lead.
Daily dose levels likely to lead to chronic poisoning are important because of the impact that contamination of the environment by industrial effluents has had. Daily dose levels likely to lead to chronic plumbism in cattle are 6–7 mg/kg BW (equivalent to 100–200 mg/kg DM of diet). This dose level must be close to the definitive point because dose levels of 100 mg/kg (in diet) may be without effect. A dose level of 15 mg/kg BW results in the loss of weight gain and normochromic anemia. In sheep, dose levels of more than 4.5 mg/kg BW are necessary to produce a toxic effect. Horses are more susceptible to the daily administration of lead, 100 mg/kg BW producing toxic effects in 28 days. A dose rate of 15–30 mg/kg BW of lead for up to 190 days is associated with toxicity and some deaths, and deaths are recorded on pastures carrying 100–300 mg/kg on foliage. Pigs appear to be more resistant, and daily doses of 33–66 mg/kg BW are required for periods of up to 14 weeks to produce fatal effects, a more serious end-point than for the other dose rates quoted.
PATHOGENESIS
Regardless of the chemical form of the ingested lead, only a small proportion is absorbed because of the formation in the alimentary tract of insoluble lead complexes which are excreted in the feces. For example, only 1–2% of lead ingested as lead acetate or carbonate is absorbed from the alimentary tract of sheep. Of the lead absorbed, some is excreted in the bile, milk, and urine and the blood levels of lead provide a reliable indication of the lead status of the animal. Urine levels may not be as reliable. Deposition in tissues occurs, particularly in the liver and renal cortex and medulla in acute poisoning and in the bones in chronic poisoning. The deposition of lead in the brain is not high compared to other tissues but deposited lead is gradually liberated from tissues into the bloodstream and excreted via the bile and urine. Consideration must be given to these aspects of lead metabolism when assessing the results of chemical analyses of tissues.
Although acute lead poisoning usually develops rapidly there may be a delay of several days after toxic material has been ingested before clinical signs appear.
Toxic effects of lead
The toxic effects of lead are manifested in three main ways:
In general, acute nervous system involvement occurs following the ingestion of large doses in susceptible animals such as calves, alimentary tract irritation following moderate doses, and peripheral nerve lesions following long-term ingestion of small amounts of lead. The nervous signs of encephalopathy and the lesions of peripheral nerve degeneration are due to the degenerative changes of nervous system tissue. Lead localizes principally in the cytoplasm of capillary endothelial cells and these localizations are later associated with the development of edema. The basic lesion is likely to be vascular with a basic change in transport mechanisms between the blood and brain.24 Gastroenteritis is associated with the caustic action of lead salts on the alimentary mucosa. Ruminal atony occurs in cattle and sheep and initially is associated with scant feces, followed later in some cases by diarrhea due to gastroenteritis. The rumen protozoa in cattle with acute lead poisoning are commonly absent or inactive. Peripheral nerve degeneration occurs principally in horses.
The lesions, including degeneration of the liver and kidney, vary in their severity with the tissue levels of lead attained. Lead does not remain in tissues for long periods except in bone where it is deposited in an inert form, but from which it can be liberated at a later date in sufficient quantities to be associated with chronic lead poisoning. This is particularly likely to occur during periods of acidosis.
The blue ‘lead-line’ at the gum–tooth junction, which is seen in man and the dog, does not commonly occur in ruminants because of failure to form tartar but may be present in the horse. The ‘lead-line’ is a deposit of lead sulfide formed by the combination of lead with sulfide from the tartar.
Lead is transferred across the placental barrier and high liver levels occur in the lambs of ewes fed more than normal amounts of lead. Calves born from cows experimentally poisoned with lead have elevated levels of lead in bone, kidney, and liver. In a naturally occurring case of lead poisoning in a pregnant heifer, the blood and liver concentrations in the fetus were 0.425 ppm and 4.84 ppm, respectively, which was 72% and 84% of the same tissue lead concentrations of the dam.25 Hepatic lysosomes of the fetus contained metallic electron densities which may have been lead.
The pathogenesis of the osteoporosis in young lambs with chronic lead poisoning has not been explained, nor has the paresis and paralysis of lambs which occur in the same circumstances. The paralysis in the former condition is caused by compression of the spinal cord by collapsed lumbar vertebrae.
Anemia may occur in chronic lead poisoning. The erythrocytes are microcytic and hypochromic, and reticulocytosis and basophilic stippling may be observed. However, basophilic stippling is non-specific and probably does not correlate well with levels of lead exposure. The basophilic stippling of erythrocytes is usually an indication of bone marrow response to anemia, although it can occur, rarely, in chronic lead poisoning. It may be related to the effects of lead on pyrimidine nucleotidase activity. The anemia in chronic lead poisoning is associated with two basic defects: a shortened erythrocyte lifespan and impairment of heme synthesis. Lead is associated with an increased concentration of protoporphyrin by inhibiting heme synthetase, the enzyme which combines protoporphyrin and iron to form heme. The measurement of free erythrocyte porphyrin is considered to be a sensitive indicator of chronic lead poisoning in calves. Lead is also associated with an inhibition of the enzyme delta-aminolevulinic acid dehydratase (ALA-D), resulting in a failure of utilization of delta-aminolevulinic acid which is excreted in increased quantities in the urine.
CLINICAL FINDINGS
Cattle
Both acute and subacute poisoning occurs in cattle. The acute form is more common in calves and the subacute form in adults.
In the acute form there is usually a sudden onset of signs and a short course of 12–24 hours so that many animals, especially those at pasture, are found dead without signs having been observed. Staggering, and muscle tremors particularly of the head and neck, with champing of the jaws (chewing gum fits) and frothing at the mouth are obvious. Snapping of the eyelids, rolling of the eyes and bellowing are common. Blindness and cervical, facial and auricular twitching are consistent in acute lead poisoning of cattle. The animal eventually falls and intermittent tonic–clonic convulsions occur and may continue until death. Pupillary dilatation, opisthotonos and muscle tremor are marked and persist between the convulsive episodes. There is hyperesthesia to touch and sound, and the heart and respiratory rates are increased. In some cases, particularly in adults, the animal remains standing, is blind, maniacal, charges into fences, attempts to climb or jump over walls, and head-presses strongly against walls or fences. Frenzy is common and some animals appear to attack humans but the gait is stiff and jerky and progress is impeded. Death usually occurs during a convulsion and is due to respiratory failure.
In the subacute form the animal remains alive for 3–4 days. There is dullness, total anorexia, blindness, and some abnormality of gait including incoordination and staggering, and sometimes circling. The circling is intermittent and not always in the same direction and usually occurs when the animal is confined in a small space like a box stall. Muscle tremor and hyperesthesia are common but not as pronounced as in the acute form. Grinding of the teeth is common, excessive salivation may occur, and mild abdominal pain may be seen occasionally. Alimentary tract dysfunction is one of the most common abnormalities. Ruminal atony is accompanied by constipation in the early stages. Later a fetid diarrhea occurs in most cases.
The animal presents a picture of extreme dullness, will not eat or drink, and stands immobile for very long periods. Death frequently occurs by misadventure, the animal walking blindly into a waterhole or being trapped in a fence or between trees. In other circumstances the animal becomes recumbent and dies quietly. In both the acute and subacute forms, the palpebral eye preservation reflex is absent or markedly diminished. This is a useful distinguishing feature from polioencephalomalacia in which this reflex is usually normal. Edema of the optic disc may be present but is not common.
Experimental lead poisoning in young milk-fed calves, initially is characterized by severe depression and hypoglossal paresis which interferes with sucking. Within the next 12–24 hours, the calves become unsteady, ataxic, and exhibit muscular tremors of the head and forelimbs and finally convulsions, opisthotonos; they die in respiratory failure during status epilepticus.
Sheep
Lead poisoning in sheep is usually manifested by a subacute syndrome similar to that seen in adult cattle. There is anorexia and scant feces followed by the passage of dark, foul-smelling feces. Weakness and ataxia follow, often with abdominal pain, but there is no excitement, tetany, or convulsions. Polyuria occurs when the intake of lead is small but with large amounts there is oliguria.
Although ruminants are relatively resistant to chronic lead intoxication, two syndromes of posterior paresis have been described in young lambs in old lead-mining areas and tissue levels of lead are abnormally high in both instances. In both syndromes there is impairment of the gait. Osteoporosis is present in one but in the other there is no suggestion of skeletal changes. In the osteoporotic disease the signs occur only in lambs 3–12 weeks of age and never in adults. There is stiffness of gait, lameness, and posterior paralysis. Affected lambs are unthrifty and the bones, including the frontal bones, are very fragile. The paralysis is caused by lesions of the vertebrae, usually affecting one or more of the lumbar bones, and resulting in compression of the spinal cord. In the other form, gait abnormalities occur in the same lamb age group and are manifested initially by incomplete flexion of the limb joints so that the feet drag while walking. In a later stage the fetlocks are flexed, the extensor muscles paretic, and the lamb soon becomes recumbent. Recovery is common, although many lambs die of intercurrent disease.
Chronic ingestion of metallic lead by pregnant sheep can be associated with abortion and transitory infertility.26
Horses
Horses are not commonly affected by lead poisoning, although the chronic form occurred occasionally in the vicinity of lead mines and processing works.27 The clinical findings are extremely variable.7 A roughened hair coat, pharyngeal dysfunction, and weight loss were the most common clinical findings in 10 case reports involving a total of 68 animals.7 Some horses died without any previous clinical illness but where clinical signs are apparent they were usually distinct and dramatic rather than subtle. Inspiratory dyspnea associated with paralysis of the recurrent laryngeal nerve is the most common finding. This may be accompanied by pharyngeal paralysis in which recurrent choke and regurgitation of food and water through the nostrils occur. Aspiration pneumonia may result after inhalation of ingesta through the paralyzed larynx. Paralysis of the lips occasionally accompanies the other signs. General muscle weakness and stiffness of the joints occur commonly and the hair coat is usually harsh and dry. When chronic poisoning with both lead and zinc occurs the signs in zinc poisoning predominate despite high lead levels in liver and kidney. In experimental chronic lead poisoning in horses, there is noisy breathing constantly, but no lesions in the pharynx or larynx. Muscle fasciculations over the triceps are prominent in some horses and recumbency and convulsions may occur7.
When large amounts of lead are ingested by horses a syndrome similar to that of the subacute form in cattle occurs. There is complete anorexia, severe nervous depression, partial paralysis of the limbs followed in most cases by complete paralysis and recumbency. Mild-to-severe abdominal pain and clonic convulsions may also occur. The response to experimental lead poisoning in the horse is highly variable.7 The dose–response effect is highly variable and unpredictable.
Pigs
Early signs include squealing as though in pain, mild diarrhea, grinding of the teeth, and salivation. The disease is usually a prolonged one and listlessness, anorexia, and loss of weight develop followed by muscle tremor, incoordination, partial or complete blindness, enlargement of the carpal joints, and disinclination to stand on the front feet. Convulsive seizures occur in the terminal stages.
Subclinical lead poisoning
Because of the present concern about environmental pollution, the effects of the chronic low-level intake of lead have been examined and defined. In cattle, at intake levels below those which are associated with clinical signs, there are metabolic changes and changes in blood variables accompanied by a decreased rate of growth.28 One concern is that continuous low-level consumption by pregnant females will result in teratogenic effects in the newborn. Trials to detect this manifestation in ewes have shown no effect on their lambs.
CLINICAL PATHOLOGY
In the living animal which has ingested lead, the element can be detected in blood, feces, urine, and milk.
Blood lead
The estimation of blood levels is generally useful for determining the lead status of the animal and is used most frequently to support or refute a clinical diagnosis of lead poisoning. Bovine blood lead reference materials are available and have been certified for many years.29 Whole blood levels of lead in normal ruminants are usually below 0.05–0.25 ppm; poisoned animals usually have levels above 0.35 ppm and deaths begin at 1.0 ppm. Interestingly, buffalo may have blood levels above 1.0 ppm and still survive, which suggests that they have a higher tolerance level than cattle. However, when used alone, blood lead concentrations do not permit evaluation of length of exposure, amount of lead deposition in the body or the effects of lead on physiological systems. Blood lead concentrations also fluctuate markedly after administration of lead and consequently the clinical importance of blood lead concentrations is often questionable and a diagnosis based on this single determinant is equivocal. Blood lead concentration also has limited value for assessing the effectiveness of therapy for lead poisoning. Blood level concentrations may change rapidly during chelation therapy, often decreasing by 50% or more within 24 hours after initiation of treatment despite certain body tissues still containing high concentrations of lead. Thus the evaluation of biochemical indicators such as ALA-D may be useful. The blood and liver levels of fetuses from pregnant cattle with lead poisoning may be higher than what are considered toxic levels in adults which suggests concentration in the fetus.25
Representative values of lead for normal and poisoned animals are summarized in Table 32.2. The levels found in the liver and the kidneys are presented under necropsy findings.
Table 32.2 Lead levels in blood and feces of normal and poisoned animals
| Lead levels (ppm) | ||
|---|---|---|
| Specimen | Normal | Poisoned |
| Whole blood (ruminants and horses) | 0.05–0.25 | More than 0.35 (deaths commence at 1.0) |
| Whole blood (pigs) | 0.05–0.25 | 1.2 |
| Feces (dry matter) (cattle) | 1.5–35 | Up to 1000 |
| Pasture | 350 | |
Milk lead
Only limited information is available on the concentrations of lead which occur in cattle affected with field cases of lead poisoning. Lead levels of 0.13 mg/L of milk have occurred in natural cases with a half-life of 4.6 days.12 The regulatory limit for lead in bovine milk in the Netherlands is 0.05 mg/L milk. In acute lead poisoning in lactating buffalo pastured near smelters in India, the lead concentrations in milk were 1.13 ppm compared to 0.24 ppm in the milk from buffaloes in unpolluted areas.8 The mean lead concentrations in the forage of poisoned animals were 706 ± 73.0 ppm, compared to the unpolluted area of 78 ± 12 ppm.
Fecal lead
Fecal levels of lead represent unabsorbed and excreted lead deriving from the bones, and are of limited value unless considered in conjunction with blood levels because ingested lead may have been in an insoluble form and harmless to the animal. When fecal levels are high it can be assumed that the lead has been ingested in the preceding 2–3 weeks but high blood levels may be maintained for months after ingestion. Thus high blood and low fecal levels indicate that the lead was taken in some weeks previously but high blood and high fecal levels suggest recent ingestion and significant absorption.
Urinary lead, ALA-D
Urine lead levels are variable, rarely high (0.2–0.3 mg/L), and although elevated urine levels are usually associated with high blood levels, this relationship does not necessarily hold.
Because of some of the limitations of blood lead, other indirect measurements of lead poisoning, such as the levels of delta-aminolevulinic acid dehydratase (Delta-ALA-D) in blood, are being used to supplement blood lead determinations.30 For example, the best method of detecting the presence of lead poisoning in its early stages, except in the horse, is the estimation of ALA-D in the blood. At dietary intakes as low as 15 mg/kg DM of lead in cattle there are detectably lowered levels of ALA-D. At the same time, the urinary levels of delta aminolevulinic acid (Delta-ALA) are increased. Delta-ALA-D is important in the synthesis of heme and is probably the most sensitive enzyme in the heme pathway. Inhibition of the enzyme results in a block in the utilization of delta-ALA, a subsequent decline in heme synthesis and a marked increase in the urinary excretion of delta-ALA. In cattle, sheep, and pigs affected with chronic lead poisoning, the plasma levels of delta-ALA-D are decreased and the urinary levels of delta-ALA are increased before clinical signs are detectable. In sheep, erythrocyte delta-ALA-D is recommended as the most sensitive diagnostic test available.
The disadvantages of the assay for blood delta-ALA-D include age-related variations particularly in calves; the methods used for analysis are not yet uniform and blood must be collected in polystyrene or polyethylene tubes rather than glass tubes and an anticoagulant other than EDTA must be used. The levels of delta-ALA-D increase in calves from birth to 10 weeks of age and age-matched controls should be evaluated simultaneously when conducting the test in calves of under 6 months of age.30 In cattle under 1 year of age, delta-ALA-D values of less than 200 mmol of porphobilinogen (PBG)/mL of RBC/hour should raise suspicion of their having ingested lead. In this same age range values below 100 mmol would confirm ingestion of lead. In cattle equal to or less than 2 years of age, values of delta-ALA-D of less than 100 mmol of PBG/mL of RBC/hour would indicate ingestion of lead. Severe inhibition of delta-ALA-D occurs rapidly in calves given 1 mg of lead/kg BW per day or 5 mg of lead/kg BW/d. Inhibition of delta-ALA-D will reach approximately 50% of pre-exposure levels when blood lead concentrations are above 0.5 mg/kg, and if the initial dose of lead increases blood lead concentration above 0.5 mg/kg the delta-ALA-D becomes maximally depressed and remains so with continued exposure. The delta-ALA-D is so sensitive to lead that it remains inhibited even after lead exposure has ceased. Following treatment with a chelating agent the blood lead levels will often decline giving a false indication of a positive treatment effect. If the delta-ALA-D levels do not decrease following therapy, it indicates that there is sufficient lead present to continue to depress the enzyme. In summary, the evaluation of delta-ALA-D and blood lead concentrations together can assist in resolving diagnostic situations in which the blood lead concentration is in the questionable range of 0.25–0.35 ppm.
Erythrocyte protoporphyrin
The levels of free erythrocyte zinc protoporphyrin increase in lead poisoning and this is indicative of the chronic metabolic effect of lead on the erythroid cells being released from bone marrow into the peripheral circulation.5 A mean value of 21.56 μg coproporphyrin/100 mL of erythrocytes has been determined. It may be of some value along with determinations of blood lead and delta-ALA-D. The use of delta-ALA-D activity and erythrocyte protoporphyrin content as cumulative lead exposure indicators in cows environmentally exposed to lead is recommended.31 Plasma exposed to ultraviolet light may fluoresce due to high concentrations of porphyrins, and this may be a useful early diagnostic test.
Environmental lead
Because of the frequency with which lead appears in the environment as a pollutant, there is often concern for the validity of the normal values for establishment of a diagnosis. In the average city-polluted atmosphere it seems that lead intake will be significantly elevated. The lead content of hair of cattle and horses, and of the wool of sheep, is reported to be raised significantly in poisoned animals but hair is not routinely used in diagnosis of acute poisoning. However, the lead content of hair when cattle are exposed to long-term ingestion as a result of industrial contamination can reach as high as 88 mg/kg (in a clean environment comparable figures are of the order of 0.1 mg/kg). There is likely to be a seasonal variation in deposition and intake of lead. Hair is also a valuable source of information on environmental pollution with cadmium, copper, and zinc.
Hematology
In chronic lead poisoning, hematological examination may reveal a normocytic, normochromic anemia in some and, although basophilic stippling does not occur often enough to be diagnostic, it is recorded in some experimental poisonings. It is recorded as occurring in lead-exposed pigs and a horse. In some, poikilocytosis and anisocytosis were marked.22 The cerebrospinal fluid (CSF) is approximately normal with slightly elevated leukocyte numbers but no increase in protein or other biochemical components.
NECROPSY FINDINGS
In most acute cases there are no gross lesions at necropsy. In cases of longer standing there may be some degree of abomasitis and enteritis, diffuse congestion of the lungs and degeneration of the liver and kidney. Epicardial hemorrhages are common. Congestion of meningeal and cerebral vessels may also be observed and hemorrhages may be present in the meninges. An increase in cerebrospinal fluid is often recorded but is of minor degree in most cases. In chronic cases gross lesions are recorded in cattle.24 These include cerebrocortical softening, cavitation and yellow discoloration with most severe lesions in the occipital lobes. Histological lesions were most severe at the tips of the gyri. Similar lesions were produced experimentally. Acid-fast inclusion bodies deep in the renal cortex have diagnostic significance. Examination of the contents of the reticulum in ruminants for particulate lead matter is essential. Flakes of paint, lumps of red lead, or sheet lead usually accumulate in this site. Their absence is not remarkable especially if animals have licked fresh paint but their presence does give weight to the provisional diagnosis.
Liver and kidney lead
The submission of alimentary tract contents and tissues for analysis forms an important part of the diagnosis of lead poisoning but results must be interpreted with caution.
Cattle: 25 mg/kg of lead WW (wet weight) in kidney cortex is diagnostic and is a more reliable tissue for assay than liver which may contain 10–20 mg/kg WW. The concentrations in kidney are always much higher than in liver.13,23 A diagnostic laboratory found mean levels in livers of poisoned cattle of 93.3 μg/g WW weight, and 437.7 μg/g in kidneys.1 Tissue lead levels in cattle from industrial areas are significantly higher (liver 0.23 mg/kg WW, kidney 0.42 mg/kg WW) than in cattle from clear air zones (liver and kidney less than 0.1 mg/kg WW). Tissues which have been fixed in formalin are useful when they are the only tissues available.
Horses: Levels of 4–7 mg/kg WW of lead have been found in the livers of horses dying of chronic lead poisoning but 25–250 mg/kg are more likely, and 40 mg/kg WW may occur in the livers of affected pigs. Mean levels in livers of poisoned horses are 5.5 μg/g WW.1
In all cases, much importance must be attached to the possibility of access to lead and the environmental circumstances which may arouse suspicion of other poisonings or errors in management. Estimation of the lead content of blood and feces should be carried out at the earliest opportunity and tissues form necropsy specimens submitted for analysis.
Cattle
Lead poisoning in cattle must be differentiated from the common diseases of the nervous system of cattle and other diseases in which neurological signs occur. The differential diagnosis of brain dysfunction and lead poisoning in cattle is summarized in Table 32.3. The diseases which closely resemble bilateral blindness are as follows:
Rabies characterized by normal eyesight, incoordination, gradual ascending paralysis, inability to swallow, bellowing and death in 4–7 days.
Polioencephalomalacia characterized by sudden onset of blindness, normal palpebral reflexes to touch, head pressing, tremors of head and neck, nystagmus, normal ruminal contractions.
Hypovitaminosis-A characterized by blindness, dilated and fixed pupils, optic disc edema, convulsions followed by recovery.
Ophthalmitis characterized by blindness due to lesions of the eyes but normal mentation.
Other diseases in which blindness is not characteristic but tremors, ataxia, convulsions, and bizarre behavior occur include: hypomagnesemic tetany, nervous acteonemia, arsenic poisoning, Claviceps paspali toxicity, memingoencephalitis.
Horses
Lead poisoning in horses must be differentiated from diseases causing loss of body weight, muscle fasciculations, incoordination, roughened hair coat, laryngeal and pharyngeal dysfunction. The differential diagnoses of diseases of the nervous system of the horse are summarized in Table 22.1. The diseases most closely resembling lead poisoning in horses include:
Viral encephalomyelidites, including West Nile virus
Hepatoencephalopathy due to hepatotoxic plants
Equine degenerative myeloencephalopathy
TREATMENT
Sedation and care
The case fatality rate of acute lead poisoning of cattle is high because of their high susceptibility and the nature of the material ingested. Sedation by IV injection of anesthetic doses of pentobarbital sodium in calves and chloral hydrate in adults temporarily relieves the convulsions.
Calcium versenate
Calcium versenate (calcium disodium ethylenediamine tetra-acetate, CaEDTA) has been used successfully in cases of lead poisoning produced experimentally in calves and in natural cases in cattle. CaEDTA is available as a 6.6% solution for IV administration. The manufacturer’s recommendations are to use 1 mL/kg BW per day given in divided doses 2–3 times daily over a period of 3–5 days. Based on the treatment of experimentally induced lead poisoning in calves, the optimum conditions for lead mobilization in calves are provided by concentrations of about 135 μmol EDTA/mL, or higher, maintained for 10–12 hours. This is attained by the IV infusion of calcium EDTA at a dose of 110–220 mg/kg BW over 12 hours, which is approached by rapid IV injections of two doses of 110 mg/kg BW weight, 6 hours apart. This can be done daily for 3–5 days.
CaEDTA removes lead directly from bone-sensitive sites and not from parenchymatous organs because cell membranes form a barrier to the therapeutic removal of intracellular lead. The lead is removed from soft tissues by equilibration with bone. The process takes time and thus necessitates multiple treatment. Thus it is recommended that calcium versenate be given on alternate days to allow redistribution of lead from soft tissues to available bone sites. An increase in the heart and respiratory rates and the development of muscle tremors during injection indicates a toxic reaction but can be avoided by slow administration. Recovery may take 5–15 days and parenteral or stomach tube alimentation may be required. Blindness may persist for several days after general recovery and may continue indefinitely. Dramatic improvement has also been reported in cases of chronic lead poisoning in horses after the use of calcium versenate.
Thiamin hydrochloride
In combination with CaEDTA, thiamin is now being used for the treatment of lead poisoning.32 Thiamin hydrochloride reduced the deposition of lead in most tissues especially liver, kidney, and the central and peripheral nervous system of experimentally poisoned calves. However, the levels of erythrocyte delta-aminolevulinic acid dehydratase (ALA-D) activity were decreased by 70% from pretreatment levels which indicated that thiamin had no protective effect on the ability of lead to inhibit the enzyme.33 In experimental lead poisoning in mature dairy cows, the use of thiamin was not successful in reducing blood lead concentration, but treatment with disodium CaEDTA and thiamin was effective.33 The use of thiamin also induced a remission of clinical signs of lead poisoning in cattle. Thus thiamin increases the elimination of lead from the body and may be beneficial in chelation therapy.
In naturally occurring cases in cattle, the use of thiamine and CaEDTA increases urinary lead output nearly a thousand times the untreated urinary level.24 The blood and urinary lead half-lives with CaEDTA and thiamine therapy were 2.08 and 1.38 days, respectively.25
In experimental lead poisoning in calves, thiamin at 25 mg/kg BW SC BID cured 50% of affected calves.34 The same dose of thiamin combined with 110 mg/kg BW of calcium versanate IV BID cured 100% of affected calves which had been given lead acetate at 5 mg/kg BW orally until clinical signs occurred. CaEDTA chelates lead from blood and bone while thiamine chelates lead in soft tissues and restores lead-induced biochemical alterations.34
In experimental lead poisoning in laboratory rats and mice, thiamin at a dose of 25 or 50 mg/kg BW along with CaEDTA at 50 mg/kg BW was more effective than the respective individual treatments alone.31,35 Thiamin alone decreased the blood, liver, and kidney concentrations of lead, and thiamin at 50 mg/kg BW reduced the tissue concentrations in tissues more effectively than 25 mg/kg BW.
In experimental lead poisoning in sheep, the use of thiamin at 75 mg/kg BW SC along with CaEDTA at 100 mg/kg BW IV, increased the excretion of lead via the bile and urine. Overall, thiamin, CaEDTA, and thiamin and CaEDTA increased lead excretion by 72%, 595%, and 842%, respectively over basal levels.36 It appears that thiamin can mobilize intracellular lead into blood and increase lead excretion via bile and urine.
Rumenotomy
Rumenotomy to remove the ingested lead has been used but may be unsatisfactory because of the difficulty of removing particulate material from the recesses of the reticular mucosa. However, it may be appropriate when a valuable animal is affected and it is known that the animal ingested a certain compound of lead which may be removable from the reticulum and rumen. Oral dosing with small amounts of magnesium sulfate has been used on the basis that soluble lead salts will be precipitated as the insoluble sulfate and excreted in the feces. However, the lead is often present in large quantities and in the form of particles which are only slowly dissolved.
Public health aspects of lead in meat and milk
A major concern with the treatment of lead poisoned animals, particularly food-producing animals, is the assurance that the edible tissues of recovered animals do not contain toxic levels of lead. The length of time required after successful treatment of cattle with typical clinical lead poisoning before such animals can be sent to slaughter or before the milk can be used safely is not known. It is suggested that treated animals should be appropriately identified and blood lead levels determined once or twice monthly for several months. When the blood lead levels have dropped to background levels for three consecutive samplings at least 2 weeks apart, the animals are assumed to be safe for slaughter. Undocumented field observations suggest that at least 6 months are necessary for background levels to be achieved. Recently, a study in dairy cattle determined a blood lead half-life that ranged from 48 to 2507 days. This was presumed due in part to exposure to batteries which may have been associated with prolonged retention of large pieces of metallic lead in the rumen or reticulum.21 Although this study did not determine milk concentrations of lead, the low ratio of milk lead to blood lead may allow marketing of the milk. Decisions about reaching acceptable residue levels will depend on national or local regulations as well as the economics of maintaining a herd for long periods without sales of milk or meat, and appropriate food safety and public health officials should be consulted in this decision. The lead concentrations in blood and milk from periparturient heifers 7 months after an episode of acute lead poisoning revealed no lead in the milk. Animals which had been severely affected by lead poisoning experienced a transient increase in whole blood lead concentration at parturition which was not high enough to be considered toxic.6
CONTROL
The following practices are recommended to reduce the incidence of lead poisoning:
• Adequate nutrition and consistent feeding practices will minimize pica or abnormal feeding behavior in livestock
• Garbage should always be dumped at a single, isolated, fenced-off location, and preferably buried and burned if appropriate. Pastures are unsuitable sites for garbage
• Used lead batteries and crankcase oil should be stored and disposed of safely, without spillage, and confined to areas where animals have no access
• Vehicle service and machinery storage areas should be separate from areas used by livestock
• Holding of animals in farm yards should be minimized, because such yards tend to be multipurpose areas with high risk for contamination
• Only lead-free paints should be used on surfaces and fixtures to which livestock have access
• All pastures should be inspected before cattle are introduced to them.
1 Hoff B, et al. Can Vet J. 1998;39:39.
2 Blakley BR. Can Vet J. 1984;25:17.
3 Blakeley BR, Brockman RP. Aust Vet J. 1976;17:16.
4 Preece BE. Vet Rec. 1995;136:475.
5 Wada Y, et al. Vet Human Toxicol. 1993;35:393.
6 Galey FD, et al. J. Vet Diagn Invest. 1990;2:222.
7 Sojka JE, et al. J Vet Int Med. 1996;10:420.
8 Dee S, et al. Vet Rec. 1996;138:336.
9 Vet Invest. Service Vet Rec. 1991;128:143.
10 Yonge KS, Morden BB. Aust Vet J. 1989;30:42.
11 Wubenga A, et al. Tyd voor Diergenes. 1992;117:78.
12 Baars AJ, et al. Food Add. Contam. 1992;9:357.
13 McEvoy JD, McCoy M. Vet Rec. 1993;132:89.
14 Stair EL, et al. J Am Vet Med Assoc.. 1995;207:341.
15 Takla PG, et al. Vet Rec. 1989;124:300.
16 Milhaud GE, Mehennaqui S. Vet Human Toxicol. 1988;30:513.
17 Mennaqui S, et al. Vet Human Toxicol. 1988;30:550.
18 Edwards WC, et al. Vet Toxicol. 1976;18:70.
19 Brams E, et al. J Environ Qual.. 1989;18:317.
20 Oskarsson A, et al. Sci Total Environ.. 1992;111:83.
21 Rumbieha WK, et al. J Vet Diagn Invest. 2001;13:373.
22 Carmichael DT, et al. Cornell Vet. 1987;77:277.
23 Brar RS, et al. Vet Res Commun. 1994;18:109.
24 Christian RG, Tryphonas L. Am J Vet Res. 1971;32:203.
25 O’Hara TM, et al. J Vet Diagn Invest. 1995;7:531.
26 Sharma RM, Buck WB. Vet Toxicol. 1976;18:186.
27 Knight HD, Burau RG. J Am Vet Med Assoc.. 1973;162:781.
28 Lynch GP, et al. J Dairy Sci.. 1976;59:1490.
29 Cox DH, et al. J Anal Toxicol. 1989;30:204.
30 Bratton GR, et al. Am J Vet Res. 1986;47:2068.
31 Telisman S, et al. Toxicol Lett.. 1990;52:347.
32 Kim JS, et al. Can J Vet Res.. 1992;56:256.
33 Coppock RW, et al. Am J Vet Res.. 1991;52:1860.
34 Dey S, et al. Vet Human Toxicol. 1995;37:230.
ARSENIC POISONING
Etiology Insecticidal dipping fluids, sprays; herbicides; wood preservatives, pharmaceuticals, feed additives. Inorganic compounds most toxic; organic arsenicals least.
Epidemiology Outbreaks due to accidental access to source, or due to use of excessive amounts as a dose rate or over time. Most cases result from ingestion but percutaneous absorption also possible.
Clinical signs Enteric form a highly fatal gastroenteritis with diarrhea, dehydration. Nervous form with incoordination and blindness, or a syndrome of incoordination, restlessness, squealing, convulsions.
Clinical pathology High levels of arsenic in feces, urine, milk for 5 days (organic arsenicals), 10 days (inorganic arsenic). Chronic cases best assayed in hair or skin.
Necropsy lesions Gastroenteritis in enteric form, no lesions in nervous form.
Diagnostic confirmation Higher than normal levels of arsenic in body fluids or tissues.
ETIOLOGY
Arsenic compounds likely to be encountered by large animals are as follows:
Inorganic compounds used as insecticidal dips or as herbicides
• Pharmaceuticals, e.g. cacodylic and phenylarsonic acids
• Weedicides, e.g. monosodium, and disodium methanearsonates (MSMA & DSMA).
Aromatic organic arsenicals, used as pharmaceuticals:
• Trivalent phenylorganic arsenicals, e.g. thiacetarsamide and arsphencomplexamine
• Pentavalent phenylorganic arsenicals, e.g. arsanilic acid, roxarsone (4-hydroxy-3-nitrophenylarsonic acid), nitarsone (4-nitrophenylarsonic acid).
Relative toxicities
Inorganic and aliphatic organic compounds.
The organic pharmaceuticals are the least toxic, while the insoluble oxides of medium toxicity and the trivalent inorganic compounds are associated with the most severe syndrome. Toxic oral doses may range from 1 to 25 mg/kg for the arsenite, 30–100 mg/kg for the arsenate, cacodylic acid 25 mg/kg daily for 8–10 days, and 10–25 mg/kg for 5–6 days for the methanearsonates.1,2
Aromatic organic arsenicals are toxic when the recommended cumulative dose is exceeded by 2–4 times the recommended dose, delivered by either exceeding the recommended percentage in the feed or feeding it for too long. Seven to 10 days feeding of Arsanilic acid at 500 mg/kg diet or 3-nitro,4,hydroxyphenylarsonic acid at 250 mg/kg diet will be associated with toxicosis in swine; approximately twice these concentrations will result in poisoning of poultry.1
EPIDEMIOLOGY
Occurrence
Arsenic is less commonly associated with the poisoning of livestock nowadays because of the displacement of arsenic from almost all phases of farming activity.
Source of toxin
Arsenic is indestructible and remains in the environment permanently so that the source may not be recorded in contemporary history.
Fluids used for dipping and spraying of animals to control ectoparasites are the commonest source. Animals may swallow the solution while in the dip or in the draining yards after dipping. Animals that are not allowed to drain completely and faulty disposal of drainage from yards and dips may contaminate the pasture. Opened containers of dipping solutions or powders may accidentally contaminate feed or be mistakenly applied as a skin dressing. Appreciable amounts of arsenic are absorbed through the skin after dipping in sodium arsenite. The absorption is increased if the animals are dipped when hot, if the fleece is long, if they are crowded too tightly in draining yards or driven too soon after dipping. However, in most outbreaks of poisoning some ingestion appears to occur and supplements the cutaneous absorption. There is some danger in dipping rams at mating time when erythema of the skin of the thighs and scrotum is present. Dipping immediately after shearing and jetting at too high pressure or with excessively strong solutions may also be associated with increased absorption.
Herbicides include sodium or potassium arsenite, arsenic pentoxide, and monosodium or disodium acid methanearsonate sprays used to kill potato haulms prior to mechanical harvesting.
Insecticidal sprays used in orchards and pasture contaminated by calcium arsenate applied to kill Colorado beetle grubs are sources. In most instances poisoning occurs when animals accidentally gain access to recently sprayed areas, although drifting of windblown spray may result in accidental contamination of pasture. Grass clippings from lawn areas treated with arsenical herbicides 6 months earlier may carry 15 000 mg/kg arsenic. With lead arsenate the major effects are usually ascribed to the effects of the lead but this does not always appear to be so.
Insect baits may contain Paris green (cupric acetoarsenite) mixed with bran and when these are laid over large areas of land in an attempt to control grasshopper plagues they constitute a major hazard to livestock.
Wood preservatives especially arsenic-copper-chromium are used to treat pine in wooden calf pens. The compound has a salty taste and is licked avidly. Ashes from burned treated pine posts are also palatable to cattle.
Some metal-bearing ore deposits including iron, arsenic pyrites in volcanic soils, gold and copper ores contain large quantities of arsenic which may be licked in situ, or carried off in the fumes from smelters and contaminate surrounding pastures and drinking water supplies.
Pharmaceuticals and growth stimulants including arsanilic acid and sodium arsanilate, and phenylarsonic acid preparations such as roxarsone, nitarsone, are used both as feed additives and in the control and treatment of vibrionic dysentery in animals, and as antidotes to selenium poisoning. Overdosing with them can occur accidentally by carrying on the administration for too long or when there is an error in mixing a batch of feed. The toxicity of feed containing arsanilic acid depends to a certain extent on the intake of drinking water but moderate water restriction does not make normal dose rates dangerous.
Route of poisoning
Arsenic poisoning usually occurs after ingestion of the toxic substance but percutaneous absorption can occur especially if the skin is abraded or hyperemic and percutaneous toxic dose is much lower (probably one-tenth) of the oral toxic dose.
Poison risk factors
Soluble salts are highly poisonous; arsenic trioxide and sodium arsenate are much less soluble and thus less toxic than sodium arsenite. Organic chemicals used as weedicides are as poisonous as the arsenite but organic arsenicals used as growth stimulants are less toxic, although they are absorbed rapidly.
Importance
In cases in which gastroenteritis is the predominant lesion, the case fatality rate approximates 100%. In cases characterized by nervous system involvement the illness is incidental and losses minimal if access to the poison denied, but residues become a problem.
Meat and milk residues reduce the safety of the products for human consumption. Arsenic is excreted rapidly after absorption, chiefly in the urine, and after the ingestion of non-toxic amounts by the cow there is no detectable excretion in the milk. When much larger doses are taken arsenic may be excreted in the milk, as well as in urine and feces, but the concentration is still low. The biological half-life of arsenic taken orally in the form of arsanilate is 4.2 days in liver, 5.7 days in kidney, and 15 days in muscle. In pigs fed arsanilic acid at 200 mg/kg in the feed the level of arsenic in muscle is still more than the admissible level of 0.1 mg/kg 18 days after withdrawal. The usual recommendation is to withdraw arsanilic acid 5–7 days before slaughter. This is adequate at normal dose levels.
PATHOGENESIS
Arsenic is a general tissue poison. The inorganic salts and the enteric-oriented organic compounds exert their toxic effects by combining with and inactivating the sulfhydryl groups in tissue enzymes. Trivalent arsenicals are most toxic because of their greater affinity for these sulfhydryl groupings. The efficiency of sulfur-containing compounds such as BAL (dimercaptopropanol) as antidotes depends on the ability of these compounds to compete with sulfur containing compounds of enzyme systems for the available arsenic.
Although all tissues are affected, deposition and toxic effects are greatest in those tissues which are rich in oxidative enzyme systems. Thus alimentary tract wall, liver, kidney, spleen, and lung are most susceptible to the general depression of metabolic activity which results.
Alimentary tract lesions produce the most obvious clinical signs due to the extensive damage to capillaries causing increased permeability and exudation of serum into tissue spaces. The mucosa lifts from the underlying muscle coat and is shed with the resulting loss of large quantities of body fluids. Arsenic does not precipitate protein and there is no direct local effect on alimentary tract mucosa; this is indicated by the fact that the parenteral injection of arsenic produces lesions in the gut wall which are identical with those associated with ingestion.
Because arsenic does not precipitate protein it does not limit its own absorption, and there is a considerable time lag after ingestion before clinical signs appear; corrosive substances produce lesions and signs immediately.
Arsenic absorbed from the skin may be associated with local necrosis without systemic signs if the peripheral circulation is poor or the concentration of arsenic is excessively high, but if the cutaneous circulation is good, the arsenic is quickly carried away and is associated with a systemic disease without skin necrosis.
The chronic toxicity of arsenic at low levels of intake is due to its accumulation in particular organs, especially the liver, kidney, alimentary tract wall, epidermis, spleen, and lung.
The nervous signs associated with organic arsenicals are the result of inhibition of dehydrogenase enzyme systems (e.g. pyruvate and alpha-ketoglutarate systems), causing degenerative changes in peripheral nerves. These appear as demyelination and axonal degeneration in prolonged cases. Animals recumbent longer than 7 days are unlikely to recover and will remain paralyzed until death from other associated conditions. In poisoning with the arsanilic acid compounds the lesions are mostly in the optic nerves, causing blindness. In poisoning with the phenylarsonic acid group the nerves to the limbs appear to be affected most.2
CLINICAL FINDINGS
Ruminant gastroenteritis syndromes
Acute cases are the commonest syndromes in ruminants; the onset of signs of illness is delayed 20–50 hours from the intake of the poison, the length of time varying with the fullness of the forestomachs. Distress develops suddenly, commencing with severe abdominal pain, restlessness, groaning, an increased respiratory rate, salivation, grinding of the teeth, complete ruminal stasis, and vomiting, even in cattle. A fluid and fetid diarrhea develops later. The heart rate is greatly increased, the pulse small in amplitude; dehydration, and oliguria are marked.
Peracute cases show little except depression and prostration and die before signs of enteritis develop. A fluid sound in the abdomen can be elicited by shaking the animal. Death occurs 3–4 hours after commencement of the illness and is usually preceded by clonic convulsions and diarrhea.
Subacute cases give the same signs as acute cases but the course may extend over 2–7 days. Nervous signs of muscle tremor, incoordination, and clonic convulsions are followed by terminal coma.
Commonly observed signs include low body weight, a dry, staring coat which is easily shed, loss of vigor and spirit, capricious appetite, bouts of indigestion, conjunctival and mucosal erythema, eyelid edema and conjunctivitis. Buccal mucosal ulceration may extend to the muzzle. Milk yield is seriously reduced and abortions and stillbirths may occur. Local skin lesions include initial hyperemia followed by necrosis and sloughing leaving indolent lesions which are extremely slow to heal.
Horses
Signs include marked congestion of the mucosae and a very sudden onset of severe colic which passes off in a few hours in horses which survive. Severe diarrhea may be followed by a period of complete stasis of the alimentary tract with diarrhea recurring just before death.
Nervous syndromes in pigs and lambs
Chronic poisoning resulting from overdosing with arsanilic acid is manifested by incoordination and blindness appearing about 7 days after the compound is first fed. Consciousness, body temperature, and appetite are unaffected. If feeding is continued the signs gradually worsen but disappear within a few days if the feed is changed. Some pigs remain permanently blind or paralyzed.
In chronic poisoning with roxarsone and nitarsone the emphasis is on restlessness, frequent urination, and defecation, incoordination due to loss of balance, frequent shrill ‘screaming’, tremor, and convulsions, all of which are stimulated by rousing the pig. If it is left alone in a recumbent position it may appear normal.
CLINICAL PATHOLOGY
Arsenic can be detected in the urine, feces, and milk for periods of up to about 10 days, beginning shortly after the toxic material is ingested. The rate of excretion is faster with organic compounds than with inorganic arsenic and urine levels may be back to normal in 5 days. The most satisfactory material for laboratory examination from a living animal is a large volume (about 1 L) of urine in which arsenic levels may be as high as 16 mg/kg. Levels in milk are low. Normal levels of up to 0.25 mg/kg in cows’ milk may be elevated to 0.34–0.47 mg/kg in cases of acute poisoning and to 0.8–1.5 mg/kg in the milk of normal cows which graze arsenic-contaminated pasture for long periods. Deposition in the hair occurs and the arsenic persists there until the hair is shed, making possible the detection of prior arsenic ingestion in the absence of arsenic from the blood and feces. The hair of animals not exposed to arsenic should contain less than 0.5 mg/kg, but that of normal, exposed animals may contain as much as 5–10 mg/kg. Estimations of amounts of arsenic present in suspected materials are mandatory, but delay in sampling of herbage after a contaminating incident may distort results because of leaching of soluble compounds.
NECROPSY FINDINGS
In acute and subacute cases of inorganic arsenic poisoning there are pronounced hyperemia and patchy submucosal hemorrhage in the stomach, duodenum, and cecum. Hemorrhage and multifocal ulceration of the cecum and large colon have been observed in horses.3 In ruminants the forestomachs are unaffected but typical lesions are present in the abomasum. The gut contents are very fluid, and contain much mucus and shreds of mucosa. Profuse subendocardial hemorrhages are common and ulceration of the gallbladder mucosa is often observed in sheep. Macroscopic lesions may be minimal in cases which die after a very short course. Histologically, most of the hemorrhages can be attributed to the necrosis of capillaries, although damage to the walls of larger vessels may sometimes be found.3 Severe intravascular hemolysis has been observed in sheep. Degenerative changes are common in the liver and kidney of animals suffering from arsenic toxicosis and these changes become more pronounced if the disease course is prolonged. In some cases of chronic poisoning, loss of myelin may be observed in the peripheral nerves, with secondary neural degeneration in the central nervous system.
The liver is the best organ for assay of acute arsenic poisoning, while kidney may contain high levels in subacute or chronic poisoning. Levels of over 10–15 mg/kg wet matter of arsenic trioxide in the kidney or liver are considered to be diagnostic of arsenic poisoning. However, it is probable that many animals die of arsenic poisoning when their hepatic levels are much lower than this. Maximum concentrations of arsenic in tissues occur about 8 hours after ingestion and animals which survive for 2–3 days may have levels as low as 3 mg/kg. Conversely, normal animals which are dipped routinely in arsenical dips may have hepatic levels of the element as high as 8 mg/kg. Levels of 1–3 mg/kg are obtained in cattle dying from arsenic poisoning after percutaneous exposure and levels of over 10 mg/kg in cattle which ingest arsenical dip. The toxic dose in ingesta varies widely but averages about 36 mg/kg. Assay of the arsenic level in hair may be useful in chronically poisoned animals.
Animals poisoned with organic arsenicals show no significant gross pathological changes. Histologically, degeneration of the optic nerves, optic tracts, and peripheral nerves is apparent.3 The animals maintain tissue levels of arsenic for as long as exposure continue, although the levels fall rapidly during the first 7 days after feeding of the arsenic ceases, and normal levels are not reached until a further 7 days. Levels of about 6 mg/kg arsenic trioxide in liver and kidney on a fresh, wet-matter basis indicated poisonous levels of intake. Arsenic levels in brain, spinal cord, and peripheral nerves are retained longer after poisoning than are liver and kidney values. Because the stomach and intestinal wall appear to attain maximum concentrations of arsenic most rapidly after poisoning, the use of these tissues for quantitative assay has been recommended. Therefore a part of the upper alimentary tract including contents should accompany the liver and kidney samples submitted for analysis. However, animals that are no longer exposed to arsenic or are recovering after exposure stops will have relatively little arsenic retained after the initial acute episode.
Diagnostic confirmation in all kinds of arsenic poisoning is by detection of toxic levels of arsenic in tissues and fluids of the patient.
Acute inorganic arsenic poisoning presents a clinical syndrome of gastroenteritis with minor signs of nervous system involvement, which is common in other diseases.
• Lead poisoning: the emphasis is on nervous system signs with gastroenteritis an inconstant accompaniment
• Bovine malignant catarrh develops in somewhat the same manner especially in the alimentary tract form but there are diagnostic lesions in the eyes and buccal mucosa
• Mucosal disease is also characterized by erosions in the buccal and nasal mucosae
• Salmonellosis is often confused with arsenic poisoning especially when the disease is seen in the later stages and the fever has subsided
• Poisonous plants which are associated with nervous signs and gastroenteritis include bracken, mustards, and a miscellaneous group in which specific toxins have not been identified
Chronic inorganic arsenical poisoning is associated with a syndrome of diarrhea and weight loss in areas around mining and smelting plants. It may be confused with:
Organic arsenical poisoning is characterized by dramatic but mild nervous signs, but a normal appetite and no fever.
TREATMENT
In acute cases treatment is of little value because of the large amounts ingested and the delay between ingestion and the appearance of illness, but affected animals are unsuitable for human consumption so that treatment is not usually undertaken.
Primary treatment
Compounds containing sulfur are theoretically the best antidotes and of these BAL (2,3-dimercaptopropranol) is an efficient antidote for poisoning by organic arsenicals, but is often disappointing in cases of poisoning by inorganic salts, unless therapy is begun before clinical signs appear. Dosing at 4-hourly intervals is necessary and the oily injection is associated with some local pain. Although BAL has a general beneficial effect and is recommended as a treatment, the drug is quite toxic itself and in the doses required may be associated with deaths in sheep. It also is associated with a reaction at the injection site sometimes serious enough to warrant the animal’s destruction.
Sodium thiosulfate is a practicable and frequently used treatment. The compound is almost completely non-toxic and can be given in large amounts and without accurate measurement. Intravenous injection is desirable as an initial treatment using 15–30 g of the salt in 100–200 mL of water and this should be followed by oral dosing of 30–60 g at 6-hour intervals. Treatment should be continued until recovery occurs which may require 3–4 days.
A comparison of these treatments in experimentally poisoned cattle shows little benefit from sodium thiosulfate administration and most effect with a combination of BAL and thioctic acid.1 Dimercaptosuccinate is a water soluble analog of dimercaprol, is less toxic than BAL, is available in the USA and should be more effective than BAL. The antioxidants zinc, methionine, and cysteine, used with chelation therapy, have been reported to enhance excretion of arsenic in experimental poisoning. Their use may be helpful as adjuncts to recommended chelation therapy.5
Supportive treatment
Attempts should be made to adsorb the residual arsenic in the gut by administering charcoal (1–4 g/kg BW per os), and then removed by the administration of an oil demulcent, or osmotic aperient like magnesium sulfate. Drastic purgatives should be avoided. Several products are used in an attempt to precipitate arsenic in the gut lumen. Ferric hydrate is most commonly used but has little apparent effect on the course of the disease.
Severe dehydration occurs and supportive treatment must include the provision of ample fluids preferably by parenteral injection. An adequate supply of drinking water containing electrolytes should be provided and the animals should be disturbed as little as possible and provided with shelter from the sun. Astringent preparations given by mouth may help to reduce the loss of body fluids. Recovering animals should receive a bland diet and high-quality protein.1
Control
Arsenical preparations must be handled and stored with care and contamination of feed and pasture avoided. Therapeutic preparations containing arsenic should be labelled ‘Poison’ and strict instructions given on dosage, particularly the length of time for which administration should continue. Animals to be dipped in arsenical solutions should be allowed to cool off before dipping, to drain properly afterwards and to dry before being driven. They should be watered before dipping to prevent them drinking the dip. Much mortality has occurred when instructions for mixing dip solutions were not closely followed. Dipping solutions containing more arsenic than is safe usually occur when tanks which have lost water by evaporation are reconstituted by guesswork. The maximum safe concentration of arsenic trioxide in a dip for cattle is 0.20%.
SELENIUM POISONING
Etiology Ingestion of or injection with excessive amounts of selenium.
Epidemiology Enzootic disease where soils and pasture contain toxic amounts of selenium. Outbreaks after errors in feed supplementation or oral or injection doses.
Clinical signs
Acute: dyspnea, diarrhea, prostration, short course, death.
Chronic: emaciation, rough coat, stiff gait, lame, hoof deformity.
Clinical pathology: toxic levels of selenium in body tissues and fluids.
Necropsy lesions: All forms – liver necrosis.
Diagnostic confirmation: High selenium levels in body fluids and tissues.
Treatment: Nil. Avoid toxic pasture. Care with medication and feed additives containing selenium.
ETIOLOGY
Selenium poisoning is associated with the ingestion of organic or inorganic selenium compounds as follows:
• Organic selenocompounds (selenocysteine, selenocystine) occurring in pasture plants
• Inorganic selenium compounds administered as feed supplements
• Pharmaceutical preparations administered orally, frequently combined with vitamin E, for prophylaxis or by injection in treatment.
Discrepancies exist in the toxic doses quoted in the literature and the following information is subject to that limitation. Daily intakes of 0.25 mg/kg BW are toxic for sheep and cattle; feed containing 44 mg/kg selenium for horses and 11 mg/kg for pigs is associated with poisoning. The daily intake of a diet containing 2 mg/kg of selenium can be marginally toxic for sheep. Toxic single oral doses (as mg/kg BW) are 2.2 for horses and sheep, 9 for cattle, and 15 for pigs. An oral dose of 10–15 mg of selenium has been known to kill lambs. Recommended limits for seleniumin feed are given under Control, and for injection in the section on treatment.
EPIDEMIOLOGY
Occurrence
• Pastoral. Selenium poisoning occurs in restricted areas in North America, Ireland, Israel, Canada, Australia, and South Africa where the soils are derived from particular rock formations containing a high content of selenium. No authoritative reason has been advanced to explain why the reported occurrence of the disease in these areas is much less now than it used to be
• Dosing errors. Substantial losses due to selenium poisoning also occur because of misunderstanding about the dose rates of selenium compounds used therapeutically or prophylactically.
Source of toxin
The effective selenium is contained in the top 60–90 cm of the soil profile, selenium at lower levels than this not being within reach of most plants. Selenium poisoning may occur on soils containing as little selenium as 0.01 mg/kg, but some soils may contain as much as 1200 mg/kg. Most pasture plants seldom contain selenium in excess of 100 ppm, but a number of species, the so-called converter or indicator plants, take up the element in such large quantities that selenium levels may reach as high as 10 000 ppm. Included in this category are Acacia cana, Artemisia canescens, Aster spp., some of the Astragalus, Atriplex and Castilleja spp., Comandra pallida, Descurainia pinnata, Grindelia spp., Machaeranthera ramosa, Morinda reticulata, Neptunia amplexicaulis, Oonopsis, Penstemon and Sideranthus spp., Stanleya pinnata and Xylorrhiza spp.
These plants tend to grow preferentially on selenium-rich soils and are thus ‘indicator’ plants. They are in general unpalatable because of a strong odor so that an acute syndrome is unlikely but heavy losses have been attributed in the past to two chronic forms of the disease known as blind staggers and alkali disease. Much of these data are now discounted and losses due to chronic selenosis in cattle in the western USA in recent years have been small.
Industrial deposition, e.g. fly ash from soft coal deposited in fields has been shown to be associated with increased selenium levels in tissues from sheep grazing there.
Dietary supplement is used in the prevention of known deficiency syndromes such as white muscle disease in lambs, as a non-specific growth stimulant, and as a prophylactic for a large number of other vague syndromes. It is not surprising that the careless use of selenium compounds has induced selenium poisoning on a wider scale.
It is now common practice to combine a selenium compound with an anthelmintic drench or injection and, if the mixture is not thoroughly shaken, poisoning may occur. Concurrent administration of monensin and selenium also increases the toxicity of the selenium being fed.1 There are many case reports of unexpected illness and mortality in animals dosed with selenium preparations and it is apparent that not all of the factors affecting selenium toxicity are known. Identified factors include the cobalt and protein status of the animal, deficiencies of either causing increased susceptibility.
Risk factors
Selenium poisoning in animals grazing plants growing on seleniferous soils may be restricted to very distinct areas as small as individual fields. A low rainfall predisposes to selenium poisoning because soluble, available selenium compounds are not leached out of the topsoil and lack of competing forage may force animals to eat large quantities of indicator plants.
Organic selenium compounds, especially those occurring naturally in plants, are generally considered to be much more toxic than inorganic compounds but this difference may not be apparent in ruminants because of alterations in ingested compounds produced by digestive processes in the rumen. Selenite is more toxic than selenate and both are more damaging than selenium dioxide.
PATHOGENESIS
Acute oral poisoning is associated with chemical erosion of alimentary tract mucosa. Poisoning with a single injection, e.g. in pigs, is associated with sudden death due to vasogenic circulatory failure, a vasculogenic shock.2
The mechanism forming the basis of chronic poisoning has not been identified. Selenium occurs in plants in analogs of the sulfur-containing amino acids, e.g. selenocysteine, and a possible mechanism of intoxication is by interference with enzyme systems which contain these amino acids. Selenium reduces the sulfur and protein content of sheep’s liver and high protein diets have a protective effect against selenium poisoning. Selenium is deposited in greatest concentration in the liver, kidney, and hair. It has a marked dystrophic effect on skeletal musculature and is associated with a marked rise in SGOT levels after subcutaneous administration.
CLINICAL FINDINGS
The terms blind staggers and alkali disease, used previously in this section have been discontinued as misleading and being based on misunderstandings about the etiological agents involved.
In naturally occurring and experimental poisoning there is severe respiratory distress, restlessness, complete anorexia, salivation, watery diarrhea, fever, tachycardia, abnormal posture and gait, prostration, and death after a short illness. Mildly affected pigs show posterior ataxia, walking on tiptoe, difficulty in rising, sternal recumbency, tremor, and vomiting in some. Extreme cases assume a posture of lateral recumbency.
Chronic poisoning is manifested by dullness, emaciation, rough coat, lack of vitality, stiffness, and lameness. In cattle, horses, and mules the hair at the base of the tail and switch is lost and in pigs, goats, and horses there may be general alopecia. There are hoof abnormalities including swelling of the coronary band, and deformity or separation and sloughing of the hooves in all species. Lameness is severe. Congenital hoof deformities may occur in newborn animals. Hemorrhagic lesions on the proximal wall and soles of claws on all four feet may accompany these deformities.3 Chronic poisoning in pigs on rations containing 20–27 mg/kg is also associated with a syndrome of reduced feed intake, paraplegia and quadriplegia4 due to poliomyelomalacia. Pigs on marginal levels of intake of selenium (10 mg/kg) develop necrosis of the coronary band, low conception rates, and increased neonatal mortality.
CLINICAL PATHOLOGY
Selenium can be detected in the urine, milk, and hair of affected animals. Clinical illness is evident at blood levels of 3 mg/kg and at urine levels of more than 4 mg/kg of selenium. Normal serum levels of 140–190 ng/mL are elevated to the 1500 ng/mL level.
Critical levels of selenium in hair include the following:
• Less than 5.0 mg/kg suggests that chronic selenosis is unlikely
• From 5.0 to 10.0 mg/kg suggests that borderline problems will occur
A moderate anemia occurs in acute and chronic poisoning and a depression of hemoglobin levels to about 7 g/dL is one of the early indications of selenium poisoning.
NECROPSY FINDINGS
In confirmed cases of natural or experimentally produced5 acute selenium poisoning most of the macroscopic findings can be attributed to cardiovascular compromise. There is pulmonary edema and congestion, petechiation of the thoracic viscera, and congestion of the liver, kidneys, and gastrointestinal tract. In parenterally overdosed lambs and piglets there is usually hydrothorax, hydropericardium, and ascites6. Histologic lesions may be minimal if the clinical course is brief. Changes which may be observed in animals surviving more than 24 hours include a serous effusion within pulmonary alveoli, mild hyaline or granular degeneration of skeletal muscle fibers, hydropic degeneration in renal tubular epithelial cells and periacinar degeneration and necrosis of hepatocytes. Cardiac myocytes may appear swollen and contain areas of cytoplasmic granularity and lysis.
The causal link between selenium intoxication and blind staggers is weak and the lesions described in accounts of this largely historical disease are in dispute. It is probable that this clinical entity is the result of other etiologic factors.7 In animals suffering from subacute to chronic selenium poisoning there is a skeletal and cardiac myopathy. Deformities of the feet and skin are usually apparent, as described under Clinical findings. Atrophy and dilatation of the heart and pulmonary edema, cirrhosis and atrophy of the liver, glomerulonephritis, mild gastroenteritis, and erosion of articular surfaces have also been recorded.2 Symmetrical poliomyelomalacia has been identified in both natural and experimental settings in pigs fed excessive selenium. The areas primarily affected are the ventral horns of the cervical and lumbar enlargements, with lesser damage in brainstem nuclei. The microscopic appearance of affected spinal cord includes vacuolation of the neuropil and sometimes of the cytoplasm of neurons. Neuronal chromatolysis, axonal swelling, and endothelial cell swelling and proliferation are consistently present.
Samples for confirmation of diagnosis
• Toxicology – 50 g liver, kidney; 500 g of suspect feed (ASSAY (Se))
• Histology – formalin-fixed skeletal muscle, heart, liver, kidney, +/− spinal cord from cervical and lumbar enlargements (LM).
In chronic selenosis in sheep hepatic and renal levels of selenium are about 20–30 mg/kg and levels in wool are in the range of 0.6–2.3 mg/kg. In horses hair levels of more than 5 mg/kg are recorded.
The diagnosis of selenium poisoning rests largely on the recognition of the typical syndromes in animals in areas where the soil content of selenium is high, or when there has been administration of selenium as medication or as a feed additive. The clinical and necropsy lesions associate with the poisoning cover a wide range of signs and lesions and are not easily summarized. Diagnostic confirmation depends on an assay of toxic levels of selenium in body tissues or fluids.
TREATMENT
A number of substances have been tried in the treatment of selenium poisoning, including potassium iodide, ascorbic acid, and beet pectin but without apparent effect. BAL is contraindicated.
CONTROL
The manufacturers’ advice on dose rates for pharmaceutical and feed additive preparations should be followed at all times. As a further guide to safe dose levels which avoid toxicity the following guidelines are provided:
• In general, the ratio between acute toxic and therapeutic doses is 50–100:1 and dosing accidents should not be common
• Sheep. The subcutaneous injection of selenium, as sodium selenite, is associated with poisoning in sheep at doses of 0.8 mg/kg BW and doses of 1.6 mg/kg are lethal. A single injection of 5 mg of selenium may kill some lambs and the toxic level for single injections in lambs has been reported as 455 μg/kg BW
• Cattle. Lethal doses by injections are 1.2 mg/kg BW
• Pigs. Lethal doses are between 1 and 2 mg/kg BW
• Ponies. Only relatively large doses, e.g. 6–8 mg/kg BW is associated with fatality.
Selenium in feeds should not exceed 5 mg/kg dry matter if danger is to be avoided and feeding on pasture containing 25 mg/kg dry matter for several weeks can be expected to be associated with chronic selenium poisoning. Pasture may contain as much as 2000–6000 mg/kg of selenium and is associated with the acute form of the disease when fed for a few days.
Protection against the toxic effects of selenium in amounts up to 10 mg/kg in the diet has been obtained by the inclusion in the ration fed to pigs of 0.01–0.02% of arsanilic acid or 0.005% of 3-nitro-4-hydroxyphenyl arsonic acid. In cattle 0.01% arsanilic acid in the ration or 550 mg/day to grazing steers gives only slight protection. The addition of linseed oil to the ration improves the efficiency of this protection. A high protein diet also has a general protective effect. Pretreatment with copper is also known to be an effective preventive measure in all species. The mechanism of this protection is unknown. A single oral dose of 20–40 mg/kg of copper given 24 hours before administration of selenium protects ponies.
PHOSPHORUS POISONING
Phosphorus is rarely used as a rodent poison nowadays and this is the only likely source of phosphorus for animals. Toxic effects are increased when the phosphorus is finely divided and mixed with oils or fats which facilitate its absorption. Phosphorus used for military purposes is associated with extensive contamination of pasture.
Phosphorus has a local caustic action and on ingestion is associated with severe irritation of the alimentary mucosa with signs of gastroenteritis appearing within an hour or two. Some phosphorus may be absorbed and is associated with acute hepatic necrosis but signs do not appear for several days.
CLINICAL FINDINGS
These include severe diarrhea, acute abdominal pain, salivation, and intense thirst. Pigs vomit violently and the vomitus is luminous and has a garlic odor. The patient often dies of acute shock during this stage. Survivors show jaundice, weakness and anorexia, oliguria and hematuria. Death may occur in coma or be accompanied by convulsions. Phosphorus can be detected in the vomitus and feces of affected animals.
NECROPSY FINDINGS
Macroscopically there is congestion and hemorrhage of the alimentary mucosa. The carcass is often jaundiced and the liver is swollen and pale. Histologically there is fatty degeneration of both the liver and kidney, sometimes accompanied by hepatic necrosis. The acute stages of phosphorus poisoning may appear similar to acute stages of inorganic arsenic, mercury, or selenium poisoning.
Samples for confirmation of diagnosis
Diagnostic confirmation requires evidence of access to the poison and the detection of large amounts of it in the alimentary tract.
An emetic or hydragogue purgative should be given immediately. Supportive treatment includes the administration of astringents to allay the gastroenteritis and parenteral electrolyte solutions to relieve the dehydration. Hypotension and shock as well as coagulopathy may occur and should be treated supportively as needed.
MERCURY POISONING
Etiology Rarely inorganic mercury, commonly organic preparations.
Epidemiology Inorganic salts in preparations used as rubifacients. Organic preparations used in seed grain fed accidentally to livestock.
Clinical signs
Inorganic salts: Acute – vomiting, diarrhea, abdominal pain; Chronic – weight loss, depression, alopecia, scabby dermatitis, long course.
Organic preparations: blindness, incoordination, paralysis.
Clinical pathology High levels of mercury in all tissues and fluids; high blood urea nitrogen, urine alkaline phosphatase in cases of nephrosis.
Necropsy lesions
• Inorganic salts: Acute – gastroenteritis; Chronic – nephrosis.
• Organic preparations: neuronal necrosis in brain and spinal nerves.
• Poisoning by mercury is associated with inflammation of the alimentary mucosa and damage to the kidneys. It is manifested clinically by gastroenteritis and terminally by signs of uremia.
• Diagnostic confirmation. High blood, urine, tissue levels of mercury.
ETIOLOGY
Mercuric chloride and mercury biniodide are highly poisonous, the toxic dose for horses and cattle being about 8 g and for sheep 4 g. Organic mercury taken regularly in the diet at a level of 1 mg/kg is associated with chronic poisoning in pigs. A level of 6 mg/kg is associated with deaths in pigs within 5 days.
EPIDEMIOLOGY
Source of toxin
Mercury poisoning in farm animals occurred in the past almost exclusively as a result of accidental feeding of grain, pellets, or concentrate mixtures treated with organic mercurial antifungal agents.
Because of the availability of fungicidal agents other than mercury it is possible to limit the use of mercuric agents by legislation to those excreted rapidly by animals, the phenylmercury compounds, and prohibiting those which are retained in animal tissues, the ethyl and methyl compounds. Worldwide use of mercurial fungicides has declined and poisoning is much less common than in the past. The commonest agents, when used, are dusts of 5.25% methoxyethylmercury silicate or methylmercury dicyandiamide. These and ethylmercuric chloride are toxic when fed to pigs at the rate of 0.19–0.76 mg of mercury per kg BW per day for 60–90 days. Methylmercury dicyandiamide fed to pigs at the rate of 5–15 mg/kg is associated with illness, and 20 mg/kg is associated with some deaths with a delay of 3 weeks between dosing and illness.
Treated seed is usually not harmful if it comprises only 10% of the ration and must be fed in large amounts for long periods before clinical illness occurs. A single feeding even of large amounts of grain is thought to be incapable of causing mercury poisoning in ruminants but horses may be susceptible.
Accidental administration of medicines containing mercury, licking of skin dressings (e.g. mercuric oxide), and absorption from liberally applied skin dressings or combined with dimethyl sulfoxide may be associated with sporadic cases as occurs in horses after application of mercury containing ‘blisters’.1 Inorganic mercury salts contaminating lakes or other anaerobic ecological areas can be reduced and converted to methyl mercury and serve as a source of organic mercurial poisoning or food contamination through accumulation in fish or fish meal.2
Risk factors
The toxicity of mercury compounds depends on their solubility and the susceptibility of the animals. Cattle are highly susceptible, toxicosis occurring on an average daily intake of mercury, in organic mercury form, of 10 mg/kg per day, while toxic effects are only obtained in sheep with intakes of 17.4 mg/kg BW per day. In horses, feeding inorganic mercury at the rate of 0.4 mg/kg BW produces only mild signs of poisoning and feeding methylmercury chloride (10 g over 10 weeks) is associated with serious illness.
PATHOGENESIS
Inorganic mercury compounds are associated with coagulation of the alimentary mucosa, leading to the rapid development of gastroenteritis. Animals that survive the alimentary tract disorder and absorb mercury may show signs of damage to peripheral capillaries especially those at the sites where mercury is excreted, in the kidney, colon, and mouth. Nephrosis, colitis, and stomatitis may result.
Organic mercurials in small doses liberate their mercury slowly into tissues and are associated with degenerative changes in brain, segmental degeneration in peripheral nerves, and degeneration in kidney. Extensive subcutaneous hemorrhages and a bleeding tendency occur in some cases of phenylmercuric acetate poisoning.
CLINICAL FINDINGS
Acute mercurialism occurs when large amounts of inorganic mercury are ingested, there is an acute gastroenteritis with vomiting of blood-stained material and severe diarrhea. Death occurs within a few hours due to shock and dehydration. In less acute cases the patient survives several days and the syndrome includes salivation, a fetid breath, anorexia, oliguria, tachycardia, hyperpnea and, in some cases, posterior paralysis and terminal convulsions.
Chronic mercurialism occurs when small amounts of inorganic mercury are ingested over long periods. The syndrome includes depression, anorexia, emaciation, a stiff, stilted gait which may progress to paresis, alopecia, scabby lesions around the anus and vulva, pruritus, petechiation and tenderness of the gums and shedding of the teeth, persistent diarrhea, weakness, incoordination, and convulsions.
Chronic organic mercurial poisoning is associated with neurological syndromes. In pigs blindness is accompanied by staggering, continuous walking, and inability to eat, although the appetite is good. Cattle poisoned in this way show a staggery gait, standing on tiptoe, and paresis, lying down most of the time; they appear normal in other respects, often eating well. Clinical signs may not develop until 30 days after feeding is commenced. Cattle poisoned experimentally show more severe nervous signs including incoordination, head-pressing, muscle tremor with twitching of the eyelids, tetanus-like spasms on stimulation, excessive salivation, recumbency, and inability to eat or drink, followed by tonic–clonic convulsions with opisthotonos.
CLINICAL PATHOLOGY
Mercury can be detected at higher levels than normal in the blood, feces, and urine of affected animals and in the toxic source material. Nephrosis can be diagnosed by examination of blood and urine; the earliest and most accurate indicators are the urinary concentrations of alkaline phosphatase and gamma-glutamyl transpeptidase. Less than 0.2% of ingested mercury is excreted in cow’s milk.3
NECROPSY FINDINGS
In acute cases there is severe gastroenteritis with edema, hyperemia, and petechiation of the alimentary mucosa. The liver and kidneys are swollen and the lungs are congested and show multiple hemorrhages. There may be an accompanying catarrhal stomatitis. A crusting focus of dermatitis may be identified if exposure was percutaneous.
Histologically the renal tubular epithelial cells are swollen and vacuolated, and proteinuria is evident. An ulcerative colitis may also be visible. In chronic mercurialism associated with organic mercury compounds there are also degenerative changes in nerve cells in the cortex of cerebrum, brain stem, and spinal cord. The lesions include neuronal necrosis, neuronophagia, cortical vacuolation, and gliosis. Fibrinoid necrosis of leptomeningeal arterioles may be seen. Other common microscopic changes include degeneration of granular cells of the cerebellar cortex and of Purkinje cells of the myocardium.
Mercury reaches its greatest concentration in kidney and this tissue should be submitted for assay. Levels of 100 mg/kg may be present in the kidney of animals poisoned with inorganic mercury. With chronic organic mercurial poisoning in swine levels of mercury up to 2000 mg/kg may be present in the kidney.
Acute mercury poisoning is rare but should be suspected in animals which are exposed to inorganic mercury compounds and which show signs of gastroenteritis and nephritis. Diagnostic confirmation depends on a positive tissue assay for mercury.
When there are indications of gastroenteritis or nephrosis the syndrome resembles:
Pigs poisoned by organic mercury compounds and showing nervous signs of blindness and incoordination resemble:
TREATMENT
In acute cases large amounts of coagulable protein such as eggs should be given by mouth immediately, followed by mild purgatives to facilitate removal from the gut before digestion and absorption occur. In acute and chronic cases treatment with sodium thiosulfate as described in arsenic poisoning is recommended. BAL can be used but has the same limitations here as in arsenic poisoning, and delay in treatment of any sort is likely to be fatal. An injection of BAL (6.5 mg/kg BW) should be given every 4 hours. Dimercapto succinic acid (DMSA) at 10 mg/kg B.W. t.i.d. is reported effective in hastening the elimination of mercury in urine.4
Suppportive treatment includes astringents given orally to control the gastroenteritis and fluids given parenterally to correct the dehydration.
1 Guglick MA, et al. J Am Vet Assoc. 1995;206:210.
2 Osweiler GD, et al. Clinical and Diagnostic Veterinary Toxicology. Dubuque, IA: Kendall-Hunt, 1985.
3 Neathery MW, Mille WJ. Dairy Science. 1975;58:1767.
4 Murphy M. A Field Guide to Common Animal Poisons. Ames, IA: Iowa State University Press, 1996.
FLUORINE POISONING
Etiology Inorganic or organic (in plants) fluorides.
Epidemiology Associated with continuous ingestion of small but toxic amounts of fluorine in the diet or drinking water.
Clinical signs Dental fluorosis – mottling and erosion of permanent teeth. Osteofluorosis – lameness and unthriftiness.
Clinical pathology High blood, urine, and tissue levels of fluorine.
Necropsy lesions Osteoporosis, widespread exostoses. Dental enamel and dentine hypoplasia. High bone content of fluorine.
Diagnostic confirmation Assay of food, water, bones, teeth for fluorine content.
Primary: aluminum salts orally, calcium salts parenterally but little chance of improvement.
Supportive: parenteral glucose solutions.
Control Drinking water held in tanks, fluorine precipitated by aluminum salts; applicable only for small water volumes.
ETIOLOGY
The severity of the poisoning depends on the amount ingested, the solubility of the fluorine compound, and the animal’s age.
Dose rate
The most satisfactory measure is the concentration in the total dry matter consumed. Levels in excess of 100 ppm of dry ration are likely to be associated with disease in cattle, sheep, and pigs when the fluorine is contained in rock phosphate or cryolite. At this or lower levels minor teeth lesions may occur but not to such a degree that they will affect the animal’s well-being during a commercial life span. Fluorine in the form of calcium fluoride or sodium fluorosilicate is much less poisonous; intakes of 400 mg to 2 g/kg BW are necessary for fatal effects. Sodium fluoride is approximately twice as toxic as rock phosphate and a general level of 50 mg/kg of dry ration should not be exceeded.
In experimentally induced fluorosis in cattle mottling of the tooth enamel occurs at intakes of 27 mg/kg in the diet but there is no pitting until levels of 49 mg/kg are fed. Bony lesions are slight at intakes of 27 mg/kg, moderate at 49 mg/kg, and marked at 93 mg/kg, and milk production in dairy cows is supposed not to be affected by intakes of 50 mg/kg of fluorine in the diet until about the fourth lactation. A more recent view is that the existing tolerance level for dairy cows of 40 mg/kg is too high and will lead to serious loss of production and some dental fluorosis in high producing cows.
EPIDEMIOLOGY
Occurrence
Fluorine intoxication has been observed in most countries, usually in association with specific natural or industrial hazards. In Europe and Great Britain losses are greatest on summer grazing of pastures contaminated by industrial fumes, including dust from factories converting rock phosphate to superphosphate as well as effluent from aluminum smelters. Iceland and parts of the southern Andes mountains are extensively affected by contamination from volcanic ash. Drinking water from deep wells, industrial contamination of pasture and the feeding of fluorine-bearing phosphatic supplements are the common associations in North America. Deep wells also are an important source in Australia and South America. In Africa the important association is the feeding of phosphatic rock supplements.
Sources
Fluorine occurs naturally in rock, particularly in association with phosphate, and these rocks, the soils derived from them and surface water leaching through the soils, may contain toxic quantities of fluorine. In such areas the soil content of fluorine may be as high as 2000–4000 mg/kg even up to 12 000 mg/kg and the levels in water up to 8.7 mg/kg; soil fluorine varies in its solubility from 10 to 20%. Levels of fluorine likely to be toxic to animals are not usually encountered in natural circumstances, interference by man being necessary in most instances to increase fluorine ingestion above the critical level.
Plants, with few exceptions, do not absorb appreciable quantities of fluorine. Two exceptions are Camellia spp., the decorative camellias, and Thea chinensis (tea) plant. Major outbreaks of intoxication occur as the result of the ingestion of pasture contaminated with fluorine, and drinking water and mineral supplements which contain excessive amounts of fluorine.
Contamination from industrial factories by smoke, vapor, or dust may produce pasture containing 20–50 mg/kg of fluorine. Factories producing aluminum by the electrolytic process, iron and steel with fluorine-containing fluxes, superphosphate, glazed bricks, copper, glass and enamels are likely to be potent sources and may be associated with toxic levels of contamination as far as 14 km downwind from the factory. Dust from factories manufacturing superphosphate from rock phosphate may contain as much as 3.3% fluorine. Industrial plants engaged in the calcining of ironstone have also been incriminated as sources of fluorine.
Contamination by effluent is a complex problem because of variation in the form of the contaminating compound. Grass can absorb and retain gaseous fluoride from the ambient air but physical deposit of liquids and dust is the critical form of contamination. Two of the common effluent substances are hydrofluoric acid and silicon tetrafluoride, both of which are as toxic as sodium fluoride, and dental lesions occur in 100% of young ruminants on an intake of 14–16 mg/kg dry matter of these substances. Severe cases occur on pasture or hay containing more than 25 mg/kg dry matter and similar lesions develop much more rapidly on pasture containing 98 mg/kg dry matter.
Fluoracetamide is also known to be a toxic factory effluent.
Dust and gases from volcanic eruptions may also be associated with acute fatal fluorine intoxication in the period immediately after the eruption, and contamination of pasture may be sufficient to be associated with subsequent chronic intoxication in animals eating the herbage, although the fluorine content of the contaminated materials decreases very rapidly if rain falls. Iceland is particularly afflicted with fluorine intoxication deriving from this source.
Top-dressing of pasture with phosphatic limestone is commonly associated with fluorosis. Most phosphatic limestones, particularly those from North Africa, are rich in fluorine (0.9–1.4%). Non-phosphatic limestones contain insignificant amounts.
Supplementary feeding of phosphates.
The common occurrence of phosphorus deficiency in animals has led to the search for cheap phosphatic materials suitable for animal feeding. Rock phosphates are commonly used and many deposits contain dangerous amounts of fluorine (3–4%). The fluorine content of the mineral can be reduced but the cost encourages the use of marginally safe material.
The major occurrence of water-borne fluorine intoxication is from water obtained from deep wells or artesian bores. The available data suggest that, although minor teeth lesions occur at 5 mg/kg of fluorine it is not until levels of 10 mg/kg are exceeded that excessive tooth wear occurs and the nutrition of the animal is impaired. More serious systemic effects do not occur until the water contains 30 mg/kg.
Miscellaneous sources of fluorine include the ingestion of superphosphate itself, but a supernatant liquid of a suspension of the fertilizer will contain no fluorine. Some wood preservatives may contain large quantities of fluoride which may be associated with acute poisoning in some circumstances.
RISK FACTORS
Host factors are age and species. Daily intakes of 0.5–1.7 mg/kg of fluorine as sodium fluoride produce dental lesions in growing animals without affecting general well-being. Intakes equal to twice these amounts are consumed by adult animals without ill-effect. In heifers a continuous intake of 1.5 mg/kg BW per day is sufficient to be associated with severe dental fluorosis without affecting growth rate or reproductive function. However, extensive osteofluorosis and periods of severe lameness will occur. The fluorine content of the bones of newborn calves depends on the dam’s intake of fluorine in the last 3–4 months of pregnancy and not on her own bone composition.
Most recorded occurrences of fluorosis are in cattle. Sheep are less susceptible than cattle. A continuous intake of 1 mg/kg BW is the maximum safe limit for ruminants, an intake of 2 mg/kg BW produces clinical signs. In pigs an intake of 1 mg/kg BW added fluorine for long periods has no deleterious effect.
Importance
Death losses are rare and restricted largely to acute poisoning, the major losses taking the form of unthriftiness associated with chronic fluorosis. Although it is possible for animal tissues to contain amounts of fluorine in excess of permissible amounts this is not usually so in chronic fluorosis. The fluorine content of milk in these circumstances is below that permitted in fluoridated drinking water (1 mg/L).
PATHOGENESIS
Fluorine is a general tissue poison; its exact mode of action does not appear to have been closely examined.
Acute intoxication, due to the ingestion of large amounts of soluble inorganic fluorides, is characterized by the immediate development of gastrointestinal irritation due to the formation of hydrofluoric acid in the acid medium of the stomach. Nervous signs including tetany and hyperesthesia, and inhibition of blood clotting, may follow as a result of the fixation of serum calcium to form physiologically inactive calcium fluoride in the blood plasma. Death occurs quickly.
Organic fluorides, including sodium fluoracetate, also known as compound 1080, and fluoracetamide, are associated with sudden death by poisoning the enzyme aconitase, leading to the accumulation of diagnostically significant levels of citrate in tissues and permanent damage to myocardium.
Metabolic effects due to the ingestion of small amounts of inorganic fluorides over long periods are associated with an initial marked reduction in the activity of ruminal infusoria, a reduction in food intake, and a decreased production of fatty acids. The level of fluorine intake is critical and intakes of 150 mg/kg or less have no effect on food intake. At intakes of 150–200 mg/kg there is a depressing effect on milk production, and at 200 mg/kg the intake of grain is reduced.
Detoxication by deposition of fluorine occurs in association with phosphate in the teeth and bones. Deposition in bone occurs throughout life but in teeth only in the formative stages. Fluorides inhibit the action of ameloblasts and odontoblasts during tooth formation, resulting in failure of the developing tooth to accept minerals.1 In bones, fluorides alter mineralization and remodeling of bone by replacing hydroxyapatite in the bone crystalline structure.2 The degree of deposition varies, being greatest on the periosteal surface of the long bones where exostoses commonly develop. Thus teeth lesions occur only if the intake is high before the teeth have erupted but bone lesions occur at any stage. When the tissue levels of fluorine are moderate, characteristic lesions due to hypoplasia of the enamel appear in the teeth. At higher levels the storage capacity of these organs is exceeded and blood and urine levels rise. General signs of toxicity thus appear in tissues at the same time as bone lesions develop. The bone lesions of osteomalacia, osteoporosis, and exostosis formation, with accompanying pathological fractures, are associated with excessive mobilization of calcium and phosphorus to compensate for their increased urinary excretion in conjunction with fluorine.
The other tissues particularly prone to fluorine intoxication and in which degenerative changes occur are bone marrow, kidney, liver, adrenal glands, heart muscle, and central nervous system. A severe anemia may rarely occur as a result of toxic depression of bone marrow activity, although this is not a constant or expected sign. The facility of storage in bone explains the long latent period which occurs in animals subjected to chronic intoxication.
There has been controversy about whether fluorine passes the placental barrier in significant amounts. Although the current view is that placental passage is infinitesimal in amount,3 cases of neonatal dental fluorosis have been identified in cattle.4
Fluorine does not occur in significant quantities in the milk or colostrum of poisoned cows.
Detoxification by leaching of fluorine from bones and teeth occurs after a decrease in the intake of fluorine leads to lowering of blood levels and mobilization from deposits commences. This is of importance when interpreting urine and blood levels of the element.
CLINICAL FINDINGS
Acute intoxication
The syndrome includes dyspnea, complete anorexia, vomiting, and diarrhea in pigs, and ruminal stasis with constipation or diarrhea in ruminants. Vomiting acts as a protective mechanism and toxic doses in pigs may be eliminated in this way without the development of other signs. Nervous signs are characteristic and include muscle tremor and weakness, a startled expression, pupillary dilatation, hyperesthesia, and constant chewing. Tetany and collapse and death follow within a few hours.
Chronic intoxication-fluorosis
Because of the distinct clinical separation between animals with dental lesions and those which have, in addition, signs of lameness and general ill-health it is customary to refer to two forms of the disease: dental fluorosis and osteofluorosis. Lesions of the teeth and bones are characteristic and the signs are largely referable to these lesions. Teeth changes are the earliest and most diagnostic sign but may not produce clinical effects until other signs have developed. Consequently, they are often missed until other clinical findings suggest that the teeth be examined. Severe dental fluorosis results in excessive dental wear, inability to graze properly, difficulty mastication and lapping of water due to tooth pain. Continued impaired dentition leads to reduced milk production and poor weight gain or actual weight loss. Reproductive function may be reduced due to locomotor dysfunction and poor nutriton as a result of reduced feed intake.
Osteofluorosis
Lameness most marked in the loins, hip joints, and hind limbs and unthriftiness in animals of any age are the signs usually observed first. The occurrence of hip lameness or fractures of the third phalanx on a herd scale in cattle is thought to be diagnostic of fluorosis. Pain is evinced on pressure over limb bones and particularly over the bulbs of the heels. The bones may be palpably and visibly enlarged. This is most readily observed in the mandible, sternum, metacarpal, and metatarsal bones and the phalanges. This overall thickness may be subsequently replaced by well-defined exostoses. The bones are subject to easy fracture. These well-defined lesions occur only in advanced cases and are often accompanied by extensive tooth lesions in young animals. In addition to the cases affected by generalized lameness there are cases which show a sudden onset of very severe lameness, usually in a forelimb, associated with transverse fracture of the third phalanx.
Dental fluorosis
Temporary teeth of animals poisoned while in utero and permanent teeth exposed to intoxication before eruption will be affected. The earliest and mildest sign is mottling with the appearance of pigmented (very light yellow, green, brown, or black) spots or bands arranged horizontally across the teeth. Occasional vertical bands may be seen where pigment is deposited along enamel fissures. Mottling and staining occur on incisors and cheek teeth and are not evident when the affected tooth erupts and in fact may not appear until some months later. The cheek teeth are usually worse affected than the incisors but are very difficult to examine clinically. If the period of exposure to intoxication has been limited only some of the teeth may be affected but the defects will always be bilateral.
Mottling may not progress any further but if the intoxication has been sufficiently severe defective calcification of the enamel leads to accelerated attrition or erosion of the teeth, usually in the same teeth as the mottling. The mottled areas become pits and the teeth are brittle and break and wear easily and unevenly. Patterns of accelerated attrition are dependent upon the chronological occurrence of the intoxication and the eruption time of the teeth. Uneven and rapid wear of the cheek teeth makes proper mastication impossible. Infection of the dental alveoli and shedding of teeth commonly follow. The painful condition of the teeth and the inability to prehend and masticate seriously reduce the food intake and are associated with poor growth in the young and unthriftiness and acetonemia in adults. Affected cattle may lap cold drinking water to avoid the discomfort occasioned by normal drinking. Eruption of the teeth may be abnormal, resulting in irregular alignment.
A standard for the classification of fluorosis has been proposed based on the degree of mottling, pitting, and rate of wear of the teeth. The effects of dental mottling, pitting, and excessive wear of incisors can be used to estimate the lifetime exposure periods of cattle at risk during dentition.5 The additional clinically apparent abnormalities include delayed eruption of permanent incisor teeth, necrosis of alveolar bone resulting in recession of bone and gingiva, oblique eruption of permanent teeth, hypoplasia of teeth, wide spaces between teeth, and rapid development of any dental lesions.
Other effects
Reproduction, milk yield, and wool growth are not usually considered to be adversely affected except indirectly by the reduced food intake. Severely lame animals may have lowered reproductive performance indirectly due to physical dysfunction that interferes with mating.
Additional signs including diarrhea and anestrus and other forms of infertility in cattle, diarrhea in sheep, and polydipsia and polyuria in pigs are recorded in the naturally occurring disease but cannot be considered as constant or pathognomonic.
In animals that are grazed for only part of the year on pasture contaminated by factory effluent during the summer, there may be considerable clinical improvement during the winter and an annual recrudescence of signs when the animals are outside.
Horses with chronic fluorosis have lameness, dental lesions including excessive molar abrasion, and hyperostotic lesions of the metatarsus, metacarpus, mandible, and ribs.
CLINICAL PATHOLOGY
Normal cattle have blood levels of up to 0.2 mg fluorine per mg/dL of blood and 2–6 mg/kg in urine. Cattle on fluorine intakes sufficient to cause intoxication may have blood levels of 0.6 mg/dL, and urine levels of 16–68 mg/kg, although blood levels are often normal. Such high levels may not be an indication of high intakes immediately preceding the examination, as heavy deposits in bones may be associated with abnormally high blood and urine fluorine levels for some months after the intake has been reduced to normal. Urine levels should be corrected to a specific gravity of 1.040. Serum calcium and phosphorus levels are usually normal and there is a significant correlation between the amount of fluoride fed and the concentration of alkaline phosphatase in the serum. The increase in phosphatase activity is probably related to the abnormal formation of bone. The increased SAP activity may be three to seven times the normal level.
Radiographic changes of bones containing more than 4000 mg/kg of fluorine include increased density or abnormal porosity, periosteal feathering, and thickening, increased trabeculation, thickening of the compact bone, and narrowing of the marrow cavity. Spontaneous rib fractures show incomplete union. Good data are available for fluorine concentrations in rib bones, and estimations of fluorine content in biopsy samples of ribs have been used in the clinicopathological study of the disease. Samples of tail bone and the spongiosa of the tuber coxae have also been used for these purposes.
Organic fluorides are difficult to assay in excretions and tissues, and even in contaminated feed sources. In affected animals indirect measurement based on tissue concentrations of citrate may be necessary. An additional suggested procedure is the administration of an aqueous extract of suspected poisoned tissues or feed material to guinea-pigs and the measurement of tissue levels of citrate in them.
NECROPSY FINDINGS
Severe gastroenteritis is present in acute poisoning. In chronic fluorosis the bones have a chalky, white appearance, are brittle and have either local or disseminated exostoses, particularly along the diaphyses. Intra-articular structures are not primarily affected, although there may be some spurring and bridging of the joints. Histologically there is defective and irregular calcification of newly formed trabecular bone and active periosteal bone formation. Hypoplasia of the enamel and dentine are consistent physical and histological defects in the teeth of affected young animals. Young animals may also develop thickened growth plates and widened metaphyses that are grossly similar to rachitic changes. Degenerative changes in kidney, liver, heart muscle, adrenal glands, and central nervous system have been reported in severe cases. Degeneration of the bone marrow and consequent aplastic anemia also occur.
Chemical examination of necropsy specimens is valuable in the diagnosis as the fluorine content of bones from poisoned animals is greatly increased. Levels of up to 1200 mg/kg are observed in normal animals but may be increased up to 3000 mg/kg in animals exposed to fluorine and showing only mottling of the teeth. Animals showing severe clinical signs have levels greater than 4000 mg/kg of bone on a dry, fat-free basis and after prolonged heavy feeding levels may be as high as 1.04%. Care must be taken in selecting the bone samples because of the great variation in the concentration of fluorine which occurs between different bones. Good data are available for comparison between metacarpal, metatarsal, rib, pelvic, and mandibular bones and antlers of deer.3 Mandibles usually show the greatest concentrations and in the long bones the distal and proximal quarters are more sensitive indicators than the center half.
Soft tissues are unreliable as a criterion for fluorosis because of their low levels of fluorine. In bone and teeth, ash levels of 0.01–0.15% fluorine are found in normal animals. Levels up to 1.5% fluorine indicate excessive intake but are not usually accompanied by anatomical changes. Where clinical signs of intoxication appear there is usually up to 2% fluorine in bone ash and 1% in teeth ash.
Samples for confirmation of diagnosis
• Toxicology – mandible/metacarpal/metatarsal; rib, vertebrae for evidence of osteofluoriosis. Urine from affected animals for evidence of recent exposure (ASSAY (F))
• Histology – formalin-fixed metacarpal/metatarsal/mandible (LM).
Diagnostic confirmation depends on fluorine assay of food and water, of blood and urine of affected animals, and bones and teeth at necropsy.
TREATMENT
Primary treatment, apart from removing the animals from the source of fluorine, is largely impractical. Acute cases require gastrointestinal sedatives.
Supportive treatment to neutralize residual fluorine in the alimentary tract and calcium salts intravenously is recommended. Aluminum salts act as neutralizers of the hydrofluoric acid produced in the stomach and because of their insolubility they are safe even in large quantities (30 g of aluminum sulfate daily for prevention, more for treatment). The calcium salts given intravenously should be given to effect, using the disappearance of tetany and hyperesthesia as a guide. This treatment will probably have to be repeated. The parenteral administration of glucose solutions is recommended because of the interference by fluorine with glucose metabolism. Irrespective of treatment used, no improvement in dental or osseous lesions can be anticipated but there may be amelioration of the other clinical signs.
CONTROL
Fluorine content of feed
Phosphatic feed supplements should contain not more than 0.2% fluorine for milking or breeding cattle or 0.3% for slaughter cattle, and should not comprise more than 2% of the grain ration if the fluorine content is of this order. In spite of this recommendation the feeding of rock phosphate containing 1–1.5% fluorine to cattle for long periods is maintained in some areas without major deleterious effects on health. Some deposits of rock phosphate have much higher contents of fluorine than others and commercial defluorination makes these toxic deposits safe for animal feeding.
Bone meal in some areas may contain excessive quantities of fluorine and should be checked for its fluorine content. Access to superphosphate made from rock phosphate with high fluorine content should be avoided. Water from deep wells and artesian bores should be assayed for fluorine content before use.
Nutritional management
Where fluorine levels are marginal careful husbandry, including the watering of young, growing stock on fluorine-free supplies, and permitting only adults to be watered on the dangerous supplies, and rotating the animals between safe and dangerous waters at 3-month intervals may make it possible to utilize land areas otherwise unsuitable for stock raising. In some areas dairy herds may have to be maintained by the purchase of replacements rather than by the rearing of young stock. In areas where long-term ingestion of fluorine is likely to occur the aim should be to provide a diet of less than 50 mg/kg of the total diet of dairy cows. Adequate calcium and defluorinated phosphorus intakes should be insured as these reduce bone storage of fluorine.
Detoxication
Aluminum salts are the principal substances used to detoxicate food and water. They are relatively ineffective, reducing the accumulation of fluorine in bone by only 20–30%, and are thus referred to as ‘alleviators’. The sulfate and phosphate have been used but all the salts are unpalatable and can only be administered daily to animals being hand-fed relatively large amounts of concentrates. It is presumed that highly insoluble aluminum fluoride is formed in the alimentary canal.
Extensive field trials of aluminum as an alleviator have not justified its use as a practicable control measure in average circumstances. Best results are obtained by improvement in nutrition of the animals and better grassland management. If effective control measures are introduced it will be some years before the affected teeth have erupted and become visible and affected animals culled.
The fluorine content of drinking water can be reduced (from 10 to 0.95 mg/kg) by adding freshly slaked lime to the water; 500–1000 mg/kg should be added and the water must be allowed to settle for 6 days. The method requires the use of large storage tanks.
Legislation to control fluoride emission from factories is now general but the usual limitation of not more than 1 μg/m3 does not completely avoid danger and serious losses can still occur at these emission levels. Prevailing tolerances for pasture contamination also appear to be incompletely protective.
ALUMINUM POISONING
Aluminum is one of the potentially toxic elements introduced into the diets of animals by the deposition of soluble salts in acid rain, or by powder particles in factory effluent. Absorption by plants may also be a factor. Overt effects are rare but at high levels of intake, the aluminum suppresses the absorption of phosphorus and may be assocated with the patient being in negative phosphorus balance. Retarded growth and hypophosphatemia have been produced experimentally in pigs.1
MOLYBDENUM POISONING
Etiology Ingestion of toxic amounts of molybdenum.
Epidemiology Enzootic in areas where soil, especially peat soils, contain high levels of molybdenum. Epizootics when molybdenum used in excessive amounts as an agricultural chemical.
Clinical signs Signs of secondary hypocuprosis (weight loss, hair coat depigmentation, anemia) plus persistent, debilitating diarrhea.
Clinical pathology High blood levels of molybdenum, low levels of copper.
Necropsy lesions No significant lesions.
Diagnostic confirmation High levels of molybdenum in feed and blood.
ETIOLOGY
A daily intake of 120–250 mg of molybdenum has proved to be toxic for cattle, although the toxic dose varies widely with the intake of sulfate, copper, and possibly other factors.
EPIDEMIOLOGY
Occurrence
The major occurrence of molybdenum poisoning is on pasture growing on molybdenum-rich soils, usually derived from particular geological formations, e.g. the ‘teart’ pastures of Somerset (UK), the USA, and Canada, marine black shales in the UK and pastures containing excess molybdenum intake with or without a marginal deficiency of copper in New Zealand, Canada, Ireland, and Australia.
Source
Toxic intakes
Soil molybdenum levels in problem areas vary between 10 and 100 mg/kg. Illness may occur on pasture containing 3–10 mg/kg. Although levels of less than 3 mg/kg are usually considered to be safe, signs of toxicity may occur at levels as low as 1 mg/kg if the sulfate intake is high and the copper status low; the trigger level of molybdenum at which the interference with the metabolism of copper may occur is 2.4 mg/kg dry matter in the diet.
Forage containing 10 mg/kg must be considered dangerous at all times and, on pasture affected by aerial contamination levels of 10–200 mg/kg may be encountered. Such intakes can be provided by:
• Industrial fallouts of 5–40 ng/m3 of air or 2 mg/m2 per month on pasture
• Contamination of pasture by motor oil containing molybdenum as an additive1
• Aerial contamination by fumes from aluminum and steel alloy factories and oil refineries using molybdenum is associated with secondary copper deficiency
• The use of molybdenum in fertilizer mixtures to increase nitrogen fixation by legumes may lead to excessive amounts of molybdenum in soils.
Drinking water may not be as toxic as the same amount in fresh forages. For calves, the minimum toxic concentration in drinking water is between 10 and 50 mg/kg when dietary copper and sulfur intake in the diet is normal.
Risk factors
Sheep and cattle are clinically affected in field outbreaks of the disease and signs are most marked in young growing animals. Cattle are much more susceptible than sheep. Horses also appear to be susceptible.2
The concentration of molybdenum in forage varies with the season, being highest in the spring and autumn and with the plant species, legumes, particularly alsike clover, taking up molybdenum in much greater quantities than grasses.
PATHOGENESIS
An extended discussion of the role of molybdenum in copper metabolism is provided in the section on secondary deficiency. Excess molybdenum intake is associated with an increased formation of thiomolybdates, important enzyme inhibitors which interfere with the hepatic storage of copper and produce a state of copper deficiency.3 This situation is exacerbated by a high intake of sulfur or a low intake of copper. The syndrome of molybdenum intoxication resembles that of copper deficiency and treatment and prevention by the administration of copper is effective.
Some of the signs of molybdenum poisoning, particularly diarrhea, are not characteristic of copper deficiency, and may represent a specific toxic effect of molybdenum. An identified specific toxic effect is that of causing the development of exostoses and hemorrhages about the long bones, and separation of the great trochanters of the femur in some sheep fed molybdenum experimentally. The lesions appear to be due to defects in connective tissue at muscle insertion points, and to defects in the epiphyseal growth plates.
Experimental feeding of molybdenum, and its intravenous injection,4 produce a syndrome identical with that seen in the naturally occurring disease in cattle but liver and plasma levels of copper may not be depressed as is usual in naturally occurring cases. Experimental feeding of a large dose, up to 40 g, of molybdenum may be associated with only transient diarrhea. Most of the molybdenum is rapidly absorbed and excreted, 90% in the first week.
CLINICAL FINDINGS IN CATTLE
• Persistent scouring commences within 8–10 days of the animals having access to affected pasture
• Emaciation and a dry, staring coat develop and there is profound depression of milk production
• Depigmentation of black hair causes a red or gray tinge to appear. This may be particularly noticeable around the eyes, giving a bespectacled appearance. Intense craving for copper supplement has been noted
• Young cattle (3 months to 2.5 years) also show abnormalities of locomotion including marked stiffness of the legs and back, difficulty in rising, and great reluctance to move. The gait is suggestive of laminitis but the feet appear normal. The lameness may be due to the periosteal lesions described above. The appetite remains good
• Rare cases in horses2 show diarrhea and impaction colic and a high mortality rate.
CLINICAL PATHOLOGY
Blood copper levels are reduced from the normal of 1.0 μg/mL to 0.25 μg/mL. Seasonal variations occur depending on the intake of molybdenum.
Blood Molybdenum levels in normal animals are of the order of 0.05 mg/kg and rise to about 0.10 mg/kg when excess molybdenum is ingested. Levels as high as 0.70 and 1.4 mg/kg have been recorded in cattle and horses grazing on pasture contaminated by smelter fumes. On very large intakes of molybdenum cattle which are clinically normal may have molybdenum levels of 1000 mg/kg in feces, 45 mg/kg in urine, 10 mg/kg in blood, and 1 mg/kg in milk.
NECROPSY FINDINGS
There are no gross or histological findings which characterize the disease, enteritis being conspicuously absent. The carcass is emaciated and dehydrated and there may be anemia if there is an accompanying copper deficiency. Tissue copper levels will be below normal.
Diagnostic confirmation. The most effective method is to treat affected animals orally with copper sulfate (2 g daily or 5 g weekly for adult cattle and 1.5 g for adult sheep). The diarrhea ceases in 2–3 days and improvement in the other signs is rapid.
The persistence of the diarrhea without other clinical signs, particularly in young cattle and sheep, may suggest:
• Internal parasitism, e.g. trichostrongylosis, ostertagiasis; examination of feces for worm eggs is necessary for differentiation
• Johne’s disease which affects only adults; usually only one animal in a herd shows clinical signs at any one time
• Acute enteritides including salmonellosis, winter dysentery and virus diarrhea, acute diseases accompanied by other diagnostic signs.
PRIMARY TREATMENT AND CONTROL
Molybdenum toxicity can be treated by the administration of copper, and controlled by increasing the copper content of the diet by 5 mg/kg. But the administration of copper to large numbers of animals presents a number of problems. For long-term control, the recommended ratio of Cu:Mo is 4:1 to 10:1, and a Sulfur:Mo ratio of <100:1 is considered safe vs. copper accumulation.5,6
1 Sas B. Vet Human Toxicol. 1989;31:29.
2 Ladefoged O, Sturup S. Vet Human Toxicol. 1995;37:63.
3 Mason J. Toxicology. 1986;42:99.
4 Auza N, et al. Vet. Human Toxicol. 1989;31:535.
5 Buck WB. J Am Vet Med Assoc. 1970;46:1078.
6 National Research Council. Nutrient Requirements of Beef Cattle, ed. 4. Washington, D.C.: National Academy Press, 1996.
PRIMARY COPPER POISONING
• Primary copper poisoning is associated with the intake of excessive amounts of copper
• Secondary copper poisoning, which occurs on low intakes of copper, is dealt with on p. 1823.
Etiology Acute or chronic accidental intake of copper.
Epidemiology Usually occurs as major outbreaks. Sheep most susceptible, horses least. Significant differences in breed susceptibility. Copper originates from copper-rich soils, industrial contamination of pasture or copper preparations used pharmaceutically, as a feed ingredient, or as an agricultural chemical.
Pathogenesis Acute poisoning due to ingestion of a single large dose is associated with alimentary tract mucosal necrosis and fatal shock. Acute intravenous or chronic oral intake is associated with fatal hemolytic anemia.
Clinical signs
Clinical pathology Chronic oral poisoning: very high blood and liver copper levels, low PCV, hemoglobinuria, elevated liver enzymes in serum.
Necropsy lesions Acute oral poisoning: severe gastroenteritis.
Chronic oral poisoning: high tissue levels of copper, tissue jaundice, swollen liver, kidneys, spleen, hemoglobinuria.
Diagnostic confirmation High copper levels in tissues.
Treatment Primary: edetate or molybdate parenterally. Supportive: blood transfusion if practicable.
Control Removal from source, prophylactic administration of molybdate.
ETIOLOGY
Acute oral poisoning is associated with the accidental ingestion of large amounts of copper salts at one time. Acute systemic poisoning and chronic oral poisoning are associated with the accumulation of small amounts of copper ingested over a long period.
EPIDEMIOLOGY
Occurrence
Sporadic outbreaks of primary copper poisoning occur in many circumstances. In both acute and chronic cases the mortality rate approximates 100%.
Acute or chronic oral poisoning
• Accidental administration of large quantities of soluble copper salts, e.g. as parasiticide drench
• Contamination of plants with fungicidal sprays
• Contamination of drinking water during snail eradication
• Grazing pasture too soon after it has been top-dressed with:
• Grazing pasture growing on soils rich in copper
• Grazing pasture contaminated by smelter fumes or by drippings from overhead power cables made of copper but corroded by the constituents of an industrially polluted area
• Feeding of seed grain which has been treated with antifungal agents containing copper
• Feeding mineral or salt licks or mixtures containing excessive amounts of copper
• Salt or mineral mixtures containing copper ingested accidentally by salt-hungry livestock
• Copper-enriched concentrate rations fed as growth supplements to poultry or pigs in excessive quantities or fed to ruminants. Pigs can survive rations providing up to 250 mg/kg; these levels are toxic for ruminants in which 20 mg/kg is maximum copper content. For sheep, continued ingestion of cattle diets that contain elevated copper and limited molybdenum results in copper accumulation.
• Miscellaneous other sources of copper causing poisoning are palm oil cake, treated pine lumber containing arsenic, copper, and chromium.
Acute parenteral poisoning
Prophylactic injections of copper salts, especially the more soluble, aqueous preparations, are very toxic if recommended dose rates are exceeded. This procedure is being increasingly used to prevent copper deficiency in grazing ruminants when other cheaper methods are not applicable. The paste preparations, usually copper glycinate, appear to be non-toxic but the soluble preparations, e.g. copper edetate, when given at recommended doses, can be associated with heavy mortalities in sheep and calves.1 Copper as the diethylamine oxyquinoline sulfonate has also been associated with deaths in sheep after injection of recommended dose rates. The absorption is fast and the blood levels high with these toxic compounds but not with copper methionate which has a good safety record. Deaths commence 24 hours after injection and continue for up to 7 days. Postmortem findings include hepatic centrilobular necrosis, nephrosis, pleural and peritoneal effusions.
Toxic doses
There is a great deal of published anecdotal evidence about the amount of copper in specific feeds fed to specific species which was associated with illness or deaths but almost no evidence of MD50s or similar levels for the several animal species. The following toxic dose rates are provided as a rough guide:
• Single doses of 20–110 mg of copper per kg BW produce acute copper poisoning in sheep and young calves
• The status of goats generally is uncertain
• In cattle a dose rate of 220–880 mg/kg BW is necessary to cause death
• Chronic poisoning occurs in sheep and calves with daily intakes of 3.5 mg of copper/kg BW, 25 ppm being the maximum tolerated concentration in the feed. Even lower concentrations (15 ppm) may poison sheep if adequate molybdenum and sulfate are not present in the diet.
None of these data on toxic intakes come with information on competing and contributory dietary factors such as sulfate, molybdenum, and zinc and these are critical in determining the toxic effects of the copper intake.
Risk factors
Environmental factors
Both acute and chronic copper poisoning occurs under field conditions. Acute poisoning usually occurs because of the accidental administration of large quantities of soluble copper salts, but chronic poisoning occurs principally as a result of ingesting feed containing or contaminated by copper derived from the soil or by its application to the diet as an agricultural chemical or feed supplement.
The toxicity of the copper ingested in this way is governed not only by the absolute amount of copper but also by the interaction of a number of factors including the amount of molybdenum and of sulfate present in the diet and the presence or absence of specific plants and the level of protein in the diet. In fact either copper deficiency or copper poisoning can occur on soils with apparently normal copper levels, the syndrome depending on the particular conditioning factors present. High molybdenum and sulfate levels in the rumen lead to the microbiological synthesis of non-absorbable thiomolybdates, and a high sulfate diet also leads to lower retention of copper in tissues.
There is also a competitive relationship between copper and zinc in the internal metabolism of ruminants, a high level of zinc in the diet reducing the intake of copper.
Host factors
Horses are the least susceptible with a tolerance to levels of 800 ppm in the diet. Cattle will usually tolerate 100 ppm and swine 250 ppm, but lethal hemolysis has occurred in cattle fed a low copper level mineral supplement (38 mg/kg BW for lactating cows) for 2 years.2 Goats are not featured in the literature on this subject and sheep criteria are recommended as guides. Sheep are the most susceptible species, tolerating as little as 25 mg/kg BW; they are peculiar in the way in which copper is handled metabolically. Increased absorption is not easily achieved but abnormally high excretion is more difficult still, so that there is the general tendency for copper to accumulate.
Sheep, e.g. the Texel and the Finnish Landrace, are more resistant while others, e.g. the Ronaldsay and the Orkney, are much more susceptible. The Angora goat appears to be most susceptible and Nubian goats appear to be more resistant than sheep. Angus cattle are much more susceptible than Charolais.3
Importance
Many deaths due to copper poisoning are followed by deaths from general debility in sheep in poor condition. Dairy cows, especially those lactating at the time, fail to produce well, and special care is needed to bring them back to full production.4
PATHOGENESIS
Soluble copper salts in high concentrations are protein coagulants. The ingestion of large quantities is associated with intense irritation of the alimentary mucosa and profound shock. Severe intravascular hemolysis occurs if the animal survives long enough. When excessive amounts of copper are injected the response is rapid and animals begin to die the next day and with a peak of mortality about the third day after dosing. Early deaths appear to be due to severe hepatic insufficiency and later deaths to renal failure due to tubular necrosis. There appear to be no renal lesions in sheep affected with chronic copper poisoning, unless a hemolytic crisis occurs, in which case there is a hemoglobinuric nephrosis.
The frequent ingestion of small amounts produces no ill-effects while copper accumulates in the liver. When maximum hepatic levels are reached after periods of exposure often as long as 6 months, the copper is released into the bloodstream and the animal dies of acute intravascular hemolysis. Thus there is really no such thing as ‘chronic’ copper intoxication; syndromes so called are fatal as acute hemolytic crises. One of the dangers of cumulative copper poisoning is that the animal shows normal health until the hemolytic crisis, when it becomes acutely ill and dies very quickly. Death is ascribed to acute anemia and hemoglobinuric nephrosis.
Two other abnormalities have been observed during and after the hemolytic crisis. One is the occurrence of methemoglobinemia; the other is the presence of degenerative lesions in the white matter of the brain. The accumulated copper can lead to the occurrence of a hemolytic crisis after ingestion of copper has ceased, and recurrent attacks can therefore occur in sheep that survive the attacks. There are a number of explanations for the development of hemolysis. One is that the erythrocytes in affected sheep become immunogenic as a result of the copper accumulation. It is suggested that this immunogenicity leads to the development of an autoantibody and the final result of an autoimmune hemolytic anemia. Alternatively, copper may act as an oxidant on the red cell membrane, leading to membrane damage and acute hemolysis as a result of oxidant injury. This is consistent with the formation of methemoglobin, a result of oxidant effects during the acute hemolytic crisis.
The liberation of the hepatic copper is incompletely understood, but the favored hypothesis is that the accumulation of copper ions in the liver cells is associated with the accumulation of electron-dense lysozymes in the hepatocytes and their eventual necrosis.5 Various stresses including a fall in plane of nutrition, traveling, and lactation, are considered to precipitate the liberation. Complex mechanisms relating to disorders of cell membranes, a marked change in hemoglobin composition, including the development of methemoglobinemia and an increase in the oxidative status of the sheep, are described as occurring during the critical stages. During the prehemolytic stage of several weeks before the crisis there is hepatic necrosis and an elevation of levels of liver-specific enzymes. A much more serious necrosis of liver occurs at the time of the hemolytic crisis.
Sheep on a selenium-deficient diet and with low blood levels of glutathione peroxidase are more susceptible to chronic copper poisoning. Some sheep are also conditioned by inheritance to have low blood glutathione levels in spite of a normal dietary intake of selenium. They also have low glutathione peroxidase blood levels and may be more susceptible for this reason.
There is also a difference between breeds in their capacity to reduce copper absorption in response to the administration of zinc, Texels being much more responsive than Friesians and North Ronaldsays are also known to be highly susceptible. These sheep normally subsist on seaweed which has a very low content of copper and molybdenum. When the sheep are fed on terrestrial herbage containing normal levels of copper and molybdenum and high levels of zinc they develop copper poisoning.
CLINICAL FINDINGS
Acute intoxication
Severe gastroenteritis occurs accompanied by abdominal pain and severe diarrhea and vomiting in some species. The feces and vomitus contain much mucus and have a characteristic green to blue color. Vomiting occurs in the pig and dog and intense thirst is apparent. Severe shock with a fall in body temperature and an increase in heart rate is followed by collapse and death usually within 24 hours. If the animal survives for a longer period, dysentery and jaundice become apparent.
Acute poisoning associated with the injection of copper salts is manifested only by anorexia, depression, and dehydration. In calves that survive the illness for 3 days or more massive ascites, hydrothorax, hydropericardium, hemoglobinuria and massive hemorrhages, dyspnea, head-pressing, aimless wandering, circling, and ataxia occur. Lambs similarly poisoned and with similar postmortem lesions die within 24 hours of injection.
Chronic intoxication
In ruminants anorexia, thirst, hemoglobinuria, pallor, and jaundice appear suddenly. There is no disturbance of alimentary tract function. Depression is profound and the animal usually dies 24–48 hours after the appearance of signs. In pigs signs of illness are uncommon, most pigs being found dead without premonitory signs, although dullness, anorexia, poor weight gain, melena, weakness, pallor, hyperesthesia, and muscle tremor may be observed occasionally.
CLINICAL PATHOLOGY
Levels of copper in the blood and liver are markedly increased in chronic copper poisoning. In acute intoxications several days are required after ingestion before these levels rise appreciably. Fecal examination may show large amounts (8000–10 000 mg/kg) of copper. Liver biopsy is a satisfactory diagnostic technique and serves a most useful purpose in the detection of chronic copper poisoning as blood levels do not raise appreciably until the hemolytic crisis occurs just before death. Because of the greater concentration of copper in the caudate lobe as compared to other parts of the liver, an autopsy specimen is to be preferred.
Blood levels of copper during the hemolytic crisis are usually of the order of 78–114 μmol/L (4.9–7.2 ppm), compared with about 15.7 μmol/L (1 ppm) in normal animals. Normal liver levels of less than 5.5 mmol/kg dry matter (349 ppm) rise to above 15.7 mmol/kg (997 ppm) in the latter stages of chronic copper poisoning in sheep, to 95 mmol/kg in pigs, and to 30 mmol/kg in calves. In sheep, liver values greater than 7.85 mmol/kg and kidney values of greater than 1.25–1.57 mmol/kg dry matter are diagnostic. After a massive single dose it is important to include kidney among specimens submitted for copper assay, because levels may be high (more than 25 mg/kg dry matter) while liver copper levels have not yet risen. When comparing normal and toxic values it should be remembered whether results are expressed as dry weight basis or wet weight basis. Assuming approximately 20% dry matter in tissue, a wet weight value of 1.5 ppm copper is actually 7.5 ppm on a dry weight basis, comparable to the toxic range reported for blood above. Thus, a commonly observed toxic value of 200 ppm copper in liver (wet weight basis) would be reported as 1000 ppm copper on a dry weight basis.
The packed cell volume of the blood decreases sharply, from 40 down to 10% in 48 hours, during an acute hemolytic episode. Methemoglobinemia may be present and the urine should be checked for hemoglobin.
Serum enzyme activity is greatly increased just before the hemolytic episode, and there is a significant reduction in the rate of bromosulfalein clearance during this period in sheep and in calves poisoned experimentally. In sheep the aspartate amino transferase (AST) levels may rise as high as 880 SF units per mL up to 6 weeks before obvious clinical signs appear, and the test is regarded as a suitable monitor of copper poisoning in this species. Plasma aspartate aminotransferase and sorbitol dehydrogenase levels in blood are elevated at 60 days after the copper feeding begins, but diethyl succinate carboxylesterase levels rise within 7 days and are therefore the better indicator. The hepatic enzymes GGT and AST were determined in one experimental study to be the best enzymes to assess copper-load in sheep during the pre-hemolytic phase. GGT increases were evident 28 days before the hemolytic crisis and AST concentrations increased from 14 days prior to onset of acute copper toxicosis.6
NECROPSY FINDINGS
Acute copper poisoning via oral exposure is uncommon in ruminants but gross changes include severe gastroenteritis with erosion and ulceration particularly in the abomasum. Macroscopic changes in calves poisoned by injected solutions of copper salts include hepatomegaly with an enhanced zonal pattern and massive fluid accumulations in body cavities. Characteristic microscopic findings in such acute copper toxicoses include extensive periacinar hepatic necrosis and a variable amount of renal tubular nephrosis.
In chronic copper poisoning, jaundice and hemoglobinuria are usual but not constant findings. The liver is swollen, yellow and may contain hemorrhagic foci. The spleen is enlarged with a soft pulp and the kidneys are swollen and have a dark gunmetal color. The hemolytic crisis typical of ovine copper toxicosis results in massive acute hepatocellular necrosis, which masks most of the chronic hepatic damage. These changes include hepatocellular vacuolation and degeneration, increased single cell necrosis of hepatocytes, a variable amount of periportal fibrosis, and proliferation of cholangiolar cells. These chronic lesions are more easily identified in cattle suffering from copper poisoning. Granular casts are often present in the renal tubules, especially in affected sheep. Hemosiderin deposits are increased in the liver and spleen.
Details of the critical copper levels of tissues are provided in the clinical pathology discussion. Although the lesions described above do occur in some outbreaks of the disease in pigs, they are not as pronounced as in ruminants and they are often accompanied by pulmonary edema and by severe hemorrhage from ulcers in the pars esophagea or large intestine.
Diagnostic confirmation is by demonstration of high blood and liver levels of copper plus histological evidence of liver damage. The history and the examination of feedstuffs and pastures are valuable aids in determining the cause.
Acute hemolytic diseases which may be mistaken for chronic copper poisoning include:
• Postparturient hemoglobinuria
• Plant poisoning including SMCO in rape, kale, etc., hepatoxins in Pithomyces chartarum
• Red maple (Acer rubrum) toxicosis
• Excessive intake of onions (Allium sp.)
• Many other associations with hemolytic anemia
• Some cases of acute pasteurellosis. The bacterial infections are usually accompanied by fever and toxemia but rape poisoning and postparturient hemoglobinuria can only be diagnosed tentatively by an examination of the environment and consideration of the history.
The differential diagnosis list for acute copper poisoning includes other associations with gastroenteritis. It can usually be identified by the blue-green color of the ingesta.
TREATMENT
For chronic copper poisoning daily oral treatment of lambs with 100 mg ammonium molybdate and 1 g anhydrous sodium sulfate significantly reduces the copper content of tissues and appears to prevent deaths in lambs known to have toxic amounts of copper. The mode of action is by increasing the fecal excretion of copper. Under experimental conditions injection intravenously or subcutaneously has an ameliorating effect on copper poisoning by reducing the capacity of circulating copper to enter erythrocytes and cause their lysis. Injection of ammonium tetrathiomolybdate (three to six times intravenously at 2–3-day intervals at a dose rate of 2.7 mg/kg BW is also effective.3 So too is the daily intravenous injections of sodium calcium edetate (70 mg/kg BW) for 2 days to calves. Different countries may have particular restrictions on the form of molybdenum approved for use, so locally available approved therapy should be determined and be a part of the veterinary pharmacy before acute hemolytic crises are encountered.
Supportive treatment should include blood transfusion.
In acute cases gastrointestinal sedatives and symptomatic treatment for shock are recommended.
CONTROL
When chronic intoxication is occurring or appears probable the provision of additional molybdenum in the diet as described under the control of phytogenous chronic copper poisoning should be effective as a preventive measure. Ferrous sulfide is effective but difficulty is usually encountered in getting the animals to eat it. In pigs and sheep the administration of iron and zinc reduces the risk of copper poisoning on diets supplemented by this element and a diet high in calcium encourages the development of copper poisoning, probably by creating a secondary zinc deficiency. A lick which contains dicalcium phosphate, sulfur, and zinc sulfate has been used to advantage as a prophylactic.7
Gooneratne SR, et al. Review of copper deficiency and metabolism in ruminants. Can J Anim Sci. 1989;69:819.
Mason J. Thiomolybdates: mediators of molybdenum toxicity and enzyme inhibitors. Toxicology. 1986;42:99-102.
Odell BL. The concept of trace-element antagonism; the Cu-Mo-S triangle. J Nutr Supp. 1997;5:S1045.
van Saun RJ. Copper toxicosis in sheep. Mod Vet Pract. 1988;69(1):1-5.
Soli NE. Chronic copper poisoning in sheep. Nord Vet. Med. 1980;32:75-77.
Solaiman WG, et al. Effects of high copper supplements on performancee, health, plasma copper and enzymes in goats. Small Ruminant Research. 2001;41:127.
SECONDARY COPPER POISONING (‘TOXEMIC JAUNDICE’ COMPLEX)
Copper poisoning is a complex problem because of the many factors that influence the intake, metabolism, and excretion of the element. Consequently secondary copper poisoning (‘toxemic jaundice’) can occur on intakes of copper which are, in other dietary circumstances, non-toxic. The common syndromes include the following:
• Phytogenous chronic copper poisoning is a condition in which relatively small amounts of copper are ingested but excessive retention occurs because of the presence of specific plants which have no apparent association with liver damage
• Hepatogenous chronic copper poisoning results from excessive retention of copper from the ingestion of specific plants which are associated with liver damage
• One of the plants that commonly contribute in this way is Heliotropium europaeum which is also capable of causing uncomplicated toxipathic hepatitis without abnormality of copper metabolism.
The ‘toxemic jaundice’ group of diseases includes all of these forms of secondary copper poisoning and toxipathic hepatitis assocated with Heliotropium europaeum.
Phytogenous chronic copper poisoning
This occurs in sheep grazing pasture containing normal amounts of copper. Although the copper intake may be low, liver copper levels are high and a hemolytic crisis typical of chronic copper poisoning occurs. The predominant association is the domination of the pasture by subterranean clover (Trifolium subterraneum) which may contain lower than normal quantities of copper (15–20 mg/kg). British breeds of sheep and their crosses with Merinos are most susceptible.
Control of this disease is by encouragement of grass growth in the pastures. Outbreaks can also be avoided if sheep are prevented from grazing lush, clover-dominant pastures in the autumn. Avoidance of stress, particularly malnutrition, is also important. The daily administration of molybdenum in the feed (7 mg/kg molybdenum) has been shown to greatly reduce the uptake of copper by lambs on diets of high copper content and this has been used as a practical preventive measure. Molybdenized superphosphate (70 g/ha), and molybdenized licks or mineral mixtures (86 kg salt, 63 kg finely ground gypsum, 0.45 kg sodium molybdate) are suitable alternatives. When an outbreak occurs, the administration of ammonium molybdate (50–100 mg/head per day) together with sodium sulfate (0.3–1.0 g/head per day) will stop further deaths in sheep within 3 days. Solutions of the above salts may be sprayed onto hay and administration should be continued for several weeks.
Hepatogenous chronic copper poisoning
This form of the disease occurs most commonly following the ingestion of sufficient quantities of the plant Heliotropium europaeum, (Senecio spp. and Echium plantagineum) over a period of 2–5 months to produce morphological and biochemical changes in liver cells without major impairment of liver function. After ingestion of these plants the liver cells have an increased affinity for copper and abnormally high amounts accumulate in the liver with an increased risk of a hemolytic crisis. Sheep grazed on H. europaeum and then on subterranean clover are particularly prone to this form of the disease. Control depends upon preventing the ingestion of hepatotoxic plants and restricting copper retention by the methods described above.
Poisoning by Heliotropium europaeum
Heliotrope contains hepatotoxic alkaloids and continued ingestion of the plant is associated with liver damage. If a high copper storage occurs, hepatogenous chronic copper poisoning may develop. On the other hand, if the sheep’s copper status remains normal liver damage proceeds until the animal suffers from simple toxipathic hepatitis. The effects of the plant are cumulative and grazing for one season may be associated with little apparent harm but further grazing in the subsequent year may be associated with heavy mortality. Control must aim at eradication of the plant.
SODIUM CHLORIDE POISONING
Etiology Ingestion of excessive amounts of sodium chloride OR normal intake but limited water intake.
Epidemiology Source of salt includes:
• Artesian bore water used as drinking water
• Water accumulating in pasture containers of salt mixture or lick
Clinical signs
Very large doses: vomiting, diarrhea, dehydration.
Average doses in cattle: opisthotonos, Nystagmus, blindness, convulsions, death.
Continuous intake in pigs: blindness, deafness, aimless wandering, head pressing, regularly periodic convulsions.
Continuous intake in ruminants: bawling, restlessness, anorexia.
Clinical pathology High blood levels of sodium and sodium chloride. High salt content in water, feed. Eosinopenia only in pigs.
Necropsy lesions
Very large doses: gastroenteritis.
Average doses: eosinophilic meningitis in pigs; polioencephalomalacia in cattle. High tissue levels of sodium and chloride.
Diagnostic confirmation Elevated sodium and chloride content of tissues. Cerebrospinal fluid sodium exceeds serum sodium. Elevated aqueous or vitreous humor sodium.
Treatment
Primary: remove source of salt, restrict water intake and allow acclimation to water slowly.
Supportive: gastroenteric sedatives and fluid/electrolyte replacement for gastroenteritis form; sedative and cerebral decompressant in convulsive form.
Control Limit intake of salt-rich water, whey, concentrate mixes; insure adequate drinking water supply at all times.
ETIOLOGY
Feed and water containing excessive quantities of salt are unpalatable to animals but excessive quantities of salt are sometimes ingested, especially in saline drinking waters. Specific details about the degree of salinity of drinking water that is compatible with health in animals are difficult to provide, because of the variation in the kinds of salts which occur in natural saline waters.
Toxic doses quoted for acute sodium chloride poisoning are for pigs, horses, and cattle 2.2 g/kg BW and for sheep 6 g/kg. The toxicity of salt is significantly influenced by the age and body weight of the subject. For example, dose rates which kill pigs of 6.5–10 kg BW have little effect on pigs of 16–20 kg.
It is probable that the pathophysiological disturbance described here is one of water intoxication rather than salt poisoning in the absolute sense. High salt intakes are extensively used in sheep to restrict food intake during drought periods and in the control of urolithiasis in feeder wethers but salt poisoning does not occur if there is free access to water. Rations containing up to 13% of sodium chloride have been fed to ewes for long periods without apparent ill-effects, although diets containing 10–20% and water containing 1.5–2% sodium chloride do reduce food consumption. This may be of value when attempting to reduce feed intake but can be a disadvantage when sheep are watered on saline artesian water.
EPIDEMIOLOGY
Occurrence
Salt poisoning will occur wherever bore water is used for livestock drinking. It is reported principally from Australia, North America, and South Africa.
Sources of toxin
• Saline drinking water, especially after a change from fresh water, and especially if the animals are thirsty
• Water accumulating in salt troughs during drought periods
• Animals previously deprived of salt may eat excessive amounts if suddenly allowed access to unlimited quantities
• In animals kept in barns and small yards when prepared feeds contain too much salt, or salt is provided only intermittently, when trough space is limited and animals tend to gorge on swill or concentrate
• Swill fed to pigs may contain excessive amounts of salt when it contains dough residues from bakeries, brine from butchers’ shops, salt whey from cheese factories, or salted fish waste
• Excessive sodium sulfate given to pigs as treatment for gut edema if the water intake is restricted
• Environmental pollution by oil wells. Cattle are attracted to oil residues of their salty flavor and may ingest toxic amounts
• Temporary restriction of the water supply to pigs of 8–12 weeks of age, lambs and calves fed prepared feeds containing the standard recommendation of 2% salt. Poisoning occurs when the animals are again allowed access to unlimited water. Similarly pigs brought into new pens where drinking water is supplied in automatic drinking cups may not be accustomed to their use and fail to drink for several days until they learn to operate the cups. Feeder lambs and calves may also be deprived of water when their troughs are frozen over.
Risk factors
Host factors
Sheep, beef cattle, and dry dairy cattle appear to be less susceptible than dairy cows in milk, which are in turn less susceptible than horses. Heavy milking cows, especially those in the early stages of lactation, are highly susceptible to salt poisoning because of their unstable fluid and electrolyte status.
Toxin factors
Saline waters often contain a mixture of salts and those containing high levels of magnesium or fluorine may be quite toxic. Water containing 0.2–0.5% magnesium chloride may be associated with reduced appetite and occasional diarrhea in sheep, especially if the sodium chloride content is also high, but water containing similar quantities of sodium sulfate does not have any harmful effect.
Variation between bore waters includes differences in the relative proportions of the acid radicals, particularly sulfates, carbonates, and chlorides.
Environment factors
Environmental temperatures have an effect on toxicity, signs occurring in the summer on water containing levels of salt which appear to be non-toxic in the winter. Australian recommendations are that the maximum concentration for sodium chloride or total salts in drinking water should not exceed 1.3% for sheep, 1% for cattle, and 0.9% for horses. South African and Canadian recommended levels are much lower but there does not appear to be any proof that such low levels of total and individual salts are necessary.
Importance
Many animals may be clinically affected and the mortality rate may be high where animals are kept under range conditions and have to depend on saline water supplies for drinking purposes. In animals kept under intensive conditions salt poisoning occurs only sporadically but most affected animals die and heavy losses may occur in groups of pigs.
PATHOGENESIS
Acute poisoning
When excessive amounts of salt are ingested gastroenteritis occurs because of the irritating effects of the high concentrations of salt. Dehydration results and is exacerbated by the increased osmotic pressure of the alimentary tract contents. Some salt is absorbed and may be associated with the involvement of the central nervous system as in chronic poisoning.
Chronic poisoning
Where the defect is one of decreased water but normal salt intake, there is an accumulation of sodium ions in tissues, including the brain, over a period of several days. An initial high sodium accumulation may inhibit anaerobic glycolosis, preventing active transport of sodium out of the cerebrospinal compartment. When water is made available in unlimited quantities, it migrates to the tissues to restore normal salt-water equilibrium. This is associated with acute cerebral edema and the appearance of signs referable to a sudden rise in intracranial pressure. The response is the same in all species but in pigs there is, in addition, an accumulation of eosinophils in nervous tissue and the meninges. The sodium ion is the one that accumulates in the tissues, identical syndromes being produced by the feeding of sodium propionate or sodium sulfate. It has also been observed that the feeding of soluble substances such as urea, which are excreted unchanged by the kidney, may be associated with anhydremia and an increase in the sodium ion concentration in brain tissue and the development of encephalomalacia.
This form of salt poisoning is chronic only in the sense that the sodium ion accumulates gradually. The clinical syndrome is acute in much the same way as the syndrome is acute in chronic copper poisoning. There is an apparent relationship between this form of salt poisoning and polioencephalomalacia in all species. Many outbreaks of this latter disease occur in circumstances which suggest chronic salt poisoning. Sheep become adapted to a continuous high salt intake (up to 1.3% sodium chloride in the drinking water) by significant changes in numbers of microflora in the rumen but this is not usually accompanied by any change in total metabolic activity. The same level of intake in sheep is associated with some mortality, chronic diarrhea and reduction in fertility, weight gain and wool growth.
CLINICAL FINDINGS
Acute salt poisoning in cattle
With very large doses the clinical signs are vomiting, diarrhea with mucus in the feces, abdominal pain, and anorexia. The more common syndrome, accompanying the dose of salt usually encountered, includes opisthotonos, nystagmus, tremor, blindness, paresis, and knuckling at the fetlocks. There may be a nasal discharge and polyuria occurs constantly. A period of recumbency with convulsions follows and affected animals die within 24 hours of first becoming ill. Sheep show similar signs. In swine the signs include weakness and prostration, muscle tremor, clonic convulsions, coma and death after a course of about 48 hours.
Chronic salt poisoning
In pigs this is ushered in by the appearance of constipation, thirst and pruritus 2–4 days after exposure. A characteristic nervous syndrome follows within 12–24 hours. Initially there is apparent blindness and deafness, the pig remaining oblivious to normal stimuli and wandering about aimlessly, bumping into objects and pressing with the head. There may be circling or pivoting on one front leg. Recovery may occur at this stage or epileptiform convulsions begin, recurring at remarkably constant time intervals, usually 7 minutes, accompanied by tremor of the snout and neck. Clonic contractions of the neck muscles may be associated with jerky opisthotonos until the head is almost vertical causing the pig to walk backwards and assume a sitting posture. This may be followed by a clonic convulsion in lateral recumbency, with jaw champing, salivation, and dyspnea. Death may occur due to respiratory failure or the pig relaxes into a state of coma for a few moments, revives and wanders about aimlessly until the next episode occurs. The pulse and temperature are normal except in convulsive pigs when both may be elevated. The course is variable and death may occur in a few hours or not for 3–4 days after the first appearance of illness.
Subacute poisoning
This syndrome in cattle and sheep on saline drinking water includes depression of appetite, thirst, constant bawling, especially in calves, loss of body weight, dehydration, hypothermia, weakness, and occasional diarrhea. Incoordination, collapse, and tetanic convulsions with frothing from the mouth and nose may occur if the animals are forced to exercise. Acetonemia may be a complication in lactating cows.
Eosinophilic dermatitis has been observed at meat inspection in pigs transported in trucks salted to prevent slipping. The condition can be reproduced experimentally by rubbing salt in the skin.
Lower levels of intake can suppress food intake and growth without overt clinical signs. This occurs in heifers drinking water containing 1.75% sodium chloride; the animals only maintain weight at a salt level of 1.5% and show suboptimal weight gains when the water contains 1.25% sodium chloride. Drinking water containing 0.25% salt significantly reduces the milk yield of high-producing dairy cows.
CLINICAL PATHOLOGY
In pigs serum sodium levels are elevated appreciably above normal levels (135–145 mmol/L), to about 170– 210 mmol/L during the severe stage of chronic sodium salt poisoning.1 Also polydipsia is recorded at blood serum levels of sodium chloride of 900 mg/dL, typical signs of salt poisoning at 1300 mg/dL, and death when levels exceed 1500 mg/dL. An eosinopenia is also evident during this stage and a return to normal levels usually indicates recovery. In cattle the same changes occur but there is no eosinopenia. Samples of feed and drinking water should be collected for salt assay.
NECROPSY FINDINGS
In acute salt poisoning of cattle there is marked congestion of the mucosae of the omasum and abomasum. The feces are fluid and dark. Animals that have survived for several days show hydropericardium and edema of the skeletal muscles. The blood appears to be thinner than normal. Gastroenteritis may be evident in some pigs poisoned with large doses of salt but in chronic poisoning there are no gross lesions. Histologically the neurologic lesions of acute poisoning are restricted to expansion of perivascular spaces in the brain. In contrast, the microscopic changes in chronic salt poisoning in pigs are quite diagnostic. The expansion of perivascular spaces typical of acute cerebral edema is accompanied by meningitis featuring large numbers of eosinophils, which extend along Virchow–Robin spaces into the brain tissue. In pigs that survive there may be residual polioencephalomalacia, especially of the cerebral cortex. Chemical estimation of the amount of sodium and chloride in tissues, especially brain, may be of diagnostic value. Levels exceeding 150 mg/kg of sodium in the brain and liver, and of chlorides in excess of 180 mg in the brain, 70 mg in muscle and 250 mg/kg in the liver are considered to indicate salt poisoning. Brain sodium concentrations from 2230 to 4250 μg/g tissue have been recorded in water deprivation/salt intoxication of cattle.2 Aqueous humor sodium values in these circumstances have ranged from 172 to 218 meq/L.
The appearance of typical signs in pigs which have been recently moved to new quarters, or subjected to a change of ration during the preceding week, or which have not had access to water at all times, should suggest sodium salt poisoning. Diagnostic confirmation depends on the detection of elevated levels of sodium chloride in blood or in tissues and fluids, and in pigs on the presence of aggregations of eosinophils in the brain.
Differential diagnosis list
Diseases which have a similar clinical profile to salt poisoning include:
• Pseudorabies which is restricted in its occurrence to young sucking pigs
• Polioencephalomalacia in pigs and ruminants is almost identical with chronic salt poisoning and occurs in many instances under the same set of circumstances
• Gut edema occurs in rapidly growing pigs in the same age group as chronic salt poisoning. There are some differences in the clinical syndromes as they occur in the field, particularly the periodicity of the convulsive episodes in salt poisoning and the altered squeal in gut edema, but in many instances it will be impossible to decide on the diagnosis without reference to the history of salt and water intake
• Mulberry heart disease may be accompanied by nervous signs similar to those of salt poisoning but the disease is usually restricted to older pigs and deaths occur quite suddenly.
Gastroenteritis associated with excessive ingestion of saline drinking water has few diagnostic features and diagnostic confirmation from other causes of enteritis depends upon identification of a high salt intake.
TREATMENT
Primary treatment of both acute and chronic salt poisoning is the immediate removal of the toxic feed or water. Serum sodium levels should not be reduced by more than 0.5 mEq/hour.3 If possible, serum sodium should be measured and a formula used to calculate the free water deficit as follows:
Free water deficit (L) = 0.6 × body weight (kg) × ([current serum sodium concentration/reference range serum sodium concentration] – 1)4
Initially access to fresh water should be restricted to small amounts at frequent intervals; unlimited access may be associated with a sudden increase in the number of animals affected. In advanced cases animals may be unable to drink and water may have to be administered by stomach tube.
Supportive treatment includes alimentary tract sedatives when gastroenteritis is present and administration of isotonic fluids when dehydration has occurred. When there is evidence of cerebral edema it may be necessary to administer a sedative, and cerebral decompression may be attempted by the use of diuretics or hypertonic solutions injected parenterally.
CONTROL
Drinking water for all classes of livestock should not contain more than 0.5% sodium chloride or total salts, although sheep and beef cattle can survive on water containing as much as 1.7% sodium chloride or total salts. Waters containing a high concentration of fluoride or magnesium are particularly dangerous to livestock. Both salt and water should be freely available at all times. Diets fed to pigs should not contain more than 1% salt. The way in which whey is fed to pigs – with minimum water intake – makes prevention difficult unless the whey can be kept free of salt at the cheese factory.