Adverse Effects of Acetaminophen and NSAIDs
WITH few adverse effects at doses less than the recommended maximum adult dose of 4 gm per day, acetaminophen is widely considered one of the safest and best tolerated analgesics (Burke, Smyth, Fitzgerald, 2006; Schug, Manopas, 2007). Like other nonopioids, chronic use does not result in tolerance or physical dependence, and carries no risk of respiratory depression. The risk of adverse effects associated with conventional doses of acetaminophen is less than that associated with the other nonopioids (Box 6-1).
Although numerous studies have established the potential for acetaminophen toxicity on diverse systems, the likelihood of clinically-relevant adverse effects when this drug is used in appropriate doses is very low. Overall, the risk of adverse effects is greater during treatment with NSAIDs. The positive aspects of NSAID therapy—good effectiveness in many types of common pain syndromes, lack of tolerance or physical dependence, no risk of respiratory depression, and very low risk of typical CNS adverse effects such as somnolence—must be balanced by the potential for serious adverse effects. (See Chapter 8 for a discussion of adverse effects associated specifically with perioperative acetaminophen and NSAID use.)
Adverse Effects of Acetaminophen
The most serious complication associated with acetaminophen use is hepatotoxicity (liver damage) as a result of overdose. In the healthy adult, a maximum daily dose below 4 gm is only rarely associated with liver toxicity (American Pain Society [APS], 2003; Bolesta, Haber, 2002; Laine, White, Rostom, et al., 2008; Rumack, 2002; Watkins, Kaplowitz, Slattery, et al., 2006). However, repeated doses exceeding this amount can result in hepatotoxicity (Daly, O’Malley, Heard, et al., 2004; United States Food and Drug Administration [U.S. FDA], 2009), and very large single doses can lead to fulminant hepatic failure.
Acetaminophen should be administered cautiously, at lower maximum doses, or not at all in situations that pose an increased risk of acetaminophen-associated hepatic injury. Factors that increase this risk include preexisting liver disease, the concurrent use of potentially hepatotoxic medications, fasting or poor nutrition, and regular consumption of alcohol (AGS, 2009; APS, 2003; Burke, Smyth, Fitzgerald, 2006; Larson, Polson, Fontana, et al., 2005; Miaskowski, Cleary, Burney, et al., 2005). Although some research shows that 4 gm per day can be safely taken in individuals who regularly drink alcohol (Graham, Scott, Day, 2005; Kuffner, Dart, 2001), the APS recommends no more than 2.5 gm per day in individuals who consume more than 2 ounces of alcohol daily because of the elevated risk of hepatotoxicity (Simon, Lipman, Caudill-Slosberg, et al., 2002). The U.S. FDA requires acetaminophen product labeling to warn consumers of the increased risk of liver damage when acetaminophen is taken by those who consume three or more alcoholic drinks per day (U.S. FDA, 2009). Acetaminophen at doses less than the maximum recommended amount may be problematic with alcohol intake as well. One study showed 65% of the subjects with acute liver failure who reported taking less than 4 gm per day were alcohol abusers (Larson, Polson, Fontana, et al., 2005). The American Geriatrics Society (AGS) lists chronic alcohol abuse/dependence as a relative contraindication to using acetaminophen (AGS, 2009).
Most experts recommend a reduction in daily dose in individuals who are at high risk for hepatotoxicity (Burke, Smyth, FitzGerald, 2006). For example, the AGS recommends a 50% to 75% reduction in dose in older individuals with hepatic insufficiency (AGS, 2009). Recommendations vary, however. Some authors recommend avoiding acetaminophen entirely in patients with hepatic insufficiency (Bannwarth, Pehourcq, 2003), whereas others suggest that it should be used as the optimal analgesic for patients with stable chronic liver disease (Graham, Scott, Day, 2005). Liver function tests should be performed every 6 to 12 months in any individual at high risk for hepatotoxicity who is taking acetaminophen (Bannwarth, 2006; Miaskowski, Cleary, Burney, et al., 2005; Simon, Lipman, Caudill-Slosberg, et al., 2002) (see Chapter 7 for more on acetaminophen dosing).
Acetaminophen overdose is a common cause of liver failure. A 6-year prospective study of 662 patients with acute liver failure showed that 42% of the cases resulted from acetaminophen hepatotoxicity, and 48% of those were from unintentional acetaminophen overdose (Larson, Polson, Fontana, et al., 2005). A review of the United Network for Organ Sharing data revealed acetaminophen alone or in combination with other drugs accounted for 49% of the drug-related liver transplants performed in the United States between 1990 and 2002 (Russo, Galanko, Shrestha, et al., 2004).
A retrospective review of 1543 patients hospitalized for acetaminophen overdose revealed that 4.5% developed hepatotoxicity despite antidote treatment (n-acetylcysteine) in 38% (Myers, Shaheen, Li, et al., 2008) (see Chapter 10 for more on overdose treatment). While the occurrence of hepatotoxicity was low in this review, the patients who did develop it were 2.5 times more likely to be admitted to the intensive care unit (ICU) and 40 times more likely to die in the hospital than those without liver damage. This led researchers to conclude that the nature of most acetaminophen overdoses is relatively benign, but the clinical impact for those who do develop hepatotoxicity is significant. This review also reinforced the impact of known risk factors—34% were alcohol abusers, 13% overdosed accidentally, and 3% had underlying liver disease (Myers, Shaheen, Li, et al., 2008). A significant finding was that the 13% who accidentally overdosed represented 49% of the cases of hepatotoxicity in this study. Co-morbidities were common (82%) in the individuals who overdosed; in addition to liver disease and alcohol abuse, 55% suffered depression. Older age was also identified as a risk factor.
The wide availability of formulations that combine acetaminophen with other ingredients for the treatment of a variety of conditions, ranging from the common cold to pain, increases the chances of exceeding recommended daily doses (Myers, Shaheen, Li, et al., 2008). For example, one of the aforementioned studies (Larson, Polson, Fontana, et al., 2005) noted that, of the 48% who had acute liver failure following unintentional overdose, 38% had taken two or more acetaminophen products simultaneously and 63% had taken opioid-acetaminophen formulations.
The risk of unintentional overdose from acetaminophen mandates that patient teaching be done when prescribing an acetaminophen-containing drug and that this discussion describes safe maximum doses and the types of OTC analgesics and medications, such as cold remedies and sleep aids, that should be avoided (Bataller, 2007; Scharbert, Gebhardt, Sow, et al., 2007) (see Patient Medication Information Form III-4 on pp. 256-257; see Form III-5 on pp. 258-259 for aspirin). In 2009, the U.S. FDA required label changes for acetaminophen and products containing acetaminophen to reflect an increased risk of liver damage under certain circumstances (e.g., maximum daily dose is exceeded, daily intake of three or more alcoholic drinks, preexisting liver disease, concomitant use of other drugs containing acetaminophen) (U.S. FDA, 2009). In addition to product labeling changes, reformulation of acetaminophen-containing opioid analgesics has been proposed to reduce the rising incidence of this preventable form of liver injury (Fontana, 2008).
There may be racial or ethnic differences in the pharmacokinetics of acetaminophen and the development of hepatotoxicity following overdose. A small study comparing acetaminophen in Chinese (N = 11) and Caucasian (N = 9) subjects showed the Chinese more rapidly absorbed a single dose of acetaminophen and tended to produce reduced amounts of cysteine and mercapturic acid conjugates, compounds that may help protect against hepatotoxicity following acetaminophen overdose (Critchley, Critchley, Anderson, et al., 2005). Further research is needed to describe these and other sources of individual variation in the risks associated with this drug.
Renal Effects
The risk of chronic renal failure also has been linked to long-term acetaminophen use (Bannwarth, 2006) (see Chapter 8 for renal effects in the perioperative setting). The Nurses’ Health Study, established in 1976, utilized questionnaires to evaluate 121,700 female registered nurses for a wide variety of health-related conditions (Colditz, 1995). A cohort (N = 1697) of this study was evaluated later for the effect of acetaminophen, aspirin, or other NSAID use on renal function, as measured by glomerular filtration rate (GFR) (Curhan, Knight, Rosner, et al., 2004). High acetaminophen, but not NSAID or aspirin use, was associated with an increased risk of decline in renal function. Women who used more than 100 gm of acetaminophen over the 11-year collection period had a GFR decline of at least 30%.
Other epidemiologic studies also have shown an association between acetaminophen use and chronic renal failure. For example, a study of nearly 1000 individuals with newly diagnosed early-stage chronic renal failure and nearly 1000 individuals without renal failure observed that aspirin and acetaminophen were used regularly by 37% and 25%, respectively, of the patients with renal failure, and by 19% and 12%, respectively, of the control group (Fored, Ejerblad, Lindblad, et al., 2001). Regular use (twice weekly for 2 months) of either drug alone was associated with a 2.5 increase in risk of chronic renal failure, and the relative risk rose more with increasing cumulative lifetime doses of acetaminophen than aspirin.
None of these epidemiologic studies confirm causality. Although the renal disease may be directly related to long-term acetaminophen use, it is possible that renal insufficiency caused by other disorders leads to pain and self-medication with acetaminophen, or that both renal dysfunction and acetaminophen use are related to a third factor. It is prudent to consider chronic renal failure as a potential complication of this drug, but recognize the need for additional research to define the relationship better.
Although acetaminophen may cause kidney disease, it usually is preferred over NSAIDs as a treatment for mild to moderate pain in patients with preexisting renal insufficiency (Bannwarth, 2006; Bannwarth, Pehourcq, 2003). This conventional view has been justified by the perceived lack of effect on platelet aggregation (which may be overstated; see discussion later in the chapter) and the low incidence of GI adverse effects (Kurella, Bennett, Chertow, 2003; Launay-Vacher, Karie, Fau, et al., 2005). Dose adjustment also is not necessary in the presence of preexisting renal failure, as it is when acetaminophen is prescribed to those with liver disease.
Cardiovascular (CV) Effects
Although CV adverse effects may be increased by acetaminophen, studies of this phenomenon are minimal and the underlying mechanisms are unclear (Chan, Manson, Albert, et al., 2006). Phenacetin, the precursor to acetaminophen, is associated with increased CV morbidity and mortality (Chan, Manson, Albert, et al., 2006), and there are data suggesting a relationship between acetaminophen dose and CV risk. A prospective study that evaluated the incidence of nonfatal and fatal coronary heart disease, and nonfatal and fatal stroke in nearly 71,000 female NSAID or acetaminophen users found that the relative risk of these outcomes was 1.38 in women who regularly (22 days or more/month) consumed acetaminophen, compared with 1.44 in those with frequent use of NSAIDs (Chan, Manson, Albert, et al., 2006). A dose-dependent risk was evident with a relative risk of 1.86 and 1.68 for those who took 15 tablets or more/week of NSAIDs or acetaminophen, respectively, compared with those who took lower doses.
Acetaminophen use may also contribute to the incidence of hypertension. Curhan and colleagues conducted a prospective study of 80,020 women who participated in the Nurses’ Health Study (Colditz, 1995) and had no previous history of hypertension to examine the effect of acetaminophen, aspirin, or other NSAID use on BP (Curhan, Willett, Rosner, et al., 2002). Compared with nonusers, the relative risk of hypertension was 1.2 and 2.0 for those taking acetaminophen for 1 to 4 days/month and 22 days/month, respectively. This means that those taking acetaminophen 22 days/month were twice as likely to develop hypertension as those who did not use acetaminophen. Other analysis has yielded similar elevated risk (Dedier, Stampfer, Hankinson, et al., 2002). An analysis of younger (N = 3220) and older (N = 1903) age cohorts of the Nurses’ Health Study revealed a correlation between relative risk of hypertension and dose of acetaminophen (Forman, Stampfer, Curhan, 2005). Dose but not age was associated with increased risk in those who took more than 500 mg/day; relative risk was 1.99 and 1.93 for younger and older women, respectively.
A later study of analgesic use in 16,031 non- hypertensive male health care professionals had similar results (Forman, Rimm, Curhan, 2007). A review of detailed information about the use of acetaminophen, aspirin, and other NSAIDs by these men revealed a relative risk of 1.34 in those who took acetaminophen 6 to 7 days per week compared with those who used no acetaminophen. The relative risk was 1.38 for NSAIDs and 1.26 for aspirin.
In contrast, a large prospective cohort study of 8229 males without hypertension at the start of a 5.8 year (mean) follow-up period found that, compared with those who never used acetaminophen, NSAIDs, or aspirin, there was no significant increase in risk of subsequent hypertension in the men who took at least 2500 pills of any of the analgesics. A small to moderately increased risk cannot be excluded from observational studies (Kurth, Hennekens, Sturmer, et al., 2005), and all observational studies must be interpreted cautiously given the potential for recall and other sources of bias (Vardeny, Solomon, 2008).
Hematologic Effects and Anticoagulant Therapy
Acetaminophen has long been used as an analgesic in patients receiving anticoagulation therapy because it was thought to have no effect on platelet aggregation (Thijssen, Soute, Vervoort, et al., 2004). Nonetheless, there are conflicting observations, which together suggest that acetaminophen may in fact interfere with platelet aggregation and potentiate the anticoagulant effect of warfarin (Coumadin) (Gebauer, Nyfort-Hansen, Henschke, et al., 2003; Mahe, Bertrand Drouet, et al., 2006; Munsterhjelm, Munsterhjelm, Niemi, et al., 2005; Ornetti, Ciappuccini, Tavernier, et al., 2005; Parra, Beckey, Stevens, 2007; Thijssen, Soute, Vervoort, et al., 2004). If indeed this is a clinically relevant effect, it may be related to the weak inhibition of COX-1 produced by acetaminophen (Munsterhjelm, Munsterhjelm, Niemi, et al., 2005) and the production of metabolites that may interfere with the enzymes involved in vitamin K–dependent coagulation factor synthesis (Mahe, Bertrand, Drouet, et al., 2005, 2006; Thijssen, Soute, Vervoort, et al., 2004).
A small double-blind, crossover study of 13 healthy male volunteers with normal platelet function was conducted to evaluate the dose-dependent effect of acetaminophen on platelet function (Munsterhjelm, Munsterhjelm, Niemi, et al., 2005). Compared with those who received placebo, the men who received IV acetaminophen demonstrated a dose-dependent increase in concentration of arachidonic acid, which was thought to cause the anticoagulant effect seen in the volunteers. In addition, acetaminophen inhibited the release of thromboxane B2, the stable metabolite of thromboxane A2, released during platelet aggregation. This effect was also dose dependent. Another small (N = 11) double-blind, crossover study randomized patients to receive 14-day regimens of acetaminophen (4 gm) or placebo with a 14-day wash-out period between (Mahe, Bertrand, Drouet, et al., 2005). The International Normalized Ratio (INR), a measure of coagulation, was significantly elevated in patients who received acetaminophen compared with those who received placebo.
The effect of 2 to 4 gm/day of acetaminophen on patients receiving warfarin anticoagulation therapy was evaluated in a randomized, placebo-controlled trial of 36 adults at an anticoagulation clinic (Parra, Beckey, Stevens, 2007). The patients were receiving warfarin and had stable INRs at the start of the study. The study was terminated after 15 patients demonstrated a significant dose-dependent increase in INR. There was also a significant increase in mean serum alanine aminotransferase (ALT) level (liver function indicator) in the patients who received 4 g/day of acetaminophen compared with those who received placebo. Another randomized, placebo-controlled study (N = 20) showed similar results (Mahe, Bertrand, Drouet, et al., 2006). Again, patients were stabilized on warfarin anticoagulation therapy. The mean INR increased quickly and significantly within 1 week of acetaminophen (4 gm/day) use. There were also significant reductions in vitamin K–dependent clotting factors.
These studies support the view that acetaminophen can have clinically relevant hematologic effects. It also is true, however, that acetaminophen inhibition of thromboxane A2 is less than that of the nonselective NSAIDs (Munsterhjelm, Niemi, Syrjala, et al., 2003), and studies have shown that surgical bleeding as a result of perioperative acetaminophen intake is low (Ashraf, Wong, Ronayne, et al., 2004; Munsterhjelm, Munsterhjelm, Niemi, et al., 2005) (see Chapter 8 for effect of perioperative nonopioid use on surgical site bleeding). It is probable that the risk of bleeding associated with acetaminophen use is low, but given the extant data, close monitoring of patients receiving acetaminophen and anticoagulation therapy is prudent (Mahe, Bertrand Drouet, et al., 2005, 2006; Ornetti, Ciappuccini, Tavernier, et al., 2005; Parra, Beckey, Stevens, 2007). In 2009, the U.S. FDA required label changes for acetaminophen to advise those who are taking warfarin to discuss the use of acetaminophen with a pharmacist or physician prior to taking acetaminophen (U.S. FDA, 2009).
Gastrointestinal (GI) Effects
The most common adult daily dose of acetaminophen is 1000 mg, a dose that is thought to produce less GI toxicity than most NSAIDs (Burke, Smyth, Fitzgerald, 2006). However, daily doses of more than 500 mg have been shown to diminish gastric mucosal protection (Rahme, Pettitt, LeLorier, 2002), and epidemiologic studies show doses of more than 2000 mg/24 h produce increased risk of severe upper GI adverse effects (Bannwarth, 2006). A study of analgesic overdose by patients with suicidal intent reported endoscopic gastric damage (lesions) from acute high-dose acetaminophen to be similar to that caused by acute high-dose NSAID ingestion (Soylu, Dolapcioglu, Dolay, et al., 2008). This is likely due, at least in part, to peripheral COX-1 inhibition (associated with poorer gastric safety). Indeed, acetaminophen has been shown to be more COX-1 selective than several NSAIDs, including naproxen (Naprosyn, Aleve), diclofenac (Voltaren), and ibuprofen (Advil, Motrin), but less than piroxicam (Feldene) and tolmetin (Tolectin) (Rahme, Pettitt, LeLorier, 2002).
A large cohort study of individuals aged 65 and older was undertaken to compare the rates of GI adverse events occurring with higher versus lower doses of acetaminophen (Rahme, Pettitt, LeLorier, 2002). Data from patients who had received a prescription for acetaminophen (N = 21,000) or a nonaspirin NSAID (N = 27,000) were examined. Unadjusted rates of hospitalization, ulcer, and dyspepsia were higher for patients taking acetaminophen compared with those taking NSAIDs, and the acetaminophen GI adverse events were dose related. After adjustment of risk susceptibility, patients receiving higher acetaminophen doses (more than 3250 mg/day) had higher rates of GI events compared with those receiving lower doses (650 mg or less/day).
Adverse Effects of NSAIDs
The most common adverse effects of the NSAIDs involve the GI system. The associated clinical conditions are heterogeneous, and the most serious concern involves GI ulceration. Ulcers and their complications, including hemorrhage and perforation, can occur in both the upper and lower GI tract (Hayashi, Yamamoto, Kita, et al., 2005). These serious events may be difficult to detect and can be fatal (Simon, 2007).
NSAID-induced GI events are blamed for an estimated 100,000 hospitalizations and 16,500 deaths annually in the United States (Bombardier, Laine, Reicin, et al., 2000). Studies have demonstrated that the risk for serious GI complications is 3- to 5-fold higher in those who take NSAIDs than in those who do not (Wilcox, Allison, Benzuly, et al., 2006) and that gastric or duodenal ulceration may occur in 15% to 30% of patients receiving long-term therapy with NSAIDs classified as nonselective COX-1/COX-2 inhibitors (Bombardier, Laine, Reicin, et al., 2000; Cryer, 2004; Laine, 2001). A review of 361 patients with peptic ulcer bleeding revealed that one-half of the cases was associated with NSAID use (Ramsoekh, Van Leerdam, Rauws, et al., 2005). An autopsy study of 713 individuals revealed gastric and duodenum ulceration in 22% of nonselective NSAID users compared with 12% of NSAID nonusers; small bowel ulcers were found in 8.4% of nonselective NSAID users compared with 0.6% of NSAID nonusers (Simon, 2007).
The primary underlying mechanism of NSAID-induced ulceration is thought to be inhibition of COX-1, which leads to reduction in GI-protective prostaglandins (Burke, Smyth, Fitzgerald, 2006; Simon, Weaver, Graham, et al., 1999). This is a systemic effect and can occur regardless of the route of administration of the NSAID (Laine, 2001). This means that GI adverse effects resulting from prostaglandin inhibition are possible when NSAIDs are taken orally, rectally, or parenterally. Systemic effects may be compounded by local processes produced by direct contact between GI mucosa and the drug. With the exception of nabumetone (Relafen), all of the nonselective NSAIDs are highly lipophilic and can easily penetrate the gastric mucosal barrier, which is a hydrophobic mucous layer along the stomach lining (Simon, 2007). This penetration is thought to result in oxidative uncoupling of cellular metabolism, producing cell death and localized tissue injury (Simon, 2007).
NSAID-induced adverse GI effects manifest as distressing symptoms alone, asymptomatic ulceration, or the more serious complications of GI bleeding, perforation, or obstruction (Cryer, 2004; Laine, 2001). Even though dyspepsia and other upper GI symptoms are commonly associated with the use of NSAIDs and often a reason for discontinuing NSAID therapy (Goldstein, Eisen, Burke, et al., 2002), these symptoms do not appear to predict the development of a more serious GI event in patients with no or low GI risk (Laine, 2001; Lanas, Bajador, Serrano, et al., 2000; Mellemkjaer, Biot, Sorensen, 2002; Simon, Fox, 2005). However, endoscopic evaluation is recommended in patients with high GI risk who experience significant NSAID treatment-induced dyspepsia (Chan, Hung, Suen, et al., 2004).
NSAID-induced adverse effects are related to the dose and duration of treatment. The higher the NSAID dose and the longer the duration of NSAID use, the higher the risk of cumulative GI toxicity (Chan, Graham, 2004). This cumulative toxicity also includes a period of relatively higher risk at the start of treatment (Wilcox, Allison, Benzuly, et al., 2006). Serious complications are most frequent during the first 3 months of NSAID administration (Gabriel, Jaakkimainen, Bombardier, 1991), and there is an 8% incidence of ulcer development within 1 week of regular NSAID use (Laine, 2001).
GI Risk Factors
The greatest risk factor for NSAID-associated GI events is the presence of prior ulcer disease with ulcer complications (Chan, Graham, 2004). The AGS lists current peptic ulcer disease as an absolute contraindication and history of peptic ulcer disease as a relative contraindication to the use of NSAIDs in older adults (AGS, 2009). Other risk factors include age at or older than 60 years, CV disease and other co-morbidities, severe rheumatoid arthritis (RA), and concomitant treatment with corticosteroids or anticoagulants (Bhatt, Scheiman, Abraham, et al., 2008; Chan, Graham, 2004; Laine, 2001; Simon, 2007; Wilcox, Allison, Benzuly, et al., 2006). Helicobactor pylori (H. pylori) infection, excessive alcohol consumption, and cigarette smoking generally are considered independent and modifiable risk factors, though the magnitude of their effects is unclear (Burke, Smyth, Fitzgerald, 2006; Wilcox, Allison, Benzuly, et al., 2006). In 2009, the U.S. FDA required label changes for NSAIDs to reflect an increased risk of gastric bleeding, particularly in certain populations, including older adults and individuals taking anticoagulants, steroids, or other NSAID-containing products, or those consuming three or more alcoholic drinks daily (U.S. FDA, 2009). Table 6-1 provides a summary of risk factors for the development of NSAID-induced GI adverse events.
Table 6-1
Risk Factors for NSAID-Induced Adverse GI Events
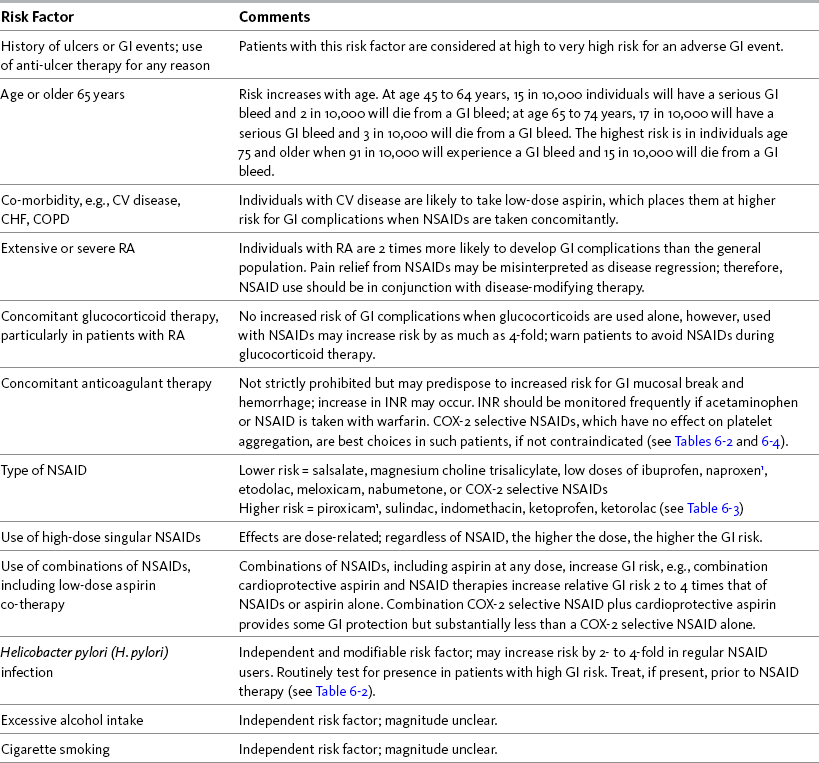
CHF, Congestive heart failure; COPD, chronic obstructive pulmonary disease; COX, cyclooxygenase; CV, cardiovascular; GI, gastrointestinal; INR, International Normalized Ratio; NSAID, nonsteroidal antiinflammatory drug; RA, rheumatoid arthritis.
1Avoid full-dose naproxen, piroxicam, and oxaprozin in older adults because of long half-life and increased risk of GI toxicity.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 192, St. Louis, Mosby. Data from Agency for Healthcare Research and Quality (AHRQ). (2007). Choosing non-opioid analgesics for osteoarthritis. Clinician’s guide. Available at http://effectivehealthcare.ahrq.gov/. Accessed July 24, 2008; American Geriatrics Society (AGS) Panel on Pharmacological Management of Persistent Pain in the Older Persons. (2009). The pharmacological management of persistent pain in older persons. J Am Geriatr Soc, 57(8), 1331-1346; Chan, F. K. L., & Graham, D. Y. (2004). Prevention of non-steroidal anti-inflammatory drug gastrointestinal complications—Review and recommendations based on risk assessment. Aliment Pharmacol Ther, 19(10), 1051-1061; Chan, F. K. L., Hung, L. C. T., Suen, B.Y., et al. (2004). Celecoxib versus diclofenac plus omeprazole in high-risk arthritis patients: Results of a randomized double-blind trial. Gastroenterology, 127(4), 1038-1043; Fick, D. M., Cooper, J. W., Wade, W. E., et al. (2003). Updating the Beers criteria fo potentially inappropriate medication use in older adults. Arch Intern Med, 163(22), 2716-2724; Gabriel, S. E., Jaakkimainen, L., Bombardier, C. (1991). Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. A meta-analysis. Ann Intern Med, 115(10), 787-796; Hanlon, J. T., Backonja, M., Weiner, D., et al. (2009). Evolving pharmacological management of persistent pain in older persons. Pain Med, 10(6), 959-961; Kurata, J. H., & Nogawa, A. N. (1997). Meta-analysis of risk factors for peptic ulcer: Nonsteroidal anti-inflammatory drugs, Helicobacter pylori, and smoking. J Clin Gastroenterol, 24(1), 2-17; Kuritzky, L., & Weaver, A. (2003). Advances in rheumatology: Coxibs and beyond. J Pain Symptom Manage, 25(2S), S6-S20; Laine, L. (2001). Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology, 120(3), 594-606; Laine, L., White, W. B., Rostom, A., et al. (2008). COX-2 selective inhibitors in the treatment of osteoarthritis. Semin Arthritis Rheum, 38(3), 165-187; Simon, L. S. (2007). Risks and benefits of COX-2 selective inhibitors. Available at http://www.medscape.com/viewprogram/6872/. Accessed April 5, 2007; Simon, L. S., & Fox, R. I. (2005). What are the options available for anti-inflammatory drugs in the aftermath of rofecoxib’s withdrawal? Medscape Rheumatology, 6(1). Available at http://www.medscape.com/viewarticle/500056. Accessed April 16, 2005; Solomon, D. H., Glynn, R. J., Rothman, K. J., et al. (2008). Subgroup analyses to determine cardiovascular risk associated with nonsteroidal anti-inflammatory drugs and coxibs in specific patient groups. Arthritis Care Res, 59(8), 1097-1104; Wilcox, C. M., Allison, J., Benzuly, K., et al. (2006). Consensus development conference on the use of nonsteroidal anti-inflammatory agents, including cyclooxygenase-2 enzyme inhibitors and aspirin. Clin Gastroenterol Hepatol, 4(9), 1082-1089. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
The First International Working Party on GI and CV Effects of NSAIDs and Anti-platelet Agents used a comprehensive series of clinical vignettes and possible scenarios to rate the appropriateness of NSAIDs. This group predefined high GI risk as age at or older than 70 years, prior upper GI event, and concomitant use of aspirin, corticosteroids, anticoagulants, or other antiplatelet drugs (Chan, Abraham, Scheiman, et al., 2008). In addition, risk was determined to be higher with specific NSAIDs and with the combination of an NSAID and low-dose aspirin (Laine, 2001; Simon, 2007; Silverstein, Faich, Goldstein, et al., 2000; Vardeny, Solomon, 2008). In a consensus guideline, the American College of Cardiology Foundation (ACCF), American College of Gastroenterology (ACG), and the American Heart Association (AHA) stated that the use of cardioprotective aspirin (81 mg) is associated with a 2- to 4-fold increase in upper GI adverse events (Bhatt, Scheiman, Abraham, et al., 2008) (see pp. 204-205 for more on cardioprotective aspirin). It is also important to note that any dose of aspirin can cause upper GI adverse events, and because the primary mechanism underlying GI toxicity is systemic rather than local, buffered and enteric-coated formulations do not decrease the incidence (Bhatt, Scheiman, Abraham, et al., 2008; Laine, 2001).
The risk of GI toxicity and death from a GI bleed increases with age at a rate of about 4% per year, and age is an extremely important consideration when starting NSAID therapy (APS, 2003; Wilcox, Allison, Benzuly, et al., 2006). At age 45 to 64 years, 15 in 10,000 individuals will have a serious GI bleed during long-term therapy, and 2 in 10,000 will die from a GI bleed; however, at age 65 to 74 years, 17 in 10,000 will have a serious GI bleed and 3 in 10,000 will die. The highest risk is in individuals age 75 and older; at this age, 91 in 10,000 will experience a GI bleed and 15 in 10,000 will die (AHRQ, 2007). This increased risk may be due to the combined effect of other risk factors associated with aging, such as medical comorbidities and concomitant use of drugs such as aspirin or anticoagulants, as well as an age-related decrease in protective GI prostaglandin concentrations (Wilcox, Allison, Benzuly, et al., 2006).
H. pylori is a gram-negative bacterium that can reside chronically in the stomach and elicit a local inflammatory response. NSAIDs produce a similar destructive effect on the gastric mucosa through COX-1 inhibition of prostaglandins in the protective lining of the gut. Although the data are conflicting, there is growing evidence of an interaction between the presence of H. pylori infection and risk of GI complications in NSAID users (Chan, To, Wu, et al., 2002; Huang, Sridhar, Hunt, 2002; Laine, 2001; Wilcox, Allison, Benzuly, et al., 2006). This interaction was suggested in a meta-analysis of 463 studies, which revealed that one-third of patients receiving long-term NSAID therapy had gastric or duodenal ulcers irrespective of the presence of H. pylori infection, but peptic ulcer disease was more common in H. pylori-infected NSAID users than in noninfected NSAID users (Huang, Sridhar, Hunt, 2002). A 2- to 4-fold increase in risk of upper GI complications in regular NSAID users who are H. pylori infected has been reported elsewhere (Chan, Graham, 2004). Eradication of H. pylori with antibiotic therapy decreases the incidence of peptic ulcers in patients who begin taking NSAIDs, but this does not seem to extend to patients with a previous history of ulceration (Wilcox, Allison, Benzuly, et al., 2006).
These data support the conclusion that H. pylori infection is an independent and modifiable risk factor for GI complications in long-term NSAID users (Chan, To, Wu, et al., 2002; Huang, Sridhar, Hunt, 2002; Kurata, Nogawa, 1997; Laine, 2001; Wilcox, Allison, Benzuly, et al., 2006). Although it may be difficult to justify routinely ruling out H. pylori infection prior to initiating NSAID therapy in patients with low risk, testing should be done routinely in patients with high GI risk (Chan, To, Wu, et al., 2002; Chan, Graham, 2004; Wilcox, Allison, Benzuly, et al., 2006). If H. pylori infection is present, it should be eradicated prior to initiating NSAID therapy (Chan, Graham, 2004; Wilcox, Allison, Benzuly, et al., 2006) (see Table 6-1).
NSAID Selection with Consideration of GI Risk
The expert panel of the First International Working Party on GI and CV Effects of NSAIDs and Anti-platelet Agents endorsed risk stratification based on a thorough patient assessment as a key step in the selection of NSAID therapy. This panel viewed the selection of a nonselective NSAID as appropriate in patients with average GI risk, including those younger than 70 years of age with no prior GI event and no concurrent use of corticosteroids, antithrombotic agents, or anticoagulants (Chan, Abraham, Scheiman, et al., 2008). The American Gastroenterological Association (AGA) (Wilcox, Allison, Benzuly, et al., 2006) and other researchers (Chan, Graham, 2004) also have provided recommendations that guide decision making with regard to NSAID selection and patient management during NSAID therapy. Table 6-2 provides a summary of NSAID treatment strategies correlated with GI risk.
GUIDELINES Table 6-2
NSAID Treatment Strategies Correlated with Gastrointestinal (GI) Risk Levels
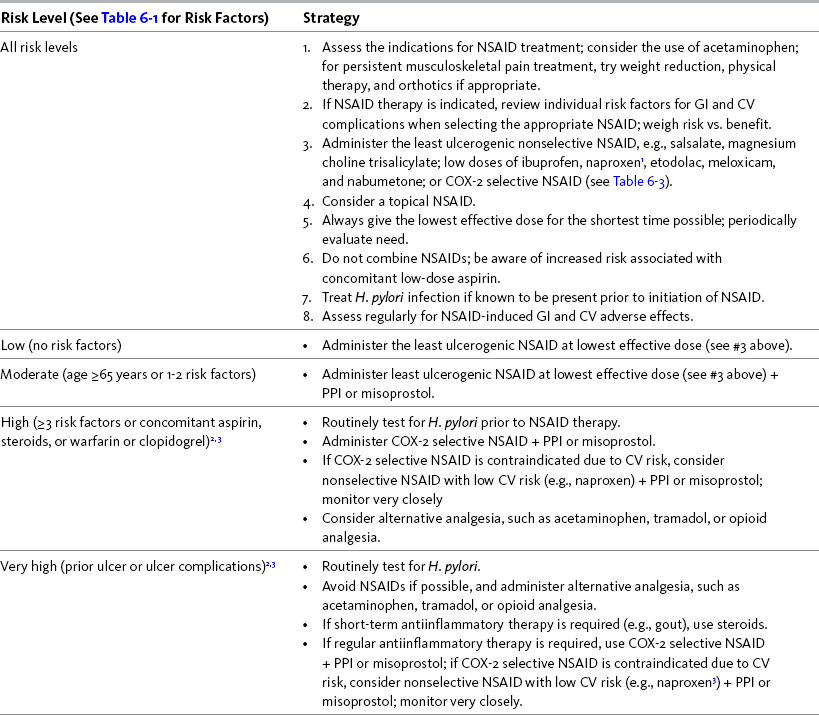
COX, Cyclooxygenase; CV, cardiovascular; GI, gastrointestinal; H. pylori, helicobacter pylori; NSAID, nonsteroidal antiinflammatory drug; PPI, proton pump inhibitor
1Avoid full-dose naproxen, piroxicam, and oxaprozin in older adults because of long half-life and increased risk of GI toxicity.
2Routinely test for and eradicate H. pylori infection, if present, prior to NSAID therapy.
3Further research is needed for consensus recommendations in this risk group. Carefully evaluate safety of COX-2 selective NSAIDs, particularly in patients taking concomitant aspirin or warfarin therapy as they may also have CV conditions in which COX-2 selective NSAIDs are contraindicated.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 194, St. Louis, Mosby. Data from American Geriatrics Society (AGS) Panel on Pharmacological Management of Persistent Pain in the Older Persons. (2009). The pharmacological management of persistent pain in older persons. J Am Geriatr Soc, 57(8), 1331-1346; Bhatt, D. L., Scheiman, J., Abraham, N. S., et al. (2008). ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of anti-platelet therapy and NSAID use. Am J Gastroenterol, 103(18), 2890-2907; Chan, F. K. L., & Graham, D. Y. (2004). Prevention of non-steroidal anti-inflammatory drug gastrointestinal complications—Review and recommendations based on risk assessment. Aliment Pharmacol Ther, 19(10), 1051-1061; Chan, F. K. L., To, K. F., Wu, J. C. Y., et al. (2002). Eradication of helicobacter pylori and risk of peptic ulcers in patients starting long-term treatment with non-steroidal anti-inflammatory drugs: A randomised trial. Lancet, 359(9300), 9-13; Chan, F. K. L., Wong, V. W., Suen, B. Y., et al. (2007). Combination of a cyclo-oxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: A double-blind, randomised trial. Lancet, 369(9573), 1621-1626; Cryer, B., Hochberg, M. C., Hennekens, C. H., et al. (February 15, 2006). Changing patterns of coxibs/NSAIDs prescribing: Balancing CV and GI risks. CME program. Available at http://www.medscape.com/viewprogram/5060. Accessed August 8, 2008; Fick, D. M., Cooper, J. W., Wade, W. E., et al. (2003); Updating the Beers criteria for potentially inappropriate medication use in older adults. Arch Intern Med, 163(22), 2716-2724; Hanlon, J. T., Backonja, M., Weiner, D., et al. (2009). Evolving pharmacological management of persistent pain in older persons. Pain Med 10(6), 959-961; Laine, L. (2001). Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology, 120(3), 594-606; Simon, L. S. (2007). Risks and benefits of COX-2 selective inhibitors. Available at http://www.medscape.com/viewprogram/6872/. Accessed August 5, 2007; Simon, L. S., & Fox, R. I. (2005). What are the options available for anti-inflammatory drugs in the aftermath of rofecoxib’s withdrawal? Medscape Rheumatology 6(1). Available at http://www.medscape.com/viewarticle/500056. Accessed April 16, 2005; Vonkeman, H. E., & van de Laar, M. A. F. J. (2008). NSAIDs: Adverse effects and their prevention. Semin Arthritis Rheum. Advanced access published on September 29, 2008; Wilcox, C. M., Allison, J., Benzuly, K., et al. (2006). Consensus development conference on the use of nonsteroidal anti-inflammatory agents, including cyclooxygenase-2 enzyme inhibitors and aspirin. Clin Gastroenterol Hepatol, 4(9), 1082-1089. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Avoiding NSAIDs altogether is the best way to prevent an NSAID-related complication (Chan, Graham, 2004), and when NSAID therapy is indicated, the lowest effective NSAID should be used for the shortest time needed (APS, 2003; U.S. FDA, 2007). If pain is mild to moderate, a trial of acetaminophen should be tried. Patients with more severe pain who are at relatively high risk for NSAID complications should be considered for a trial of an opioid or another centrally-acting analgesic. The AGS specifically noted that opioids may be safer than NSAIDs in some older patients (e.g., those with high GI and CV risk factors) (AGS, 2002; AGS, 2009), an observation that may be underappreciated by clinicians.
NSAID-induced GI toxicity, like antiinflammatory activity, is believed to correlate with COX-1 inhibition (Chan, Graham, 2004; Simon, 2007). Some NSAIDs (e.g., piroxicam [Feldene], indomethacin [Indocin]) achieve adequate analgesia at doses associated with high antiinflammatory activity, and appear to have a relatively greater risk of GI toxicity (Chan, Graham, 2004). These drugs generally are not first-line for this reason. In addition, NSAIDs with longer half-lives (e.g., sulindac [Clinoril], piroxicam, indomethacin) may expose GI mucosa to the drug for longer periods at a higher concentration and also have been linked to greater GI toxicity (Wilcox, Allison, Benzuly, et al., 2006). These are the reasons cited for avoiding full-dose piroxicam, oxaprozin, and naproxen in older adults (Fick, Cooper, Wade, et al., 2003; Hanlon, Backonja, Weiner, et al., 2009)
Some of the nonselective NSAIDs exhibit relatively selective COX-2 inhibition and less COX-1 inhibition, particularly at lower doses; these drugs are considered safer in terms of GI toxicity and often are preferred as a result (Simon, 2007). For example, ibuprofen, which is one of the most commonly used NSAIDs in the United States, is considered a relatively safe choice from a GI perspective when doses are kept below full antiinflammatory effect (less than 2.4 gm/day) (Chan, Graham, 2004).
In general, a lower risk of GI toxicity characterizes all nonacetylated salicylates, including salsalate (Disalcid) and magnesium choline trisalicylate (Trilisate), and several of the nonselective, nonsalicylate NSAIDs. The latter group includes etodolac (Lodine), meloxicam (Mobic), nabumetone, and low doses of ibuprofen or naproxen (Laine, 2001; Simon, 2007; Wilcox, Allison, Benzuly, et al., 2006). It is reasonable to consider these drugs as first-line options for NSAID therapy, although, as mentioned, full-dose naproxen is not recommended in older adults because of its long half-life (Fick, Cooper, Wade, et al., 2003; Hanlon, Backonja, Weiner, et al., 2009). As mentioned, nabumetone also has the advantage of relatively low lipophilicity, leading to less penetration of the gastric mucosal barrier (Bannwarth, 2008; Hedner, Samulesson, Wahrborg, et al., 2004; Simon, 2007). Table 6-3 shows the various NSAIDs and their associated risk for GI events.
Table 6-3
NSAID Agents and Associated Risk for GI Events
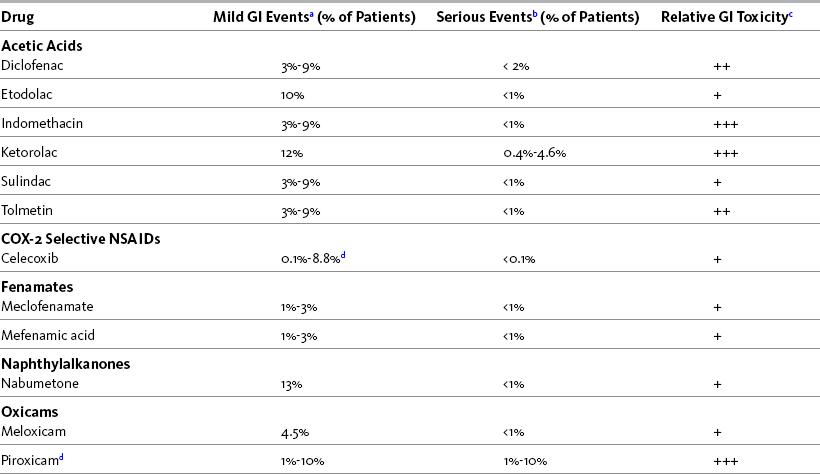
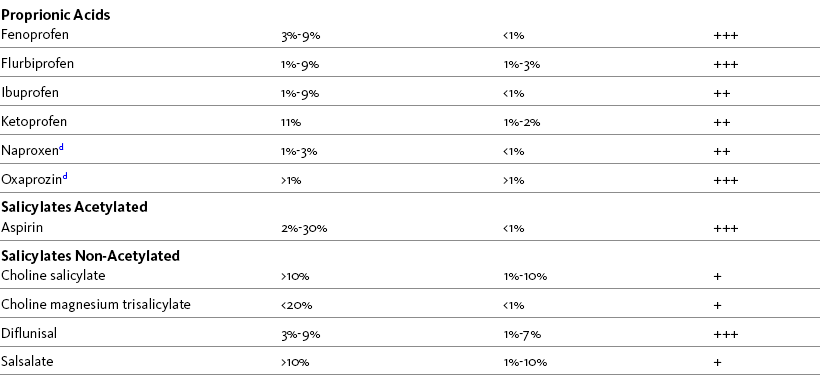
Note: Incidence may be much lower since many of the patients selected had previous history of GI risk.
aDyspepsia, nausea, abdominal pain
bUlceration, perforation, obstruction, bleeding ulcer
c+ = low risk; ++ = moderate risk; +++ = high risk
dAvoid full doses in older adults because of long half-life.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 195, St. Louis, Mosby. Data from Clinical Pharmacology Online; Fick, D. M., Cooper, J. W., Wade, W. E., et al. (2003); Updating the Beers criteria for potentially inappropriate medication use in older adults. Arch Intern Med, 163(22), 2716-2724; Hanlon, J. T., Backonja, M., Weiner, D., et al. (2009). Evolving pharmacological management of persistent pain in older persons. Pain Med, 10(6), 959-961; Gold Standard, Inc. Available at http:// clinicalpharmacology.com. Accessed August 10, 2008. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
The so-called COX-2 selective NSAIDs (e.g., celecoxib [Celebrex]) also carry a relatively lower risk of GI adverse effects, at least when taken by patients who are not also using cardioprotective aspirin therapy (Chan, Graham, 2004; Dajani, Islam, 2008; Jacobsen, Phillips, 2004; Laine, White, Rostom, et al., 2008; Silverstein, Faich, Goldstein, et al., 2000; Simon, 2007; Singh, Fort, Goldstein, et al., 2006). The COX-2 selective NSAIDS, like all NSAIDs, vary in the degree to which they affect COX-2 over COX-1. The ratio of inhibition associated with the best safety profile overall is not known. At the present time, only one drug in this category is available in the United States—celecoxib.
According to one review (Laine, White, Rostom, et al., 2008), COX-2 selective NSAIDs are associated with a 75% relative risk reduction for gastroduodenal ulcers and a 61% relative risk reduction for ulcer complications compared with nonselective NSAIDs. These benefits decline or disappear during concurrent therapy with cardioprotective aspirin (Vardeny, Solomon, 2008). Although the combination of a COX-2 selective NSAID with cardioprotective aspirin may be somewhat safer than the combination of aspirin and a nonselective NSAID, the risks are still substantially higher than during treatment with a COX-2 selective NSAID alone (Simon, Fox, 2005). Nevertheless, a cohort study using government databases of patients age 65 and older concluded that celecoxib may be safer from a GI perspective than other NSAIDs in older patients receiving cardioprotective aspirin (Rahme, Bardou, Dasgupta, et al., 2007).
Although the results have not been uniform, most randomized trials support a relatively better GI risk profile for the COX-2 selective NSAIDs. One study showed no significant difference in gastroduodenal ulcers between celecoxib and naproxen (Simon, Weaver, Graham, et al., 1999), but another showed celecoxib produced a lower incidence of upper GI ulcers and adverse effects compared with diclofenac (Emery, Zeidler, Kvien, et al., 1999). The Celecoxib Long-term Arthritis Safety Study (CLASS), which compared high-dose celecoxib (400 mg twice daily), ibuprofen (800 mg 3 times daily), and diclofenac (75 mg twice daily) in patients with osteoarthritis (OA) or RA observed that celecoxib caused significantly fewer symptomatic ulcers and ulcer complications (Silverstein, Faich, Goldstein, et al., 2000).
As expected, newly developed COX-2 selective NSAIDs, such as lumiracoxib (Prexige) and etoricoxib (Arcoxia), also have a relatively lower risk of GI toxicity. The Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), which evaluated over 18,000 patients, showed a 3- to 4-fold reduction in ulcer complications with the use of lumiracoxib compared with naproxen or ibuprofen (Schnitzer, Burmester, Mysler, et al., 2004). A later study of healthy volunteers who were randomized to receive lumiracoxib, naproxen plus omeprazole (Prilosec), or placebo revealed that lumiracoxib yielded an incidence of small bowel mucosal breaks that was lower than naproxen plus omeprazole and similar to placebo (Hawkey, Ell, Simon, et al., 2008).
The Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) program demonstrated fewer uncomplicated upper GI adverse events with etoricoxib than diclofenac; however, there was no difference in complicated events (perforation, obstruction, and complicated bleeding) (Laine, Curtis, Cryer, et al., 2007; Laine, Curtis, Langman, et al., 2008). Prior GI event and age 65 years and older were identified as significant risk factors for GI complications. Another study showed similar efficacy and lower GI adverse effects with etoricoxib compared with diclofenac in patients with OA over a 52-week evaluation period (Curtis, Bockow, Fisher, et al., 2005). An international multicenter study of 997 patients with OA compared long-term use of etoricoxib and naproxen and found the drugs had similarly satisfactory efficacy and tolerability, but there was a lower incidence of GI adverse effects and higher incidence of CV adverse effects in patients who took etoricoxib compared with those who took naproxen (Reginster, Malmstrom, Mehta, et al., 2007).
The latter study highlights the observation that diminished GI risk should not be interpreted as lower risk overall; CV risk remains a concern and continues to be evaluated in the newer COX-2 selective NSAIDs (Medscape Medical News, 2005; Topol, Falk, 2004; Wood, 2007) (see pp. 197-204 this chapter for discussion of CV risk). An assessment of risk factors (risk-benefit analysis) prior to NSAID therapy must include factors that go beyond GI risk (Tannenbaum, Bombardier, Davis, et al., 2006; Wilcox, Allison, Benzuly, et al., 2006). If the primary concern is GI risk, selection of an NSAID should focus either on a COX-2 selective drug or a nonselective drug with low GI risk (Wilcox, Allison, Benzuly, 2006) (see p. 204 for discussion of NSAID selection in the presence of both CV and GI risk).
Gastroprotective Co-Therapy
Gastroprotective co-therapy can reduce GI risk during NSAID therapy (Steen, Nurmohamed, Visman, et al., 2008; Wilcox, Allison, Benzuly, et al., 2006). The ACCF/ACG/AHA consensus guideline recommends gastroprotective co-therapy, preferably proton pump inhibitors (PPPs), in individuals who are taking a nonselective or COX-2 selective NSAID in conjunction with low-dose aspirin (Bhatt, Scheiman, Abraham, et al., 2008). The expert panel of the First International Working Party on GI and CV Effects of NSAIDs and Anti-platelet Agents also recommended co-therapy when NSAIDs are used in individuals with increased GI risk (Chan, Abraham, Scheiman, et al., 2008).
Three classes of gastroprotective agents are suggested as options for co-therapy: (1) the prostaglandin analogues such as misoprostol (Cytotec); (2) histamine receptor type-2 (H2) antagonists such as cimetidine (Tagamet), famotidine (Pepcid), and ranitidine (Zantac); and (3) PPIs such as esomeprazole (Nexium), lansoprazole (Prevacid), and omeprazole (Prilosec). A Cochrane Collaboration Review of 33 clinical trials concluded that all three classes of drugs reduced the incidence of both gastric and duodenal NSAID-induced ulcers (Rostom, Dube, Wells, et al., 2002); however, their effectiveness varies. Following is an overview of key research related to the three classes of gastroprotective agents.
Misoprostol is a synthetic prostaglandin that has been studied extensively in trials of ulcer prevention (Wilcox, Allison, Benzuly, et al., 2006). A Cochrane Collaboration Review concluded that doses of 800 mcg/day of misoprostol were superior to 400 mcg/day for prevention of gastric ulcers; however, no dose-related response was noted for duodenal ulcers (Rostom, Dube, Wells, et al., 2002). A prospective, double-blind study of long-term NSAID users with a history of gastric ulcer found patients who received 800 mcg/day of misoprostol remained ulcer free longer than those who received placebo or 15 or 30 mg of the PPI lansoprazole; however, a higher number of patients in the misoprostol group reported adverse effects and withdrew early from the study (Graham, Agrawal, Campbell, et al., 2002). The high incidence of adverse effects and study withdrawals led the researchers to conclude that misoprostol is clinically equivalent to PPIs. All doses of misoprostol can produce a high incidence of adverse effects, such as nausea, abdominal cramps, and diarrhea, and may not be well tolerated, particularly by the older patient (Chan, Graham, 2004; Rostom, Dube, Wells, et al., 2002; Wilcox, Allison, Benzuly, et al., 2006). Adherence to the regimen can be another barrier due to misoprostol dosing requirements that include taking the drug 4 times daily with food.
Whereas standard doses of H2-antagonists are reported to be effective in prevention of duodenal but not gastric ulcers in patients taking NSAIDs, double doses have been shown to be effective against both types of ulcers (Rostom, Dube, Wells, et al., 2002). However, a review of nearly 40 years of randomized controlled trials revealed that misoprostol and PPIs were more effective than H2-antagonists in reducing the risk of clinically significant GI adverse effects associated with NSAID use (Jacobsen, Phillips, 2004). Further, it has been suggested that any major benefit from H2-antagonists may be limited to patients with H. pylori infection (Chan, Graham, 2004).
The ASTRONAUT study of 541 NSAID users showed that the PPI omeprazole prevented and healed all types of ulcers more effectively than the H2-antagonist ranitidine (Yeomans, Tulassay, Juhasz, et al., 1998). A larger double-blind study (N = 935), called the OMNIUM study, randomly assigned NSAID users with a history of ulcers (gastric or duodenal or both) to receive omeprazole or misoprostol for 4 weeks or, in the absence of healing, 8 weeks (Hawkey, Karrasch, Szczepanski, et al., 1998). Rates of successful ulcer treatment were similar between the two drugs; however, omeprazole was associated with a lower relapse rate and was better tolerated than misoprostol. Other PPIs have shown similar results. A prospective, double-blind study of 353 chronic NSAID users with active gastric ulcers found better healing rates after 8 weeks of treatment with lansoprazole than with the H2-antagonist ranitidine (Agrawal, Campbell, Safdi, et al., 2000). AGA consensus guidelines recommend misoprostol if tolerated or a PPI in patients at high risk for GI complications (Wilcox, Allison, Benzuly, et al., 2006) (see Table 6-2). The AGS recommends a PPI or misoprostol in older adults taking either a nonselective NSAID or a COX-2 selective NSAID with low-dose aspirin (AGS, 2009).
The use of a COX-2 selective NSAID may be as effective as gastroprotective co-therapy in some populations (Chan, Graham, 2004; Jacobsen, Phillips, 2004; Schnitzer, Burmester, Mysler, et al., 2004; Laine, White, Rostom, et al., 2008; Silverstein, Faich, Goldstein, et al., 2000; Simon, 2007; Simon, Fox, 2005). Healthy, lesion-free subjects were randomized to receive celecoxib 200 mg twice daily or naproxen 500 mg twice daily plus omeprazole 20 mg once daily, or placebo for 2 weeks (Goldstein, Eisen, Lewis, et al., 2005). Those taking celecoxib exhibited significantly fewer small bowel mucosal breaks than those who took naproxen plus omeprazole as determined by video capsule endoscopy; those taking placebo had the fewest breaks. A later study by these researchers reported similar findings—celecoxib 200 mg twice daily was associated with significantly fewer small bowel mucosal breaks than ibuprofen 800 mg three times daily plus omeprazole 20 mg once daily, or placebo for 2 weeks (Goldstein, Eisen, Lewis, et al., 2007). There were no significant differences between celecoxib and placebo in this study.
A COX-2 selective NSAID with gastroprotective co-therapy may be most beneficial to patients with a high or very high risk for GI complications. A prospective, double- blind trial (N = 441) recruited long-term nonselective NSAID users with arthritis admitted to the hospital with upper GI bleeding. After ulcer healing, all patients were given 200 mg of celecoxib twice daily and were randomly assigned to receive the PPI esomeprazole or placebo twice daily for 12 months. Combination therapy with the PPI was found to be more effective in preventing GI adverse effects than celecoxib alone, leading the researchers to recommend a COX-2 selective NSAID and a PPI in patients at high risk for recurrent GI bleeding who need to take an NSAID (Chan, Wong, Suen, et al., 2007).
Greater attention to identification of those who might benefit from gastroprotective therapy and surveillance of patient adherence to gastroprotective therapy are warranted. A review of good-quality meta-analyses and large observational studies found that patients with GI risk factors often do not receive a prescription for gastroprotection and that those who do are often nonadherent to the treatment plan (Moore, Derry, Phillips, et al., 2006).
Cardiovascular (CV) Effects
CV homeostasis is among the key processes influenced by the effects of COX-1 and COX-2 on arachidonic acid (Rodriguez, 2001) (see Section I and Figure I-3 on p. 6). COX-1, which is present (constitutive) in most tissues, mediates the production of thromboxane A2, which in turn promotes platelet aggregation, vasoconstriction, and smooth muscle proliferation (Bennett, Daugherty, Herrington, et al., 2005; Segev, Katz, 2004; Vardeny, Solomon, 2008). Conversely, COX-2 is induced at sites of inflammation where it is involved in the production of the prostaglandin prostacyclin from endothelial cells; prostacyclin is a potent vasodilator and antagonizes platelet aggregation, counteracting the effects of thromboxane A2 (Bennett, Daugherty, Herrington, et al., 2005; Rodriguez, 2001; Segev, Katz, 2004; Vardeny, Solomon, 2008).
Presumably, any drug that inhibits COX-2 (thereby reducing levels of prostacyclin) will have prothrombotic effects, and those that inhibit COX-2 to a much greater extent than COX-1 will promote thrombosis more than others because of a disturbance in the physiologic balance between prostacyclin and thromboxane A2 (Rodriguez, 2001; Scheiman, 2006; Segev, Katz, 2004). Because all NSAIDs inhibit COX-2 to some extent (Antman, DeMets, Loscalzo, 2005; Rodriguez, Patrignani, 2006; Vane, Warner, 2000; Warner, Mitchell, 2004, 2008; Vardeny, Solomon, 2008; Zhang, Ding, Song, 2006), all have prothrombotic effects; those classified as COX-2 selective NSAIDs and those nonselective NSAIDs with relatively greater effects at COX-2 probably have more intense prothrombotic effects than others (Bennett, Daugherty, Herrington, et al., 2005; FitzGerald, 2004; Pratico, Dogne, 2005; Scheiman, 2006; Segev, Katz, 2004; Vardeny, Solomon, 2008). The variability of COX-1 and COX-2 selectivity of a given NSAID is known as the selectivity index or the ratio of COX-1 selectivity to COX-2 selectivity (Patrono, Patrignani, Garcia, 2001; Warner, Mitchell, 2004) (Figure 6-1), and it is possible that this index provides a guide to the CV risk profile of the various NSAIDs. This hypothesis summarizes the so-called Mechanism-Based FitzGerald Hypothesis (Figure 6-2).
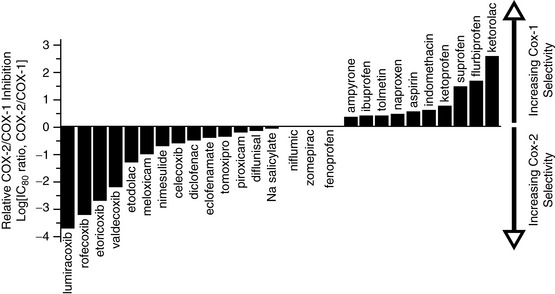
Figure 6-1 NSAID COX-1 and COX-2 selectivity. From Antman, E. M., DeMets, D., & Loscalzo, J. (2005). Cyclooxygenase inhibition and cardiovascular risk. Circulation, 112(5), 759-770. Modified from Warner, T. D., & Mitchell, J. A. (2004). Cyclooxygenases: New forms, new inhibitors, and lessons from the clinic. FASEB J, 18, 790-804.
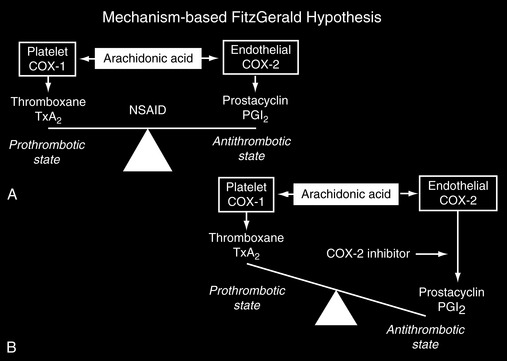
Figure 6-2 Mechanism-Based Fitzgerald Hypothesis. From Scheiman, J. M., Goldstein, J. L., Sung, J. J. Y, et al. (2006). The power of a new perspective on nonsteroidal anti-inflammatory drug-related gastrointestinal complications: Strategies for endoscopic and medical therapy. Medscape Sep 15. Data from FitzGerald, G. A. (2004). Coxibs and cardiovascular disease. N Engl J Med, 351(17), 1709-1711; FitzGerald, G. A., & Patrono, C. (2001). The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med, 345(6), 433-442; Rodriguez, L. A. (2001). The effect of NSAIDs on the risk of coronary heart disease: Fusion of clinical pharmacology and pharmacoepidemiologic data. Clin Exp Rheumatol, 19(6 Suppl 25), S41-S44.
This disruption in the balance between platelet thromboxane A2 and endothelial prostacyclin may explain, at least in part, the relatively high rate of CV adverse events that occurred with the COX-2 selective NSAIDs rofecoxib and valdecoxib. This toxicity led to their eventual withdrawal from the U.S. market (Linton, Fazio, 2002; FitzGerald, 2004; Segev, Katz, 2004).
Although the hypothesis linking the CV effects of COX isoenzymes to their varied effects on thromboxane A2 and prostacyclin provides a strong mechanism-based explanation for the observed toxicities, it is likely that other mechanisms also are involved (Joshi, Gertler, Fricker, 2007). For example, inhibition of prostacyclin by COX-2 also reduces the ability of the epithelium to defend against hypertension and atherosclerosis (FitzGerald, 2004; Psaty, Furberg, 2005) and may increase the risk of CV adverse effects on this basis.
The selectivity index also may explain some other differences in the incidences of adverse effects associated with each of the NSAIDs. For example, as Figure 6-1 shows, most of the nonselective NSAIDs have a tendency toward greater COX-1 than COX-2 selectivity, which accounts for their associated greater risk of GI toxicity and increased bleeding times. Presumably, additional studies of newer, highly selective COX-2 NSAIDs will demonstrate that these drugs reduce the risk of GI toxicity while simultaneously having a relatively higher risk of prothrombotic effects. Again, however, it must be emphasized that all NSAIDs, including those classified as nonselective NSAIDs (COX-1/COX-2 inhibitors), have some degree of COX-2 inhibition and associated risk of adverse CV events (Antman, DeMets, Loscalzo, 2005).
CV Risk Factors
All patients who are being considered for ongoing NSAID therapy should be carefully assessed for preexisting risk factors for vascular disease and closely monitored for CV adverse events during NSAID use, regardless of the type of NSAID administered (Andersohn, Suissa, Garbe, 2006; Brophy, Levesque, Zhang 2007; Crofford, Oates, McCune, et al., 2000; FitzGerald, Patrono, 2001; Gislason, Jacobsen, Rasmussen, et al., 2006). For example, Solomon and colleagues (2008) found that individuals with prior CV disease, hypertension, RA, chronic renal disease, chronic obstructive pulmonary disease, and those ≥ 80 years old all are at increased risk of CV events when taking NSAIDs. Assessment of these and other factors, and interventions to reduce risk and monitor over time, are important recommendations for NSAID therapy (Antman, Bennett, Daugherty, et al., 2007; Chan, Abraham, Scheiman, et al., 2008).
The First International Working Party on GI and CV Effects of NSAIDs and Anti-platelet Agents predefined high CV risk as the presence of established CV disease (e.g., prior myocardial infarction (MI), stroke, or angina) or an estimated 10-year CV risk of greater than 20% in patients without CV disease (Chan, Abraham, Scheiman, et al., 2008). The AHA and the American Stroke Association categorize CV risk factors as nonmodifiable, modifiable, or potentially modifiable (Goldstein, Adams, Alberts, et al., 2006). This provides a good format for consideration of CV risk prior to initiating NSAID therapy (see Table 6-4).
Table 6-4
Risk Factors for NSAID-Induced Adverse Cardiovascular Events
| Risk Level | Examples |
| Non-modifiable1 | Advanced age; sex; low birth weight; race/ethnicity; genetic factors4 |
| Modifiable2 | Noncerebrovascular atherosclerotic vascular disease (e.g., coronary heart disease, cardiac failure, symptomatic peripheral arterial disease)4; other cardiac conditions (e.g., previous MI, valvular heart disease, cardiomyopathy) 4; hypertension4; diabetes4; obesity4; cigarette smoke4; atrial fibrillation; dyslipidemia; asymptomatic carotid stenosis; sickle cell disease; rheumatoid arthritis; postmenopausal hormone replacement therapy; diet; nutrition; physical inactivity |
| Potentially modifiable3 | Metabolic syndrome; oral contraceptive use; sleep disordered breathing; migraine; hyperhomocysteinemia; elevated lipoproteins; inflammation; infection; toxins (alcohol or drug abuse, chemotherapy, some medications including NSAIDs)1 |
MI, myocardial infarction; NSAID, nonsteroidal antiinflammatory drug.
1Risk factors that are beyond the person’s ability to change
2Risk factors that are well documented as contributing to adverse CV events and within the individual’s ability to change or control effectively enough to significantly reduce risk
3Risk factors that can be changed or controlled but are less well documented in terms of their association with adverse CV events
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 199, St. Louis, Mosby. Data from Bhatt, D. L., Scheiman, J, Abraham, N. S., et al. (2008). ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of anti-platelet therapy and NSAID use. Am J Gastroenterol, 103(18), 2890-2907; Goldstein, L. B., Adams, R., Alberts, M. J., et al. (2006). Primary prevention of ischemic stroke. A guideline from the American Heart Association/American Stroke Association Stroke Council: Cosponsored by the Atherosclerotic Peripheral Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation, 113(24), e873-e923; Pearson, T. A., Blair, S. N., Daniels, S. R., et al. (2002). AHA guidelines for primary prevention of cardiovascular disease and stroke: 2002 update. Circulation, 106(4), 388-391; Schoken, D. D., Benjamin, E. J., Fonarow, G. C., et al. (2008). Prevention of heart failure. A scientific statement from the American Heart Association Councils on Epidemiology and Prevention, Clinical Cardiology, Cardiovascular Nursing, and High Blood Pressure Research; Quality of Care and Outcomes Research Interdisciplinary Working Group; and Functional Genomics and Translational Biology Interdisciplinary Working Group. Circulation, 117(19), 2544-2565; Solomon, D. H., Glynn, R. J., Rothman, K. J., et al. (2008). Subgroup analyses to determine cardiovascular risk associated with nonsteroidal anti-inflammatory drugs and coxibs in specific patient groups. Arthritis Rheum 59(6):1097-1104; Solomon, D. H., Karlson, E. W., Rimm, E. B., et al. (2003). Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation, 107(9), 1303-1307. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Nonmodifiable risks are those beyond the person’s ability to change, such as age, sex, and race. Advanced age is a major contributor, with the risk of stroke doubling with each decade after the age of 55 years. It is expected that the aging of the “baby boomer” population will become the biggest factor boosting the prevalence of heart failure (Schoken, Benjamin, Fonarow, et al., 2008). The aging process also brings other conditions that increase CV risk, including hypertension, diabetes, and renal insufficiency (McDermott, Fried, Simonsick, et al., 2000; Schoken, Benjamin, Fonarow, et al., 2008) (see renal effects later in this chapter). The AGS lists heart failure as an absolute contraindication and hypertension as a relative contraindication to the use of NSAIDs in older adults (AGS, 2009).
Other nonmodifiable risk factors, like age, are strongly influenced by related conditions and type of CV event. For example, stroke is more common in men than in women (Goldstein, Adams, Alberts, et al., 2006), whereas mortality from coronary heart disease is higher among women (Jousilahti, Vartiainen, Tuomilehto, et al., 1999). Genetic factors, including genetic disorders such as sickle cell anemia (James, 2000) also are important, both in terms of risk factors and CV events themselves. Although nonmodifiable risk factors cannot be changed, they help identify individuals who are at highest risk and who might benefit from aggressive disease prevention or treatment, as well as close monitoring during NSAID therapy (Goldstein, Adams, Alberts, et al., 2006).
Modifiable risk factors are those that are well documented as contributing to adverse CV events and are within the individual’s ability to change or control effectively enough to significantly reduce risk. Preexisting CV disease, hypertension, diabetes, and dyslipidemia are among the modifiable factors (Goldstein, Adams, Alberts, et al., 2006). Obesity, sedentary lifestyle, and diet interact with other physiologic abnormalities such as insulin resistance and a pro-inflammatory state to produce the metabolic syndrome, which has been separately identified as an important risk factor for CV events (Goldstein, Adams, Alberts, et al., 2006). Frank diabetes with poorly controlled glycemia is a potentially major factor (Fox, Coady, Sorlie, et al., 2007), as is hypertension alone (Schoken, Benjamin, Fonarow, et al., 2008). Hypertension, for example, causes a 2- to 3-fold increase in risk of heart failure, as well as an increased risk of coronary artery disease, and early detection and control of BP is perceived to be a key strategy for prevention of CV disease and catastrophic CV events (Pearson, Blair, Daniels, et al., 2002).
The role of hypertension as a mediator of both heart failure and coronary artery disease underscores the need for a careful assessment of CV risk factors in those considered for NSAID therapy. NSAIDs increase the risk of hypertension or worsen preexisting hypertension (Bombardier, Laine, Reicin, et al., 2000; Curhan, Willett, Rosner, et al., 2002; Dedier, Stampfer, Hankinson, et al., 2002; Simon, 2007; Whelton, Lefkowith, West, et al., 2006; Whelton, White, Bello, et al., 2002; White, Kent, Taylor, et al., 2002), and BP monitoring, with adjustment of antihypertensive therapy if needed, is essential to reduce NSAID-associated CV risk that is mediated through BP abnormalities (see pp. 205-206 for more discussion about NSAIDs and hypertension).
Cigarette smoking is another extremely well-documented, potent modifiable CV risk factor; both first-hand and second-hand smoke increase risk (Goldstein, Adams, Alberts, et al., 2006; Hermann, Krum, Ruschitzka, 2005; McAdam, Byrne, Morrow, et al., 2005). NSAID users who smoke have a higher incidence of CV adverse events than NSAID users who do not smoke (Chan, Manson, Albert, et al., 2006).
Some potentially modifiable factors are less well documented in terms of their association with adverse CV events, including obstructive sleep apnea and migraine headache (Goldstein, Adams, Alberts, et al., 2006). In addition to NSAIDs, several drugs have been implicated in heart disease, such as some chemotherapies and hormonal agents, including oral contraceptives; IV drug abuse and alcohol abuse also may predispose to heart disease (Schoken, Benjamin, Fonarow, et al., 2008).
Unfortunately, diseases with an inflammatory component that may benefit from NSAID therapy, such as RA and systemic lupus erythematosus (SLE), have also been shown to produce elevated CV risk (D’Cruz, 2006; de Carvalho, Bonfa, Borba, 2008; Solomon, Karlson, Rimm, et al., 2003). A large cohort study of over 114,000 women who participated in the Nurses’ Health Study (Colditz, 1995) found that those with RA had an increased risk for MI (Solomon, Karlson, Rimm, et al., 2003). This led the researchers to recommend aggressive CV disease-prevention strategies in individuals with RA. CV risk factors for individuals with SLE are many and include dyslipoproteinemia with low HDL (high-density lipoprotein) cholesterol and increased triglycerides (de Carvalho, Bonfa, Borba, 2008; Thiagarajan, 2001) as well as an association with autoantibodies to oxidized LDL (low-density lipoprotein) cholesterol (Svenungsson, Jensen-Urstad, Heimburger, et al., 2001).
The link between CV risk and inflammatory diseases is explained, at least in part, by evidence that shows inflammation is a prominent feature of atherosclerotic plaque formation (Solomon, Karlson, Rimm, et al., 2003; Svenungsson, Jensen-Urstad, Heimburger, et al., 2001; Vasan, Sullivan, Roubenoff, et al., 2003), such that atherosclerosis has been called an inflammatory condition of the vessel wall (Thiagarajan, 2001). A variety of inflammatory factors associated with increased CV risk, such as elevated C-reactive protein concentration, are also elevated in RA and SLE (Svenungsson, Jensen-Urstad, Heimburger, et al., 2001).
Potentially modifiable CV risk factors commonly co-exist and may potentiate each other, further highlighting the need to have a comprehensive medical assessment as part of safe NSAID prescribing. For example, women who smoke and use oral contraceptives are at 1.3 times greater CV risk than nonsmoking women who use oral contraceptives (Goldstein, Adams, Alberts, et al., 2006). Alcohol intake may increase CV risk by increasing BP (Schoken, Benjamin, Fonarow, et al., 2008). Obese individuals often experience multiple comorbidities that represent elevated risk including sleep-disordered breathing, physical inactivity, and diabetes (Schoken, Benjamin, Fonarow, et al., 2008).
CV Effects of COX-2 Selective NSAIDs
Shortly after the release of rofecoxib in 1999, the 12-month Vioxx Gastrointestinal Outcomes Research (VIGOR) trial, which was designed to compare GI adverse effects associated with naproxen and rofecoxib, showed a 50% reduction in serious GI events in the subjects who were given rofecoxib (Bombardier, Laine, Reicin, et al., 2000). However, an unexpected and disturbing finding was a 5-fold increase in thromboembolic events, such as acute MI, in the rofecoxib group. The VIGOR trial did not have a placebo control group, which lent plausibility to the frequently-offered explanation that naproxen, a nonselective NSAID, was cardioprotective and thus produced fewer CV events than rofecoxib (Graham, 2006; Graham, Campen, Hui, et al., 2005). Later analysis, however, suggested that naproxen is not cardioprotective (Graham, Campen, Hui, et al., 2005; McGettigan, Henry, 2006; Ray, Stein, Hall, et al., 2002; Salpeter, Gregor, Ormiston, et al., 2006), at least at typical doses (Chan, Abraham, Scheiman, et al., 2008). Graham (2006) proposed that a more accurate interpretation of the VIGOR findings would have been that rofecoxib increased CV risk by 5-fold rather than that the CV risk associated with naproxen was one-fifth that of rofecoxib.
The Adenomatous Polyp Prevention On Vioxx (APPROVe) study was undertaken to determine if long-term (3 years) rofecoxib treatment would reduce the risk of recurrent adenomatous polyps in patients with a history of colorectal adenomas (Bresalier, Sandler, Quan, et al., 2005). Over 2500 patients were randomized to receive either rofecoxib or placebo. During the first 18 months of treatment, the CV adverse event rates were similar among the two groups; however, after 18 months, data suggested a greater number of MIs and ischemic cerebrovascular events occurred in the rofecoxib group. A later systematic review contradicted the suggestion that vascular risk with rofecoxib occurred only after months of treatment (McGettigan, Henry, 2006). Increasing evidence of elevated CV risk prompted the withdrawal of rofecoxib from the worldwide market in September 2004 (Graham, 2006).
Valdecoxib, which is the active metabolite of the parenteral COX-2 selective NSAID parecoxib (Dynastat) available in Europe, was withdrawn from the U.S. market in 2005 following research showing CV events similar to rofecoxib (Vardeny, Solomon, 2008). Studies showing an increase in CV events in patients who were given parecoxib and valdecoxib perioperatively (Nussmeier, Whelton, Brown, et al., 2005; Ott, Nussmeier, Duke, et al., 2003) led to recommendations against the use of any COX-2 selective NSAIDs following high-risk open heart surgery (U.S. FDA, 2007). (See Chapter 8 for a discussion of perioperative NSAID use.) Valecoxib was withdrawn from the market on the basis of the CV risk as well as reported cases of cutaneous hypersensitivity (see pp. 207-208 for discussion of nonopioid hypersensitivity).
Celecoxib currently is the only COX-2 selective NSAID in the United States. The so-called CLASS study randomized 8059 patients with OA or RA to receive a 6-month trial of celecoxib, ibuprofen, or diclofenac (Silverstein, Faich, Goldstein, et al., 2000). Patients were allowed to continue cardioprotective therapy during the study period. No difference in CV events was found among the three NSAIDs, and new-onset or aggravation of preexisting hypertension was lowest in the celecoxib-treated group. Other studies have shown similar results (Hudson, Rahme, Richard, et al., 2007; Solomon, Schneeweiss, Glynn, et al., 2004; Steinbach, Lynch, Phillips, et al., 2000; Whelton, White, Bello, et al., 2002). These data questioned whether the risk of CV toxicity identified during rofecoxib and valdecoxib therapy, which suggested a class effect (i.e., associated with the so-called COX-2 selective drugs), was indeed this, or rather a risk that attended some drugs and not others.
Numerous other studies followed the early efficacy trials and were intended to clarify the risk of CV events for individual drugs and the classes they occupy. Although some of the data are conflicting, a broad picture of the CV risk associated with NSAID therapy has emerged.
Studies of insurance claims data provide one source of evidence pertaining to this issue. An examination of data of Medicare beneficiaries, which consisted of new users of COX-2 selective NSAIDs (N = 76,082), new users of nonselective NSAIDs (N = 53,014), and nonusers of any NSAIDs (N = 46,558), suggested that most NSAIDs, when taken at standard doses, do not significantly increase the risk of CV events (Solomon, Glynn, Rothman, et al., 2008). These data showed that rofecoxib was associated with the highest CV event rates and that naproxen was associated with the lowest—even lower than nonusers. Certain patient characteristics (prior CV disease, hypertension, RA, chronic renal disease, chronic obstructive pulmonary disease, and age 80 years or older) were associated with elevated risk as well. Other large database studies (Ray, Stein, Hall, et al., 2002; Solomon, Schneeweiss, Glynn, et al., 2004; Graham, Campen, Hui, et al., 2005) suggested that the incidence of acute MI with celecoxib use was actually lower than that of other NSAIDs (Simon, 2007).
An extensive systematic review of observational studies that investigated CV events associated with a variety of NSAIDs found that celecoxib at doses of 200 mg/day or less was not associated with CV increased risk and that naproxen was not associated with any reduced CV risk (McGettigan, Henry, 2006). The highest risk among the nonselective NSAIDs was with commonly used doses of diclofenac (e.g., 150 mg daily). This finding may be notable in light of data indicating a relatively higher COX-2 selectivity of this drug than other nonselective NSAIDs (Graham, 2006; McGettigan, Henry, 2006; Vardeny, Solomon, 2008) (see Figure 6-1). Meloxicam, another long-acting nonselective NSAID that preferentially inhibits COX-2, at least in lower prescribed doses, has shown similar efficacy and a better GI profile than the comparable nonselective NSAIDs piroxicam and diclofenac and less peripheral edema and weight gain when compared with diclofenac (Smith, Baird, 2003). Studies are needed to evaluate this and other nonselective NSAIDs that possess relatively higher levels of COX-2 inhibitory activity.
Data from randomized controlled trials of celecoxib and other NSAIDs have yielded mixed results. A meta-analysis of randomized clinical trials evaluated the incidence of CV events in nearly 7500 patients exposed to celecoxib 200 to 800 mg daily; nearly 14,000 patients treated with diclofenac, ibuprofen, naproxen, ketoprofen (Orudis), or loxoprofen (available outside of the United States); and over 4000 patients who were given placebo (White, West, Borer, et al., 2007). The analysis failed to demonstrate a difference in the incidence of CV events associated with celecoxib compared with other NSAIDs or compared with placebo for up to 1 year of treatment exposure. Use of aspirin or presence of CV risk factors did not alter the findings.
In contrast, placebo-controlled trials designed to study a potential role for celecoxib in the prevention of colorectal adenoma revealed an increased CV risk in patients who received celecoxib. The Prevention of Colorectal Sporadic Adenomatous Polyps (PreSAP) trial demonstrated a reduced recurrence of colorectal adenomas in patients who received a single 400-mg dose of celecoxib daily; however, CV events occurred more often in patients who received celecoxib (2.5%) than in those who received placebo (1.9%) (Arber, Eagle, Spicak, et al., 2006). More concerning was the Adenoma Prevention with Celecoxib (APC) trial, which randomized patients to receive celecoxib 200 mg or 400 mg twice daily or placebo (Bertagnolli, Eagle, Zauber, et al., 2006). As in the PreSAP trial, a reduction in the recurrence of colorectal adenomas was noted; however, serious adverse CV events occurred in 18.8%, 20.4%, and 23% of those who received placebo, 200 mg of celecoxib twice daily, and 400 mg of celecoxib twice daily. This increase in CV events led the researchers to caution against the routine use of celecoxib for prevention of colorectal adenomas. Celecoxib administration in several long-term trials, including the APC and PreSAP trials, was halted as a result of CV events (Solomon, Wittes, Finn, et al., 2008). (See Chapter 7 for more on NSAIDs and prevention of cancer.)
The PreSAP and APC trials elucidated interesting findings related to the impact of both dose and dosing regimens on the level of CV risk associated with celecoxib (Solomon, Pfeffer, McMurray, et al., 2006; Solomon, Wittes, Finn, et al., 2008; Vardeny, Solomon, 2008). Vardeny and Solomon (2008) pointed out that the evidence for CV risk in the PreSAP trial, which administered a single 400-mg celecoxib dose daily, was less compelling than that of the APC trial, which showed significant risk associated with a 200-mg or 400-mg twice-daily dosing regimen. This was demonstrated in an independent review of combined data from the APC and PreSAP trials that found a hazard ratio (estimate of relative risk) for death from MI, stroke, or heart failure of 1.3 in patients taking 400 mg once daily (PreSAP), 2.6 in those taking 200 mg twice daily (APC), and 3.4 in those taking 400 mg twice daily (Solomon, McMurray, Pfeffer, et al., 2005). Similarly, an analysis of pooled data from nearly 8000 patients in six placebo-controlled trials, which included the APC and PreSAP data, assessed CV risk associated with three dosing regimens of celecoxib and found the lowest, intermediate, and highest risk associated with dosing regimens of 400 mg daily, 200 mg twice daily, and 400 mg twice daily, respectively (Solomon, Wittes, Finn, et al., 2008). Patients with highest baseline risk had an increased relative risk for CV events.
Notwithstanding the conflicting data, these studies suggest that there is some CV risk associated with celecoxib treatment, as there is with other NSAIDs. The finding of dose-dependency is relatively strong evidence of a pharmacologic effect. It is curious, however, that two studies observe that lower risk is associated with once-daily dosing compared with twice-daily dosing at the same overall daily dose (400 mg). This also may have a pharmacologic explanation in that the maximum plasma concentration of celecoxib is reached in 90 minutes and the half-life is just 1.5 hours (Paulson, Hribar, Liu, et al., 2000). Because prostacyclin levels require approximately 12 hours to recover following a single oral dose of celecoxib (McAdam, Catella-Lawson, Mardini, et al., 1999), it has been proposed that once-daily dosing (but not twice-daily dosing) of celecoxib may allow enough prostacyclin recovery and normalization to attenuate the prothrombotic effect (Grosser, Fries, FitzGerald, 2006; Vardeny, Solomon, 2008). Although further research is needed, the findings in other studies that utilized once-daily dosing support this theory (Sowers, White, Pitt, et al., 2005; Whelton, Fort, Puma, et al., 2001).
The evidence overall suggests that there is CV risk during all NSAID therapies, however the link with COX-2 inhibition suggests that drugs with COX-2 selectivity must be particularly scrutinized for this effect. This has been done with newer COX-2 selective NSAIDs, such as etoricoxib and lumiracoxib (Andersohn, Suissa, Garbe, 2006; Cannon, Curtis, FitzGerald, et al., 2006; Farkouh, Kirshner, Harrington, et al., 2004; Laine, Curtis, Cryer, et al., 2007; Schnitzer, Burmester, Mysler, et al., 2004). These drugs were not available in the United States at the time of publication (Medscape Medical News, 2005; Wood, 2007).
The TARGET study randomized over 18,000 patients with OA to receive lumiracoxib, naproxen, or ibuprofen (Farkouh, Kirshner, Harrington, et al., 2004). Patients were allowed to take low-dose aspirin during the study. The incidence of adverse CV events was low and did not differ among the NSAID groups, irrespective of aspirin use. Though not statistically significant, there were more nonfatal MIs in the lumiracoxib group than in the naproxen nonaspirin group. The lack of a placebo arm in this study makes it difficult to ascertain actual CV risk associated with lumiracoxib.
The MEDAL program evaluated pooled data from three trials involving 34,701 patients with OA and 9787 with RA who were given etoricoxib or diclofenac at recommended doses for a mean duration of 18 months (Cannon, Curtis, FitzGerald, et al., 2006). Patients in the two groups were reported to have almost identical rates of thrombotic CV events (320 patients in the etoricoxib group; 323 patients in the diclofenac group). This study has been criticized for the lack of a placebo arm and the use of diclofenac as the comparator NSAID (Graham, 2006; Hughes 2006; Rodriquez, Patrignani, 2006). Given the suggestion that diclofenac has a relatively high CV risk (Hernandez-Diaz, Varas-Lorenzo, Rodriguez, 2006; Kearney, Baigent, Godwin, et al., 2006; McGettigan, Henry, 2006), comparability between this drug and etoricoxib does not establish the safety of the latter drug (Graham, 2006). Although diclofenac is widely prescribed and does not appear to interfere with the antiplatelet effects of low-dose aspirin (Cannon, Curtis, FitzGerald, et al., 2006) (see later discussion regarding NSAID interference with cardioprotective aspirin), its use in this study complicates interpretation of the results.
Class Effect.: As noted, an important controversy surrounding the emerging data on NSAID CV risk relates to the most useful way to categorize the findings in terms of drug effects vs. drug class effects (Helstrom, Rosow, 2006; Porter, 2005; Pratico, Dogne, 2005). Some researchers claim there is enough evidence showing a “cardiovascular hazard” with three of the COX-2 selective NSAIDs—rofecoxib, celecoxib, and parecoxib—to support the hypothesis that the adverse CV events associated with these drugs are related to a class effect (Pratico, Dogne, 2005). Others view this as problematic given the conflicting data about celecoxib and the evidence that at least some of the drugs classified as nonselective NSAIDs have risk.
In an effort to address this controversy, a meta-analysis was performed that included data from 114 trials involving 116,094 patients who received the COX-2 selective NSAIDs rofecoxib, celecoxib, etoricoxib, lumiracoxib, or valdecoxib plus parecoxib. This analysis yielded insufficient evidence to claim a COX-2 selective class effect (Zhang, Ding, Song, 2006), a conclusion supported by others (Hudson, Rahme, Richard, et al., 2007). Despite some conflicting data that suggest minimal to no risk among the nonselective NSAIDs (NIH, 2004; Salpeter, Gregor, Ormiston, et al., 2006), a recent meta-analysis of randomized trials again showed that COX-2 selective NSAIDs, which as a group impose a 2-fold increased risk of MI compared with placebo and naproxen, did not increase risk compared with non-naproxen NSAIDs (Laine, White, Rostom A, et al., 2008).
Despite these meta-analyses and large epidemiologic studies, the debate surrounding CV risk as drug effect vs. COX-2 selective class effect continues. More head-to-head comparisons between the various NSAIDs are required to better address this ongoing controversy (Porter, 2005). At the present time, the most prudent conclusion continues to be that CV risk is inherent in the capacity to inhibit COX-2, and thereby reduce tissue levels of prostacyclin. Because both nonselective NSAIDs and COX-2 selective NSAIDs have COX-2 effects, and because the various so-called COX-2 selective drugs vary in the extent to which COX-2 is blocked relative to COX-1, it is best to assume that all NSAIDs have CV risk and that the risk varies across drugs, even within classes. The evidence of elevated risk prompted the U.S. FDA to issue almost identical product label warnings for all NSAIDs, irrespective of class (Dajani, Islam, 2008).
NSAID Selection with Consideration of CV Risk
This conclusion obligates clinicians to be aware of evidence that supports different risk profiles for different NSAIDs and to consider relative risks when recommending a specific drug in clinical practice. The First International Working Party on GI and CV Effects of NSAIDs and Anti-platelet Agents suggested that either nonselective NSAIDs or COX-2 selective NSAIDs could be appropriate in patients with no evidence of elevated GI risk and average CV risk; naproxen was preferred in patients with no GI risk but high CV risk (Chan, Abraham, Scheiman, et al., 2008). The panel rated the use of COX-2 selective NSAIDs in patients with high CV risk as uncertain or inappropriate (see later discussion of NSAID selection in the presence of both GI and CV risk). As mentioned, an extensive review of Medicare beneficiary data confirmed the safety of naproxen from a CV perspective as well (Solomon, Glynn, Rothman, et al., 2008).
The AHA proposes a “stepped care” approach for the treatment of musculoskeletal pain in individuals with known CV disease or risk factors for ischemic heart disease (Antman, Bennett, Daugherty, et al., 2007). Others have incorporated similar recommendations into their guidelines for the management of high-risk cardiac patients (Anderson, Adams, Antman, et al., 2007). The first step of the AHA approach recommends acetaminophen, aspirin, tramadol, or short-term opioid analgesics. Nonacetylated salicylates, such as choline magnesium trisalicylate or salsalate (Disalcid), are a second-step option. If this does not control pain, a nonselective NSAID with low COX-2 selectivity such as naproxen or ibuprofen is recommended. The use of a COX-2 selective NSAID is suggested as a last choice. Of note is a review of the literature that suggested the use of a COX-2 selective NSAID with cardioprotective aspirin in patients with CV risk is preferential to nonselective NSAIDs (Strand, 2007). However, some experts have questioned the rationale of using a COX-2 selective NSAID with aspirin at all in patients with high CV risk citing a lack of research showing aspirin can sufficiently mitigate the absolute CV risk imposed by COX-2 selective NSAIDs (Hughes, 2007).
Other general AHA recommendations when NSAIDs are used are to select patients at low risk for thrombotic events, prescribe the lowest dose required to control symptoms, add cardioprotective aspirin (with a PPI) to patients at increased risk of thrombotic events, and regularly monitor for sustained hypertension, edema, worsening renal function, and GI bleeding. The AHA cautions that effective pain relief with the use of NSAIDs may come at the cost of an increase in risk for CV or cerebrovascular complications (Antman, Bennett, Daugherty, et al., 2007).
NSAID Selection in Presence of Both CV and GI Risk
Patients who are at risk for both GI and CV adverse effects present special challenges in determining how to proceed with NSAID therapy. Although the use of naproxen or ibuprofen combined with nonaspirin antiplatelet therapy (e.g., clopidogrel [Plavix]) has been suggested (Hughes, 2006), there are few data that support this approach.
The expert panel of the First International Working Party on GI and CV Effects of NSAIDs and Anti-platelet Agents suggests that the initial choice of an NSAID should depend on the patient’s CV risk, whereas the addition of gastroprotective therapy depends on the number and severity of GI risk factors (Chan, Abraham, Scheiman, et al., 2008). The panel rated naproxen plus a PPI or misoprostol as appropriate, regardless of patient age, in patients with both CV and GI risks. Non-naproxen NSAIDs and COX-2 selective NSAIDs, with or without gastroprotective therapy, were rated as uncertain or inappropriate in such patients. Some researchers recommend avoiding full doses of NSAIDs with a long half-life, such as naproxen (Fick, Cooper, Wade, et al., 2003).
A retrospective cohort study using government databases including patients 65 years and older concluded that celecoxib may be safer from a GI perspective than other NSAIDs in older adults receiving cardioprotective aspirin to minimize CV risk (Rahme, Bardou, Dasgupta, et al., 2007). Others have suggested a low-dose COX-2 selective NSAID plus low-dose aspirin plus a PPI if NSAID therapy is used in patients with major GI and CV risk factors (Laine, White, Rostom, et al., 2008).
It is important to remember that avoiding NSAIDs altogether may be the best approach for high-risk patients (Laine, White, Rostom, et al., 2008). In fact, the expert panel of the First International Working Party on GI and CV Effects of NSAIDs and Anti-platelet Agents recommends avoidance of NSAID therapy entirely in patients with high CV risk and multiple GI risk factors (e.g., patients older than age 70 who are receiving concomitant corticosteroids, low-dose aspirin, and other antiplatelet agents or anticoagulants) (Chan, Abraham, Scheiman, et al., 2008). Opioid analgesics are among the safest analgesics from a GI and CV perspective and should be considered in patients with pain who present with significant risk for NSAID-induced adverse effects.
Hematologic Effects and Cardioprotective Aspirin
NSAIDs have the potential to produce cardioprotection through COX-1 inhibition of thromboxane; however, 95% suppression of COX-1 is required to inhibit thromboxane-induced platelet aggregation (Salpeter, Gregor, Ormiston, et al., 2006). Aspirin reaches this level of suppression, producing an irreversible effect on platelets and a reported increase in bleeding time for up to 7 days after the last dose (i.e., until the platelets are replaced with new) (Sun, Crowther, Warkentin, et al., 2005). Nonselective NSAIDs inhibit thromboxane within a range of 59% to 95%, and their inhibition is reversible and not maintained over the dosing interval (Salpeter, Gregor, Ormiston, et al., 2006). As a result, these drugs should not be used in an effort to provide a therapeutic antiplatelet effect. The AHA recommends immediate-release, nonenteric coated aspirin at cardioprotective doses (i.e., 81 mg) for patients at increased risk of thrombotic events (Antman, Bennett, Daugherty, et al., 2007).
Ibuprofen (400-mg doses) competes with aspirin’s binding site on the platelet and has been found to interfere with aspirin’s cardioprotective (antiplatelet) effect; therefore, patients taking ibuprofen concomitantly with cardioprotective aspirin should be instructed to take the ibuprofen 30 minutes to 2 hours after aspirin intake (Catella-Lawson, Reilly, Kapoor, et al., 2001; Cryer, Berlin, Cooper, et al., 2005; Hong, Gengo, Rainka, et al., 2008) or at least 8 hours before (U.S. FDA, 2006). The AGS recommends that older adults taking cardioprotective aspirin avoid ibuprofen (AGS, 2009).
Although acetaminophen, diclofenac (Antman, Bennett, Daugherty, et al., 2007; Catella-Lawson, Reilly, Kapoor, et al., 2001), and celecoxib (Wilner, Rushing, Walden, et al., 2002) have been shown not to interfere with platelet aggregation induced by low-dose aspirin, a subgroup analysis from a 5-year randomized, placebo-controlled trial (N = 22,071 males) indicated that regular NSAID use (more than 60 days/year), but not intermittent NSAID use (1-59 days/year), interfered with the clinical benefits of aspirin (325 mg every other day) in the prevention of first MI (Kurth, Glynn, Walker, et al., 2003. One study of naproxen found that this drug interfered with the inhibitory effect of aspirin on platelet COX-1 activity (Capone, Sciulli, Tacconelli, et al., 2005), but another did not (Rauscher, 2007). Further studies are needed to determine if other NSAIDs interact with cardioprotective aspirin. See Chapter 8 for a discussion of preoperative discontinuation of aspirin therapy.
NSAIDs with Minimal Effect on Platelet Aggregation
Although NSAIDs do not block platelet function sufficiently to be useful as antithrombotic agents, most of the nonselective NSAIDs do interfere enough to increase bleeding time. For this reason, these drugs may carry relatively higher risk for patients with bleeding disorders, such as hemophilia. In such populations, the least risky drugs classified as nonselective NSAIDs are presumably those that are least likely to exert a COX-1-mediated inhibition of platelet function, such as nabumetone, meloxicam, choline magnesium trisalicylate, magnesium salicylate (Arthriten, Backache, Doan’s), and salsalate (Disalcid). The COX-2 selective NSAIDs (e.g., celecoxib) have no effect on bleeding time and should be considered as well if not contraindicated (Visser, Goucke, 2008) (see Chapter 8 for perioperative NSAID use).
Renal Effects and Hypertension
The COX isoenzymes play an important role in renal function through their effects on prostaglandin formation (Barkin, Buvanendran, 2004; Brater, 2002; DeMaria, Weir, 2003). COX-2, the isoenzyme that is largely inducible in response to inflammation, is constitutively expressed in the human kidney (Brune, 2003; Zhang, Ding, Song, 2006). It is present in the ascending limb of the loop of Henle, macula densa, and afferent arteriole and produces prostaglandins that inhibit sodium reabsorption and have diuretic effects (DeMaria, Weir, 2003). COX-1 is located in the glomerulus and afferent arteriole where it produces prostaglandins that affect renal homeostasis and promote dilation of the renal vascular bed (DeMaria, Weir, 2003).
The four categories of NSAID-induced renal toxicity are acute ischemic renal insufficiency (most commonly associated with perioperative use of NSAIDs), acute interstitial nephritis, analgesic associated nephropathy, and progressive hypertensive nephropathy (Forrest, Camu, Greer, et al., 2002). The most common adverse physiologic effects are reduced renal blood flow, glomerular filtration rate (GFR), and sodium and potassium excretion. These changes potentially can result in fluid retention, edema, hypertension, and hyperkalemia (DeMaria, Weir, 2003; Laine, White, Rostom, et al., 2008).
Endogenous renal prostaglandin synthesis does not have a significant role in maintaining optimal GFR and renal blood flow in individuals with normal CV, hepatic, endocrine, and renal function, as long as adequate volume and sodium stores exist (Helstrom, Rosow, 2006; Launay-Vacher, Karie, Fau, et al., 2005). In these healthy individuals, NSAID-induced renal effects are usually minor and transient (Forrest, Camu, Greer, et al., 2002; Helstrom, Rosow, 2006; Lee, Cooper, Craig, et al., 2007). There appears to be no difference in these effects between nonselective and COX-2 selective NSAIDs (Helstrom, Rosow, 2006).
Overall, NSAID-induced adverse renal effects occur in approximately 1% to 5% of NSAID users (DeMaria, Weir, 2003). Large epidemiologic studies provide some detail. For example, a cohort study of data from the 14-year Physicians’ Health Study (N = 11,032) revealed no increased risk of renal dysfunction with long-term moderate use (3 to 4 pills/week) of acetaminophen, aspirin, or NSAIDs in men who had no preexisting renal impairment (Rexrode, Buring, Glynn, et al., 2001). In contrast, a large case-control study of 72,114 men age 45 and older (Verhamme, Dieleman, Van Wijk, et al., 2005) showed that current use of NSAIDs was associated with a 2-fold increased risk of acute urinary retention. The latter study used the primary health care database in the Netherlands to compare cases with 10 age- and time-matched controls and found that the highest risk was in men who had recently started taking NSAIDs and in those who were taking a dose equal to or higher than the recommended daily dose; past use of NSAIDs and low-dose aspirin use were not associated with risk, and potential confounding factors, such as concomitant medication use (including opioids) and history of urinary tract infection, did not modify the results.
A variety of conditions may increase the risk of nephrotoxicity associated with NSAID use. In the setting of acute or chronic volume depletion, prostaglandin synthesis is necessary to maintain adequate renal blood flow (“prostaglandin dependence”) (Helstrom, Rosow, 2006). NSAID inhibition of prostaglandin synthesis in such patients can cause acute renal ischemia and acute renal failure (ARF) (Helstrom, Rosow, 2006). It is important to remember that, although NSAID-induced ARF is most often discussed in the literature as a perioperative adverse event, it can develop from both short- and long-term NSAID administration (McQuay, Moore, 2004) (see Chapter 8 for discussion of ARF).
Adverse renal effects also are more likely to occur in patients who have co-morbidities, such as diabetes, hypertension, cirrhosis, ascites, or congestive heart failure (Brater, 2002; Brune, 2003; Launay-Vacher, Karie, Fau, et al., 2005; Solomon, Schneeweiss, Levin, et al., 2004). Some experts have suggested NSAIDs be avoided entirely in patients with cirrhosis, congestive heart failure, or renal insufficiency (Laine, White, Rostom, et al., 2008). The AGS lists heart failure and chronic kidney disease as absolute contraindications and hypertension as a relative contraindication to the use of NSAIDs in older adults (AGS, Panel on Pharmacological Management, 2009). Because NSAIDs do not adversely affect GFR in individuals with normal renal function (Baker, Cotter, Gerrard, et al., 2005), including older adults, age alone is not a risk factor for NSAID-induced renal impairment (Brater, 2002); however, older adults are more likely to have risk factors such as volume depletion and chronic medical illness, and those who are prescribed NSAIDs should be regularly assessed for adverse renal effects (Barkin, Buvanendran, 2004).
Studies have evaluated class-selective and drug-selective risk of NSAID-induced renal toxicity. Although a small placebo-controlled study (N = 67) of older adults showed no significant differences between the nonselective NSAID naproxen and the COX-2 selective NSAIDs rofecoxib and celecoxib in terms of renal adverse effects (Schwartz, Vandormael, Malice, et al., 2002), most research has shown a higher risk for adverse renal effects with rofecoxib than other NSAIDs (Solomon, Schneeweiss, Levin, et al., 2004). Shortly after the release of the first COX-2 selective NSAIDs, an analysis of a safety database found that celecoxib, ibuprofen, and diclofenac had similar cardiorenal safety profiles, but rofecoxib possessed significantly greater renal toxicity (water retention, abnormal renal function, renal failure, cardiac failure, and hypertension) (Zhao, Reynolds, Lejkowith, et al., 2001). Similar findings were revealed in a case-controlled (4:1) study of 3914 patients with preexisting hypertension (Solomon, Schneeweiss, Levin, et al., 2004). Rofecoxib was associated with a significant risk of new hypertension, but there was no association between celecoxib use and the development of hypertension. In addition, relative risk was twice as high in patients with co-morbidities and advanced age taking rofecoxib compared with celecoxib. A nested, case-control study of individuals hospitalized for ARF found that the risk of ARF for all NSAIDs was highest during the first 30 days of NSAID use and was comparable for rofecoxib, naproxen, and nonselective, non-naproxen NSAIDs but was borderline lower for celecoxib (Schneider, Levesque, Zhang, et al., 2006).
Other research of patients with preexisting hypertension further suggests that celecoxib is associated with a relatively low risk of renal adverse effects. A study was conducted to evaluate the cardiorenal effects of COX-2 selective NSAIDs in 810 older patients (≥ 65 years old) with OA who had well-controlled hypertension and were taking antihypertensive medication (Whelton, Fort, Puma, et al., 2001). The patients were randomized to receive either celecoxib (200 mg once daily) or rofecoxib (25 mg once daily). Both groups exhibited significant peripheral edema, but twice as many of the rofecoxib-treated patients than celecoxib-treated patients did so, and 17% and 11% of those receiving rofecoxib and celecoxib, respectively, experienced a significant increase in systolic BP. Another study showed new-onset or worsening edema with weight gain occurring in 7.7% and 4.7% of individuals taking antihypertensive medications who received rofecoxib and celecoxib, respectively (Whelton, White, Bello, et al., 2002). Neither celecoxib nor rofecoxib caused significant increases in BP in patients receiving calcium channel antagonists or diuretic monotherapy; however, rofecoxib caused the greatest increase in systolic BP in patients receiving angiotensin-converting enzyme (ACE) inhibitors or beta blockers. A randomized, placebo-controlled study found celecoxib (200 mg twice daily) was not associated with statistically or clinically significant destabilization of BP in patients treated with the ACE inhibitor lisinopril (Zestril) (White, Kent, Taylor, et al., 2002). Similarly, a randomized-controlled study of patients with medication-stabilized hypertension, OA, and type-2 diabetes demonstrated that rofecoxib but not celecoxib or naproxen induced a significant increase in BP (Sowers, White, Pitt, et al., 2005). Although destabilization of existing hypertension occurred to some extent in all three groups, it was most prominent in those taking rofecoxib. These studies underscore the likelihood of drug-selective differences and the importance of careful monitoring of BP and renal function in patients receiving NSAIDs while being treated for hypertension (Barkin, Buvanendran, 2004).
Other studies have shown that celecoxib has a relatively low risk of cardiorenal adverse effects. A meta-analysis of 114 randomized trials involving 116,094 patients compared the cardiorenal adverse effects among the various COX-2 selective NSAIDs rofecoxib, celecoxib, etoricoxib, lumiracoxib, and valdecoxib plus parecoxib (Zhang, Ding, Song, 2006). Rofecoxib was associated with all of the adverse renal endpoints in a time- and dose-related manner with increased risk of peripheral edema and renal dysfunction. Celecoxib, on the other hand, was associated with a lower risk of cardiorenal dysfunction than the controls in this analysis. A study evaluating patients with arthritis showed a higher incidence of adverse renal effects in patients taking diclofenac (34.8%) than in those taking celecoxib (24.3%) (Chan, Hung, Suen, et al., 2004). The COX-2 selectivity of diclofenac is similar to celecoxib, notwithstanding its labeling as a nonselective NSAID (see Figure 6-1). The database from the previously mentioned CLASS study (Silverstein, Faich, Goldstein, et al., 2000) (N = 8059) was analyzed to assess the cardiorenal effects of higher-than-clinically-indicated doses of celecoxib (400 mg twice daily) and standard doses of ibuprofen (800 mg 3 times daily) and diclofenac (75 mg twice daily) (Whelton, Lefkowith, West, et al., 2006). The incidence of serious cardiorenal complications was reported to be low in all groups, and the celecoxib-treated patients experienced the lowest risk of cardiorenal adverse effects, particularly those with prerenal compromise.
The previously discussed APC and PreSAP trials underscore that both dose and dosing regimens of celecoxib influence BP (Solomon, Pfeffer, McMurray, et al., 2006). Whereas there were no significant BP changes in patients who took a single 400-mg dose of celecoxib once daily in the PreSAP trial, the twice-daily treatment groups in the APC trial demonstrated a pattern of BP elevation. Patients in the APC treatment group who received celecoxib in doses of 200 mg twice daily experienced an increase in BP of 2.0 mm Hg and 2.6 mm Hg at 1 and 3 years, respectively, and those in the group that received 400 mg twice daily experienced BP elevations of 2.9 mm Hg and 5.2 mm Hg at 1 and 3 years, respectively (see pp. 200-203 for explanation of lower risk associated with once-daily dosing compared with twice-daily dosing of the same overall total daily dose of celecoxib). Solomon and colleagues (2008) reinforce that the timing of BP measurements may be important for interpretation of study results since once-daily dosing may result in less sustained BP elevations than twice-daily dosing (Solomon, Wittes, Finn, et al., 2008).
Hepatic Effects
NSAID-related liver injury is much less common than other adverse effects. NSAIDs were implicated in approximately 10% of the cases of drug-induced liver injury and 1% of the liver transplants reported in European and United States literature (Andrade, Lucena, Fernandez, et al., 2005; Bjornsson, Olsson, 2005; Laine, White, Rostom, et al., 2008). In comparison, acetaminophen alone or in combination with other drugs was associated with 49% of the drug-related liver transplants performed in the United States between 1990 and 2002 (Russo, Galanko, Shrestha, et al., 2004) (see earlier in this chapter for more on acetaminophen-induced hepatotoxicity). Mortality from NSAID-related hepatotoxicity is less than 1 per 100,000 (Laine, White, Rostom, et al., 2008).
A systematic review concluded there is a possibility of a small increase in the risk of hepatotoxicity with NSAID use, and many experts do not recommend NSAID use in individuals with cirrhosis (Laine, White, Rostom, et al., 2008). Large-population epidemiologic studies are required to confirm that such a risk exists (Rubenstein, Laine, 2004). Significant increases in clinical liver events have not been noted with the introduction of the COX-2 selective NSAIDs, although a trend in increased episodes of liver injury with lumiracoxib led to its removal from the Australian market (Laine, White, Rostom, et al., 2008). A review of randomized-controlled trials on a variety of NSAIDs revealed that diclofenac and rofecoxib were associated with elevated aminotransferase levels, but no elevations were noted with naproxen, ibuprofen, celecoxib, valdecoxib, or meloxicam (Rostom, Goldkind, Laine, 2005).
Cognitive Effects
Mild dizziness and drowsiness are relatively common and transient effects of both acetaminophen and NSAIDs (Burke, Smyth, FitzGerald, 2006; McQuay, Moore, 2004). Cognitive dysfunction with symptoms of decreased attention span and loss of short-term memory may also be an adverse effect of NSAIDs. This has been noted in older individuals in response to naproxen and ibuprofen, even when doses are within the accepted range. Other CNS adverse effects are headache, lowered seizure threshold, and adverse mood effects (Burke, Smyth, FitzGerald, 2006; Drug Facts and Comparisons, 2006).
Hypersensitivity to Nonopioids: Respiratory and Cutaneous Reactions
Allergy or (more accurately) hypersensitivity to nonopioids is fairly common (Leimgruber, 2008). Patients may erroneously report an allergy to a nonopioid simply because they have experienced adverse effects such as heartburn or nausea after NSAID use. However, true hypersensitivity involves far more serious symptoms. Most of the research on the incidence of nonopioid hypersensitivity is found in the pediatric literature, where hypersensitivity to aspirin, ibuprofen, and acetaminophen was found to be 60%, 76.5%, and 23.2%, respectively (Hassani, Ponvert, Karila, et al., 2008).
The pathogenesis of NSAID hypersensitivity may be nonimmunologic and related to altered arachidonic acid metabolism and excessive production of leukotriene (inflammatory response mediator) (Babu, Salvi, 2000; Forster, Olze, 2008; Leimgruber, 2008). This is thought to lead to a decrease in PGE2 (the antiinflammatory prostaglandin) synthesis, tilting the balance in favor of inflammation (Babu, Salvi, 2000). When hypersensitivity to one NSAID occurs, cross-sensitivity to others frequently occurs, further supporting the logic that most reactions result from a nonallergy hypersensitivity to the pharmacologic properties of the drug (Hassani, Ponvert, Karila, et al., 2008).
Although the COX-2 selective NSAIDs theoretically should be a safer alternative to nonselective NSAIDs in terms of hypersensitivity reactions because they preserve PGE2 (Babu, Salvi, 2000), both nonselective and COX-2 selective NSAIDs are known to produce various skin reactions, including the rare but potentially fatal Stevens-Johnson Syndrome (SJS) and toxic epidermal necrolysis (TEN) (Simon, 2007). SJS produces inflammatory eruptions on the skin and mucous membranes; TEN presents with large areas of bloody-looking lesions and can advance to epidermal detachment.
One study showed the highest risk of skin reactions from nonselective NSAIDs to be with piroxicam and tenoxicam and the lowest with ibuprofen, diclofenac, and ketoprofen (Simon, 2007). A retrospective review of prescription-event monitoring data in England revealed that the incidence of cases of SJS related to the use of the COX-2 selective NSAIDs rofecoxib, celecoxib, etoricoxib, and valdecoxib was very low at 0.08% for all four combined (Layton, Marshall, Boshier, et al., 2006).
Nevertheless, all of the COX-2 selective NSAIDS have been associated with cutaneous adverse reactions of some sort, and a class effect for skin reactions has been suggested (Atzori, Pinna, Pau, et al., 2006). The COX-2 selective NSAID valdecoxib was implicated in several case reports of serious cutaneous adverse reactions (Glasser, Burroughs, 2003; Knowles, Phillips, Wong, et al., 2004; Ziemer, Wiesend, Vetter, et al., 2007), which contributed to the manufacturer’s voluntary withdrawal of valdecoxib from the U.S. market in 2005 (U.S. FDA, 2005). The presentation of severe cutaneous reactions associated with COX-2 selective NSAIDs has been described as different from that of SJS and TEN, specifically in terms of a “widespread, erythematous, to some extent targetlike, or purpuric skin eruption” (Ziemer, Wiesend, Vetter, et al., 2007, p. 715). Adverse cutaneous reactions, including a case of fatal vasculitis (Schneider, Meziani, Chartier, et al., 2002), have been reported following administration of celecoxib (Atzor, Pinna, Pau, et al., 2006; Drago, Brusati, Desirello, et al., 2004).
Sodium lauryl sulfa is an inactive ingredient of both celecoxib and valdecoxib, and although sulfa allergy is not always identified as the cause of reported cutaneous reactions, it is important to remember that sulfa-based drugs like celecoxib should not be given to individuals who are hypersensitive to sulfonamides. Although no evidence of cross-reactivity between celecoxib and sulfonamide antimicrobials was found in 28 patients with a history of allergy to sulfonamide antimicrobials (Shapiro, Knowles, Weber, et al., 2003), the risk of cross-sensitivity is presumed to exist, and all patients should be routinely asked about previous allergic reactions, and specifically about past reactions to sulfa-based drugs, prior to treatment (Schneider, Meziani, Chartier, et al., 2002).
Respiratory reactions also signal NSAID hypersensitivity and are the second most common NSAID adverse effect (Bannwarth, 2006). Individuals with asthma and chronic urticaria are especially vulnerable to these reactions (Leimgruber, 2008) and must be watched closely during NSAID therapy. As many as 20% of asthmatics are sensitive to aspirin and other NSAIDs and may develop rhinoconjunctivitis, sinusitis, nasal polyps, and asthma symptoms that can progress to bronchoconstriction following ingestion of even small amounts of aspirin or NSAID (Babu, Salvi, 2000).
Cross-sensitivity between the various NSAIDs is common in patients who are hypersensitive to NSAIDs, but only 7% of these individuals experience cross-sensitivity to acetaminophen (Bannwarth, 2006), and cross-sensitivity between NSAIDs may not extend to the COX-2 selective NSAIDs in all cases. A study of 36 adults showed a high incidence of urticaria with naproxen but none with celecoxib in aspirin-sensitive patients with chronic idiopathic urticaria (Zembowicz, Mastalerz, Setkowicz, et al., 2003). Other research and case reports also describe safe use of celecoxib and other COX-2 selective NSAIDs in asthma patients who are hypersensitive to nonselective NSAIDs (Andri, Falagiani, 2007; Dahlen, Szczeklik, Murray, 2001; Marks, Harrell, Fischer, 2001; El Miedany, Youssef, Ahmed, et al., 2006; Szczeklik, Nizankowska, Bochenek, et al., 2001). Just 1 hypersensitivity reaction to celecoxib occurred in a study of 29 patients who were hypersensitive to acetaminophen and nonselective NSAIDs (Liccardi, Cazzola, De Giglio, et al., 2005). The authors proposed celecoxib as a safe alternative in individuals who cannot tolerate acetaminophen. Following an extensive literature review, researchers recommended that NSAID treatment in individuals with aspirin/NSAID sensitivity should include avoidance of aspirin and other nonselective NSAIDs, use instead of COX-2 selective NSAIDs, acetaminophen in adult doses less than 1000 mg, and desensitization techniques (Knowles, Drucker, Weber, et al., 2007). Daily doses of 300 mg of aspirin are recommended for optimal desensitization (Rozsasi, Polzehl, Deutschle, et al., 2008). The use of other antiplatelet agents, such as clopidogrel (Plavix) or ticlopidine (Ticlid), is recommended in patients with true aspirin hypersensitivity who require cardioprotection (Ramanuja, Breall, Kalaria, 2004).
Conclusion
There is an abundance of evidence to support the use of nonopioid analgesics for a wide variety of acute and persistent (chronic) pain states. Prevention of the adverse effects of acetaminophen and NSAIDs whenever possible is a basic pain management principle. This is accomplished by individualizing the choice of nonopioid and taking into consideration the type of pain and the patient’s previous nonopioid use and risk factors.