Recombinant Dna Techgenetic And Genetic Engineering
Development of recombinant DNA technology is a major breakthrough in molecular biology. New combination of unrelated genes can be constructed in laboratory by means of the DNA manipulating techniques, which also make it possible to isolate and determine base sequences of individual genes, to manipulate them in any desired way, and to transfer genes from one species to another. The transferred genes can be placed in a functional role in many different types of cells to produce the protein it codes for in whatever amount is required. The associated technologies can be used in medical diagnosis of genetic diseases. The general biological applications are almost unlimited.
About 33,000-44,000 genes are scattered among billions of base pairs (3.1 × 109) that make up the human genome. To identify a given gene from this vast expanse of base pairs, separate it, and produce multiple identical copies of it, has been a challenging task for the researchers, it resembles not so much as finding a needle in a haystack, as finding a particular piece of hay in a haystack. The molecular biology changed it all, making it possible to isolate and amplify a specific gene. The student is introduced to this fascinating and ever expanding field in this chapter.
After going through this chapter, the student should be able to understand.
Uses and mechanism of action of various enzymes that are employed to manipulate nucleic acids (restriction enzymes, terminal transferases, ribonuclease H, etc.); role of cloning vectors (plasmids, phages and cosmids) in transferring the gene of interest to host cell; and application of techniques of genetic engineering in isolating and amplifying a gene of interest.
Difference between two types of DNA libraries: genomic and complementary.
Polymerase chain reaction (PCR based DNA cloning).
Specific methods used in analysis of DNA.
Special applications of recombinant DNA technology in medical diagnosis and treatment (gene therapy).
Current status and future goals of the Human Genome Project.
The term recombinant refers to recombining different segments of DNA, and cloning refers to production of multiple identical copies of the recombinant DNA, which are therefore of the same origin. The essence of cloning involves separating a specific gene or a segment of DNA from a large chromosome, attaching it to a carrier (vector), and introducing it into a suitable cell where it is propagated. In addition to such cell-based cloning, another method is enzyme-based (represented by polymerase chain reaction), which is a simpler, quicker and a more robust technique for mass DNA amplification.
It is worthwhile to first examine the properties of some of the enzymes and be familiar with some techniques used in DNA cloning.
I Techniques and Enzymes Used in Manipulation of DNA
It has been possible to manipulate DNA because of three key factors: the annealing properties of nucleic acids and availability of certain enzymes that act on nucleic acids in a specific manner. In addition, cloning vectors (vehicles) play the vital role of carrying DNA into the suitable host cell.
A Annealing Properties
Any two segments of a single-stranded DNA or RNA, having continuous complementary sequences of 20 or more bases in common, form complementary base pairs to form duplex structure. Duplex may be formed between DNA and DNA, RNA and RNA, or between DNA and RNA to generate a hybrid.
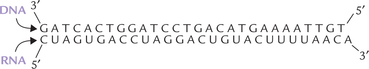
An application of annealing properties is construction of nucleic acid probes which provide useful means for detecting genes and mRNAs.
B Enzymatic Reactions
The commonly used enzymes in manipulation of DNA are enumerated here.
Restriction Endonucleases
These are bacterial enzymes that cleave specific palindromic sequences in double stranded DNA (Fig. 25.1 ). Werner Arber (Nobel Prize 1978) showed that entry of phages into host bacteria is restricted by such bacterial enzymes. Therefore, they are named as restriction endonucleases (RE), or simply restriction enzymes.
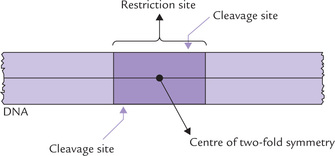
More than 800 types of REs are known; and more than 400 of them are available commercially. The restriction enzymes serve as the most “exquisite scalpels” cleaving the double-stranded DNA very selectively at specific sites, called the restriction sites. Each restriction site is a short sequence of 4-6 base pairs, and is palindromic, showing a two-fold rotational symmetry. As shown in Table 25.1, the top strand of each palindromic sequence has the same nucleotide sequence (from 5’ to 3’ end) as the bottom strand, when read in the 5’ →3’ direction. Therefore, if the structure is rotated 180 degrees, it remains the same; for example, turning the page upside down will not affect the above structures.
Table 25.1
Specificity of some restriction endonucleases
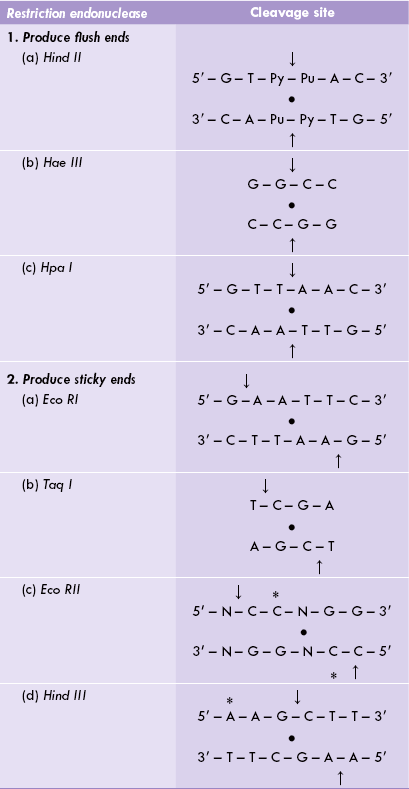
Arrows (→) indicate cleavage sites, • indicates the axis of two-fold rotational symmetry, * indicates methylation sites in the parent organism.
Some restriction enzymes can recognize the sites that are relatively small, e.g. four nucleotides such as Hae III, or larger sites, e.g. six nucleotides in the case of Eco RI. Enzymatic digestion of a DNA molecule by different REs produces several different fragments of varying sizes depending upon the cutting frequency of the RE. Each RE cuts DNA into different fragment sizes, which is not necessarily the same size as those cut by another enzyme (Fig. 25.2 ). If a piece of DNA from a species is exposed to a specific RE, a characteristic array of DNA fragments is produced; this is called restriction map.
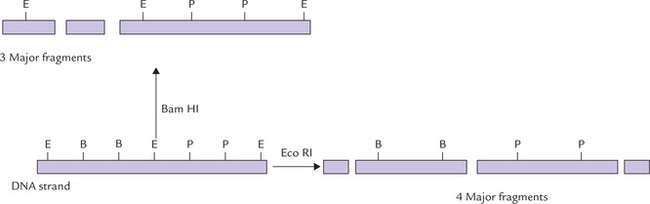
Fig. 25.2 Restriction enzyme digestion of DNA by different restriction enzymes may result in different restriction fragments (E = Eco RI site, B = Bam HI site, P = Pst I site).
Nomenclature
The nomenclature of the restriction enzymes is based on the bacteria from which they are isolated. It consists of a three-letter abbreviation: the first letter comes from the genus of the bacterial source, the second and the third letters are derived from the name of the bacterial species. For example, the restriction enzyme Hae is isolated from the bacterium, Haemophilus aegypticus. An additional Roman numeral indicates the order in which the enzyme was discovered in a particular organism. Hae III indicates that this is the third enzyme isolated from H. aegypticus, whereas Hae I is the first enzyme so isolated from this organism. Likewise, Taq I refers to the first RE isolated from Thermus aquaticus and Hind II refers to the second enzyme from H. influenzae (Table 25.1).
Most REs do not cleave in the centre of their recognition sequence but rather one or two base pairs away from the symmetry axis on both strands of DNA. As a result, a double-stranded DNA with short, single-stranded ends is produced. Such single-stranded ends are called sticky ends (or cohesive ends). Examples of such REs include Eco RI and Taq I (Table 25.1). Other REs, such as Hae III or Hpa I, cleave vertically the palindromic sequence, leaving two ends that do not have any overhanging nucleotides; these are blunt ends or flush ends.
Restriction Enzyme Cutting Frequency
Each restriction enzyme cleaves its own cutting site; variation in just one nucleotide adjacent to the cutting site may be sufficient to eliminate endonuclease activity for that particular enzyme. Depending on how many sites for a given enzyme are present in DNA, fragments of varying sizes are produced when that DNA is digested by a restriction enzyme. The frequency of cutting sites for various different enzymes varies considerably from enzyme to enzyme, and accordingly the REs are divided into two groups: (a) the frequent cutters—enzymes whose recognition site is very common throughout DNA, e.g. Pvu II and Hae III; and (b) the rare cutters—enzymes whose recognition sites are uncommon, often involving extended DNA sequence, e.g. Not I.
Size of the cut fragments (termed restriction fragments) differs in the two groups: the frequent cutters generating many small fragments and rare cutters generating fewer but larger restriction fragments. These differences serve as indispensable tools when it is intended to map the location of certain genes to chromosomal locations.
Biological Functions of REs
Bacterial cells have restriction enzymes to destroy invading foreign DNA, for example of the lambda phage which injects its DNA into an E. coli cell. The bacterial enzymes recognize the restriction sites and promptly cut the DNA to prevent phage infections: it restricts the ability of foreign DNA to infect the cell. A question arises as to why the restriction enzyme does not recognize the same restriction site on the bacterial DNA (a tetramer sequence recognized by Hae III for example must be occurring many times in DNA—statistically every 44 base pairs) and cleave at that site. The cell guards against cutting its own DNA by methylation: it adds a methyl group to one of the bases in all of the recognition sequences on each new DNA strand soon after it is synthesized. This does not interfere with the base pairing or gene function, but the restriction enzyme no longer recognizes the methylated sequence and the cell’s own DNA is, therefore, protected from attack by that enzyme.
This system, however, is not absolutely reliable because occasionally the intruding virus encounters the methylating enzyme first; and once methylated, it is resistant to the RE.
Terminal Transferases
They catalyze the addition of homo-oligonucleotide sequences (poly dN) to the 3’ end of double-stranded DNA molecule (Fig. 25.3a ). No template is required for these reactions. The 3’ ends of DNA serve as primers. A given enzyme can catalyze addition of many copies of a particular nucleotide only, and there are separate enzymes for each of the four deoxyribonucleotides. For example, the enzyme catalyzing addition of the poly-dA sequence is different from that catalyzing addition of the poly-dT sequence.
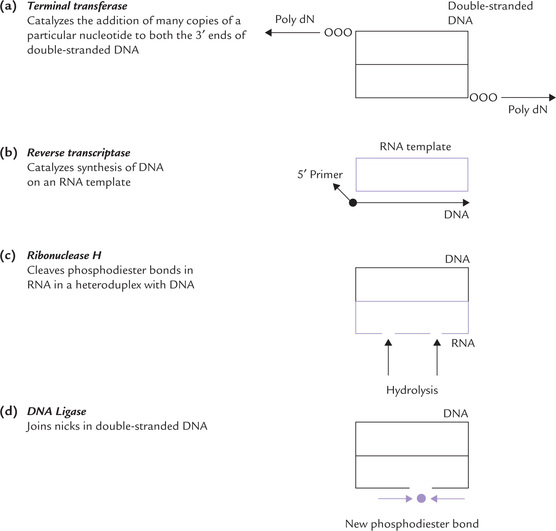
Reverse Transcriptase (RT or RNA-Dependent DNA Polymerase)
RT is an enzyme encoded in the genome of RNA-containing retroviruses. It catalyzes formation of DNA on an RNA template using deoxyribonucleoside triphosphates as substrates (Fig. 25.3b). Primer is required by RT. The DNA thus synthesized has a base sequence complementary to that of the RNA template and is called the complementary DNA or cDNA. For details regarding action of RT refer to Boxes 25.1 and 25.2.
Ribonuclease H
It is an endonuclease that catalyzes random hydrolysis of phosphodiester bonds of RNA, but only when the latter forms a hybrid duplex structure with DNA (Fig. 25.3c).
Box 25.1 Reverse Transcriptase
Copying of RNA template to synthesize DNA by reverse transcriptase was initially considered antithetical to the central dogma of molecular genetics. However, there is no thermodynamic prohibition to the RT action and, in fact under certain conditions, DNA polymerase can also copy RNA templates. RT is an essential enzyme of retroviruses which are the RNA-containing viruses, such as human immunodeficiency virus (HIV; the causative agent of AIDS). The enzyme was independently discovered in 1970 by Howard Temin and David Baltimore.
RT catalyzes the first step in the conversion of the single-stranded RNA genome of virus to a double-stranded DNA. After the virus enters the cell, its RT synthesizes complementary DNA from the viral RNA template to yield an RNA-DNA hybrid helix. (The DNA synthesis is primed by a host cell tRNA whose 3’-end unfolds to base pair with a complementary segment of viral RNA.) The viral RNA strand is then nucleolytically degraded by an RNase H (an RNase activity that hydrolyzes the RNA of an RNA-DNA heteroduplex). The single-stranded DNA is now left. This DNA acts as a template for the synthesis of its complementary DNA by DNA polymerase.
Finally, action of SI nuclease, a single-strand endonuclease, yields a double-stranded DNA that is then integrated into the host cell chromosome.
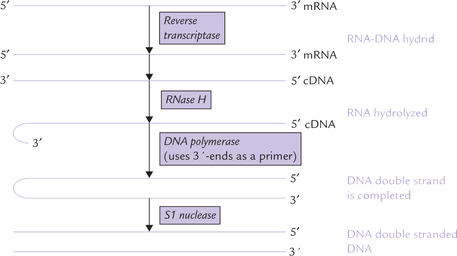
RT is a useful tool in genetic engineering because of its ability to transcribe mRNAs to complementary DNA strand. The latter can be used, for example, to express eukaryotic structural genes in E. coli. Since the bacterial cell cannot splice out the intron segments, the use of genomic DNA to express a eukaryotic structural gene in E. coli would require prior excision of the introns. This is a difficult feat and highly unfavourable thermodynamically.
DNA Ligases
These enzymes catalyze ATP or NAD+-dependent joining of nicks in double-stranded DNA (Fig. 25.3d).
C Cloning Vectors
The cloning vector is a self-replicating entity which is capable of carrying DNA into the host cell. Essential
properties of a cloning vector are as here.
A cloning vector may be a plasmid, bacteriophage, or a cosmid. An essential technique in genetic engineering is to introduce the human gene into a cloning vector. Covalent binding of the two results in production of chimeric DNA or recombinant DNA, which is then carried into a suitable host cell (Fig. 25.4 ). The inserted gene is replicated, transcribed and translated in the new setting, thus undergoing mass amplification. The name ‘chimeric’ derives from chimera, a mythological creature with the head of a lion, the body of a goat and the tail of a serpent.
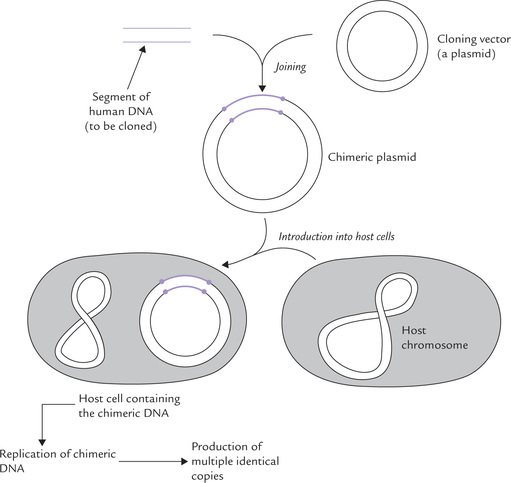
Fig. 25.4 Role of cloning vector plasmid in carrying chimeric DNA into the suitable host cell where it is amplified.
 Cloning vector, a self-replicating entity, helps in the in vivo propagation of DNA by effectively introducing it into a bacterium or some other suitable host cell.
Cloning vector, a self-replicating entity, helps in the in vivo propagation of DNA by effectively introducing it into a bacterium or some other suitable host cell.
Plasmids
Plasmids are circular, extrachromosomal DNA molecules of 2000-200,000 base pairs, present in some prokaryotic cells. Most plasmids are very small and contain only a few genes, compared with the prokaryotic chromosomal DNA which contain thousands of genes. In some cells, however, plasmids may be quite large. A single cell may contain 10-20 copies of plasmids. They undergo replication with each cell cycle, which may or may not be synchronized to the chromosomal DNA.
Plasmids usually carry genes that impart resistance to antibiotics. Thus, a bacterium having a plasmid, which has genes for ampicillin resistance, will be resistant to this antibiotic. Plasmids serve as efficient means of delivering a desired DNA segment into a host cell; the process is called transfection. Two remarkable properties of plasmids enable them to perform the stated function:
1. They can pass from one cell to another and also from one species of bacteria to another; for instance, some plasmids readily move from the E. coli cell to the Salmonella typhimurium cell. This explains the observation that mixing of the S. typhimurium cells with an ampicillin resistant strain of E. coli makes the former cells also resistant to ampicillin. The genes for ampicillin resistance, present in a plasmid of E. coli are transmitted to the S. typhimurium cells in this case.
2. Second important property is that foreign genes can be inserted into plasmids quite easily and may then be carried into a bacterium where they become part of the host-cell genome. Technical details about the role of plasmid in gene manipulation will be described later in this chapter.
Plasmid vectors suffer from a serious disadvantage: they can accommodate only small pieces of DNA, that is, up to about 5000 base pairs (5 kb). Larger inserts tend to be deleted randomly during replication of the plasmid.
Bacteriophages
Lambda (λ) phage is another choice vector. In view of the disadvantage in case of plasmids (the plasmids cannot carry large fragments of DNA) there is a need of other cloning vectors capable of carrying larger fragments. Lambda (λ) phages were found to be capable of carrying much larger DNA inserts, as large as 40 kb.
The genomic material of λ phage comprises a linear, double-stranded DNA of 48,513 base pairs (Fig. 25.5 ). It is packaged into the capsid protein within the host cell. The genome has two distinctive properties which permit easy insertion of foreign DNA into it:
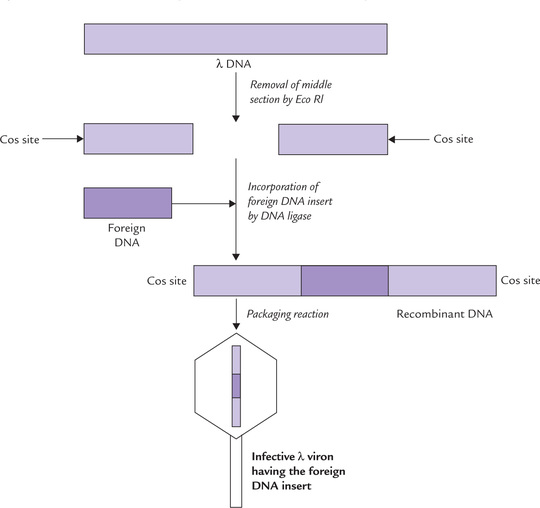
1. About one-third of the λ phage genome (lying in the middle section) is not essential for integration into the host-cell chromosome. This nonessential DNA can be replaced by an insert DNA, without affecting properties of the phage genome.
2. The DNA fragments present at the ends (referred to as cos sites) form packaging signals. They are essential for packaging the DNA into the phage particles.
Insertion of Foreign DNA into Bacteriophage
It involves the following steps:
• The middle section is removed first by certain restriction endonucleases (such as Eco RI). Nearly one-third of the phage DNA is thus removed, leaving behind two pieces having a combined length equal to approximately 70% of the unit genome (Fig. 25.5).
• Incorporation of a suitably long DNA insert is done next, to create a recombinant DNA. The DNA insert should be of nearly the same size as the middle-section (~15 kb).
• The insert DNA is then linked covalently at its ends by DNA ligase to create a recombinant DNA.
• Packaging reactions follow, wherein the recombinant DNA is mixed with capsid proteins and the infective phage particles are assembled by in vitro packaging. This results in the formation of λ virions, harbouring the foreign DNA.
An advantage of using the λ virions is that it “injects” its DNA into the bacterium with an efficiency much higher than that of plasmids. The injected DNA is either (i) incorporated into the bacterial chromosome to replicate during mitotic division, or (ii) it replicates within the cell independent of the bacterial chromosome. Such independent replication leads to synthesis of new viral particles, which can then lyze the host cell and then infect adjacent cells.
II DNA Amplification and Cloning
Production of multiple copies of a gene from the same origin (cloning) is central to the study of molecular biology and genetics. The cloned DNA can be used for many purposes; the human genome project, for example, used cloned DNA to sequence the human genome. Moreover, several techniques in molecular biology require visualization or quantification of the DNA and this requires mass amplification.
Two methods for the mass amplification of DNA are introduced in the following section.
• Cell-based cloning: DNA is amplified in vivo by a cellular host, such as a replicating bacteria (commonest), baker’s yeast, frog oocytes or cultured hamster’s ovary cells. The desired DNA template is introduced into a cellular host. As the number of replicating host cells increases (by cell divisions), the number of copies of the desired DNA template also increases correspondingly.
• Enzyme-based cloning: This method, represented by polymerase chain reaction (PCR), involves in vitro DNA amplification. It concentrates on the targeted increase in the number of copies of a specific DNA sequence.
A Cell-based Cloning
This method has been introduced earlier (Fig. 25.4). It uses recombinant DNA in the replicating bacteria, and is based on the ability of the latter to sustain presence of the recombinant DNA. The term recombinant refers to any DNA molecule that is artificially constructed from two pieces of DNA not normally found together. One of these pieces will be the target DNA that is to be amplified and the other will be the replicating vector or replicon (a sequence capable of initiating DNA replication).
This method requires separation of target DNA, its insertion into vector to produce chimeric (recombinant) DNA, and introduction into host cell where it is amplified. It is a complex process which can be divided in the following major steps:
• Isolation of the target DNA.
• Introduction of target DNA into replicon.
Step 1: Isolation of the Target Gene
Isolation of a specific gene of interest is a difficult task because a given gene represents a very small part of the total genomic material. There are three general approaches for obtaining the target gene:
• Reverse transcription of mRNA: This procedure involves isolation of mRNA complementary to the target gene and obtaining cDNA by reverse transcription of the mRNA. Though it is an indirect approach, this method is considered simpler because of the relative ease with which mRNA and then cDNA is obtained.
Note that the cDNA contains all coding regions of the target gene (without any introns).
• Shotgun approach: It involves (i) fragmentation of the entire genome, (ii) joining of all the DNA fragments with plasmids opened by the same restriction enzyme, and (iii) selection of the plasmids containing the desired gene. These aspects are described later in this chapter (see genomic library and Southern blotting).
• Chemical synthesis of the gene: The DNA that codes for a small protein may be chemically synthesized. However, since genetic code is degenerate, it is not possible to know the precise DNA sequence corresponding to the protein. More DNAs than one have to be synthesized.
Step 2: Introduction of Target DNA into Replicon
The target DNA is introduced into a replication vector (mostly plasmid vector) using restriction enzyme to cut the target and the replicon DNA. The enzyme used is the one that produces sticky ends. The plasmid, when digested with such an enzyme, for example Eco RI, becomes linear DNA strand with sticky ends complementary to the target (Fig. 25.6 ). Annealing of the two yields a new circular chimeric (recombinant) plasmid with the target gene inserted.
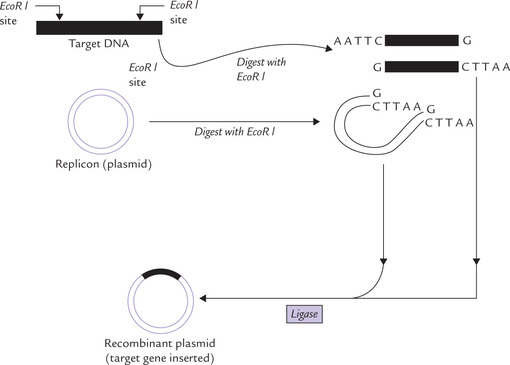
Fig. 25.6 Formation of a recombinant plasmid containing a target gene for cloning. Both the plasmid and the target DNA are cut with the same restriction enzyme and mixed together. The cohesive ends of each DNA reanneal and are ligated together to form the recombinant plasmid molecule which would be introduced into bacterial host cells.
Creation of sticky ends by homopolymer tailing can also be used to construct recombinant plasmid with the enzyme terminal transferase (Fig. 25.3a). A poly(A) tail may be attached to one DNA fragment and poly(T) to another, so that they can base pair to yield recombinant DNA.
Step 3: Transformation of the Host Cell
This refers to cellular uptake of plasmid DNA. This step (also called transfection) is critical in cell-based cloning because each of the successfully transformed cells would act as individual replication unit, capable of amplifying the target DNA in huge amount (Fig. 25.7 ). However, the cell membrane of bacteria is selectively permeable, restricting passage of large molecules such as DNA into the cell. Efficiency of transformation is very low: only 1-2% of cells may take up plasmid DNA, and often only a single plasmid is introduced during transformation. However, the permeability of the cell membrane can be increased by factors such as electric currents (electroporation) and high-solute concentration (osmotic stress), so that DNA can move into the cell more freely. Such processes are said to render the cells more competent, i.e. they can take up foreign DNA from extracellular fluid.
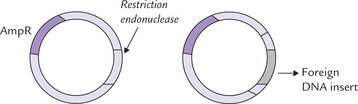
Fig. 25.7 Structure of a plasmid containing (a) genes conferring resistance to antibiotics ampicillin (AmpR), (b) the inserted DNA within the plasmid. The antibiotic resistance genes are useful for selection of the transformed cells.
The recombinant DNA has to be brought into the host bacterial cell by transformation. Transformation is rather inefficient but it can proceed with reasonable yield if the recombinant plasmid is mixed with the bacteria in presence of high solute concentration of if electric current is passed.
Selection of transformed cells
Following transformation, the next step is to identify/select the cells that have been successfully transformed. Such cells contain the recombinant plasmids, with target DNA inserted. Their selection makes use of the antibiotic resistance gene(s) present in the plasmid vector. If the vector contains an antibiotic resistance gene the transformed host cells will become resistant to this antibiotic. Plating on medium containing the relevant antibiotic will allow only the transformed host cells to grow. The non-transformed cells will be destroyed by the antibiotic. The following example will clear this point.
• The plasmid shown in Figure 25.7 contains genes that confer resistance to ampicillin.
• After transformation, the bacteria are cultured in a medium that contains ampicillin.
• The transformed cells will not be destroyed by this antibiotic because the gene for ampicillin resistance are present in them.
• The non-transformed bacteria (not containing the plasmid DNA), will however be destroyed by ampicillin.*
Cell-based cloning uses recombinant DNA in replicating bacteria or other host cells. Selectable markers can identify the transformed host-cells.
As shown in the Figure 25.8, this plasmid contains genes for ampicillin resistance and tetracycline resistance. Several restriction endonucleases (BamHI in this example) cleave the plasmid at unique sites within either the ampicillin resistance (amp-R) gene or the tetracycline-resistance (tet-R) gene. In the application shown in Figure 25.8, the restriction enzyme cleaves the plasmid at the gene for tetracycline resistance. Thus, the tet-R gene is destroyed while the amp-R gene remains intact. Bacteria transformed by this recombinant plasmid can grow in the presence of ampicillin but not of tetracycline. They are grown in suitable medium to form colonies.
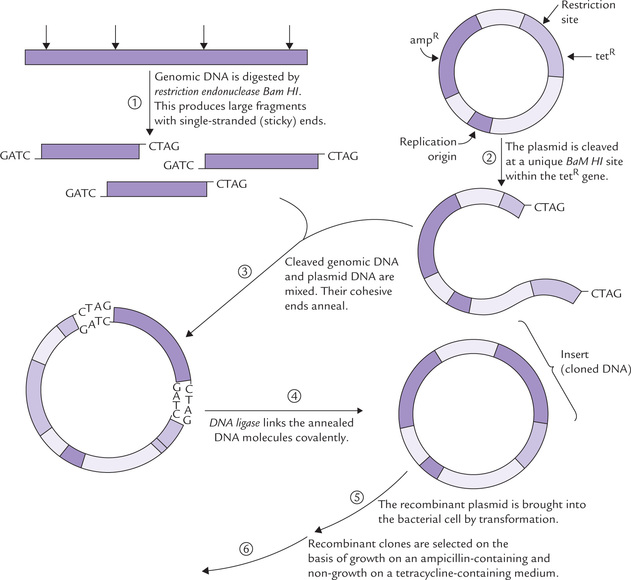
Fig. 25.8 The use of plasmid pBR322. This plasmid has been constructed specifically as a cloning vector.
Further selection of colony having the desired DNA
The cells containing the chimeric-plasmids are grown to form colonies. However, there will be many more colonies also. This is because the bacteria containing other plasmids (not the desired chimeric plasmid) also grows to form distinct colonies. In this way we obtain a large collection of bacterial clones, each containing a random piece of DNA insert. Identification of the cells that contain the desired DNA insert is the only problem left now. This requires the process of screening done by labelled DNA probe complementary to the desired DNA insert. The probe may be an isolated DNA fragment or a synthetic oligonucleotide (discussed later).
Step 4: Isolation of DNA Insert or its Protien Product
Once a clone with the desired DNA insert has been identified, the vector can be recovered from the bacterial clone and the insert can be excised with the same restriction endonuclease that had been used for the construction of the recombinant plasmid. In order to isolate large quantities of protein product of the inserted DNA, the cells are grown under conditions that are favourable for production of proteins. The protein is then isolated from these cells by usual methods. Several medically important proteins are thus obtained (Box 25.3).
Usually the recombinant DNA technology successfully produces human proteins using enzymatic machinery of bacteria. However, bacterial RNA polymerase may not recognize the human promoter regions, and so expression of human genes may be minimal. Use of expression vectors—these include bacterial plasmids into which appropriate transcriptional and translational signals have been added— permit effective expression of human genes in bacterial cells (Box 25.4).
The recombinant DNA methodology suffers from a major drawback, which is—the cloned genes can be rapidly propagated, but they cannot be effectively expressed in bacterial cells. Hence this methodology is not effective for generating the protein products of the cloned genes. This is a major pitfall because an important goal of recombinant DNA studies is to propagate a foreign gene in bacteria and obtain the product in a biologically active form. To achieve this goal, expression vectors, which facilitate expression of the DNA inserts, have been developed. The following conditions must be met during their construction:
1. Since bacteria can express the recombinant eukaryotic gene only when properly positioned transcriptional and translational control sequences are present, a strong bacterial promoter region is added to the upstream of the eukaryotic gene. It is then inserted into the vector.
2. The mRNA transcript of a recombinant eukaryotic gene must contain the bacterial ribosome binding sequence (the Shine-Dalgarno sequence) required to properly orient it with functional bacterial ribosome.
3. Only complementary DNA inserts (not genomic DNA) must be used. This is because the complementary DNA can be effectively expressed in bacterial cell (bacteria cannot remove intron sequences from primary transcript).
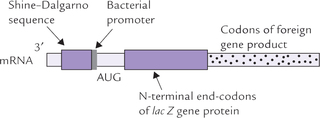
The whole of the cloned complementary DNA is inserted within a bacterial genome, commonly in the lac Z gene. The mRNA transcript of the recombinant DNA contains the following elements in that order; lac Z-Shine-Dalgarno sequence, the bacterial promoter, the start codon, and the codons for first few amino acids of the bacterial protein (encoded by a portion of the 3’-end of the lac Z gene). This is followed by codons of the foreign gene product.
The engineered protein product is a fusion protein that contains few N-terminal amino acids of the lac Z gene protein followed by the complete amino acid sequence of the cloned complementary DNA.
The aforementioned discussion centres around plasmid vectors, which appear to represent ideal replicon for DNA amplification. They undergo intracellular replication, are passed vertically from the parent cell to each daughter cell, and (unlike chromosomal DNA) are copied many times during each cell division. Comparative features of other vectors, such as lambda phage, cosmid, BACs and YACs, have been discussed earlier.
III DNA Library
A Genomic Library
Genomic library refers to a large collection of bacterial cells, each containing a random piece of human genomic DNA. For constructing a gene library, the entire DNA of cell is cleaved into small pieces by using different REs. All these cut fragments are then introduced into appropriate vectors. This forms a large collection of different recombinant clones, which are then introduced into the host bacterial cells to form the gene library. All the genes of an organism are represented in the gene library. In order to produce a complete gene library for E. coli, about 1500 fragments are required, whereas about 10 lakhs fragments are required for human gene library.
B cDNA Library
In a genomic library, all the genomic material is represented but only about 3% of the cloned DNA codes for proteins. If a collection of only expressed DNAs were to be made, then RNA and not DNA would be the starting point.
cDNA library is a collection of all the expressed DNA of a particular cell type or tissue. For example, a cDNA from pancreatic β-cell contains clones with cDNA for proinsulin. On the other hand, a cDNA library from bone marrow cell contains many clones with cDNA for α- and β-chains of haemoglobin. Thus, for a cDNA library the tissue of origin is important. For a genomic library, the tissue of origin is unimportant because the genomic material is same in all cell types of an organism.
Building of cDNA library: The mRNA is extracted from a specific tissue. It is used as a template for synthesis of complementary DNA strand; the enzyme catalyzing this synthesis is reverse transcriptase, which yields a single-stranded cDNA (Fig. 25.3). Double-stranded cDNA is then obtained from it by adding the DNA polymerase. The latter is incorporated in plasmid, λ phage or cosmid and introduced into host bacterial cell.
Genomic libraries contain all the DNA of an organism, and cDNA libraries contain only expressed DNA.
Screening of genomic library: It is possible to screen the genomic library and obtain a gene of interest from it. Although a daunting task, use of nucleotide probes has made it possible.
IV Nucleotide Probes
To search a desired DNA sequence of interest from a vast array of DNA fragments present in a genomic library, a reagent is needed which would react only with the correct fragment and ignore the rest. This type of reagent is called a nucleotide-based probe. It is a single-stranded piece of DNA (sometimes RNA), which can range in size from as little as 15 bp to several hundred kilobases (Table 25.2 ). It can identify, through base pairing, a specific DNA fragment of the library, which contains complementary sequence. A probe DNA, for example, will form complementary base pairing with another DNA strand (termed template) if the two strands are complementary and a sufficient number of hydrogen bonds are formed.
Table 25.2
Characteristics of nucleic acid probes
| Probe type | Size | Origin |
| DNA | 0.1-100 kb | Cell-based DNA cloning, PCR |
| RNA (or riboprobe) | 1-2 kb | RNA transcription from plasmid (or phage vectors) |
| Oligonucleotide | 15-50 nucleotides | Chemical synthesis |
An effective molecular hybridization between the probe and the template requires that both must be single stranded. Likewise, a mRNA can be used as a probe: it will bind to the DNA fragment that contains exon sequences of its gene. RNA probe, termed riboprobe, can be produced by in vitro transcription of cloned DNA inserted into a plasmid vector. Synthetic oligonucleotide probes, constructed by chemical methods, are most commonly used. The probe has to be at least 15-18 nucleotides long because shorter sequences may be present, by chance, at multiple sites in the genome (e.g. a trinucleotide every 43 base pairs). Oligonucleotides of this size can be easily synthesized.
A Probes must have a Label to be Identified
To render them recognizable, probes are labelled with the radioisotopes, such as 32P or tritium. These probes can be detected by autoradiography, which involves placing the sample in direct contact with the photographic material, usually an X-ray film. Alternatively, end-labelling probes with fluorescent tags can be used. The latter are visible under the UV lamp.
B Techniques for Labelling Probes
There are two general ways in which a labelled nucleotide can be incorporated into the structure of the probe:
1. End-labelling: Addition of a labelled group to one terminal of the probe is done, for example, by exchanging a labelled γ-phosphate from ATP with a phosphate from the 5’-terminal on (single or double-stranded) DNA.
2. Polymerase-based labelling: Using a DNA polymerase, multiple-labelled-nucleotides are incorporated into the probe during DNA synthesis. Such a reaction requires dNTPs, and it is customary to have one of them to be labelled, e.g. dGTP. Because on an average 25% of the nucleotides incorporated are labelled, this type has a higher specific activity than the end-labelling where only terminal nucleotide is labelled.
C Uses of Nucleotide Probes
1. To search specific DNA sequences of DNA library, as discussed.
2. In Southern and Northern blot techniques, probes are used to identify DNA or RNA fragments respectively, as described later.
3. In diagnosis of genetic disorders, such as sickle cell anaemia, thalassaemia, cystic fibrosis, etc.
V Blotting Techniques
These are standard techniques for the identification of a specific DNA, an RNA or a protein from a vast expanse of others. The technique for DNA identification is termed Southern blot, whereas Northern blot is for RNA and Western blot for protein identification.
A Southern Blot Technique
This technique, used for the identification of a specific DNA sequence, is named after its originator, E.M. Southern, who developed the method in 1970s. Supposing it is proposed to identify a restriction fragment that carries a particular DNA sequence, for example, exon of a protein coding gene (11-γ-hydroxylase; Fig. 25.9 ), the method would involve the following steps:
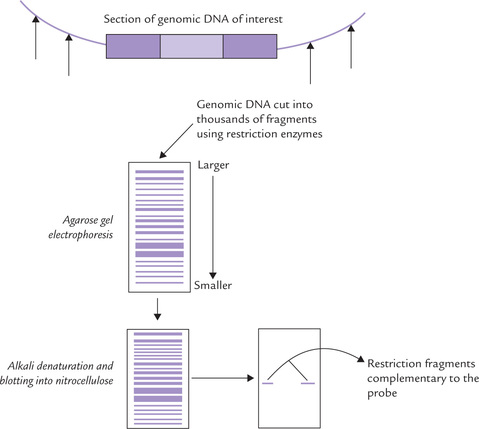
1. Extraction of genomic DNA from the cells, and cutting into fragments by restriction endonuclease to prepare a restriction digest. The latter is likely to contain thousands of double-stranded restriction fragments.
2. Electrophoretic separation of different fragments on the basis of their size in a cross-linked agarose or polyacryl amide gel; the smallest fragments move furthest, whereas the larger ones are retarded by smaller ‘gaps’ in the gel This method can separate fragments ranging in size from 100 bases to approximately 20 kb in length (above 40 kt resolution is minimal). Furthermore, the method is sufficiently powerful to separate even those restriction fragments that may differ in length by only one or a few nucleotides (refer to Box 25.5 for more information or electrophoretic separation).
3. Denaturation of the DNA fragments to render them single-stranded by soaking the gel slab in a strong alkali solution. This step is needed because the nucleotide probe (single-stranded oligonucleotide that is complementary to the desired DNA sequence on a restriction fragment) can identify only a single-stranded DNA by hybridization.
4. Transfer of the single-stranded DNA to a sheet of nitrocellulose filter (or nylon membrane); the process of transfer involves the passage of solute through the gel and into the filter, passively, carrying the DNA. Transfer of DNA from the semisolid phase of a gel to a solid phase in this manner is referred to as blotting (hence the name blot).
This step is important because the restriction fragments are not tightly bound to the semisolid gel and therefore at risk of being easily leached out of the gel slab. Nitrocellulose or nylon immobilizes the single-stranded DNA to form a replica of the DNA from the gel, which is still accessible to hybridization reaction with probes.
5. Addition of nucleotide-based probe (RNA or single-stranded DNA) by dipping the nitrocellulose filter into a neutral solution of the probe and holding at a suitable temperature and time period to allow hybridization of the probe with the matching DNA fragment. The filter is then washed to remove the excess unbound probes. The radioactively labelled probes are generally used.
6. Autoradiography or fluorescent scanning determines the position of the labelled probe, base paired with the sequence of interest.
Southern blotting is prototype of permanent DNA storage, wherein a replica of the gel slab with its separated restriction fragments is made on the nitrocellulose filter. The replica can act as template for any number of different probes.
The DNA transferred from the semisolid to the solid phase, if preserved properly, forms a permanent record of the digested DNA and then acts as a template for any number of different probes. Either DNA or RNA probes can be used. Subsequently, analogous techniques were developed for the transfer of RNA and proteins, and adopting the theme of direction, they were termed Northern and Western blots, respectively (Table 25.3 ).
B Northern Blot Technique
This technique (which is similar to the Southern blot technique) is used for identification of RNAs by electrophoresis, blot-transfer and probing with radioactive RNA or single-stranded DNA probes (Table 25.3).
C Western Blot Technique
It is a method for (electrophoretic) separation and identification of a specific protein from a mixture. In contrast with the Southern and Northern blots, which use nucleotide-based probes, this technique relies upon the ability of a monoclonal antibody to bind to a protein that it has been raised against.
A combination of some of the techniques described above permit analysis of long stretches of DNA from genomic library. The method used is chromosome walking (refer to Box 25.6 for details).
In situ Hybridization
It is a modified version of hybridization in which labelled probes are used to detect complementary DNA sequences in tissue sections. The term in situ is applied because the DNA is not extracted in this process. The following steps are involved:
• Histology slides are prepared from the tissue under study and examined by traditional staining techniques.
• DNA is then denatured with NaOH and specific probe is layered over it.
• The probe would hybridize, on the microscope slide, with the complementary sequences of the denatured DNA in tissue sections (or whole cell).
• After the probe is properly fixed, it is autoradio-graphed; hybridization complexes are seen as silver grains on photographic film.
The advantage of this technique is that it allows correlation of findings of cytopathological examination with the presence of an intracellular viral pathogen, as shown by use of probe.
This technique has emerged as an important diagnostic tool and is particularly useful for detection of intracellular viral pathogens. Some modified versions of in situ hybridization are as below:
1. Fluorescent in situ hybridization (FISH): It is a non-isotopic in situ hybridization (NISH) technique where fluorescent labelled probes are used. It is faster, safer, more sensitive, and shows greater resolution compared to the ordinary in situ work discussed above. Probes used in FISH are short, of about 20-25 nucleotides long. The slide is visualized using ultraviolet light. Under the ultraviolet light, the probes emit fluorescence, intensity of which depends on the extent of hybridization.
2. Use of riboprobes: Riboprobes are RNA probes produced by in vitro transcription of cloned DNA inserted into plasmid (or phage) vectors (Table 25.2). The labelled radioprobes can be used to detect complementary RNA in tissue sections. To maximize the efficiency of the hybridization, cRNA probe is preferred. cRNA is single-stranded and is complementary to the transcribed mRNA of the gene. Therefore, it hybridizes with any mRNA present in the tissue section with high efficiency, and serves as a useful diagnostic tool (Case 25.1).
VI Applications of Recombinant DNA Technology
The knowledge of genes and techniques for manipulating them is revolutionizing medicine. The genes for several genetic diseases have been mapped and sequenced already, and molecular methods for diagnosis of these diseases have been developed. Treatment based on manipulation of gene expression was no more than an intellectual pastime, but is a reality now. A variety of proteins are being produced by genetically engineered micro-organisms, and widely used as therapeutic agents. However, treatment based on transfer of intact gene into the cells of patients with genetic diseases is still in experimental stage.
The major applications of recombinant DNA technology are in:
• Commercial preparation of proteins and hormones
• Construction of useful organisms
• Basic applications, and several others described in this section.
A Medical Diagnosis
The DNA-based diagnostic tests can identify structural variants (point mutations) as well as dysfunctional and absent genes. They are also used to identify a recessive-disease-gene in unaffected carriers, or even in healthy people to estimate disease risk. Prospective parents may be screened, and if identified as carriers of a severe, debilitating disease, may be advised not to have a child or to go for prenatal or preimplantation diagnosis.
Cloned genes are currently being used as hybridization probes for the detection of cancer and genetic diseases. Even infectious diseases can be diagnosed by specially designed probes which identify DNA of pathogen. Some representative procedures and applications of DNA-based diagnosis to investigate a possible mutation include the following (a far from exhaustive list):
Sickle Cell Mutation Analysis
Sickle cell disease is caused by a single base substitution (A to T transversion) in the β-chain gene of haemoglobin. This mutation can be easily recognized by a procedure based on restriction endonuclease cleavage and Southern blotting as it changes length of a restriction fragment. This is because the mutation occurs within a palindromic sequence recognized by a restriction endonuclease, Mst-II. The mutation results in obliterating this cleavage site so that it is not cleaved by Mst-II; Example:
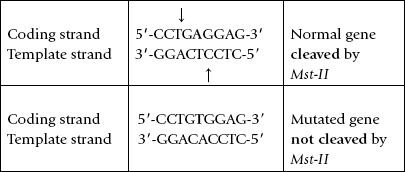
In the Mst-II restriction digest, the size of the restriction-fragment produced from the mutated gene will be different from that produced from the normal gene (Fig. 25.10 ). This can be exploited for the molecular diagnosis of the disease by Southern blotting, using a probe for a sequence 5’ of the mutation.
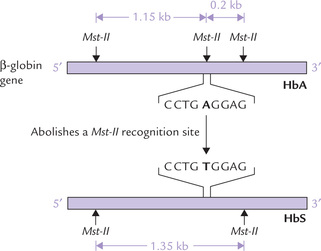
Fig. 25.10 Molecular diagnosis of sickle cell mutation by elec-trophoresis of an Mst-II restriction digest, transfer to nitrocellulose filter and analysis with a probe.
• In the absence of mutation a 1150 base pair (1.15 kb) fragment is produced and identified by the probe.
• If the mutation is present, Mst-II is unable to cleave the mutated site and so a larger restriction fragment (1350 base pair) is generated. The probe sequence will therefore be present on the 1350 bp (1.35 kb) fragment.
The above method can be used for diagnosis of various other gene abnormalities where a mutation may, rather than obliterating or deleting pre-existing cleavage site, creates a new one.
Analysis with Allele-specific Probes
Some mutations do not obliterate (or create) a restriction site, therefore the method shown in Figure 25.10 cannot be used in their diagnosis. A different diagnostic approach, which aims to directly identify a sequence variation, is thus needed for discriminating between the normal and the mutated DNA. It involves Southern blotting and analysis with two different probes: one specific for the normal sequence and the other specific for the mutated sequence.
Thus, two probes to identify the two sequence variants ( alleles) of the gene are used. Note that the probes identify sequence variants in this case, rather than identifying restriction fragments of different lengths. The probes may have to distinguish between sequences that differ in only a single base, for example, in sickle cell anaemia, and therefore, require rigorous adjustment of the conditions during annealing (base-pairing). Since annealing is favoured by elevated temperature and low ionic strength, these conditions must be present during the assay.
This method is applicable only when molecular nature of the mutation is known.
Allele-specific probes are small oligonucleotides that are used for rapid diagnosis of point mutations by probing the blotted DNA template under highly stringent conditions, e.g. low ionic concentration and high temperature.
Allelic heterogeneity: A number of different mutations in the same gene may disrupt the normal function of the protein product, and so lead to the same disease. This phenomenon, called allelic heterogeneity, is observed in most single gene disorders, for example β-thalassaemia. In this disorder, approximately 100 different mutations in the β -chain gene, all leading to the same disease, are known. Each individual mutation requires its own pair of allele-specific probes, which is not an easy feat. Thus, allelic heterogeneity is an important limitation to the use of allele-specific probes.
Dot-blot Analysis
It is a rapid and relatively inexpensive procedure for detection of a mutation, where crude DNA is examined directly without any prior amplification or electrophore-sis. The extracted DNA is denatured, applied directly to nitrocellulose filter and then probed with allele-specific oligonucleotide probes. Evidently, the procedure is fast, relatively inexpensive, and is suitable for testing large number of people, such as during population screening.
Steps
1. Sample DNA is extracted from different individuals and denatured.
2. Every sample of the denatured DNA is applied onto two different nitrocellulose filters, each in a single dot (dotting).
3. One filter is dipped into a solution with a fluorescent-labelled probe for the normal sequence, and the other into a solution with a probe for the mutant sequence.
4. Excess probe is washed off and the bound probe is visualized under the UV lamp.
5. In normal individuals, probe for the normal sequence will bind, whereas in the individuals carrying the mutation, the probe for the mutant sequence will bind.
Uses
1. Population screening for single mutation: Dot blotting is less costly and nearly instantaneous as compared to more expensive Southern blotting based procedures (takes few days), and PCR based procedures (require few hours). It is more suitable for screening populations for a single mutation or polymorphism.
2. Multiplex genetic testing: DNA of a single individual is dotted on different filters and each of these filters is analyzed with a different probe. By this approach it is possible to detect different mutations, and so identify individuals who carry susceptibility genes for such multifactorial diseases as osteoporosis or Alzheimer’s disease.
A number of newer techniques for the clinical analysis of DNA are being developed, and there is a shift from experimental DNA analysis to diagnostic services in clinical settings.
B Gene Therapy
Advances in the field of DNA recombinant technology have made it possible to ameliorate or cure a number of inherited disorders as well as diseases caused by somatic mutations. The goal of gene therapy is to replace (or repair) the defective, disease-causing genes by normal functioning copies. It involves the following approaches:
1. Gene replacement, which involves removal of a defective sequence and its replacement with a normal one.
2. Gene correction, wherein a pathological change in the nucleotide sequence is repaired.
3. Gene augmentation (most commonly used), defined as introduction of genetic material into cells without any attempt to delete or modify the endogenous genetic material.
In essence, gene therapy uses the capacity of a patient’s cells to synthesize the therapeutic agent from the introduced gene.
Gene therapy may be either somatic cell gene therapy which targets the patient’s somatic cells; or the germline gene therapy which targets the germline cells. However, at present only somatic gene therapy is permitted in humans. It does not attempt to change all somatic cells, but the gene is delivered only to the affected tissue. For example, in Duchenne muscular dystrophy, an intact dystrophin gene is targeted specifically to muscle tissue. Likewise, in cystic fibrosis the intact gene for the cystic fibrosis (CF) chloride channel is targeted to the lung epithelium.
Germline gene therapy is considered unethical as it carries the risk of transmitting genetic modifications to the offspring, and so not permitted.
The procedure of gene therapy involves isolation of the gene, its incorporation into a suitable carrier, followed by its delivery into the target cell.
Delivery Methods of Therapeutic Gene into Target Cell
Several techniques are currently available for delivery of therapeutic genes into host cell. However, none of these techniques is efficient. There are two major obstacles to effective gene delivery: first, the large therapeutic genes do not cross the biological membrane easily; and second, the foreign DNA after entering the cell is rarely integrated in chromosome. Normally, they remain extrachromosomal, do not replicate and are degraded by nucleases. As a result, they are diluted with successive mitotic divisions.
Some of the commonly practiced methods for gene delivery are:
Physical Methods
The gene is encapsulated in liposome, an artificial vesicle in which aqueous core is surrounded by lipid bilayer. The bilayer fuses with the plasma membrane of the target cell and the enclosed gene is released into the cytoplasm. Exposure to high voltage enhances cellular uptake of exogenous DNA into the cell (electroporation).
Receptor-mediated Endocytosis
The foreign gene is covalently linked to a ligand that is taken up by receptor mediated endocytosis. Only the cells possessing the receptor (for the ligand) are transformed by this method. An undesirable outcome is that most of the foreign DNA is directed to lysosomes for degradation. To prevent this, endosomes are disrupted with adenovirus.
Retroviruses
The most advanced technique for targeting therapeutic genes to mammalian cells involves retroviral vectors (Fig. 25.11 ). Retroviral vectors are artificial and disabled constructs obtained by modification of certain retroviruses:

• The key viral genes (pol, gag and env) are replaced by the foreign (therapeutic) gene.
• The long terminal repeat (LTR) are retained (LTR are required for integration of the viral DNA in the host chromosome). The ψ sequences, required to pack viral RNA in viral protein coat, are also retained.
To assemble virus with the above recombinant genetic information, however, poses a major problem. This is because the retroviral vector DNA cannot synthesize viral proteins (recall, it lacks gag, pol and env genes). To solve this problem the retroviral vector DNA is introduced into tissue culture, infected with a helper virus, which contains the key viral genes (pol, gag and env). Therefore, within the culture cell, a cooperative action of the two types of viruses (recombinant virus and helper virus) ensures that the recombinant DNA is transcribed and the RNA packaged into the virus particle. (Note that the retrovirus particle so produced also contains reverse transcriptase and integrase.)
After the retrovirus enters the host cell, the following events follow:
• A cDNA copy of the recombinant RNA genome is produced (by reverse transcriptase).
• The cDNA is integrated into host chromosome (by integrase).
• Because of its inability to synthesize viral proteins and produce infectious viral particles, the engineered RNA genome becomes a permanent part of the host chromosome. Instead of producing viral particles, it is expressed to generate the desired protein product.
Because of the reason that viral proteins are not coded by them, the retroviruses have little or no cytotoxicity and are, therefore, called “clean vectors”.
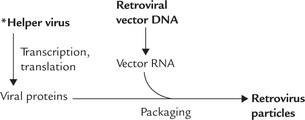
Use of retroviral vector, though considered a highly advanced technique, suffers from two disadvantages. First, the retroviral gene transfer known technically as transfection is not a particularly efficient process. In most experiments, less than 10% of the cells are transfected. Second, the retroviruses are not able to infect non-dividing cells (with sole exception of AIDS virus) and this is true for the retroviral vectors as well, which are therefore, poorly suited for gene therapy of neurons and muscle fibres.
DNA Viruses
DNA viruses have also been explored as potential gene transfer vectors for gene therapy. Strategies have been developed for incorporating normal genes into the adenoviral genome. Adenoviruses, the large (35 kb) double-stranded DNA viruses which are minor respiratory pathogens in humans, have been used to transfer genes of cystic fibrosis transmembrane conductance regulator (CFTR) protein to the respiratory epithelium in patients with cystic fibrosis (respiratory epithelium is the usual host cell for adenoviruses). In the adenoviral vectors, one of the viral genes is replaced by the therapeutic CFTR gene. However, the viral proteins can still be synthesized by the vector. This gives rise to cytotoxicity and immunological responses. Also, the therapeutic benefits of the adenoviral vector are transient because it rarely, if ever, incorporates its DNA into the host cell genome. However, these vectors can be applied in higher titers than the retro-viral vectors, and they are also capable of infecting the non-dividing cells. Therefore, they can be effectively used for gene therapy of neurons and also muscle fibres.
Achievements and Limitations of Gene Therapy
In addition to the conditions discussed above, gene therapy has been attempted in various other disorders.
Haemophilia B (Deficiency of Factor IX)
A clotting disorder, is treated by retroviral transfer of factor IX gene into hepatocyte. Although the intracellular level of factor IX reaches only 0.1% of normal after this treatment, the clinical manifestations of the defective gene are mitigated. Haemophilia A, caused by deficiency of factor VII is also treated by retroviral transfer of this factor in patient’s fibroblast, followed by implantation of the latter under the patient’s skin.
Severe Combined Immunodeficiency (SCID)
The first success for gene therapy was reported in the year 2000 by a French medical team, who succeeded in curing SCID, a disease caused by deficiency of adenosine deaminase (ADA). The molecular defect is a mutation located on chromosome 20. The ADA deficiency results in accumulation of adenosine, deoxyadenosine and dATP. All the three compounds are toxic to lymphocytes. The immune responses, both cell-mediated and humoral, are suppressed and so the patient is susceptible to repeated infections. Treatment involves isolation of the ADA genes from normal healthy cells and their introduction into retroviruses. These retroviruses are then used to introduce the gene into lymphocytes of the patient in vitro. The cells so transformed are introduced back into the patient. These cells produce ADA normally. Furthermore, no side effects have been observed. Therefore, this line of treatment holds promise for the future. However, one of the problems is that the introduced gene may not express at the desired level in the new environment.
Non-genetic Conditions
Some non-genetic conditions such as rheumatoid arthritis and cancers are also treated by gene therapy. Rheumatoid arthritis is an inflammatory process mediated by the cytokine interleukin-I (IL-1). The transfected gene encodes a receptor-blocking protein that prevents the interaction of IL-1 with its receptor. The vector carrying the gene is injected directly into the inflammed joint. For the treatment of cancers, the tumour cells are transfected with cytokines such as IL-2 and tumour necrosis factor (TNF). The aim is to stimulate immune response against tumour cells.
Other diseases under study for somatic cell gene therapy are factor VIII deficiency, HGPRT deficiency, pyrimidine nucleotide phosphorylase deficiency, etc.
Germline Gene Therapy
Studies on germline gene therapy have been conducted on transgenic mice. The fertilized mouse eggs are harvested and injected with desired DNA molecules. These are then reimplanted in the pseudo-pregnant female mouse. The technique has been used in murine gene disorders, such as growth hormone deficiency and γ-globin deficiency. Though beneficial effects have been observed in animals presently, this therapy appears to have no application in human genetic disorders (see transgenesis).
Current Status of Gene Therapy
Gene therapy is still in the experimental stage and at present only a few diseases are treated by it. There are several reasons for its limited use, such as lack of consistent results and lack of ideal vector. Most non-viral vectors lack precise targeting ability, whereas use of viral vectors carries risk of carcinogenesis.
However, in spite of several technical difficulties, recent advances in molecular and cellular biology raise hope that gene therapy will soon start playing an increasingly important role in clinical practice and have applications in several fields of medicine.
A new approach for treatment of hereditary disorders aims at increasing expression of an endogenous gene. For example, hydroxyurea enhances expression of the γ-globin gene. In patients with sickle cell anaemia, this approach increases synthesis of nonsickling haemoglobin. Antisense technology is also emerging as an effective mode of treatment (refer to Box 25.7 for details).
C Commercial Preparation of Proteins and Hormones
Many human proteins can be used as therapeutic agents. These include hormones such as insulin and growth hormones; blood-clotting and fibrinolytic proteins such as clotting factors VIII and IX and tissue type plasminogen activator; and humoral mediators of immune response such as the interferon and interleukins. Traditionally, these proteins have been obtained from human tissue. But this method suffers from a serious drawback in that transmission of serious diseases may occur. Many recipients have been reported to have contracted AIDS virus after treatment with contaminated proteins. Moreover, the yield from the human tissue is very poor and the price of production is very high.
The cloning strategies described above are sometimes not useful: they may be able to propagate DNA, but effective transcription and translation of the latter do not occur. Consequently, the genes are not expressed. The use of expression vectors, is therefore, favoured.
D Construction of Useful Organisms
Some bacteria that produce increased amounts of industrial chemicals such as methanol, ether, and butanol from biological wastes have been genetically engineered. Marine bacteria capable of metabolizing petroleum have been constructed. These are useful in clearing oil spills. The future possibilities of these methods are enormous and highly promising. For example, attempts are being made to introduce the nitrogen fixation system into non-nitrogen fixing bacteria and plants. If successful, these measures will reduce the dependence on nitrogen fertilizers.
E Basic Applications
Site-directed Mutagenesis
This is a technique that can introduce controlled alterations of selected regions of a DNA molecule. It may involve the insertion or deletion of selected DNA sequences, or replacement of a specific nucleotide with a different base. It can also be accomplished by replacing the appropriate section of the isolated gene with a synthetic oligonucleotide whose base sequence is such that it codes for the altered amino acid. The altered activity of the mutant protein helps us to know the function of the particular amino acid in a protein.
Knockout Mice
These are useful animal models developed by inducing gene disruptions. They are proving to be very useful animal models for the study of human genetic diseases. They also help to ascertain the biological significance of proteins by observing the abnormalities (if any) when the functional protein is missing.
F Analytic Techniques
The recombinant DNA technology is used in a number of analytic techniques, such as polymerase chain reaction, restriction fragment length polymorphism (RFLP), DNA typing, Northern blotting, Southern blotting and Western blotting.
G Transgenesis
It refers to the process of introducing exogenous genes into germline of an organism, which changes its phenotypic characteristics and which would be passed on to the successive generations. Some success has been achieved in the transgenesis experiments in mice. The rat growth hormone gene was introduced into the germline of mice, and the resulting transgenic mice grew to more than twice the normal size. Because the gene of interest must be inherited in the germ line, it is essential to use vectors (containing the gene of interest) with appropriate regulatory elements for introducing the gene into the nucleus of the fertilized eggs. The eggs are then transplanted into the uterus of receptive females for the development of the potential transgenic off-springs.
H Agricultural and Livestock Industries
The process of transgenesis is being utilized in both the plant and livestock industries, with an aim to develop robust plants and animals that are more resistant to diseases and infections. Some transgenic animals are developed to obtain high levels of expression of therapeutically important proteins. For example, a desired human protein can be produced in sheep by attaching the isolated human gene to the signaling part of the milk-protein-gene. This results in secretion of that (desired) protein into milk. This allows us to obtain large amounts of human protein from the milk of transgenic animals.
I Enhancement Engineering
Genes targeted to germline can permanently change genetic makeup of a species. For example, humans lack the enzyme gulonolactone oxidase required for synthesis of vitamin C. The transfer of a gene for this enzyme would restore our ability to synthesize vitamin C and push scurvy into oblivion. This is an example of enhancement engineering. In contrast to gene therapy, which is aimed at repair/replacement of the defective genes, the goal of enhancement engineering is to improve the genetic constitution in general.
J Other Uses
Several vaccines are prepared using recombinant DNA technology (refer to Box 25.8 to know the process of DNA vaccine preparation). The first vaccine prepared by this technology was hepatitis B vaccine. The engineered proteins are used as food additives to enhance the nutritional value of food.
VII Restriction Fragment Length Polymorphism
Restriction endonuclease cleaves the DNA only at some specific recognition sequence, the restriction site. In case a restriction site is disrupted, either by a pathologic change in the DNA sequence resulting in a disease (a mutation), or a naturally occurring variation in the DNA sequence (polymorphism), it may create new sites or obliterate the existing sites. This may result in fragments of different lengths following digestion by restriction enzyme—restriction fragment length polymorphism (RFLP).
Such RFLP can be used either to identify disease-causing mutations or to study variations in non-coding DNA, which can be used in the study of genetic linkage.
A Origin of RFLP
The following sequence variations—mutations or polymorphism—can change the pattern of restriction-sites to generate RFLP:
1. Base substitution: A single base substitution may create a new restriction site or delete a pre-existing one.
2. Insertion or deletion mutations: Bases are added to or removed from a DNA segment between two restriction sites. This changes length of DNA between the two cleavage sites, resulting in the polymorphism.
3. Variable number of tandem repeats: Tandem repeats are short sequences of DNA—between two to a dozen base pairs—repeated over and over in tandem arrangement (next to each other) in the DNA molecule. There is much variation between individuals in number of repeats of these short sequences. This hypervariability is referred to as variable number of tandem repeats (VNTR); for example, a given sequence may be repeated less than 5 times in some people and more than 20 times in others.
B Clinical Applications
1. Diagnosis of genetic defects: RFLP can be used to identify a disease-causing mutation because of a single point mutation, creating or abolishing a restriction site. This has been discussed earlier with reference to sickle cell anaemia (Fig. 25.10) A more recent use is to map and isolate genes for hereditary diseases in which the gene responsible (for the disease) is unknown.
2. Linkage studies: If the polymorphism is located close to a defective gene on the same chromosome, the two are transmitted together from one generation to the next (simply for being on the same DNA molecule). Therefore, inheritance of the defective gene can be traced by tracing the inheritance of the associated restriction fragments. The advantage of RFLPs is that they can be used even if the sequence of the defective gene is unknown. Moreover, allelic heterogeneity, probably the most serious obstacle to DNA-based diagnosis, does not affect the use of RFLPs.
3. Study of genetic individuality: RFLPs, which are inherited and conserved in a Mendelian manner, are highly conserved in different individuals and therefore, allow study of genetic individuality at molecular level. The fact that probability of obtaining identical pattern (of restriction fragments) in two randomly selected individuals is only 1 in 1019 makes this technique applicable to problems of genetic individuality.
4. Detection of changes in DNA sequence: RFLPs can also be used to detect larger pathological changes in the DNA sequence, either deletions or duplications. In case of gene deletion, a restriction site may be abolished, leading to disappearance of a fragment on Southern blot. In case of gene duplication a new gene may be formed having a different pattern of restriction sites, which allows its detection.
5. Others: Value of RFLP in detecting the presence of the disease allele in heterozygotes (carriers) is obvious. It is also useful as a diagnostic test for detecting the individuals in which the disease has not yet manifested.
VIII Polymerase Chain Reaction (PCR)
It is a technique for adjective amplifying a single template DNA. It permits generation of up to 109 copies of the target nucleotide sequence in a matter of few hours. PCR can be used to amplify DNA sequences from any type of source: bacterial, viral, plant or animal.
A Requirements for PCR
A Standard PCR requires the following:
1. DNA template—the piece of target DNA that is to be amplifi ed.
2. Amplimers—small oligonucleotide primers that would hybridize with the complementary DNA sequences upstream of the template and act as a starting point for the synthesis of the newly amplified DNA strands.
3. Polymerase enzyme—an enzyme that would catalyze the formation of DNA during the amplification reaction. A thermostable DNA polymerase of bacterial origin is most commonly used.
4. dNTPs (deoxynucleoside triphosphates)—these are the substrate molecules for the new DNA synthesis by the polymerase.
B Steps
The average PCR involves about 30-35 cycles of reactions that provide sufficient DNA, about 109 copies of the original DNA. Each cycle comprises three steps: denaturation, annealing and elongation (Fig. 25.12 ).
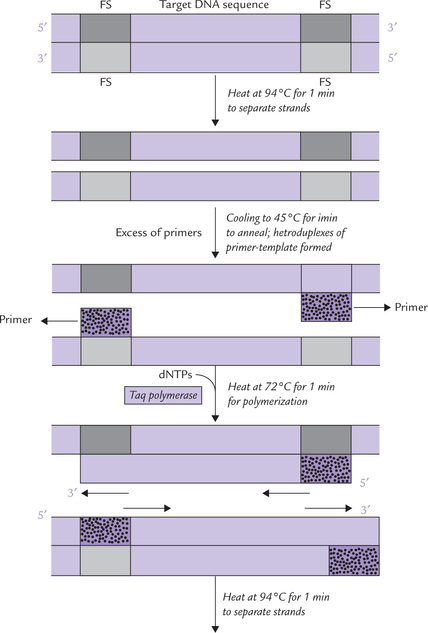
Fig. 25.12 The polymerase chain reaction for amplification of DNA sequences of interest. dNTPs (deoxynucleoside triphosphates) = Substrate molecules for new DNA synthesis.  and
and  = Flanking sequences (FS),
= Flanking sequences (FS),  = Primer.
= Primer.
1. Denaturation: The isolated duplex DNA containing the target sequence is heated to separate the two strands. Generally, heating at 94°C for one minute is enough to ensure that the template (as also the primers) are single stranded.
2. Annealing of primers: The mixture is cooled to allow formation of heteroduplexes of primer and template. The temperature for this (45°C) is based on the Tm of the expected duplex (usually 3°-5°C below Tm). The primer sequence is complementary to a short nucleotide sequence in the regions flanking the target sequence. A large excess of 108 molar excess primers must be added to ensure that they promptly anneal with the flanking sequences.
3. Elongation: Deoxyribonucleoside triphosphate precursors (dATP, dGTP, dCTP and dTTP) are then incorporated into the growing DNA chain by the polymerase enzyme. The process of PCR has been greatly facilitated by the discovery of thermostable enzymes, which can withstand the heat required during the denaturation step and still function as polymerase. The most commonly used enzyme, Taq polymerase is isolated from Thermus aquaticus, a thermophile bacterium that was isolated originally from a hot spring in Yellowstone National Park. The other such enzyme, Pfu I, also functions effectively at higher temperatures and can survive repeated heating to 94°C.
The polymerase chain reaction is a novel in vitro method of copying a single template DNA in vitro. PCR is used widely in all aspects of biomedical research, in particular the human genetics.
These three steps—denaturation, primer annealing and primer extension—can be carried out repetitively just by changing temperature of the reaction mixture in a suitable thermocycling device.
• When temperature is raised to 94°-100°C, denaturation occurs.
• Lowering the temperature to about 45°C optimizes annealing.
• A temperature of 72°C permits polymerization. The thermostable nature of the Taq I polymerase makes it feasible to carry out PCR in a closed container. Further addition of reagent is not required after the first cycle.
PCR is a simple and quick technique to amplify target DNA, but it requires prior knowledge of the nucleotide sequences in the regions flanking the target sequences. If these flanking sequences are known, then it is possible to construct the well-designed, specific primers, which will only amplify the DNA of interest. Primers that are non-specific, will amplify sequences other than the target DNA to produce haphazard results. If, for example, the 3’-primer contains sequences that contain tandem repeats of a base, the specificity of the amplification may be reduced. This is because such repetitive sequences are widely distributed throughout the genome, and so unwanted sequences may be amplified.
Reverse transcriptase PCR
cDNA is generated from mRNA template by reverse transcription. The cDNA is then amplified by PCR to produce large amount of cDNA. This overall process of producing cDNA from mRNA is called reverse transcriptase PCR (RT-PCR). By this technique, a huge amount of cDNA may be generated, which can be used to synthesize useful proteins.
C PCR-based DNA Cloning versus Cell-based Cloning
Advantages
PCR has three principal advantages over cell-based cloning:
1. Cost and time: Compared to cell-based cloning, PCR is less expensive and quicker. The denaturation step takes 1 minute, the annealing step takes 3-5 minutes and elongation step 1-5 minutes. Thus, 30 cycles would take 3-6 hours to cause 1 lakh-fold amplification. The cell-based cloning can be expected to yield such results in several days or weeks.
2. Sensitivity: PCR can amplify a DNA template to usable amounts even if a few nanograms of target DNA is available. The primers are added to the DNA in about 108 molar excess, together with a generous amount of the enzyme and substrates. After that, the only additional requirement is a thermal cycling device (heat to denature and cool for annealing), which causes exponential increase in DNA.
3. Robustness: PCR is able to amplify DNA that is often badly degraded, or is present in inaccessible sites, e.g. formalin-fixed tissue.
Disadvantages
Following are the disadvantages of PCR-based over cell-based cloning:
1. Inaccuracy: The Taq polymerase has no proofreading activity, so it has an error rate on the order of one base mismatch every few thousand base pairs.
2. Small target DNA amplification: PCR effectively amplifies small target sequences of 200-1000 bases, but cannot reliably amplify larger sequences. However, use of long PCR methods allows sequences up to 20 kb or more to be amplified.
3. Prior information of flanking sequences: This is essential for synthesizing flanking primers, and may require prior cell-based cloning to derive the flanking nucleotide information.
D Applications of PCR
The major use of PCR is to provide massive quantity of DNA for prenatal diagnosis, pre-implantation diagnosis, diagnosis of infectious diseases, forensic medicine studies among others.
PCR in Prenatal Diagnosis
PCR can be used for the prenatal diagnosis of genetic defects early in pregnancy. Sample DNA is extracted from the fetal cells, obtained by either chronic villus sampling at 9th week of gestation or by aminocentesis at approximately 15th week, and is amplified (by PCR). The PCR products are electrophoresced, denatured, transferred on nitro-cellulose filter and then probed with complementary nucleic acid sequence for specific detection of a gene abnormality.
For example, mutation in about 70% of the cases of cystic fibrosis consists of deletion of three successive base pairs that code for the amino acid phenylalanine in position 508 of the polypeptide chain (Phe508 mutation). A short section of DNA around the site of mutation is amplified. Since the mutated sequence yields a PCR product three nucleotides shorter than normal, the two products can be easily separated by polyacrylamide gel electrophoresis. This method can be employed for all small insertion/deletion mutations.
In case a crippling mutation is detected by this technique, selected abortion of the affected fetus may be carried out. However, an ethical dilemma is associated with this approach.
PCR in Pre-implantation Diagnosis
In view of the above ethical considerations, the DNA sample is obtained from an alternate source instead of the growing fetus. In vitro fertilization is carried out and the embryo is grown in a test tube to the 8- or 16-cell stage, when a single cell is removed for extracting its DNA. Removal of a single cell does not impair further growth of the embryo.
Further analysis is carried out by electrophoretic separation of PCR products, detection of genetic abnormality, if any, by radioactive probes. Typically, 5-8 ova are fertilized in vitro at the same time, and all developing embryos are subjected to the test. Only disease-free embryos are re-implanted.
If only small amount of DNA is available, for example, in prenatal and pre-implantation diagnosis, the DNA of interest has to be amplified by PCR, before it can be analyzed.
A major limitation of the above method is that the quantity of DNA from a single cell is too small to be amplified reliably even by “standard PCR”. A more sensitive method—PCR with nested primers, is required to accomplish this feat. A relatively large section of the target DNA is initially amplified through a 20- to 30-cycle PCR to generate approximately 106 molecules. These molecules are then subjected to a second round of PCR (20-30 cycles) with a new pair of primers, which yields more than 1010 molecules of the PCR products.
Application to Forensic Medicine
PCR amplification serves as a potent tool for investigating cases of assault or rape, and answering simple questions such as is this man father of this child? Because PCR requires only small quantities of DNA, a blood stain or drop of semen provides sufficient DNA for DNA fingerprinting, a technique that relies on a high degree of polymorphism of microsatellite repeats, discussed earlier (Box 25.9).
Linkage Studies of Disease Trait
The high degree of polymorphism makes microsatellites very useful for the study of genetic linkage, particularly of complex disorders such as diabetes mellitus. Study of the PCR-amplified microsatellites has successfully established the linkage of disease traits, for example type 1 diabetes mellitus, to certain areas of the human genome.
Diagnosis of Infectious Diseases
Bacterial, viral or protozoal DNA is amplified for diagnosis of diseases before they manifest clinically. As few as 10 tubercle bacilli (the bacteria causing tuberculosis) per million human cells, for example, can be detected using PCR amplification.
Archaeology and Paleontology
PCR is opening new fields of molecular archaeology and molecular paleontology because of its potential in cloning DNA fragments from mummies and the remains of extinct animals. Amplifications of the rare, surviving fragments of the ancient DNA samples is providing useful insight into evolution. Comparisons of DNA sequences of the extinct and living species can reveal missing links in evolution.
E PCR-based Methods in Advanced DNA Research
Several methods used in DNA research are based on use of PCR. Some of the more important ones are given here.
Restriction Site Polymorphism (RSP)
The restriction fragments obtained after digestion with appropriate restriction enzymes are PCR amplified prior to their detection. Thus, RSP is considered a small scale equivalent of RFLP.
Mutation Detection by Allele-Specific PCR
Standard PCR uses two primers to amplify both strands of the DNA, and relies on the fact that each primer will amplify the target strand. However, PCR can be performed using allele-specific primers, which will amplify one allele of the target gene. Their use is based on the principle that amplification of target DNA relies upon the presence of complete hybridization of primer and template: polymerization will be most effective when the primer and target are 100% complementary. If, in particular, the base at the 3’ end of the primer does not match the base in its target strand, the new DNA synthesis is severely impaired.
Application of allele-specific primers to detect single base substitution in sickle cell anaemia, is discussed here.
Allele-specific primers are constructed whose 3’-terminal base corresponds to the site of mutation. One of these primers (the s-primer) contains a sequence, the extreme 3’ base of which recognizes the (HbS) mutation. Therefore, this primer will hybridize only with the mutant (sickle cell) DNA, and amplify it. The extreme 3’ base of the other primer (the a-primer) detects normal allele, and therefore it amplifies only the normal allele.
← s-primer (recognizes the sickle cell allele; amplifies mutated gene).
← a-primer (recognizes the normal allele; amplifies normal gene).
PCR is performed separately with the two allele-specific primers, the PCR products electrophoresed and then identified.
Detection of Deletions in Genes Causing Diseases
Some genetic diseases are caused by large deletions of DNA segments that remove one or several exons, or occasionally whole of the gene. PCR is used to identify such deletions by a technique termed deletion scanning, which is performed using primers that generate PCR products of different sizes (Fig. 25.13 ). Its application in the diagnosis of Duchenne muscular dystrophy (DMD) is discussed here.
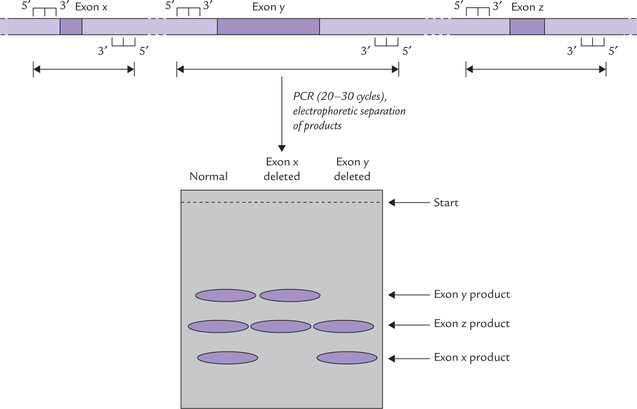
Fig. 25.13 The principle of deletion scanning with the PCR. Note that the primer pairs are designed to generate PCR products of different lengths that can be separated from each other by polyacrylamide gel electrophoresis (  = exons;
= exons;  = primer).
= primer).
DMD is a severe X-linked disorder in which the deletions affect the gene for the muscle protein, dystrophin. It is the largest of all human genes, with 79 exons scattered over a total length of more than 2 million base pairs. One or several exons are deleted in DMD; but some exons are more likely to be deleted than others and are termed deletion prone exons. Specific amplification of some of these exons using primers, which generate PCR products of different sizes for each exon, can be used to screen quickly for the presence of a disease-causing deletion in an affected individual (or determine if the fetus of a carrier possesses the deletion). Such PCR is carried out in a single reaction using a single template and 5-10 different primer pairs, each generating a separate amplification product. This form of PCR is called multiplex PCR.
Detection of Microsatellite Repeats
Microsatellite repeats are the short nucleotide sequences that are present in non-coding regions, either intergenic or within introns. A typical microsatellite contains tandem repeats of a simple dinucleotide, trinucleotide or tetranucleotide (see Chapter 24). Amplification of the DNA including the microsatellite repeat is performed by PCR using a radio-labelled primer and the products are then size-fractionated onto a polyacrylamide gel to aid resolution.
One important feature of these microsatellite repeats is that they are highly pleomorphic, i.e. the number of tandem repeats in a particular microsatellite may vary among different individuals. For example, a sequence may be repeated only 5 or 6 times in some people and more than 20 times in others. Indeed, we can expect different repeat numbers not only in different people but also on the two homologous chromosomes that carry the repeat in the diploid cells. As a result of this high degree of polymorphism, microsatellite repeats are of great value in the study of genetic linkage and the forensic application of DNA fingerprinting.
IX Human Genome Project: Current Status and Future Goals
Human Genome Project (HGP) is a large multicentric, international collaborative venture, the main aim of which is to obtain the DNA sequence of the entire human genome. It is a gigantic effort considering the fact that human genome (with 33,000-44,000 genes; 3.1 × 109 base pairs) is 25 times as large as any previously sequenced genome and eight times as large as sum of all such genomes. Ever since its initiation a decade earlier, it has been marked by relentless coordinated drive involving at least 20 groups from USA, UK, France, Japan, Germany and China, thus reflecting the scientific community at its best. The main objectives set out early in history of the project include:
1. To obtain complete sequence of pooled DNA extracted from cells donated by several anonymous donors, so as to determine the sequence of DNA in each chromosome.
2. To construct genetic map for facilitating genetic linkage studies.
3. To discover all human genes to allow further study of human genetic diseases.
4. To develop simplified and automated technology for DNA sequencing process.
HGP was initially a public research effort, but subsequently a private company, Celera Genomics (Rockville, MD) undertook the sequencing task. In a joint announcement by the US Government and Celera Genomics, a successful completion of the project was declared in June, 2000. About 90% of the gene rich portion of the genome (euchromatin) was declared to be sequenced and assembled; the latter term used to describe the process of using computers to join up bits of sequences into a larger whole. There were, however, gaps and errors in the sequence and the two groups (Cellera Genomics & US Government) agreed to continue cooperation to complete the sequencing task as soon as possible.
By independent methods the two groups declared the following results:
• The total number of genes in the human genome is in the range of 33,000 to 44,000.
• Approximately 75% of these genes have the same DNA sequence in all individuals, except for those with rare mutations.
• Any two human genomes are approximately 99.9% identical in sequence. The apparently insignificant difference of 0.1% has a highly significant effect on personality, behaviour, intelligence, disease susceptibility and other traits.
Building upon these achievements, the goals for the HGP have been expanded, as discussed below (Source: Collins et al., New goals for the US HGP: Science, 1998; 282: 683):
1. Establish the complete human genome sequence and to make it freely accessible.
2. Improve the sequencing technology by developing new and more effective methods.
3. Analyze sequence variations in the human genome, such as single nucleotide polymorphisms (SNPs) and other DNA sequence variations.
4. Develop technology for functional genomics. It includes: development of additional cDNA resources and technology for detailed analysis of gene expression; comprehensive study of functions of non-protein coding sequences; and encourage development of technology for global protein analysis.
5. Study comparative genomics by completing the genome sequence of some model organisms (e.g. Drosophilia, mouse, etc.) which would enhance our understanding of the human genome.
6. Consider ethical, legal and social implications of the vastly expanding knowledge base. It is anticipated that clash of this new and advanced knowledge with the pre-existing ethical and philosophical perspectives may result in undesirable consequences, which have to be taken care of.
7. Develop bioinformatics and computational biology, to impart advanced training to young scientists and encourage establishment of academic careers in genomic research.
The medical implications of the huge amount of genetic information obtained from the HGP are tremendous. It would serve as resource for identification of the human disease genes. For example, the oncogenic sequence changes in cancer cells can be directly identified by comparing cancer genome sequences against draft genome. Advancement in biotechnology with regard to development of useful genes would expand the scope of gene therapy and open new ways of combating disease (Collins FS. Positional cloning moves from periditional to traditional. Nature Genetics 1998; 9: 347). It may even initiate new fields such as pharmacogenomics which would individualize therapies depending on genetic make up of the patient.
Exercises
Essay type questions
1. What are the steps involved in isolating a particular clone from a genomic library?
2. How does a restriction enzyme differ from a pancreatic DNase? What is meant by blunt ends and sticky ends as applied to DNA molecules?
3. What is meant by genomic clone and cDNA clone, and how do they differ from each other? Give one example each to give their specific applications in genetic engineering.
4. Enumerate the importance and principle of the polymerase chain reaction, specifying the requirements for it to be used.
5. What is Human Genome Project? State its implications in the study of health and disease.
6. Write briefly about reverse transcriptase, its biological significance and usefulness in recombinant DNA technology.
*To learn further about these aspects, the student may refer Figure 25.8 gives a detailed account (of use of the plasmid pBR322), which the undergraduates may skip.