Molecular Biology IV: Eukaryotic Chromosome And Gene Expression
Size of human genome is much larger and its organization is much more complex than that of prokaryotes. It contains about 1000 times more DNA than E. coli (3.1 ×; 109 base pairs vs 4.2 ×; 106 base pairs). The genomic material of a human cell is packaged in 23 pairs of chromosomes. Each chromosome contains only one duplex DNA, which may be several centimeters long. In contrast to the circular shape of the bacterial DNA, the eukaryotic DNA is linear. A unique feature of the latter is the presence of vast expanse of non-coding DNA sequences. These sequences separate some 1000–6000 genes present in a single DNA molecule. According to the findings by Human Genome Project, human beings contain 33,000–44,000 genes in their genome.
The total length of all the DNA in a single human cell is about 2 m (in E. coli it is 1.4 mm only). The DNA is tightly packaged in association with proteins called histones. Structural features of the complex between the two, referred to as nucleohistone, have been described earlier (Chapter 21).
The processes of DNA replication, transcription and translation in eukaryotes are fundamentally similar to those of prokaryotes. However, they are more complex, and have certain distinctive features. Specialized features of eukaryotic gene and its expression will be discussed in this chapter.
After going through this chapter, the student should be able to understand:
• Differences in the structural organization of genome of eukaryotes (from prokaryotes) and know their implications for gene control.
• Role of eukaryotic DNA polymerases; steps of eukaryotic cell cycle and biochemical events in each step; details of steps of transcription (i.e. binding of polymerase, initiation, elongation and termination); and post-transcriptional modification and processing of RNAs (mRNA, rRNA and tRNA).
• Fundamental differences between the eukaryotic and the prokaryotic protein biosynthesis and various levels at which control of gene expression can be exercised in eukaryotes.
I Replication has Multiple Sites of Origin
DNA replication has thousands of origins in humans. Approximately one origin exists for every 10,000 to 100,000 base pairs. Bidirectional replication from these origins, termed replication bubbles, proceeds semicon-servatively until the daughter strands are complete. The new duplexes then separate; each contains a parent and a daughter strand (Fig. 24.1 ).
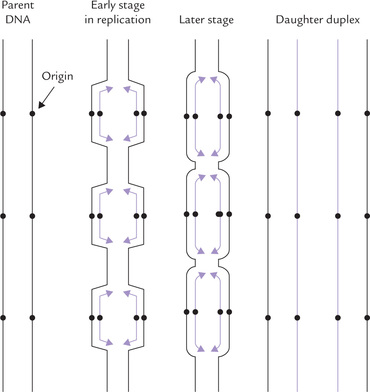
Fig. 24.1 Replication of the eukaryotic chromosomal DNA starts from multiple origins, one every ten thousand to one lakh base pairs (••• indicates sites of origin).
Multiple origins make the replication process sufficiently fast so as to complete replication of a DNA molecule in about 9 hours. It is noteworthy that eukaryotic (DNAPs) are much less efficient when compared to their bacterial counterparts. The α-DNAP (a eukaryotic enzyme) can add only 100 base pairs per second, 10 times slower than the prokaryotic DNAP III. This may be because of more complicated arrangements of DNA in eukaryotic chromosomes. Considering the large size of eukaryotic chromosome, it would take long time (up to 3 weeks) to replicate it fully if there were only one pair of replication fork per chromosome. Multiple sites of origin ensures that replication of total genome takes less time than replication of a bacterial chromosome.
 The eukaryotic DNA replication starts from several origins—approximately one in every ten thousand to one lakh base pairs, and therefore replication does not take much longer in humans compared to bacteria.
The eukaryotic DNA replication starts from several origins—approximately one in every ten thousand to one lakh base pairs, and therefore replication does not take much longer in humans compared to bacteria.
All regions of a chromosome are not replicated simultaneously. Rather, many replication bubbles (consisting of two replication forks moving in opposite directions) will be found in one region of the chromosome, but none in the other. The ends of chromosomes, called telomeres play a vital role in eukaryotic replication.
A Eukaryotic DNA Polymerases
Five distinct eukaryotic DNA polymerases are known, which are designated in order of their discovery. These are: DNA polymerases α, β, y, δ and ε (Table 24.1 ). The eukaryotic DNAPs are not as well characterized as their bacterial counterparts.
Table 24.1
Properties of eukaryotic DNA polymerases
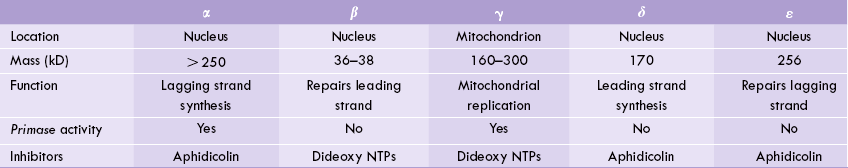
All DNAPs except α and β possess 3’ to 5’ exonuclease activity.
Polymerase α is composed of four distinct subunits with different catalytic activities. One of its subunits constitutes a primase enzyme, capable of synthesizing short RNA transcript that can serve as primer for DNA chain elongation. Polymerase α is a moderately processive enzyme: it can add about 100 nucleotides per binding event. Its activity correlates with cellular proliferation, being high when there is rapid cell division and dropping to a low level in quiescent cells. Evidently, this enzyme has a direct role in chromosomal DNA synthesis.
It was believed that polymerase α is the polymerase for the synthesis of the leading strand. However, recent evidence suggests that polymerase δ may be the key enzyme for the leading strand, whereas polymerase α is primarily for the lagging strand. This appears beneficial as the enzyme with moderate processivity is more suited for the enzyme synthesizing the lagging strand. The polymerase δ possesses proofreading ability (3’–5’ exonuclease activity), which contributes to the fidelity of replication.
Polymerase β and ε are for repairing the damaged DNA like δ-DNAP; ε-DNAP also has high processivity and possesses 3’ to 5’ exonuclease activity. Polymerase γ most likely copies the mitochondrial DNA. It possesses 3’ to 5’ exonuclease activity though it is not significant. Therefore, error rate during mitochondrial replication is much higher than that during chromosomal replication. The polymerase γ has some primase activity.
B Role of Telomeres in Eukaryotic Replication
The ends of mammalian chromosomes are formed by the telomeres (Greek: telos = end) which consist of the repeat sequence (TTAGGG)n. This sequence is repeated in tandem (one after another) between 500 and 5000 times in each telomere. Telomeres play a crucial role in eukaryotic DNA replication. Without them a linear chromosome would become progressively shorter with each cell division and the cell’s descendants would ultimately die from loss of essential genes. This is because of the inability of DNA polymerase to synthesize the extreme 5’ end of the lagging strand.

As diagrammed above, the DNA polymerase can only extend an RNA primer that is paired with the 3’ end of a template strand.
Removal of this RNA primer would leave a gap that cannot be filled by DNA polymerase (recall that DNA polymerase operates only in 5’ to 3’ direction; it can only extend an existing primer, and the primer must be bound to its complementary strand; Fig. 21.13). Consequently, in the absence of a mechanism for completing the lagging strand, DNA molecule would be shortened by the length of an RNA primer with each round of replication.
Mechanism must exist to prevent such shortening, referred to as telomere erosion. This is accomplished by telomerase, the enzyme which synthesizes and maintains the telomeric DNA. It adds tandem repeats of the telomeric sequence TTGGGG at the extreme 5’ end of the lagging strand. This telomeric sequence now acts as primer: the DNA polymerase adds an okazaki piece at its 3’ end to initiate a new round of replication.
Telomerase acts without benefit of template. Therefore, it provides the template itself. The template is 150-nucleotide RNA which is an integral part of the enzyme. Thus telomerase is a ribonucleoprotein (complex of protein and RNA).
Telomerase, Aging and Cancer
Telomerase is required for mortality. This is evident from the following observations:
1. Germline cells which express telomerase are immortal.
2. Somatic cells that are not capable of expressing telomerase have only a finite life span. Lack of telomerase activity may contribute to cell aging and ultimate death.
3. Besides germline cells, cancer cells express telomerase and are immortal.
Though lack of telomerase activity in somatic cell appears to be a disadvantage, does it offer some selective advantage to multicellular organisms? A possibility is that mechanism of cellular aging and death protects multicellular organisms from cancer.
Because telomerase apparently stabilizes even short telomeres, inhibition of telomerase should eventually lead to complete loss of telomeres and hence cessation of cell division. This hypothesis makes telomerase inhibitors an attractive target for anti-tumour drug development.
C Cell Cycle
In eukaryotic cells, replication of DNA and cell divisions occur at different times, separated by gap phases. These processes ultimately lead to the formation of a pair of daughter cells from a parental eukaryotic cell, and are together referred to as the cell cycle.
Total duration of the cell cycle ranges from 12 to 24 hours. This period is mainly occupied by two active phases: the synthetic phase (S-phase) and the mitotic phase (M-phase), as shown in Figure 24.2 . Intervening between these two active phases are gap phases termed G1 and G2.
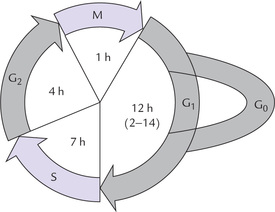
Fig. 24.2 Diagrammatic representation of the cell cycle. Mitotic (M) and synthetic (S) are the active phases, while G1 and G2 are the intervening gap phases. G0 is the silent phase wherein cells are not involved in the cell cycle, but can enter the cycle as and when required.
• During the M-phase, growth and division of the cell (mitosis) occurs. Duration of this phase is about one hour.
• During the S-phase, duplication of DNA occurs. New DNA strand is synthesized based on the formation in the pre-existing DNA template to form two daughter DNA molecules. A copy of the daughter DNA would subsequently, pass into each of the daughter cells. The S-phase occupies about 7 hours.
• The G1-phase, which precedes the S-phase, is the preparatory phase for replication. In this phase, the enzymes necessary for replicating the genome are produced and the genomic DNA is checked for integrity. Its duration is variable (12 hours: range, 2–14 hours).
• In the G2-phase, which follows the S-phase, the completion of replication of genome is confirmed. It occupies 4 hours.
Finally, at the end of a cycle the duplicate daughter chromosomes are segregated, the nuclear membrane is disrupted and the daughter chromosomes are distributed equally on either side of the parental cell.
The S-phase refers to the time when DNA synthesis occurs, and the M-phase refers to the time of mitosis. G1 and G2 are gap phases, before and after DNA synthesis respectively.
G0 phase
The events of the cell cycle are ordered, as shown in Figure 24.2. However, the cells leaving the M-phase are not absolutely committed to entering the G1 phase; they may enter a silent phase called G0 phase. Cells in the G0 phase are said to be viable but not in the cycle; they are capable of entering the cell cycle under appropriate circumstances. Duration of this phase shows great variation: few hours in most cells to several years in neurons. The duration may be subject to availability of growth factors.
Regulation
Various phases of the cell cycle are meticulously controlled. A number of regulatory enzymes are involved to ensure correct operation of the cell cycle. The regulatory process is very complex, but a simplified account is presented below. The regulatory enzymes are protein kinases which are commonly referred to as the cell cycle kinases. A cyclic variation in catalytic activities of these enzymes is observed as the cell cycle proceeds from one phase to another. Activities of protein kinases in turn depend on regulatory subunits, called cyclins. The cyclins make periodic appearance during the cell cycle. Although they are synthesized throughout the cell cycle, they are abruptly destroyed during mitosis.
Accumulation of the cyclins during cell cycle promotes phosphorylation of the protein kinases. This results in activation of the latter (these enzymes being active in the phosphorylated state), and the activated enzyme causes phosphorylation of chromosomal scaffold proteins, his-tones and nuclear lamins. These changes signal onset of mitosis. This is followed by appearance of proteases which remove the cyclins. The result is M-phase to G1-phase transition.
II Untranscribed Human DNA
The organization of eukaryotic DNA is strikingly different from prokaryotic DNA. E. coli has 4.2 million base pairs, most of which encode for proteins. A total of approximately 2000 polypeptides are encoded by the bacterial genome. In contrast, most of the human DNA is not involved in protein synthesis. Only about 3–4% is active in this regard and the rest of the 96–97% DNA is non-coding and remains untranscribed. This can be estimated from the fact that the size of human genome (3.1 X 109 base pairs) is nearly a thousand times as large as that of the E. coli genome, which is sufficient to code for more than 15 lakh polypeptides. But the number of polypeptides it actually encodes is much smaller (about 100,000); it is about 50-fold, rather than a 1000-fold, greater than the number of polypeptides encoded by the E. coli genome. Thus, it is far less compact (Box 24.1). Most of the non-coding sequences are present between the adjacent genes, while a few are present within the gene. Since no obvious function is performed by the transcriptionally inactive DNA, it is referred to as the junk DNA.
What is the function of the non-coding DNA which exceeds several billion base pairs? A convincing answer is yet to come. No doubt, this has been a mysterious scenario, it is challenging as well to elucidate the function of this seemingly useless DNA.
III Gene Distribution Along DNA
Genes are transcribed as continuous sequences, but only some segments of the resulting mRNA molecule contain information that codes for the protein product of the gene. These sequences are called exons. The regions between exons are called introns, which are the untranslated, intervening sequences lying within the gene. Thus the exons are interspersed with long stretches of introns. The introns are transcribed along with exons, but the intron sequences are subsequently removed by specific enzyme systems. After removal of these sequences, a continuous coding sequence is produced (Fig. 24.3 ). Most human genes have few to several dozens of introns. The total length of introns may exceed that of exons by several times.
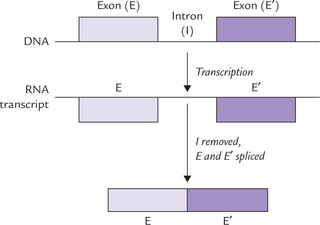
Exons are those parts of the gene that are represented in mature RNA, whereas introns do not appear in the functional RNA product.
A Repetitive Sequences in Human Genome
A vast majority of DNA in human genome consists of unique or nearly unique sequences. Other sequences are repeated hundreds or thousands of times and are scattered throughout the genome. They constitute approximately 30–40% of the human DNA. Two major classes of the repetitive sequences have been recognized: highly repetitive sequences (also called simple sequence DNA) and moderately repetitive sequences.
Highly Repetitive Sequences
These are present at > 106 copies per genome. They are clusters of nearly identical oligonucleotide sequences, each between two and a dozen base pairs, that are repeated over and over in tandem arrangement (next to each other) in a DNA molecule. Highest concentrations of such sequences are present (i) in centromere, (ii) in telomere regions of eukaryotic chromosomes, and (iii) smaller agglomerations are located outside these, in junkn DNA.
(i) Centromeres, the region attached to the microtubular spindle during mitosis, help align chromosomes and facilitate recombination. They contain short oligonucleotide sequences that are repeated more than a million times in the genome.
(ii) Telomeres, as already stated, form ends of mammalian chromosomes and consist of a repeat sequence (TTAGGG)n. This sequence is repeated in tandem between 500 and 5000 times in each telomere.
(iii) Outside telomere and centromere, the repetitive simple sequence DNA is present in junk DNA of introns or untranscribed spacers. Each simple repeat unit consists of 2–5 base pairs. A repeat unit occurs from few to 50 times in a given location and the same repeats may be present in a number of different locations. The most abundant repeat is a dinucleotide repeat (AC)n that occurs at about 50,000 to 100,000 different sites. Repeats of 3, 4, or 5 bases are also common, although they are not as numerous as the dinucleotide repeats.
At least a dozen human neurological diseases result from excessively repeated trinucleotide sequences (Table 24.2, Box 24.2).
Table 24.2
Some diseases associated with trinucleotide repeats
| Disease | Repeat | Site of repeat |
| Fragile X syndrome | GGG or CCG | 5’ UTR |
| Myotonic dystrophy | CTG | Upstream region, 3’ UTR |
| Friedrich’s ataxia | GAA | Intron |
| Spingobulbar muscular atrophy | CAG | Exon |
| Huntington’s disease | CAG | Exon |
Satellites, Minisatellites and Microsatellites
The DNA in centromeric and telomeric regions is called satellite DNA. After partial endonuclease digestion, when the mixture of pieces is analyzed by a density gradient method, most of the DNA runs in one band, but the repetitive DNA runs as a ’satellite’ (separate from the main band). Likewise, the repetitive DNA fragments outside centromeric and telomeric regions present as microsatellites (total length below 1000 nucleotides) or as minisatellites (total repeat length more than 1000 nucleotides).
Moderately Repeated Sequences
Such sequences (< 106 copies per haploid genome) occur in segments of 100 to several thousand base pairs that are interspersed with larger blocks of unique DNA. The best characterized one of these sequences belongs to Alu family: human genome contains 300,000–500,000 widely distributed Alu sequences that are on average 80–90% homologous with their consensus sequence (Alu is named so because most of its ~300 base pair long segments contain a cleavage site for the restriction endonuclease Alu I).
The Alu sequences and similar sequences measure up to few hundreds. Bases are referred to as short interspersed repeat elements (SINEs). In addition, there are long interspersed repeat elements (LINEs), which are relatively longer sequences of about 5000 base pairs. They may occur up to 10,000 times per cell. Like SINEs, the LINEs are also transcriptionally active and are usually mobile segments of DNA.
No definite function has been assigned to the moderately repetitive sequences, which are therefore termed as selfish DNA. This otherwise harmless DNA appears to be a molecular parasite that, over many generations, has disseminated itself throughout the genome through transposition. Since it puts additional metabolic burden on the replication machinery of the cell, theory of natural selection would predict its elimination. Yet this has been avoided probably because relative disadvantage of replication of additional base pairs of selfish DNA in a billion base pairs genome is so slight that its rate of elimination would be balanced by its rate of propagation.
The repetitive sequences are produced by the duplication of sequences, as discussed under gene families and pseudogenes in Chapter 21. This causes formation of additional copies of the genes which have no effect on the phenotypes.
B Exon Shuffling
Exons from different locations are believed to have combined to yield new functional genes. This is important in view of evolution of genes. Exons appear to be the building blocks from which various new genes have been constructed in course of the evolution. For example, fibronectin, a connective tissue protein, consists of repetitive modules; some of these modules have close relatives in the genes for some proteins of the clotting system.
Relation of Introns to Evolution
Though no specific function has been unravelled for introns, they are believed to be important for the evolution of eukaryotic genes. During evolution, a new trait is produced by development of new genes. Mutations in the existing gene produces new gene. However, mutation in the functional gene may have deleterious consequences for the organism. Hence the non-functional genes are mutated to produce new traits. Simply stated, the introns may be prototype of the future functional genes.
IV Transcription
Transcription is more complex in eukaryotes. It occurs mainly in the nucleus, where only one of the two DNA strands, i.e. the antisense (–) strand is copied by RNA polymerases. Transcription also occurs in mitochondria to a limited extent where both strands are copied, but one is subsequently degraded. Not more than 3–4% of the human DNA codes for proteins, and the remaining 96–97% consists of the non-coding sequences. Most of the non-coding sequences are transcriptionally inactive and lie between the adjacent genes.
Other non-coding sequences are present within the genes. These non-coding sequences (called introns) are transcribed, but they do not contain information that codes for the gene’s protein product. They are removed by nuclear enzymes before the mRNA reaches ribosomes.
Most of the human genes are patchwork of exons (whose mRNA transcripts are processed to mature RNA) and the untranslated intervening sequences, the introns. After removal of introns, the exons form a single, continuous coding sequence (Fig. 24.3).
Some genes have only 1–2 introns, others may have several dozens, whereas some are intronless (e.g. histone genes).
A Types of RNA Polymerases
In contrast to a single RNA polymerase (RNAP) in prokaryotes, in eukaryotes there are four RNA polymerases— three nuclear and one mitochondrial (Table 24.3 ). This was demonstrated by studying the action of α-amanitin (a toxin from mushroom, Amanita phalloides) which inhibits different RNAPs to different extents. Molecular weight of these enzymes range from 500,000 to 600,000.

RNAP I is found in the nucleolus. It synthesizes a large 45S RNA precursor which is subsequently cleaved to three smaller molecules (5.8S, 18S and 28S rRNAs). RNAP II is the principal enzyme which synthesizes almost all mRNA precursor, also known as heteronuclear (hn) RNA. It is present in nucleoplasm and is inhibited by low concentration of α-amanitin. RNAP III is responsible for the formation of small species of RNAs, such as 5S rRNA and tRNAs. It is moderately sensitive to inhibitory action of α-amanitin, being inhibited only by high concentration of the latter. It also transcribes the gene for the 7S RNA associated with signal recognition particle (SRP). The latter is involved in the translocation of proteins across the endo-plasmic reticulum membrane.
B Chemistry of mRNA Synthesis
The basic processes involved in eukaryotic mRNA production are the same as in prokaryotes. The process occurs in three stages: initiation, elongation and termination.
Initiation
About 105 initiation sites are present in the entire DNA. RNAP recognizes these and moves along the DNA rapidly, incorporating about 1000 base pairs per second in the new RNA.
How does RNAP recognize the correct DNA strand and initiate RNA synthesis at the beginning of a gene? RNAP binds to its initiation site through the base sequence known as the promoter—the basic recognition unit signalling that there is a gene nearby and can be transcribed. A promoter lies on the same strand as the gene being transcribed and is commonly referred to as cis-acting element. Other cis-acting elements include response elements and enhancers, which (together with promoters) influence specificity and efficiency of transcription. Although the promoters are essential for the initiation of transcription, they are not alone in influencing the efficiency of transcription.
Promoters
These are perhaps the most important and fundamental elements required for initiation of transcription. Promoters are nucleotide sequences in front (upstream) of the start site, located up to –200 base pairs (Table 24.4 ). They ensure correct positioning of the RNA polymerase II at the transcription start site (i.e. the first base copied into RNA), which is designated as number 1. Promoters of different genes look quite different, but some basic key elements occur with great regularity in all of them. These elements may be present in varying combinations, some elements being present in one gene but absent in another. However, some form of promoter element is virtually always present.
Table 24.4
Some promoter elements in eukaryotes
| Element | Location | Sequence |
| TATA box | Centred at –25 bp | TATAA |
| GC box | Between –40 bp and –110 bp | GGGCGG |
| CAAT box | Between –40 bp and –110 bp | GGCCAATCT |
| Ig octamer | Up to –200 bp | ATTTCGAT |
bp = base pairs.
Some of the common elements found within the promoter region are: initiator region, TATA box, and the upstream elements such as GC and CAAT boxes.
• Initiator region: The start site itself has a recognizable short sequence between nucleotides –3 and +5, called initiation (Inr). It is the simplest form of promoter known to be recognizable by RNAP II, and its structure varies from gene to gene. However, in general, it has the nucleotide sequence of Py2CAPy5 (P = pyrimidine base, C = cytosine, A = adenine).
• TATA Box (Hogness Box): About 80% genes have a TATA box centred at about – 25 bp whose sequence is reminiscent of the Pribnow box in the prokaryote promoter. It has an 8 bp consensus sequence that consists entirely of adenine-thymine base pairs, although very rarely a guanine-cytosine pair may be present.
• Upstream elements: Further upstream elements, within about 40–200 or so base pairs of the start site are elements or boxes. These are short DNA sequences that are recognized by specific proteins called transcription factors. Three common upstream elements are the CAAT box, the GC box, and an eight base pair octamer box; these are mostly located at sites within the –100 to –200 region in different genes. They occur less regularly than TATA box; for example, CAAT and GC consensus sequences occur only in 10–15% of eukaryotic promoters.
Table 24.4 summarizes location and consensus sequence cis-acting elements seen within promoters.
Note that boxes and elements are accepted terms, but these short stretches of bases are not in any sense boxes nor do they have anything to do with atomic elements.
Enhancers and silencers
These are regulatory DNA sequences, which respectively increase or decrease the rate of transcription by binding to regulatory proteins. It is the bound proteins that increase or decrease the rate of transcription, thereby acting as specific transcription factors.
Positive control of transcription by enhancer elements is far more common than negative control by silencer sequences.
Some remarkable features of the enhancers are:
• They can be located far away from the gene—even thousands of base pairs away.
• Their orientation in the DNA can be reversed without impairing their function.
• Enhancers often contain a number of elements that are recognized by transcription factors.
Enhancers may lie upstream or even downstream of the start-point of transcription. For example, in the immunoglobulin gene the enhancers may be present downstream, located within the intron of the gene being actively transcribed.
Response elements
These elements are nucleotide sequences that allow specific stimuli, such as steroid hormones, cyclic AMP, or insulin-like growth factors to control gene expression. They are often found within 1 kilobase of the start point, and a single gene may possess, any number of different response elements.
An interesting and somewhat disturbing aspect of eukaryote gene control is its apparent haphazard nature. No one element is essential for transcription and different genes have different combinations of elements. Twenty per cent of genes lack TATA boxes, some lack other elements and some have multiple copies of an element. It is quite different from the comparatively orderly situation in prokaryotes.
Mechanism of Initiation
Most of the promoter elements promote transcription only if they are located on the correct (non-template) strand of the double helix and in the correct 5’ to 3’ orientation. The sequence-specific DNA-binding proteins that recognize and bind to promoter elements are called general transcription factors. They assemble on promoter to form a complex, termed basal transcription complex, which enables RNAP to initiate transcription. In addition to these general transcription factors, the eukaryotic cells contain other regulatory proteins, which act as specific transcription factors, regulating transcription of some, but not all genes.
The basal initiation complex
In prokaryotes, the sigma subunit of RNAP recognizes the correct binding site on a promoter and binds directly to the DNA. In eukaryotes, there is no functional equivalent of the sigma subunit; instead a whole retinue of proteins, called general transcription factors (GTFs), assemble into a complex on the promoter. The RNA polymerase attaches to this complex, termed the basal initiation complex, and this is needed for the transcription of all type-II genes.
Figure 24.4 shows formation of the complex. First a general transcription factor, universally present in eukaryote cells, called TATA-bindingprotein (TBP), attaches to TATA box (Fig. 24.4). Eight or more other proteins called TAFs (TBP associated factors) then get associated with this, forming a complex known as TFII-D. This is followed by attachment of other general transcription factors to DNA in the region of the promoter; to from the basal initiation complex. The GTFs serve three important roles: (1) Facilitates attachment of RNAP II to the promoter at the correct nucleotide for initiation, (2) destabilizes (helicase activity) the DNA at the promoter and (3) initiates transcription.
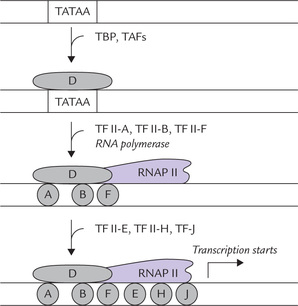
Fig. 24.4 Transcription factors assemble on promoter to form a complex which enables RNAP to initiate transcription. Placement of various factors in this diagram is arbitrary; their exact position is not shown (TBP = TATA-binding protein, TAFs = TBP associated factors; TFII-D = Transcription factor D for polymerase II [likewise TFII-B, TFII-F, TFII-E, TFII-H and TFII-J refer to the respective transcription factors; D, A, B...J are abbreviations for these transcription factors]).
A description of functions of all GTFs is beyond the scope of this book; however, some have been outlined in Table 24.5 .
Table 24.5
The General transcription factors
| Factor | Subunits | Function |
| TFII-D | 13 | Recognizes TATA box; recruits TFII-B (composed of factors TBP and TAFs) |
| TFII-A | 3 | Stabilizes TFII-D binding |
| TFII-B | 1 | Orients RNAPII to start site |
| TFII-E | 2 | Recruits TFII-H (helicase) |
| TFII-F | 2 | Destabilizes non-specific RNAPII-DNA interactions |
| TFII-H | 9 | Promotes promoter melting by helicase activity |
* RNAPII contains nine subunits.
Histones inhibit initiation of transcription
When tightly bound by histones, the DNA of native chromatin is unavailable for transcription. A histone may be bound over the promoter or enhancer of a gene, thereby inhibiting initiation of transcription. During transcription, the regular packaging of DNA into nucleosomes is altered so that the decondensed chromosome is accessible to tran-scriptional proteins. Apparently, histones have to be displaced from promoters and enhancers by competitive binding of certain protein factors, which thereby disassemble chromosomes. A distinct class of protein factors has been identified, for example, RSF (remodelling and spacing factor), which facilitates transcription initiation on chromatin templates in vitro. Another one is FACT (facilitates chromatin transcription) that promotes elongation of RNA chains through nucleosomes. Together, RSF and FACT disassemble nucleosomes and permit transcription initiation to occur.
GTFs that control the initiation of transcription at the TATA box and the protein factors that disassemble nucleosomes, together with specific DNA sequences are important for initiation of transcription.
The given description makes it clear that the initiation stage is more complex in eukaryotes and is better controlled as per cellular requirements. The next two stages— elongation and termination—are remarkably similar to those in prokaryotes.
C Transcription Factors
General characteristics
Transcription of a human gene is initiated and regulated by a number of transcription factors. These factors must bind to double-stranded DNA for transcription to occur. The site-specific binding results from the ability of a relatively small area of the protein to come into close contact with the double helix of the DNA. The regions of these proteins that contact the DNA are called DNA-binding regions or motifs, and are highly conserved between species. These proteins are active in a dimeric state, either as homodimers of two identical subunits or as heterodimers of slightly different subunits. The response elements of these proteins show a dyad symmetry (dyad means two units treated as one, or a group of two) that matches the symmetry of the dimers.
The binding of proteins with DNA involves weak hydrogen bond formation between amino acid side chains of the protein and the DNA bases, though additional stabilizing bonding to the sugar-phosphate-sugar backbone may occur. Individually a hydrogen bond is weak, but the average DNA-binding protein may have 20 or more sites of contact, which considerably increases the strength and specificity of the contact.
Structural elements
The following structural elements have been recognized in the DNA-binding proteins:
1. DNA-binding domain: It is the protein recognition motif that makes contact with the DNA bases, predominantly in the major groove of the double helix, where the edges of the bases are exposed. Contact is made by an α-helix, 20–40 amino acid long, having a high content of basic amino acid residues. Positive charges on these amino acids permit interaction with the negative DNA bases. The DNA-binding domain is highly conserved among different species.
2. Dimerization domain: It allows the protein to assume the active dimeric state. The dimerization is effected by an amphipathic α-helix that forms a two-stranded coil.
3. Transcription activating (or inhibiting) domain: It is required for increasing (or decreasing) the strength of transcription.
Major classes
Four major classes of DNA-binding domains in transcription factors have been recognized (Fig. 24.5 ). Salient features together with examples of each are as follows:
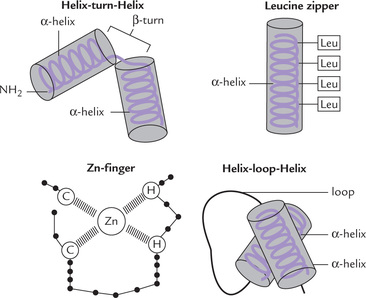
1. Helix-turn-helix proteins: This was the first type to be identified.
• Structural features: It has two α-helices linked by a β-turn (Fig. 24.5). One of these two (called the recognition helix) grips DNA by fitting into the major groove of the double helix. This allows precise alignment of the transcription factor in relation to the DNA sequence recognized.
• Examples: Proteins regulating embryonic development, e.g. homeodomain proteins and lambda repressor.
• Structural features: The whole molecule is an α-helix, having a (i) DNA-binding basic region, and (ii) a dimerization helix which has hydrophobic leucine residues consistently on one face of the helix. The regularly spaced leucine residues form a hydrophobic phase, which allows two such subunits to interact by hydrophobic forces between the leucine side chains. This brings about dimerization of the two subunits. The term “zipper” is a misnomer resulting from the initial belief that the leucine residues were interdigitated.
• Examples: Growth regulators, proto-oncogenes, protooncogene products, e.g. c-Myc, c-Fos, c-Jun.
• Structural features: In this motif, there are loops or fingers of amino acids with a zinc ion at their core. Each finger consists of approximately 12 amino acids. The zinc atom attached to four amino acid residues (either 4 cysteines, or two cysteines and two histidines) stabilizes the finger-like structure.
• Examples: Intracellular receptors to which steroid hormones bind have variants of the zinc-finger theme.
Functions
Specific interaction of the DNA-binding proteins with response elements is required not only during development, but also within tissues of the mature organism for a number of important functions. These include:
1. Differential expression of genes in tissues: Though DNA compositions from all cells of the same organism are same, some genes are optimally expressed in certain tissues and remain silent in others. Genes for transthyretin (a plasma protein), for example, are expressed in liver only. About 10 upstream binding sites have been recognized for at least five different DNA-binding proteins. These proteins are present in much higher concentration in hepatocytes than in any other cell type, and so transthyretin synthesis occurs only in liver.
2. Growth and development: Cell growth, differentiation of stem cells into fully differentiated cells, and embryogenesis are controlled by transcriptional regulator proteins. Sequential appearance or disappearance of these proteins in course of growth and differentiation effects regulation of transcription.
3. Activation of hormonal response elements: The receptors for steroid hormones, thyroid hormones, retinoic acid and calcitriol are transcription factors (zinc-finger proteins). Formation of hormone-receptor complex activates the latter, allowing it to bind response elements and stimulate transcription.
4. Expression of viral genes: Several viral genes can be expressed only after the cellular transcription factors bind to the promoters or enhancers of the viral genes.
Regulation of activity
Transcription factors are regulated at various levels by following means:
1. Covalent modification: Some transcription factors are phosphoproteins, and are regulated by phospho-dephosphorylation, effected respectively by protein kinases and phosphatases. Second messengers of the water-soluble hormones regulate transcription by inducing phosphorylation of transcription factors.
2. Allosteric effects: Some small molecules have been observed to bind transcription factors reversibly, and act as positive or negative modulators of their activities.
3. Several transcription factors are regulated at the level of their own synthesis.
D Post-transcriptional Processing
All three classes of RNA are synthesized as larger primary transcripts, also known as heterogeneous nuclear (hetero-nuclear, hn) RNAs. hnRNAs are usually much larger than the RNAs found in the cytoplasm and require processing to a small size before they can carry out their functions.
The extent and type of processing are different for each type of RNA. Processing of tRNA and rRNA is similar to that in prokaryotes, as described in Chapter 22. However, eukaryotic mRNA undergoes extensive modifications, in contrast to prokaryotes where it is rarely subject to any post-transcriptional processing.
Processing of mRNA
Three processing steps occur almost uniformly with eukaryotic mRNA, as summarized in Figure 24.6 .
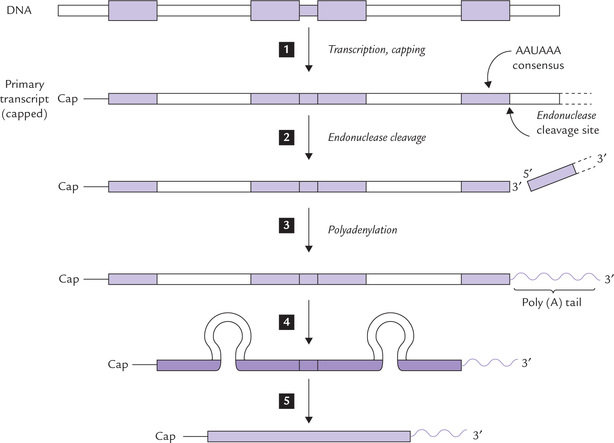
Fig. 24.6 Post-transcriptional processing of mRNA in eukaryotes. All processing steps occur in the nucleus. The mature mRNA is transported into the cytoplasm, where it is translated by the ribosomes. Steps  = exon.
= exon.
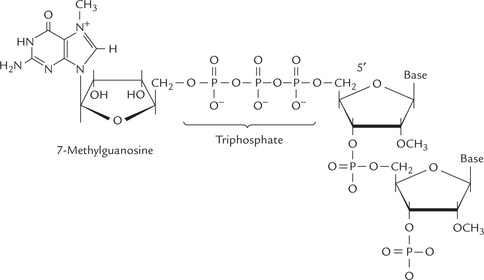
The 5’ end of the mRNA is capped
Shortly after initiation of mRNA synthesis, a guanosine residue is attached at the 5’ terminus in an unusual 5’-5’ triphosphate linkage. The N-7 of the terminal guanine is then methylated by S-adenosylmethionine. This unit 7MeG-5’PPP-5’G is called a cap (Fig. 24.7 ). The cap is thought to protect the 5’mRNA from the action of 5’-exonucleases, to facilitate its binding to the ribosome and is important for subsequent splicing reactions.
The 3’ end of the mRNA receives a polyadenylate tail
Most eukaryotic mRNAs contain a long (up to 250 residues) poly(A) tail at the 3’ end, which is added to the mRNA before it can leave the nucleus. The polyadenylation signal resides in a highly conserved AAUAAA consensus sequence, lying near 3’ end of the primary transcript. This sequence is recognized by a specific endonuclease that cleaves the RNA approximately 20 nucleotides downstream. The newly created 3’ terminus, however, serves as primer for the enzymatic addition of up to 250 adenine residues.
The polyA tail is believed to associate with proteins that retard action of 3’-exonucleases.
The removal of introns and rejoining of exons
The eukaryotic genes are patchwork of exons, which are represented in the mature mRNA and introns, which do not appear in the mature mRNA as they are spliced out of the primary transcript. The removal of introns requires nuclear enzyme complexes, called spliceosomes.
Mechanism of Action of Spliceosomes
The spliceosome contains five small RNAs (U1, U2, U4, U5 and U6) each between 106 and 185 nucleotide long. These are associated with proteins to form small nuclear ribonucleoprotein particles (mRNPs or “snurps"). The individual small RNAs apparently recognize specific conserved nucleotide sequences in the mRNA precursor by RNA-RNA base pairing. As shown in Figure 24.8, these conserved sequences are present at:
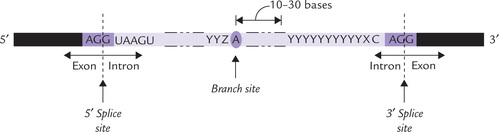
• The 5’ exon-intron boundary (5’ splice site).
• The 3’ intron-exon boundary (3’ splice site).
• A branch site within the intron, approximately 30 bases from the 3’ end.
Roles of snRNAs are summarized in Table 24.6 .
Table 24.6
| SnRNA | Size (no of nucleotides) | Function |
| U1 | 165 | Binds 5’ splice site |
| U2 | 185 | Binds the branch site within an intron |
| U4 | 145 | Helps assemble the spliceosome* |
| U5 | 116 | Binds 3’ splice site |
| U6 | 106 | Helps assemble the spliceosome* |
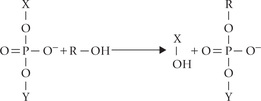
The removal of an intron and rejoining of two exons is based on two transesterification reactions. In this, a phosphodiester bond is transferred to a different –OH group, as diagrammed below.
If X-Y were an RNA chain, it would be broken. Note that energy is not required in the process and there is no hydrolysis.
The removal and rejoining can be considered to occur in the following steps (Fig. 24.9 ):
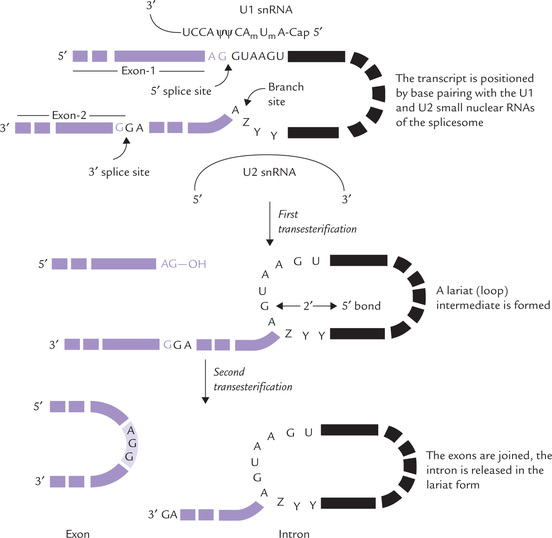
Fig. 24.9 Splicing of introns from the transcripts of protein-coding genes in eukaryotes. The lariat intermediate has a 2’ → 5’ (A→G) phosphodiester bond (Z = purine, Y = pyrimidine, ψ = pseudouridine, Am and Um = 2-O-methylated adenosine and uridine).
1. Positioning: The transcript is positioned by base pairing with the U1- and U2-snRNA, which respectively bind to the 5’ splice site and the branch site within the intron.
2. The first transesterification: The 2’-OH of an adenine residue at the branch site within the intron plays a crucial role in the first transesterification. This 2’-OH group attacks the phosphate bond of a guanosine residue at the 5’ splice site, forming a lariat structure (named so because of its resemblance to cowboy’s lariat). This breaks the chain at the 3’ end of the exon 1.
3. The second transesterification: Breaking of chain at 3’ end of exon 1 produces a free 3’-OH which attacks and cleaves the 5’ end of exon 2, thus joining the two exons. The intron is released in the lariat form.
It is evident that RNA itself is able to catalyze cutting and splicing reactions, hence it is referred to as self-splicing catalytic RNA (Box 24.3).
Removal of introns takes place in the nucleus and requires ribonucleoprotein particles called spliceosomes.
Note
Mutations at a conserved site may interfere with the correct removal of introns from the precursor mRNA. Abnormal splicing may result in diseases, for example β-thalassaemia, where production of ß-haemoglobin chains is greatly reduced.
Systemic lupus erythematosus, an autoimmune disorder, results due to production of antibodies against the snRNPs. It is a serious condition, often fatal.
The most remarkable feature of eukaryotic gene expression is the extensive post-transcriptional processing of mRNA by nuclear enzyme systems. The processing may involve specific exo-and endonucleolytic cleavages to cut out unwanted sequences from the primary transcript, or removal and rejoining of segments of the transcript. A number of other modifications also occur, which include 3’- and 5’-additions, base and sugar modifications, changes in tertiary conformations, etc.
Processing of rRNAs
The genes for 18S, 5.8S and 28S rRNAs are clustered in a unit that is tandemly repeated many times. This cluster of three genes is transcribed by RNAP in the nucleolus to yield a 45S RNA. This large precursor (13 kb) is enzymat-ically modified and cleaved to yield the mature 18S, 5.8S and 28S rRNAs (Fig. 22.6). The modification occurs through methylation of over 100 of its 14,000 nucleo-tides, mostly on the 2’-hydroxyl groups of their ribose units. The methylated 45S RNA then undergoes a series of enzymatic cleavages and trimming brought about by nucleases. These changes ultimately yield the 18S, 5.8S and 28S rRNAs.
Processing of tRNA
Transcription by RNAP III produces tRNA. A distinctive feature of its transcriptional activity is that it recognizes promoters, located within the coding region of the gene. Initial modifications in the primary transcript include cleavages at the 5’ and 3’ ends, and removal of introns. Modifications of some nucleotide units occur to produce the unusual bases of the tRNA such as pseudouridine (ψ), ribothymidine (T) and dihydrouridine (D). Addition of the sequence CCA to the 3’ end of tRNA also occurs. The reaction is catalyzed by the enzyme nucleotidyl transferase; CTP and ATP serve as substrates for the enzyme.
V Translation
Translation in eukaryotes is remarkably similar to that in prokaryotes. However, there are four notable differences:
1. The first amino acid that is incorporated in the eukar-yotic polypeptide is not N-formylmethionine, but methio-nine. A special initiator tRNA is required for initiation, which is distinct from that used in elongation.
2. No Shine-Dalgarno sequence (which defines start site in prokaryotes) has been detected in the eukaryotic mRNA. Instead, a group of protein factors attach to the cap (the methylated guanine at 5’ end), which enable specific initiation of transcription at the AUG codon nearest to the 5’ end.
3. Eukaryotic ribosomes are larger (80S with 60S and 40S subunits) and have extra rRNAs and proteins.
4. Instead of binding with the rRNA, the eukaryotic mRNA binds with proteins in the small ribosomal subunit.
Knowledge of the last difference (eukaryotic mRNAs do not bind rRNA) has important implications in genetic engineering. Because of their inability to bind rRNA, the eukaryotic mRNAs cannot be translated by bacterial ribosomes. The only way to make it possible is to artificially fuse the coding sequence of the eukaryotic mRNA with such a sequence, that is capable of binding with the bacterial ribosome (the sequence is fused in the 5’-untranslated region of the eukaryotic mRNA).
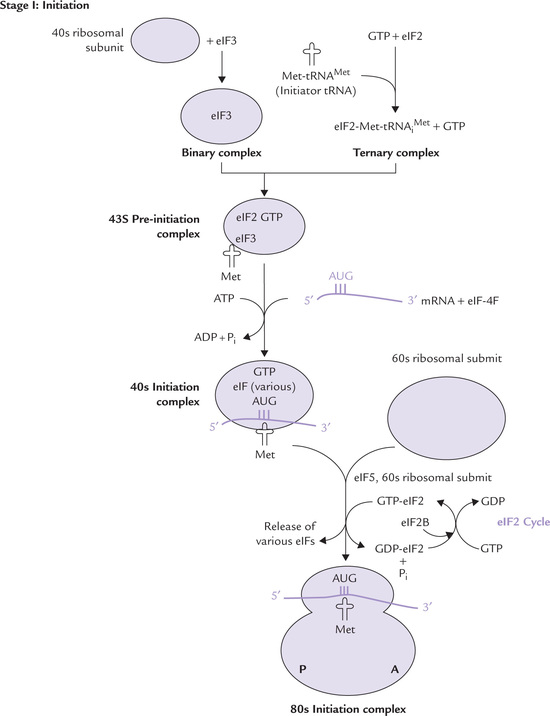
Though the process of protein synthesis in prokar-yotes and eukaryotes is similar, it is far more elaborate and complicated in the latter and needs participation of a number of protein factors (Fig. 24.10 ).
A Process of Translation
Initiation
Initiation of protein synthesis takes place when a ribo-some (both large and small subunits) has assembled on the mRNA and the P site is occupied by the initiator codon. This complex is formed by the action of proteins known as initiation factors. In eukaryotic cells there are at least 12 different initiation factors, which help promote the association of the small ribosomal subunit with the mRNA and a charged initiator tRNA (Table 24.7 ).
Table 24.7
| Name | No. of subunits | Functions |
| elF1 | 1 | mRNA binding to 40s, prevents association of 40s and 60s ribosomal subunits |
| elF2 | 3 | met-tRNA binding to 40s |
| elF3 | 9 | Prevents reassociation of ribosomal subunits |
| elF4A | 1 | Stimulates, helicase binds simultaneously with eIF4F |
| elF4G | 1 | mRNA binding. Acts as a scaffold for assembly of eIF4E and eIF4A in the eIF4F complex |
| elF4F | 4 | mRNA binding |
| elF4E | 1 | Recognition of mRNA 5’ cap |
| elF5 | 1 | Association of 40s and 60s subunits; release of eIF2 and eIF3, ribosome-dependent GTPase |
eIF = Eukaryotic initiation factors.
Specific initiation of translation requires four steps:
• Dissociation of ribosome into its 40s and 60s sub-units.
• Formation of a 43s pre-initiation complex, consisting of the initiator tRNA, GTP, eIF-2 and the 40s subunit.
• The mRNA is bound to the pre-initiation complex, forming the 40s initiation complex.
• Joining of the 60s ribosomal subunit to form the 80s initiation complex.
Details of these steps are as follows:
Step 1: Dissociation of Ribosome
The initiation factors eIF1 and eIF3 bind to the 40S ribosomal subunit, which favours its dissociation from the 60S subunit.
Step 2: Formation of 43s Pre-initiation Complex
Binding of GTP to eIF2 occurs. It gets attached to the activated initiator tRNA charged with methionine (Met-tRNAMet), resulting in formation of a ternary complex (Fig. 24.10). Simultaneously, the 40s ribosomal, subunit is complexed with eIF3 (and probably eIF1 also) to form binary complexes.
The binary and the ternary complexes then join together, forming the 43s pre-initiation complex. This complex is stabilized by earlier association of eIF1 and eIF3 to the 40s subunit.
Step 3: Formation of 40s initiation complex
Binding of mRNA to the pre-initiation complex occurs next. This is effected by the eIF4F, which is a complex of three proteins: eIF4E, 4A and 4G.
• eIF4E physically recognizes and binds to the 5’-cap structure of the mRNA.
• eIf4A binds and hydrolyzes ATP and exhibits RNA helicase activity. (Unwinding of the mRNA secondary structure is necessary to allow access to the ribosomal subunits.)
• eIF4G aids in delivering the mRNA to the 43s pre-initiation complex. It also ensures proper alignment of the mRNA on the 43s complex, resulting in formation of the 40s initiation complex.
The initiation complex so formed is at some distance away from the initiation codon, AUG. By an ATP-driven mechanism, it moves along the mRNA till it encounters the first AUG triplet. This scanning mechanism has two interesting repercussions:
1. There can be only one initiation site per mRNA molecule, so eukaryotic mRNAs are monocistronic (code for single polypeptide) as contrasted to prokaryotic mRNAs, which are often polycistronic with Shine-Dalgarno sequence provided for the initiation of translation of each cistron.
2. Efficiency of translation is decreased in improperly capped mRNAs.
Step 4: Formation of 80s Initiation Complex
The 40s initiation complex, having migrated to AUG, now joins 60s subunit to form 80s complex. The joining is facilitated by eIF5. The energy needed to stimulate formation of the 80s complex is provided by hydrolysis of GTP bound to eIF2. The GDP bound form of eIF2 then binds to eIF2B, which stimulates the exchange of GTP for GDP on eIF2. This is termed as the eIF2 cycle (Fig. 24.10). It ensures availability of the GTP bound to eIF2 (GTP-eIF2) for additional rounds of translation to occur.
With formation of the 80s complex, all initiation factors are released. This completes the initiation phase and the protein synthesis is now ready to begin. At this stage, the initiator tRNA (charged with methionine) is bound to the mRNA within the P site (peptide site) of ribo-some. The other site within the ribosome to which incoming charged tRNAs bind is termed the A site (for amino acid site).
It is noteworthy that assembly of initiation complex was driven by hydrolysis of GTP and the movement of the complex down the mRNA was driven by hydrolysis of ATP.
Elongation
The elongation process is similar to that of prokaryotes, involving sequential addition of amino acids to the car-boxyl end of the growing polypeptide chain by formation of peptide bonds (Fig. 24.10). A notable difference is that the elongation factors involved in the process are different. These are termed eEF1 and eEF2, the former consists of two subunits, which function in a manner analogous to prokaryotic EF-Tu and EF-Ts, bringing the charged tRNA molecule to ribosome. The eEF2 functions like the pro-karyotic EF-G, moving the ribosome one codon down the mRNA (i.e. translocation). Though they are functional equivalents of each other, the prokaryotic and the eukar-yotic elongation factors are not interchangeable. eEF2 is a specific target for inactivation by diphtheria toxin.
Once the correct charged tRNA molecule has been delivered to the A site of the ribosome, peptidyl transferase catalyzes the formation of a peptide bond between the amino acids in the A site. Translocation occurs next because the A site needs to be freed in order to accept the next aminoacyl-tRNA. As described in Chapter 22, during translocation the ribosome is moved along the mRNA such that the next codon of mRNA resides under the A site. Following translocation, the eEF2 is released and the whole process is begun again for addition of next amino acid. (For additional information on peptidyl transferase refer to Box 24.4). The elongation process is remarkably similar in prokaryotic cells, but the factors are different, which explains the utility of antibiotics that specifically target the prokaryotic factors (Chapter 22).
Termination
Termination of protein synthesis in both prokaryotes and eukaryotes is accomplished when the A site of the ribosome reaches one of the stop codons of the mRNA. A eukaryotic releasing factor (RF) recognizes these codons and causes the protein that is attached to the last tRNA molecule in the P site to be released. This process resembles that in prokar-yotes, but there are three RFs in prokaryotes (Chapter 22).
After termination, the ribosomal subunits, tRNA and mRNA dissociate from each other. The eIF3 associates with the small ribosomal subunit, and this prevents its reassociation with the 60S subunit in the absence of mRNA. Finally, eIF2 binds to the small ribosomal subunit, setting the stage for translation of another mRNA.
Post-translational modification and antibiotic inhibitors of translation have been described in Chapter 22.
Diphtheria Toxin Inactivates eEF2
Diphtheria is a bacterial infection of the upper respiratory tract, caused by a toxic protein secreted by Corynebacterium diphtheriae. The toxin causes death (necrosis) of the muco-sal cells, leading to severe pathologic changes and airway obstruction. Initially, it interacts with a receptor on the surface of a sensitive cell, and is proteolytically cleaved to yield two fragments, an A fragment and a B fragment. The B fragment facilitates the penetration of the A fragment through the cell membrane, where it inactivates elongation eEF2. It does so by catalyzing the ADP ribosylation (transfer of an ADP-ribose moiety from NAD+) of a single unusual amino residue present in eEF2 (diphthamide), which is formed by post-translational modification of his-tidine. This ADP ribosylation irreversibly blocks the capacity of eEF2 to carry out the translocation step of protein chain elongation, there by blocking the protein synthesis. A single molecule of the fragment A is sufficient to kill the cell because of its capacity to cause enzymatic inactivation of thousands of eEF2 molecules. Function of diph-thamide in eEF2 is not known, but it is clearly critical.
Interestingly, the gene for diphtheria toxin does not naturally reside in the bacterial genome. It is carried as a prophage (viral DNA that integrates into the bacterial chromosome). Only lysogenic strains (the ones harbouring prophage) of bacteria are pathogenic; the non-lysogenic strains are peaceful members of the normal bacterial flora on our skin and mucous membrane.
B Protein Folding and Prion Diseases
As discussed in Chapter 4, protein folding may start and make considerable headway while translation is still in progress, suggesting that the amino acid sequence of a polypeptide is an important determinant of its final native conformation. However, it is not the only determinant. The constituent amino acids can associate with one another in a number of ways and accordingly the polypeptide chain can fold in various shapes. It is theoretically possible that the polypeptide tries out every conceivable internal association pattern until it arrives at the correct folding association and thereby assume the native 3-dimensional structure—the one having biological activity. However, such process would require millions of years to find the folded form, while in the cell it all has to happen over in a time-scale of a minute or so. Evidently, protein folding is not entirely an automatic unaided process, but requires participation of other factors. Two classes of proteins have been described: one class is of molecular chaperones and the other is of conventional enzymes or proteins.
Molecular Chaperones
These proteins aid production of correct spatial arrangement. They are concerned with kinetics of folding and not with the nature of the final folded form, which is determined by the amino acid sequence (Chapter 4). They can recognize and bind to partially folded proteins (or more appropriately, partially unfolded proteins), and prevent improper associations that may occur; for example, between hydrophobic patches. Such associations could prevent the formation of correct folding. Binding of chaperones to certain reactions of polypeptides is believed to stabilize the partially folded molecule until a stage is reached when correct associations can occur.
Chaperones ensure proper associations; of course, the word chaperone literally means elderly lady entrusted with responsibility of looking after unmarried girls during social occasions probably to ensure correct associations. Chaperones have enormous requirement for ATP energy.
Heat shock proteins
These are examples of chaperones that are produced in response to heat, and other forms of stress, such as toxins, heavy metals, free radicals, radiations, bacteria, etc. (Chapter 22). Some of the major families of heat shock proteins (HSP) are HSP-10, HSP-60 (called chaperonin), HSP-70 and HSP-90; the numbers represent the molecular weight in kD.
Enzymes/Proteins Involved in Protein Folding
These include protein disulphide isomerase, proline isomer-ase, and immunoglobulin binding protein.
• Protein disulphide isomerase (PDI): This enzyme shuffles the disulphide bonds in the polypeptide chains to ensure its correct positioning. If an incorrect disulphide bond were to be formed, PDI cleaves it and reforms a correct one, so that the folding can correct itself.
• Peptidyl proline isomerase (PPI): Wherever a proline residue occurs in a peptide linkage, the configuration can be cis or trans. PPI plays the role of shuffling proline residues between the configurations so as to permit the whole protein to assume the correct configuration.
• Immunoglobulin-binding protein: It is more punishing in its action, being able to recognize the incorrectly folded immunoglobulins and cause their destruction.
Prion Diseases
The prion diseases (also known as spongiform encepha-lopathies) are a group of transmissible, fatal neurological degenerative diseases that affect humans and animals. These include Creutzfeldt-Jacob disease and Kuru in humans. The disease in sheep and goats is designated as scrapie and in cows as spongiform encephalopathy (mad cow disease). It is not clear whether the diseases are genetic or infectious.
The diseases are characterized by the accumulation of altered forms of normal proteins. These proteins, commonly termed "prion proteins" have certain changes in 3-dimensional structures. The major change is replacement of α-helices by ß-sheets. This forms an abnormal isoform, called prion protein-cellular form (PrPC) in affected brains. The scrapie form of the prion protein (PrPSc) has a high α-helical content and is devoid of ß-pleated sheets.
It is believed that the abnormal conformation of the protein makes it resistant to action of proteolytic enzymes. These proteins are highly infectious in nature. Moreover, the progression of infectious prion disease appears to involve an interaction between PrPC and PrPSc, which induces a conformational change of the α-helix rich PrPC to the ß-pleated sheet-rich conformer of PrPSc.
C Translation in Mitochondria
Most of the cellular DNA resides in the nucleus, but a small fraction (less than 1%) resides in mitochondria. The mito-chondrial DNA is a circular duplex with a length of less than 20,000 base pairs in mammals (16,569 base pairs in humans), and there can be five to ten copies in each mitochondrion. The mtDNA is genetically active. The protein coding genes are transcribed by mitochondrial RNA poly-merase and translated by mitochondrial ribosomes that are structurally different from those in cytoplasm.
Though the mitochondrion is a self-replicating organelle with its own DNA and protein synthesizing machinery, it is far from self-contained because its DNA codes for only a small fraction of mitochondrial proteins (the same is true of chloroplasts). The nuclear DNA of the cell codes for the rest of the mitochondrial proteins, which are synthesized in the cytoplasm and transported into the mitochondria. mtDNA codes for a total of13 polypeptides, most of them being components of electron transport chain, using a code slightly different from the virtually universal code.
It is not known with certainty why mitochondria have their own DNA, though it is believed that mitochondria evolved from engulfed prokaryotic cells that became symbiotic (a similar theory holds for the chloroplasts). Most of the original prokaryotic genes were transferred to the nucleus, leaving the mitochondria with a rudimentary but still functional protein synthesizing system. This endosymbiont hypothesis thereby suggests that our genome is partly descended from a primordial eukaryote and partly from a symbiotic prokaryote, and is therefore of hybrid nature (Box 24.5).
VI Regulation of Eukaryotic Gene Expression
The regulatory mechanisms for controlling expression of eukaryotic genes operate at various levels. Some of these involve certain changes in genes such as gene loss, gene rearrangement, gene amplification, etc. Others operate at the level of gene transcription, which has overriding importance in control of gene expression, both in eukar-yotes and prokaryotes.
Several additional mechanisms are also present in eukaryotes (not in prokaryotes), which operate at the post-transcriptional level at any or all of the steps from RNA to protein. These steps include RNA processing (using different promoters and polyadenylation sites, and alternative splicing), selection of mRNAs to be transported from the nucleus to the cytoplasm, selective stabilization or degradation of specific mRNA molecules in the nucleus or cytoplasm. By control over these mechanisms, gene expression may be altered more than 1000-folds.
A Changes in Genes
Gene Amplification
By this mechanism, repeated initiation of DNA synthesis occurs. Additional copies of the gene may be produced in this manner. The drug methotrexate causes production of hundreds of copies of the gene responsible for the enzyme dihydrofolate reductase. In electron microscopy, it is seen as a big replication bubble (Fig. 24.11 ).
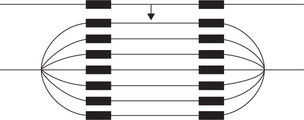
Gene Rearrangement
A segment from the DNA moves from one location to another on the genome. This forms new combination of genes so that several different proteins are encoded. For example, various portions of the antibody-producing genes lie at distant location of the DNA of the germ line cell. By gene rearrangement, DNA segments are brought together and combined to produce the active gene.
Production of the active heavy chain gene of immunoglobulin by gene rearrangement is shown in Figure 24.12 .
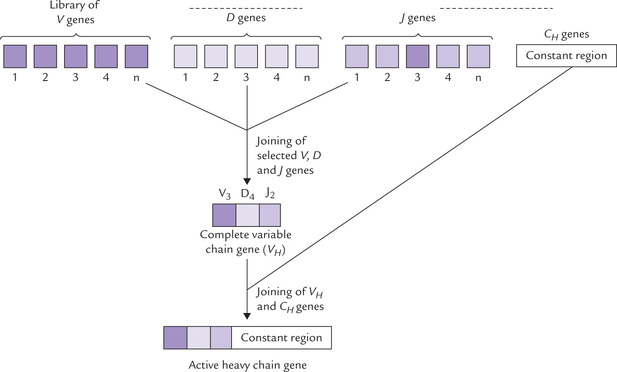
• The heavy chain consists of a variable region (VH) and a constant region (CH). The genes coding for these two regions that lie far apart in genome of an immu-nocyte, are brought close and joined to yield the active heavy chain gene.
• Furthermore, the complete VH gene (the functional form) is formed by assembly of three different types of genes: the variable gene, V; the diversity gene, D; and the joining gene, J. These genes are scattered throughout the genome of the immunocyte. They can be transposed next to one another and joined to yield the complete VH gene.
Various components of the VH originate from different DNA segments that lie far apart in the genome.
The VH gene is then joined with the constant region gene (CH) to yield the heavy chain antibody gene (Fig. 24.12).
Gene Loss
Certain genes may be lost from a cell so that the functional protein encoded by them are not produced. The gene loss may be partial or even complete. For example, during differentiation of red blood cells, the nuclei are extruded resulting in complete loss of all genes. Sometimes the gene may not be lost but its expression may stop (Box 24.6).
Modification of DNA
Modification of certain bases affects transcriptional activity of the gene. When cytosine is methylated at its fifth position to methylcytosine, reduction of the transcriptional activity of the concerned gene results. More the methylation, less is the frequency of transcription of the affected gene.
Most of the methylcytosine is present in the sequences, 5’-CG-3’ throughout the genome. In fact, more than 70% of the susceptible sites are methylated in this way. In a double-stranded DNA, the complementary cytosine is also methylated (because the methylating system uses the hemimethylated sequence as a preferred substrate), giving rise to a palindromic sequence:
So much is control of methylation on the rate of transcription, that the highly methylated areas of DNA are transcriptionally inactive.
B Regulation at the Level of Transcription
Regulation of eukaryotic gene expression occurs principally at the level of transcription. Various genetic elements, e.g. promoters, enhancers, etc. are involved in regulating the transcriptional activity, as already described in this chapter.
C Regulation at Post-transcriptional Level
Use of Alternative Promoters and Different Polyadenylation Sites
Some genes have evolved a series of promoters that confer tissue-specific expression. Use of these alternative promoters in different tissues result in transcripts differing in the 5’ portion and the translated polypeptides differing in the N-terminal sequence. The best example of the use of alternative promoters in humans is the gene for dystrophin, the muscle protein that is deficient in Duchenne muscular dystrophy. This gene uses different promoters, each associated with its own first exon, so that each mRNA and its protein has a tissue specific N-terminal sequence. Thus, brain, muscle, and retinal specific proteins differ in N-terminal amino acid structures.
In other genes, alternative polyadenylation sites can be used, and this produces transcripts with different 3’ ends. These complexities suggest that genes be redefined (Box 24.7).
Tissue-specific Splicing
In some primary transcripts, sequences that are treated as introns by some cells are treated as exons by others. This leads to formation of different mRNAs and different proteins from a single gene.
mRNA Editing
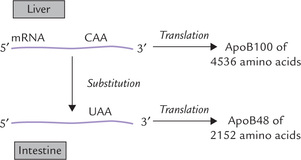
RNA editing involves the enzyme-mediated alteration of RNA in the cell nucleus before translation. The process may involve insertion, deletion or substitution of nucleotides in the RNA molecule. The substitution of one nucleotide for another, for example, results in alteration of a codon to yield tissue-specific transcripts. Apolipo-protein B (apoB) illustrates this. The gene for this protein encodes a 14.1 kb mRNA transcript in the liver and a 4536 amino acid protein product, apoB100 (Fig. 24.13 ). In the small intestine, the editing of nucleotide 6666 of apoB100 mRNA, by changing cytidine to uracil, generates a stop codon in the intestinal mRNA. The translation product of this edited mRNA (7 kbp) is therefore shorter (2152 amino acids) than the unedited mRNA.
Regulation by Nuclear RNase
Several nuclear RNase extensively degrade the primary transcriptor mRNAs before the latter can emerge into the cytoplasm as mature, translatable RNAs. This may prevent inappropriate expression of genes, and also protects the cell from viruses whose mRNAs are degraded in the nucleus.
The mRNA Stability
The survival time of mature mRNA in the cytoplasm is regulated by relative activities of nucleases that degrade the mRNA and by mRNA-binding proteins that prevent nuclease attack. An average eukaryotic mRNA molecule has a lifetime of 3 hours. In certain highly differentiated cells, it is prolonged so that production of a large amount of a single type of protein by its translation is made possible. For example, cells of the chicken oviduct which makes ovalbumin, contain only a single copy of the ovalbumin gene per haploid set of chromosomes, but the cellular mRNA is long lived.
D Translational Regulation
This refers to the number of times a finished mRNA molecule is translated. Regulation of gene of ferritin, an iron-binding protein, in iron deficiency and iron excess illustrates this mode of regulation.
In conditions of iron excess (haemochromatosis) there is an increase in the synthesis of ferritin; and in iron deficiency there is a decrease in the synthesis of ferritin and increase in the synthesis of the transferrin receptor protein (Chapter 19). The mRNAs for both ferritin and transferrin receptor mRNA contain a specific sequence known as the iron-response element (IRE), to which a specific IRE-binding protein can bind. When iron is deficient the IRE-binding protein binds the ferritin mRNA and prevents translation of ferritin. It also binds the transferrin receptor mRNA and prevents its degradation. Thus in iron deficiency, ferritin concentrations falls low and transferrin receptor concentration rises. A reverse series of events occurs in iron excess and translation of ferritin mRNA increases, whereas transferrin receptor mRNA undergoes degradation.
Various regulatory mechanisms act cooperatively to give rise to tissue-specific gene products (Box 24.8).
E Differential Expression of a Parental Allele of a Gene
There are 23 pairs of chromosomes, each of which has genes that are present on both chromosomes: they are biallelic. One of the alleles is inherited from father and the other from mother, and normally both are identical. Under normal circumstances, either of the two alleles has equal chance for expression without preference being given to either. However, in humans certain genes have been identified, in which only one allele—either maternal or paternal—is preferentially expressed. This occurs despite the fact that both are perfectly normal or identical. As a result of such restriction of the expression of biallelic genes, only 50% of the product is produced. The mechanisms for such a restriction are: allelic exclusion, X chromosome inactivation and genomic imprinting.
Allelic exclusion implies tissue-specific expression of a single allele product (for example, synthesis of a single immunoglobulin chain in a B cell from one allele only); X chromosome inactivation implies switching off an entire X chromosome, thereby inactivating all X-linked genes located on it. Genomic imprinting refers to effect of methylation of RNA (refer to Box 24.9 for details).
F Need for Elaborate System of Gene Regulation in Eukaryotes
Finally, it is worthwhile to consider as to why such an elaborate system of gene regulation is required in eukar-yotes and not in prokaryotes. Compared to prokaryotes where most, if not all, genes are expressed at the same time in eukaryotes there is need to express different genes in different tissues and at different times. Though all cells contain identical sets of genes, one cell type (for example muscle) expresses only certain genes and never expresses the liver-specific genes. Moreover, during embryonic development certain genes are required at an early stage of the differentiation process and others later in the development of the specific tissue. Thus, gene control needs are much more complex in differentiated eukaryotes.
Exercises
Essay type questions
1. Describe the transcription and post-transcriptional modifications in eukaryotes.
2. At which points in protein synthesis do fidelity mechanisms operate? Explain why the mechanism of initiation of translation in eukaryotes is not compatible with polycistronic mRNA.
3. Describe the process of protein synthesis in eukar-yotes. Explain the participation and, where known, the role of GTP in protein synthesis.
4. Describe the regulation of gene expression in eukar-yotes.
5. Explain the role of chaperones and other proteins in folding of polypeptide chains. What diseases may be associated with improper protein folding?