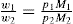The boiling point diagram for an ideal mixture corresponding to
Fig. 16.8 is shown in
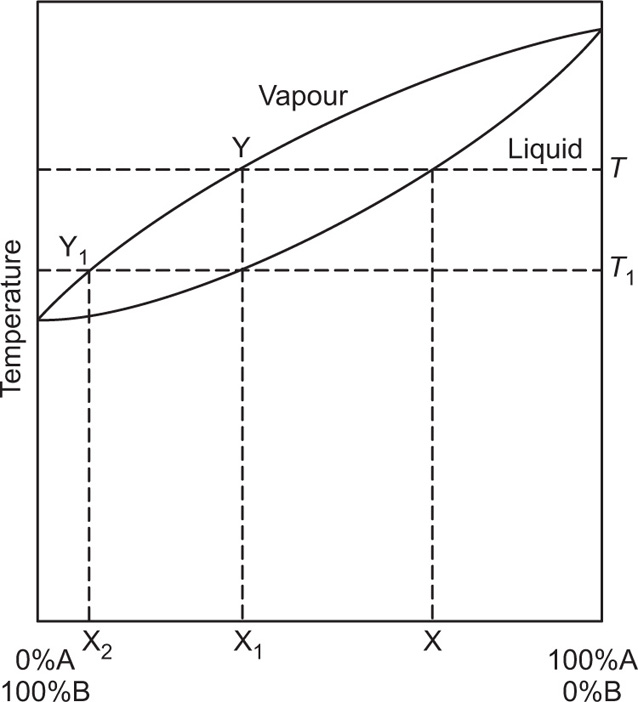
Fig. 16.9. The lower curve shows the manner in which the boiling point of the mixture changes with composition; the upper curve relates the composition of the vapour in equilibrium with the liquid at the same temperature. Hence the boiling point of the mixture X is
T. The vapour at T will have a composition fixed by point Y, which corresponds to a mixture richer in B than in A. If this vapour is condensed, a liquid having the same composition (X
1) will be obtained. The boiling point of the mixture X
1 is T
1 and when it is boiled, vapour having the composition Y
1, which yields a liquid X
2 on condensation, is obtained. X
2 is nearly pure B. Hence in this example, extensive fractionation has been achieved by boiling the mixture and condensing the vapour in equilibrium with the liquid and repeating the process with the condensed vapour. The volume of distillate (composition X
2), obtained will be small, since as the vapour is drawn off, the liquid remaining in the still
gradually becomes poorer in component B and its boiling point rises. Fractional distillation is based on these principles.
Azeotropic Mixtures
An azeotropic mixture or
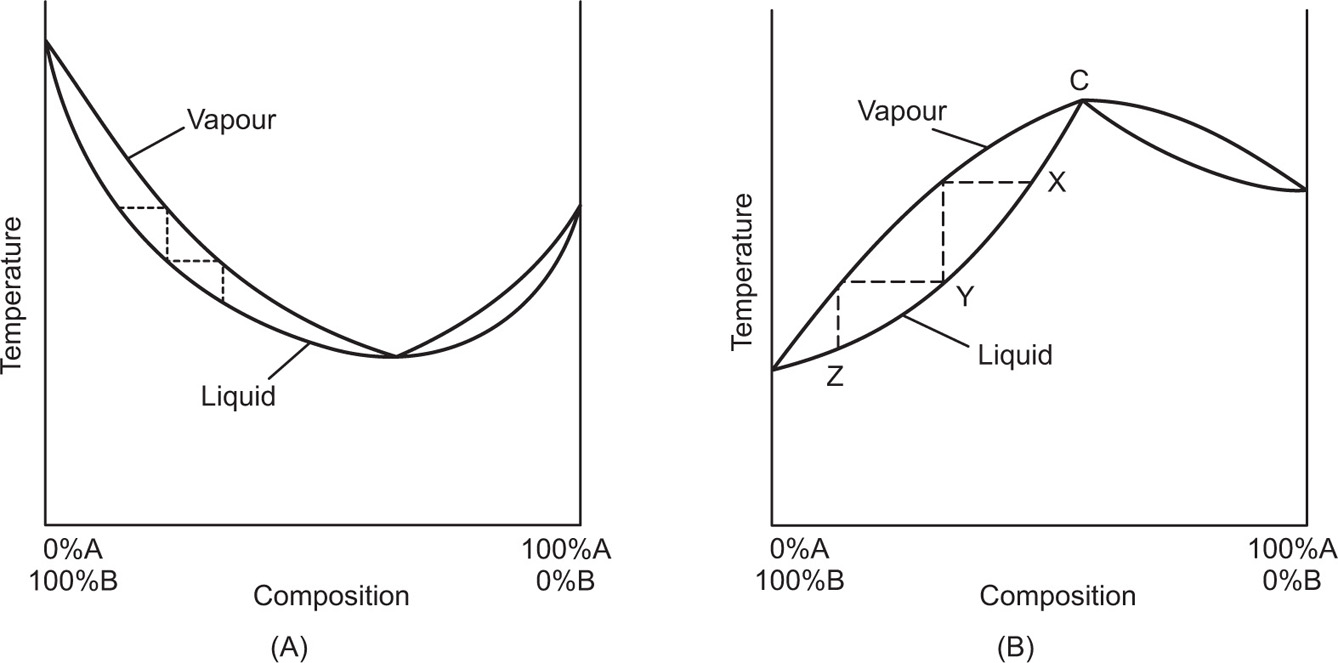
constant boiling mixture is one in which the composition of the liquid and the vapour in equilibrium with it is the same. Thus the mixture behaves like a pure liquid in so far as it distils without change in composition or boiling point. Miscible binary liquids form azeotropic mixtures when the vapour pressure curve of the mixture exhibits a maximum or minimum. Such mixtures cannot be separated into their pure components by distillation. It is possible to separate them by distillation only into one component and a constant boiling mixture. Their behaviour on distillation is most easily followed by referring to the boiling point versus composition curves as shown in
Fig. 16.10; (A) represents a mixture which possesses a maximum vapour pressure, i.e. low boiling point azeotrope; (B) represents a mixture with a minimum vapour pressure, i.e. high boiling point azeotrope. In this figure the concentration changes arising during distillation using a fractionating column equivalent to two theoretical plates are shown, i.e. X to Z. From (A) it can be seen that repeated fractionation will produce a distillate tending to the composition of the azeotropic mixture represented by point C. The material left in the still will be richer in B than it was originally; eventually pure B will be left after the whole of A has been distilled off in the form of the azeotropic mixture. If the original composition lies to the right of C, then by similar reasoning A will be left in the still. Mixtures of minimum boiling point are more common than mixtures exhibiting a maximum boiling point, e.g. alcohol and water, alcohol and benzene, alcohol and chloroform. Alcohol, boiling point 78.3°C, and water boiling point 100°C form a mixture of minimum boiling point 78.15°C, containing 95.57 per cent w/w alcohol. The composition of mixtures of minimum boiling point varies with pressure as, for example, water and alcohol can be completely separated by distilling at 28°C under 7
cm pressure.
On distilling a mixture of maximum boiling point, the distillate will be richer in the component A or B depending on whether the original concentration lies to the right or left of C.
Fig. 16.10 (B) shows the original concentration X lying to the left of C and the distillate thus becomes richer in B.
Ternary Mixtures
Mixtures of three components, which do not form azeotropes, may be separated by fractional distillation in the same way as binary mixtures. Azeotropic ternary mixtures of maximum vapour pressure (minimum boiling point) are important. Water,
boiling point 100°C, alcohol, boiling point 78.3°C and benzene, boiling point 80.2°C, form a ternary azeotropic mixture, boiling point 64.85°C, containing 18.5 per cent of alcohol, 7.4 per cent of water and 74.1 per cent of benzene; the boiling point of this mixture is lower than the boiling point of any binary mixture of any of the components. Fractionation of this ternary mixture is used on the large scale for the production of absolute alcohol. Absolute alcohol cannot be obtained by normal fractionation of dilute alcohol since a constant boiling mixture of 95.57 per cent w/w is formed. Benzene is added to the alcohol–water azeotrope and when distilled the mixture yields first the ternary water–alcohol–benzene azeotrope, boiling point 65.85°C, until all the water is removed from the system. Next a binary alcohol–benzene azeotrope, boiling point 68.2°C distils over and finally absolute alcohol, boiling point 78.3°C. Trichloroethylene may be used instead of benzene in this process. Solutions in a solvent such as benzene may be dried by azeotropic distillation; benzene forms a constant boiling mixture with water, boiling point 69.3°C. All the water present in the benzene is removed when about 10 per cent of the solvent has been distilled.
Fractionating Columns
If a mixture of chloroform, boiling point 61.2°C and benzene, boiling point 80.2°C, is distilled, the vapours first evolved will be richer in the more volatile component, chloroform; as this is distilled off, the vapours will become gradually richer in benzene, and the temperature of distillation will gradually rise. By changing the receiver when the temperature recorded on the thermometer in the head of the still has risen from 61 to 63°C, a fraction is obtained which is richer in chloroform than in benzene. Other fractions, which are increasingly rich in benzene, may be collected over the ranges 63–68°C, 68–73°C, 73–78°C, 78–80°C. Repeated separate distillations of the intermediate fractions, fractions of the same boiling range being combined, will ultimately result in a separation of the liquid into two main fractions, boiling point 61–63°C and 78–80°C, which represent a rough separation of the two constituents of the mixture. This process is very tedious and the same or a better effect can be achieved in a single distillation through a fractionating column. It should be remembered that complete fractionation by distillation is possible only with liquids which do not form azeotropic mixtures.
A fractionating column, which is inserted between the still and the condenser, acts by bringing about repeated distillations throughout the length of the column. The action of the column is partially to condense the vapours rising from the boiling liquid; this condensate will be richer in the more volatile component than the original liquid and it is vapourized again by the condensation of more ascending vapours; the vapours so produced will be still richer in the more volatile component, and when condensation and vapourization takes place further up the column, further enrichment in the more volatile component will be effected. Under ideal conditions this will result in the lower boiling point component arriving at the top of the column and the higher boiling point component being left at the bottom of the column. Thus a temperature gradient will be established along the column when distillation is in progress, and ultimately the vapour passing out of the column will be very rich in the low boiling point component. This series of events may be easily visualized by considering the action of a bubble-cap column which is used in large-scale distillation plant. The column consists of a number of plates mounted above one another, over which flows the condensed liquid (reflux) and through which the ascending vapour is made to bubble.
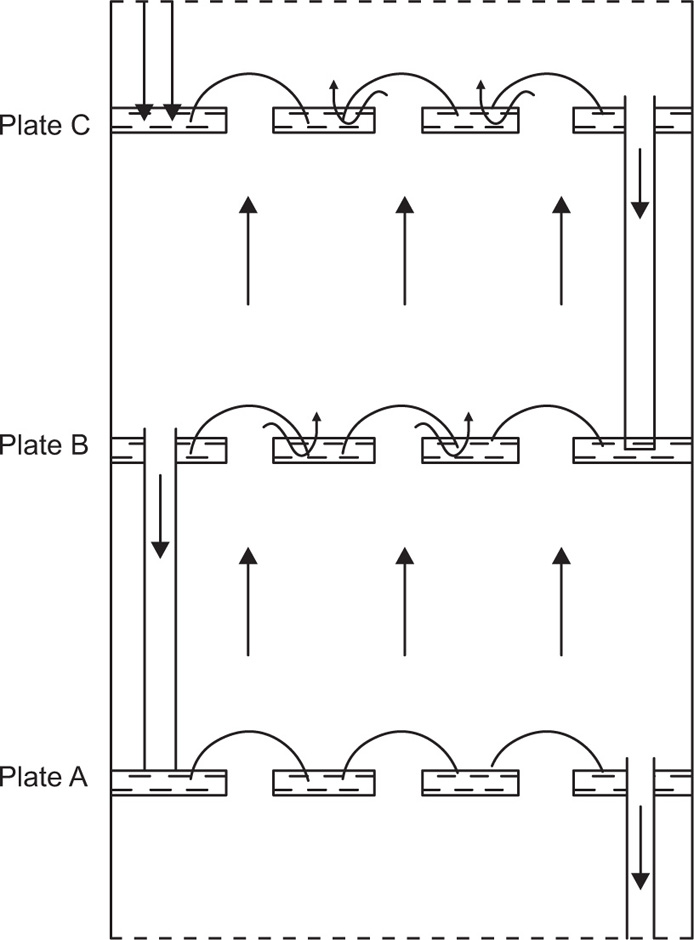
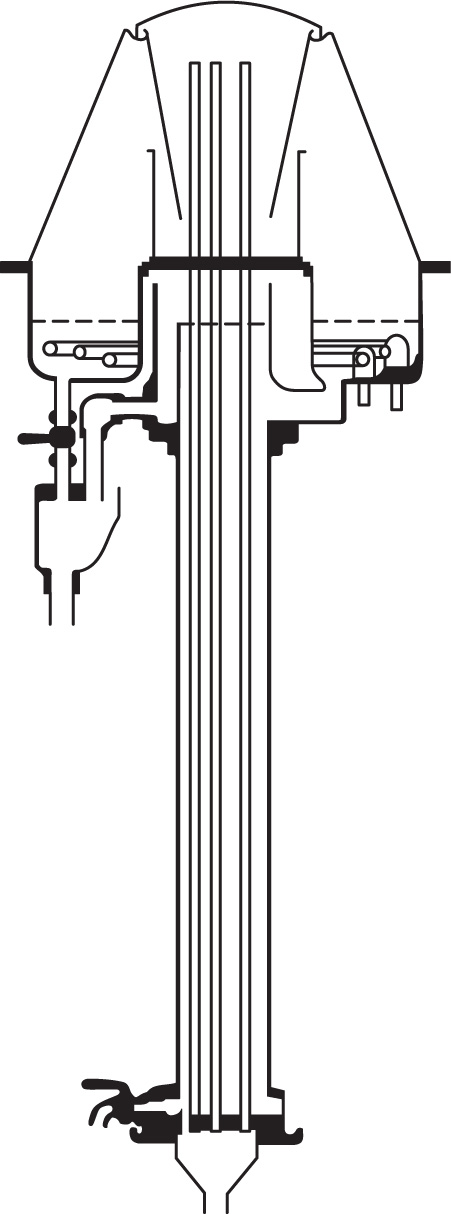
Fig. 16.11 shows three bubble-cap plates in section. If we assume that at each plate the vapour and liquid reach equilibrium conditions, i.e. each plate is theoretically perfect the change in composition of liquid on passing from plate A to plate C will be equivalent to that produced by three theoretical plates. Ascending vapour from the still passes through the bubble caps on plate A and the vapour rising from it will be richer in the more volatile component. This vapour passes through the liquid on B, condenses, and the heat of condensation partially vapourizes the liquid. The process of condensation and vapourization will be repeated at C and so on, all the way up the column. In this simplified consideration of column action, each bubble-cap plate has the same effect as a separate still. The changes in composition at each plate can be estimated from the boiling point versus composition curves.
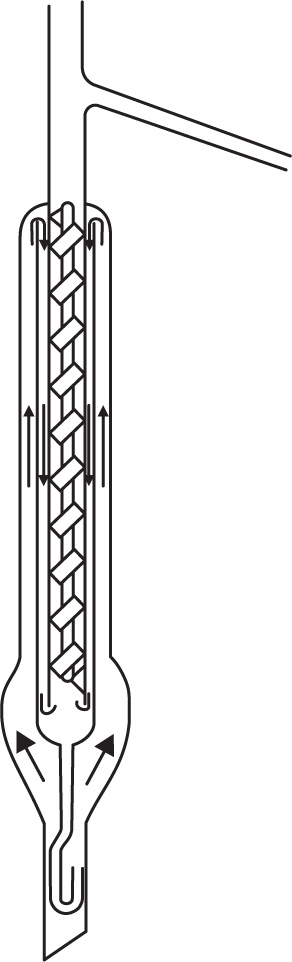
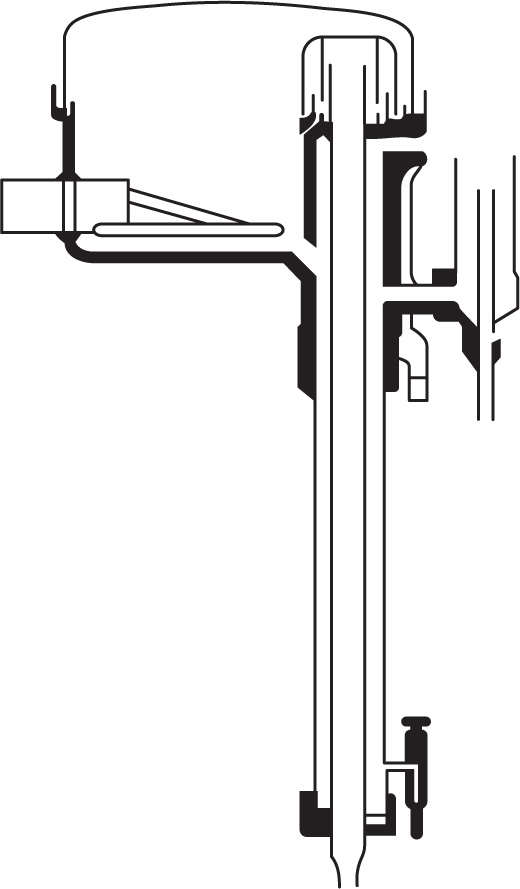
Design of fractionating columns: The purpose of a fractionating column is to achieve an extensive liquid–vapour interface so that equilibrium between ascending vapour and reflux can be rapidly attained. In the laboratory, packed columns are frequently used; the packing may consist of single turn helices (spirals) of wire or glass, glass rings, cylindrical glass beads, stainless steel rings, etc. The Vigreux column which in the best types has indentations in the walls, spirally arranged and occupying most of the interior, acts as a packed column. In the Widmer column (
Fig. 16.12)
fractionation occurs in the central spiral which is kept warm by the vapour around it. Condensed liquid is returned to the still by a trap.
The separation of a mixture in a column depends upon the proportion of vapours traversing the column which are condensed and returned to the still—the
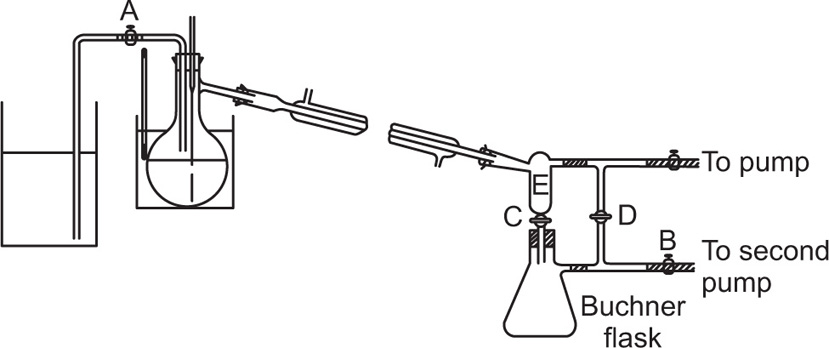
reflux ratio. A column operating under total reflux will not yield distillate; a state of dynamic equilibrium is reached and a maximum degree of separation of the components is obtained along the length of the column. If the quantity of distillate removed at a time is small, this will minimize disturbance of equilibrium conditions and the concentration gradient along the column will remain reasonably stable. For columns operating above about 60°C, heat loss should be prevented by insulation, e.g. asbestos cord, silver vacuum jacket. For temperatures above 100°C the column is surrounded by a heating jacket which is generally adjusted to the temperature of the vapour that emerges from the top of the column. Heat loss will cause excessive condensation within the column which may result in flooding and also will disturb the steady temperature gradient along the column. Under adiabatic conditions the temperature gradient is determined by the vapour–liquid equilibria in the column. For efficient separation it is essential to use a high reflux ratio and collect distillate slowly so that the column is operating under equilibrium conditions the reflux ratio is con veniently controlled by means of a suitable still head.
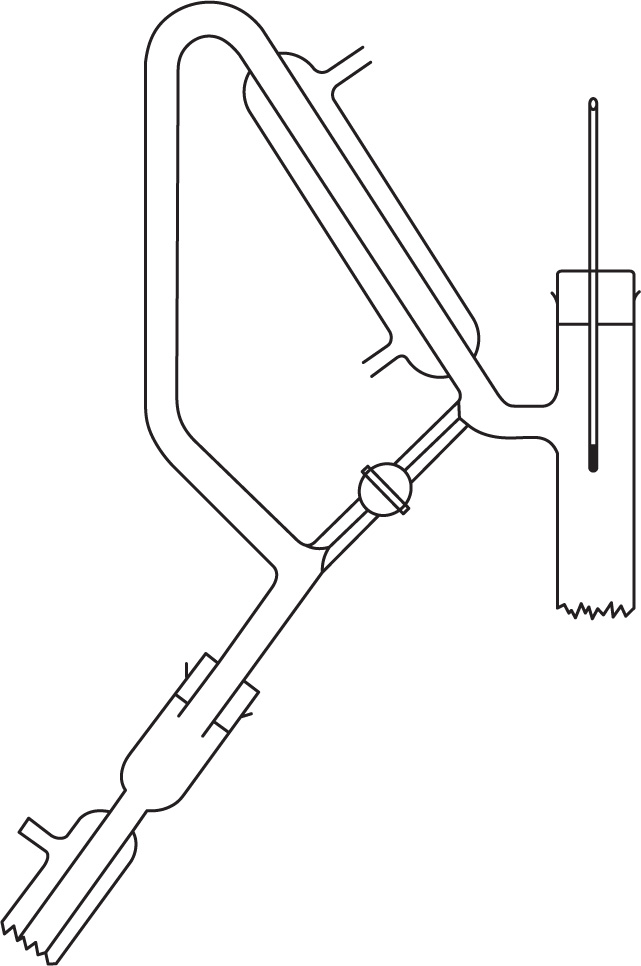
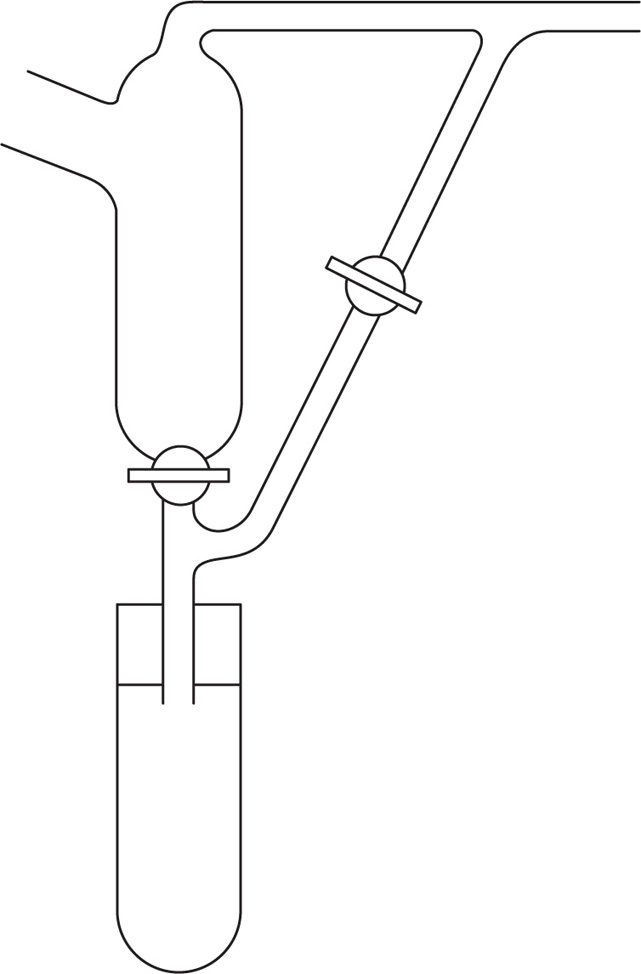
Fig. 16.13 shows a total condensation variable take-off still head. In operation the vapours are refluxed with the tap closed until equilibrium and the resulting optimum separation are attained; the tap is opened and a small quantity of distillate is taken off; the tap is closed, the column again operated under total reflux and more distillate is then run off. By this means very sharp separations are achieved both at atmospheric and under reduced pressure. Heat input to the still should be controlled—if too little, the packing is insufficiently wetted by reflux and if too much, the vapour velocity may be too great for equilibrium to be attained.
Efficiency of fractionating columns: The efficiency of a column is commonly measured by the height equivalent to a theoretical plate (HETP). At a theoretically perfect plate, the vapours are brought into equilibrium with the liquid through which they pass; the length of a column required to achieve this is defined as the HETP, and vapour drawn off from the top of such a length of column is in equilibrium with the liquid at the bottom. The HETP value is obtained by analysis of the liquid in the still, the vapour at the head of the column and from data for the composition of liquid and vapour in equilibrium with one another.
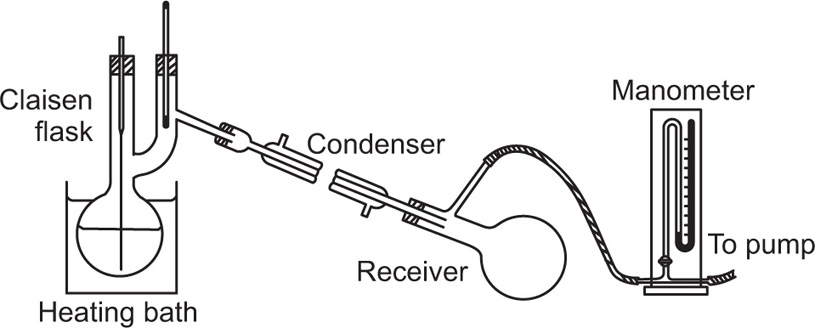
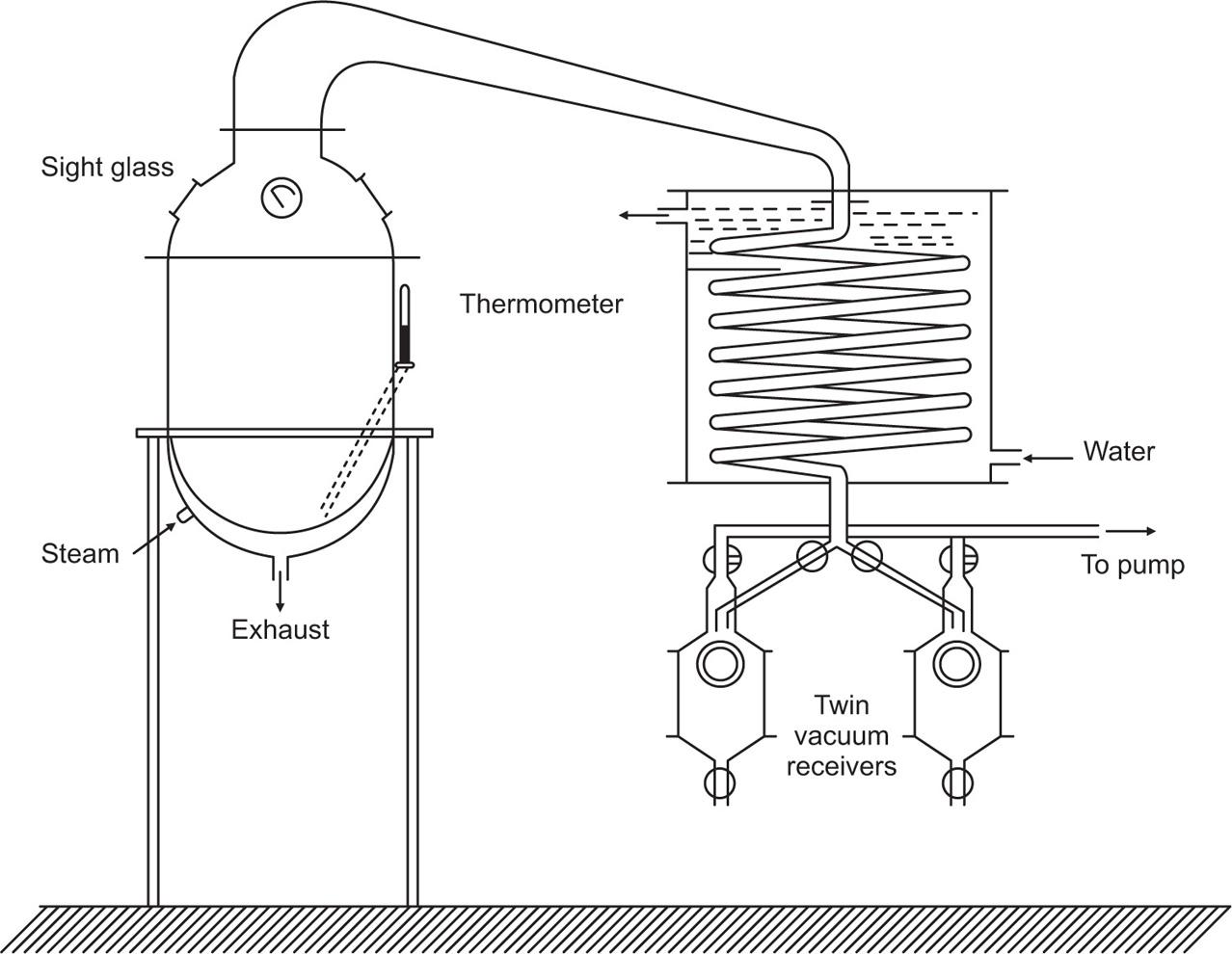
Molecular Distillation
In molecular distillation or evaporative distillation the pressure of the vapour above the liquid is much lower than the pressure of a saturated vapour in equilibrium with the liquid. The distance between the evaporating surface and the condenser is about equal to the mean free path of the vapour molecules at that very low pressure. Molecules leaving the surface of the
liquid are therefore more likely to hit the condenser surface than to collide with other molecules and little or no recondensation at the surface of the liquid takes place. In ordinary distillation at atmospheric pressure and under reduced pressure, conditions approaching equilibrium are maintained and a considerable amount of recondensation (refluxing) occurs. Molecular distillation is usually carried out at 0.001–0.00001
mm pressure. There is no well-defined temperature at which distillation commences, unlike ordinary distillation procedures. An extensive review of the principles and techniques has been given by Hickman (1944). This technique has been applied with great success to the purification of vitamin A. For this purpose the fish liver oil is fed continuously as a thin film on to heated discs rotating in a high vacuum.

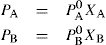
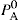 and
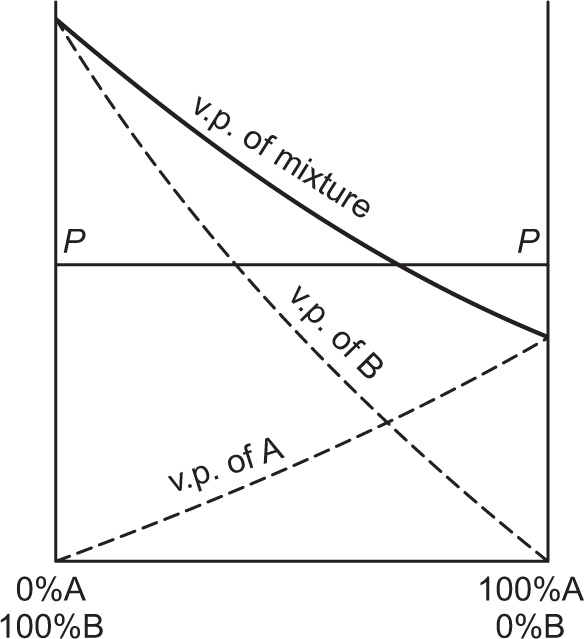
and 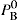 The total vapour pressure P of the system is then PA + PB (Fig. 16.8).
The total vapour pressure P of the system is then PA + PB (Fig. 16.8).