Emerging Trends
An interesting pursuit in tumor biology would be studying the biologic behavior of neoplasms in general and follow the subtle differences that exists with individual tumors. Among the many variables would be consideration of the site, size, histologic grades, etiologic factors and stage of progression of individual neoplasms. This newer approach of carefully monitoring tumors comprise a separate area of ‘biologic staging’, which is currently gaining acceptance in clinical practice (Table 2-10). Another interesting trend currently being investigated is the use of the polymerase chain reaction (PCR) to determine if surgical margins obtained at the time of surgery that are histopathologically free of tumor contain a small amount of histologically undetectable tumor cells. Specifically, the use of PCR to detect specific p53 mutations identified in the primary tumor in the histopathologically negative surgical margin could be very useful, as these mutations would indicate the presence of residual (histopathologically undetected) tumor cells. It will be very important to establish whether the presence of submicroscopic tumor cells contributes to prognosis and clinical outcome. The recent development of in situ PCR will allow amplification of both DNA and RNA directly in tissue section; this technique should be extremely helpful in the future to localize tumor cells containing altered oncogenes or tumor suppressor genes. An improved understanding of the molecular biology of head and neck cancer may also contribute to future therapeutic improvements. Finally, molecular probes may be used to facilitate the early detection of secondary malignancies.
Table 2-10
Biomarker predictors in oral precancerous and cancerous lesions
| Marker method | Detection | Target |
| Proliferation | ||
| PCNA, Ki67, BrU | IHC | Cycling cells |
| Histone | mRNA ISH | |
| AgNORs | Silver stain | |
| Genetic | ||
| Ploidy | FC | Aneuploid cells |
| Oncogenes | ||
| C-myc | IHC | Cycling cells |
| Tumor suppressor | ||
| p53 mutations | IHC, PCR | Cycling cells |
| Cytokeratin 8/19 | IHC | Anaplasia |
| Blood group antigens | IHC | Anaplasia |
| Integrins/ECM ligands | IHC | Invasion and metastatic potential |
IHC: immunohistochemistry, FC: flowcytometry, PCR: polymerase chain reaction, ISH: in-situ hybridization.
Under the current management, the strategy for head and neck cancer includes specialists from many disciplines and their concerted effort will promote the use of increasingly refined laboratory techniques for improved diagnosis and therapy. A good deal of current laboratory and clinical research is focusing on identifying the relative contributions of certain oncogenes and tumor suppressor genes on carcinogenesis, tumor stage, and clinical outcome. Although abnormalities of some oncogenes and tumor suppressor genes have been identified, their relative contribution and optimal use in diagnosis, prognosis, or treatment remain unknown. The same might be said for the role of Epstein-Barr, hepatitis, and herpes simplex viruses; further clinicolaboratory studies will be needed to define which are clinically relevant and when they should be investigated. Genetic analysis at a molecular/chromosomal level is emerging as a science that may aid in identifying risk and possibly prevention as well. Finally, a reliable and predictable histopathology grading system should be developed to include, in addition to differentiation of tumor cells, such factors as basement membrane protein expression and invasion patterns, perineural invasion, and immunologic responses.
Carcinoma of Lip
Epidermoid carcinoma of the lip is a disease that occurs chiefly in elderly men. Furthermore, the lower lip is involved by this neoplasm far more commonly than the upper lip. Great numbers of cases have been reported in the literature, and the data of Cross, Guralnick and Daland on 563 patients with lip cancer may be cited to illustrate certain typical characteristics. In this large series, it was found that 98% of the patients were men and that the age of these patients at the onset of the disease ranged from 25–91 years, with the greatest incidence between 55 and 75 years and a mean of 62 years of age. Of the total group of lip cancers, 88.3% occurred on the lower lip, 3.3% on the upper lip and 8.3% on the labial commissures. The right and left sides were affected with equal frequency. In 636 cases reviewed by Schreiner, similar findings were reported. In his series 97% of the lesions occurred in men, and 96% were found on the lower lip. Nearly one-third of his cases occurred between the ages of 60 and 70 years.
Etiology
A number of possible etiologic factors have been suggested by the review of the records of many patients. One of the most common of these has been the use of tobacco, chiefly through pipe-smoking. The data of Cross and his coworkers indicate that 64% of their patients with lip cancer were pipe smokers, while a total of 94% habitually used tobacco in some form. These observations are in agreement with the data of Widmann, who, in a review of 363 cases of lip cancer, estimated an incidence of approximately 40% pipe smokers. Schreiner reported that 87% of his lip cancer patients were tobacco users. Although no conclusions may be drawn from such data because of the widespread use of tobacco in the general population, it appears suggestive that the heat, the trauma of the pipe stem and possibly the combustion end-products of tobacco may be of some significance in the etiology of lip cancer.
Syphilis is probably not as significant an etiologic factor in lip cancer as in certain other oral sites, since the incidence of lip cancer has been found to be low in syphilitic patients: 7.2% by Cross and his associates, 8% by Widmann and 3.6% by Schreiner. As indicated previously; however, the data of Wynder and his group indicated that syphilis was of etiologic significance in lip cancer.
Sunlight is generally considered to be important, as discussed previously, because the alterations which occur in skin and lips as a result of prolonged exposure to sun are characterized as preneoplastic. Ju has discussed this problem and constructed a profile of the lip cancer patient.
Poor oral hygiene is an almost universal finding in patients with lip cancer. Cross and his group pointed out that only approximately 8% of their entire series of patients had good or even fair oral hygiene. In addition, some patients indicate a history of trauma before the appearance of a lesion. They report not only a single traumatic experience, such as a cigarette burn or a cut, but also chronic trauma from jagged teeth and so forth. Unfortunately, it is difficult to assess scientifically the role of such factors in the etiology of cancer.
Leukoplakia has often been associated with the development of carcinoma. Since clinical leukoplakia is a fairly common lesion of the lip, it was only natural that the relationship should be investigated. Schreiner found leukoplakia present in only 2.4% of his 636 cases of lip cancer, while Cross and coworkers found leukoplakia associated with carcinoma in 14.5% of the cases in their series. This would indicate that the simultaneous occurrence of the two conditions is probably due to chance and that leukoplakia is not a common predecessor of lip cancer. However, in light of more recent studies dealing with the frequency of preneoplastic and neoplastic transformation in lesions of leukoplakia of the lip (q.v.), the interpretation of the above findings is uncertain.
Clinical Features
There is considerable variation in the clinical appearance of lip cancer, depending chiefly upon the duration of the lesion and the nature of the growth. The tumor usually begins on the vermilion border of the lip to one side of the midline. It often commences as a small area of thickening, induration and ulceration or irregularity of the surface. As the lesion becomes larger it may create a small crater like defect or produce an exophytic, proliferative growth of tumor tissue. Some patients have large fungating masses in a relatively short time, while in other patients the lesion may be only slowly progressive.
Carcinoma of the lip is generally slow to metastasize, and a massive lesion may develop before there is evidence of regional lymph node involvement. Some lesions; however, particularly the more anaplastic ones, may metastasize early. When metastasis does occur, it is usually ipsilateral and involves the submental or submaxillary nodes. Contralateral metastasis may occur, especially if the lesion is near the midline of the lip where there is a cross drainage of the lymphatic vessels.
Histologic Features
Most lip carcinomas are well-differentiated lesions, often classified as grade I carcinoma. This type of cancer tends to metastasize late in the course of the disease. In the series of Widmann, the following approximate distribution of graded lesions was found: grade I, 60%; grade II, 26%; grade III, 13%; grade IV, 2%.
Treatment and Prognosis
Carcinoma of the lip has been treated by either surgical excision or X-ray radiation with approximately equal success, depending to some degree upon the duration and extent of the lesion and the presence of metastases. Interestingly, in the series of Cross the overall cure rate of patients with lip cancer treated by surgery was approximately 81%, while in the series of Widmann the cure rate of patients with the same type of neoplasm treated by X-ray radiation was approximately 83%. This would indicate that either form of therapy, in skilled hands, will produce equally good results.
Many factors may influence the success or failure of treatment of lip carcinoma. The size of the lesion, its duration, the presence or absence of metastatic lymph nodes and the histologic grade of the lesion must all be considered carefully by the therapist in planning his/her approach to the neoplastic problem.
Carcinoma of Tongue
Cancer of the tongue comprises between 25 and 50% of all intraoral cancer. It is less common in women than in men except in certain geographic localities, chiefly the Scandinavian countries, where the incidence of all intraoral carcinoma in women is high because of the high incidence of a preexisting Plummer–Vinson syndrome. In a series of 441 cases of tongue cancer reported by Ash and Millar, 25% occurred in women and 75% in men, with an average age of 63 years. In a series of 330 cases of cancer of the tongue reported by Gibbel, Cross and Ariel the average age of the patient was 53 years, with a range of 32–87 years. Thus it is essentially a disease of the elderly, but it may occur in relatively young persons. To exemplify this latter point, a series of 11 patients less than 30 years of age, four of them less than 20 years, with carcinoma of the tongue has been reported by Byers. This group of patients represented approximately 3% of all patients seen at MD Anderson Hospital with epidermoid carcinoma of the tongue between 1956 and 1973 (418 cases).
Etiology
A number of causes of cancer of the tongue have been suggested, but in our present state of knowledge no precise statements can be made. A definite relation does appear to exist; however, between tongue cancer and certain other disorders. Many investigators have found syphilis, either an active case or at least a past history of it, coexistent with carcinoma of the tongue. In the series of Gibbel et al, 22% of patients with lingual cancer demonstrated a positive complement fixation or Kahn reaction, while in the general admission at their hospital only 5% of the patients had a positive reaction for syphilis. Martin reported that 33% of his patients with cancer of the tongue also had syphilis. The relationship can be explained on the basis of a chronic glossitis produced by the syphilis, chronic irritation long being recognized as carcinogenic under certain circumstances. This explanation implies a local effect of syphilis rather than a generalized or systemic effect. It should be pointed out that some studies do not confirm the theory of a relationship between syphilis and tongue cancer. Wynder has confirmed this relationship, but questioned whether the neoplasm might be related to the arsenic therapy, the treatment of choice before the advent of antibiotics, rather than to the syphilis itself. Meyer and Abbey have also questioned this relationship since they found only 15 patients (6%) who showed positive evidence of a history of syphilis in a survey of 243 cases of primary carcinoma of the tongue.
Leukoplakia is a common lesion of the tongue which has been observed many times to be associated with tongue cancer. Martin noted that 46% of his series of cancer patients had leukoplakia of the tongue, while Gibbel et al, found only a 10% incidence of leukoplakia in his series. It is not unusual to see typical lesions of carcinoma in leukoplakic areas; on the other hand, many lesions of leukoplakia appear to persist for years without malignant transformation, and many cases of carcinoma of the tongue develop without evidence of preexisting leukoplakia.
Other factors which have been thought to contribute to the development of carcinoma of the tongue include poor oral hygiene, chronic trauma and the use of alcohol and tobacco. Poor hygiene and the use of alcohol and tobacco are so prevalent as nearly to preclude the possibility of drawing conclusions about a possible cause and effect relation. A considerable number of cases have been observed in which cancer of the tongue developed at a site exactly corresponding to a source of chronic irritation such as a carious or broken tooth or an ill-fitting denture. The work of Wynder and his group; however, suggests that these findings may be fortuitous.
Clinical Features
The most common presenting sign of carcinoma of the tongue is a painless mass or ulcer, although in most patients the lesion ultimately becomes painful, especially when it becomes secondarily infected. The tumor may begin as a superficially indurated ulcer with slightly raised borders and may proceed either to develop a fungating, exophytic mass or to infiltrate the deep layers of the tongue, producing fixation and induration without much surface change (Fig. 2-27 A–C).
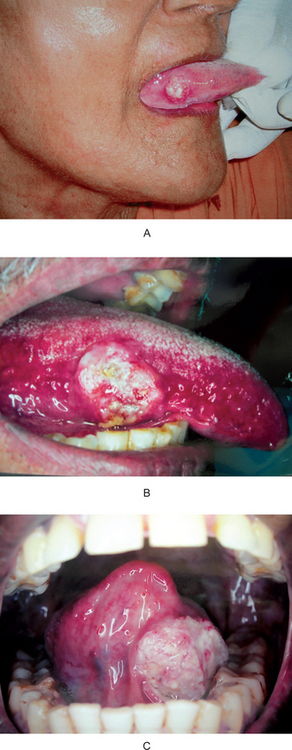
The typical lesion develops on the lateral border or ventral surface of the tongue. When, in rare cases, carcinoma occurs on the dorsum of the tongue, it is usually in a patient with a past or present history of syphilitic glossitis. In a series of 1,554 cases of carcinoma of the tongue reported by Frazell and Lucas, only 4% occurred on the dorsum. The lesions on the lateral border are rather equally distributed between the base of the tongue, the anterior third and the mid portion, although in the above series 45% of cases occurred on the middle third. Lesions near the base of the tongue are particularly insidious, since they may be asymptomatic until far advanced. Even then the only presenting manifestations may be a sore throat and dysphagia. The specific site of development of these tumors is of great significance, since the lesions on the posterior portion of the tongue are usually of a higher grade of malignancy, metastasize earlier and offer a poorer prognosis, especially because of their inaccessibility for treatment.
Metastases occur with great frequency in cases of tongue cancer. In 302 patients on whom information was available, Gibbel and his group reported that cervical metastases from tongue lesions were present in 69% at the time of admission to the hospital. The metastatic lesions may be ipsilateral, bilateral, or because of the cross lymphatic drainage, contralateral in respect to the tongue lesion.
Treatment and Prognosis
The treatment of cancer of the tongue is a difficult problem, and even now no specific statements can be made about the efficacy of surgery in comparison to that of X-ray radiation. As in other areas, it will probably be found that the judicious combination of surgery and X-ray will be of greatest benefit to the patient. Many radiotherapists prefer the use of radium needles or radon seeds to X-ray radiation because they are able with these devices to limit the radiation to the tumor, sparing adjacent normal tissue. Metastatic nodes are highly complicating factors, but treating them without controlling the primary lesion is useless.
The prognosis of cancer in this location is not good. Although statistics vary between series, the five-year cure rate is generally conceded to be below 25%. Martin reported a 22% survival in 556 patients with tongue cancer, while Gibbel et al, found only a 14% survival among 213 patients. Frazell and Lucas reported an overall five-year cure rate of 35% on a series of 1,321 patients with tongue cancer.
The most significant factor affecting prognosis of these patients is the presence or absence of cervical metastases. Thus the studies of Gibbel and his associates showed an 81% survival rate if no metastases ever developed, 43% if no metastases were present at the time of hospital admission, and only 4% if metastases were present at the time of admission or developed subsequent to admission. The necessity for early diagnosis thus becomes obvious, and the role of the dentist in recognizing cancerous lesions is, of course, of paramount importance.
Carcinoma of Floor of Mouth
Carcinoma of the floor of the mouth represents approximately 15% of all cases of intraoral cancer and occurs in the same age group as other oral cancers. The average age of the patient was 57 years in the series of Tiecke and Bernier, 67 years in the series of 110 cases reported by Ash and Millar and 63 years in the group of 100 cases reported by Ballard and his associates. In the latter report, 81% of the lesions occurred in men, while 93% of the patients were men in the series of Ash and Millar.
Smoking, especially a pipe or cigar, has been considered by some investigators to be important in the etiology of cancer in this location. In the series of Ballard and his coworkers, 50% of the patients were classified as heavy smokers, 33% as heavy drinkers and 28% as heavy smokers and heavy drinkers. Nevertheless, little evidence has been gathered to suggest an obvious cause-and-effect relation with regard to tobacco or other factors such as alcohol, poor oral hygiene or dental irritation. Leukoplakia does occur in this location and there is evidence to indicate that epithelial dysplasia and malignant transformation in the leukoplakia occur here with greater frequency than in other oral sites.
Clinical Features
The typical carcinoma in the floor of the mouth is an indurated ulcer of varying size situated on one side of the midline. It may or may not be painful. This neoplasm occurs far more frequently in the anterior portion of the floor than in the posterior area. Because of its location, early extension into the lingual mucosa of the mandible and into the mandible proper as well as into the tongue occurs with considerable frequency (Fig. 2-28). Carcinoma of the floor of the mouth may invade the deeper tissues and may even extend into the submaxillary and sublingual glands. The proximity of this tumor to the tongue, producing some limitation of motion of that organ, often induces a peculiar thickening or slurring of the speech.
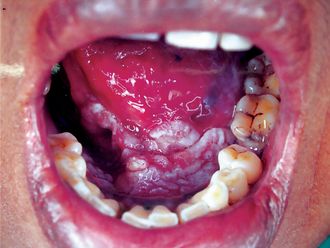
Metastases from the floor of the mouth are found most commonly in the submaxillary group of lymph nodes, and since the primary lesion frequently occurs near the midline where a lymphatic cross drainage exists, contralateral metastases are often present. Of 95 cases of carcinoma of the floor of the mouth reviewed by Ash and Millar, 21% presented lymph node involvement at the time of admission, while an additional 23% subsequently developed lymphadenopathy. Thus the total incidence of metastasis was 44%. This corresponds to the incidence of 42% metastatic lymph node involvement in the series reported by Tiecke and Bernier and 58% by Martin and Sugarbaker. Fortunately, distant metastases are rare.
Treatment and Prognosis
The treatment of cancer of the floor of the mouth is difficult and all too frequently unsuccessful. Large lesions, because of the anatomy of the region, usually are not a surgical problem. Even small tumors are apt to recur after surgical excision. For this reason, X-ray radiation and the use of radium often give far better results than surgery. The problem is complicated, however, if there is concomitant involvement of the mandible.
The prognosis for patients with carcinoma of the floor of the mouth is fair. The net five-year survival of 86 patients with cancer in this location reviewed by Ash and Millar was 43%. All patients in this series were treated by some form of radiation. Martin and Sugarbaker reported a five-year survival rate of 21% in their series of 103 patients.
Carcinoma of Buccal Mucosa
Reported studies of carcinoma of the buccal mucosa reveal exceptional variation in incidence, the widest differences being accounted for by studies from countries other than the United States. In the series of Krolls and Hoffman, cancer of the buccal mucosa comprised 3% of the total cases of intraoral carcinoma. Like most cancers of the oral cavity, cancer of the buccal mucosa is approximately 10 times more common in men than in women and occurs chiefly in elderly persons. In the study of Tiecke and Bernier, the average age at occurrence of carcinoma of the buccal mucosa was 58 years.
Etiology
The etiology of carcinoma of the buccal mucosa is no better understood than that of carcinoma of other areas of the oral cavity. Several factors appear to be of indisputable significance, however; these include the use of chewing tobacco and the habit of chewing betel nut, which is widespread in many countries in the Far East. It is a fairly common clinical observation that carcinoma of the buccal mucosa develops in the area against which a person has habitually carried a quid of chewing tobacco for years while the opposite cheek may be normal, the patient never having rested the tobacco there. Although this is only presumptive evidence of a cause-and-effect relation; it has been recognized so frequently that it appears to be more than a coincidental finding. A special form of neoplasm known as ‘verrucous carcinoma’ (q.v.) occurs almost exclusively in elderly patients with a history of tobacco chewing. Since the betel nut quid contains tobacco as well as other substances, including slaked lime, the high incidence of cancer in persons addicted to its use may be explained on a similar basis.
Leukoplakia is a common predecessor of carcinoma of the buccal mucosa. It is usually of extremely long duration and may or may not necessarily be associated with the use of tobacco. Chronic trauma in the form of cheek biting and dental irritation such as that from jagged teeth does not appear to be associated with the development of carcinoma, although focal areas of leukoplakia sometimes occur when these conditions exist.
Clinical Features
There is considerable variation in the clinical appearance of carcinoma of the buccal mucosa. The lesions develop most frequently along or inferior to a line opposite the plane of occlusion. The anteroposterior position is variable, some cases occurring near the third molar area, others anteriorly towards the commissure.
The lesion is often a painful ulcerative one in which induration and infiltration of deeper tissues are common. Some cases, however, are superficial and appear to be growing outward from the surface rather than invading the tissues. Tumors of this latter type are sometimes called exophytic or verrucous growths.
The incidence of metastases from the usual epidermoid carcinoma of the buccal mucosa varies considerably, but is relatively high. Tiecke and Bernier reported that 45% of the patients in their study exhibited metastases at the time of presentation for treatment. This is similar to the incidence of approximately 50% reported by Richards. The most common sites of metastases are the submaxillary lymph nodes.
Treatment and Prognosis
The treatment of carcinoma of the buccal mucosa is just as much of a problem as that of cancer in other areas of the oral cavity. In early cases, it is probable that similar results may be obtained by either surgery or X-ray radiation. The combined use of these two forms of treatment undoubtedly also has a place in the therapy of this tumor.
The prognosis of this neoplasm depends upon the presence or absence of metastases. The findings of Modlin and Johnson indicate that the five-year survival rate for patients with cancer of the buccal mucosa approximates 50%, but in another series Martin reported only a 28% survival.
Carcinoma of Gingiva
Carcinoma of the gingiva constitutes an extremely important group of neoplasms. The similarity of early cancerous lesions of the gingiva to common dental infections has frequently led to delay in diagnosis or even to misdiagnosis. Hence institution of treatment has been delayed, and the ultimate prognosis of the patient is poorer.
Martin reported that approximately 10% of all malignant tumors of the oral cavity occur on the gingiva. In the study of Krolls and Hoffman, 11% of the intraoral carcinomas occurred on the gingiva, while Tiecke and Bernier found a similar incidence of 12% in their series. In the group reported by Martin, one patient was only 22 years old, but the average age of the patients was 61 years. This is essentially a disease of elderly persons, since only 2% of the tumors occurred in patients under the age of 40 years. In the same group of patients, 82% were men and only 18% were women. This is similar to the gender distribution found in oral cancer in other locations.
Etiology
The etiology of carcinoma of the gingiva appears to be no more specific or defined than that in other areas of the oral cavity. Syphilis does not appear to be as significant a factor here as it is in carcinoma of the tongue, and the relation to the use of tobacco is indefinite. Since the gingiva, because of calculus formation and collection of microorganisms, is in nearly all persons the site of a chronic irritation and inflammation lasting over a period of many years, one may speculate the possible role of chronic irritation in the development of cancer of the gingiva. Occasionally, cases of gingival carcinoma appear to arise after extraction of a tooth. If such cases are carefully examined, however, it may usually be ascertained that the tooth was extracted because of a gingival lesion or disease or because the tooth was loose. In fact, the tooth was extracted because of the tumor, which, at the time of surgery, went unrecognized or undiagnosed.
An unusual situation arises in some instances after extraction of a tooth in that a carcinoma appears to develop rapidly and proliferate up out of the socket. Those cases which appear to represent such a phenomenon probably are due to carcinoma of the gingiva growing down along the periodontal ligament and then proliferating suddenly after the dental extraction.
Clinical Features
It is generally agreed that carcinoma of the mandibular gingiva is more common than the involvement of the maxillary gingiva, although the distribution of cases varies considerably between different series. In 47 cases reported by Tiecke and Bernier, 81% of the tumors were found on the mandibular gingiva and only 19% on the maxillary gingiva. Martin, however, reported a more equal distribution of 54% on the mandibular gingiva and 46% on the maxillary gingiva. Data on the exact position in the dental arch at which carcinoma is most apt to develop are insufficient to draw valid conclusions.
Carcinoma of the gingiva usually is manifested initially as an area of ulceration which may be a purely erosive lesion or may exhibit an exophytic, granular or verrucous type of growth (Fig. 2-29). Many times, carcinoma of the gingiva does not have the clinical appearance of a malignant neoplasm. It may or may not be painful. The tumor arises more commonly in edentulous areas, although it may develop in a site in which teeth are present. The fixed gingiva is more frequently involved primarily than the free gingiva.
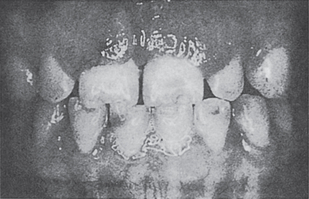
Figure 2-29 Carcinoma of the gingiva. The mild overgrowth of the gingiva over the maxillary central incisors in this 25-year-old girl resembled inflammatory gingival hyperplasia. Microscopic examination of the gingivectomy specimen revealed epidermoid carcinoma. This is an unusual clinical appearance, age and location for this neoplasm, but many early cases are difficult to recognize (Courtesy of Harold R Schreiber and Charles A Waldron. J Periodontol, 29: 196, 1958).
The proximity of the underlying periosteum and bone usually invites early invasion of these structures. Although many cases exhibit irregular invasion and infiltration of the bone, superficial erosion arising apparently as a pressure phenomenon sometimes occurs. In the maxilla, gingival carcinoma often invades into the maxillary sinus, or it may extend onto the palate or into the tonsillar pillar. In the mandible, extension into the floor of the mouth or laterally into the cheek as well as deep into the bone is rather common. Pathologic fracture sometimes occurs in the latter instance (Fig. 2-30).
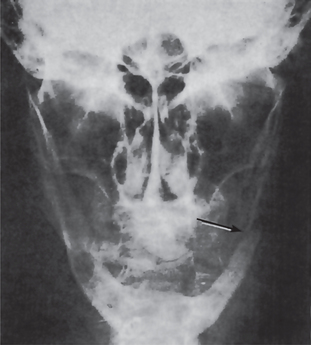
Figure 2-30 Pathologic fracture of mandible caused by the invasion of epidermoid carcinoma arising on the alveolar ridge.
Metastasis is a common sequela of gingival carcinoma. Cancer of the mandibular gingiva metastasizes more frequently than cancer of the maxillary gingiva. In most series of cases, metastases to either the submaxillary or the cervical nodes eventually occur in over 50% of the patients regardless of whether the involvement is maxillary or mandibular.
Treatment and Prognosis
The use of X-ray radiation for carcinoma of the gingiva is fraught with hazards because of the well-known damaging effect of the X-rays on bone. In general, treatment of carcinoma in this location is a surgical problem.
The prognosis of cancer of the gingiva is not particularly good. In the series of 105 cases reported by Martin, only 26% of the patients were alive and free of the disease five years after treatment. It is of great significance that in this same series there were no five-year survivals if the patient presented lymph node metastases at the time of admission. This again illustrates the great need for early diagnosis of these neoplasms.
Carcinoma of Palate
Epidermoid carcinoma of the palate is not a particularly common lesion of the oral cavity. It exhibits approximately the same percentage of occurrence as carcinoma of the buccal mucosa, floor of the mouth and gingiva. The palate was the primary site of 9% of the intraoral epidermoid carcinomas reported by Krolls and Hoffman. In a study by Tiecke and Bernier, of 38 palatal tumors in which the site was specific, 53% occurred on the soft palate, 34% on the hard palate and 13% on both. Ackerman, however, stated that epidermoid carcinoma of the hard palate is a rare finding. New and Hallberg found an incidence of only 0.5% of cases of epidermoid carcinoma of the hard palate among approximately 5,000 cases of intraoral carcinoma. (Accessory salivary gland tumors of the hard palate appear to be three to four times more common than epidermoid carcinoma).
Clinical Features
Palatal cancer usually manifests itself as a poorly defined, ulcerated, painful lesion on one side of the midline (Fig. 2-31). It frequently crosses the midline, however, and may extend laterally to include the lingual gingiva or posteriorly to involve the tonsillar pillar or even the uvula. The tumor on the hard palate may invade into the bone or occasionally into the nasal cavity, while infiltrating lesions of the soft palate may extend into the nasopharynx.
The epidermoid carcinoma is almost invariably an ulcerated lesion, whereas the tumors of accessory salivary gland origin, even the malignant lesions, are often not ulcerated, but are covered with an intact mucosa. This fact may be of some aid in helping to distinguish clinically between these two types of neoplasms.
Metastases to regional lymph nodes occur in a considerable percentage of cases, but there is little evidence to indicate whether such metastases are more common in carcinoma of the soft palate or in that of the hard palate.
Treatment and Prognosis
Both surgery and X-ray radiation have been used in the treatment of epidermoid carcinoma of the palate. Few large series of cases are available for analysis to aid in determining which form of therapy may be expected to give the greatest survival. Nor are any significant series of purely palatal carcinomas available to aid in the determination of overall survival rate of patients with this lesion. It does appear that the prognosis is somewhat comparable to that of carcinoma of the gingiva.
Carcinoma of Maxillary Sinus
Antral carcinoma is an exceedingly dangerous disease. Although the actual incidence of the disease in respect to intraoral carcinoma cannot be determined, it does appear to be considerably less frequent than any other form of oral cancer. Seelig presented an excellent 10-year survey of the literature and described the chief findings in 624 cases of antral carcinoma; a review of this disease has also been reported by Chaudhry and his associates. Although nothing is known of the etiology of this particular neoplasm, Ackerman stated that chronic sinusitis does not seem to predispose to the development of carcinoma of the maxillary sinus. It might be pointed out here that, although most cases of carcinoma of the maxillary sinus are of the epidermoid type, occasional cases of adenocarcinoma occur, apparently originating from the glands in the wall of the sinus.
Clinical Features
One of the features which contribute to the deadly nature of this disease is that it is often hopelessly advanced before the patient is conscious of its presence. The dentist must be fully aware of the potentialities of this neoplasm and the various ways in which it may manifest itself clinically.
Available studies indicate that carcinoma of the antrum is somewhat more common in men and that, though it is chiefly a disease of elderly persons, occasional cases occur in young adults.
The first clinical sign of antral carcinoma is frequently a swelling or bulging of the maxillary alveolar ridge, palate or mucobuccal fold, loosening or elongation of the maxillary molars or swelling of the face inferior and lateral to the eye (Fig. 2-32). Unilateral nasal stuffiness or discharge is sometimes a primary complaint. In edentulous patients wearing maxillary dentures, loosening of or inability to tolerate the prosthetic appliance may occur before there is any visible clinical evidence of the disease.
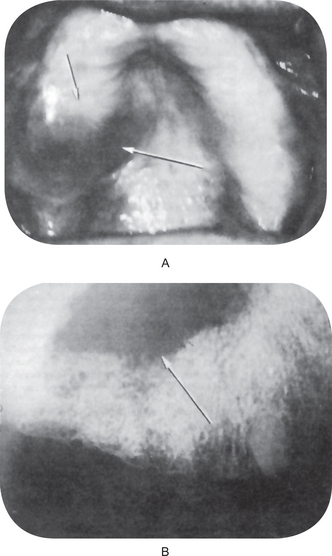
Figure 2-32 Epidermoid carcinoma of the maxillary sinus. (A) The alveolar ridge shows thickening, reddening and deformity, although there is no ulceration of the mucosa. (B) The radiograph reveals raggedness of the maxillary sinus and obvious bony alteration.
The actual spread of the neoplasm which determines the clinical manifestations of the disease is reflected by the extent of involvement of the various walls of the antrum. In some cases, only the floor of the sinus is invaded so that the manifestations of the disease are associated solely with oral structures. If the medial wall of the sinus is involved, nasal obstruction may result. The involvement of the superior wall or roof produces displacement of the eye, while invasion of the lateral wall produces bulging of the check. Ulceration either into the oral cavity or on the skin surface may occur, but only late in the course of the disease.
Metastases usually do not occur until the tumor is far advanced, but when they appear they involve the submaxillary and cervical lymph nodes. The lack of metastasis does not indicate a favorable course, since many patients die from local infiltration alone.
Treatment and Prognosis
Both surgery and X-ray radiation have been used to treat this form of neoplastic disease. If the cancer is confined to the antrum and inferior structures, hemimaxillectomy gives favorable clinical results in some cases. Radiation treatment frequently takes the form of radium needles inserted into the antrum or the tumor mass. This has proved effective in some cases, even though considerable invasion of adjacent structures has occurred.
The overall prognosis of patients with antral carcinoma is not good. In the series of Chaudhry and his associates, only 10% of 49 patients with carcinoma of the antrum lived for more than five years.
Verrucous Carcinoma
A warty variant of squamous cell carcinoma characterized by a predominantly exophytic overgrowth of well-differentiated keratinizing epithelium having minimal atypia and with locally destructive pushing margins at its interface with underlying connective tissue.
Verrucous carcinoma was originally described as a distinct entity on account of its clinical and microscopic features and its mode of behavior. Well differentiated, hyperplastic stratified squamous epithelium is organized into bulbous rete-ridges that exhibit little or no cytological atypia or mitotic activity. There may be a significant endophytic component and the invading margin is usually below the level of the surrounding mucosa. Deep surface invaginations are filled with keratin. The advancing epithelial border is broad and the basement membrane is generally intact. There is usually a heavy inflammatory cell reaction in the adjacent connective tissue. Local destruction of connective tissue occurs in advance of the deep epithelial border. Growth is generally slow and metastatic spread occurs late, if at all. There is a view that verrucous carcinomas may become more aggressive if irradiated.
Although most verrucous carcinomas can be distinguished from squamous cell carcinomas on the basis of their mode of growth, infrequent dysplasia and absence of metastases, there are occasionally foci of conventional squamous cell carcinomas within a verrucous carcinoma. Such lesions should be classified and treated as squamous cell carcinomas. Thorough sectioning of specimens is therefore, necessary to eliminate this possibility. Another hazard in diagnosis occurs when the extremely thick layers of keratin and hyperplastic epithelium are biopsied at insufficient depth to include underlying connective tissue.
The term verrucous hyperplasia describes an exophytic overgrowth of well differentiated keratinizing epithelium that is similar to verrucous carcinoma but without the destructive pushing border at its interface with the underlying connective tissue. Areas of verrucous hyperplasia may be encountered in association with verrucous carcinoma, squamous cell carcinoma or proliferative verrucous leukoplakia.
Exophytic papillary lesions that show epithelial dysplasia, possibly even carcinoma in situ, and relatively inconspicuous areas of invasive squamous cell carcinoma separate from the surface epithelium, should be distinguished from verrucous carcinomas and classified as papillary squamous cell carcinomas (Pindborg JJ et al, 1997).
Clinical Features
Verrucous carcinoma is generally seen in elderly patients, the mean age of occurrence being 60–70 years, with nearly 75% of the lesions developing in males, according to a review by Shafer of nearly 300 reported cases. The vast majority of cases occur on the buccal mucosa and gingiva or alveolar ridge, although the palate and floor of the mouth are occasionally involved.
The neoplasm is chiefly exophytic and appears papillary in nature, with a pebbly surface which is sometimes covered by a white leukoplakic film. The lesions commonly have rugae-like folds with deep clefts between them. Lesions of the buccal mucosa may become quite extensive before the involvement of deeper contiguous structures. Lesions on the mandibular ridge or gingiva grow into the overlying soft tissue and rapidly become fixed to the periosteum, gradually invading and destroying the mandible (Fig. 2-33 A–C). Regional lymph nodes are often tender and enlarged, simulating metastatic tumor, but this node involvement is usually inflammatory. Pain and difficulty in mastication are common complaints, but bleeding is rare.
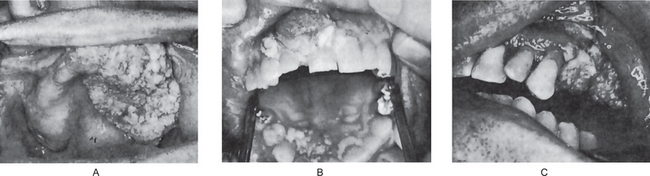
Figure 2-33 Verrucous carcinoma. The exophytic, verrucous nature of the lesion is evident (A and B, Courtesy of Dr Charles A Waldron, and C, of Dr George G Blozis and Dr Mirdza E Neiders).
The term oral florid papillomatosis has been used by dermatologists to describe a lesion with not only a clinical and microscopic appearance similar to verrucous carcinoma but also a similar biologic behavior. For this reason, many authorities now believe that oral florid papillomatosis and verrucous carcinoma represent one and the same disease with no justification for continued use of the former term, since it fails to imply the neoplastic nature of the disease.
It is consistently reported that a very high percentage of patients with this disease are tobacco chewers. A small number of patients give no such history but, instead, use snuff or smoke tobacco heavily. Occasional patients deny the use of tobacco and these usually have ill-fitting dentures.
Histologic Features
The histologic features may be extremely deceptive, and many cases have been diagnosed originally as simple papillomas or benign epithelial hyperplasia because of the orderly and harmless appearance of the specimen. There is generally marked epithelial proliferation with downgrowth of epithelium into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and shows little mitotic activity, pleomorphism or hyperchromatism. Characteristically, cleft like spaces lined by a thick layer of parakeratin extend from the surface deeply into the lesion. Parakeratin plugging also occurs extending into the epithelium. The parakeratin lining the clefts with the parakeratin plugging is the hallmark of verrucous carcinoma. Even though the lesion may be very extensive, the basement membrane will often appear intact. When the lesions become infected, focal intraepithelial abscesses are often seen. Significant chronic inflammatory cell infiltration in the underlying connective tissue may or may not be present (Fig. 2-34).
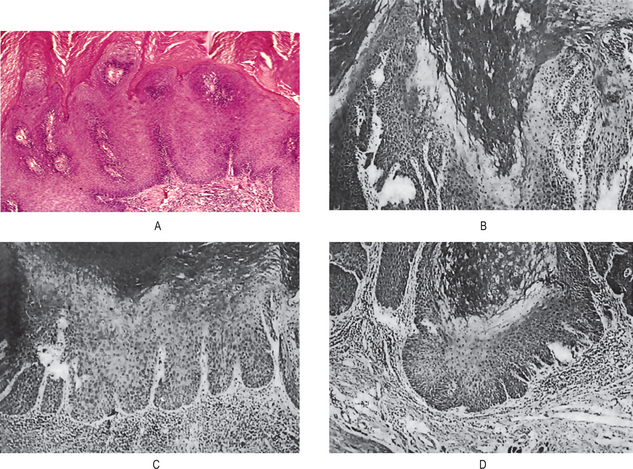
Figure 2-34 Verrucous carcinoma. The tumor is exophytic and has a papilliferous surface. It is composed of proliferating sheets and groups of stratified squamous epithelium, which exhibit few cytological features of malignancy. There is a round cell infiltrate in the fibrous tissue stroma. It may turn invasive at a later stage. (A, Courtesy of Dr Hari S, Noorul Islam College of Dental Science, Trivandrum)
Unfortunately, the diagnosis of verrucous carcinoma is often difficult even when the biopsy specimen is generous, and the pathologist will sometimes request a second biopsy.
Treatment and Prognosis
Verrucous carcinoma has been treated in several ways in the past, usually by surgery, X-ray radiation or a combination of the two. However, there have been some reports of anaplastic transformation of lesions occurring in patients treated by ionizing radiation. While the radiation appears to be the triggering mechanism, other factors contributing to or related to the transformation are unknown. Even though such an occurrence is uncommon, many investigators believe the treatment should be entirely surgical. Since the lesion is so slow-growing and late to metastasize, many cases can be treated by relatively conservative excision without a mutilating procedure. The prognosis is much better than for the usual type of oral epidermoid carcinoma.
Spindle Cell Carcinoma (Carcinosarcoma, pseudosarcoma, polypoid squamous cell carcinoma, Lane tumor)
A carcinoma within which there are some elements resembling a squamous cell carcinoma that are associated with a spindle cell component.
In a true spindle cell carcinoma, the malignant spindle shaped cells should be demonstrably of epithelial origin and derived from the squamous cell component of the carcinoma. This must be distinguished both from a squamous cell carcinoma that has provoked a reactive fibroblastic stromal proliferation and from a carcinosarcoma in which a squamous cell carcinoma is accompanied by a sarcoma of fibroblastic or other connective tissue cell type. Care should also be taken not to confuse a spindle cell carcinoma with a spindle cell malignant melanoma or with sarcomas of various types. Often the squamous cell carcinoma component in a spindle cell carcinoma is inconspicuous and multiple sections or blocks may be necessary to find it. Most of the neoplasm comprises of thin elongated cells amongst which there may be occasional pleomorphic cells. Mitotic figures, including abnormal forms, are usually not difficult to find. The behavior is similar to that of the more frequent and usual type of squamous cell carcinoma (Pindborg JJ et al, 1997).
Clinical Features
A series of 59 cases of the oral cavity has been reported by Ellis and Corio, who noted a predilection for occurrence in males, although this finding was somewhat biased because of the number of military cases involved. The mean age of occurrence of the lesion was 57 years, with a range of 29–93 years. The lesions developed with the greatest frequency on the lower lip (42%), tongue (20%) and alveolar ridge or gingiva (19%) with the remainder scattered at other sites.
The most common presenting findings were swelling, pain and the presence of a nonhealing ulcer. The initial lesion appeared either with a polypoid, exophytic or endophytic configuration. It is perhaps significant that 13 patients in this series were known to have a history of prior therapeutic radiation to the region where the tumor subsequently developed. The time interval from radiation to diagnosis of the tumor ranged from 1.5–10 years with a mean of about seven years.
Histologic Features
Spindle cell carcinoma is a bimorphic or biphasic tumor which, although almost always ulcerated, will show foci of surface epidermoid carcinoma or epithelial dysplasia of surface mucosa, usually just at the periphery and often quite limited. Proliferation and ‘dropping-off ’ of basal cells to spindle cell elements is a common phenomenon. The tissue patterns making up the bulk of the tumor have been categorized as either fasciculated, myxomatous or streaming. The cells, particularly in the fasciculated form, are elongated with elliptical nuclei, although pleomorphic cells are also common. The number of mitoses may vary from few to many. Giant cells, both benign-appearing of the foreign body type and the bizarre, pleomorphic, atypical cells may be found. Finally, an inflammatory cell infiltrate is often present. Osteoid formation within the tumor component is sometimes seen. Microscopic invasion of subjacent structures is evident, as it is with most epidermoid carcinomas of the oral cavity.
Numerous ultrastructural studies of the spindle cell carcinoma, such as those of Leifer and his associates, have been carried out in clarification of the histogenesis and pathogenesis of this lesion.
Treatment and Prognosis
Surgical removal of the tumor, with or without radical neck dissection, alone or in combination with radiation, or radiation therapy alone all have been used in the treatment of this disease. In the series reported by Ellis and Corio, those treated by surgery had the best survival rate although only nine of 18 patients treated in this fashion were alive and well. Radiation therapy appears ineffective. The presence of metastasis signals a poor prognosis, since 81% of the patients with recorded metastases in this study died of their disease. The value of chemotherapy is not known.
Adenoid Squamous Cell Carcinoma (Adenoacanthoma, pseudoglandular squamous cell carcinoma)
A squamous cell carcinoma containing pseudoglandular spaces or lumina is an interesting tumor of the skin which also occurs with considerable frequency on the lips. It was originally described by Lever in 1947. This variant is produced as a result of acantholysis and degeneration within islands of a squamous cell carcinoma. The result is a pseudoadenocarcinomatous appearance, but there is no evidence of glandular differentiation or of secretory activity or products. There are insufficient reported cases to establish likely behavior.
Clinical Features
Adenoid squamous cell carcinoma is reported to occur as early as 20 years of age, although in the series of Johnson and Helwig, nearly 75% of the patients were 50 years of age or older. In their series, only 2% of the patients were females. Significantly, they found that 93% of the lesions were in the head and neck region.
The lesions on the skin appear as simply elevated nodules that may show crusting, scaling or ulceration. Sometimes there is an elevated or rolled border to the lesion.
A series of 15 cases of adenoid squamous cell carcinoma of the lips (11 lower, three upper, one unstated) has been reported by Jacoway and his associates, while Tomich and Hutton have reported two cases and discussed the lesion in detail. These lip lesions often appear clinically similar to epidermoid carcinoma, being described frequently as ulcerated, hyperkeratotic or exophytic.
There have been four cases of this lesion reported intraorally, two on the gingiva. The latter includes the first case reported from India by Sivapathasundharam B and Rohini S. However three of these lesions behaved in a very aggressive fashion, metastasized in at least two instances and the patient died in all three cases as a result of the tumor. Because of the aggressive nature of the lesions, these intraoral cases may not be identical to those of the skin and lips.
Histologic Features
There is a proliferation of surface dysplastic epithelium into the connective tissue as in the typical epidermoid carcinoma. However, the lateral or deep extensions of this epithelium show the characteristic solid and tubular ductal structures which typify the lesion. These duct like structures are lined by a layer of cuboidal cells and often contain or enclose acantholytic or dyskeratotic cells.
Treatment and Prognosis
The adenoid squamous cell carcinoma is generally treated by surgical excision. On only rare occasions does it metastasize or cause death of the patient. However, recurrence is relatively common (38% in the series of lip lesions reported by Jacoway and his coworkers, although it is possible that some of these may have been second lesions, since multiple adenoid squamous cell carcinomas in the same patient often occur).
Basaloid Squamous Cell Carcinoma
Basaloid squamous cell carcinoma is a form of carcinoma with a mixed composition of basaloid and squamous cells. This is a form of oral carcinoma in which the basaloid component comprises small cells with hyperchromatic nuclei and scant cytoplasm that are crowded together into lobulated sheets or strands focally connected to the surface epithelium. Cells at the periphery of the lobules are often palisaded; more centrally there may be cystic spaces, sometimes containing material resembling mucin, and focal squamous differentiation. Mitotic figures, including abnormal forms, and areas of necrosis are commonly seen. There is often hyalinization of the surrounding stroma and chronic inflammatory cell infiltration is variable. Confusion with ameloblastoma and adenoid cystic carcinoma is to be avoided; a focal squamous cell carcinoma component among the basaloid areas is the most important distinguishing feature. Most cases have been described in the larynx, hypopharynx and base of the tongue (Pindborg JJ et al, 1997).
Adenosquamous Carcinoma
A malignant tumor with histological features of both adenocarcinoma and squamous cell carcinoma. This tumor may arise from the ducts of minor salivary glands or from the overlying surface epithelium. The component identified as squamous cell carcinoma may be in situ or invasive, and the adenocarcinomatous component comprises glandular structures lined by basaloid, columnar or mucin-secreting cells. A distinction between adenosquamous carcinoma and high-grade mucoepidermoid carcinoma may be difficult, though in the former the glandular and squamous components are generally more distinct. Care must also be taken to distinguish adenosquamous carcinoma from adenoid squamous cell carcinoma (Pindborg JJ et al, 1997).
Undifferentiated Carcinoma
Undifferentiated carcinoma is carcinoma that lacks evidence of squamous, glandular or other types of differentiation. Accurate diagnosis is almost certainly dependent upon the use of adjunctive diagnostic techniques.
Lymphoepithelioma and Transitional Cell Carcinoma
There is an unusual group of malignant neoplasms exhibiting many features in common which involves nasopharynx, oropharynx, tongue, tonsil and anatomically associated structures such as the nasal chamber and paranasal sinuses. These tumors arise from the mucosa of these areas, exhibit a relatively specific histologic pattern and react in a rather atypical fashion to X-ray radiation. This group of neoplasms consists of the lymphoepithelioma, transitional cell carcinoma and undifferentiated squamous cell carcinoma.
Regaud, and later Schminke as well as Ewing, described the lymphoepithelioma as a lesion occurring chiefly in the nasopharynx of young or middle-aged persons. It was found to be usually a small lesion which often did not manifest itself clinically before regional lymphadenopathy was apparent. Death of the patient was the frequent outcome of the disease even though the lesion was found to be radiosensitive.
Under the term ‘transitional cell epidermoid carcinoma’, Quick and Cutler reported a series of cases in which the lesions arose chiefly from the tonsil, base of the tongue and nasopharynx. It is in these areas that a transitional type of stratified epithelium is found, the schneiderian membrane. These tumors, then, occurred in areas similar to the sites of the lymphoepithelioma. It was noted, however, that this transitional cell carcinoma was extremely malignant, running a rapid clinical course, metastasizing widely and causing very early death.
Clinical Features
The primary lesion of the lymphoepithe-lioma or transitional cell carcinoma is usually very small, often completely hidden, usually slightly elevated and either frankly ulcerated or presenting a granular, eroded surface. The tumor is indurated and, in some instances, appears as an exophytic or fungating growth. Since the primary lesion usually remains small, the patient may not seek advice until metastasis to the regional lymph nodes has already occurred.
Scofield carried out an excellent study of 214 cases of malignant nasopharyngeal lesions, comprising transitional cell carcinoma, lymphoepithelioma and undifferentiated squamous cell carcinoma. He found that swelling of the regional lymph nodes was the most common presenting symptom of the observed patients, followed by sore throat, nasal obstruction, defective hearing or ear pain, headache, dysphagia, epistaxis and ocular symptoms. Differences in the median age of the patients were found, patients with transitional cell carcinoma averaging 44 years of age, those with lymphoepithelioma averaging only 26 years and those with undifferentiated squamous cell carcinoma, 56 years. Bloom published a review of cancer of the nasopharynx with particular reference to the significance of the histopathology of the lesions.
Histologic Features
The diagnosis of these neoplasms and their differentiation depends solely upon their microscopic structure.
Transitional cell epidermoid carcinoma consists of cells growing in solid sheets or in cords and nests. The individual cells are moderately large, round or polyhedral, and exhibit a lightly basophilic cytoplasm and indistinct cell outlines. The nuclei appear large and round, and they exhibit varying degrees of mitotic activity. Although a slight degree of intercellular bridging may be present, keratinization and pearl formation are completely lacking. The stroma exhibits little or no lymphocytic infiltration.
The lymphoepithelioma is made up of cells growing in a syncytial pattern with the stroma infiltrated by varying numbers of lymphocytes. The individual cells are large and polyhedral with indistinct outlines. The cytoplasm stains lightly eosinophilic. The nuclei appear large, oval and vesicular, and characteristically contain one or two large eosinophilic nucleoli.
Treatment and Prognosis
Because of the general inacces-sibility of the majority of these lesions and their unusual property of being highly radiosensitive, X-ray radiation has been the most commonly accepted treatment. The response of this tumor to radiation is different from that of the epidermoid carcinoma found in these locations. Regional lymph node metastases also respond well to X-ray radiation. The complicating factor lies in the relative inability to treat the widespread metastases in the various organs.
The outlook for patients with these forms of neoplastic disease is poor. Since widespread metastases frequently occur before there is any clinical manifestation of disease, the unfavorable prognosis can be readily understood. In the series of Scofield the probability of five-year survival was calculated. He found that, after onset of symptoms, only 30% of treated patients suffering from transitional cell carcinoma or lymphoepithelioma would be alive in five years, while only 11% with squamous cell carcinoma in these areas would survive.
Nasopharyngeal Carcinoma
A tumor of the nasopharynx involving squamous epithelium, malignant in nature is widely prevalent in parts of south China (Cantonese) (98 per 100,000 of the population); where it is the commonest tumor in men and the second commonest in women. The tumor is rare in most parts of the world, though pockets occur in north and central Africa, Malaysia, Alaska, and Iceland. The most undifferentiated form of the tumor is always associated with EBV whereas the rarer, more differentiated forms are not consistently so. The evidence that EBV is involved in the pathogenesis is based on detection of multiple copies of the EBV genome can be detected in the malignant cells of 100% of undifferentiated NPC. All the malignant cells express EBNA-1. Furthermore, infectious EBV particles can be recovered from NPC cell lines. And also 100% of sera from undifferentiated NPC patients have high-titer antibodies to EB-viral antigens.
Clinical Features
NPC is a tumor proven to have a genetic background mainly restricted to southern China, with intermediate frequency in some Negro and Mongoloid races and rare in Caucasians. Studies have shown that first-generation immigrants from south China retain the high incidence of the disease, with the later generations showing a decline in incidence. This suggests that environmental as well as genetic factors are involved. NPC is especially associated with certain HLA haplotypes, e.g. HLA A2. More genetic linkage studies demonstrated the presence of NPC susceptibility genes near the MHC locus. Environmental factors are thought to play a role, particularly the consumption of salted fish and foods containing nitrosamines.
The EBV associated undifferentiated type arises mainly in younger patients whereas the more differentiated types occur in older patients and constitute the bulk of the sporadic cases. The tumor most commonly arises in the posterior wall of the nasopharynx in the fossa of Rosenmuller, where it often remains silent and metastasize to the local lymph nodes. The most common presentation of NPC is bilateral enlargement of the glands in the neck. The primary tumor may be very small and difficult to locate. Less frequently, the patient may present with the symptoms of invasion by the primary tumor, e.g. nasal obstruction, postnasal discharge, epistaxis, partial deafness and cranial nerve palsies. If untreated, the disease is rapidly fatal due to the development of laryngeal and pharyngeal obstruction.
Histologic Features
Three types of NPC are recognized on histological appearance:
Serum antibodies to EBV antigens can be used to confirm the diagnosis and monitor the progress of the disease.
Treatment
NPC is difficult to treat surgically because of the early metastasis to regional lymph nodes. The tumor is resistant to chemotherapy, and radiotherapy is the treatment of choice. However, because the tumor usually presents late, the prognosis is poor with a five-year survival rate of 20%. It may be possible to prevent the development of NPC with the use of an EBV vaccine at an early age.
Malignant Melanoma
Malignant melanoma is a neoplasm of epidermal melanocytes. It is one of the more biologically unpredictable and deadly of all human neoplasms. Although it is the third most common cancer of the skin (basal and squamous cell carcinomas are more prevalent), it accounts for only 3% of all such malignancies. However, it results in over 83% of all deaths due to skin cancer in the United States.
Cutaneous melanoma is increasing in incidence. The frequency of its occurrence is closely associated with the constitutive color of the skin, and depends on the geographical zone. Incidence among dark skinned ethnic groups is 1 per 100,000 per year or less, but among light-skinned Caucasians up to 50 and higher in some areas of the world. The highest incidence rates have been reported from Queensland, Australia with 56 new cases per year per 100,000 for men and 43 for women. In contrast, for Africans and Asians the annual incidence rate of malignant melanoma is only 0.2–0.4 per 100,000 population, affecting mainly the palms, soles, and mucous membranes. Cutaneous malignant melanoma is the most rapidly increasing cancer in whites. The annual increase in incidence rates has been estimated to be between 3 and 7%. These estimates suggest a doubling of rates every 10–20 years. These epidemiologic studies have supported the belief that sunlight is an important etiologic factor in cutaneous melanoma.
For many years, it was believed that many melanomas developed in preexisting pigmented nevi, particularly junctional nevi. However, Clark and his colleagues are of the opinion that junctional nevi are not histogenetically related to melanomas. It is quite possible that lesions which were interpreted as junctional nevi were, in fact, premalignant melanocytic dysplasias of some type, thus leading to the erroneous concept of malignant transformation of nevi. In support of this is a study by Jones and his colleagues in which 169 cases diagnosed as junctional nevi were studied retrospectively. Only 74 were actually junctional nevi, whereas 41 were actually melanomas in various phases of growth. The remainder were nevoid and non-nevoid pigmented lesions of various types. Melanomas may develop in or near a previously existing precursor lesion or in healthy-appearing skin. A malignant melanoma developing in healthy skin is said to arise de novo, without evidence of a precursor lesion. Certain lesions are considered to be precursor lesions of melanoma, including the common acquired nevus, dysplastic nevus, congenital nevus, and cellular blue nevus.
The following environmental and genetic factors are described in the etiology of malignant melanoma.
Environmental Factors
The highest incidence of melanoma has been reported from areas with long hours of sunlight throughout most of the year. Studies reported lower risk for melanoma among people who resided in a low ultraviolet environment in childhood compared with those who resided in a high UV environment. Intermittent exposure to high intensity UV seems to be more detrimental than continual exposure in its causation. Exposure in childhood appears to be particularly important. Recreational activity leading to sunburns in adulthood, such as sailing, has also been incriminated as an etiological factor.
Several case-control studies of melanoma risk and tanning lamp use have demonstrated a positive relation, suggesting that longer wave artificial UVA may play a part in the etiology of melanoma in addition to exposure to natural sunlight. The association of melanoma with PUVA (combination of psoralen (P) and long wave ultraviolet radiation (UVA)) therapy has also been reported. This topic is still controversial, though, and further studies are needed.
Several studies have reported that melanoma is more prevalent in those of high socioeconomic status. An explanation of this finding may be the fact that the latter can better afford holidays in areas of high UV intensity, as well as expensive outdoor hobbies like sailing, which increase the risk of melanoma due to intermittent intense sun exposure.
Genetic Factors
Between 2 and 5% of melanoma patients have a positive family history of melanoma in at least one first-degree relative. In approximately 30% of melanoma patients abnormalities on chromosome 9p21 are seen.
In this genetically determined disorder, defective DNA repair mechanisms lead to excessive chronic UV damage and subsequent development of different sun-related skin tumors, including melanoma, in sun-exposed areas.
Risk factors for oral mucosal melanomas are unknown. These melanomas have no apparent relationship to chemical, thermal, or physical events (e.g. smoking, alcohol intake, poor oral hygiene, irritation from teeth, dentures, or other oral appliances) to which the oral mucosa is constantly exposed. Although benign, intraoral melanocytic proliferations (nevi) occur and are potential sources of some oral melanomas; the sequence of events is poorly understood in the oral cavity. Currently, most oral melanomas are thought to arise de novo.
In 1975, Clark and his coworkers presented an interesting concept regarding the developmental biology of cutaneous melanoma. They documented two phases in the growth of melanoma: the radial-growth phase and the vertical-growth phase.
The radial-growth phase is the initial phase of growth of the tumor. During this period, which may last many years, the neoplastic process is confined to the epidermis. Neoplastic cells are shed with normally maturing epithelial cells and although some neoplastic cells may actually penetrate the basement membrane, they are destroyed by a host-cell immunologic response. The vertical-growth phase begins when neoplastic cells populate the underlying dermis. This takes place because of increased virulence of the neoplastic cells, a decreased host-cell response, or a combination of both. Metastasis is possible once the melanoma enters the vertical-growth phase. It is recognized that not all melanomas have both radial-and vertical-growth phases. Nodular melanoma (q.v.) exists only in the vertical-growth phase.
Cutaneous melanoma has been classified into a number of types. However, the most common types are: superficial spreading melanoma; nodular melanoma; lentigo maligna melanoma (Hutchinson’s freckle); and acral lentiginous melanoma.
Clinical Features
Superficial spreading melanoma is the most common cutaneous melanoma in Caucasians. It accounts for nearly 65% of cutaneous melanomas. It exists in a radial-growth phase which has been called premalignant melanosis or pagetoid melanoma in situ. The lesion presents as a tan, brown, black or admixed lesion on sun-exposed skin, especially the back. It also occurs on the skin of the head and neck, chest and abdomen and the extremities. The radial-growth phase may last for several months to several years. The vertical-growth phase is characterized by an increase in size, change in color, nodularity and, at times, ulceration.
Nodular melanoma accounts for approximately 13% of cutaneous melanomas. It apparently has no clinically recognizable radial-growth phase, existing solely in a vertical-growth phase. It presents as a sharply delineated nodule with varying degrees of pigmentation. They may be pink (amelanotic melanoma) or black. They have a predilection for occurrence on the skin of the back and head and neck skin of men. In other cutaneous sites, there is an even gender distribution.
Lentigo maligna melanoma accounts for approximately 10% of cutaneous melanomas. It exists in a radial-growth phase which is known as lentigo maligna or melanotic freckle of Hutchinson. The melanotic freckle has been recognized as a clinicopathologic entity for nearly 100 years. However, the concept that it represents a melanoma in a radial-growth phase is much more recent. The lesion occurs characteristically as a macular lesion on the malar skin of middle-aged and elderly Caucasians. It occurs more often in women than in men. In an extensive series of 85 cases, Wayte and Helwig found an average age of 58 years in men and 55 in women. In a series studied by Clark and his colleagues, the median age was 70 years. Both studies showed a female gender predilection. The lesion can remain in the radial-growth phase for years. In Wayte and Helwig’s study, the average duration in which an accurate history was possible was 14 years. Clark and Mihm have documented a lentigo maligna for 50 years prior to the development of a vertical-growth phase. Nearly 53% of the lesions evolved into lentigo maligna melanoma, the vertical-growth phase of this form of melanoma.
Melanoma may occur as a primary lesion not only on the skin but also in the eye and on mucous membranes. It has also been reported as a primary lesion in the parotid gland, although melanomas in this site are usually metastatic to lymph nodes in the parotid region.
Melanoma developing on the palms and soles, as well as on toes and fingers, represents only 10% of cases in whites, but over 50% of all melanomas on Black and Asian skin. The tumor is characterized by a macular, lentiginous pigmented area around a nodule. Mechanical stress may lead to erosion and ulceration. Subungual melanomas present as pigmentations of the nail bed and are often mistaken for subungual hematomas, and are thus commonly diagnosed at a late stage in development. They are extremely aggressive, with rapid progression from the radial to vertical growth phase.
Mucosal lentiginous melanomas develop from the mucosal epithelium that lines the respiratory, gastrointestinal, and genitourinary systems. These lesions account for approximately 3% of the melanomas diagnosed annually and may occur on any mucosal surface, including the conjunctiva, oral cavity, esophagus, vagina, female urethra, penis, and anus. Noncutaneous melanomas are commonly diagnosed in patients of advanced age. When compared to cutaneous melanomas, mucosal lentiginous melanomas appear to have a more aggressive course, although this may be because they are commonly diagnosed at a later stage of disease than the more readily apparent cutaneous melanomas.
Amelanotic melanoma presents as an erythematous or pink, sometimes eroded, nodule. This tumor is often confused for other tumors, and only the histological examination provides the right diagnosis.
The following criteria aid clinical diagnosis of melanoma (ABCDE-rule):
• Asymmetry—in which one half does not match the other half
• Border irregularity—with blurred, notched, or ragged edges
• Color irregularity—pigmentation is not uniform. Brown black, tan, red, white, and blue—can all appear in a melanoma
• Diameter—greater than 6 mm. Growth in itself is also a sign
Oral Manifestations
Malignant melanoma is an uncommon neoplasm of the oral mucosa. Pliskin reviewed the literature on oral melanoma and found that they accounted for 1.6% of over 7,500 reported melanomas. Other authors have reported rates of 0.2–8%. Conley and Pack reported 26 cases of primary oral melanoma and McCaffrey, Neel and Gaffey reported on the 10 cases treated at the Mayo Clinic. Of epidemiologic interest is the fact that melanoma of the oral mucosa is one of the most common sites for the neoplasm in Japanese. Melanomas in Blacks are seldom found in the skin yet occur on mucous membranes and on the plantar skin.
Primary oral melanoma is nearly twice as common in men as in women. The overall age of occurrence is approximately 55 years, with most cases occurring between 40 and 70 years.
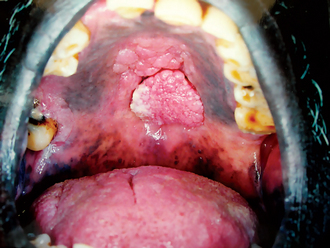
The oral melanoma exhibits a definite predilection for the palate and maxillary gingiva/alveolar ridge. Seventy-seven% of the cases in Pliskin’s review occurred in these two sites. Cases are also recorded on the buccal mucosa, mandibular gingiva, tongue, lips and floor of the mouth. The lesion usually appears as a deeply pigmented area, at times ulcerated and hemorrhagic, which tends to increase progressively in size (Fig. 2-35A–C). Amelanotic melanoma accounts for 5–35% of oral melanomas which appear as a white, mucosa-colored, or red mass.
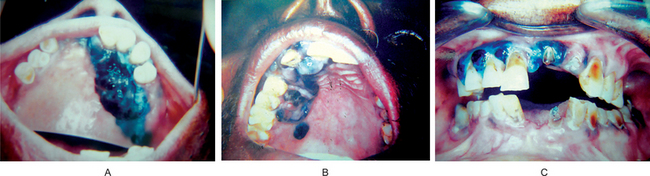
Figure 2-35 Malignant melanoma. Typical lesions involve the plate (A) and (B), and the alveolar ridge (C) (Courtesy of Sivakumar G, Sivapathasundharam B, Karthiga KS. Malignant melanoma of the oral cavity–case reports and review of literature. Indian J Dent Res, 2004, 15(2): 70–73).
Significantly, focal pigmentation preceding the development of the actual neoplasm frequently occurred several months to several years before clinical symptoms appeared. For this reason it has been suggested that the appearance of melanin pigmentation in the mouth and its increase in size and in depth of color should be viewed seriously.
Although clinicopathologic correlation is well established for cutaneous melanomas, it is unfortunate that such correlation does not exist for oral melanomas. It is now apparent that melanomas of the oral mucosa can exist in radial-and vertical-growth phases but only a few such cases have been reported. Takagi and his coworkers were able to document a preexisting or concurrent melanosis in 62 of 94 cases of oral melanomas which they studied. Regezi, Hayward and Pickens evaluated three cases of oral melanoma in accordance with the established clinicopathologic parameters of cutaneous melanomas. They were able to classify one as a superficial spreading melanoma which had a preexisting melanosis for 11 years. The other two had histologic features consistent with lentigo maligna melanoma. However, since these lesions behaved much more aggressively, they were termed acral-lentiginous melanomas as suggested by Clark and his colleagues and by Reed.
Most dermatopathologists recognize the existence of lentigo maligna (melanotic freckle of Hutchinson) only on sun-damaged skin and do not accept its occurrence intraorally. It follows that lentigo maligna melanoma (melanoma arising in melanotic freckle of Hutchinson) cannot occur on oral mucosa, although such a case has been reported by Robinson and Hukill. In light of present knowledge, such a case would be called acral-lentiginous melanoma because of the clinicopathologic similarity of the oral lesion to those on palmar and plantar skin (Table 2-11).
Table 2-11
Differential diagnosis for melanoma
| Condition | Distinguishing characteristics |
| Seborrheic keratosis | “Stuck-on” appearance, symmetric, often multiple |
| Traumatized or irritated nevus | Returns to normal appearance within 7–14 days |
| Pigmented basal cell carcinoma | Waxy appearance, telangiectasias |
| Lentigo | Prevalent in sun-exposed skin, evenly pigmented, symmetric |
| Blue nevus | Darkly pigmented from dermal melanocytes, no history of change from melanoma |
| Angiokeratoma | Vascular tumors, difficult to distinguish |
| Traumatic hematoma | May mimic melanoma but resolves in 7–14 days |
| Venous lake | Blue, compressible, found on ears and lips |
| Hemangioma | Compressible, stable |
| Dermatofibroma | Firm growths of fibrous histiocytes, ‘button-hole’ when pinched |
| Pigmented actinic keratosis | Sandpapery feel; sun-exposed area |
Source: Beth G Goldstein, and Adam O Goldstein. American Academy of Family Physicians, News and Publications, 2001.
Thus, oral melanomas may be expected to exist in superficial spreading, acral-lentiginous and nodular types. Batsakis and his associates and Hansen and Buchner have recently discussed the current concepts of oral mucosal melanomas relative to their cutaneous counterparts.
Histologic Features
The malignant cells often nest or cluster in groups in an organoid fashion; however, single cells can predominate. The melanoma cells have large nuclei, often with prominent nucleoli, and show nuclear pseudoinclusions due to nuclear membrane irregularity. The abundant cytoplasm may be uniformly eosinophilic or optically clear. Occasionally, the cells become spindled or neurotized in areas. This finding is interpreted as a more aggressive feature, compared with findings of the round or polygonal cell varieties.
The intraepithelial component (radial-growth phase) of superficial spreading melanoma is characterized by the presence of large, epithelioid melanocytes distributed in a so-called ‘pagetoid’ manner (Figs. 2-36, 2-37 A). This pagetoid spread within the epidermis is sometimes known as ‘buckshot scatter’. As long as the malignant cells are confined to the epithelium, there is no host-cell response in the underlying connective tissue. When melanocytes penetrate basement membrane, a florid host-cell response of lymphocytes develops. Macrophages and melanophages may be present. The tumor cells are often destroyed by this cellular response. The vertical-growth phase is characterized by the proliferation of malignant epithelioid melanocytes in the underlying connective tissues (Fig. 2-37 B). The cells may be arranged singly or in clusters. Melanin pigment is usually scanty.
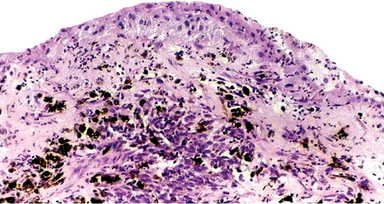
Figure 2-36 Vertical growth phase characterized by malignant melanocytes invading the underlying connective tissue (Courtesy of Dr Hari S, Noorul Islam College of Dental Science, Trivandrum).
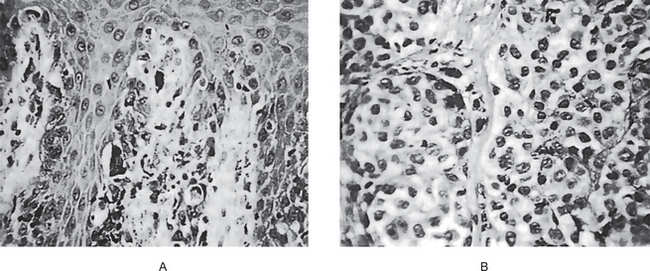
Figure 2-37 Malignant melanoma. (A) The radial-growth phase or premalignant melanosis is characterized by atypical melanocytes within the epithelium. (B) The vertical-growth phase is characterized by malignant epithelioid melanocytes invading the underlying connective tissue.
Nodular melanoma also is characterized by large, epithelioid melanocytes within the connective tissue. However, small ovoid and spindle-shaped cells may be present. Melanin pigment is usually but not invariably present. The tumor cells may invade and ulcerate the overlying epithelium and penetrate the deep soft tissues.
Lentigo maligna (melanotic freckle of Hutchinson) has well-defined histologic features which have been discussed by Wayte and Helwig and by Clark and Mihm. The lesion is characterized by increased numbers of atypical melanocytes within the basal epithelial layer. The epithelium is generally atrophic and the dermal collagen shows the effects of sun-damage (basophilic degeneration). If skin appendages are present, they are often involved with atypical melanocytes as well. In time, cords and nests of atypical melanocytes may be evident. Lentigo maligna melanoma is characterized by invasive spindle-shaped cells into the underlying dermis. A lymphohistiocytic infiltrate is usually present.
The acral-lentiginous melanoma is histologically similar to lentigo maligna melanoma. Coleman and his coworkers have studied 35 cases and have pointed out the salient histologic features. These are:
• A lentiginous radial-growth phase
• A deep vertical-growth phase composed predominantly of spindle-shaped cells
• Psoriasiform epidermal hyperplasia
• An intense host-cell response
• A prominent desmoplasia associated with the vertical-growth phase.
Although the majority of melanomas are diagnosed by routine light microscopy, ultrastructural study can be of value in diagnosing and distinguishing between various types of melanoma. As pointed out by Klug and Gunther, the three main types of cutaneous melanoma differ ultrastructurally in the number and size of melanosomes. Regezi, Hayward and Pickens studied an oral acral-lentiginous melanoma ultrastructurally and found premelanosomes and melanosomes similar to those found in normal melanocytes and nevus cells.
Less common histologic variants of melanoma such as desmoplastic, neurotropic, spindle cell and balloon cell melanomas exist. These have been discussed by Ainsworth and her colleagues.
The lymph node metastasis is identified by using lymphoscintigraphy and a radioactive tracer (technetium-labeled sulfur colloid or human serum albumin).
Treatment and Prognosis
The treatment of cutaneous malignant melanoma is surgical excision. Although regional lymph node dissection is indicated when nodes are involved, prophylactic lymph node dissection is very controversial. The decision of the surgeon relative to elective node dissection should be based upon the thickness of the lesion and its specific anatomic location. In this regard, tumors greater than 0.75 millimeters in thickness and located in the so-called BANS (back, arm, neck and scalp) sites have a greater tendency to metastasize. On the other hand, melanomas of the skin of the face have a much more favorable prognosis.
Chemotherapy, immunotherapy and radiation therapy have been used in the treatment of cutaneous melanoma. The role of these modalities is discussed in the monograph on melanoma edited by Clark, Goldman and Mastrangelo.
The treatment of oral melanoma has been and still is surgical excision. Jaw resection and lymph node dissection is indicated in cases involving bone and regional lymph nodes. Other forms of therapy such as cryosurgery, radiation, chemotherapy and immunotherapy have been employed on occasion but none have significantly improved the dismal prognosis of melanoma in the oral cavity.
There are both clinical and histologic factors which are of great prognostic significance in cutaneous melanomas. According to McGovern, clinical features with prognostic significance are the gender and age of the patient and the site of the lesion. Women have a much better survival rate up to the age of 50 years and then the rate declines.
Histologic features which are of prognostic significance are histologic type and depth of invasion. Nodular melanoma and superficial spreading melanoma have much poorer prognosis than lentigo maligna melanoma. In fact, McGovern believes that lentigo maligna melanoma should be considered as a separate entity because of its better prognosis. In 1999, Clark and his colleagues related prognosis to the anatomic level of invasion at the time of diagnosis. In 1970, Breslow proposed that the actual thickness of the tumor as measured in millimeters by an ocular micrometer correlated well with prognosis. It is now recognized that tumors less than 0.75 mm in thickness rarely metastasize or cause death, regardless of the location on skin.
Unfortunately, oral mucosal melanomas have a far worse prognosis than cutaneous melanomas. According to Pliskin, the five-year survival rate for such tumors is approximately 7%. Batsakis and his colleagues have pointed out that establishment of prognostic indicators for oral mucous membrane melanomas has not kept pace with those which have been established for cutaneous melanomas.
The level of tumor invasion is another important indicator of the prognosis of MM. The Clark system is generally used to grade tumor invasion based on the deepest histologic cutaneous structure the tumor infiltrates. Table 2-12 depicts five-year survival (FYS) rates for MM based on the Breslow and Clark grading systems.
Table 2-12
Malignant melanoma classification and five-year survival (FYS) rates
| Clark system | FYS rate (percentage) |
| Level I: Tumor in situ Tumor is conined to the epidermis and is entirely above the basement membrane | 100–98 |
| Level II Invasive cells are only present in the papillary dermis. The tumor is usually still considered to be in the radial growth phase | 96–72 |
| Level III Tumor cells are found throughout the papillary dermis with impingement on the reticular dermis. The tumor has entered the vertical growth phase | 90–46 |
| Level IV Tumor cells are clearly seen between the collagen bundles of the reticular dermis | 67–31 |
| Level V Tumor cells show invasion of the subcutaneous fat. | 48–12 |
| Breslow system* | FYS rate (in percentage) |
| 0.00–0.76 mm | 98–99 |
| 0.76–1.49 | mm 85 |
| 1.50–2.49 mm | 84 |
| 1.50–2.49 mm | 84 |
| 4.00 mm | 44 |
*Total tumor thickness as measured from the outermost layer of the stratum granulosum to the deepest identiiable point of tumor invasion in the dermis.
Benign Tumors of Connective Tissue Origin
Oral Fibroma and Fibromatoses Fibroma (Irritation fibroma, focal fibrous hyperplasia)
This connective tissue tumor is the most common benign soft tissue neoplasm occurring in the oral cavity. Most fibromas represent reactive focal fibrous hyperplasia due to trauma or local irritation. Although the term focal fibrous hyperplasia more accurately describes the clinical appearance and pathogenesis of this entity, it is not commonly used. It is intimately related to fibrous hyperplasia and, in many instances, is histologically indistinguishable from it.
Clinical Features
A fibroma may occur at any oral site, most commonly it is seen on the buccal mucosa along the plane of occlusion. Other frequent sites are the gingiva, buccal mucosa, the tongue, lips and the palate.
The fibroma appears as an elevated nodule of normal color with a smooth surface and a sessile or, occasionally, pedunculated base. The tumor may be small or, in rare instances, may range up to several centimeters in diameter. Projecting above the surface, the tumor sometimes becomes irritated and inflamed and may even show superficial ulceration or hyperkeratosis. It is nearly always a well-defined, slowly growing lesion that occurs at any age, but is most common in the third, fourth and fifth decades. Females are affected twice as frequently as males.
The clinical differential diagnosis of a fibroma includes giant cell fibroma, neurofibroma, peripheral giant cell granuloma, mucocele, and benign and malignant salivary gland tumors.
Histologic Features
The fibroma consists of bundles of interlacing collagenous fibers interspersed with varying numbers of fibroblasts or fibrocytes and small blood vessels. The surface of the lesion is covered by a layer of stratified squamous epithelium which frequently appears stretched and shows shortening and flattening of the rete pegs (Fig. 2-38). If trauma to the tissue has occurred, vasodilatation, edema and inflammatory cell infiltration are variably present. Areas of diffuse or focal calcification or even ossification are found in some fibromas, chiefly those occurring on the gingiva, and these lesions have sometimes been called ‘peripheral ossifying fibroma’, ‘ossifying fibroid epulis’, ‘peripheral cementifying fibroma’ or ‘peripheral odontogenic fibroma’ (q.v.).
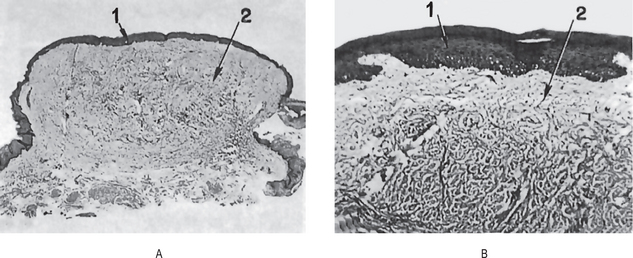
Figure 2-38 Fibroma. (A) The low-power photomicrograph illustrates the typical sessile base, the thin surface epithelium (1) and coarse bundles of collagen (2) comprising the bulk of the tumor. (B) The high-power photomicrograph shows the relative acellularity of the connective tissue.
It is interesting to note that the fibroma, a true neoplasm of connective tissue origin, is microscopically similar to the condition known as inflammatory hyperplasia, an increased bulk of connective tissue which forms as part of an inflammatory reaction. In few situations is the distinction between the two general processes, hyperplasia and neoplasia, as poorly defined as it is here. Hyperplasia is usually considered to be a self-limiting process which is not etiologically related to neoplasia. Both processes, however, are typified by an increase in the number of cells brought about by increased mitotic activity. Hyperplastic tissue sometimes, but not invariably, regresses after the removal of the stimulus or irritant. Neoplastic tissue shows no such regression.
This distinction between hyperplasia and neoplasia is not clearcut, and cases occur intraorally in which there is focal or diffuse proliferation of tissue obviously due to irritation which does not regress after removal of the irritant. The tissue appears identical with that in other cases which do regress when the irritant is removed. This suggests that the processes of hyperplasia and neoplasia may not be as completely dissociated as previously considered and that a true oral neoplasm may result from chronic irritation.
Many persons think, however, that a great number of the lesions of the oral cavity diagnosed as fibromas are, in reality, simply examples of focal or localized hyperplasia, resulting from inflammation, and that the true fibroma is much rarer than is presently believed. There is no doubt that the pyogenic granuloma (q.v.), if left untreated, will undergo eventual healing by sclerosis and will then microscopically resemble the fibroma.
Fibromatoses
Fibromatoses, sometimes referred to as juvenile or aggressive fibromatoses, represent a group of infiltrating fibrous proliferations with a biologic behavior and microscopic appearance intermediate between those of benign lesions and fibrosarcomas.
Clinical Features
The head and neck region, particularly the submandibular area, is a common site of involvement. However, intraoral lesions are rare. The age of affected patients varies from 0–51 years, but in 74% of patients, fibromatosis is diagnosed before they are aged 10 years. No significant gender predilection is apparent.
A desmoplastic fibroma is considered to be the intraosseous counterpart of the soft-tissue fibromatosis. A desmoplastic fibroma appears as a firm, painless, poorly demarcated mass that is either rapidly or slowly growing. The mass is locally aggressive, blends into surrounding structures, and causes resorption of the underlying bone when it is present.
Fibromatosis has to be differentiated clinically from low-grade fibrosarcoma, nodular fasciitis, reactive fibrous hyperplasia, fibrous histiocytoma, and neurofibroma.
Histologic Features
Histologically, fibromatosis is characterized by a poorly delineated, infiltrating cellular proliferation of mature spindle cells arranged in streaming and interlacing fascicles. Collagen production is prominent. Infiltration of the adjacent structures is common at the periphery, but cellular atypia is not present.
Giant Cell Fibroma
Giant cell fibroma is an oral tumor first described in 1974 by Weathers and Callihan as a distinctive entity. The distinctive histologic appearance sets it apart from a conventional fibroma.
Clinical Features
It appears as an asymptomatic sessile or pedunculated nodule that is less than 1 cm in diameter. Often, it has a bosselated or somewhat papillary surface. Most cases are diagnosed in persons aged 10–30 years, and no gender predilection exists. The most common site is the mandibular gingiva, followed by the maxillary gingiva, the tongue, and the palate.
The clinical differential diagnoses include squamous papilloma, irritation fibroma, pyogenic granuloma, and peripheral giant cell granuloma.
Histologic Features
Microscopically, a giant cell fibroma is an unencapsulated mass of loose fibrous connective tissue that contains numerous characteristic large, plump, spindle-shaped and stellate fibroblasts, some of which are multinucleated. These cells are easily observed in the peripheral areas of the lesion, whereas the more central areas contain typical fusiform fibroblasts. The surface epithelium is corrugated and atrophic; in contrast to an irritation fibroma, a giant cell fibroma has thin, elongated rete ridges.
The origin of stellate and multinucleate cells is not well known. Few studies showed positive immunostaining for vimentin. This suggests that the stellate and multinucleate cells of GCF have a fibroblast phenotype.
Myofibroma and Myofibromatosis
The terms myofibroma (if solitary) or myofibromatosis (if multicentric) are applied to the tumors which show predominant myofibroblasts. Myofibroblasts have features of both fibroblasts and smooth muscle cells.
Clinical Features
The tumors are benign and similar to fibromatosis but less aggressive. Tumors of the myofibroblasts may occur in either gender and in patients of all ages, with a mean patient age of 26.6 years.
Myofibroblastic tumors are most common in the head and neck region, including oral and perioral sites, than in other areas. Intraoral lesions occur mainly on the tongue, lips, and buccal mucosa. Tumors have also been described in the dermis, soft tissues, viscera, and bone. Jaw lesions, usually mandibular lesions, generally appear as well-defined unilocular or multilocular radiolucencies. Oral lesions appear as firm submucosal nodules or exophytic masses with a diameter of 0.3–5.0 cm. Although lesions are most often asymptomatic, they may be tender or even painful.
The clinical differential diagnosis for oral myofibroma includes irritation fibroma, peripheral giant cell fibroma, neurofibroma, leiomyoma, and benign and malignant neoplasms of the minor salivary glands.
Histopathologic Features
Histologically, all cases showed a biphasic pattern that consisted of fascicles of spindle cells with abundant eosinophilic cytoplasm that resembled smooth muscle, in addition to a population of more primitive spindled cells associated with a hemangiopericytoma like vascular pattern.
Peripheral Ossifying Fibroma (Peripheral odontogenic fibroma, peripheral cementifying fibroma, calcifying or ossifying fibroid epulis, peripheral fibroma with calcification)
There are numerous histologically different types of focal overgrowths which may occur on the gingiva, such as the peripheral giant cell granuloma, the giant cell fibroma, the pyogenic granuloma, the simple fibroma (which may be simply a healed pyogenic granuloma in many cases) and the present lesion, which in the past has been known by a variety of names indicated above. The terms most frequently used have been the ‘peripheral ossifying fibroma’ and the ‘peripheral odontogenic fibroma’. In as much as the latter term has been used for a lesion described by the World Health Organization in their classification of odontogenic tumors as a totally different entity, the term peripheral ossifying fibroma will be used here for that relatively common gingival lesion characterized by a high degree of cellularity usually exhibiting bone formation, although occasionally cementum-like material or rarely dystrophic calcification may be found instead.
Some investigators believe that the lesion is nevertheless odontogenic in origin, being derived from the periodontal ligament, especially since it only occurs on the gingiva and may contain oxytalan fibers. At the present time, however, its exact derivation is still uncertain. Despite the similarity in terminology, it is not considered to be the extraosseous counterpart of the central ossifying fibroma. An attempt at clarification of the terms ‘peripheral ossifying fibroma’ and ‘peripheral odontogenic fibroma’ has been published recently by Gardner.
Clinical Features
The peripheral ossifying fibroma can occur at any age, although it appears to be somewhat more common in children and young adults. In a study of 365 cases by Cundiff, 50% of the lesions occurred between the ages of five and 25 years with the peak incidence at 13 years, while the mean age was 29 years. Most reported series of cases show a predilection for occurrence in females by a ratio ranging from 2 : 1 to 3 : 2. In addition, the lesions are approximately equally divided between the maxilla and the mandible. In the series reported by Cundiff, over 80% of the lesions in both jaws occurred anterior to the molar area. A series of 185 cases of ‘peripheral fibroma with calcification’ were also reported by Bhaskar and Jacoway with very similar clinical data.
The clinical appearance of the lesion is characteristic but not pathognomonic. It is a well-demarcated focal mass of tissue on the gingiva, with a sessile or pedunculated base (Fig. 2-39A). It is of the same color as normal mucosa or slightly reddened. The surface may be intact or ulcerated. It most commonly appears to originate from an interdental papilla.
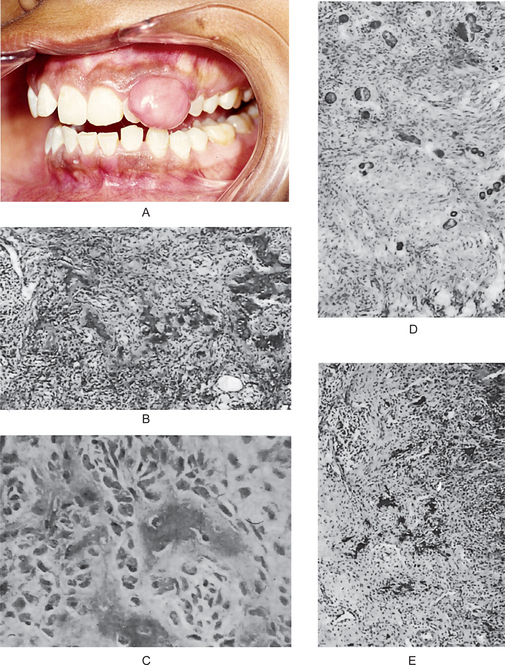
Figure 2-39 Peripheral ossifying ibroma. The sessile lesion on the gingiva (A) is histologically a very cellular lesion which also exhibits most frequently irregular trabeculae of osteoid or bone ([B] in low power, [C] high power), but occasionally presents cementum-like droplets (D) or even dystrophic calcification (E). (A, Courtesy of Dr Vandhana KL, Department of Periodontics, College of Dental Sciences, Davangere).
Radiographic Features
In the vast majority of cases, there is no apparent underlying bone involvement visible on the radiograph. However, on rare occasions, there does appear to be superficial erosion of bone.
Histologic Features
The surface of the peripheral ossifying fibroma exhibits either an intact or, more frequently, an ulcerated layer of stratified squamous epithelium. The bulk of the lesion is composed of an exceedingly cellular mass of connective tissue comprising large numbers of plump proliferating fibroblasts intermingled throughout a very delicate fibrillar stroma. The lesion is quite characteristic in its high degree of cellularity in contrast to the usual simple fibroma. In addition, vascularity is not nearly as prominent a feature of this lesion as in the pyogenic granuloma. Several forms of calcification occur in this particular lesion and will vary in amount from case to case. The calcification may be in the form of single or multiple interconnecting trabeculae of bone or osteoid (either mature lamellar bone or immature cellular bone), although less commonly globules of calcified material closely resembling acellular cementum, or a diffuse granular dystrophic calcification may be found (Fig. 2-39B, C, D, and E). Significantly, the degree of cellularity of the lesions is usually greatest in the areas of the bone cementum of calcification.
On occasion, areas will be found containing multinucleated giant cells which, with the surrounding tissue, bear considerable resemblance to some areas of the peripheral giant cell granuloma.
Treatment and Prognosis
The lesions should be surgically excised and submitted for microscopic examination for confirmation of diagnosis. The extraction of adjacent teeth is seldom necessary or justified. However, the lesions do recur with some frequency and, in fact, repeated recurrences are not uncommon. In the series of Cundiff, 16% of the cases recurred, while in a series of 50 cases reported by Eversole and Rovin, the recurrence rate was 20%.
Central Ossifying Fibroma of Bone (Central fibro-osteoma)
The ossifying fibroma of bone is a central neoplasm of bone which has caused considerable controversy because of confusion of terminology and criteria of diagnosis. It now appears that this represents a definite entity which should be separated from fibrous dysplasia of bone and the other fibro-osseous lesions which do not represent true neoplasms. This concept has been discussed by Pindborg, by Waldron and by many others.
There is a remarkable similarity in clinical features between this lesion and the central cementifying fibroma, a tumor accepted by most investigators as being odontogenic in origin. There is also considerable similarity and even overlap in the histologic features of these two lesions. For these reasons, it has been suggested that:
• These are two separate benign tumors, identical in nature except for the cell undergoing proliferation, the osteoblast with bone formation in one case, or the cementoblast with cementum formation in the other case; or
• These represent simply two facets of the same basic tumor. Further investigation will be necessary to clarify the relationship, or lack of it, between the central ossifying fibroma and the central cementifying fibroma.
Clinical Features
The central ossifying fibroma may occur at any age, but is far more common in young adults. The age range of occurrence in a series of 31 cases presented by Shafer was nine to 52, with a mean of 33 years of age. Langdon and his associates reported similar findings. Either jaw may be involved, but there appears to be a predilection for the mandible. In the series of Shafer, there were 26 cases in the mandible but only five in the maxilla. A marked predilection for occurrence in females was also noted: 26 cases compared to only five in males. In addition, there was an unusually high incidence of lesions in blacks; 13 cases contrasted to 18 cases in white patients.
The lesion is generally asymptomatic until the growth produces a noticeable swelling and mild deformity; displacement of teeth may be an early clinical feature. It is a relatively slow growing tumor and may be present for some years before discovery. Because of the slow growth, the cortical plates of bone and overlying mucosa or skin are almost invariably intact.
Radiographic Features
The neoplasm presents an extremely variable radiographic appearance depending upon its stage of development. Yet despite the stage of development, the lesion is always well circumscribed and demarcated from the surrounding bone, in contrast to fibrous dysplasia.
In its early stages, the central ossifying fibroma paradoxically appears as a radiolucent area with no evidence of internal radiopacities (Fig. 2-40 A). As the tumor bone apparently matures, there is increasing calcification, so that the radiolucent areas becomes flecked with opacities until, ultimately, the lesion appears as a relatively uniform radiopaque mass. Displacement of adjacent teeth is common, as well as impingement upon other adjacent structures.
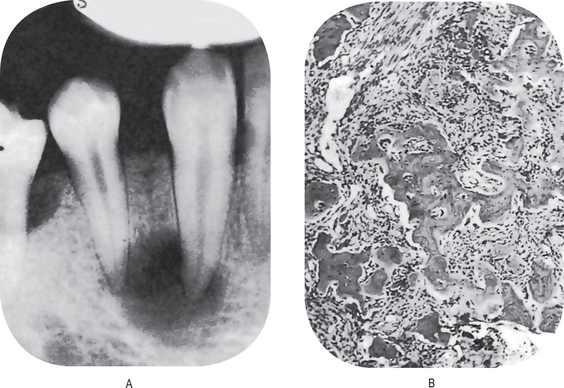
Histologic Features
The lesion is composed basically of many delicate interlacing collagen fibers, seldom arranged in discrete bundles, interspersed by large numbers of active, proliferating fibroblasts. Although mitotic figures may be present in small numbers, there is seldom any remarkable cellular pleomorphism. This connective tissue characteristically presents many small foci of irregular bony trabeculae (Fig. 2-40B) which may bear some similarity to the bizarre Chinese-character shape of the bony trabeculae in fibrous dysplasia of bone (q.v.).
As the lesion matures, the islands of ossification increase in number, enlarge and ultimately coalesce. This, with the probable increase in degree of calcification, accounts for increasing radiopaqueness of the lesion on the radiograph.
Peripheral Giant Cell Granuloma (Peripheral giant cell epulis, peripheral giant cell reparative granuloma)
Peripheral giant cell granuloma has an unknown etiology, with some dispute as to whether this lesion represents a reactive or neoplastic process. However, most authorities believe peripheral giant cell granuloma is a reactive lesion. Since the lesion does not appear to be truly a ‘reparative’ one, this term reparative granuloma has been deleted from the name in the past few years.
The use of the term ‘epulis’ in connection with this or any other oral tumor is to be deplored. By definition, the word means only a growth on the gingiva and is entirely nonspecific. Since it gives no hint as to the true nature of a lesion, the term should be discarded.
The etiology of peripheral giant cell granuloma is unknown, but local irritation due to dental plaque or calculus, periodontal disease, poor dental restorations, ill-fitting dental appliances, or dental extractions has been suggested to contribute to the development of the lesion.
Clinical Features
Although the lesion may be found in very young children as well as in dentulous or edentulous elderly persons, most patients were in fourth to sixth decades of life and the mean age of patients at the time of diagnosis is typically 38–42 years. It was found that females were affected almost twice as frequently as males — 65% compared to 35%. A similar gender predilection has been found for the central giant cell granuloma. Lesions are generally asymptomatic and have a relatively rapid growth rate, often attaining a size of 1 cm within a few months.
Peripheral giant cell granuloma may vary considerably in clinical appearance. It always occurs on the gingiva or alveolar process, most frequently anterior to the molars, and presents itself as a pedunculated or sessile lesion that seems to be arising from deeper in the tissue than many other superficial lesions of this area such as the fibroma or pyogenic granuloma, either of which it may resemble clinically. Thus it seems to originate from either periodontal ligament or mucoperiosteum. The lesion also varies widely in size, but usually is between 0.5 and 1.5 cm in diameter. It is most often dark red, vascular or hemorrhagic in appearance, and it commonly exhibits surface ulceration.
In the edentulous patient, the lesion may appear sometimes as a vascular, ovoid or fusiform swelling of the crest of the ridge, seldom over 1–2 cm in diameter (Fig. 2-41 A). Or there may be a granular mass of tissue which seems to be growing from the tissue covering the slope of the ridge. The color of these lesions varies, but is usually similar to that of lesions in dentulous patients. Ulceration is less common in the edentulous situation. A slight predilection for the mandible is observed in most reported series.
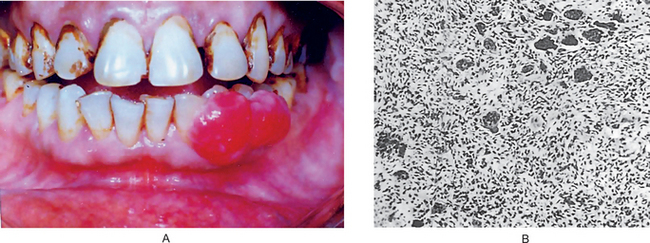
Histologic Features
The microscopic appearance of the giant cell lesion is unique. It consists of a nonencapsulated mass of tissue composed of a delicate reticular and fibrillar connective tissue stroma containing large numbers of ovoid or spindle-shaped young connective tissue cells and multinucleated giant cells. The giant cells in some instances resemble osteoclasts and in other cases are considerably larger than the typical osteoclast. Capillaries are numerous, particularly around the periphery of the lesion, and the giant cells sometimes may be found within the lumina of these vessels. Foci of hemorrhage, with liberation of hemosiderin pigment and its subsequent ingestion by mononuclear phagocytes, as well as inflammatory cell infiltration, are also characteristic features. Spicules of newly formed osteoid or bone are often found scattered throughout the vascular and cellular fibrous lesion (Fig. 2-42 B).
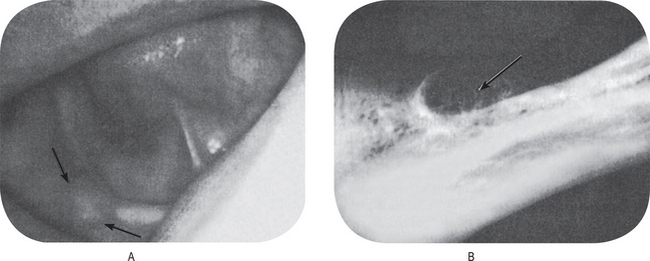
Figure 2-42 Peripheral giant cell granuloma. The lesion in edentulous patients produces a circumscribed growth, causing expansion of the alveolar ridge, (A). The radiograph shows ragged loss of bone with peripheral ‘cufing’, (B).
A histochemical study of this lesion has been reported by Shklar and Cataldo with distinct differences observed in different multinucleated giant cells with respect to the distribution of tyrosine and sulfhydryl groups.
The origin of the giant cells has never been established. Although their resemblance to osteoclasts is sometimes striking, seldom are they seen carrying out the ascribed normal resorptive function of such cells. Geschickter and Copeland suggested that the giant cells might be derived from proliferating giant cells associated with the resorption of deciduous tooth roots. Thus they suppose the lesion to be concerned with the transition from the deciduous to the permanent dentition. Unfortunately, such association of lesions is present in only a few cases even though the tumor is a rather common one in youngsters. Such a theory would account for the predominance of the lesions anterior to the permanent molars.
There has been considerable support for another theory of origin from endothelial cells of capillaries. There is some basis in fact for such an idea, the chief being the common occurrence of the giant cells within vascular channels, suggesting that they arise here through fusion of endothelial cell. This suggested to earlier workers that the tumor was a malignant metastasizing one, and they applied to it the term ‘giant cell sarcoma’ or, in some cases, ‘myeloid sarcoma’.
A recent study of the giant cells in the peripheral giant cell granuloma by electron microscope has been reported by Sapp. He found that these giant cells ultrastructurally contained a sufficient number of features in common with osteoclasts to conclude that they represent a slightly modified form of that cell. In addition, he reported that the stromal cells were structurally compatible with the various stages of differentiating osteoprogenitor cells.
Laboratory studies are generally not necessary. Serum calcium level or a parathyroid hormone assay may be indicated to rule out the rare possibility of brown tumor for lesions that are particularly large and recurrent, may be associated with systemic signs of hyperparathyroidism.
Radiographic Features
The intraoral radiograph may or may not exhibit evidence of involvement of the bone underlying the lesion. In edentulous areas the peripheral giant cell granuloma characteristically exhibits superficial erosion of the bone with pathognomonic peripheral ‘cuffing’ of the bone as seen in the radiograph (Fig. 2-41 B). When the tumor occurs in areas in which teeth are present, the radiograph may reveal superficial destruction of the alveolar margin or crest of the interdental bone, but this is by no means invariably present.
Central Giant Cell Granuloma and Giant Cell Tumor of Bone
Central giant cell granuloma (CGCG) is an uncommon, benign, and proliferative lesion whose etiology is not defined. It was Jaffe who first introduced the term central giant cell reparative granuloma to distinguish this lesion from the giant cell tumor of long bones. However, since a reparative response was quite rare and most of these lesions were found to be destructive rather than reparative, the word ‘reparative’ was omitted from that term.
Clinical Features
Though central giant cell granuloma may be seen in all age group, it is much more common in the young, especially those under 30 years of age. It is somewhat more common in females than in males. The gender distribution of 38 cases reported by Waldron and Shafer was approximately 2 to 1, females over males. A study of 34 cases by Austin, over 60% of the cases occurred before the age of 30 years. Furthermore, 60% of their cases were under the age of 20 years. Either jaw may be involved, but the mandible is affected more often. Two-thirds of their cases occurred in the mandible, and only one-third in the maxilla. The lesions are more common in the anterior segments of the jaws and, not uncommonly, cross the midline (Fig. 2-43).
Figure 2-43 Central giant cell granuloma. Distribution of sites of lesions. (Source: Waldron CA, Shafer WG: The central giant cell reparative granuloma of the jaws. Am J Clin Pathol, 45: 437, 1966).
The lesion may present no signs or symptoms and may be discovered accidentally, but sometimes, central giant cell granuloma may lead to an expansion of the cortex and perforation, mobility, displacement, and root resorption of associated tooth. The borders of the lesions may be regular or diffuse.
Depending on clinical and radiographic features, central giant cell granuloma can be classified into two types. The first type of lesion is nonaggressive, slow growing, does not show root resorption or cortical perforation, and often shows new bone formation. The second type is an aggressive type which grows quickly, shows pain, cortical perforation, and root resorption. On the other hand, although the clinical, radiographic and histologic features of CGCG have been extensively evaluated, the dimensional features of these lesions have not been clearly defined.
Radiographic Features
Central giant cell granuloma is essentially a destructive lesion, producing a radiolucent area with either a relatively smooth or a ragged border, and some-times showing faint trabeculae. Definite loculations are often present, particularly in larger lesions. The cortical plates of the bone are often thin and expanded and may become perforated by the mass. Displacement of the teeth by the lesion is seen with some frequency. The appearance of the giant cell granu-loma is not pathognomonic and may be confused with that of many other lesions of the jaw, both neoplastic and non-neoplastic (Fig. 2-44 A–F).
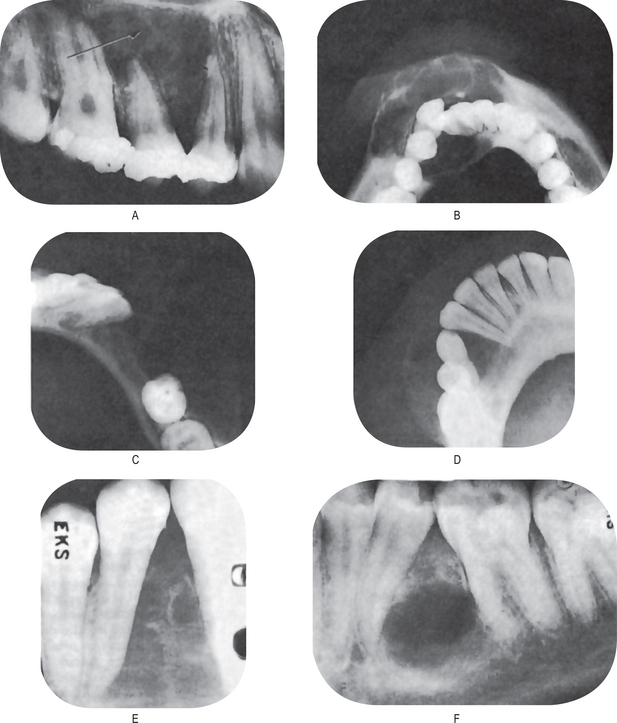
Histologic Features
Central giant cell granuloma is made up of a loose fibrillar connective tissue stroma with many interspersed proliferating fibroblasts and small capillaries. The collagen fibers are not usually collected into bundles; however, groups of fibers will often present a whorled appearance. Multinucleated giant cells are prominent throughout the connective tissue, but not necessarily abundant (Fig. 2-45). These giant cells vary in size from case to case and may contain only a few or several dozen nuclei. In addition, there are usually numerous foci of old extravasated blood and associated hemosiderin pigment, some of it phagocytized by macrophages. Foci of new trabeculae of osteoid or bone also are often seen, particularly around the periphery of the lesion.
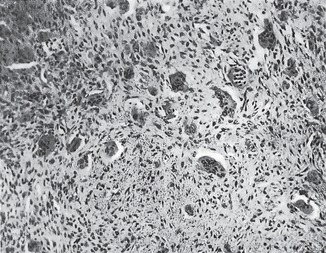
There is a debate whether the giant cells are fibroblast origin or from monocyte/macrophages. Recent study by Itonaga et al, indicate that the giant cells in CGCG of the jaw are osteoclast like and formed from monocyte/macrophage precursors which differentiate into osteoclasts.
Treatment and Prognosis
The treatment of the giant cell granuloma is curettage or surgical excision. The lesions so treated almost invariably fill in with new bone and heal with no difficulty. Occasional lesions recur, but this is seldom sufficient cause for more radical procedures. X-ray radiation is contraindicated.
Giant Cell Tumor of Bone (Osteoclastoma)
Giant cell tumor of bone is a distinctive neoplasm of undifferentiated cells. Multinucleated giant cells apparently result from fusion of the proliferating mononuclear cells, and although they are a constant and prominent part of this tumor, the giant cells are probably of less significance than the mononuclear cells. The exact cell of origin of this neoplasm is still unknown. Several immunohistochemical studies have suggested that the mononuclear cells are of histiocytic origin and that the giant cells arise from their fusion.
Malignant giant cell tumor cannot be diagnosed with assurance unless evidence of ordinary benign giant cell tumor exists within the lesion or has been demonstrated previously at the same site. If the stromal cells of a tumor that has many benign giant cells are malignant throughout with features of osteosarcoma, malignant fibrous histiocytoma, or fibrosarcoma, the tumor probably has no relationship to giant cell tumor. The benign giant cells are but an incidental and confusing component. The clinical correlative studies reported by Troup and coworkers in 1960 have fortified this concept.
Clinical Features
The 568 cases of giant cell tumor reported from Mayo Clinic files represented 5.12% and 22.7% of the benign tumors. In many series, female patients are predominant. Approximately 84% of the neoplasms occurred in patients older than 19 years, with a peak incidence in the third decade of life (Unni KK, 1996). Pain of variable severity is almost always predominant symptom. More than three-fourths of the patients had noted swelling of the affected region. Less common symptoms were weakness, limitation of motion of the joint and pathologic fracture.
Histologic Features
The basic proliferating cell has round-to-oval or even spindle-shaped nucleus in the field that is diagnostic of true giant cell tumor. The nucleus is surrounded by an ill-defined cytoplasmic zone, and discernible intercellular substance is absent. Mitotic figures can be found, sometimes numerous. Mitotic activity has no prognostic significance. The giant cells are usually scattered uniformly throughout the lesion. They usually contain 40–60 nuclei. Areas of infarctlike necrosis are common in giant cell tumors. Some tumors are almost completely necrotic. The necrosis is not associated with an inflammatory response. Small collections of foam cells are common. Grading of giant cell tumors has no prognostic significance (Fig. 2-46).
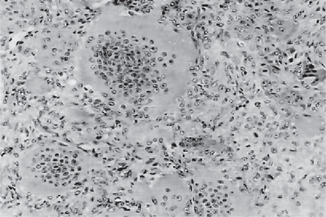
Aneurysmal Bone Cyst
The aneurysmal bone cyst is an interesting solitary lesion of bone which was separated as a distinct entity in 1942 by Jaffe and Lichtenstein. Since their original account, many cases have been reported in the literature, although the first cases occurring in the jaws were not described until 1958.
Features that make it logical to exclude aneurysmal bone cyst from the neoplastic category include the observation that the lesion has been known to regress after incomplete removal. The cause of this strange process in bone is unknown but several examples apparently arose after a fracture. It is similar to and probably related to other reactive non-neoplastic processes, including giant cell reparative granuloma of the jaws, traumatic reactions in periosteum and bone and even florid heterotopic ossification. Aneurysmal bone cyst may arise de novo in bone; that is, a definite preexisting lesion cannot be demonstrated in the tissue. Rarely malignant tumors of bone contain such benign areas as well. Obviously, recognition of an underlying process is important.
Clinical Features
The aneurysmal bone cyst is generally a lesion of young persons, predominantly occurring under the age of 20 years, with no significant predilection for either gender. The lesion can be encountered in adults. A history of traumatic injury preceding development of the lesion may often be obtained.
Cases of aneurysmal bone cyst have been observed in nearly every part of the skeleton, although over 50% of all cases occur in the long bones and vertebral column. Lesions are also seen frequently in the clavicle, rib, innominate bone, skull and bones of the hands and feet as well as other sites.
The lesions are usually tender or painful, particularly upon motion, and this soreness may limit movement of the affected bone. Swelling over the area of bone involvement is also common.
Gross findings at the time of operation are characteristic. Upon entering the lesion, excessive bleeding is encountered, the blood ‘welling up’ from the tissue. The tissue has been described as resembling a blood-soaked sponge with large pores representing the cavernous spaces of the lesion. In manometric studies by Biesecker and his associates, some of the cysts had elevated vascular pressures as high as arteriolar levels.
Four phases of pathogenesis are recognized, as follows:
• Active growth phase, which is characterized by rapid destruction of bone and a subperiosteal blow out pattern.
• Mature stage, also known as stage of stabilization, which is manifested by the formation of a distinct peripheral bony shell and internal bony septa and trabeculae that produce the classic soap-bubble appearance.
• Healing phase, with progressive calcification and ossification of the cyst and its eventual transformation into a dense bony mass with an irregular structure.
Oral Manifestations
The aneurysmal bone cyst occurs with some frequency in the jaws, although many probably are misdiagnosed as some other bone lesion (Fig. 2-47 A). In a review of the literature, Neumann-Jensen and Praetorius found 26 reported cases in the jaws, 17 in the mandible and nine in the maxilla. Of these, the average age of occurrence was 18 years with a range of 6–69 years, although only two patients were over 22 years of age. There was a slight predilection for occurrence in females, 14–19, where the gender was known.
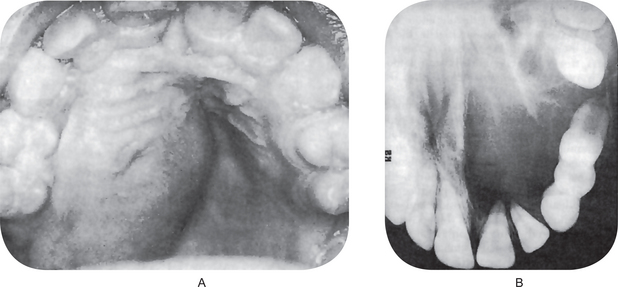
Radiographic Features
The radiographic picture of the lesion is often distinctive. The bone is expanded, appears cystic with a honeycomb or soap-bubble appearance in many cases, and is eccentrically ballooned (Fig. 2-47 B). The cortical bone may be destroyed, and a periosteal reaction may be evident.
Histologic Features
The aneurysmal bone cyst consists of a fibrous connective tissue stroma containing many cavernous or sinusoidal blood-filled spaces. These spaces may or may not show thrombosis. Young fibroblasts are numerous in the connective tissue stroma, as well as multinucleated giant cells with a patchy distribution similar to that in the giant cell granuloma. But in the latter lesion the cavernous spaces are not found. Varying amounts of hemosiderin are present and, invariably, new osteoid and bone formation (Fig. 2-48 A–D).
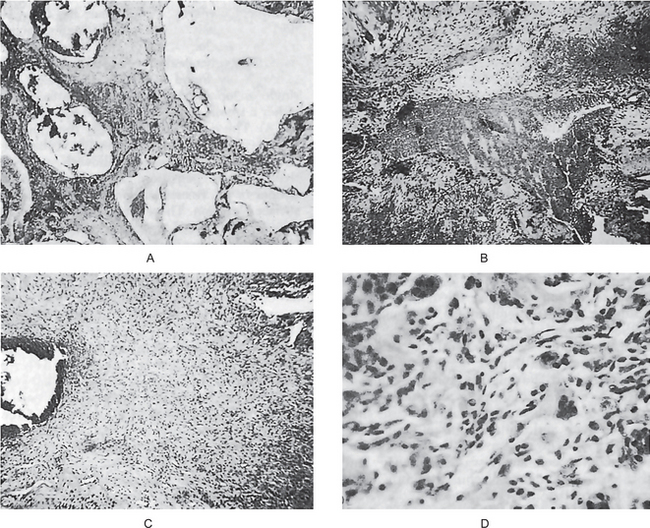
Pathogenesis
The nature of this lesion is still controversial despite the many cases that have been studied and reported. Lichtenstein has proposed that the aneurysmal bone cyst arises as a result of a persistent local alteration in hemodynamics, leading to increased venous pressure and subsequent development of a dilated and engorged vascular bed in the transformed bone area. Resorption of bone then occurs, to which the giant cells are related, and this is replaced by connective tissue, osteoid and new bone.
An alternative explanation of the lesion is that it represents an exuberant attempt at repair of a hematoma of bone, similar to the central giant cell granuloma. But in the case of the aneurysmal bone cyst, it is postulated that the hematoma maintains a circulatory connection with the damaged vessel. This would produce a slow flow of blood through the lesion and account for the clinical ‘welling’ of blood when the lesion is entered. Thus the only real difference between this aneurysmal cyst and the giant cell granuloma is that in the latter lesion damaged blood vessels fail to retain a circulatory connection with the lesion.
It is most significant that Biesecker and his associates have reported that 32% of their series of aneurysmal bone cysts had an accompanying benign primary lesion of bone. On this basis, they have proposed a new hypothesis for the etiology and pathogenesis of this lesion—namely, that a primary lesion of bone initiates an osseous, arteriovenous fistula and thereby creates, via its hemodynamic forces, the secondary reactive lesion of bone, the aneurysmal bone cyst.
This occurrence of aneurysmal bone cyst secondary to or in association with other osseous lesions has been confirmed by other investigators. For example, Levy and his associates reported 57 cases of aneurysmal bone cyst with which the most commonly associated lesion was the solitary or unicameral bone cyst (16 cases), the osteoclastoma or benign giant cell tumor (14 cases) and osteosarcoma (12 cases). Other associated lesions included the nonosteogenic fibroma, benign osteoblastoma, hemangioendothelioma and hemangioma of bone. Five aneurysmal bone cysts were secondary to fracture or other bone trauma.
Hillerup and Hjrting-Hansen have summarized the different theories on the etiology and pathogenesis of the aneurysmal bone cyst, simple bone cyst and central giant cell granuloma of the jaws. They suggested that these three lesions have a common dysvascular etiology and that local environmental factors within the bone may differentiate the pathogenesis.
Treatment and Prognosis
Surgical curettement or excision is the treatment of choice, although low doses of radiation have also been used. The possibility of radiation sarcoma is always a potential hazard, however, and the propriety of treatment of benign lesions by radiation has been seriously questioned on this basis. Recurrence in bones other than the jaws has been reported variously in between 21 and 59% of cases. However, no lesion in the jaws is known to have recurred.
Lipoma
It is a relatively rare intraoral tumor, although it occurs with considerable frequency in other areas, particularly in the subcutaneous tissues of the neck.
This is a benign, slow-growing neoplasm composed of mature fat cells. It appears that the cells of the lipoma differ metabolically from normal fat cells even though they are histologically similar. Thus a person on a starvation diet will lose fat from normal fat depots in the body, but not from the lipoma. Furthermore, fatty acid precursors are incorporated at a more rapid rate into lipoma fat than into normal fat while lipoprotein lipase activity is reduced.
The lipoma is a very common benign tumor of adipose tissue, but its presence in the oral and oropharyngeal region is relatively uncommon, with a prevalence rate of only 1/5,000 adults. The first description of an oral lesion was provided in 1848 by Roux in a review of alveolar masses; he referred to it as a ‘yellow epulis’. While most lesions are developmental anomalies, those which occur in the maxillofacial region usually arise late in life and are presumed to be neoplasms of adipocytes, occasionally associated with trauma. Few lipomas show rearrangement of 12q, 13q, 6p chromosomes.
Clinical Features
It is usually found in adults and there is no gender predilection. In most cases the size of the lesion is less than 3 cm at the time of diagnosis, but can increase up to 5–6 cm over a period of years. The tumor has been reported to occur in a variety of locations, including the tongue, floor of the mouth, buccal mucosa, gingiva, and mucobuccal or labial folds. Morphologically intraoral lipomas can be classified as diffuse form affecting the deeper tissues, and a superficial and encapsulated form.
Superficial form appears as a single or lobulated, painless lesion attached by either a sessile or a pedunculated base (Fig. 2-49 A). There is a yellow surface discoloration and well-encapsulated, lipoma are freely movable beneath the mucosa. The epithelium is usually thin, and the superficial blood vessels are readily visible over the surface. The lipoma is yellowish, and relatively soft to palpation. Lesions in deeper tissues produce only a slight surface elevation which tend to be more diffuse than the superficial type of lipoma. When palpated, the diffuse form feels like fluid, sometimes leading to a mistaken tentative diagnosis of ‘cyst’. Since this diffuse form often occurs in areas in which some fat is normally present, the diagnosis of lipoma depends upon the recognition of simply an overabundance of this tissue. Thus the diagnosis is essentially a clinical one.
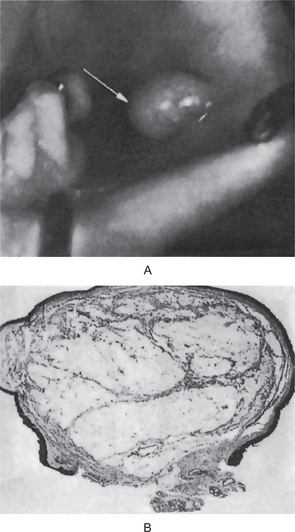
Figure 2-49 Lipoma.
The circumscribed mass on the buccal mucosa (A) is composed chiely of adult fat cells with occasional connective tissue septa coursing through the lesion (B).
Multiple head and neck lipomas have been observed in neurofibromatosis, Gardner syndrome, encephalocraniocutaneous lipomatosis, multiple familial lipomatosis, and Proteus syndrome. Generalized lipomatosis has been reported to contribute to unilateral facial enlargement in hemifacial hypertrophy.
Histologic Features
The lipoma is composed predomi-nantly of mature adipocytes, possibly admixed with collagenic streaks, and is often well demarcated from the surrounding connective tissues. A thin fibrous capsule may be seen and a distinct lobular pattern may be present. Quite often, however, lesional fat cells are seen to ‘infiltrate’ into surrounding tis-sues, perhaps producing long, thin extensions of fatty tissue radiating from the central tumor mass. When located within striated muscle this infiltrating variant is called intramuscular lipoma (infiltrating lipoma), but extensive involvement of a wide area of fibrovascular or stromal tissues might best be termed lipomatosis.
Occasional lesions exhibit excessive fibrosis between the fat cells (fibrolipoma), excess numbers of small vascular channels (angiolipoma), a myxoid background stroma (myxoid lipoma, myxolipoma), or areas with uniform spindle-shaped cells interspersed between normal adipocytes (spindle cell lipoma). When spindle cells appear somewhat dysplastic or mixed with pleomorphic giant cells with or without hyperchromatic, enlarged nuclei, the term pleomorphic lipoma is applied. When the spindled cells are of smooth muscle origin, the term myolipoma may be used, or angiomyolipoma when the smooth muscle appears to be derived from the walls of arterioles.
Rarely, chondroid or osseous metaplasia may be seen in a lipoma (osteolipoma, ossifying lipoma, chondroid lipoma, ossifying chondromyxoid lipoma). When bone marrow is present, the term myelolipoma is used. Also on rare occasions, isolated ductal or tubular adnexal structures are scattered throughout fat lobules, in which case the term adenolipoma is applied. Perineural lipoma has also been reported.
On occasion, lipoma of the buccal mucosa cannot be distinguished from a herniated buccal fat pad, except by the lack of a history of sudden onset after trauma. Otherwise, lipoma of the oral and pharyngeal region is not difficult to differentiate from other lesions, although spindle cell and pleomorphic types must be distinguished from liposarcoma. When metaplastic calcified tissue is present, the lesion may be confused with soft tissue chondroma or soft tissue osteoma.
The benign neoplasm of brown fat, hibernoma, has been reported in the oral/pharyngeal region only rarely.
Lipoblastoma
This is a rare related lesion originally described by Vellios and his associates in 1958. It is probably not a true neoplasm but rather a continuation of the normal process of fetal fat development carried into postnatal life. It is characterized clinically by the occurrence in infants of solitary or multiple soft-tissue masses developing at various sites such as the buttock, chest, axilla or neck. The lesions are benign but may recur if inadequately excised. Diffuse lipoblastoma involving deeper soft tissue is called lipoblastomatosis.
Histologically, the central core of mature adipocytes and a peripheral envelope of cells containing variably sized fat vacuoles are noticed. Affected cells are smaller than normal, with 1–4 vacuoles, with a light, wispy cytoplasm between vacuoles. Some cells have nuclei centrally located, as seen in the moderately-sized cells of hibernoma, while others show the nucleus to be pushed toward the cytoplasmic membrane (signet-ring cell). Mitotic activity is extremely rare and fibrous septa separate fat lobules in this tumor. An abnormality of the long arm of chromosome 8q11-13 is a rather consistent finding in the lesional cells.
Verruciform Xanthoma (Verrucous xanthoma, inflammatory papillary hyperplasia with foam cell response)
Verruciform xanthoma is an uncommon lesion that usually occurs on the oral mucosa of unknown etiology and uncertain nature, first described as an entity by Shafer in 1971. Numerous cases have now been reported in the literature, including on the vulva, penis, on the skin of the thigh and perineum. Many authors consider it to be a reactive process rather than a true neoplasm.
The etiopathogenesis of verruciform xanthoma is unknown. It is thought that damage to the squamous cells with increased epithelial cell turnover, leading to the deposition of epithelial cell debris that is engulfed by macrophages in the corium may lead to the development of this lesion. An immunologic etiology has also been proposed.
Most commonly, the lesion has a verruciform appearance, but it may appear polypoid, papillomatous, or sessile. Rare lesions have been found to occur in cysts. The chief distinguishing feature of verruciform xanthoma is the presence of large numbers of lipid-laden foamy histiocytes in, and essentially limited to, the connective tissue papillae in the lesion.
Clinical Features
Verruciform xanthoma is uncommon, accounting for 0.025–0.05% of all pathology cases. The age of occurrence varies widely from 2 to 89 years. But most oral cases occur in middle-aged persons with mean age of 40–50 years, and no gender predilection is apparent. Most cases are reported in whites, but blacks are also affected. Verruciform xanthoma may occur at any site but its most preferred site is the alveolar ridge, or gingiva, followed by the buccal mucosa and then the palate, floor of the mouth, lip and lower mucobuccal fold with equal frequency.
Verruciform xanthoma occurs as a solitary lesion, either normal or reddish in color but sometimes pale or ‘hyperkeratotic’, with a rough, pebbly surface and either a sessile or pedunculated base. It may be as small as 2 mm or as large as 1.5 cm in diameter, and is invariably asymptomatic. In the oral cavity lesions can be found in association with lichen planus, pemphigus vulgaris, oral bullae, carcinoma in situ, or frank squamous cell carcinoma.
Depending on the nature of the individual lesion, it may clinically resemble any verrucous, papillary, or lichenoid oral lesion, particularly any such lesion that is also hyperkeratotic. It is frequently misdiagnosed at clinical examination as a papilloma.
Verruciform xanthoma outside oral mucosa is usually associated with other conditions like lymphoedema, epidermal nevi and congenital hemidysplasia with ichthyosiform erythroderma and limb defect (CHILD) syndrome.
Histologic Features
Verruciform xanthomas may appear verrucous, papillary, or cauliflower like, or they may show a lichenoid pattern histologically. A mixture of the above patterns may also be observed. A variable degree of parakeratosis is observed that is usually more marked in verrucous and papillary lesions, with hyperparakeratosis present in the crypts between papillae. The rete pegs are extremely elongated and uniform, although no increased mitoses or pseudoepitheliomatous hyperplasia are found. The connective tissue papillae are of variable length and thickness; they often extend close to the surface. Rarely, the entire process may occur in a cyst like structure.
The most striking and characteristic feature is the presence of large foam cells in the connective tissue papillae between the epithelial pegs. These are confined to the papillae and do not extend into the dermis beneath the pegs. Almost all of the papillae are involved with these cells, which occasionally may also be seen in the epithelium (i.e. epidermis, mucosa).
A slight inflammatory infiltrate may be present beneath the lesion. Occasional lesions have been associated with a massive or lichenoid infiltrate. No granulomatous change has been described other than the presence of the foamy macrophages. Microorganisms are not characteristically present. Although bacteria and fungal hyphae are occasionally described, they appear to play no role in the etiopathogenesis. Attempts to incriminate a viral role have been counterproductive.
Ultrastructurally, most studies have concluded that the foam cells in verruciform xanthoma are fat-laden macrophages, although other cell types, including Langerhans cells and even fibroblasts have been proposed.
Oral Hemangiomas and Vascular Malformations (Oral vasoformative tumor)
Hemangiomas are lesions that are not present at birth. They manifest within the first month of life, exhibit a rapid proliferative phase, and slowly involute to nonexistent. Hemangiomas are tumors identified by rapid endothelial cell proliferation in early infancy, followed by involution over time; vascular malformations result from anomalous development of vascular plexuses. The malformations have a normal endothelial cell growth cycle that affects the veins, the capillaries, or the lymphatics, and they do not involute. Vascular malformations are more stable and fail to regress.
Hemangiomas of the oral cavity are not common pathologic entities, but the head and neck are common sites. Most true hemangiomas involute with time, but 10–20% of true hemangiomas incompletely involute and require postadolescent ablative treatment.
Hemangiomas are associated with the following syndromes:
• Rendu-Osler-Weber syndrome (autosomal dominant in-heritance, multiple telangiectasias, occasional GI tract in-volvement, occasional CNS involvement).
• Sturge-Weber-Dimitri syndrome (noninherited and non-familial, port-wine stain, leptomeningeal angiomas).
• Kasabach-Merritt syndrome (thrombocytopenic purpura associated with hemangioma, consumptive coagulopathy, microangiopathic hemolysis, intralesional fibrinolysis).
• Maffucci syndrome (hemangiomas of the mucous mem-branes, dyschondroplasia).
• von Hippel-Lindau syndrome (genetic transmission vari-able, hemangiomas of the cerebellum or the retina, cysts of the viscera).
• Klippel-Trenaunay-Weber syndrome (port-wine stain, an-giomatosis of the extremities).
In 1982, Mulliken and Glowacki classified the vasoformative tumors into two broad groups: hemangiomas and vascular malformations (Table 2-13). Vascular malformations can be further subdivided into venous, capillary, arteriovenous and lymphatic malformations.
Table 2-13
Classiication of vasoformative tumors
| Vasoformative tumor | New nomenclature | Old nomenclature |
| Hemangiomas | Capillary hemangioma Cavernous hemangioma Mixed hemangioma | Strawberry hemangioma Juvenile hemangioma Parotid hemangioma |
| Vascular malformations | Venous malformation | Cavernous hemangioma Hemangiomatosis |
| Intramuscular venous malformation | Intramuscular hemangioma | |
| Capillary malformation | Capillary hemangioma Port-wine stain Arteriovenous hemangioma Arterial angioma | |
| Arteriovenous malformation | Arteriovenous aneurysm Cirsoid angioma Red angioma Serpentine aneurysm Capillary lymphangioma Cavernous lymphangioma | |
| Lymphatic malformation | Lymphangioma Cystic hygroma |
Source: Randall Wilk. eMedicine Speciaities. Diseases of the Oral Mucosa, 2003.
A simple classification is that proposed by Watson and McCarthy based upon a series of 1,308 blood vessel tumors, and is as follows: (1) capillary hemangioma, (2) cavernous hemangioma, (3) angioblastic or hypertrophic hemangioma, (4) racemose hemangioma, (5) diffuse systemic hemangioma, (6) metastasizing hemangioma, (7) nevus vinosus, or port-wine stain, and (8) hereditary hemorrhagic telangiectasia.
Clinical Features
By their definition, hemangiomas occur in infants and children. The incidence of hemangiomas increases to 23% in premature infants with a birthweight of less than 1,000 gm. The peak incidence of central hemangiomas of the jaws is in the second decade of life. But vascular malformations have a much broader range of incidence.
Hemangioma affects as many as 12% infants in whites, but it rarely occurs in darker-skinned individuals. Vascular malformations are also more common in whites. Hemangiomas are about three times more common in females than in males. But for venous malformations gender ratio is reported to be 1:1.
The most commonly affected facial bones are the mandible, the maxilla, and the nasal bones. Intraosseous lesions affect the mandible more often than the maxilla, with a ratio of 2:1. Involvement of the zygoma is rare. Intramuscular hemangiomas in the oral region are most commonly seen in the masseter, comprising 5% of all intramuscular hemangiomas.
Patients with multiple lesions have rates of resolution similar to those with single lesions; however, separate lesions in the same individual do not necessarily grow or involute simultaneously.
Vascular malformations are present at birth and continue to grow with the child. The growth may become accelerated when the patient undergoes puberty or pregnancy, with the attendant hormonal changes.
Oral Manifestations
The hemangioma of the oral soft tissue is similar to the hemangioma of the skin and appears as a flat or raised lesion of the mucosa, usually deep red or bluish red and seldom well circumscribed. They are readily compressible and fill slowly when released. The most common sites of occurrence are the lips, tongue, buccal mucosa and palate (Fig. 2-50). The tumor often is traumatized and undergoes ulceration and secondary infection.
Figure 2-50 Hemangioma (A) Congenital bilateral hemangioma of the face and lip. The lesion follows the distribution of the beard. (B) The hemangioma of the tongue is supericial but diffuse.
Certain tiny vascular formations of lip vessels called ‘microcherry’, ‘glomerulus’ and ‘venous lake’ have been described by Gius and his associates as lesions encountered with increasing frequency in the later decades of life and occurring with greater frequency in patients with gastric and duodenal ulcers.
The intramuscular hemangioma is one special form of hemangioma which is quite rare in the head and neck region. It arises within normal skeletal muscle, comprises less than 1% of all hemangiomas and is important chiefly because of the problem in differential diagnosis and of treatment of the lesion. Intramuscular hemangiomas represent a challenge on diagnosis because they exhibit few signs on clinical examination. Often times, the extent of the lesion is not clinically apparent on examination, and imaging studies frequently define more extensive lesions than suspected.
Central hemangiomas of the maxilla or mandible occasionally occur and frequently present difficulty in differential diagnosis. Lund and Dahlin reviewed 35 cases of hemangioma of the jaws and found that over 50% of the cases occurred in the first two decades of life and that two-thirds of the lesions were located in the mandible. Here the tumor is a bone-destructive lesion which may be of varying size and appearance, but is often suggestive of a cyst (Fig. 2-51). Root resorption of the teeth has been reported in 30% of cases, but the vitality of the teeth is usually not affected. Some central hemangiomas present a honeycombed appearance on the radiograph, sometimes with radiating spicules at the expanded periphery forming a ‘sunburst’ appearance reminiscent of that effect seen in osteosarcoma. Radiographic appearance is also similar to that of a giant cell lesion or an ameloblastoma.
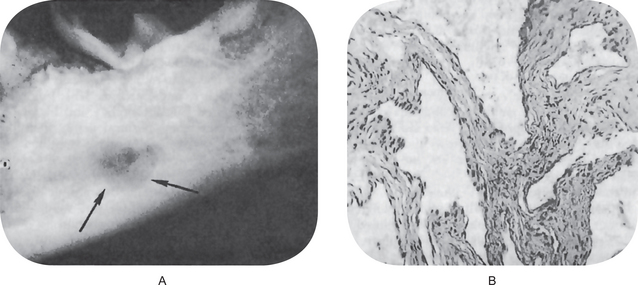
The attempt at radical surgical excision of such central lesions has often resulted in severe loss of blood, which has occasionally led to exsanguination of the patient to the point of death. In such bony lesions, it is always advisable to attempt to aspirate fluid contents through a needle before surgically opening the area. When the hemangioma is entirely central within bone, Gamez-Araujo and his associates have stated that the prognosis is excellent with adequate surgical intervention, in contrast to those lesions where soft tissue is also involved by capillary hemangioma. In these latter instances, the lesions become aggressive, invade locally and often recur if not totally eradicated.
The arteriovenous aneurysm, or arteriovenous fistula, is an uncommon lesion which often has been mistaken clinically for a hemangioma. It represents a direct communication between an artery and a vein through which the blood bypasses the capillary circulation. The arteriovenous aneurysm may be congenital or acquired, the latter usually being traumatic in origin. It may occur either in the soft tissues, such as in the case of the palate reported by Ingram and Coker, and on the alveolar ridge as reported by McComb and Trott, or central in the jaw as reported by Cook and Zbar. These aneurysms typically are classified as:
• A cirsoid aneurysm which is a tortuous mass of small ar-teries and veins linking a larger artery and vein.
• A varicose aneurysm which consists of an endothelium-lined sac connecting an artery and a vein
• An aneurysmal varix representing a direct connection between an artery and a dilated vein
The arteriovenous aneurysm has been reviewed in detail by Gomes and Bernatz, while Orbach has discussed the entire gamut of congenital arteriovenous malformations of the head and neck. He has emphasized the histogenesis and pathogenesis of these abnormalities, stressing the fact that they range from the most trivial angioma to the large-bore arteriovenous fistula.
Radiographic Features
Imaging should be considered to determine their extent and flow characteristics. Angiography is considered the most definitive of the studies, although the angiographic appearance of intraosseous lesions is less well defined than that of soft tissue lesions. Ultrasonography can be used to determine angiomatous in nature (i.e. hemangioma, lymphangioma), but it cannot differentiate a hemangioma from a lymphangioma. Contrast-enhanced MRI can be used to differentiate a hemangioma from a lymphangioma in the oral cavity. MRI appears to be highly reliable for lesions of either soft tissue or bone. Radiographically, intraosseous hemangioma shows honeycombed pattern that is well demarcated from normal bone. CT scans often show an expansile process with a high-density amorphous mass that may be suggestive of fibrous dysplasia.
Histologic Features
The usual hemangioma is com-posed of many small capillaries lined by a single layer of endothelial cells supported by a connective tissue stroma of varying density. It bears considerable resemblance to young granulation tissue and is nearly identical with some cases of pyogenic granuloma. Some cases show rather remarkable endothelial cell proliferation (Fig. 2-52). In fact, one com-mon form is referred to as a juvenile hemangioendothelio-ma because it occurs in very early life and is characterized by an extremely cellular pattern. It is generally believed that this lesion is an immature stage of a capillary hemangioma and that, in time, it will either develop into a simple hem-angioma or regress.
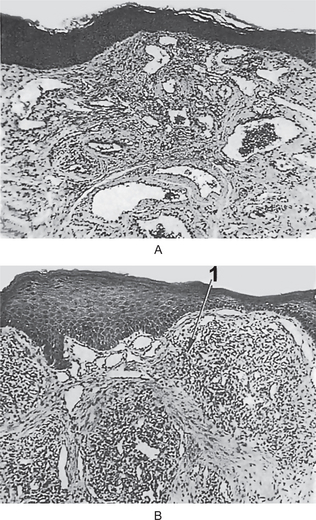
Figure 2-51 Hemangioma. Some lesions (A) are characterized by the presence of many small dilated capil-laries and a minimum of endothelial cells, while others (B) show large numbers of proliferating endothelial cells (1), but only moderate numbers of patent capillaries.
The cavernous form of hemangioma consists of large dilated blood sinuses with thin walls, each showing an endothelial lining. The sinusoidal spaces usually are filled with blood, although an admixture with occasional lymphatic vessels occurs in some instances.
Intravascular angiomatosis, also referred to in the litera-ture most commonly under the terms hemangioendotheliome vegetant intravasculaire of Masson, Masson’s pseudoangiosar-coma, and intravascular papillary endothelial hyperplasia, is actually an unusual form of an organizing thrombus, although it is often mistaken for a vascular tumor, especially a malignant one such as angiosarcoma. This lesion has been discussed by McClatchey and his associates, who described perioral cases, including two on the lips which clinically resembled mucoceles, and emphasized that angiosarcoma could best be excluded as a diagnosis by the intravascular localization, the absent to rare mitoses and necrosis, and the rare solid cellular areas without vascular differentiation.
Treatment and Prognosis
Many congenital hemangiomas have been found to undergo spontaneous regression at a relatively early age. Cases which do not show such remission or those which arise in older persons have been treated in a variety of ways, including: (1) surgery, (2) radiation therapy (external radiation or radium), (3) sclerosing agents, such as sodium morrhuate or psylliate, injected into the lesion, (4) carbon dioxide snow, (5) cryotherap and (6) compression. Each form of treatment has its proponents and antagonists, but it appears that in skilled hands each has its proper place.
The prognosis of the hemangioma is excellent, since it does not become malignant or recur after adequate removal or destruction.
Hereditary Hemorrhagic Telangiectasia (Osler-Weber-Rendu syndrome, Rendu-Osler-Weber syndrome, heredofamilial angiomatosis, familial hemorrhagic angiomatosis, Osler's disease)
Osler-Weber-Rendu syndrome, also known as hereditary hemorrhagic telangiectasia, is an autosomal dominant disorder identified typically by the triad of telangiectasia, recurrent epistaxis, and a positive family history for the disorder. The major cause of morbidity and mortality due to this disorder lies in the presence of multiorgan arteriovenous malformations (AVMs) and the associated hemorrhage that may accompany them.
Clinical Features
The disease most commonly occurs in white patients, but it has been described in patients of Asian, African and Arabic descent. The spider-like telangiectases are occasionally present at or shortly after birth, although in the majority of cases they do not become conspicuous until after puberty. They appear to increase in number and prominence as the patient ages. The skin lesions are most common on the face, neck, and chest, although any area may be involved (Fig. 2-53). Involvement of the oral mucous membrane constitutes an important feature of the disease, the most commonly affected areas being the lips, gingiva, buccal mucosa and palate, as well as the floor of the mouth and the tongue.
One of the earliest signs of the disease, often occurring in childhood and preceding the appearance of telangiectasia, is epistaxis as well as bleeding from the oral cavity, both of which may be difficult to control. The diagnosis may be established if a history of epistaxis dating from childhood, the presence of the telangiectatic areas and the familial history are ascertained. The main areas of involvement are the nasal mucosa, skin, the GI tract, the pulmonary vasculature, and the brain.
Similar telangiectases of skin and oral mucous membranes may occur in a variety of other situations, however, and these include progressive systemic sclerosis (scleroderma) and the CREST syndrome (calcinosis cutis, Raynaud’s phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia), lupus erythematosus, sarcoid and other rarer diseases. The differential diagnoses are important and have been discussed by Donaldson and Myall.
Histologic Features
The disease is due primarily to defects in the small blood vessels of the skin and mucosa. Hashimoto and Pritzker have shown, in an electron microscope investigation, that the actual cause of the hemorrhage is either a primary intrinsic defect of the endothelial cells permitting their detachment, or a defect in the perivascular supportive tissue bed which weakens the vessels, rather than a lack of elastic fibers as was thought at one time. Both clotting and bleeding times are normal, as are the blood elements, although with severe bleeding mild anemia and thrombocytopenia may result.
Treatment and Prognosis
The treatment of the disease is varied, depending upon its severity. The spontaneous hemorrhages may be controlled by pressure packs, particularly the nasal bleeding. The angiomatous or telangiectatic areas are sometimes cauterized, treated with X-ray radiation or surgically excised.
The disease is seldom so severe that life is endangered. Nevertheless a considerable number of deaths from severe hemorrhage have been reported.
Encephalotrigeminal Hemangiomatosis (Sturge-Weber syndrome)
Sturge-Weber syndrome is a rather uncommon congenital condition. It consists of congenital hamartomatous malformations that may affect the eye, the skin, and the central nervous system at different times, characterized by the combination of a venous angioma of the leptomeninges over the cerebral cortex with ipsilateral angiomatous lesions of the face, and sometimes, of the skull, jaws and oral soft tissues. Sturge-Weber syndrome (SWS) belongs to a group of disorders collectively known as the phakomatoses (mother-spot diseases—Fig. 2-54).
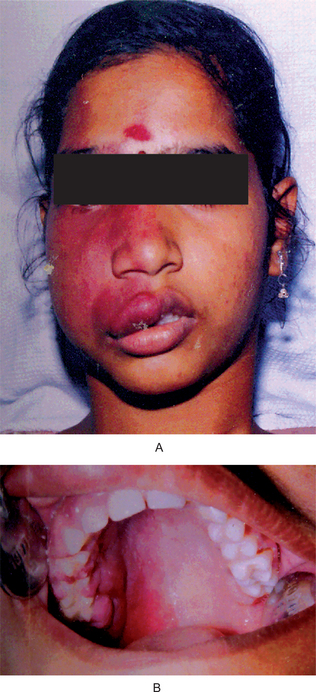
Figure 2-54 Sturge-Weber syndrome. (A) The unilateral distribution of angiomatous malformations which never cross the midline is pathognomonic of the syndrome. (B) Unilateral involvement of palate with angiomatous malformation.
Etiology
The clinical manifestations of SWS have a common embryological basis. The primary defect is a developmental insult affecting precursors of tissues that originate in the promesencephalic and mesencephalic neural crest. Then, these affected precursors give rise to vascular and other tissue malformations in the meninges, the eye, and the dermis.
Although the exact nature of the insult is unknown, it has been postulated that a somatic mutation in these precursors may lead to overproduction of an angiogenic factor. Others have suggested that SWS may be due to a lethal gene surviving by mosaicism. The influence of heredity in SWS has not been documented. To date, no gene defect has been associated with the syndrome. Several types of chromosomal abnormalities have been reported, but most patients with SWS have normal karyotypes. Most patients with SWS have a sporadic, nonfamilial disease.
Clinical Features
The facial cutaneous capillary venous angiomas (or port-wine nevi) are usually the first component of the syndrome to be observed, at birth, and are confined almost exclusively to the skin area supplied by the trigeminal nerve. A second common feature is the presence of typical intracranial convolutional calcifications discernible in cranial radiographs. Ocular involvement occurs in some patients, consisting generally of glaucoma, exophthalmus, angioma of the choroid or other abnormalities.
Neurologic manifestations are among the most characteristic features of the disease and consist of convulsive disorders and spastic hemiplegia with or without mental retardation. These manifestations are directly related to the leptomeningeal angioma and calcifications, the latter being also related to the vascular disturbance.
Nasopharyngeal Angioibroma (Juvenile nasopharyngeal fibroma)
The nasopharyngeal angiofibroma is a relatively uncommon benign neoplasm occurring almost exclusively in the nasopharynx of adolescent males. Occasional cases have extended to involve the oral cavity, however, and for this reason the entity deserves consideration here.
Hippocrates described the tumor in the 5th century BC, but Friedberg first used the term angiofibroma in 1940. Other terms (e.g. nasopharyngeal fibroma, bleeding fibroma of adolescence, fibroangioma) also have been used.
Etiopathogenesis
A hormonal theory has been suggested due to the lesion’s occurrence in adolescent males. Other theories include a desmoplastic response of the nasopharyngeal periosteum or the embryonic fibrocartilage.
Clinical Features
Nasopharyngeal angiofibroma accounts for 0.05% of all head and neck tumors. A frequency of 1: 5,000–1: 60,000 in otolaryngology patients have been reported. It occurs exclusively in males. Females diagnosed with nasopharyngeal angiofibroma should undergo genetic testing. Onset most commonly happens in the second decade; the range is 7–19 years, and it is rare in patients older than 25 years.
The lesion is generally manifested by nasal obstruction (80– 90%), epistaxis (45–60%), sinusitis (25%), and facial swelling (10–18%). On examination, nasal mass (80%), orbital mass (15%), proptosis (10–15%) may be noticed. Other findings may include serous otitis due to eustachian tube blockage. Zygomatic swelling and trismus denote spread of the tumor to the infratemporal fossa. Decreasing vision due to optic nerve involvement has rarely been reported. Care must be taken to differentiate this lesion from ordinary nasal polyps, which it may resemble superficially. As the tumor mass enlarges, inferior depression of the palate and facial deformity may occur.
Oral Manifestations
The oral manifestations of the na-sopharyngeal angiofibroma are generally those of a palatal or tonsillar mass, often with nasal obstruction. Occasionally, however, lesions of the posterior portion of the maxilla and even of the mandible have been seen which are microscopi-cally identical with the nasopharyngeal lesions, and they may be considered similar in nature.
Radiographic Features
Radiograph of sinuses may dem-onstrate nasopharyngeal polyp, bowing of the posterior wall of the maxillary sinus and maxillary sinus opacification is very suggestive of JNA. CT scan can demonstrate extent of the tumor. Magnetic resonance imaging (MRI) is indicated to delineate and define the extent of the tumor, especially in cases of intracranial involvement.
Histologic Features
On gross examination, the tumor is usually sessile, lobulated, rubbery, and red-pink to tangray in appearance. In rare cases, the tumor is polypoid or pedunculated.
The tumor is encapsulated and consists essentially of two basic and characteristic components: a vascular network and a connective tissue stroma. The vessels comprising the vascular network are of varying caliber, irregular in shape and generally consisting of a simple endothelial lining. The vascular element is generally most pronounced at the periphery of the lesion where active growth is occurring. Thrombosis and occlusion are frequently seen, usually in association with vasculitis.
The connective tissue stroma consists of both fine and coarse collagen fibrils, usually with an irregular, unoriented pattern, interspersed with plump, stellate cells arranged in a haphazard fashion (Fig. 2-55). An abundance of mast cells in the stroma and a lack of other inflammatory cells exist. Hyalinized foci are sometimes present, as well as areas resembling myxomatous degeneration. When the stromal cells are abundant, the resemblance to the sclerosing hemangioma may be pronounced. Multinucleated stromal cells are sometimes present, and with the occasional atypical nuclear and cellular changes that may be found, the lesion may be mistaken for a sarcoma.
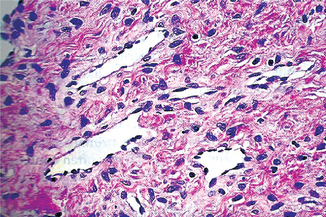
Figure 2-55 Nasopharyngeal angioibroma. The dense fibrous quality of the stroma and the numerous thin-walled vessels are characteristic of this entity (Courtesy of Dr Juan Rosai).
When examined under electron microscope, stromal cells mostly are fibroblasts and show intensive immunostaining for vimentin. However, myofibroblasts may occur focally in connection with fibrotic areas and are characterized by the coexpression of vimentin and smooth muscle actin.
Treatment
Spontaneous regression is rare. Surgical resection with preoperative embolization is the preferred treatment. The testosterone receptor blocker flutamide was reported to reduce the size of the tumor in early stages. External beam irradiation is most often reserved for intracranial, unresectable, or recurrent disease. Complications are related to intracranial extension, uncontrolled hemorrhage and death, iatrogenic injury to vital structures. Postoperative radiographic surveillance is important due to high rates of recurrence (6–60%). Recurrences can occur as early as three or four months after surgery.
Lymphangioma
The lymphangioma is a benign hamartomatous hyperplasia of lymphatic vessels, with three-fourths of all cases occurring in the head and neck region. While occasional adult-onset cases occur, this tumor is thought to be a developmental malformation of vessels which have poor communication with the normal lymph system. Diagnosed cases are typically superficial but may extend deeply into underlying connective tissues. Rarely, multiple lesions are seen in infancy and childhood in lymphangiomatosis, the lymphatic counterpart to angiomatosis of blood vessels and a potentially life-threatening disease when visceral involvement occurs.
A classification of the lymphangiomas has been suggested by Watson and McCarthy based upon their study of 41 cases. In this classification the following divisions are proposed: (1) simple lymphangioma, (2) cavernous lymphangioma, (3) cellular or hypertrophic lymphangioma, (4) diffuse systemic lymphangioma, and (5) cystic lymphangioma or hygroma. Very large cystic spaces may be seen in lesions proliferating in loose connective tissues and fascial spaces.
Clinical Features
The majority of cases of lymphangioma are present at birth, according to Watson and McCarthy, in whose series 95% of the tumors had arisen before the age of 10 years. In the series of 132 patients reported by Hill and Briggs, 88% of the lesions had developed by the end of the second year of life. In contrast to that of the hemangioma, the gender distribution of the lymphangioma is nearly evenly divided. The head and neck area in the series of Watson and McCarthy was the site of the tumor in 52% of the cases.
The most common head and neck location is the lateral neck, where this lesion typically contains large cystic spaces and is commonly called cystic lymphangioma or cystic hygroma.
Oral Manifestations
The intraoral lymphangioma most commonly occurs on the tongue, but is seen also on the palate, buccal mucosa, gingiva and lips (Fig. 2-56). The superficial lesions are manifested as papillary lesions which may be of the same color as the surrounding mucosa or of a slightly redder hue. The deeper lesions appear as diffuse nodules or masses without any significant change in surface texture or color. In some cases relatively large areas of tissue may be involved. If the tongue is affected, considerable enlargement may occur, and to this clinical feature the term ‘macroglossia’ may be applied. A series of 46 cases of lymphangioma of the tongue has been reviewed by Litzow and Lash. They pointed out that the anterior dorsal part of the tongue was most frequently affected. The irregular nodularity of the surface of the tongue with gray and pink projections is the commonest sign of the disease, and when associated with macroglossia, is pathognomonic of lymphangioma.
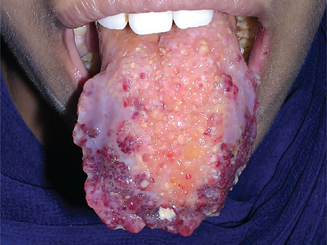
Figure 2-56 Lymphangioma of tongue. (Courtesy of Dr Ravindra Shetty, Dr G Sriram, Dr Vaishali Natu, Department of Oral Pathology, Nair Hospital Dental College, Mumbai).
Lip involvement and its attendant deformity are referred to as macrocheilia. The cystic hygroma is a common and distinct entity that is not manifested in the oral cavity but occurs in the neck as a large, deep, diffuse swelling. This has been discussed in particular by Bill and Sumner and by Paletta.
An unusual form of lymphangioma termed lymphangioma of the alveolar ridge in neonates has been reported by Levin and his associates. They found small blue domed fluid-filled lesions on the alveolar ridges in 55 (3.7%) of 1,470 normal black newborns; none were found in whites. Histologically, those biopsied were lymphangiomas. The natural history of this lesion is unknown, although spontaneous regression was noted in several cases.
Occasional cases of central lymphangioma of bone are also known to occur such as that in the tibia reported by Bullough and Goodfellow, as well as in the jaw.
Histologic Features
The lymphangioma consists of multi-ple, intertwining lymph vessels in a loose fibrovascular stroma. The lymphangioma, of which the cavernous type is the most common, consists of numerous dilated lymphatics, single layer of endothelial cells with flattened, occasionally plump, nuclei and containing lymph (Fig. 2-57). Those vessels just beneath the surface epithelium tend to fill or replace the con-nective tissue papillae, perhaps producing a papillary surface change. It is not unusual for a superficial lesion to have little or no fibrous stroma separating it from the overlying epithe-lium. Occasional channels may be filled with blood, a mixed hemangiolymphangioma. Occasional lesions demonstrate proliferation of lymphatic channels with another connec-tive tissue component, primarily smooth muscle cells called lymphangiomyoma.
Figure 2-57 Lymphangioma of buccal mucosa. (Courtesy of Dr Ravindra Shetty, Dr G Sriram, Dr Vaishali Natu, Department of Oral Pathology, Nair Hospital Dental College, Mumbai).
No encapsulation of even the tumors which appear well-circumscribed clinically. Deeper lesions show vessels interspersed between adipocytes and striated muscle bundles.
Lymphangioma can be subclassified into four categories:
1. Lymphangioma simplex (capillary lymphangioma, lymphangioma circumscriptum), composed of small, thin-walled lymphatics.
2. Cavernous lymphangioma, comprised of dilated lymphatic vessels with surrounding adventitia.
3. Cystic lymphangioma (cystic hygroma), consisting of huge, macroscopic lymphatic spaces with surrounding fibrovascular tissues and smooth muscle.
4. Benign lymphangioendothelioma (acquired progres-sive lymphangioma), lymphatic channels appear to be dissecting through dense collagenic bundles.
These categories are somewhat artificial and many lesions are combinations of categories.
Treatment and Prognosis
The treatment of the lymphan-gioma is considerably different from that of the hemangioma. Surgical excision is probably the treatment of choice, since the lymphangioma is more radioresistant and insensitive to scle-rosing agents, such as sodium morrhuate, than the hemangio-ma. Spontaneous regression, according to most studies is rare. Because of the nonencapsulated and ‘infiltrating’ nature of the lymphangioma, complete removal is often impossible without excessive removal of surrounding normal structures. Surgical debulking of the tumor is, the typical treatment provided, and additional debulking procedures will most likely be required as the affected child grows.
Myxoma
Myxoma is a heterogeneous group of soft tissue tumors which have a common histologic appearance of abundant myxoid ground substance. This is composed of mucopolysaccharides, mainly hyaluronic acid. The myxoma of the soft tissues is a tumor which has been described by Stout as a true neoplasm made up of tissue resembling primitive mesenchyme. Thus it is composed of stellate cells arranged in a loose mucoid stroma which also contains delicate reticulin fibers. The lesion is benign and does not metastasize, although it frequently infiltrates adjacent tissues.
Clinical Features
Most soft-tissue myxomas are deeply situated lesions, occurring in the skin or the subcutaneous tissues, the genitourinary tract, the gastrointestinal tract or in organs such as the liver, the spleen, or even the parotid gland.
This tumor may make its appearance at any age, approximately equal numbers of cases having been reported in every decade of life. There is no definite gender predilection of this neoplasm. A detailed study of 58 patients with 64 tumors has been reported by Ireland and his associates.
Oral Manifestations
The intraoral soft-tissue myxoma is an extremely rare lesion. The majority of oral cases undoubtedly represent only myxomatous degeneration in a fibrous tumor, and these cannot be considered true myxomas, although Elzay has presented 15 intraoral cases which he accepted as bona fide examples.
Other cases occurring centrally within the bone of the jaws have been classified, particularly in the dental literature, as odontogenic myxomas, i.e. as tumors derived from odontogenic tissue. Even though such tumors are sometimes associated with missing or displaced teeth, their dental origin is difficult, is not impossible, to establish. Nevertheless, for the sake of convenience, the central myxoma of bone is discussed under the odontogenic myxoma in the odontogenic tumor section (q.v.).
The nerve sheath myxoma is a benign tumor thought to arise from perineural cells of peripheral nerves and is characterized by the occurrence of stellate cells in a prominent mucoid matrix. A few cases have been reported in the oral cavity on the tongue, buccal mucosa and retromolar area and these have been reviewed by Sist and Greene. Their ultrastructural studies, along with those of Webb, supported the perineural origin of these lesions. They are treated by local excision and do not recur.
Oral focal mucinosis, the oral counterpart of a dermal lesion known as cutaneous focal mucinosis and/or cutaneous myxoid cyst, is the lesion most commonly misdiagnosed as an intraoral soft tissue myxoma, according to the study of Tomich. In fact, he has stated that most, if not all, cases diagnosed as oral soft-tissue myxomas are in reality lesions of this entity. He reported eight cases of oral focal mucinosis, including the histologic and histochemical findings, postulating that the lesion develops because of a fibroblastic overproduction of hyaluronic acid due to an unknown stimulus.
Histologic Features
The soft-tissue myxoma is characteris-tically a loose-textured tissue containing moderate numbers of delicate reticulin fibers and mucoid material, probably hyaluron-ic acid. Oral focal mucinosis lacks this reticulin network. Inter-spersed throughout are varying numbers of stellate cells which occasionally assume a spindle shape. The tumor is not encapsu-lated and may invade surrounding tissue. This invasiveness is not characteristic of oral focal mucinosis.
Tomich has listed the ‘myxoid’ appearing lesions to be considered in the differential diagnosis as including: soft-tissue myxoma, odontogenic myxoma, myxomatous degeneration in a fibrous lesion, oral focal mucinosis, nerve sheath myxoma, myxoid neurofibroma, mucous retention phenomenon, and localized myxedema.
Treatment and Prognosis
The treatment of the myxoma is essentially surgical, since X-ray radiation is of little benefit. Recurrence is common, but this is not of grave concern, since the tumor does not metastasize. The attempt to avoid recurrence may necessitate the sacrifice of an appreciable amount of apparently uninvolved surrounding tissue, although this is generally not true of the intraoral lesion. In contrast, oral focal mucinosis has no tendency to recur.
Chondroma
The chondroma seldom develops in membrane bones, particularly if no vestigial cartilaginous rests are present, but since both the maxilla and mandible may contain such remnants, the tumor certainly may occur in these bones. Miles pointed out that areas of ‘secondary cartilage’, cartilage not related to recognized parts of the chondrocranium, occur in the mandible in the mental region, coronoid process and condyle. At least in the embryo, cartilage is found in the maxilla on the lateral aspects near the malar process close to the molar teeth, as well as in the premaxillary area.
Clinical Features
This neoplasm may develop at any age and shows no apparent gender predilection. The chondroma usually arises as a painless, slowly progressive swelling of the jaw which, like many other neoplasms, may cause loosening of the teeth. The overlying mucosa is seldom ulcerated. The anterior portion of the maxilla is the most frequent site of involvement by this tumor because it is here that vestigial cartilage rests are found, particularly in the midline lingual to or between the central incisors. In the mandible the most common site of occurrence is posterior to the cuspid tooth, involving the body of the mandible, or the coronoid or condylar processes. It is also documented in the nasal septum and nasal spine and has been discussed here by Tomich and Hutton.
Occasional peripheral cases, outside bone, have been reported, such as that on the soft palate described by Gardner and Paterson or the relatively common chondroma or osteochondroma of the tongue. However, these may represent only islands of chondroid metaplasia or even a choristoma rather than a true neoplasm. Thus, they would be analogous to osteoma mucosae (q.v.). The relationship of these intraoral peripheral chondromas, especially those of the tongue, to the well-recognized chondroma of soft parts, reviewed by Chung and Enzinger, is not clearly established.
Radiographic Features
The radiograph shows an irregular radiolucent or mottled area in the bone. The chondroma is a destructive lesion and, in addition, has been shown to cause root resorption of teeth adjacent to it.
Histologic Features
The distinction between chondroma and chondrosarcoma is a narrow one and may offer a serious problem to the pathologist confronted with only a small biopsy specimen. The chondroma is made up of a mass of hyaline cartilage which may exhibit areas of calcification or of necrosis. The cartilage cells appear small, contain only single nuclei and do not exhibit great variation in size, shape or staining reaction. Cartilaginous tumors vary considerably in appearance from area to area, so that some malignant lesions present regions of apparent benignity. For this reason, care must be exercised in the unequivocal diagnosis of such a tumor from a small biopsy specimen.
Treatment and Prognosis
The treatment of the chondroma is surgical, since the tumor is resistant to X-ray radiation. The fact that sarcomatous change is not an unlikely occurrence suggests that somewhat more than a conservative enucleation should be carried out, although radical resection cannot be justified unless the tumor is of unusual size. Because of the dearth of reported cases, the prognosis of this disease in the jaws is not known, but to judge from cases in other parts of the body, it is probably good.
Benign Chondroblastoma ((Epiphyseal chondromatous giant cell tumor, Codman’s tumor)
The benign chondroblastoma of bone, named by Jaffe and Lichtenstein in 1942 but described earlier by Ewing in 1928 and Codman in 1931, is a distinct entity usually involving long bones but sometimes occurring in the cranial bones. Of 13 such cranial tumors reviewed by Al-Dewachi and his coworkers, nine occurred in the temporal bone, one in the parietal, two in the mandible and one in the maxilla.
Clinical Features
This benign, primary central bone tumor occurs predominantly in young persons, nearly 90% of a series of 69 cases reported by Schajowicz and Gallardo ranging between the ages of five and 25 years. Identical findings were reported in a series of 125 cases by Dahlin and Ivins. Males are involved more than females, usually in a ratio of about 2 to 1. The vast majority of cases involve the long bone of the upper and lower limbs. However, cases involving the mandibular condyle have been reported by Goodsell and Hubinger and by Dahlin and Ivins, while a case involving the anterior maxilla of a 13-year-old girl has been reported by Al-Dewachi and his associates. In addition, an extraskeletal chondroblastoma of the ear has been reported by Kingsley and Markel.
Histologic Features
The tumor is composed of relatively uniform, closely packed, polyhedral cells with occasional foci of chondroid matrix. A scattering of multinucleated giant cells may be found, usually associated with areas of hemorrhage, necrosis or calcification of the chondroid material. Formation of bone and osteoid also occurs, and as pointed out by Gravanis and Giansanti, is probably more common than is generally accepted. In addition, occasional cases have histologic features overlapping those of the chondromyxoid fibroma of bone.
Treatment
Conservative surgical excision is the usually accepted treatment, although recurrence is not uncommon. In a series of 25 cases reported by Huvos and his associates, an aneurysmal bone cyst was found engrafted on the primary bone lesion in 6–24% of the cases, and recurrence rate was higher in this group than in the group without the associated aneurysmal cysts.
Chondromyxoid Fibroma
The chondromyxoid fibroma is an uncommon benign tumor of cartilage derivation first described as an entity in 1946 by Jaffe and Lichtenstein.
Clinical Features
This central bone tumor has a predilec-tion for occurrence in young persons, approximately 75% of patients being under the age of 25 years. There is no definite gender predilection. The majority of cases occur in long bones but it is also found in the small bones of the hands and feet, in the pelvic girdle and elsewhere sporadically. A case in the anterior mandible extending on either side of the symphysis of a 10-year-old girl has been reported by Schutt and Frost. It is extremely rare here, however, considering the relatively large series of cases of chondromyxoid fibroma in other bones reported, such as the 189 cases by Feldman and his associates, the 32 cases by Schajowicz and Gallardo, and the 76 cases by Rahimi and his associates. The possible cases in the jaws have been reviewed by Grotepass and his associates, adding an ad-ditional case, as have Davis and Tideman as well as Browne and Rivas.
Pain is the outstanding clinical characteristics of the lesion. Evident swelling is uncommon but does occur.
Histologic Features
This tumor characteristically exhibits lobulated myxomatous areas, fibrous areas and areas having a chondroid appearance, i.e. cells resembling chondroblasts and chondrocytes in lacunae in a chondroid matrix. In addition, foci of calcification are sometimes found. Care must be taken not to make a mistaken diagnosis of chondrosarcoma or mesenchymal chondrosarcoma.
Treatment
Conservative surgical excision is the preferred treatment for this benign tumor. However, recurrence of this tumor in other bones is not uncommon, particularly in young patients in whom the lesions appear to act somewhat more aggressively. Too few cases in the jaws have been reported to draw valid conclusions regarding biologic behavior here.
Osteoma
The osteoma is a benign neoplasm characterized by a proliferation of either compact or cancellous bone, usually in an endosteal or periosteal location. In many parts of the body it is not difficult to establish an incontrovertible diagnosis of osteoma. In the jaws, however, where infection is common it is not always possible to differentiate a bony mass induced by irritation or inflammation from one of a true neoplastic nature. In addition, the so-called exostoses and enostoses further complicate the picture, since they may produce a similar clinical, radiographic and histologic picture. Although their nature is unknown, they might more properly be considered developmental lesions.
Clinical Features
The osteoma is not a common oral lesion. Although it may arise at any age, it seems to be somewhat more common in the young adult. The lesion of periosteal origin manifests itself as a circumscribed swelling on the jaw producing obvious asymmetry, but it must not be confused with Garre’s nonsuppurative sclerosing osteomyelitis or proliferative periostitis. The osteoma is a slow-growing tumor, and so the patient does not usually become alarmed. The osteoma of endosteal origin is slower to present clinical manifestations, since considerable growth must occur before there is expansion of the cortical plates (Fig. 2-58 A and B). There is seldom any pain associated with this tumor. Multiple osteomas of the jaws, as well as of long bones and skull, are a characteristic manifestation of Gardner syndrome (q.v.).
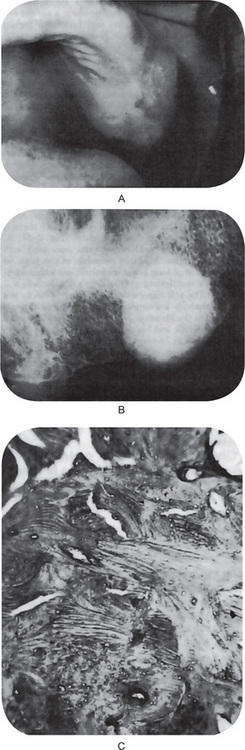
Figure 2-58 Osteoma. The clinical expansion of the alveolar ridge, (A) is seen on the radiograph, (B) to be due to a well-circumscribed radiopaque mass. Microscopically, this was composed entirely of dense, compact bone (C).
The soft-tissue osteoma of the oral cavity is a relatively uncommon lesion, a total of 25 cases from the literature being reviewed and reported by Krolls and his coworkers. The lesion is also known as ‘osteoma mucosae,’ analogous to the well-recognized dermal lesion ‘osteoma cutis’ and as ‘osseous choristoma’ after Krolls. These lesions occur almost exclusively in the tongue, although occasional cases are found in the buccal mucosa. They occur at any age and present as a firm nodule ranging up to 2 cm in diameter. Of the 25 reported cases, 18 occurred in females and only seven in males but the significance of this is not known. The bone itself is normal, well-circumscribed lamellar bone, usually compact but sometimes showing fatty marrow (Fig. 2-59B).
Radiographic Features
The central lesion usually appears within the jaw as a well-circumscribed radiopaque mass which is indistinguishable from scar bone (Fig. 2-58 B). Sometimes this osteoma is diffuse, but it must be differentiated from chronic sclerosing osteomyelitis. The periosteal form of the disease also is manifested as a sclerotic mass.
Histologic Features
The osteoma is composed either of extremely dense, compact bone or of coarse cancellous bone. In any given area the bone formed appears normal (Fig. 2-58 C). The lesion is most often well circumscribed, but not encapsulated. In some tumors foci of cartilage may be found, in which case the term ‘osteochondroma’ is often used. Myxomatous tissue also may be intermingled on rare occasions.
Osteoid Osteoma
The osteoid osteoma is a benign tumor of bone which has seldom been described in the jaws. The true nature of this lesion is unknown. Jaffe and Lichtenstein have suggested that the lesion is a true neoplasm of osteoblastic derivation, but other workers have reported that the lesion occurs as a result of trauma or inflammation. In some instances it has been confused with chronic sclerosing osteomyelitis, and the osteoid osteoma may actually represent a form of this osteomyelitis. A review of the literature with an additional report of 80 cases by Freiberger and his associates has supported the neoplastic theory.
Clinical Features
The osteoid osteoma usually occurs in young persons, seldom developing after the age of 30 years. Young children under the age of 10 years or even five years are frequently affected. In most series, males predominate over females by a ratio of at least 2 to 1. It has been reported most frequently in the femur or in the tibia, although other bones throughout the body have occasionally been involved. One of the chief symptoms of the condition is severe pain out of all proportion to the small size of the lesion. The pain of osteoid osteoma is described as unrelenting and sharp, worse at night. Classically, the pain is relieved by aspirin. Localized swelling of the soft tissue over the involved area of bone may occur and may be tender.
Oral Manifestations
Greene and his associates have re-viewed the literature and added one more case, bringing the total number of cases of osteoid osteoma of the jaws to seven. Of these, four have occurred in the mandible and three in the maxilla. Of the mandibular lesions, three were in the body and one in the condyle, while one maxillary lesion involved the antrum.
Radiographic Features
Radiographically, the osteoid os-teoma presents a pathognomonic picture characterized by a small ovoid or round radiolucent area surrounded by a rim of sclerotic bone. The central radiolucency may exhibit some cal-cification. The lesion seldom is larger than 1 cm in diameter, but the overlying cortex does become thickened by subperiosteal new bone formation.
Histologic Features
The microscopic appearance of the osteoid osteoma is characteristic and consists of a central nidus composed of compact osteoid tissue, varying in degree of calcification, interspersed by a vascular connective tissue. Formation of definite trabeculae occurs, particularly in older lesions, outlined by active osteoblasts. Osteoclasts and foci of bone resorption are also usually evident. The overlying periosteum exhibits new bone formation, and in this interstitial tissue collections of lymphocytes may be noted (Fig. 2-60).
Figure 2-60 Osteoid osteoma. This is the central nidus of an osteoid osteoma composed of irregular reactive new bone.
Ultrastructural investigation of five cases of osteoid osteoma by Steiner has revealed the morphology of the osteoblasts to be similar to that of normal osteoblasts although atypical mitochondria could be seen. For comparison, the osteoblasts of a benign osteoblastoma were studied and were found to be essentially identical, including the atypical mitochondria. The author concluded that his observations supported the idea that the osteoid osteoma and the osteoblastoma are closely related lesions. Unlike in osteoblastoma, neural staining techniques reveal many axons throughout an osteoid osteoma, which probably accounts for the pain (the nidus). Levels of prostaglandin E2 are markedly elevated in the nidus; this is presumably the cause of pain and vasodilatation.
Benign Osteoblastoma (Giant osteoid osteoma)
The osteoblastic nature of the tumor often results in zones similar to those of an osteoid osteoma producing a histologic resemblance that cannot be ignored. Benign osteoblastoma differs, however, in that it does not share the markedly limited growth potential of the average osteoid osteoma. Furthermore, the benign osteoblastoma frequently lacks the characteristic pain and the halo of sclerotic bone associated with osteoid osteoma. McLeod and coworkers resolved this problem by arbitrarily regarding an equivocal lesion as an osteoblastoma when the lesion was more than 1.5 cm in its greatest dimension.
The term giant osteoid osteoma, introduced several years ago, was an attempt to recognize the pathologic similarity of this lesion to osteoid osteoma, at the same time indicating a difference, especially in the size of the average tumor. Benign osteoblastoma, nevertheless, has become the most widely accepted designation for this tumor (Unni KK, 1996).
One can logically question whether benign osteoblastoma is correctly classed with true neoplasms because some osteoblastomas regress or become arrested after incomplete surgical removal. Fields within some of these tumors resemble portions of an aneurysmal bone cyst. This resemblance, coupled with the pronounced clinical similarity, suggests that both tumors may be different manifestations of a reaction to some as yet unknown agent.
There are rare well documented examples of osteoblastoma undergoing malignant change to osteosarcoma (Unni KK, 1996). The lesion called cementoblastoma, at or around the root of a tooth, is similar, and has been included with osteoblastoma.
The benign osteoblastoma was first described under the name ‘giant osteoid osteoma’ by Dahlin and Johnson in 1954 and under the presently more accepted name by Jaffe and by Lichtenstein in 1956 in separate reports. The lesion is not common but is nevertheless very important in as much as it is frequently mistaken for a malignant bone tumor even though it is actually entirely benign.
Clinical Features
This central bone tumor occurs most frequently in young persons, approximately 75% of the patients being under 20 years of age and 90% under 30 years of age. However, it does occur even in elderly adults. In most reported series, there is a definite predilection for occurrence in males. The lesion is characterized clinically by pain and swelling at the tumor site, the duration being just a few weeks to a year or more. Unlike osteoid osteoma, the pain of osteoblastoma is more generalized and less likely to be relieved by salicylates.
The most common site of occurrence is the vertebral column. Other frequently affected sites include the sacrum, long tubular bones and calvarium. The first case in the jaws was reported by Borello and Sedano in 1967, but it is now recognized that the benign osteoblastoma occurs in both the maxilla and mandible with some frequency, and a number of cases have been reported. These have been reviewed by Greer and Berman, and more recently, by Miller and his coworkers, who tabulated 26 cases: 14 mandible, 11 maxilla, and one unstated.
Radiographic Features
The lesion is not distinctive but, on the radiograph, appears rather well circumscribed. In some instances, there is purely bone destruction, while in other cases there is sufficient bone formation to produce a mottled, mixed radiolucent-radiopaque appearance (Fig. 2-61). A periosteal counterpart has been described by Goldman and such a case in the mandible is reported by Farman and his associates. The spectrum of the osteoblastoma is discussed by McLeod and his associates.
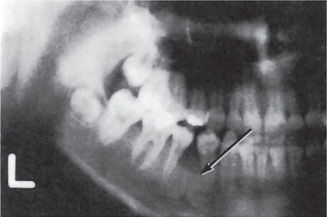
Histologic Features
There may be considerable variation in the microscopic appearance of the benign osteoblastoma. Nevertheless, the hallmark of the benign osteoblastoma consists of:
• The vascularity of the lesion with many dilated capillaries scattered throughout the tissue
• The moderate numbers of multinucleated giant cells scat-tered throughout the tissue, and
• The actively proliferating osteoblasts which pave the irregular trabeculae of new bone (Fig. 2-62).
These osteoblasts often appear so active and are present in such numbers that, in the past, mistaken diagnosis of osteosarcoma have often been rendered. In addition, some cases bear remarkable resemblance to an aneurysmal bone cyst.
The osteoblastoma has been studied ultrastructurally by Steiner who noted that, with a few exceptions, the tumor osteoblasts resembled normal osteoblasts. Comparative differences of osteosarcoma cells from osteoblastoma cells also did not appear pathognomonic, so he concluded that the final diagnosis of osteoblastic tumors rested at the light microscope level.
A malignant osteoblastoma has been described by Schajowicz and Lemos on the basis of a histologically more bizarre pattern of cells: more abundant and often plump hyperchromatic nuclei, greater nuclear atypia, and numerous giant cells. While locally more aggressive than the benign osteoblastoma, it has a better prognosis than a conventional osteosarcoma.
Finally, malignant transformation of a previously benign osteoblastoma into an osteosarcoma has also been reported, such as the case discussed by Merryweather and his coworkers.
Torus Palatinus
The torus palatinus is a slowly growing, flat-based bony protuberance or excrescence which occurs in the midline of the hard palate. Numerous theories have been suggested, but a plausible and thoroughly convincing explanation for this common oral lesion is still lacking. A study by Suzuki and Sakai offered evidence that both the torus palatinus and torus mandibularis are hereditary conditions, thought to follow a mendelian dominant pattern.
Clinical Features
The incidence of torus palatinus reported in the United States varies between 20 and 25%. Women are affected more frequently than men, the approximate ratio found by Kolas and his associates being 2 to 1. Although the palatine torus may occur at any age, including the first decade, it appears to reach its peak incidence shortly before the age of 30 years. Certain races, such as the American Indian and the Eskimo, are reported to exhibit a much higher incidence of torus palatinus than the general population in this country, including blacks.
The torus palatinus presents itself as an outgrowth in the midline of the palate and may assume a variety of shapes (Fig. 2-63). It has been classified clinically on this basis as flat, spindle-shaped, nodular or lobular. The mucosa overlying the torus is intact, but occasionally appears blanched. It may become ulcerated if traumatized. The torus itself may be composed either of dense compact bone or of a shell of compact bone with a center of cancellous bone, and thus it is often visible on an intraoral palatal radiograph.
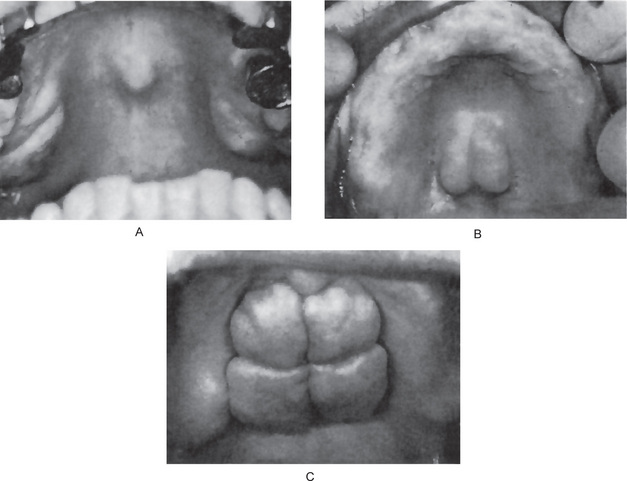
Treatment and Prognosis
There is little clinical signifi-cance attached to this lesion, since it is benign and never be-comes malignant. The torus is usually not treated, although occasionally it may be of such size and shape that it is impos-sible or impractical to construct a full or partial denture over the structure because of undercuts, the probability of trauma to the overlying mucosa or inability to seat the denture, owing to rocking. In such cases the situation must be appraised and the torus removed surgically before the construction of the prosthetic appliance.
Torus Mandibularis
The torus mandibularis is an exostosis or outgrowth of bone found on the lingual surface of the mandible. Just as in the case of the palatine torus, numerous causes have been suggested, but the etiology of the torus mandibularis is actually still unknown.
A genetic or ethnic background is suggested, for example, by the high frequency of occurrence in Mongoloid groups and a low frequency in Caucasoid groups, as pointed out by Sellevold. Supporting this idea through familial investigations, Suzuki and Sakai have commented on its apparent hereditary nature. Essentially, their studies showed that when one or both parents had either type of torus, the frequency of occurrence of a torus in the children ranged between 40 and 64%. When neither parents had a torus, the incidence of a torus in the children was only 5–8%. In contrast, some studies seem to favor an environmental background as the more important factor. For example, it has been found that groups of the same population living in different environments have different frequencies of occurrence of this torus, while different racial groups living in approximately the same environment have similar frequencies of occurrence. It has been an idea held for many years that a torus mandibularis will develop as a reinforcement of bone in this bicuspid area in response to the torsional stress created by heavy mastication.
Clinical Features
This growth on the lingual surface of the mandible occurs above the mylohyoid line, usually opposite the bicuspid teeth (Fig. 2-64). Like the palatine torus, it may vary considerably in size and shape. Although the mandibular tori are usually bilateral, they are seen as a unilateral condition in about 20% of the cases. Both unilateral and bilateral protuberances may be single or multiple, and they are frequently visible on dental periapical radiographs. There is no correlation in the frequency of simultaneous occurrence of torus palatinus and torus mandibularis, according to the studies of Kolas and coworkers, suggesting that the two conditions are not related. Suzuki and Sakai reported a highly significant correlation in the frequency of simultaneous occurrence of the two tori, however.
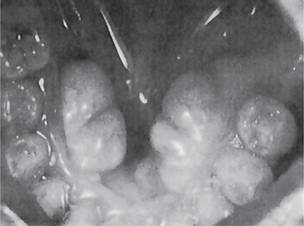
The bilateral bony growths on the lingual of the mandible may be lobed or multiple.
The reported incidence in the United States varies between 6 and 8%, with no differences between the genders noted. Some races, such as the Alaskan Eskimos and Aleuts, are reported to have a much higher incidence of mandibular tori. Among the general population in this country the torus mandibularis is infrequently seen in the first decade of life, but it usually has its onset by the age of 30 years.
Multiple Exostoses
Multiple exostoses of the jaws are somewhat less common than the maxillary and mandibular tori and are usually found on the buccal surface of the maxilla below the mucobuccal fold in the molar region. Clinically, these exostoses appear as small nodular protuberances over which the mucosa may appear blanched (Fig. 2-65).
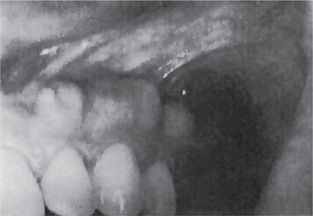
There are numerous small excrescences part of bone on the buccal surface of the maxilla above the teeth and below the mucobuccal fold.
Their etiology is unknown, and no figures are available as to their incidence or disposition. They are of no clinical significance except that, if large, they may interfere with the preparation or insertion of a prosthetic appliance.