Diseases of the Skin
 . Pemphigus
. PemphigusDermatology, the specialized study of skin diseases, has become an important subdivision of the practice of medicine not only because of the many primary diseases that affect the skin, but also because of the common cutaneous manifestations of deeper visceral or systemic diseases. The dermatologist is well aware that many primary cutaneous diseases also involve the mucous membranes throughout the body, including the oral mucosa.
It is especially important for the dentist to recognize not only that some dermatoses exhibit concomitant lesions of the oral mucous membranes, but also that manifestation of some of the diseases may be preceded by oral lesions. Thus the dentist may be in a position to establish the diagnosis of a dermatologic disease before the cutaneous lesions become apparent.
There is no universally accepted classification of these dermatologic diseases. However, several broad groups of diseases may be separated out, all of which have significant interest to dentistry, on the basis of the nature of the disease process or the nature of the lesion itself.
One large group of specific lesions which has been recognized in recent years is that known as the genodermatoses. These basically represent hereditary skin disorders, many of which are also accompanied by various systemic manifestations of different altered enzyme functions. Some of these genodermatoses are characterized particularly by alterations in the normal keratinization process and these have been specifically referred to as genokeratoses. Unfortunately, there are numerous defects and considerable overlap in even such a simple scheme. There are, for example, numerous diseases characterized by alterations in the keratinization process which are not genetically transmitted, and therefore, are not genokeratoses. There is considerable value in classifying certain dermatologic diseases as vesiculobullous diseases because of the aid provided in the differential diagnosis of a given case in which vesicles and bullae are present. However, some of the vesiculobullous diseases are genetically transmitted and thus could also be classified as genodermatoses, while others have no hereditary pattern.
Ectodermal Dysplasia: (Hereditary ectodermal dysplasia, ectodermal dysplasia syndrome)
Ectodermal dysplasia syndrome (EDS) is a large, heterogeneous group of inherited disorders, the manifestations of which could be seen in more than one ectodermal derivatives. These tissues primarily are the skin, hair, nails, eccrine glands, and teeth. Defects in tissues derived from other embryologic layers are not uncommon. The current classification of ectodermal dysplasia (ED) is based on clinical features. The disorders are congenital, diffuse, and nonprogressive. To date, more than 150 distinctive syndromes have been described with all possible modes of inheritance. The most common syndromes within this group are hypohidrotic (anhidrotic) ED and hidrotic ED. Several EDSs may manifest in association with midfacial defects, mainly cleft lip and palate.
Etiology
Ectodermal dysplasia syndrome results from aberrant development of ectodermal derivatives in early embryonic life. Genes responsible for the varied syndromes are located on different chromosomes and may be mutated or deleted.
Clinical Features
Individuals affected by EDS have abnormalities in different structures. Some EDS types are mild, while others are devastating. EDSs have been reported most often in whites, but they have also been observed in persons of other races. X-linked hypohidrotic ED has full expression only in males. Female carriers outnumber affected men, but females show little or no signs of the condition. The remaining EDSs have no gender predilection. Obvious manifestations of the disorders are not clinically apparent in newborns. Dental, hair, and nail anomalies are evident during infancy or childhood. The number of hair follicles, sweat glands, and sebaceous glands varies. Symptoms of a reduction in hair follicles vary from sparse scalp hair (usually short, fine and dry) to a complete absence of hair. Hair bulbs may be distorted, bifid, and small. Eccrine sweat glands may be absent or sparse and rudimentary, particularly in those with hypohidrotic EDS. In some cases, mucous glands are absent in the upper respiratory tract and in the bronchi, esophagus, and duodenum. The mouth may be dry from hypoplasia of the salivary glands; lacrimal glands also may be deficient. Teeth show abnormal morphogenesis or are absent. Nails are often brittle and thin or show abnormal ridging, but they may be grossly deformed. Other signs and symptoms like lack of breast development, deficient hearing or vision, cleft lip and/or palate and missing fingers or toes are also seen. The presence or absence of these abnormalities defines the different syndromes. Following are the best-defined syndromes within this group.
Hypohidrotic (anhidrotic) ED (Christ-Siemens-Touraine syndrome) is the most common phenotype in this group and is usually inherited as an X-linked recessive trait; autosomal recessive and autosomal dominant forms have been reported but are rare. It is characterized by several defects (e.g. hypohidrosis, anomalous dentition, onychodysplasia, hypotrichosis). Typical facies are characterized by frontal bossing; sunken cheeks; saddle nose; thick, everted lips; wrinkled, hyperpigmented skin around the eyes; and large, low-set ears. Because such characteristics are not obvious at birth, clinical clues for diagnosis in the neonatal period are extensive scaling of the skin and unexplained pyrexia. Dental manifestations include conical or pegged teeth, hypodontia or complete anodontia, and delayed eruption of permanent teeth. The prevalence of atopic eczema is high. Other common signs are short stature, eye abnormalities, decreased flow of tears and photophobia. Intelligence is normal.
Oral Manifestations
The oral findings are of particular interest, since patients with this abnormality invariably manifest anodontia or oligodontia, complete or partial absence of teeth, with frequent malformation of any teeth present, both deciduous and permanent dentitions (Fig. 19-1B and C). Where some teeth are present, they are commonly truncated or cone shaped. It should be pointed out that even when complete anodontia exists, the growth of the jaw is not impaired. This would imply that the development of the jaws, except for the alveolar process, is not dependent upon the presence of teeth. However, since the alveolar process does not develop in the absence of teeth, there is a reduction from the normal vertical dimension resulting in the protuberant lips. In addition, the palatal arch is frequently high and a cleft palate may be present (Fig. 19-2).
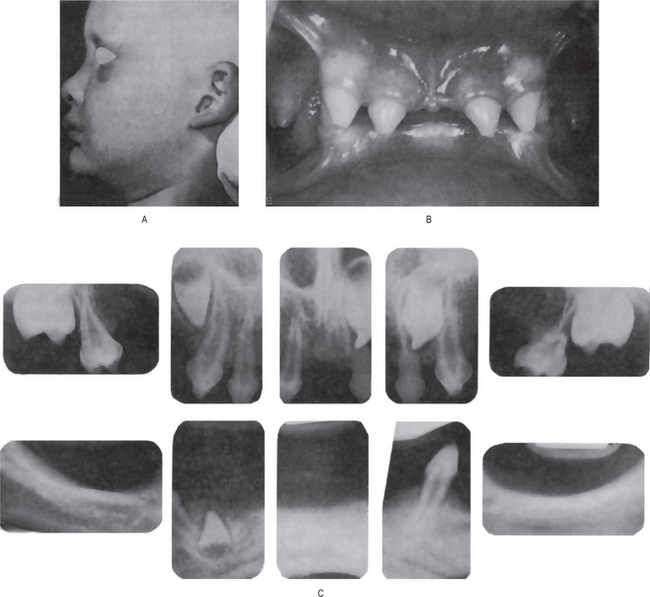
Figure 19-1 Hereditary ectodermal dysplasia.
(A)The protuberant lips, the thin, scanty hair and the saddle-nose are characteristic of the disease. (B) The teeth are cone-shaped. (C) Radiographs showing congenitally missing teeth Courtesy of Dr Ralph E McDonald.
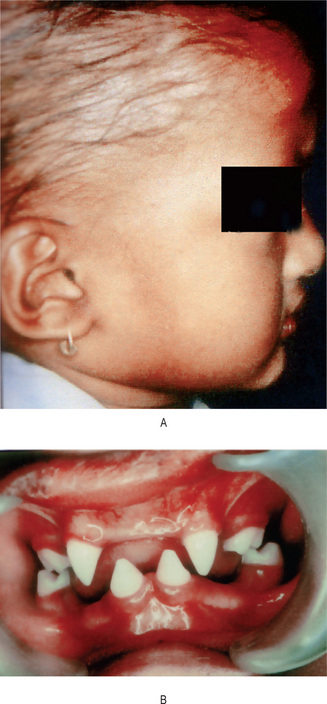
Figure 19-2 Ectodermal dysplasia.
(A) Lateral view showing frontal bossing, collapse of the middle third face, and sparse hair on the scalp. (B) Intraoral view showing peg-shaped incisors Courtesy of the Department of Oral Pathology and Microbiology, KLE's Institute of Dental Sciences, Belgaum.
According to Bessermann-Nielsen, the salivary glands, including the intraoral accessory glands, are sometimes hypoplastic in this disease. This results in xerostomia, and the protuberant lips may be dry and cracked with pseudorhagades formation. As a related phenomenon, there may be hypoplasia of the nasal and pharyngeal mucous glands which leads to chronic rhinitis and/or pharyngitis, sometimes with associated dysphagia and hoarseness.
Hidrotic ED (Clouston syndrome) is inherited in an autosomal dominant manner; the homozygous state may be lethal. Clinical features include nail dystrophy associated with hair defects and palmoplantar dyskeratosis. Nails are thickened and discolored; persistent paronychial infections are frequent. Scalp hair is very sparse, fine, and brittle. Eyebrows are thinned or absent. Patients have normal facies, normal sweating and no specific dental defect is seen.
Histologic Findings
Skin histopathology documents the reduction in the number of sweat glands, hair follicles, and sebaceous glands associated with the different syndromes. In hypohidrotic EDS, the epidermis is thin and flattened. Eccrine sweat glands are few or poorly developed or are very rudimentary. Beyond the skin, mucous glands in the upper respiratory tract and bronchi are often reduced in number. Salivary glands may show ectasia of ducts and inflammatory changes.
Chondroectodermal Dysplasia: (Ellis-van Creveld syndrome)
This uncommon disease is not classified as a dermatologic disease but is discussed here because of the similarity of certain of its features to those of hereditary anhidrotic ectodermal dysplasia. The disease appears to be inherited as an autosomal recessive characteristic with parental consanguinity in about 30% of the cases. McKusick and his coworkers reported 52 cases of this condition among 30 families in an Amish isolate.
Clinical Features
Chondroectodermal dysplasia is characterized by a number of ectodermal disturbances, including involvement of the nails and teeth as well as chondrodysplasia, polydactyly and sometimes congenital heart disease.
The nails are generally hypoplastic with marked koilonychia. The sweat mechanism has been reported to be normal in contrast to that in hereditary anhidrotic ectodermal dysplasia. The arms and legs are shortened and thickened. The bilateral polydactyly affects the hands and occasionally the feet. Many additional malformations are often present, although cardiac abnormalities are present in only about half of all cases.
Oral Manifestations
A discussion of both the systemic and oral manifestations of the disease has been published by Gorlin and Pindborg, by McKusick and his associates, and by Winter and Geddes.
The most constant oral finding is a fusion of the middle portion of the upper lip to the maxillary gingival margin eliminating the normal mucolateral sulcus. Thus, the middle portion of the upper lip appears hypoplastic.
Natal teeth, prematurely erupted deciduous teeth, frequently occur as well as congenital absence of teeth, particularly in the anterior mandibular segment. Tooth eruption is often delayed and those erupted are commonly defective, being small, cone-shaped, irregularly spaced and demonstrating enamel hypoplasia. Supernumerary teeth are also reported.
Oral Lichen Planus: (Lichen ruber planus)
Oral lichen planus (OLP) is a common mucocutaneous disease. It was first described by Wilson in 1869 and is thought to affect 0.5–1% of the world's population. The condition can affect either the skin or mucosa or both. It can cause bilateral white striations, papules, or plaques on the buccal mucosa, tongue, and gingivae. Erythema, erosions, and blisters may or may not be present. The involvement of the oral mucous membrane is so frequent and accompanies or precedes the appearance of lesions on the skin and genital mucous membrane.
Epidemiology
The overall prevalence of oral lichen planus among Indians was 1.5%; it was highest (3.7%) in those people with mixed oral habits and lowest (0.3%) in nonusers of tobacco. The annual age-adjusted incidence rate was 2.1 and 2.5 per 1,000 among men and women, respectively (Bhonsle et al, 1979). The relative risk for oral lichen planus was highest (13.7) among those who smoked and chewed tobacco.
Etiology
The data available suggests that oral lichen planus is a T-cell-mediated autoimmune disease in which cytotoxic CD8+ T-cells trigger the apoptosis of oral epithelial cells. However, the precise cause of OLP is unknown. The CD8+ lesional T-cells may recognize an antigen associated with the major histocompatibility complex (MHC) class I on keratinocytes. After antigen recognition and activation, CD8+ cytotoxic T-cells may trigger keratinocyte apoptosis. Activated CD8+ T-cells (and possibly keratinocytes) may release cytokines that attract additional lymphocytes into the developing lesion. The lichen planus antigen is unknown, although it may be a selfpeptide. The expression or unmasking of the lichen planus antigen may be induced by drugs (lichenoid drug reaction), contact allergens in dental restorative materials or toothpastes (contact hypersensitivity reaction), mechanical trauma (Koebner phenomenon), viral infection, or unidentified agents. It is interesting to note that the disease is seldom seen in carefree persons; the nervous, high-strung person is almost invariably the one in whom the condition develops. The course of the disease is long, from months to several years, frequently undergoing periods of remission followed by exacerbations which often correspond to periods of emotional upset, overwork, anxiety or some form of mental strain. Other causes suggested include traumatism (since outbreaks often develop along scratch lines), malnutrition and infection.
An interesting association of lichen planus, diabetes mellitus and vascular hypertension has been described by Grinspan, the triad being described as Grinspan's syndrome by Grupper. However, the reported associations between OLP and systemic diseases may be coincidental, because OLP is relatively common, it occurs predominantly in older adults, and many drugs used in the treatment of systemic diseases trigger the development of oral lichenoid lesions as an adverse effect.
Clinical Features
Oral lichen planus affects all racial groups, with a female-to-male ratio of 1.4:1. It predominantly occurs in adults older than 40 years, although younger adults and children can be affected. The skin lesions of lichen planus appear as small, angular, flat-topped papules only a few millimeter in diameter. These may be discrete or gradually coalesce into larger plaques, each of which is covered by a fine, glistening scale. The papules are sharply demarcated from the surrounding skin. Early in the course of the disease the lesions appear red, but they soon take on a reddish, purple or violaceous hue. Later, a dirty brownish color develops. The center of the papule may be slightly umbilicated. Its surface is covered by characteristic, very fine grayish-white lines, called Wickham's striae. The lesions may occur anywhere on the skin surface, but usually are distributed in a bilaterally symmetrical pattern, most often on the flexor surfaces of the wrist and forearms, the inner aspect of the knees and thighs, and the trunk, especially the sacral area. The face frequently remains uninvolved. In chronic cases, hypertrophic plaques may develop, especially over the shins. The primary symptom of lichen planus is a severe pruritus that may be intolerable. In patients with OLP, scalp involvement (lichen planopilaris) and nail involvement is rare.
Oral Manifestations
The majority of patients with dermal lichen planus have associated oral lesions of the disease, according to the study of Shklar and McCarthy. Conversely, in a study of 115 patients with oral lichen planus by Andreasen, only 44% had skin lesions as well.
In the oral cavity, the disease assumes a somewhat different clinical appearance than on the skin, and classically is characterized by lesions consisting of radiating white or gray, velvety, thread-like papules in a linear, annular or retiform arrangement forming typical lacy, reticular patches, rings and streaks over the buccal mucosa and to a lesser extent on the lips, tongue and palate. A tiny white elevated dot is frequently present at the intersection of the white lines, known here also as the striae of Wickham. When plaque-like lesions occur, radiating striae may often be seen on their periphery.
Shklar and McCarthy have reported the following distribution of oral lesions: buccal mucosa, 80%; tongue, 65%; lips, 20%; gingiva, floor of mouth and palate, less than 10% (Fig. 19-3). These oral lesions produce no significant symptoms, although occasionally patients will complain of a burning sensation in the involved areas.
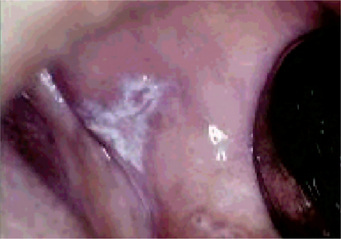
Figure 19-3 Oral lichen planus.
Plaque-like oral lichen planus on the buccal mucosa on the left side.
Vesicle and bulla formation has been reported in oral lesions of lichen planus, but this is not a common finding, and the diagnosis of lichen planus from the clinical appearance of the lesions is extremely difficult. This bullous form of lichen planus has been discussed by Shklar and Andreasen. Still another type, the so-called erosive form of lichen planus, usually begins as such and not as a progressive process from ‘nonerosive’ lichen planus. Nevertheless the vesicular or bullous form of the disease may clinically resemble erosive lichen planus when the vesicles rupture. Eroded or frankly ulcerated lesions are irregular in size and shape and appear as raw, painful areas in the same general sites involved by the simple or reticular form of the disease (Fig. 19-4). Despite the erosion of the mucosa, the characteristic radiating striae may often be noted on the periphery of the individual lesions.
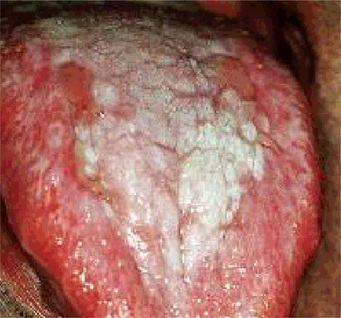
An atrophic form of lichen planus occurs with some frequency and appears clinically as smooth, red, poorly defined areas, often but not always with peripheral striae evident. The term ‘chronic desquamative gingivitis’ was at one time used to describe a red, diffuse, painful condition of the gingiva, usually found in postmenopausal women and generally quite refractory to therapy. It is now generally accepted that this is not an entity but represents a variety of conditions including the oral manifestations of several dermatologic diseases, one of which is lichen planus (usually the atrophic or erosive form), occurring on the gingiva.
A hypertrophic form of lichen planus may also occur on the oral mucosa, generally appearing as a well-circumscribed, elevated white lesion resembling leukoplakia. In such cases biopsy is usually necessary to establish the diagnosis.
The oral manifestations of lichen planus may occur weeks or months before the appearance of the skin lesions; in fact, in the clinical experience of most investigators, the great majority of patients exhibiting oral lichen planus do not have skin lesions present at the time of presentation of the oral lesions. Indeed, many patients with oral lichen planus never manifest the cutaneous form of the disease, although most patients are not followed up for a sufficiently long time to be absolutely certain of this. Other mucous membranes may be affected also, such as those of the penis, vagina and epiglottis. The genitals are involved in as many as 25% of women with OLP, compared with only 2–4% of men with OLP. Involvement of these locations may occur concomitant with or independent of oral lesions.
A variety of drugs may cause lesions that appear clinically similar to lichen planus and are termed as lichenoid lesions. Oral mucosal lichenoid lesions may occur after the administration of systemic drugs such as nonsteroidal antiinflammatory drugs (NSAIDs), sulfonylureas, antimalarials, beta-blockers, and some angiotensin-converting enzyme (ACE) inhibitors. The period between the commencement of the drug therapy and the clinical appearance of OLP-like disease varies. In rare cases, oral mucosal lichenoid lesions occur after a dental restoration is performed or after the patient starts using a denture; the lag period varies. Patients with an associated allergy to metals or components of the appliance should be evaluated by means of patch testing. In many patients, a cause for the oral lichenoid lesions cannot be identified; in these patients, the disease is called idiopathic OLP. Oral lichenoid reactions are considered to be a part of the spectrum of graft-versus-host disease.
They are present as reticular, erythematous, erosive lesions or ulceration, with whitish streak similar to that of Wickham's striae of lichen planus. Clinical manifestations of LR are very much similar to that of lichen planus. An important factor which distinguishes LR from lichen planus is its atypical location and absence of bilateral occurrence.
There is no specific test to diagnose LR. The widely accepted criterion is based on the observation of disappearance of the lesions after withdrawal of triggering agent and recurrence of the lesions when it is reintroduced.
Though histologically LR has superficial resemblance to lichen planus there are notable differences. The inflammatory infiltrate is diffuse and extends deeper into the lamina propria unlike the sharp band of infiltrate seen in lichen planus. Inflammatory infiltrate consists of plasma cells and eosinophils in addition to lymphocytes. Increased numbers of colloid or Civatte bodies may be present in LR compared to lichen planus. A perivascular chronic inflammatory cell infiltrate can be seen in drug related lichenoid lesions, which is not commonly found in lichen planus.
Proliferative verrucous leukoplakia, an unusual form of leukoplakia shares some demographic and clinical similarities with lichen planus. It occurs most commonly in older female patients and is not associated with tobacco usage. Microscopically it exhibits epithelial dysplasia with a band-like inflammatory infiltrate which on low-power can mimic lichen planus and is known as lichenoid dysplasia.
Identification and elimination of the triggering factors play a major role in the management of LR. Lichenoid lesions can take many months or longer to resolve. The malignant transformation rate is reportedly higher in oral lichenoid lesions which do not have all the typical clinical and histologic features of oral lichen planus.
Histologic Findings
Histopathologic examination of lesional tissue is the most relevant investigation in cases of OLP. Typical findings include hyperparakeratosis or hyperorthokeratosis with thickening of the granular layer, acanthosis with intracellular edema of the spinous cells in some instances, the development of a ‘saw tooth’ appearance of the rete pegs. Band-like subepithelial mononuclear infiltrate consisting of T-cells and histiocytes; increased numbers of intraepithelial T-cells; and degenerating basal keratinocytes that form colloid (Civatte, hyaline, cytoid) bodies, which appear as homogenous eosinophilic globules are consistently seen.
Degeneration of the basal keratinocytes and disruption of the anchoring elements of the epithelial basement membrane and basal keratinocytes (e.g. hemidesmosomes, filaments, fibrils) weakens the epithelial-connective tissue interface. As a result, histologic clefts (i.e. Max-Joseph spaces) may form, and blisters on the oral mucosa (bullous lichen planus) may be seen at clinical examination. B cells and plasma cells are uncommon findings (Fig. 19-5).
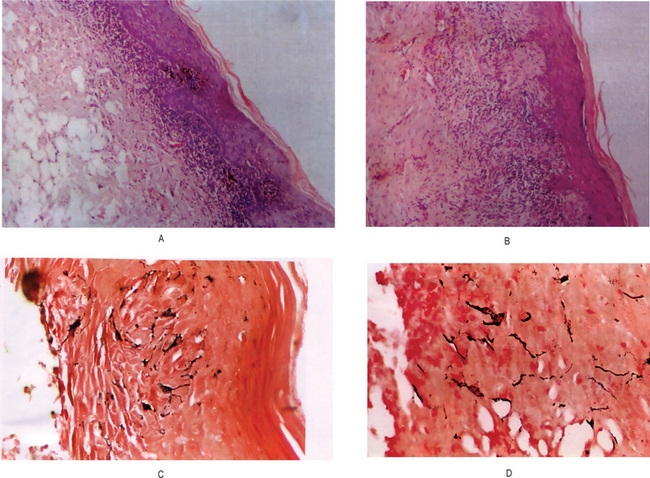
Figure 19-5 Oral lichen planus.
(A) Note the basilar degeneration and band-like infiltration of inflammatory cells in the subepithelial zone. (B) Histopathology of lichenoid mucositis (H and E x 100). Note the diffuse infiltration of inflammatory cells involving parts of submucosa. (C) Photomicrograph of Langerhans cells in lichen planus (Gold Chloride staining x400). (D) Photomicrograph of Langerhans cells in lichenoid mucositis (Gold Chloride staining x400) Courtesy of the Dept of Oral Pathology, Ragas Dental College and Hospital, Chennai.
Immunoglobulin or complement deposits are not a consistent feature of OLP. In some instances, fibrinogen and fibrin are deposited in a linear pattern in the basement membrane zone. Colloid bodies contain fibrin, IgM, C3, C4, and keratin. Laminin and fibronectin staining may be absent in areas of heavy fibrin deposition and colloid body formation; this finding suggests basement membrane damage in these areas. Direct immunofluorescent studies of lichen planus by Daniels and Quadra-White have shown that nearly all specimens from oral lesions of this disease react with antifibrinogen and exhibit an intensely positive fluorescence that outlines the basement membrane zone with numerous irregular extensions into the superficial lamina propria. This particular pattern is characteristic of both lichen planus and lupus erythematosus. This is not present in pemphigoid or erythema multiforme, in both of which the fluorescence instead tends to form a patchy linear pattern, nor is it seen in pemphigus, in which the fluorescence has a granular pattern. These workers reported a general absence of immunoglobulins in lichen planus lesions while only a few specimens exhibited a fine granular fluorescence with anti-C3 (complement) at the basement membrane zone. They concluded that the pattern of fibrinogen deposition, in the absence of fluorescence by other reagents, is sufficiently unique to be used as a diagnostic criterion for oral mucosal lichen planus.
In OLP, electron microscopy is used principally as a research tool. The ultrastructure of the colloid bodies suggests that they are apoptotic keratinocytes, and recent studies of the endlabeling method revealed DNA fragmentation in these cells. Electron microscopy shows breaks, branches, and duplications of the basement membrane in OLP.
Differential Diagnosis
It is important that lichen planus be differentiated from other lesions of the oral cavity which may present a similar clinical appearance, but which may have a different prognosis. Oral lesions which bear superficial resemblance to lichen planus include lichenoid reactions, leukoplakia, candidiasis, pemphigus, cicatricial pemphigoid, erythema multiforme, syphilis, recurrent aphthae and lupus erythematosus (q.v.). Although microscopic examination of tissue may be necessary to establish a definitive diagnosis, the clinical characteristics of these various diseases are often sufficient to differentiate one from the other.
Malignant Transformation
There is some controversy regarding its malignant potential. There seems to be a slightly higher incidence of oral squamous cell carcinoma in patients with oral lichen planus than in the general population. The actual overall frequency of malignant transformation is low, varying between 0.3 and 3%. The forms that more commonly undergo malignant transformation are the erosive and atrophic forms.
Treatment
At present there is no cure, although various agents have been tried. Due to its minimal potential for malignant transformation, these patients used to be kept on longterm follow-up. Medical treatment of OLP is essential for the management of painful, erythematous, erosive, or bullous lesions. The principal aims of current OLP therapy are the resolution of painful symptoms, the resolution of oral mucosal lesions, the reduction of the risk of oral cancer, and the maintenance of good oral hygiene. As it is an autoimmune mediated condition, corticosteroids are recommended. In patients with recurrent painful disease, another goal is the prolongation of their symptom free intervals. The main concerns with the current therapies are the local and systemic adverse effects and lesion recurrence after treatment is withdrawn. Patients should be observed periodically, particularly those with the erosive or atrophic forms and those who also have a history of alcohol and tobacco misuse, because of the risk of malignant transformation.
Psoriasis
Psoriasis is a noncontagious skin disorder that most commonly appears as inflamed, edematous skin lesions covered with a silvery white scale. The most common type of psoriasis is plaque psoriasis and is characterized by patches on the scalp, trunk, and limbs. The nails may be pitted and/or thickened. In rare instances it has been reported to manifest oral mucous membrane lesions.
Etiology
The cause of psoriasis is unknown. Patients do have a genetic predisposition for the disease; the disease has a strong association with HLA Cw6 and B57 region. Recent evidence suggests that in addition to these regions many other gene loci such as 19p13, 17q25, and 1q21 may also increase the susceptibility to this disease. The trigger event may be unknown in most cases but is likely to be an immunologic event. Significant evidence is accumulating that psoriasis is an autoimmune disease. Lesions of psoriasis are associated with increased activity of T-cells in underlying skin. Also of significance is that 2.5% of persons with HIV develop psoriasis during the course of the disease. Perceived stress can cause exacerbation of psoriasis. Some authors suggest that psoriasis is a stress-related disease and offer findings of increased concentrations of neurotransmitters in psoriatic plaques. The pathogenesis of psoriatic lesions is due to an increase in the turnover rate of dermal cells, from the normal turnover duration of 23 days to three to five days in affected areas (Fig. 19-6). As would be expected, there is also a dramatic increase in the mitotic index of psoriatic skin which is said to even surpass that of epidermoid carcinoma.
Clinical Features
Psoriasis of the skin is characterized by the occurrence of small, sharply delineated, dry papules, each covered by a delicate silvery scale which has been described as resembling a thin layer of mica. If the deep scales are removed, one or more tiny bleeding points are disclosed, a characteristic feature termed Auspitz's sign. After removal of the scale the surface of the skin is red and dusky in appearance.
The cutaneous lesions, which are painless and seldom pruritic, may be few in number or extensive in distribution. The papules enlarge at the periphery and tend to become slightly infiltrating and elevated, smaller lesions coalescing to form large plaques of irregular outline. They are roughly symmetrical and are most frequently grouped on the extensor surfaces of the extremities, particularly the elbows and knees, the scalp, back and chest, face and abdomen. Involvement of the hands and feet, with the exception of the fingernail, is uncommon.
The disease commences with the appearance of a few small papules, which gradually increase in size. New lesions slowly arise over a period of weeks, months or even years. The disease may remain static for a long time, progresses slowly to involve more and more skin area, or exhibits acute generalized exacerbations. The disease is more severe in the winter and less severe in the summer as a result of increased exposure to ultraviolet light; patients who move to a warm sunny climate usually undergo improvement in their condition. Mental anxiety or stress almost invariably appears to increase the severity of the disease or induce acute exacerbations. Arthritis is a complication in about 12% of persons with psoriasis, according to Allen. Psoriasis is uncommon in children, and seldom does a primary attack occur after the age of 45 years; it most frequently arises in the second and third decades of life. The median age at onset is 28 years. Psoriasis is slightly more common in women.
Oral Manifestations
Most authorities consider psoriatic involvement of oral mucosa extremely rare and point out that many oral lesions occurring concomitant with psoriasis of the skin are actually other diseases such as leukoplakia or lichen planus. In fact some investigators deny the existence of oral psoriasis. For example, in a study of 100 patients with dermal psoriasis, Buchner and Begleiter found none with oral lesions of the disease. However, they did note in these patients an 11% incidence of angular cheilosis, 6% incidence of fissured tongue and 5% incidence of benign migratory glossitis. Nevertheless it has been reported that in occasional cases oral lesions have exhibited all histologic features of psoriasis and in some instances have been identical with the coexisting skin lesions.
Such lesions have been reported on the lips, buccal mucosa, palate, gingiva and floor of the mouth (Fig. 19-7). Clinically, they are described as gray or yellowish-white plaques; as silvery white, scaly lesions with an erythematous base; as multiple papular eruptions which may be ulcerated; or as small, papillary, elevated lesions with a scaly surface. Reports by Goldman and Bloom and by Levin emphasized the vagaries in the clinical appearance of oral psoriasis. Psoriasis of the gingiva was reported by Brayshaw and Orban and that of the alveolar ridge by Wooten and his associates in patients without skin lesions; however, cases of mucosal involvement without skin manifestations must be viewed with caution even though the histologic sections of the lesions do present a psoriasiform pattern. White and his associates have reviewed the literature on intraoral psoriasis while reporting an additional case. Fischman and his coworkers studied an oral lesion in a patient with skin lesions of psoriasis utilizing light and electron microscopy, as well as immunologic methods, and noted in all instances that the findings in the oral lesion were similar to those in the skin lesions. They concluded that true oral lesions do occur in psoriasis.
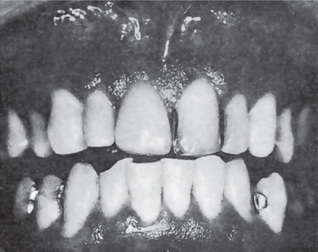
Figure 19-7 Psoriasis.
The patient manifested typical psoriasis of the skin, and in addition, presented granular gingival lesions which microscopically exhibited the characteristic psoriasiform pattern Courtesy of Dr Robert J Gorlin and Dr Frank Vellios.
The general problem of ‘psoriasiform’ lesions of the oral mucosa has been reviewed by Weathers and his associates. These lesions included psoriasis, Reiter's syndrome, benign migratory glossitis and 'ectopic geographic tongue', and the authors concluded that their exact interrelationship, if any, is still unknown.
Histologic Features
The microscopic appearance of psoriasis is characterized by uniform parakeratosis, absence of the stratum granulosum and elongation and clubbing of the rete pegs (Figs. 19-8, 19-9). The epithelium over the connective tissue papillae is thinned, and it is from these points that bleeding occurs when the scales are peeled off. Tortuous, dilated capillaries extending high in the papillae are prominent. Intraepithelial microabscesses (Monro's abscesses) are a common but not invariable finding; they are reported by Pisanty and Ship to be absent in oral psoriasis (Fig. 19-10). Mild lymphocytic and histiocytic infiltration of the connective tissue is also typical, particularly perivascular and periadnexal in location.
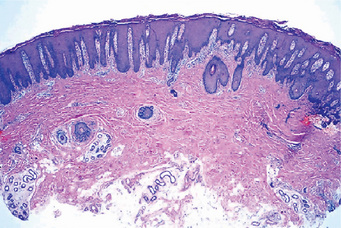
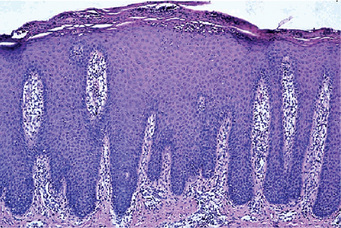
Treatment
The lesions are usually benign but a few cases may be refractory to treatment. Treatments for more general or advanced psoriasis include UV-A light, psoralen plus UV-A light (PUVA), retinoids (e.g., isotretinoin, acitretin), methotrexate (particularly for arthritis), cyclosporine, and alefacept.
Pityriasis Rosea
Pityriasis rosea (PR) is a common benign papulosquamous disease causing acute skin eruption of unknown etiology. Pityriasis denotes fine scales, and rosea implies rose-colored or pink. It can have a number of clinical manifestations and its diagnosis is important because it may resemble secondary syphilis.
Etiology
Pityriasis rosea has often been considered to be a viral exanthem. Its clinical presentation supports this concept. It has an increased incidence in individuals who are immunocompromised. As with viral exanthems, a single outbreak tends to elicit lifelong immunity. Despite these tendencies, no single virus has been proven to cause the disease. A number of viruses have been studied for a link to PR, which include picornavirus and parvovirus B19. Other recent work demonstrated human herpesvirus 7 (HHV-7) viral DNA in the lesions and the plasma of patients with PR. However, because HHV-7 is frequently found in healthy individuals, its etiologic role is controversial. Lesions are also thought to be increased in individuals with high stress levels.
Clinical Features
The disease is more common in hot, dry climate countries like Australia, Malaysia and India. PR is more common in women than in men and commonly develops in children and young adults, although any age group can be affected. Pityriasis rosea is characterized by the appearance of superficial light red macules or papules, either generalized over most of the skin surface, with the usual exception of the face and hands, or localized to certain areas such as the trunk, thighs, axillae or groin. This generalized outbreak is frequently preceded by the appearance of a ‘primary lesion’ or ‘herald spot’ 7–10 days previously. This spot is brighter red and larger (3–4 cm in diameter) than the multiple eruptions which follow its appearance. The individual exanthematous lesions are commonly ovoid, with the long axis parallel to the natural lines of cleavage of the skin, and are covered by a thin silvery scale.
The lesions often manifest mild itching sometimes accompanied by headache and low-grade fever; cervical lymphadenopathy may also be present.
Pityriasis rosea usually runs its course in three to six weeks and seldom recurs. It is interesting that the disease occurs seasonally, being far more common in the spring and autumn than at other times.
Oral Manifestations
It was pointed out by Guequierre and Wright, and confirmed by others, that involvement of the oral mucous membranes occurs with some frequency in pityriasis rosea. The oral lesions appear either concomitantly with or subsequent to the skin manifestations; they are not present throughout the clinical course of the disease, but are usually prominent during its most severe phase.
The oral lesions usually occur only on the buccal mucosa, although both tongue and palatal lesions have also been recorded. They appear as erythematous macules with or without a central area of grayish desquamation. The lesions may be single or multiple, are irregular in shape, occasionally show a raised border and vary in size from a few millimeters to 1 or 2 cm in diameter. These lesions are asymptomatic and of no clinical significance. They clear simultaneously with the skin lesions.
Histologic Features
The microscopic changes in pityriasis rosea are not pathognomonic, but consist of slight acanthosis and focal parakeratosis with microvesiculation or simply sprinkling of leukocytes within the epithelium. In addition, edema, hyperemia and perivascular infiltration of lymphocytes, plasma cells and histiocytes are prominent in the superficial connective tissue. Increased amounts of CD4 T-cells and Langhans cells are present in the dermis; this observation may indicate viral antigen processing and presentation. The histologic features suggest little more than a nonspecific dermatitis.
Treatment
The most important part of treating patients with PR is reassurance that the rash will resolve. Relief of pruritus is helpful and can be accomplished by using topical steroids, oral antihistamines, topical menthol-phenol lotions, and oatmeal baths. Systemic steroids are not recommended. Although they suppress pruritus, systemic steroids do not shorten the overall disease; in fact, they may prolong or exacerbate the disease. Ultraviolet B (UV-B) light therapy may rapidly relieve pruritus in resistant cases. One must take into consideration the possibility of postinflammatory pigmentation with light therapy. The prognosis for PR is excellent. Patients may return to work or school because they are not considered to be contagious.
Erythema Multiforme: (Stevens-Johnson syndrome, erythema multiforme major, erythema multiforme minor, herpes-induced EM major, herpes-associated erythema multiforme, drug-induced Stevens-Johnson syndrome)
Erythema multiforme (EM) is an acute self-limiting dermatitis characterized by a distinctive clinical eruption manifested as the iris or target lesion. EM may present with a wide spectrum of severity. EM minor represents a localized eruption of the skin with mild or no mucosal involvement. EM major and Stevens-Johnson syndrome (SJS) are more severe mucosal and skin diseases and are potentially life-threatening disorders. Recently, different workers have suggested that EM and SJS could be separated as two distinct clinical disorders with similar mucosal reactions but different patterns of cutaneous lesions. The clinical picture is as follows: erythema multiforme major is characterized by mucosal erosions of raised atypical target lesions. These are usually located on the extremities and/or on the face. The characteristic findings of SJS are mucosal erosions plus widespread distribution of flat atypical targets or purpuric macules. The lesions may be present on the trunk, the face, and on the extremities.
Etiology
Many suspected etiologic factors have been reported to cause EM. EM and SJS are both caused by drugs, but infectious agents are considered to be the major cause of EM. Today, EM minor is regarded as being triggered by HSV in nearly 100% of cases; many instances of idiopathic EM minor may be precipitated by subclinical HSV infection. A herpetic etiology also accounts for 55% of cases of EM major. Among the other infections, Mycoplasma infection appears to be a common cause. Drugs are reported in many documented cases of SJS and EM major. Sulfa drugs are the most common triggers.
Clinical Features
Erythema multiforme occurs chiefly in young adults, although it may develop at any age, the highest incidence is in the second to fourth decades of life and affects males more frequently than females. This disease is characterized by the occurrence of asymptomatic, vividly erythematous discrete macules, papules or occasionally vesicles and bullae distributed in a rather symmetrical pattern most commonly over the hands and arms, feet and legs, face and neck. The individual lesions may vary considerably in size even in the same patient, but are generally only a few centimeters or less in diameter. A concentric ringlike appearance of the lesions, resulting from the varying shades of erythema, occurs in some cases and has given rise to the terms ‘target’, ‘iris’, or ‘bull’s eye' in describing them (Fig. 19-11). They are most common on the hands, wrists and ankles. Mucous membrane involvement, including the oral cavity, is common. The lesions make their appearance rapidly, usually within a day or two, and persist from several days to a few weeks, gradually fading and eventually clearing. Recurrence of the disease over a period of years is common, however.
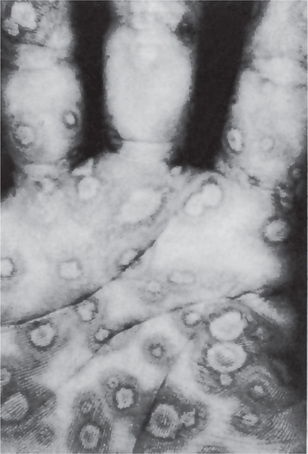
The oral mucous membrane lesions are not usually a significant feature of the disease except for the pain and discomfort they cause. The hyperemic macules, papules or vesicles may become eroded or ulcerated and bleed freely. The tongue, palate, buccal mucosa and gingiva are commonly diffusely involved (Fig. 19-12). Occasionally, mucous membrane lesions occur before the cutaneous manifestations, but oral involvement without dermal lesions has been questioned. Nevertheless, Lozada and Silverman have reported that 12 of 50 patients with erythema multiforme had oral lesions only.
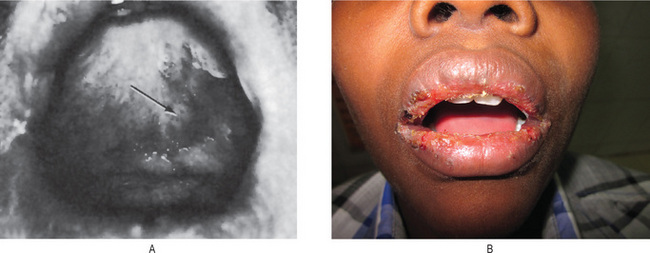
Figure 19-12 Erythema multiforme.
Lesions of the palate (A) and lip (B). B, Courtesy of Meenkashi Ammal Dental College, Chennai
At one time considered to be a separate disease, Stevens-Johnson syndrome is now recognized as simply a very severe bullous form of erythema multiforme with widespread involvement typically including the skin, oral cavity, eyes and genitalia. It commences with the abrupt occurrence of fever, malaise, photophobia, and eruptions of the oral mucosa, genitalia and skin. The cutaneous lesions in this mucocutaneous-ocular disease are similar to those of erythema multiforme, although they are commonly hemorrhagic and are often vesicular or bullous.
Oral mucous membrane lesions may be extremely severe and so painful that mastication is impossible. Mucosal vesicles or bullae occur which rupture and leave surfaces covered with a thick white or yellow exudate. Erosions of the pharynx are also common. The lips may exhibit ulceration with bloody crusting and are painful (Fig. 19-13A). The oral lesions may be the chief complaint of the patient, and understandably, have been mistaken for acute necrotizing ulcerative gingivostomatitis. Interestingly, however, it has been reported that the organisms of Vincent's infection are scarce in patients with this disease. The mucosal involvement in SJS is more severe and extensive than in EM major.
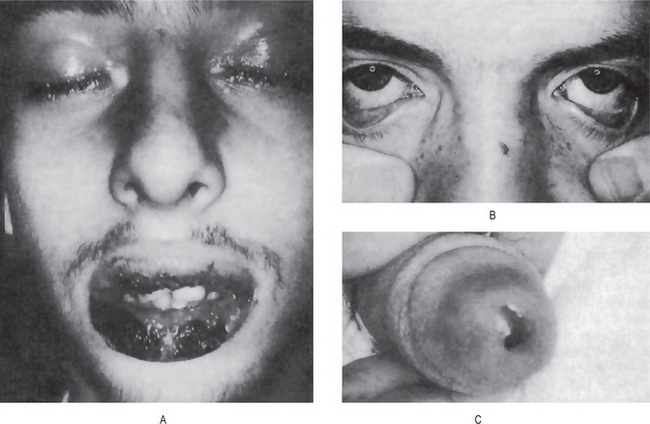
Figure 19-13 Stevens-Johnson syndrome.
Crusting ulcerated lesions of the oral cavity and lips (A), conjunctivitis (B), urethritis (C) are characteristics of the disease.
Eye lesions consist of photophobia, a characteristic of the disease referable to the conjunctivitis, corneal ulceration and panophthalmitis which may occur (Fig. 19-13B). Keratoconjunctivitis sicca also has been described. Blindness may result chiefly from intercurrent bacterial infection.
Genital lesions are reported to consist of a nonspecific urethritis, balanitis and/or vaginal ulcers (Fig. 19-13C).
Other reported complications are related to respiratory tract involvement such as tracheobronchial ulceration and pneumonia. The patients usually recover unless they succumb to a secondary infection.
Histologic Features
The microscopic appearance of erythema multiforme is not diagnostic. Although considerable variation occurs, corresponding to the variation in clinical appearance, the cutaneous or mucosal lesions generally exhibit intracellular edema of the spinous layer of epithelium and edema of the superficial connective tissue which may actually produce a subepidermal vesicle. In a study of oral lesions, Shklar has also described a zone of severe liquefaction degeneration in the upper layers of the epithelium, intraepithelial vesicle formation and thinning with frequent absence of the basement membrane. Dilatation of the superficial capillaries and lymphatic vessels in the uppermost layer of connective tissue is prominent, and a varying degree of inflammatory cell infiltration, chiefly lymphocytes, but often neutrophils and eosinophils, is also present. Similar findings were described by Buchner and his coworkers in a series of 25 cases. The findings have been confirmed in an electron microscopic study by von Bulow and his coworkers.
Differential Diagnosis
The varied nature of the disease may present difficulty in diagnosis, particularly when the occurrence of cutaneous lesions is minimal. In the presence of oral lesions, aphthous stomatitis, contact dermatitis or stomatitis and acute necrotizing gingivitis must be considered, as well as pemphigus, dermatitis herpetiformis, bullous lichen planus, herpes zoster, chickenpox and toxic epidermal necrolysis (Lyell's disease).
Toxic epidermal necrolysis is a very serious, often fatal, bullous drug eruption, so severe that large sheets of skin peel off, giving the appearance of a widespread scalding burn. Oral erosions may also occur and have been described by Giallorenzi and Goldstein. It is now considered to be a confluent form of Stevens-Johnson syndrome. Toxic epidermal necrolysis must be differentiated from the staphylococcal scalded skin syndrome, which appears clinically similar even though the latter is a milder disease with a better prognosis.
Treatment
Identification of the cause should be made if possible. If a drug is suspected, it must be withdrawn. Infections should be appropriately treated after cultures and/or serologic tests have been performed. For all forms of EM, symptomatic treatment, including oral antihistamines, analgesics, local skin care, and soothing mouthwashes, is of great importance. Topical steroids may be considered. Oral antacids may be helpful for discrete oral ulcers. The use of liquid antiseptics, such as 0.05% chlorhexidine, during bathing is preferable. Systemic corticosteroids are controversial, and some believe they may predispose to complications.
Mucocutaneous Lymph Node Syndrome: (Kawasaki disease)
An acute self-limiting febrile illness in a large number of Japanese children was first described in 1967 by Dr Tomisaku Kawasaki. Since then, Kawasaki disease (KD) has been observed worldwide in children and adult patients of all races.
Mucocutaneous lymph node syndrome, Kawasaki syndrome or KD, is a systemic vasculitis of unknown etiology and the most common cause of acquired heart disease in children in Japan and the United States. However, Asians are most commonly affected. The hallmarks of KD are fever of unknown origin for more than five days, generalized erythema and desquamation of skin, cervical nonsuppurative lymphadenopathy, and swelling of the hands and the feet.
Etiology
The cause of Kawasaki disease is unknown. Evidence suggests an abnormal inflammatory response triggered by a neoantigen or a conventional antigen from one or more etiologic agents. Several infectious causes of KD have been theorized; these include Epstein-Barr virus; retroviruses; Streptococcus pyogenes; Streptococcus viridans; Staphylococcus species; Chlamydia infections; Propionibacterium, and Pseudomonas species. However, conventional bacterial and viral cultures and serologic studies have not confirmed an infectious cause. Other postulated etiologic agents are immunization; medications; and environmental agents, such as house dust mites.
Clinical Features
The vast majority of cases have occurred in children between three months and 12 years of age, although it has also been reported in adults. KD occurs more often in boys than in girls, with a ratio of about 1.4 : 1. The most frequent symptoms of the disease are:
• Fever for five days or more, with no response to antibiotics.
• Bilateral congestion of ocular conjunctiva.
• Changes in the extremities including indurative edema, erythema of palms and soles and membranous desquamation of fingers and toes.
• Changes in lips and mouth including dryness, redness and fissuring of lips, strawberry-like reddening and swelling of tongue papillae and diffuse reddening of oral and pharyngeal mucosa, sometimes with gingival ulceration.
• Polymorphous exanthema of torso without vesicles or crusts.
• Acute, nonpurulent swelling of cervical lymph nods of 1.5 cm or more. Other less common findings include diarrhea, arthralgia, proteinuria, leukocytosis, increased sedimentation rate and positive C-reactive protein.
One of the unfortunately common complications of the disease is cardiac abnormality. While the vast majority of cases are self-limiting and nonfatal, occasional deaths do occur, almost invariably a result of the cardiac complications, usually a coronary thrombosis or vascular damage related to infantile periarteritis nodosa. The implications of dental treatment in these patients have been reviewed by Taylor and Peterson.
Differential Diagnosis
Unfortunately, there are no laboratory tests available for confirmation of the diagnosis of the disease. Therefore, its diagnosis is based entirely on clinical manifestations. It must be carefully distinguished from scarlet fever, erythema multiforme or Stevens-Johnson syndrome.
Pachyonychia Congenita: (Jadassohn-Lewandowsky syndrome, polykeratosis congenita [Touraine])
Pachyonychia congenita (PC) is a rare form of hereditary palmoplantar keratoderma (PPK), extremely uncommon in occurrence. In the dermatologic literature, PC is better known as Jadassohn-Lewandowsky syndrome. The condition is rare, but more than 250 cases have been reported. Various classifications for PC have been proposed. Currently two distinct syndromes of PC are recognized:
Etiology
PC results from mutations in the genes encoding epidermal keratinocyte keratins, specifically K6a, K6b, K16, and K17. In most cases, an autosomal dominant mode of inheritance is described; however, autosomal recessive inheritance is also mentioned in the literature. Cockayne (1933) was the first to express the opinion that the presence of an additional factor, probably a second genetic mutation, is necessary for the expression of the disease. Munro (1994) was the first to propose that the genetic defect in PC is linked to the keratin gene cluster on chromosome 17.
Clinical Features
The skin lesions of pachyonychia congenita usually occur shortly after birth; both genders are affected equally and consist of dystrophic changes in the fingernails and toenails, hyperkeratotic calluses of the palms and soles, follicular keratosis about the knees and elbows, and hyperhidrosis or excessive sweating of the hands and feet. The nail changes from which the disease derives its name consist of marked thickening, increasing toward the free border, with the nail bed becoming filled with yellowish keratotic debris, often causing the nail to project upward at the free edge. Associated sparse hair and corneal dyskeratosis producing corneal opacities have been reported also.
Oral Manifestations
Oral lesions are nearly always present, according to Gorlin and Chaudhry. They consist of either focal or generalized, white, opaque thickening of the mucosa involving the buccal mucosa, tongue or lips. These leukoplakic oral lesions should not be confused clinically with lichen planus. These have even been reported present at birth. Angular cheilosis is also reported to be commonly seen. Teeth present at birth, natal teeth, have been found on a number of occasions. Cases with typical oral lesions have been reported by Maser and by Young and Lenox.
Histologic Findings
The hyperkeratotic lesions of the skin and oral mucosa show acanthosis, hyperkeratosis, and parakeratosis. Premalignant changes are not observed. Electron microscopy shows thickened and clumped intermediate filaments, as well as enlarged keratohyalin granules. In the broadened granular layer, thick masses of tonofilaments and large, irregular keratohyalin granules are present. In the spinous layer, thick masses of tonofilaments are found at the periphery of the cells.
Keratosis Follicularis: (Darier disease, Darier-White disease)
Keratosis follicularis, also known as Darier disease (DD) or Darier-White disease, is a dominantly inherited genodermatosis that is characterized by hyperkeratotic papules in seborrheic regions and various nail abnormalities. The disease was first reported independently by Darier and White in 1889.
Etiology
Abnormal cell-cell adhesion and aberrant epidermal keratinization are the primary features of DD. Electron microscopy reveals loss of desmosomes, breakdown of desmosome-keratin intermediate filament attachment, and perinuclear aggregates of keratin intermediate filaments. These observations suggest that the molecules responsible for cellcell adhesion, such as desmosomal cadherins, desmosomal plaque proteins, or intermediate filament proteins, may be involved in the disease process. However, recently, mutations in the gene ATP2A2 (located in band 12q23-24.1) were found in patients with DD.
Clinical Features
Keratosis follicularis is usually manifested during childhood or adolescence and has an equal gender distribution. The cutaneous lesions appear as small, firm papules, which are red when they first appear, but characteristically become grayish brown or even purple, ulcerate and crust over (Fig. 19-14). Especially in the skin folds, the lesions tend to coalesce and produce verrucous or vegetating macerated, foul-smelling masses. They are generally distributed on the forehead, scalp, neck and over the shoulders, but often spread to the limbs, chest and genitalia. Palmar and plantar keratotic thickening may be so severe as to interfere with function. In severe cases, all the intertriginous areas are involved. Characteristic nail changes are also seen consisting of splintering, fissuring, longitudinal streaking and subungual keratosis.
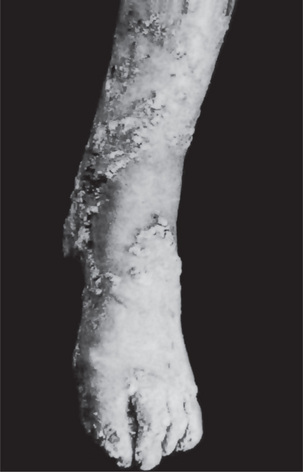
Oral Manifestations
The oral mucosa is probably more commonly involved than is generally realized, according to Gorlin and Chaudhry, who found a number of reports of oral lesions in the literature. In addition, they pointed out that other mucosal surfaces such as vulva, pharynx and larynx have also been reported as sites of the disease. Mucosal lesions have been said to be apparent only when there is extensive skin involvement. However, Weathers and Driscoll have stated that severe skin involvement is not necessary for the occurrence of oral lesions, although the severity of oral involvement did tend to parallel that of the skin. Nevertheless, oral involvement has been reported in as high as 50% of all cases.
The oral lesions appear as minute, whitish papules which feel rough upon palpation. Some cases have been described as rough, pebbly areas with verrucous white plaques or as having a cobblestone appearance as in the cases of Weather and his associates and Prindiville and Stern. These are most frequently found on the gingiva, tongue, hard and soft palates, buccal mucosa and even the pharynx (Fig. 19-15).
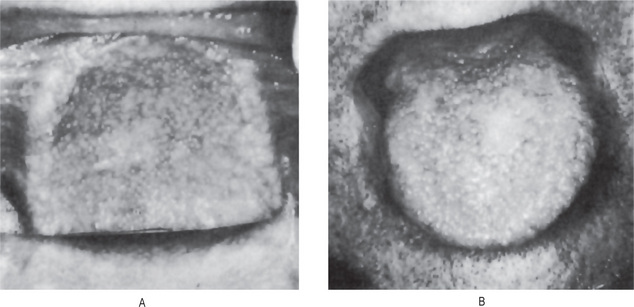
Histologic Features
The disease is misnamed, since the changes are not restricted to the hair follicles. The characteristic findings in skin lesions are hyperkeratosis, papillomatosis, acanthosis and a peculiar benign dyskeratosis. This benign dyskeratosis is characterized by rather typical cells called corps ronds and grains. The corps ronds are larger than normal squamous cells and have a round, homogeneous, basophilic nucleus with a dark eosinophilic cytoplasm and a distinct cell membrane. These are found usually in the granular layer and superficial spinous layer. The grains are small, elongated parakeratotic cells situated in the keratin layer. Both corps ronds and grains represent partially keratinized cells and are found also in the typical slit-like intradermal vesicle just above the basal layer of cells, the typical suprabasilar cleavage. Acantholytic cells are commonly found floating in the lacunae produced by this intraepithelial separation. The microscopic features of the oral lesions are identical except that the hyperkeratotic changes are generally not pronounced.
In an excellent review of keratosis follicularis, Spouge and his associates have also described a peculiar hyperplasia of the epithelial rests of Malassez in the periodontal ligament with a maturation of these individual cells into prickle cells, some of which even exhibited individual cell keratinization.
The cytologic findings in scrapings taken from the deeper portion of oral mucosal lesions have been described by Burlakow and his coworkers. They pointed out that, while some of the cells might be mistaken for malignant cells, the general cell population, the presence of grain cells and corps ronds and the ‘leafing-out’ pattern of the parabasal cells should permit the correct diagnosis from such cytologic smears.
It has been demonstrated by the electron microscope that the basic defect in epidermal synthesis, turnover and resultant keratinization is related to a defect in the desmosometonofilament complex. It is also reported that there is a sevenfold decrease in turnover time of this epithelium.
Treatment
Oral retinoids have been the most effective medical treatment for DD. However, long-term treatment with oral retinoids is needed in DD. Unfortunately, prolonged use of oral retinoids can cause significant adverse effects, and many patients have to stop taking them because of the toxicity. A few authors reported symptomatic and cosmetic improvement in DD by using surgical procedures. Overall, patients with DD have a life expectancy similar to that of the general population.
Warty Dyskeratoma: (Isolated dyskeratosis follicularis, isolated Darier's disease)
The warty dyskeratoma is a lesion which bears marked histologic similarity to keratosis follicularis but, in contrast to the latter, is usually a single isolated focus. The suggested origin of the lesion from the pilosebaceous apparatus would seem somewhat unlikely since oral lesions are known to occur.
Clinical Features
The skin lesions have occurred on the face, scalp or neck and upper chest in the majority of reported cases. They are almost invariably single lesions varying in size from only 1–10 mm in diameter. They appear as elevated nodules, somewhat umbilicated, with a raised border and varying in color from yellow or brown to gray or black. Purulent drainage as well as bleeding frequently occurs.
There were 80 males and 32 females in a group of 112 cases of this lesion reviewed by Tanay and Mehregan and the majority occurred in middle-age or in older adults. In nearly every case, careful examination will reveal a hair passing through the lesion.
Oral Manifestations
Oral lesions are rare but do occur, three cases having been reported by Tomich and Burkes and several solitary cases reviewed by Patibanda and by Danforth and Green. These lesions were described as small whitish areas of the mucosa with a central depression and were situated on the alveolar ridge and palate. The patients were aware of the presence of these lesions and in at least two of these cases discomfort was present.
Histologic Features
The microscopic findings in the skin and mucosal lesions are identical except for the absence of a pilosebaceous structure in the oral lesions. The intraoral lesions exhibit a central orthokeratin or parakeratin core beneath which the epithelium shows a suprabasilar separation resulting in a cleft-like space containing acantholytic and benign dyskeratotic cells. The connective tissue papillae are covered usually by a single layer of basal cells while the underlying connective tissue shows a nonspecific chronic inflammatory cell infiltrate.
The term focal acantholytic dyskeratosis was suggested by Ackerman in 1972 for a clinically heterogeneous group of dermatologic conditions all characterized by certain histologic features which they share in unison but with no implied common etiology or pathogenesis. The warty dyskeratoma is one of the groups fulfilling the criteria, and an intraoral case has been reported under the more inclusive generic term by Freedman and his associates.
Incontinentia Pigmenti: (Bloch-Sulzberger syndrome)
Incontinentia pigmenti (IP), sometimes termed Bloch-Sulzberger syndrome, is an X-linked dominant singlegene disorder with neurologic, ophthalmologic, and dental involvement, as well as cutaneous findings. Bloch and Sulzberger defined the condition as a clinical syndrome with a constellation of unique features, which include typical cutaneous lesions. Since then, a large number of small series and individual case reports have been published.
Etiology
The patchy distribution of the skin lesions is thought to be the result of tissue mosaicism due to random X-inactivation. Normal X chromosomes are active in unaffected skin, and mutated X chromosomes are active in skin affected with IP.
Clinical Features
The disease generally appears shortly after birth. More than 95% of reported cases occur in females, but the disease does occur in males and is characterized by the appearance of erythematous and vesiculobullous lesions on the trunk and extremities which frequently disappear, then reappear. This phase is often associated with a marked eosinophilia. These are gradually replaced by white keratotic, lichenoid, papillary or verrucous lesions which then persist for some months.
The third type of characteristic skin lesions in these infants are brownish-gray macules in a streaked, patchy distribution over the trunk and extremities, occurring subsequent to the verrucous, keratotic lesions. This pigmentation begins to fade within a few years. It is the heavy melanin pigmentation of the epithelium, dropping down into clusters of chromatophores in the upper dermis (incontinence), which gives the disease its name and is considered the hallmark of the syndrome.
A variety of associated defects are often seen in incontinentia pigmenti, including local or generalized baldness; ophthalmologic lesions including cataracts, optic atrophy, strabismus and retrolental fibroplasia; central nervous system involvement and lesions of the skeletal system.
Oral Manifestations
Oral changes in this disease have been described by Gorlin and Anderson and by Russell and Finn, among others, and appear limited to the teeth. Dental abnormalities are seen in 80% of patients. Both the deciduous and permanent dentitions may be affected. These dental changes have been described as consisting of delayed tooth eruption, peg or cone-shaped tooth crowns, congenitally missing teeth, malformed teeth and additional cusps. The cone-shaped teeth are very similar to those seen in hereditary ectodermal dysplasia.
Treatment
Treatment of the cutaneous lesions is usually not required. The vesicles of the inflammatory stage should be left intact, and the skin should be kept clean to prevent secondary bacterial infection. Oral hygiene and regular dental care is necessary, and dental restoration may be indicated. The prognosis of IP is generally good.
Porokeratosis of Mibelli
Porokeratosis of Mibelli is an uncommon genokeratosis characterized by faulty keratinization of the skin followed by atrophy. It appears to be inherited as a simple dominant characteristic, although many sporadic cases are known. There is no adequate documentation that the lesions of porokeratosis, despite the name of the disease, have their origin in the epidermal pores of sweat glands.
Clinical Features
The majority of cases begin in early childhood but the progression of the lesions is generally exceedingly slow. It appears to occur in males with greater frequency than in females. The lesions themselves consist initially of crateriform keratotic papules which gradually enlarge to form elevated plaques ranging in size from a few millimeters to several centimeters. The lesions have a predilection for the extremities, particularly the hands and feet, as well as the shoulders, face and neck, and the genitalia. The nails commonly become thickened and ridged. The central portion of the lesions ultimately becomes atrophic, leaving permanent scarring. Epidermoid carcinoma has been reported developing in this atrophic skin. Lesions of the oral cavity are said to occur with considerable frequency in patients with this disease.
Histologic Features
The elevated horny margin of the lesion exhibits hyperkeratosis and acanthosis with a deep groove filled with parakeratin and a characteristic absence of the usual underlying granular layer. This constitutes the ‘cornoid lamella’ which is characteristic of the lesion. The central portion of the lesion shows epithelial atrophy and occasionally dyskeratosis. The connective tissue beneath the cornoid lamella may exhibit a lymphocytic infiltrate.
Dyskeratosis Congenita: (Zinsser-Engman-Cole syndrome, Hoyeraal-Hreidarsson syndrome)
Dyskeratosis congenita (DKC) is a well recognized but rare genodermatosis characterized by cutaneous reticulated hyperpigmentation, nail dystrophy, premalignant leukoplakia of the oral mucosa, and progressive pancytopenia. The importance of the syndrome lies in the high incidence of oral cancer which develops in the young affected adults.
Etiology
Mutations in DKC1 have been shown to cause the X-linked form of DKC. The inheritance pattern of most cases of DKC is X-linked recessive, but autosomal dominant and recessive patterns have been reported.
Clinical Features
Dyskeratosis congenita is a rare syndrome, with approximately 180 individuals reported in the literature. Because this disorder is primarily X-linked recessive, the male-to-female ratio is approximately 10 : 1. The nail changes are usually the first manifestation of the disease, becoming dystrophic and shedding some time after the age of five years. The grayish-brown skin pigmentation appears at the same time or a few years later and is distributed over the trunk, neck, and thighs. The skin may become atrophic and telangiectatic and the face appears red.
Occasional cases have also been reported with a wide spectrum of other minor manifestations including a frail skeleton, mental retardation, small sella turcica, dysphagia, transparent tympanic membranes, deafness, epiphora and eyelid infections, urethral anomalies, small testis, dental abnormalities and, commonly, hyperhidrosis of the palm and soles.
Oral Manifestations
Mucosal leukoplakia typically occurs on the buccal mucosa and can affect the tongue and oropharynx. The leukoplakia may become verrucous, and ulceration may occur. Other mucosal sites may be involved (e.g. esophagus, urethral meatus, glans penis, lacrimal duct, conjunctiva, vagina, anus). Constriction and stenosis can occur, with the development of dysphagia, dysuria, phimosis, and epiphora. Patients have an increased incidence of malignant neoplasms, particularly squamous cell carcinoma of the skin, mouth, nasopharynx, esophagus, rectum, vagina, and cervix. These often occur within sites of leukoplakia. Patients also may have an increased incidence and severity of dental caries and tooth loss.
Histologic Findings
Skin biopsy specimens from the areas of reticulated pigmentation typically show mild hyperkeratosis, epidermal atrophy, telangiectasia of the superficial blood vessels, and melanophages in the papillary dermis. Interface changes have also been reported, with mild basal layer vacuolization and a lymphocytic inflammatory infiltrate in the upper dermis. Oral lesions have not been thoroughly studied but the leukoplakic lesions appear to be nonspecific hyperparakeratosis or hyperorthokeratosis and acanthosis. Depending on the stage of the disease, the epithelium may show dysplasia. The exact nature of the preceding vesicles and ulcers has not been described.
Laboratory Findings
Many cases have been characterized also by hematologic changes including anemia, leukopenia, thrombocytopenia and pancytopenia. Some patients have developed Fanconi's anemia. In fact, the suggestion has been made that Fanconi's syndrome, or Fanconi's familial pancytopenia, is simply a varied expression of dyskeratosis congenita.
Treatment
Short-term treatment options for bone marrow failure in patients with DKC include erythropoietin and granulocyte colony-stimulating factor; however, the only long-term, curative option is allogenic bone marrow transfer. The high frequency of malignant transformation of oral lesions would necessitate careful periodic examination of the patient for such an occurrence.
White Sponge Nevus: (Familial white folded dysplasia of mucous membrane, white folded gingivostomatitis, oral epithelial nevus, congenital leukokeratosis, Cannon's disease)
Familial white folded dysplasia is a relatively uncommon condition of the oral mucosa described by Cannon in 1935. The disease appears to follow a hereditary pattern as an autosomal dominant trait but with irregular penetrance and no definite sex predilection.
Clinical Features
This mucosal abnormality is congenital in many instances. In other cases it does not appear until infancy, childhood or even adolescence, by which time it has generally reached the full extent of its severity.
The oral lesions may be widespread, often involving the cheeks, palate, gingiva, floor of the mouth and portions of the tongue. The mucosa appears thickened and folded or corrugated with a soft or spongy texture and a peculiar white opalescent hue (Fig. 19-16). There is sometimes a minimal amount of folding present. Ragged white areas may also be present which can be removed sometimes by gentle rubbing without any ensuing bleeding (Fig. 19-17A, B). The lesions themselves are almost invariably asymptomatic. Banoczy and her associates have provided a detailed review and discussion in their report of 45 cases of this disease. In occasional cases reported in the literature, the oral lesions were accompanied by similar lesions of other mucosal surfaces, including the vagina and labia, anus, rectum and nasal cavity.
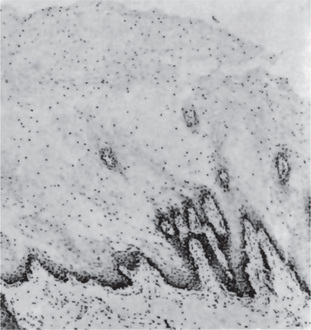
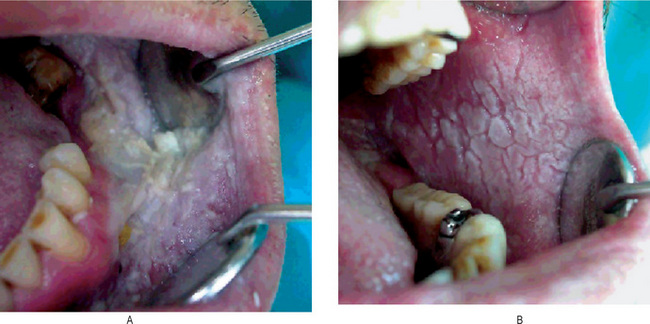
Histologic Features
The microscopic findings in familial white folded dysplasia are characteristic but not entirely pathognomonic of the disease. The epithelium is generally thickened, showing both hyperparakeratosis and acanthosis, and the basal layer is intact. The cells of the entire spinous layer, continuing to the very surface, exhibit intracellular edema (Fig. 19-16). These vacuolated cells may show pyknotic nuclei. In addition, parakeratin plugs running deep into the spinous layer are typically found. The submucosa may show a mild inflammatory cell infiltration, but this is not consistent.
Several electron microscopic investigations of the white sponge nevus have been reported, the first being that of Whitten, and subsequent studies, those of McGinnis and Turner and of Frithiof and Banoczy. There is some lack of agreement on interpretation of the ultrastructural findings in the various studies so that, until these are resolved, their discussion in explanation of the light microscopic findings should be deferred.
Hereditary Benign Intraepithelial Dyskeratosis
This unusual hereditary syndrome was discovered in 1954 in a racial isolate group of mixed Caucasian, Indian and Negro ancestry living in North Carolina. Since that time it has been thoroughly studied and described by Witkop and his coworkers. The disease appears superficially similar to familial white folded dysplasia or white sponge nevus in its hereditary pattern, although the clinical and microscopic features are different.
Clinical Features
The oral lesions of hereditary benign intraepithelial dyskeratosis appear generally as white, spongy, macerated lesions of the buccal mucosa, with or without folds (Fig. 19-18A). They are also described on the floor of the mouth, ventral and lateral surfaces of the tongue, the gingiva and palate. These lesions vary from delicate, opalescent white membranous areas to a rough, shaggy mucosa. Lesions frequently involve the corners of the mouth and appear as soft plaques with pinpoint elevation when the mucosa is stretched.
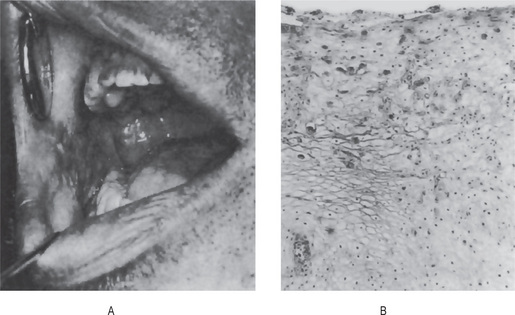
Figure 19-18 Hereditary benign intraepithelial dyskeratosis.
The white, macerated appearance of the lesions on the buccal mucosa is seen in (A). The peculiar ‘dyskeratotic’ cells are shown in (B) Courtesy of Dr Carl J Witkop.
Patients with this disease also manifest lesions of the eye characterized by superficial, foamy, gelatinous white plaques overlying the cornea, sometimes producing temporary blindness. In addition, the conjunctivae are usually intensely congested. Interestingly, these eye lesions in some cases show a seasonal variation, tending to appear or increase in severity in the spring and disappear, sometimes by spontaneous shedding of the pseudomembrane, in late summer or fall.
Histologic Features
Sections of the buccal mucosa exhibit thickening of the epithelium with pronounced hydropic degeneration. In addition, numerous round, waxy-appearing eosinophilic cells resembling minute epithelial pearls are evident, the ‘dyskeratotic’ cells (Fig. 19-18B). An excellent detailed description of the microscopic features of this disease has been provided by Witkop. Sadeghi and Witkop also have described ultrastructural differences between the mature dyskeratotic cells in this disease and in other dyskeratotic conditions of the mucous membranes.
Acanthosis Nigricans
Acanthosis nigricans (AN) is an unusual dermatosis, the first documented case was reported in 1889. Acanthosis nigricans is divided into two broad categories, benign and malignant. Patients with the benign form of acanthosis nigricans experience very few, if any, complications of their skin lesions. However, many of these patients have an underlying insulin-resistant state that is the cause of their disease. Malignant acanthosis nigricans is associated with significant complications because the underlying malignancy is often an aggressive tumor (e.g. adenocarcinomas of various internal organs, particularly the stomach or malignant lymphomas). The average survival time of patients with signs of malignant acanthosis nigricans is two years.
Etiology
The definitive cause for acanthosis nigricans has not yet been ascertained, although several possibilities have been suggested. Acanthosis nigricans is most likely caused by factors that stimulate epidermal keratinocyte and dermal fibroblast proliferation. In the benign form of acanthosis nigricans, the factor is probably insulin or an insulin-like growth factor (IGF) that stimulate the epidermal cells. In malignant acanthosis nigricans, the stimulating factor is hypothesized to be a substance secreted either by the tumor or in response to the tumor. Exogenous medications also have been implicated as etiologic factors.
Clinical Features
Acanthosis nigricans is much more common in people with darker skin. The incidence is equal in men and women. Lesions of benign acanthosis nigricans may be present at any age, including at birth, although it is found more commonly in the adult population. Malignant AN occurs more frequently in elderly persons; however, cases have been reported in children with Wilms tumor. The skin lesions in all forms of the disease are similar although the severity of the lesions and their distribution may vary from case to case. Generally, the skin lesions vary from a symmetric, mild hyperpigmentation and mild papillary hypertrophy of only small patchy areas to heavily pigmented, aggressively verrucous lesions involving much of the skin, especially the axillae, palms and soles, and face and neck (Fig. 19-19A). The verrucous lesions are often pedunculated. Generalized pruritus is also a common finding.
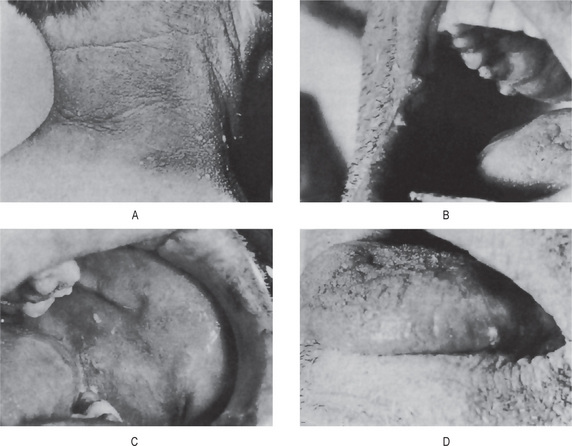
Oral Manifestations
Oral mucous membrane involvement has been reported in between 25 and 50% of all cases of acanthosis nigricans. The oral findings in the benign form have been described by Pindborg and Gorlin, and in the malignant form, by Bang. These oral findings in both forms appear essentially the same.
The tongue and lips appear to be most frequently involved and to the greatest degree. There is hypertrophy of the filiform papillae producing a shaggy, papillomatous surface to the dorsal tongue. The lips may be enlarged and covered by papillomatous growths, particularly at the angles of the mouth. The buccal mucosa is less frequently involved, but generally shows a velvety white appearance with occasional papillary lesions. Similar findings may be seen in other areas, including the palate (Fig. 19-19B, C, D). In addition, gingival enlargement has been reported, clinically resembling idiopathic fibromatosis.
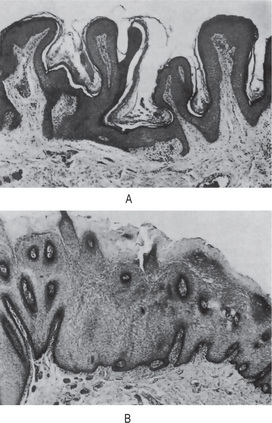
Histologic Findings
The histologic findings are characteristic but not pathognomonic. Histologic examination reveals hyperkeratosis, papillomatosis, and slight irregular acanthosis with minimal or no hyperpigmentation. The dermal papillae project upward as finger-like projections, with occasional thinning of the adjacent epidermis (Fig. 19-20A, B). Pseudohorn cysts may be present. Clinical discoloration is secondary to the hyperkeratosis and not to increased melanocytes or increased melanin deposition.
Pemphigus
Pemphigus is a serious chronic skin disease characterized by the appearance of vesicles and bullae, small or large fluid-filled blisters that develop in cycles. Pemphigus is derived from the Greek word pemphix meaning bubble or blister. Pemphigus describes a group of chronic bullous diseases, originally named by Wichman in 1791. The term pemphigus once included most bullous eruptions of the skin, but diagnostic tests have improved, and bullous diseases have been reclassified. Pemphigus includes a group of autoimmune blistering diseases of the skin and mucous membranes characterized histologically by intradermal blisters and immunologically by the finding of circulating immunoglobulin G (IgG) antibody directed against the cell surface of keratinocytes.
The three primary subsets of pemphigus include pemphigus vulgaris (PV), pemphigus foliaceus, and paraneoplastic pemphigus. Each type of pemphigus has distinct clinical and immunopathologic features. Pemphigus vulgaris accounts for approximately 70% of pemphigus cases.
Pemphigus Vulgaris
Pemphigus vulgaris (PV) is an autoimmune, intraepithelial, blistering disease affecting the skin and mucous membranes and is mediated by circulating autoantibodies directed against keratinocyte cell surfaces. Clinical and experimental observations indicate that the circulating autoantibodies are pathogenic. An immunogenetic predisposition is well established. Blisters in PV are associated with the binding of IgG autoantibodies to keratinocyte cell surface molecules. These intercellular or PV antibodies bind to keratinocyte desmosomes and to desmosome free areas of the keratinocyte cell membrane. The binding of autoantibodies results in a loss of cell-cell adhesion.
Intercellular adhesion in the epidermis involves several keratinocyte cell surface molecules. Pemphigus antibody binds to keratinocyte cell surface molecules desmoglein 1 and desmoglein 3. Patients with the active disease have circulating and tissue-bound autoantibodies of both the immunoglobulin G1 and G4 subclasses. Disease activity correlates with antibody titer in most patients. Pemphigus antibody fixes components of complement to the surface of epidermal cells. Antibody binding may activate complement with the release of inflammatory mediators and recruitment of activated T-cells.
Clinical Features
Pemphigus vulgaris affects all races, with an equal gender distribution among males and females. The mean age of onset is approximately 50–60 years; however, the range is broad, and disease onset in older individuals and in children has been described. Patients are younger at presentation in India than in western countries.
Pemphigus vulgaris is characterized by the rapid appearance of vesicles and bullae, varying in diameter from a few millimeters to several centimeters, in such numbers that large areas of the skin surface may be covered (Fig. 19-21). These lesions contain a thin, watery fluid shortly after development, but this may soon become purulent or sanguineous. When the bullae rupture, they leave a raw eroded surface identical with that seen when focal areas of epithelium slide off either under oblique pressure or spontaneously without the prior formation of a vesicle or bulla (Fig. 19-22A, B). The loss of epithelium occasioned by rubbing apparently unaffected skin is termed Nikolsky's sign. It is a characteristic feature of pemphigus and is caused by prevesicular edema which disrupts the dermal-epidermal junction. The course of pemphigus vulgaris is a variable one, the disease terminating in death or recovery within a few days or weeks, or being prolonged over a period of months or even years.
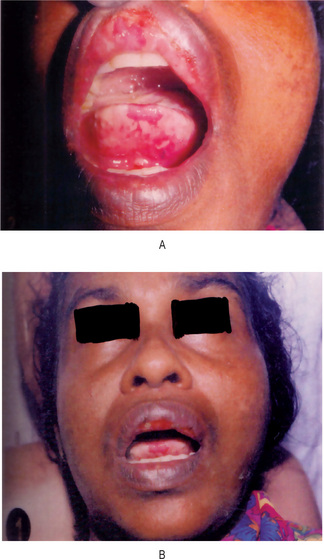
Pemphigus vegetans is an uncommon variant of pemphigus vulgaris. It occurs in 1–2% of pemphigus vulgaris cases. The median age of onset is 40–50 years. Two clinical subtypes of pemphigus vegetans exist, characterized initially by flaccid bullae and erosions (Neumann) or pustules (Hallopeau). Both subtypes subsequently develop into hyperpigmented vegetative plaques with pustules and hypertrophic granulation tissue at the periphery. Lesions are typically located at intertriginous areas and the oral mucosa. Oral involvement is present in nearly all pemphigus vegetans cases. A characteristic feature of pemphigus vegetans is the cerebriform tongue, characterized by a pattern of sulci and gyri on the dorsum of the tongue.
Oral Manifestations
Mucous membranes typically are affected first in PV. Mucosal lesions may precede cutaneous lesions by months. The disease involves mucosa in 50–70% of patients. Intact bullae are rare in the mouth. More commonly, patients have ill-defined, irregularly shaped, gingival, buccal or palatine erosions, which are painful and slow to heal (Fig. 19-23). The erosions extend peripherally with shedding of the epithelium. Erosions may be seen on any part of the oral cavity and can be scattered and often extensive. Erosions may spread to involve the larynx with subsequent hoarseness. The patient is often unable to eat or drink adequately because the lesions are so uncomfortable. Other mucosal surfaces may be involved, including the conjunctiva, esophagus, labia, vagina, cervix, penis, urethra, and anus. The oral lesions are similar to those occurring on the skin.
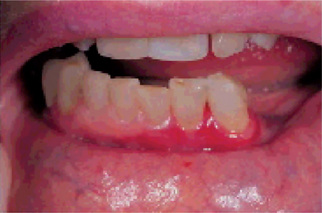
Histologic Features
Pemphigus as an entity is characterized microscopically by the formation of a vesicle or bulla entirely intraepithelially, just above the basal layer producing the distinctive suprabasilar ‘split’ (Fig. 19-24A, B). Prevesicular edema appears to weaken this junction, and the intercellular bridges between the epithelial cells disappear. This results in loss of cohesiveness or acantholysis, and because of this, clumps of epithelial cells are often found lying free within the vesicular space (Fig. 19-24A, B). These have been called ‘Tzanck cells’ and are characterized particularly by degenerative changes which include swelling of the nuclei and hyperchromatic staining. These changes are particularly obvious in cytologic smears taken from early, freshly opened vesicles (Fig. 19-25). Such smears form the basis for a rapid supplemental test for pemphigus, the ‘Tzanck test’, and these cytologic findings have been discussed in detail by Medak and his associates as well as by Shklar and Cataldo. Shklar has also reported that there is a marked increase in RNA in the cytoplasm of these acantholytic cells as well as in the epithelial cells at the floor of the vesicle.
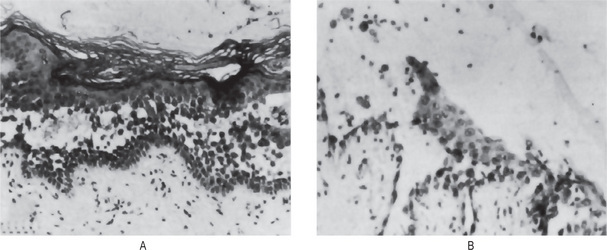
Figure 19-24 Pemphigus vulgaris.
(A) Early stage of vesiculation with suprabasilar cleavage accompanied by prominent acantholysis and numerous Tzanck cells. (B) In the bullous stage, acantholysis is still evident with occasional leukocytes in the bullous fluid Courtesy of Dr Frank Vellios and Dr Charles A Waldron
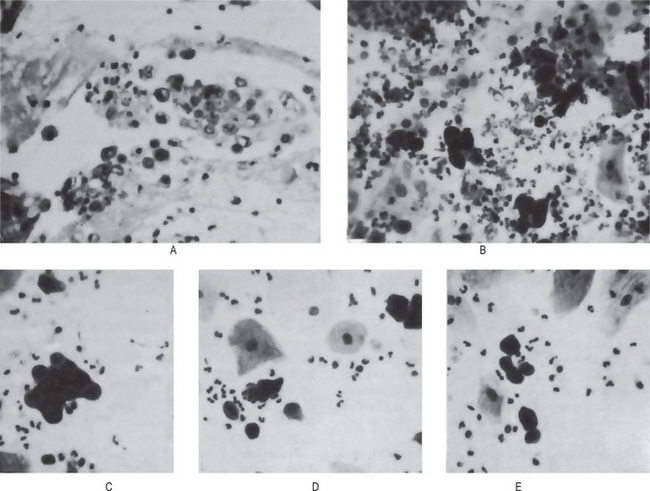
Figure 19-25 Pemphigus vulgaris.
The characteristic acantholytic cells are seen in the histologic section of the vesicle (A). These same cells are found on cytologic smear of the vesicle (B, C, D, E).
The fluid in most vesicles, particularly those more than a day or two old, contains variable numbers of polymorphonuclear leukocytes and lymphocytes. The relative scarcity of inflammatory cell infiltration; however, in both the vesicular fluid and in the connective tissue at the base of the vesicle or bulla, is suggestive of pemphigus, since most other bullous diseases manifest marked inflammation. Once secondary infection occurs, this feature is masked (Fig. 19-26).
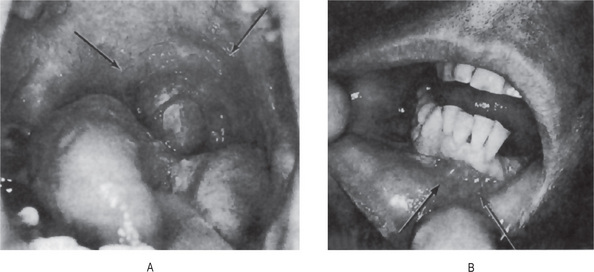
Figure 19-26 Pemphigus vulgaris.
Rupture of bullae of the oral mucous membranes results in large, denuded, painful lesions Courtesy of Dr Wilbur C Moorman.
Immunofluorescent testing has proven to be of great value in establishing the diagnosis of pemphigus, especially when the clinical or microscopic findings are inconclusive. In this test, direct immunofluorescence is used to demonstrate the presence of immunoglobulins, predominantly IgG but sometimes in combination with C3, IgA and IgM, in the intercellular spaces or intercellular substance in either the oral epithelium of the lesions or of clinically normal epithelium adjacent to the lesions. This test is carried out by incubating a biopsy specimen (either frozen section or one specially fixed in Michel solution) with a fluorescein-conjugated antiglobulin. Indirect immunofluorescence has also been used to substantiate the diagnosis of pemphigus. This is accomplished basically by incubating normal animal or human mucosa with serum from the patient suspected of having the disease and adding the fluorescein-conjugated human antiglobulin. A positive reaction in the tissue indicates the presence of circulating immunoglobulin antibodies. Daniels and Quadra-White have reported positive direct immunofluorescence for IgG in 10 biopsies of 10 patients (100%) with pemphigus vulgaris (Fig. 19-27). Six of these 10 were also positive for C3, while only one each was positive for IgA and IgM. Laskaris has also found, in testing a series of 58 patients with pemphigus vulgaris limited to the oral cavity, positive direct immunofluorescence in 57 (98%), demonstrating intercellular substance deposition of IgG, either alone or in combination with C3, IgA, and IgM. By indirect immunofluorescence, Laskaris also reported that, when normal human oral mucosa was used as the substrate, circulating intercellular substance antibodies were present in 50 of the 58 patients (86%).
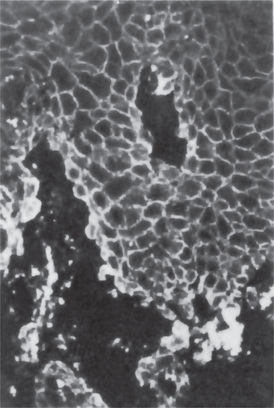
Figure 19-27 Pemphigus vulgaris.
Oral specimen showing intercellular space fluorescence throughout the epithelium with anti-IgG. At lower left there is an intraepithelial vesicle above the basal epithelial cells attached to the basement membrane zone Courtesy of Dr Troy E Daniels and CV Mosby Company. From TE Daniels, and C Quadra-White. Direct immunofluorescence in oral mucosal disease: a diagnostic analysis of 130 cases. Oral Surg, 51: 38, 1981.
Differential Diagnosis
Oral lesions constitute an important feature of pemphigus, and for this reason pemphigus must always be considered in the differential diagnosis of blisterlike eruptions of oral mucous membranes. Some difficulty may be experienced in differentiating pemphigus from other bullous diseases such as dermatitis herpetiformis, erythema multiforme bullosum, bullous lichen planus, epidermolysis bullosa and other chronic bullous dermatoses such as bullous pemphigoid and cicatricial pemphigoid. Clinical experience, however, aided by the histologic appearance of the lesions, usually suffices to separate the diseases.
Treatment
The aim of treatment in PV is the same as in other autoimmune bullous diseases, which is to decrease blister formation, promote healing of blisters and erosions, and determine the minimal dose of medication necessary to control the disease process. Therapy must be tailored for each patient, taking into account pre-existing and coexisting conditions. Patients may continue to experience mild disease activity while under optimal treatment.
Pemphigus Foliaceus: (Superficial pemphigus, fogo selvagem)
Pemphigus foliaceus (PF) is generally a benign variety of pemphigus. It is an autoimmune skin disorder characterized by the loss of intercellular adhesion of keratinocytes in the upper parts of the epidermis (acantholysis), resulting in the formation of superficial blisters. It is typified by clinical involvement of healthy-appearing skin that blisters when rubbed (the Nikolsky sign). Pemphigus foliaceus is characterized by a chronic course, with little or no involvement of the mucous membranes. It includes the following six subtypes: pemphigus erythematosus (PE), pemphigus herpetiformis (PH), endemic pemphigus foliaceus, immunoglobulin A (IgA) pemphigus foliaceus, paraneoplastic pemphigus foliaceus (PNPF), and drug-induced pemphigus foliaceus.
Superficial blisters in PF are induced by immunoglobulin G (IgG) (mainly IgG4 subclass) autoantibodies directed against a cell adhesion molecule, desmoglein 1, expressed mainly in the granular layer of the epidermis. Precipitating factors include medications and ultraviolet light radiation. It was recently suggested that both factors enhanced autoantibody epidermal binding and preferential neutrophil adhesion to UV-irradiated epidermis which contribute to acantholysis in photo-induced PF.
Clinical Features
Pemphigus foliaceus is manifested by characteristic early bullous lesions which rapidly rupture and dry to leave masses of flakes or scales suggestive of an exfoliative dermatitis or eczema. The disease may originate in this form or may develop from one of the other types of pemphigus. It is a relatively mild form of pemphigus, which is most common in older adults but may occur in young children as well.
Brazilian pemphigus (fogo selvagem or Brazilian wildfire) is a mild endemic form of pemphigus foliaceus found in tropical regions, particularly in Brazil, that often occurs in children and frequently in family groups. The course of the disease is similar to that of pemphigus foliaceus. Oral lesions in pemphigus foliaceus are rare, according to Perry and Brunsting (1965) in their extensive study of this form of the disease.
Histologic Findings
Pemphigus foliaceus begins as acantholysis of the upper epidermis. It usually enlarges and detaches without bullae formation, though a bulla may form showing acantholysis at both the roof and the floor. More established lesions may have acanthosis and mild-to-moderate papillomatosis. Hyperkeratosis and parakeratosis may also be evident, with dyskeratotic cells within the granular layer. These features may be particularly pronounced in long-standing pemphigus erythematosus. A mild dermal lymphocytic infiltrate occurs, often with the presence of eosinophils.
Treatment
Therapy for PF is usually less aggressive than that for pemphigus vulgaris because of their lower morbidity and mortality rates. Few results indicate that nonsteroidal treatment of pemphigus is possible. Mestinon may be used to slow down progression of the disease and to treat mild cases with chronic lesions on limited areas.
Paraneoplastic Pemphigus
Anhalt et al, first described paraneoplastic pemphigus in 1990. The authors reported five patients with underlying neoplasms who developed oral erosions and bullous skin eruptions. Since then, more than 60 patients with paraneoplastic pemphigus have been reported, and patients previously believed to have other diseases have been retrospectively diagnosed. Skin biopsy samples showed both suprabasal acantholysis and interface dermatitis. Immunofluorescence tests revealed intraepidermal intercellular staining with immunoglobulin G (IgG).
A summary of criteria for the diagnosis of paraneoplastic pemphigus includes the following:
• Painful mucosal erosions, sometimes with a skin eruption that eventually results in blisters and erosions, in the setting of confirmed or occult malignancy.
• Histopathologic changes of acantholysis, keratinocyte necrosis, and interface dermatitis.
• Direct immunofluorescence (DIF) typically reveals IgG and complement (C3) within the epidermal intercellular spaces as well as at the epidermal basement membrane.
• Indirect immunofluorescence (IDIF) observation of circulating antibodies specific for stratified squamous or transitional epithelia (transitional epithelium) is found.
• Immunoprecipitation of a complex of proteins with typical molecular weights.
Tumor antigens are hypothesized to evoke an immune response that leads to the development of an autoimmune response to intercellular adhesins (plakins). This autoantibody response leads to blistering in mucosa and other epithelia. Paraneoplastic pemphigus is often fatal. Mortality rates approach 90%. Causes of death include sepsis, with resultant multiorgan failure and respiratory failure due to the direct effects of the disease on the respiratory epithelium. The susceptibility to infection caused by the loss of skin integrity is exacerbated by the potent immunosuppressive medications used to treat the condition.
Clinical Features
The mean age at onset is 60 years. Patients ranging in age from 7 to 76 years have been reported. Males and females are affected equally. With the exception of a few patients, all patients with paraneoplastic pemphigus have had tumors, most of which have been malignant. The most common malignancy associated with paraneoplastic pemphigus is non-Hodgkin lymphoma. Other associated malignancies include chronic lymphocytic leukemia, Castleman tumor, giant cell lymphoma (reticulum cell sarcoma), Waldenström macroglobulinemia, thymoma, poorly differentiated sarcoma, bronchogenic squamous cell carcinoma, and follicular dendritic cell sarcoma.
Oral Manifestations
Most patients with paraneoplastic pemphigus present with oral erosions or ulcerations. Patients present with painful oral erosions, often accompanied by a generalized cutaneous eruption. The eruption can assume a wide variety of morphologies, including morbilliform, urticarial, bullous, papulosquamous, or erythema multiformelike lesions. Some patients complain of pruritus or pain. The erosions can occur anywhere in the mouth, including the buccal, the labial, the gingival, and the lingual mucosa. Erosions and subsequent crusting on the vermilion of the lips are typical and similar to that seen in Stevens-Johnson syndrome. The nose, the pharynx, and the tonsils can be affected, as can the genital mucosal surfaces. Nasal ulcers may cause epistaxis.
Histologic Findings
Vesicular lesions express the most characteristic histopathologic features. Oral and cutaneous lesions show variable epidermal necrosis, suprabasal acantholysis, dyskeratotic keratinocytes, vacuolar interface dermatitis, and lymphocytic infiltration. Oral mucosal lesions show the greatest acantholysis, while some skin lesions may not have any acantholysis at all. A distinctive feature of paraneoplastic pemphigus is dyskeratosis. Dyskeratosis is a constant feature, but the number of dyskeratotic keratinocytes is variable. Dyskeratotic keratinocytes are found at all levels in the epidermis, especially within the zones of acantholysis, and they can be found in cutaneous adnexa. The presence of dyskeratosis in a suprabasal acantholytic bullous disorder is a clue to the presence of paraneoplastic pemphigus.
Treatment
Initial care is aimed at treating superinfection, if present. Warm compresses, nonadherent wound dressings, and topical antibiotic ointments are helpful. Potent immunosuppressive agents are required to decrease blistering, but they are often ineffective. In general, skin lesions are more responsive to therapy than mucosal lesions. Other therapeutic options include plasmapheresis and immunopheresis. For solid neoplasms, curative resection should be attempted when appropriate. The prognosis of paraneoplastic pemphigus is poor.
Familial Benign Pemphigus: (Hailey-Hailey disease)
Familial benign pemphigus was originally described by Hailey brothers in 1939. It is a chronic autosomal dominant disorder with incomplete penetrance. Approximately two-thirds of patients have a family history of the disorder. A history of multiple relapses and remissions is characteristic. Many hypotheses exist concerning the pathogenesis of familial benign pemphigus. It is hypothesized to result from a genetic defect in a calcium pump protein. The pump mutation is in ATP2C1, a gene localized on chromosome 3. This gene defect is similar to the genetic defect in Darier disease, which also is a calcium pump defect. In addition to the primary gene defect in HaileyHailey disease, contributing factors like heat, friction, and infection are known to exacerbate the disease.
Clinical Features
The disease first manifests itself during adolescence or young adult life, although there are occasional exceptions. There is no apparent predilection for occurrence in either gender. The lesions themselves develop as small groups of vesicles appearing on normal or erythematous skin, which soon rupture to leave eroded, crusted areas. These lesions then appear to enlarge peripherally but heal in the center. Nikolsky's sign is present. It has been frequently noted that heat and sweating amplify the outbreak of the lesions while spontaneous remissions may occur in cold weather. The lesions themselves develop most commonly on those areas of skin which are exposed to friction, e.g. flexure surfaces of the axillae and groin, the neck and the genital area. Tender and enlarged regional lymph nodes may also be present.
It has been recognized that bacterial infection also appears to precipitate the appearance of lesions, and more recently, infection by Candida albicans has been implicated.
Oral Manifestations
Oral lesions occasionally occur in patients with familial benign chronic pemphigus, and these are similar to those occurring on the skin. The lesions develop as crops of vesicles which rapidly rupture leaving raw eroded areas.
Histologic Features
The histologic appearance of the epithelial lesions in familial benign chronic pemphigus bears remarkable similarity to that seen in pemphigus vulgaris and in keratosis follicularis or Darier's disease. However, in familial benign chronic pemphigus there is generally more extensive acantholysis than in pemphigus vulgaris and there is usually less damage to the acantholytic cells. One of the characteristic features of this disease is that occasional intercellular bridges persist so that adjacent epithelial cells still adhere to each other and are not entirely acantholytic.
This appearance has been given the classic description of the dilapidated brick wall effect. Finally, benign dyskeratotic cells similar to the corps ronds of Darier's disease may be present.
Treatment
Familial benign pemphigus waxes and wanes in intensity. Soothing compresses (aluminum acetate) followed by intermittent use of mild corticosteroid preparations and topical antibiotics (clindamycin or erythromycin) result in transient improvement. More widespread flares may require systemic antibiotics to suppress protease activation and acantholysis. Erythromycin and tetracycline are favored. Bacterial culture and sensitivity can help guide appropriate therapy.
Cicatricial Pemphigoid: (Benign mucous membrane pemphigoid, ocular pemphigus)
Cicatricial pemphigoid (CP) is an autoimmune blistering disease that predominately affects the mucous membranes, including the mouth and the oropharynx, the conjunctiva, the nares, and the genitalia. Patients with cutaneous involvement present with tense blisters and erosions, often on the head and the neck or at sites of trauma. Blisters heal with scarring and pigmentation. Sequelae of mucosal involvement include decreased vision, blindness, and supraglottic stenosis with hoarseness or airway obstruction.
Etiology
Cicatricial pemphigoid is an autoimmune blistering disease associated with autoantibodies directed against basement membrane zone target antigens. Autoantibodies of the IgG subclass, particularly IgG4, are associated with CP; however, IgA antibodies have also been detected. The two major antigens associated with CP are bullous pemphigoid antigen 2(BPAG2) and epiligrin (laminin-5).
Clinical Features
The disease occurs nearly twice as frequently in females as it does in males with the peak age of involvement being between 40 and 50 years. Typically, the vesiculobullous lesions occur on the oral mucous membranes and conjunctiva. Lesions also occur on the skin, particularly around the genitalia and near the body orifices in about 25% of cases. Typically, these lesions heal by scar formation, particularly on the conjunctiva. Other mucous membrane surfaces may be involved such as the nose, larynx, pharynx, esophagus, vulva, vagina, penis and anus.
The ocular involvement is probably the most serious complication of this disease. Following the initial conjunctivitis, adhesions develop between the palpebral and bulbar conjunctivae resulting in obliteration of the palpebral fissure, with opacity of the cornea frequently leading to complete blindness.
Oral Manifestations
The most consistent oral lesions to occur are those involving the gingivae, although ultimately other sites in the oral cavity become involved. The mucosal lesions are also vesiculobullous in nature but appear to be relatively thick-walled, and for this reason, may persist for 24–48 hours before rupturing and desquamating. Eventually their rupture does occur leaving a raw, eroded, bleeding surface. The gingivae frequently manifest a persistent erythema for weeks or even months after the original erosions have healed (Fig. 19-28A). These oral lesions rarely scar. In the past, this disease has often been diagnosed as ‘chronic desquamative gingivitis,’ a term now used only in the descriptive sense and not as a specific disease entity.
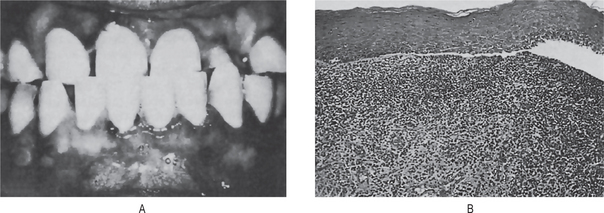
Histologic Features
The histologic findings in cicatricial pemphigoid, in contrast to pemphigus vulgaris, are nonspecific. The vesicles and bullae are subepidermal rather than suprabasilar and there is no evidence of acantholysis (Fig. 19-28B). The basement membrane structure appears to detach with the epithelium from the underlying connective tissue, as shown in the electron microscopic studies of Susi and Shklar. There is a nonspecific chronic inflammatory infiltrate in the connective tissue, chiefly lymphocytes, plasma cells and eosinophils.
Immunofluorescence studies have revealed the presence of tissue-bound basement membrane zone antibodies in most patients with this disease as well as circulating antibasement membrane zone antibodies in the serum of some patients. Daniels and Quadra-White reported the results of direct immunofluorescence examinations in 33 patients with cicatricial pemphigoid. Of these, they found that 17 (51%) showed a linear basement membrane zone pattern of IgG, 10 (33%) IgA, 12 (36%) IgM, and 32 (97%) C3 (Fig. 19-29). Laskaris and Angelopoulos also reported both direct and indirect immunofluorescent studies on 33 patients with cicatricial pemphigoid and found 32 of the 33 (97%) had a direct total positive basement membrane zone pattern (IgG 32/33; IgA 9/33; IgM 4/33; C3 26/33; fibrin 13/33), while only 12 of the 33 (36%) had an indirect positive using the patients' serum and a substrate of normal human oral mucosa (IgG only; all others negative). These authors pointed out that the immunofluorescence pattern of cicatricial pemphigoid was indistinguishable from the pattern observed in bullous pemphigoid and that this gave support to the possibility that the two diseases may represent variants of the same entity.
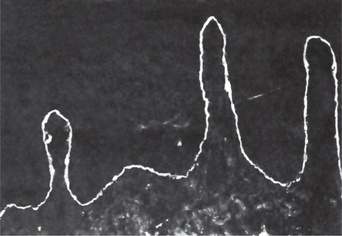
Figure 19-29 Cicatricial pemphigoid.
Oral specimen showing continuous linear fluorescence in the basement membrane zone with anti-C'3 Courtesy of Dr Troy E Daniels and CV Mosby Company. From TE Daniels, and C Quadra-White. Direct immunofluorescence in oral mucosal disease: a diagnostic analysis of 130 cases. Oral Surg, 51: 38, 1981.
Differential Diagnosis
Because of the nonspecific microscopic findings on biopsy, a variety of other diseases must be considered in the differential diagnosis. The chief of these are pemphigus vulgaris, bullous pemphigoid, erosive lichen planus, and bullous erythema multiforme.
Treatment
The goal of treatment is to suppress extensive blister formation, to promote healing, and to prevent scarring. The lowest dose of medication to suppress disease activity and to minimize the risk for the patient should be used. This disorder is extremely difficult to treat. Even with optimum control, blisters may continue to develop in some patients. The risks and the benefits of therapy must always be evaluated for each patient. Complications of CP include visual loss or blindness, airway stenosis, esophageal stricture, or cutaneous blistering with scarring and milia formation.
Bullous Pemphigoid (Parapemphigus)
Bullous pemphigoid (BP) is a chronic, autoimmune, subepidermal, blistering skin disease that rarely involves mucous membranes. It is characterized by the presence of immunoglobulin G (IgG) autoantibodies specific for the hemidesmosomal bullous pemphigoid antigens BP230 (BPAg1) and BP180 (BPAg2). IgG autoantibodies bind to the skin basement membrane and activate complement and inflammatory mediators. Activation of the complement system is thought to play a critical role in attracting inflammatory cells to the basement membrane. These inflammatory cells in turn are postulated to release proteases, which degrade hemidesmosomal proteins and lead to blister formation. Eosinophils are characteristically present, although their presence is not an absolute diagnostic criteria.
Clinical Features
Bullous pemphigoid is basically a disease of elderly persons, approximately 80% of patients being over 60 years of age. Nevertheless, it can occur earlier in life. There appears to be no gender predilection.
The cutaneous lesions begin as a generalized nonspecific rash, commonly on the limbs, which appears urticarial or eczematous and which may persist for several weeks to several months before the ultimate appearance of the vesiculobullous lesions. The vesicles and bullae arise in these prodromal skin lesions as well as in normal skin. In addition to the occurrence on the limbs, the abdomen is frequently affected. These vesicles and bullae are relatively thick-walled and may remain intact for some days. Rupture does not always occur, although when it does it leaves a raw, eroded area which heals rapidly.
Oral Manifestations
Oral lesions occur far less frequently in bullous pemphigoid than in cicatricial pemphigoid, varying from approximately 10–45% in various reported series. These oral lesions of bullous pemphigoid have been reviewed by Shklar and his associates and are usually described as vesicles and areas of erosion and ulceration. An important feature of the oral involvement is the similarity of gingival lesions to those of cicatricial pemphigoid. This gingival involvement generally involves much, if not all, of the gingival mucosa and is exceedingly painful. The gingival tissues appear extremely erythematous and may desquamate as the result of even minor frictional trauma. The vesicles and ultimate erosions may develop not only on the gingival tissues but in any other area such as the buccal mucosa, palate, floor of the mouth the tongue.
Histologic Features
The vesicles and bullae in this disease are subepidermal and nonspecific. There is no evidence of acantholysis of epithelial cells; in fact, the epithelium appears relatively normal. The vesicles contain a fibrinous exudate admixed with occasional inflammatory cells.
Electron microscopic studies have shown that, in contrast to cicatricial pemphigoid, the basement membrane remains attached to the connective tissue rather than to the overlying separated epithelium. In addition, these ultrastructural studies have shown that the primary changes in bullous pemphigoid appear to occur in the connective tissue where the blood vessels show alterations in their penetrability. The basement membrane also shows thickening, with interruption of continuity.
Bullous pemphigoid is thought to be an autoimmune disease and circulating basement membrane zone antibodies have been found by the indirect immunofluorescence technique in about 80% of patients with this disease, according to the study of 29 patients reported by Laskaris and Nicolis. Direct immunofluorescent studies of these same patients revealed tissue-bound antibasement membrane zone antibodies of the IgG class in all oral mucosal biopsies (100%) of patients who had both mucosal and skin lesions (14 patients), but in only 80% of oral mucosal biopsies in the group of patients who had only skin lesions (15 patients).
Treatment
The goal of therapy is to decrease blister formation, to promote healing of blisters and erosions. Therapy must be individualized for each patient, keeping in mind preexisting conditions and other patient-specific factors. Most patients affected with BP require therapy for 6–60 months, after which many patients experience long-term remission of the disease. However, some patients have long-standing disease requiring treatment for years.
Epidermolysis Bullosa
Epidermolysis bullosa (EB) is a group of inherited bullous disorders characterized by blister formation in response to mechanical trauma. Historically, epidermolysis bullosa subtypes have been classified according to skin manifestations. Recent discoveries of the molecular basis of EB have resulted in the development of new diagnostic tools, including prenatal testing.
Epidermolysis bullosa is classified into three major categories including:
• Epidermolysis bullosa simplex (EBS) (intraepidermal skin separation)
• Junctional epidermolysis bullosa (skin separation in lamina lucida or central basement membrane zone)
• Dystrophic epidermolysis bullosa (sublamina densa basement membrane zone separation).
Researchers have recently proposed a new category termed hemidesmosomal epidermolysis bullosa (HEB), which produces blistering at the hemidesmosomal level in the most superior aspect of the basement membrane zone. EBS is usually associated with little or no extracutaneous involvement, while the more severe hemidesmosomal, junctional, and dystrophic forms of EB may produce significant multiorgan system involvement.
Etiology
Most cases of epidermolysis bullosa simplex (EBS) are associated with mutations of the genes coding for keratins 5 and 14. The level of skin separation is at the mid-basal cell associated with variable intermediate filament clumping. Junctional epidermolysis bullosa (JEB) has a highly variable molecular etiology and represents a collection of different diseases. These diseases all cause blistering in the lamina lucida and variable hemidesmosomal abnormalities. More than one half of JEB cases are caused by one of two recurrent nonsense mutations in the LAMB3 gene, which is helpful for mutation analysis and prenatal testing. Dystrophic epidermolysis bullosa (DEB) thus far has been associated in all cases with mutations of the gene coding for type VII collagen (COL7A1). Anchoring fibrils are affected in patients with DEB, and the degree of involvement ranges from subtle changes to complete absence.
Epidermolysis Bullosa Simplex
Clinical Features
The generalized form of epidermolysis bullosa simplex is inherited as an autosomal dominant characteristic, manifests itself at birth or shortly thereafter and is characterized by the formation of vesicles and bullae, chiefly on the hands and feet at sites of friction or trauma. The knees, elbows and trunk are only rarely involved and the nails are only occasionally affected. When the blisters heal, usually within 2–10 days, it is an important feature that there is no resultant scarring or permanent pigmentation. The disease appears to improve at puberty and prognosis is good for a normal life span.
The localized form of the disease (Weber-Cockayne syndrome), which is also familial may occur early in childhood or later in life and is commonly recurrent. The bullae only develop on the hands and feet, are related to frictional trauma and tend to exacerbate in hot weather. There is no scarring upon healing.
Oral Manifestations
Bullae of the oral cavity have been reported in occasional cases of generalized epidermolysis bullosa simplex, but it is doubtful that they actually occur. In addition, the teeth are not affected.
Histologic Features
In the generalized form of epidermolysis bullosa simplex, the vesicles and bullae develop as a result of destruction of basal and suprabasal cells so that some nuclei may persist on the floor of the blister, according to Lowe. The individual cells become edematous and show dissolution of organelles and tonofibrils with displacement of the nucleus to the upper end of the cell. The PAS (periodic acid-Schiff)-positive basement membrane remains on the dermal side of the separation. The elastic, pre-elastic and oxytalan fibers in the connective tissue are normal.
In the localized form of the disease, the bullae are intraepidermal and suprabasal in location.
Junctional Epidermolysis Bullosa
It was earlier suggested by some workers that the junctional or lethal type is simply an extremely severe from of the dystrophic recessive form which is incompatible with prolonged survival. However, recent studies have proven that the two are distinctly different disorders.
Clinical Features
Three criteria have been established for the diagnosis of this form of the disease. These are:
The bullae are similar to those seen in the dystrophic recessive type except that they commonly develop spontaneously, and sheets of skin may actually be shed.
Oral Manifestations
Oral bullae are frequently very extensive, and because of their extreme fragility, produce serious feeding problems. Similar lesions also occur in the upper respiratory tract, the bronchioles, and the esophagus.
Severe disturbances in enamel and dentin formation of the deciduous teeth also occur but this is of only academic interest. These have been described by Arwill and his associates and by Gardner and Hudson in significant detail.
Epidermolysis Bullosa Dystrophic, Dominant
Clinical Features
This form of the disease may have its onset in infancy or it may be delayed until puberty. The blisters commonly develop on the ankles, knees, elbows, feet and head; healing results in scarring which is sometimes keloidal in type.
In the majority of cases, the nails are thick and dystrophic, and milia are commonly present. However, the eye is never involved. Palmar-plantar keratoderma with hyperhidrosis also may occur as well as ichthyosis and sometimes hypertrichosis.
Oral Manifestations
Bullae of the oral cavity have been described as occurring in about 20% of cases of this type, and Andreasen has described oral milia. The teeth are unaffected.
Histologic Features
The bullae in this form of the disease develop as a result of separation through the very thin, irregular PAS-positive basement membrane which becomes divided. The basal layer appears normal although flattened on the roof of the blister. The underlying connective tissue shows an absence of elastic and oxytalan fibers.
Epidermolysis Bullosa Dystrophic, Recessive
Clinical Features
This type of epidermolysis bullosa (EB) is the best known and classic form of the disease. It has its onset at birth or very shortly thereafter and is characterized by the formation of bullae spontaneously or at sites of trauma, friction or pressure (Fig. 19-30A). The typical sites of involvement are the feet, buttocks, scapulae, elbows, fingers and occiput. The bullae contain a clear, bacteriologically sterile or sometimes blood-tinged fluid. When these bullae rupture or are peeled off under trauma or pressure, they leave a raw, painful surface. These patients frequently have a positive Nikolsky's sign. The bullae heal by scar, milia and pigmentation (Fig. 19-30B). This scarring may result in afunctional club-like fists. The hair may be sparse while the nails are usually dystrophic or absent.
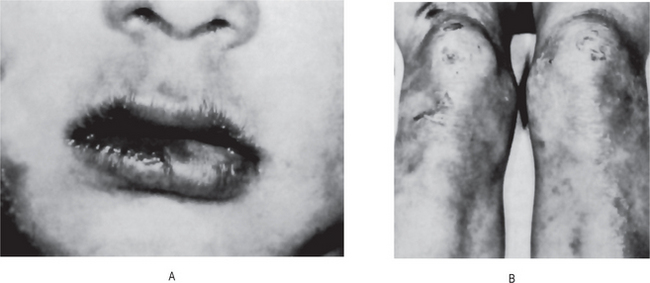
Oral Manifestations
Oral bullae are common in this form of the disease. They may be preceded by the appearance of white spots or patches on the oral mucous membrane or the development of localized areas of inflammation. The bullae may be initiated by nursing or by any simple dental operative procedure in the oral cavity. Unless great caution is used, large areas of mucous membrane may be inadvertently denuded. These bullae are painful, especially when they rupture or when the epithelium desquamates. Scar formation often results in obliteration of sulci and restriction of the tongue. Hoarseness and dysphagia may occur as a result of bullae of the larynx and pharynx. Esophageal involvement may produce serious stricture.
Dental defects have also been described, consisting of rudimentary teeth, congenitally absent teeth, hypoplastic teeth and crowns denuded of enamel. These have been discussed in detail by Arwill and his associates.
Histologic Features
The separation and bulla formation here occur immediately beneath the poorly defined PASpositive basement membrane which remains attached to the roof of the blister. Fragments of the basement membrane may adhere to the dermis, however. The basal layer of cells is normal. The pre-elastic and oxytalan fibers in the connective tissue are increased in number. Elastic fibers are also increased but appear fragmented, according to Lowe.
Treatment
This group of diseases cannot be cured so that therapy is chiefly symptomatic. The simplex form of the disease requires little treatment; the lethal form will terminate fatally in most cases regardless of management. In the dystrophic forms, prevention of trauma may reduce the incidence of bulla formation, but this is almost impossible to achieve. Antibiotics are useful in controlling secondary infection and corticosteroids have sometimes been found effective. EB is a lifelong disease. Some subtypes, especially the milder EB forms, improve with age.
Dermatitis Herpetiformis (Duhring-Brocq disease)
Dermatitis herpetiformis (DH) is a rare, benign, chronic, recurrent, immune-mediated blistering dermatologic disease with an associated, most often asymptomatic, gluten-sensitive enteropathy (GSE). Characteristic skin lesions found in patients with dermatitis herpetiformis are extremely itchy grouped vesicles most frequently located on extensor surfaces.
The pathogenesis of DH is associated with the presence of GSE; an increased expression of human leukocyte antigen1 (HLA-A1), human leukocyte antigen B8 (HLA-B8), human leukocyte antigen DR3 (HLA-DR3), and human leukocyte antigen DQ2 (HLA-DQ2) haplotypes; and granular deposition of IgA at the dermal-epidermal junction of the skin.
Clinical Features
The disease occurs chiefly between 20 and 55 years of age, although children are occasionally involved. A slight male predilection has been reported. The first manifestations of the disease are usually pruritus and severe burning, followed by the development of erythematous papules, vesicles, bullae or pustules. These occur most frequently on the extremities, trunk and buttocks as well as on the face, scalp and sometimes oral cavity. The vesicle is the most common and characteristic lesion, usually occurring symmetrically and in groups. Pigmentation of involved areas of skin ultimately develops in most cases. Patients frequently show increased severity of the disease in summer months.
Oral Manifestations
Vesicles and bullae which rupture rapidly to leave areas of superficial ulceration at any intraoral site are the characteristic finding. These have been described by Russotto and Ship.
Histologic Features
The lesions begin by accumulation of neutrophils and eosinophils in the dermal papillae producing a microabscess. The connective tissue becomes necrotic and the overlying epithelium separates, usually forming a subepithelial vesicle with destruction of basement membrane. The presence of eosinophils is generally prominent and characteristic, aiding in the differential diagnosis by excluding epidermolysis bullosa, erythema multiforme and pemphigus.
Direct immunofluorescent staining of uninvolved paralesional skin has been shown by Katz to be positive at the epidermal dermal junction. Almost invariably, IgA alone or in combination with the immunoglobulins IgG or IgM will be found in the upper dermis. When IgA deposits occur at the dermal-epidermal junction in a linear pattern (about 14% of cases) rather than as granules, the variant has been termed linear IgA disease. This has been discussed by Wiesenfeld and his colleagues, who also described the oral manifestations as being similar to those in the usual form of dermatitis herpetiformis.
Laboratory Findings
Some patients develop a blood eosinophilia of over 10%. Interestingly, these patients also show a sensitivity to the halogens (chlorine, bromine, iodine and fluorine) both by patch test and after ingestion.
Treatment
Skin lesions of dermatitis herpetiformis can be treated with dapsone, with relief of symptoms within 24–48 hours of the start of therapy. Alternatively, many patients can control the skin disease with a gluten-free diet, often without medication. Prognosis is good for patients who can tolerate dapsone and for the few who maintain a gluten-free diet.
Acrodermatitis Enteropathica: (Zinc deficiency)
Acrodermatitis enteropathica (AE) is an autosomal recessive disorder characterized by periorificial and acral dermatitis, alopecia, and diarrhea. The nature of the metabolic defect continues to be debated. However, two new fibroblast proteins that are absent in the fibroblasts of patients with acrodermatitis enteropathica have recently been discovered. These proteins may be responsible for decreased zinc uptake and abnormal zinc metabolism. Symptoms of acrodermatitis enteropathica occur within the first few months after birth and tend to appear shortly after discontinuation of breastfeeding. This phenomenon has led many to believe that human milk has a beneficial role, which bovine milk lacks.
Clinical Features
The disease begins in the first few weeks or months of life with a localized eruption of the skin, particularly near the body orifices. Shortly, there is loss of hair and gastrointestinal disturbance accompanied by diarrhea. The skin lesions are vesiculobullous in nature and tend to occur in crops. These lesions rupture and become crusted and ultimately erythematous, scaling with a psoriasiform pattern. The skin lesions, and oral lesions as well, are prone to secondary infection, especially by Candida albicans. This is probably related to the deficiency in cell-mediated immunity reportedly manifested by the children.
Oral Manifestations
The oral mucosa, chiefly the buccal mucosa, becomes erythematous and edematous with erosive desquamative lesions. These oral lesions have been described in the cases of Danbolt.
Histologic Features
Histopathologic examination reveals parakeratosis of the stratum corneum with occasional neutrophils and intracellular edema. The granular cell layer is diminished, and the upper epidermis demonstrates pallor and edema. Focal dyskeratosis is seen. The epidermis may be psoriasiform or atrophic. Occasionally, subcorneal pustules are seen.
Laboratory Findings
Plasma zinc levels are low. Determining hair, urine, and parotid saliva zinc levels as well as serum alkaline phosphatase activity (which lowers later in the disease) may be helpful. Analysis of maternal breast milk zinc concentrations may also help in differentiating AE from acquired zinc deficiency.