Bearberry
Synonyms
Arctostaphylos officinalis Wimm., Arbutus uva ursi L. (botanical synonyms), mountain cranberry, green manzanita, uva ursi (Engl), Uvae ursi folium (Lat), Bärentraube (Ger), busserole, raisin d’ours (Fr), uva d’orso, uva ursina (Ital), melbær (Dan).
What is it?
The Arctostaphylos genus contains 50 species indigenous to western North America; A. uva ursi has circumpolar distribution and is found in central and northern Europe, as well as in North America.1,2 Bearberry leaves have been used as a urinary antiseptic in the UK since the 13th century.1 It is also a traditional herb of the Native Americans, who used the leaves for ceremonial smoking. However, their main use was in the form of a tea to treat venereal disease and inflammation of the genitourinary tract.3 The berries of Arctostaphylos species have provided food, not only for wildlife such as birds and bears but also for humans. Arctostaphylos species suppress the growth of neighbouring plants due to the hydroquinone formed from the arbutin in their leaves, bark and roots.3
Traditional view
Bearberry was traditionally used for its astringent property and was considered of great value in diseases of the bladder and kidneys, strengthening and imparting tone to the urinary passages and alleviating inflammation of the urinary tract.4 Uses by the Eclectic physicians included chronic irritation of the bladder, enuresis, excessive mucus and bloody discharges in the urine, chronic diarrhoea, dysentery, menorrhagia, leucorrhoea, diabetes, chronic gonorrhoea and strangury.5
Can be used for
Indications supported by clinical trials
Cystitis, recurrent cystitis (in conjunction with other herbs).
Traditional therapeutic uses
Urinary infections such as cystitis, urethritis, prostatitis, pyelitis, lithuria, diarrhoea and intestinal irritations, and any condition requiring an astringent action including chronic diarrhoea. The specific indication listed in the British Herbal Pharmacopoeia 1983 is acute catarrhal cystitis with dysuria and highly acid urine.6
May also be used for
Extrapolations from pharmacological studies
Internally and externally as adjuvant treatment of inflammatory conditions such as contact dermatitis, inflammatory oedema and arthritis.
Other applications
As a whitening agent for the skin and may assist in the control of hyperpigmentary disorders.7
Preparations
Dried leaves as a cold infusion or liquid extract for internal or external use. Cold water extraction of powdered leaves results in better levels of arbutin and lower levels of tannins compared to hot water extraction.8
Dosage
• 3 to 12 g dried leaf per day (the latter equivalent to at least 700 mg arbutin) prepared as an infusion or cold macerate
• 4.5 to 8.5 mL of 1:2 liquid extract per day, 11 to 22 mL of 1:5 tincture per day or the equivalent in tablet or capsule form.
Some studies have found that the antimicrobial effect of bearberry is optimal when the urine has an alkaline pH. However, the majority of urinary tract infections produce acid urine. Alkalinisation of the urine may therefore be beneficial in conjunction with herbal therapy using bearberry (although the need for this has been questioned in a recent study). This can be achieved, at least in the short term, by concurrent administration of bicarbonate or a proprietary urinary alkalinising product. An alkaline-forming diet high in fruit and vegetables could also be consumed during treatment. Consumption of plenty of water during treatment is also advised.
Duration of use
Due to its high tannin content, bearberry is not suitable for prolonged internal use at higher doses.
Summary assessment of safety
There is a very low risk associated with the short-term administration of bearberry, but its use should be avoided during pregnancy and lactation.
Technical data
Botany
Arctostaphylos uva ursi is a small, evergreen, prostrate, mat-forming shrub belonging to the Ericaceae (heath) family. The leathery leaves are alternate, obovate from a wedge-shaped base, 1 to 2 cm long, dark green on the upper surface and pale green underneath. The small pink flowers with a bell-shaped corolla are arranged in drooping clusters. The fruit is shiny, small, round and scarlet-red.4,9
Adulteration
Substitution with other species of Ericaceae is relatively common in commerce. Vaccinium vitis idaea L., V. uliginosum L., V. myrtillus L. (bilberry), Gaultheria procumbens L. (wintergreen), Arctostaphylos alpinus (L.), Buxus sempervirens L. (box) have all been detected in batches of ‘bearberry leaves’.10 According to the German Pharmacopoeia, samples containing less than 6% arbutin should be considered as adulterated.
Arctostaphylos uva ursi is protected and/or has restrictions for wildcrafting in several areas of Europe.11
Key constituents
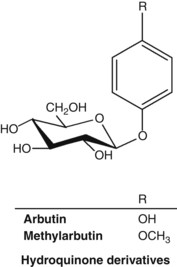
• Hydroquinone glycosides (normally between 6.3% and 9.2% – higher in autumn crops)12 including arbutin and methylarbutin13
• Polyphenols (predominantly gallotannins); phenolic acids, flavonoids, triterpenes.13
Interestingly, arbutin is found at high concentrations in some plants capable of surviving extreme and sustained dehydration.14
Pharmacodynamics
Antimicrobial activity
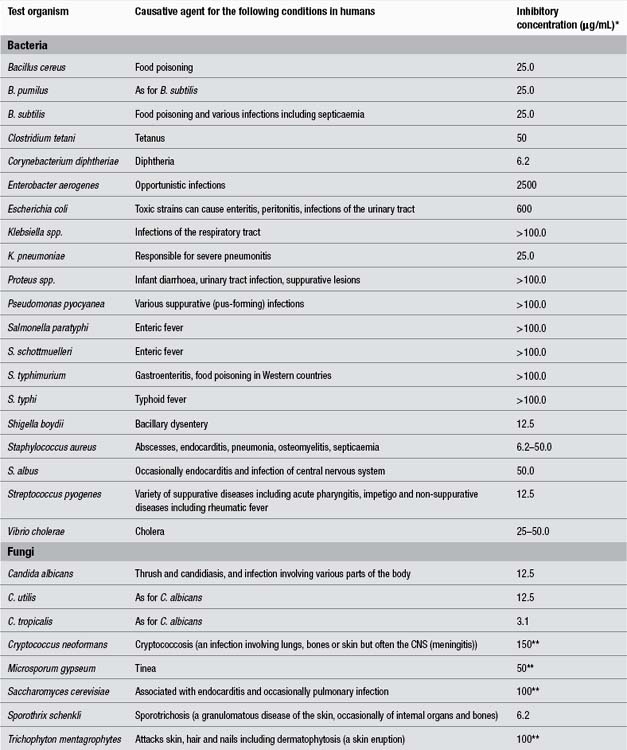
There is some debate as to whether the antimicrobial effect of bearberry is due to hydroquinone esters such as arbutin or to free hydroquinone.13 The antimicrobial activities of arbutin and an aqueous extract of bearberry were tested in vitro against bacterial strains implicated in urinary tract infections. The antibacterial activity of arbutin was directly correlated with the beta-glucosidase activity of the bacteria. (This enzyme converts arbutin into free hydroquinone.) The highest enzyme activity was found in Streptococcus, Klebsiella and Enterobacter, the lowest in Escherichia coli.15 Arbutin (128 μg/L) inhibited three of eight clinical isolates of Pseudomonas aeruginosa tested in vitro.16 Arbutin and hydroquinone inhibited the growth of Ureaplasma urealyticum and Mycoplasma hominis in vitro.17 These bacteria are associated with non-gonococcal urethritis.
Piceoside, a glucoside isolated from bearberry, did not demonstrate antibacterial activity in vitro, but its aglycone p-hydroxyacetophenone showed activity against Proteus vulgaris, Enterobacter aerogenes and Bacillus subtilis.18 Another study found antimicrobial activity for bearberry extracts in vitro against E. coli, Proteus vulgaris, Enterobacter aerogenes, Streptococcus faecalis, Staphylococcus aureus, Salmonella typhi and Candida albicans.19 The summer and autumn leaves were more potent than the winter leaves.20
The antibacterial activity of various agents was tested in vitro using 74 different strains of bacteria isolated from the urinary tract including E. coli, Proteus mirabilis, Pseudomonas aeruginosa, Staphylococcus aureus and species of Enterobacter, Citrobacter and Klebsiella. Urine was collected from healthy volunteers 3 h after oral administration of 0.1 g or 1.0 g of arbutin; several synthetic antibiotics were also tested. Of all test substances, only gentamicin, nalidixic acid and urine collected after intake of 1.0 g of arbutin and adjusted to pH 8 were active against every strain used.21
A hydroethanolic extract of bearberry was amongst the most active of 14 traditional Canadian medicinal plants tested against a wide variety of strains of Neisseria gonorrhoeae, including isolates resistant to antibiotics.22 A minimum inhibiting concentration of 32 μg/mL was demonstrated in vitro.
A study of the dry leaf extract of bearberry on the course of acute bacterial pyelonephritis (caused by E. coli) in white rats showed that bearberry extract (25 mg/kg) had a marked antibacterial and nephroprotective effect.23
Samples of both normal urine and urine collected from healthy subjects after consuming bearberry tea were compared for resistance to bacterial contamination. Urine from the bearberry tea drinkers was more bacteriostatic than normal urine samples. However, the addition of arbutin to normal urine did not result in the same bacteriostatic activity. A series of solutions were then tested for their inhibition of the growth of strains of Staph. aureus and E. coli in vitro. The solutions included hydroquinone, methylhydroquinone and arbutin, normal urine and urine from bearberry tea drinkers. They were tested at normal pH and at pH elevated to 8.0 by the addition of potassium hydroxide. Only the hydroquinone and methylhydroquinone solutions inhibited bacterial growth at normal pH. However, incubation of bacteria with bearberry tea drinkers’ urine adjusted to pH 8 also resulted in inhibition of bacterial growth. This inhibitory effect was also seen for urine adjusted to pH 8 from subjects given pure arbutin.24 The authors suggested that antibacterial activity would only occur when the excretion products of arbutin (hydroquinone paired with glucuronate and sulphate) appear in sufficiently high concentrations in an alkaline urine. It was hypothesised that at an alkaline pH these excretion products of arbutin released small amounts of free hydroquinone in the presence of bacteria, thereby conferring antibacterial activity to the urine. The maximum antibacterial effect from the hydroquinone glucuronides and sulphates formed from arbutin was obtained about 3 to 4 h after taking the herb.25 Free hydroquinone is only excreted in trace amounts, which is desirable given the toxic potential of this agent.
A recent paper has expressed a contradictory view. In this study, urine was collected from four healthy individuals after ingestion of 420 mg of arbutin.26 The samples were added to an E. coli suspension and it was noted that the concentration of hydroquinone in bacteria was 20-fold higher than a control. The authors commented that the pH of urine is unlikely to be important, as the intracellular pH of E. coli is not affected by urine alkalinisation. They concluded that deconjugating enzymes such as beta-glucuronidases found in the bacteria enrich and deconjugate hydroquinone glucuronides and/or sulphates regardless of pH. The pH of the test urine was not stated, but in the study design attempts to increase it to more than 6.5 via a vegetarian diet were instituted.
Urine produced by a healthy person consuming a meat and fish diet is typically in the pH range 4.5 to 6.0; a vegetarian diet will make urine more alkaline. A urine of pH >7 during a urinary tract infection indicates infection by a micro-organism capable of splitting urea, with the release of ammonia.27 Urea-splitting organisms include Proteus spp., Klebsiella spp., some Citrobacter spp., some Haemophilus spp., Bilophila wadsworthia, the yeast Cryptococcus neoformans and several other bacteria and fungi.28 Infection with these organisms should be particularly susceptible to treatment with bearberry if alkaline urine does enhance its activity. Alkalinisation of the urine with buffering agents (for example containing sodium bicarbonate, sodium citrate, citric acid and tartaric acid) in conjunction with bearberry intake may prove to be clinically effective for the treatment of cystitis caused by non-urea-splitting bacteria, but given the conflicting findings noted above, this requires further research.
The antibacterial effect of bearberry may also be useful in the gastrointestinal tract. An aqueous extract of bearberry was found to modulate cell surface hydrophobicity and demonstrated antibacterial effects on ten strains of Helicobacter pylori in vitro.29 The research team concluded that the hydrolysable tannins were largely responsible, as pure tannic acid produced comparable results.
Anti-inflammatory and antiallergic activities
Co-administration of arbutin (50 mg/kg, oral) and indomethacin (subcutaneous) showed an inhibitory effect on swelling in a delayed-type hypersensitivity model, which was stronger than that of indomethacin alone.30 In the same model, arbutin (10 and 50 mg/kg, oral) plus prednisolone or dexamethasone showed stronger effects than each of the anti-inflammatory drugs alone.31 Arbutin may therefore have a synergistic anti-inflammatory activity on type IV allergic reaction-induced inflammation. In the same model, oral administration of a bearberry methanolic extract (100 mg/kg) demonstrated an inhibitory effect on swelling. When administered simultaneously with subcutaneous prednisolone, the inhibitory effect was more potent than that of prednisolone alone.32
Although ointments containing 1% and 2% aqueous extract of bearberry did not inhibit the ear swelling caused by experimentally induced contact dermatitis or carrageenan-induced paw oedema in rats and mice, they did increase the anti-inflammatory effect of a steroid ointment (dexamethasone). Co-administration of bearberry did not increase the side effects of dexamethasone.33 Topical doses of bearberry might also increase the anti-inflammatory effects of other steroid-like compounds, such as plant-derived saponins.
Effect on melanin synthesis
Arbutin at a concentration of 5×10−5 M decreased melanin content to approximately 39% when compared to untreated melanoma cells in vitro, without affecting cell growth. Tyrosinase activity also dropped significantly in the arbutin-treated cells. (This enzyme is involved in melanin synthesis.) Arbutin was not hydrolysed to hydroquinone, suggesting that the observed inhibitory effect was for arbutin itself, not hydroquinone.34 Further studies have revealed that the depigmenting mechanism of arbutin in humans involves inhibition of melanosomal tyrosinase activity, rather than the suppression of expression and synthesis of tyrosinase.35
A 50% methanolic extract of bearberry inhibited melanin synthesis in vitro. Both the bearberry extract and arbutin had an inhibitory effect on tyrosinase activity and inhibited the production of melanin by both tyrosinase and autoxidation.7 Bearberry extract could have a bleaching effect on freckles and may assist in the control of hyperpigmentary disorders.
Other activity
Oral doses of arbutin (50 mg/kg) suppressed experimentally induced cough reflex. The effect of arbutin was stronger than that of the non-narcotic antitussive dropropizine and comparable to that of codeine.36
Oral administration of a bearberry infusion (3 g/L in drinking water) to healthy rats fed a standard diet containing calcium (8 g/kg) and magnesium (2 g/kg) did not induce significant diuresis, nor affect calcium or citrate concentration levels.37
Aqueous and methanolic extracts of bearberry have demonstrated in vitro molluscicidal activity against the freshwater snail Biomphalaria glabrata (the intermediate host of schistosomiasis). The methanol extract was active at a concentration of 50 ppm.38
A methanol extract of bearberry showed algicidal activity when tested in ponds. It is believed the tannins precipitated the algal proteins.39
Pharmacokinetics
Urinary excretion of phenolic metabolites after the oral administration of either bearberry leaf tea or arbutin occurs within 1 to 2 h and reaches a maximum 4 h after administration. In healthy subjects given bearberry tea, 70% to 75% of the administered dose was excreted within 24 h. Arbutin is altered after its passage through the body; it only occurs in trace amounts in urine when high doses are given. Free hydroquinone is only excreted in trace amounts, if at all.
It has been suggested that there are two possible processes for the absorption and metabolism of arbutin.40 The major process involves the absorption of intact arbutin by small intestinal enterocytes via the sodium-glucose pump. On first-pass metabolism in the liver, arbutin is deconjugated to hydroquinone and then reconjugated to sulphate and glucuronide phase II derivatives, which are then excreted via the urine. The minor process involves the conversion to free hydroquinone of arbutin in the colon by the action of bacterial beta-glucosidase. The hydroquinone is then converted by colonic enterocytes into sulphate and glucuronide derivatives which are absorbed into the bloodstream and passed into the urine.
In a crossover study involving six healthy volunteers, enteric-coated bearberry tablets demonstrated the same bioavailability within a 24-h period as an equivalent bearberry extract. The release of arbutin metabolites was retarded by at least 3 h with the tablets. In a pilot study conducted prior to this main study, no free hydroquinone was found in the urine of volunteers, although the above hydroquinone derivatives were found.41 This study was designed to compare the bioavailability of enterically coated tablets containing bearberry extract with uncoated tablets, but it does also add some support to the above metabolic pathways. Additionally, a small, randomised crossover trial in sixteen adults evaluated the bioavailability of an aqueous solution of bearberry as compared to film-coated tablets.42 The maximum mean urinary concentration of hydroquinone equivalents was marginally higher and peaked a little earlier in the tea group, although this was not statistically significant. The authors concluded there were no significant differences between the two groups in terms of metabolites or total amounts of hydroquinone equivalents excreted.
Some other studies investigating the elimination of arbutin in rats have arrived at different conclusions, which cast doubt on their relevance to humans. In these studies, orally administered arbutin was excreted unchanged in urine43 and oral administration of bearberry tea resulted in the excretion of six unidentified phenolic compounds, but no hydroquinone. No degradation products were observed after the perfusion of isolated rat liver with arbutin, thus leading to the conclusion that it was hydrolysed in the kidneys.44 As noted above, the results of these studies are not supported by the human studies. Moreover, in the case of the orally administered arbutin, the authors may have actually measured arbutin metabolites and mistakenly assigned them as arbutin.
Clinical trials
Urinary disorders
In a double blind, placebo-controlled, randomised clinical trial, 57 women who had experienced at least three episodes of cystitis during the preceding year received either herbal medicine or placebo. The herbal medicine consisted of bearberry extract (standardised for arbutin and methylarbutin content) and extract of dandelion root and leaf (dose of individual herbs not specified). Treatment for 1 month significantly reduced the recurrence of cystitis during the 1-year follow-up period, with no incidence of cystitis in the herbal group and a 23% occurrence in the placebo group (p<0.05). No side effects were reported.45
Toxicology and other safety data
Toxicology
Hydroquinone is a recognised toxic compound. However, arbutin and bearberry extracts are considerably less toxic than hydroquinone, as somewhat evidenced by the studies cited below.
The oral LD50 of hydroquinone as a 2% aqueous solution has been reported as between 320 and 550 mg/kg in various laboratory animals.46 Hydroquinone is non-mutagenic in the Ames test but induces chromosome aberrations and karyotypic effects in eukaryotic cells.47 In contrast, arbutin did not induce mutations in concentrations up to 10−2 M in a gene mutation assay. An increase in mutation frequency was observed with concentrations of 10−3 M and higher when arbutin was preincubated with beta-glucosidase. Hydroquinone, used as a positive control, also exhibited clear effects. In vivo, hydroquinone administered by intraperitoneal injection induced elevated micronucleus incidences. However, there was no induction of micronuclei in bone marrow when arbutin was administered orally (0.5 to 2.0 g/kg). This research suggests that arbutin itself is not mutagenic, but any generated hydroquinone could exert a mutagenic potential.48
Contraindications
According to the British Herbal Compendium, bearberry is contraindicated in kidney disorders13 but there is no evidence to support this, and the contraindication probably arose out of a theoretical caution.
According to the Commission E, bearberry is contraindicated in pregnancy and lactation and for children less than 12 years of age.49
Special warnings and precautions
Bearberry is not suitable for prolonged use. Use cautiously in highly inflamed or ulcerated conditions of the gastrointestinal tract.50
In principle, the prolonged use of herbs with high levels of tannins is inappropriate in constipation, iron deficiency anaemia and malnutrition.51
Interactions
Concomitant acidification of the urine (for instance by medication) may result in a reduction of efficacy,24 although this is hypothetical and of uncertain relevance to the urinary antiseptic mechanism of bearberry.
Oral or topical use of bearberry or arbutin has been observed to augment the anti-inflammatory effects of indomethacin,30 prednisolone,31,32 and dexamethasone31,33 in experimental models. The clinical relevance of these findings is uncertain.
Bearberry extract markedly potentiated the action of beta-lactam antibiotics against methicillin-resistant Staph. aureus in vitro. The constituent corilagin (a polyphenol) was responsible for the activity.52 However, whether this leads to a clinical interaction is uncertain.
The high tannin levels will cause interference with the absorption of various nutrients and drugs, especially metal ions, thiamine and alkaloids. Bearberry should be consumed at least 2 h away from oral thiamine, mineral supplements such as iron and alkaloid-containing drugs.51
Use in pregnancy and lactation
Pregnancy category C – has caused or is associated with a substantial risk of causing harmful effects on the foetus or neonate without causing malformations.10
There is a minor theoretical risk to fetal development due to the uterotonic properties of arbutin in vivo.53,54 However, arbutin also occurs in food: wheat products (1 to 10 ppm), pears (4 to 15 ppm), and coffee and tea (0.1 ppm).55
The transfer of arbutin or hydroquinone to breast milk is not advisable, and therefore the herb should be avoided in lactation.
Side effects
Hydroquinone depigmenting creams may cause exogenous ochronosis (hyperpigmentation)56 and/or allergic contact dermatitis.57 However, these side effects have not been reported for cosmetic creams containing bearberry.56
A case of bilateral bull’s-eye maculopathy has been reported in a 56-year-old woman after ingestion of bearberry tea for 3 years, dose unknown.58 While it is generally acknowledged that bearberry inhibits melanin production, which is present in ocular tissue, therapeutic doses for short periods of time are routinely considered safe.
Due to the high tannin content, internal use of high doses of bearberry may cause cramping, nausea, vomiting and constipation.
Regulatory status in selected countries
Bearberry is official in the European Pharmacopoeia (2006).
Bearberry is covered by a positive Commission E Monograph and has the following application: inflammatory disorders of the lower urinary tract.
Bearberry is on the UK General Sale List and in France the herb is accepted for the internal treatment of benign urinary infections and to promote the renal elimination of water.
Bearberry does not have GRAS status. However, it is freely available as a ‘dietary supplement’ in the USA under DSHEA legislation (1994 Dietary Supplement Health and Education Act). Bearberry has been present in the following over-the-counter (OTC) drug products: weight control drug products and orally administered menstrual drug products. The FDA, however, advises: ‘that based on evidence currently available, there is inadequate data to establish general recognition of the safety and effectiveness of these ingredients for the specified uses’.
Bearberry is not included in Part 4 of Schedule 4 of the Therapeutic Goods Act Regulations of Australia and is freely available for sale.
References
1. Mabberley DJ. The Plant Book, 2nd ed. Cambridge: Cambridge University Press, 1997. p. 53
2. Evans WC. Trease and Evans’ Pharmacognosy, 14th ed. London: WB Saunders, 1996. p. 223
3. Brinker FJ. Eclectic Dispensatory of Botanical Therapeutics, vol 2, Section 1: Native Healing Gifts. Sandy: Eclectic Medical Publications, 1995. pp. 19–23
4. Grieve MI, A Modern Herbal, New York, Dover, 1971;vol 1. pp. 89–90
5. Felter HW, Lloyd JU. King’s American Dispensatory, 18th ed., 3rd rev, vol 2, 1905. Reprinted by Eclectic Medical Publications; 1983. pp. 2038–2040.
6. British Herbal Medicine Association’s Scientific Committee. British Herbal Pharmacopoeia. West York: BHMA, 1983. pp. 29–30
7. Matsuda H, Nakamura S, Shiomoto H, et al. Yakugaku Zasshi. 1992;112(4):276–282.
8. Frohne D. Pharm Ztg. 1980;125:2582–2583.
9. Launert EL. The Hamlyn Guide to Edible and Medicinal Plants of Britain and Northern Europe. London: Hamlyn, 1981. p. 128
10. Mills S, Bone K. The Essential Guide to Herbal Safety. St Louis: Elsevier, 2005. p. 259
11. Lange D. Europe’s Medicinal and Aromatic Plants: Their Use, Trade and Conservation. Traffic International, 1998. p. III
12. Parejo I, Viladomat F, Bastida J, et al. Phytochem Anal. 2001;12(5):336–339.
13. British Herbal Medicine Association, British Herbal Compendium, Bournemouth, BHMA, 1992;vol 1. pp. 211–213
14. Oliver AE, Crowe LM, De Araujo PS, et al. Biochim Biophys Acta. 1996;1302(1):69–78.
15. Jahodar L, Jilek P, Patkova M, et al. Cesk Farm. 1985;34(5):174–178.
16. Ng TB, Ling JM, Wang ZT, et al. Gen Pharmacol. 1996;27(7):1237–1240.
17. Robertson JA, Howard LA. J Clin Microbiol. 1987;25(1):160–161.
18. Jahodar L, Kolb I. Pharmazie. 1990;45(6):446.
19. Holopainen M, Jabodar L, Seppanen-Laakso T, et al. Acta Pharm Fenn. 1988;97(4):197–202.
20. Skvortsov SS, Khan-Fimina VA. Fitontsidy Mater Soveshch. 1969;6:207–209.
21. Kedzia B, Wrocinski T, Mrugasiewicz K, et al. Med Dosw Mikrobiol. 1975;27:305–314.
22. Cybulska P, Thakur SD, Foster BC, et al. Sex Trans Dis. 2011;38(12):1–5.
23. Nikolaev SM, Shantanova LN, Mondodoev AG, et al. Rastitel’Nye Resursy. 1996;32(3):118–123.
24. Frohne D. Planta Med. 1970;18:1–25.
25. German Federal Minister of Justice. German Commission E for Human Medicine Monograph, Bundes-Anzeiger (German Federal Gazette) no. 228, dated 05.12.84 and no. 109, dated 15.06.1994.
26. Siegers C, Bodinet C, Ali SS, et al. Phytomedicine. 2003;10(suppl 4):58–60.
27. Bouchier IAD, Morris JS, eds. Clinical Skills – A System of Clinical Examination, 2nd ed, London: WB Saunders, 1982. p. 243
28. Baron EJ, Peterson LR, Finegold SM. Bailey and Scott’s Diagnostic Microbiology, 9th ed. St Louis: Mosby Year Book, 1994. p. 106
29. Annuk H, Hirmo S, Turi E, et al. FEMS Microbiol Lett. 1999;172(1):41–45.
30. Matsuda H, Tanaka T, Kubo M. Yakugaku Zasshi. 1991;111(4–5):253–258.
31. Matsuda H, Nakata H, Tanaka T, et al. Yakugaku Zasshi. 1990;110(1):68–76.
32. Kubo M, Ito M, Nakata H, et al. Yakugaku Zasshi. 1990;110(1):59–67.
33. Matsuda H, Nakamura S, Tanaka T, et al. Yakugaku Zasshi. 1992;112(9):673–677.
34. Akiu S, Suzuki Y, Asahara T, et al. Nippon Hifuka Gakkai Zasshi. 1991;101(6):609–613.
35. Maeda K, Fukuda M. J Pharmacol Exp Ther. 1996;276(2):765–769.
36. Strapkova A, Jahodar L, Nosalova G. Pharmazie. 1991;46(8):611–612.
37. Grases F, Melero G, Costa-Bauza R, et al. Int Urol Nephrol. 1994;26(5):507–511.
38. Schaufelberger D, Hostettmann K. Planta Med. 1983;48(2):105–107.
39. Ayoub SMH, Yankov LK, Hussein-Ayoub SM. Fitoterapia. 1985;6(4):227–229.
40. Garcia de Arriba S, Stammwitz U, Pickartz S, et al. Z Phytother. 2010;31(2):95–97.
41. Paper DH, Koehler J, Franz G. Pharm Pharmacol Lett. 1993;3:63–66.
42. Schindler G, Patzak U, Brinkhaus B, et al. J Clin Pharmacol. 2002;42(8):920–927.
43. Jahodar L, Leifertova I, Lisa M. Pharmazie. 1983;38(11):780–781.
44. Leifertova I, Lisa M, Jahodar L, et al. Rozvoj Farm Ramci Ved-Tech Revoluce, Sb Prednasck Sjezdu Cesk Farm Spol, 7th ed. Prague: University of Karlova, 1979. Meeting date 1977; pp. 41–43
45. Larsson B, Jonasson A, Fianu S. Curr Ther Res Clin Exp. 1993;53(4):441–443.
46. Woodard G, Hagan CE, Radomski JL. Fed Proc. 1949;8:348.
47. Devillers J, Boule P, Vasseur P, et al. Ecotoxicol Environ Saf. 1990;19(3):327–354.
48. Mueller L, Kasper P. Mutat Res. 1996;360(3):291–292.
49. German Federal Minister of Justice. German Commission E for Human Medicine Monograph, Bundes-Anzeiger (German Federal Gazette) no. 109, dated 15.06.94; no. 19, dated 28.01.1994.
50. Blumenthal M, Busse WR, Goldberg A, et al. The Complete German Commission E Monographs. Texas: American Botanical Council, 2000. pp. 224–225
51. Mills S, Bone K. The Essential Guide to Herbal Safety. St Louis: Elsevier, 2005. p. 260
52. Shimizu M, Shiota S, Mizushima T, et al. Antimicrob Agents Chemother. 2001;45(11):3198–3201.
53. Shipochliev T. Vet Med Nauki. 1981;18(4):94–98.
54. Itabashi M, Aihara H, Inoue T, et al. Iyakuhin Kenkyu. 1988;19:282–297.
55. Deisinger PJ, Hill TS, English JC. J Toxicol Environ Health. 1996;47(1):31–46.
56. Howard KL, Ferner BB. Cutis. 1990;45(3):180–182.
57. Engasser PG, Maibach HI. J Am Acad Dermatol. 1981;5(2):143–147.
58. Wang L, Del Priore LV. Am J Ophthalmol. 2004;137(6):1135–1137.
59. Stübler M, Krug E. Leesers Lehrbuch der Homöopathie, Pflanzliche Arzneistoffe II. Heidelberg: Haug-Verlag, 1988. pp. 403–406
60. Standardzulassung für Fertigarzneimittel. Pharmazeutischer Verlag, Deutscher Apotheker, Frankfurt/Main, 1987/89, Verlag.