CHAPTER 42
Basic Science of Peripheral Nerve Injury and Repair
MARY BATHEN, BS AND RANJAN GUPTA, MD
CRITICAL POINTS
▪ Neuron: The primary cell of the PNS, which is polarized and has dendrites to receive information and axons to transmit information
▪ Nerves: Bundles of axons enclosed in specialized connective sheaths
▪ Schwann cell: Glial cell that supports and myelinates PNS axons
▪ Endoneurium: Collection of collagenous tissue that surrounds each individual axons and their associated Schwann cell
▪ Perineurium: The blood nerve barrier, which is composed of concentric layers of fibroblasts that form a sheath around each fascicle
▪ Epineurium: The collagenous connective tissue layer that surrounds the outer limits of the peripheral nerve, protecting it from external stress
▪ Monofascicular: Nerve section composed of one large fascicle
▪ Oligofascicular: Nerve section composed of a few fascicles
▪ Polyfascicular: Nerve section composed of many fascicles of varying sizes
▪ Neurapraxia: Nerve injury characterized by a reduction or complete block of conduction across a nerve lesion
▪ Axonotmesis: Nerve injury characterized by conduction block and interruption of axonal continuity
▪ Neurotmesis: Nerve injury characterized by conduction block, interruption of axonal continuity, and connective tissue damage
The human nervous system arguably defines the very nature of humanity. Through this complex and intricate system, effective communication and response is possible within the body and to the environment. This system has evolved to allow complex, coordinated movements while maintaining basic reflexes. These capabilities are facilitated by a variety of cells that make up the central nervous system (CNS)—brain and spinal cord and the peripheral nervous system (PNS). The PNS, the focus of this chapter, provides the mechanism for relaying information between the CNS and the environment.
One of the central paradigms of neuroscience has long been that the PNS, in contrast to the CNS, possesses the capacity of self-regeneration after traumatic injury. Despite advances in peripheral nerve repair techniques, however, complete functional recovery is seldom achieved in adults. In order for clinicians to better understand some of the limitations associated with the current treatment protocols for neural injuries, they need to be aware of the basic science behind peripheral nerve anatomy, injury, and healing. These topics are the focus of this chapter.
Relevant Developmental and Functional Anatomy
The origin of the PNS is from the ectodermal layer of the blastocyst, with most of the PNS cells being derived from the neural crest. The primary cell of the PNS is the neuron—a polarized cell with dendrites to receive information and axons to send information. Nerves are bundles of axons enclosed in specialized connective sheaths. Within the nerve, there are several different satellite cells known as glial cells, including Schwann cells, macrophages, and fibroblasts. Peripheral nerves can thus be defined as heterogeneous composite structures composed of neurons, Schwann cells, fibroblasts, and macrophages.
In 1839, Theodor Schwann identified the cell that now bears his name. Schwann cells, derived from the neural crest, are glial cells that support and myelinate PNS axons.
A mature, myelinating Schwann cell is responsible for extending a process of its plasma membrane to wrap an axon in myelin. As such, myelin is a multilamellar, membranous sheath that surrounds the axons of CNS and PNS nerves. Myelin itself acts as an insulator around the axon, reducing the dissipation of the action potential into the surrounding medium as it propagates through the axon. The myelin sheath has discontinuities along the nerve fiber known as nodes of Ranvier, which create distinct morphologic and biochemical domains along PNS nerves (Fig. 42-1). Nodes of Ranvier contain high concentrations of voltage-gated sodium channels. The arrival of an action potential at a node depolarizes the membrane, opening the sodium channels to induce a massive influx of sodium ions into the axon. This generates an electrical pulse that is propagated down the axon to the next node of Ranvier, a process termed saltatory conduction. Saltatory conduction allows not only for an increased speed of nerve impulse transmission, but also reduces the energy requirement.
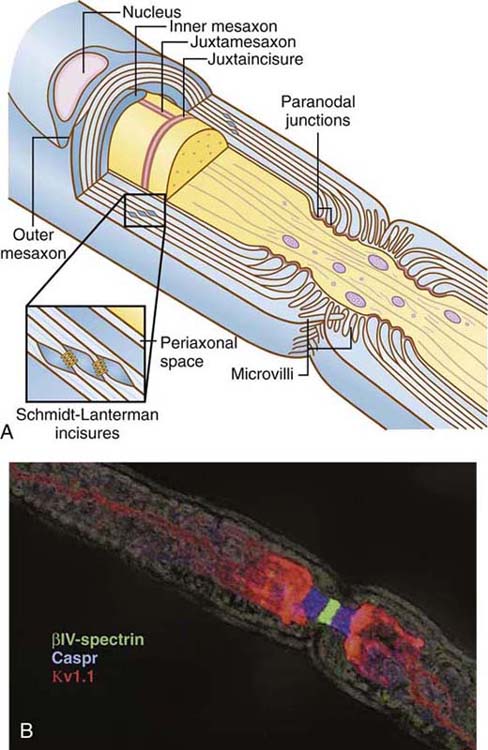
Figure 42-1 Morphology of domains of myelinated axons in the peripheral nervous system. A, Schematic organization of a peripheral myelinated nerve is shown. Cross section through a myelinated axon (gray) surrounded by two myelin sheaths is illustrated, demonstrating the node of Ranvier (red), to which Schwann cell microvilli project; the paranodal loops and junctions (green); the juxtaparanodal region (purple); Schmidt–Lanterman incisures, which form gap junctions (shown at higher magnification in the inset); compact myelin (light blue); and the inner (IM) and outer (OM) mesaxons. The axon diameter is reduced in the region of the node and paranodes, with more tightly packed neurofilaments and accumulation of membrane vesicles and mitochondria. The intranodal axonal specializations, the juxtamesaxon, and juxtaincisures are also shown. The entire structure is surrounded by a basal lamina (not illustrated). B, Confocal immunofluorescence microscopy of teased sciatic nerve fiber illustrating axonal domains. The node is stained for βIV-spectrin (green), the paranodes for Caspr (blue), and the juxtaparanodes for Kv1.1 (red); juxtamesaxonal staining of Kv channels is also seen in the lower right. The field was photographed with differential interference contrast microscopy to illustrate the overall dimensions of the myelin sheath. (Figure borrowed from Salzer JA. Polarized domains of myelinated axons. Neuron. 2003;40:297-318.)
Unmyelinated and myelinated nerves are bundled together in fascicles, with three main layers of tissue: the endoneurium, perineurium, and epineurium (Fig. 42-2). The endoneurium is a collection of collagenous tissue that surrounds each individual axon and their associated Schwann cells. The perineurium is an extension of the blood–brain barrier and is known as the blood–nerve barrier. It is composed of concentric layers of fibroblasts that form a sheath around each fascicle. Prominent basement membranes and numerous tight junctions control the intraneural environment by restricting diffusion into the fascicles. The perineurium is the neural layer most resistant to longitudinal traction and thus provides the peripheral nerve’s elastic properties. The epineurium is the collagenous connective tissue layer that surrounds the outer limits of the peripheral nerve protecting it from external stress. The epineurium is divided into two layers: the internal and external. The internal epineurium cushions the fascicles within the nerve, while the external epineurium protects the nerve from external environment.
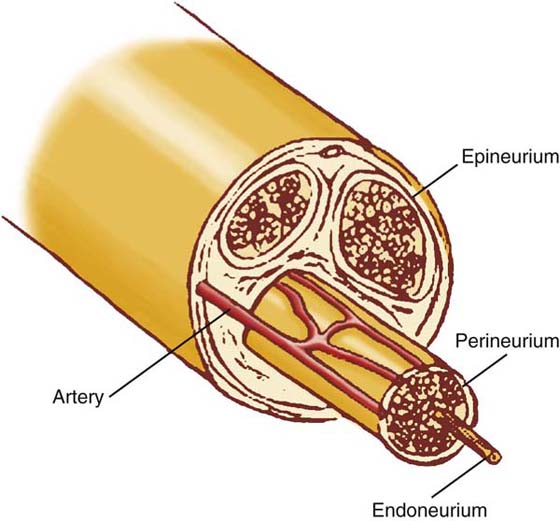
Figure 42-2 Anatomy of the nerve. Unmyelinated and myelinated axons are bundled together into fascicles. The individual axons are surrounded by the endoneurium. The entire fascicle is ensheathed by the perineurium, which forms the blood–nerve barrier. The epineurium surrounds the entire nerve, holding these structures together.
Fascicular patterns are divided into the following three types: monofascicular, oligofascicular, and polyfascicular.1 Monofascicular patterns consist of one large fascicle, whereas oligofascicular patterns consist of a few fascicles; polyfascicular patterns consist of many fascicles of varying sizes that can be arranged with or without groupings of fascicles (Fig. 42-3). Nerves found in the upper arm are routinely polyfascicular. In its course from the upper arm to the fingertips, a peripheral nerve undergoes changes from a polyfascicular pattern in the upper arm, oligofascicular in the elbow region, and monofascicular in the hand and fingers. For example, the ulnar nerve is polyfascicular as it exits the brachial plexus until just before the elbow, at which point it becomes oligofascicular. After the division into the motor branch at the wrist, the pattern is monofascicular. These patterns may help to determine which type of nerve repair is appropriate for a particular nerve injury. In surgical nerve repair, proper identification of fascicular arrangement is crucial to maximizing the chances for a successful outcome.
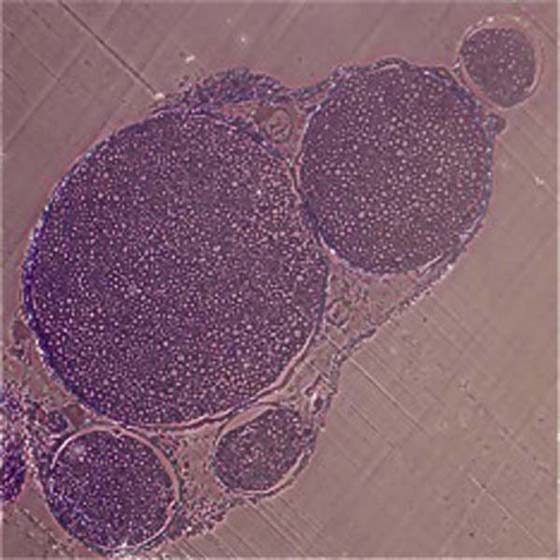
Figure 42-3 Toluidine blue–stained polyfascicular cross section of a normal murine sciatic nerve (×10).
Peripheral nerves are extensively vascularized with separate yet interconnected microvascular systems in the epineurium, perineurium, and endoneurium.2,3 The vascular pattern of the peripheral nerve is characterized by longitudinally oriented groups of vessels, with a great number of communicating anastomoses. The vasculature is composed of an intrinsic vascular system consisting of vascular plexa in the epineurium, perineurium, and endoneurium and an extrinsic system derived from closely associated vessels running with the nerve. The role of the intraneural microvascular system is vital in regard to the effects of chronic irritation, compression, mobilization, stretching, and transection, and should be considered in determining the proper surgical technique.
Classification of Nerve Injury
Nerve lesions are usually described using the standard classification schemes of Seddon4 or Sunderland5 (Table 42-1). In 1943, Seddon distinguished between three grades of damage: neurapraxia, axonotmesis, and neurotmesis.4
Table 42-1 Seddon and Sunderland’s Classification of Nerve Injuries
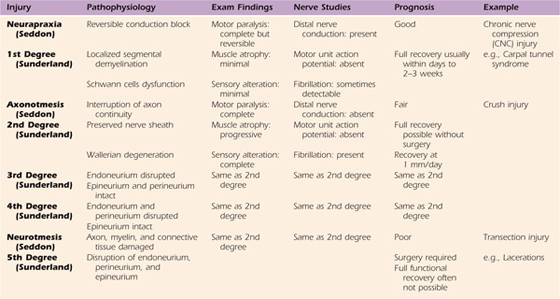
Neurapraxia is the simplest form of nerve injury and is characterized by a reduction or complete block of conduction across a nerve lesion, with preservation of conduction both proximal and distal to the lesion and conservation of axonal continuity.4 Neurapraxic conduction blocks are caused by localized segmental demyelination.4 Such demyelination is often mediated by progressive dysfunction of the Schwann cells, common in chronic nerve compression (CNC) disorders such as carpal tunnel syndrome. The prognosis for neurapraxic injuries is good.4
Axonotmesis is a more severe form of nerve injury, which is characterized by interruption of axonal continuity with preserved integrity of the nerve sheath.4 Following injury, Wallerian degeneration occurs. Nerve crush injuries exemplify axonotmetic injuries.4
Neurotmesis is the most severe form of nerve injury and occurs when the axon, myelin, and connective tissue components are damaged and disrupted or transected.4 In neurotmesis, recovery through regeneration cannot occur without surgical intervention.4 Such an injury can be created by incision, fracture, or laceration and is classified as neurotmesis.4
In 1951, Sunderland devised a five-grade classification scheme, which is now more widespread than Seddon’s three-grade system.5 Roughly, grades I and II correspond to Seddon’s neurapraxia and axonotmesis, respectively.5 Sunderland further divided Seddon’s category of neurotmesis injuries into grades III, IV, and V, based on the extent of damage to the axonal supporting structures.5 In grade III injuries, axon continuity is disrupted by loss of endoneurial tubes, but the perineurium is preserved.5 In grade IV injuries, nerve fasciculi (axon, endoneurium, perineurium) are damaged and intraneural scarring occurs, but nerve sheath continuity is preserved.5 In grade V injuries, the endoneurium, perineurium, and epineurium are completely divided, and substantial perineural hemorrhage and scarring occurs.5
Although not routinely used, additional modifications to the classification scheme have occurred more recently. In 1988, Mackinnon and Dellon presented a grade VI injury that represented a complex peripheral nerve injury, involving combinations of Sunderland’s grades of injury.1 Furthermore, in 1992, Millesi amended Sunderland classification to include the extent of associated fibrous tissue proliferation (type A fibrosis, type B fibrosis, type C fibrosis).6
Traumatic/Acute Nerve Injury
Although the preceding classification scheme is helpful to the clinician, the basic science researcher classifies nerve injuries as simply acute or chronic. Acute and chronic nerve injuries lead to disparate physiologic and histopathologic changes to the involved nerve and its adjacent tissue, including demyelination, degeneration, remyelination, and regeneration.
Acute peripheral nerve injuries have a sudden onset, as in nerve crush and transection injuries. Almost immediately, the injured nerve undergoes a process known as Wallerian degeneration (WD), whereby the damaged segment of the nerve is phagocytosed, beginning at the first intact node of Ranvier and progressing distally to allow for regrowth to begin.7 Granular disintegration of the axonal cytoskeleton, the hallmark of WD, is triggered by increased production of axoplasmic calcium and proceeds in an anterograde fashion down the axon. By 48 to 96 hours after the injury, axonal continuity is lost and impulse conduction is blocked.8 By 36 to 48 hours afterward, myelin disintegration is well advanced.8 The clearance of axonal and myelin debris creates an environment hospitable for regeneration.
Schwann cells and macrophages play pivotal, intertwined roles in degeneration and regeneration after acute peripheral nerve injury. Soon after injury, activated Schwann cells secrete chemotactic factors to recruit hematogenous monocytes to the site of injury, where they differentiate into macrophages. The macrophage is the primary phagocyte of myelin and accumulates by 72 hours. Early on, the macrophages express major histocompatibility complex class II antigen 1a and are not phagocytic.9,10 Later, the hematogenously derived macrophages penetrate the basal lamina, lose 1a expression, become phagocytic, and produce interleukin-1 (IL-1).9,10 IL-1 stimulates Schwann cells to produce nerve growth factor (NGF), which is required for regeneration of axons and myelin formation.9
Schwann cells play yet another role in regeneration. Whereas normal adult Schwann cells do not divide, Schwann cells within the distal segment divide within 24 hours of injury with peak response by 72 hours. They form Bünger’s bands, which are cytoplasmic processes that interdigitate and line up in rows under the original basal lamina of the nerve fiber. Regenerating axons travel within these channels to reach their target end-organ.
By 5 to 8 weeks after the injury, the degenerative process is usually complete; all that remains are nerve fiber remnants composed of Schwann cells within an endoneural sheath.8 Nonetheless, in more severe injuries, a more significant local reaction occurs, whereby hemorrhage and edema lead to a vigorous inflammatory response.8 Fibroblasts, glial cells that help maintain the extracellular matrix, proliferate, and a dense fibrous scar causes a fusiform swelling of the injured segment.8 In addition, interfascicular scar tissue develops, permanently enlarging the nerve trunk.8 While denervated, the endoneurial tubes begin to shrink, reaching a minimum size at 3 to 4 months after injury.11,12 Concurrently, the endoneurial sheath progressively thickens secondary to collagen deposition along the outer surface of the Schwann cell basement membrane.11,12 In severe injuries, numerous endoneurial tubes do not receive a regenerating axon, and progressive fibrosis ultimately obliterates them.11,12
Changes in the neuronal cell bodies and nerve fibers proximal to the site of injury depend on the severity of the injury and the proximity of the injured segment to the cell body.8 In extreme cases, in which the cell body degenerates, the entire proximal segment undergoes WD and is phagocytosed.8 Arguably, the cell body and axon depend on each other for recovery: the cell body does not fully recover without the reestablishment of functional peripheral connections, and the final axonal caliber greatly depends on the recovery of the cell body.8 In response to axonal injury, the nucleus of the nerve cell body undergoes a process referred to as chromatolysis, whereby the nucleus migrates to the periphery of the cell and Nissl’s granules (nerve rough endoplasmic reticulum) break up and disperse.8 It is believed that chromatolysis somehow signals the perineuronal glial cells to proliferate and extend processes that interrupt synaptic connections and isolate the neuron for recovery.8 In addition, chromatolysis reprograms the metabolic machinery to enable the cell body to increase lipid and protein production necessary for regeneration.8 The reversal of chromatolysis is one of the earliest signs of regeneration.8
In mild injuries the regenerative and repair processes begin almost immediately, but in more severe injuries nerve regeneration begins only after WD has run its course. One of the early signs of regeneration is the sprouting of new axons (growth cones) by myelinated and unmyelinated nerve fibers proximal to the injury site. Axons that successfully reinnervate the distal stump will mature. Magill and colleagues found that when the mouse sciatic nerve was crushed, 100% of motor end plates are denervated 1 week after crush, with partial reinnervation at 2 weeks, hyperinnervation at 3 and 4 weeks, and restoration of a 1 : 1 axon to motor end plate relationship 6 weeks after injury.13 Schwann cells help guide the regenerating axonal sprouts to the denervated motor end plates, reforming neuromuscular junctions.14 Depending on the size of the defect and extent of injury, axons may regenerate and begin to remyelinate at 6 to 8 weeks; however, the original thickness is never achieved.4 On average, axonal growth occurs at a rate of 1 to 2 mm/day with a decreased rate in distal regions. Nevertheless, in more serious injuries, prolonged denervation is observed, which can lead to muscle atrophy. Denervated muscle fibers atrophy quite rapidly (on average, 70% reduction of cross-sectional area by 2 months).8 Although not common, occasional dropout of muscle fibers does occur as a late phenomenon, between 6 and 12 months after denervation.8
Healing of Acute Nerve Injury: Regeneration and Remyelination
Unlike CNS nerves, peripheral nerves are capable of spontaneous regeneration after injury. Regeneration occurs when the environment is permissive and the intrinsic growth capacity of neurons is activated. Functional regeneration requires both axon regrowth and remyelination by Schwann cells.
That functional recovery is often suboptimal for a number of reasons: (1) The correct organ does not innervate in the correct location. (2) The end organ may be degenerated by the time the axon reaches the target. (3) An incorrect receptor may be innervated. (4) The receptor may be in the wrong location. (5) Axon continuity may not be maintained.
Many of these issues are related to the surgical coaptation. Even under the ideal surgical conditions, results are suboptimal. As such, most surgeons recognize that molecular mechanisms for regeneration are not effective at producing normal functional regeneration without advances in medical intervention.
Regeneration
Activation of Intrinsic Growth Capacity
The activation of the intrinsic growth capacity is best studied in the dorsal root ganglion (DRG) primary sensory neurons. DRG neurons are pseudobipolar neurons that have only one axon stemming from the cell body, which branches out to two axons: the peripheral branch innervates the sensory organs in the peripheral tissues, whereas the central branch enters the spinal cord and ascends the dorsal column to terminate in the brain. Largely due to disparate environments, the peripheral branch spontaneously regenerates after injury, while the central branch does not. However, if the peripheral branch is injured before the central branch, a phenomenon known as a conditioning peripheral lesion occurs, whereby the central branch can regenerate beyond the injury site, in the inhibitory environment of the spinal cord.15,16
The intrinsic growth capacity is controlled by a number of genes and gene products, proteins. Myelin-associated glycoprotein (MAG) is an important constituent of myelin in the PNS, where it is expressed primarily by promyelinating glial cells and plays a key role in the early stages on myelination. MAG has a biphasic function: early in development it enhances axonal growth, but in adults it inhibits axonal regeneration and neurite outgrowth. In adults, the binding of either myelin or MAG to the Nogo receptor (NgR)-p75 neurotrophic receptor complex activates the Rho pathway, which in turn inhibits cytoskeleton assembly.
In vitro experiments have shown that MAG inhibits DRG outgrowth.17 However, in vitro as well as in vivo experiments have shown that when the peripheral branch is lesioned, the DRG axons sufficiently activate the intrinsic growth capacity to overcome the inhibitory effects of MAG.18,19 Two independent studies argue that disinhibition occurs because of the increased postlesional intracellular cyclic adenosine monophosphate (cAMP) levels.18-20 cAMP is synthesized from adenosine triphosphate and is a well-known second messenger, used for intracellular signal transduction. cAMP’s significance was discovered when injection of dibutyryl-cAMP (a cAMP analog) into a nonlesioned DRG resulted in regeneration of the central branch of DRG neurons.18-20
The action of cAMP proceeds through protein kinase A (PKA). PKA refers to a family of enzymes whose activity is dependent on cAMP levels. Experiments have shown that if PKA is blocked, lesion-induced growth of neurons that have been plated on MAG is blocked.15,19 The effects of both cAMP and PKA are transcription-dependent and are regulated by the cAMP response element binding (CREB) proteins.21,22 CREB proteins are transcription factors that bind to certain sequences of DNA (Cre elements) to increase or decrease transcription of certain genes. When PKA triggers gene expression through CREB, the transcription of regeneration-related genes, such as Arginase I, is upregulated.21 Arginase I, the liver isoform of arginase, is an enzyme involved in polyamine synthesis (the urea cycle), and it has been discovered that cAMP disinhibition of MAG is ineffective when polyamine synthesis is blocked.21 It is hypothesized that polyamines may induce expression of other regenerative genes or may directly affect cytoskeleton organization.21 In sum, cAMP increases the transcription of Arginase I through PKA activation of CREB (Fig. 42-4).
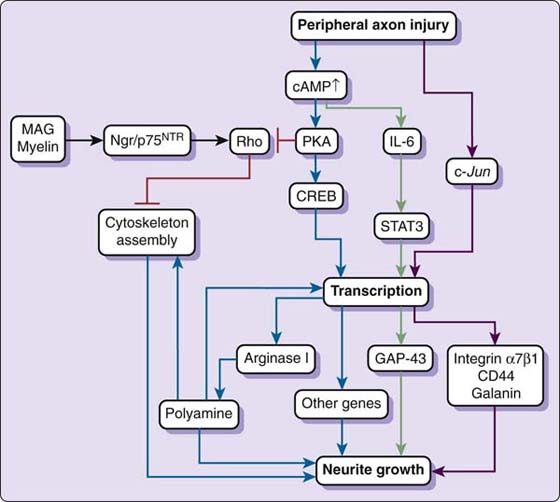
Figure 42-4 Activation of intrinsic growth capacity by peripheral nerve injury. Peripheral nerve injury elevates intracellular cyclic adenosine monophosphate levels, which activates protein kinase A (PKA). PKA triggers gene expression through cAMP response element binding (CREB), resulting in transcriptional up-regulation of regeneration-related genes such as Arginase I. Arginase I promotes the synthesis of polyamines, which may directly regulate cytoskeleton assembly or further induce gene expression necessary for regeneration. Activation of PKA also inhibits Rho antagonizing myelin-associated glycoprotein (MAG) or myelin-induced Rho activation and inhibition of neurite growth. Elevated cAMP levels also up-regulate interleukin-6 (IL-6), which, through signal transducers and activators of transcription-3 (STAT-3), induces regeneration-related genes such as the one that produces growth-associated protein-43 (GAP-43). Peripheral injury additionally induces c-Jun transcription factor-dependent regeneration-related gene expression, such as for integrin-α7β1 CD44 and galanin. Activation of the intrinsic growth capacity is regulated mainly at transcriptional level. (Figure borrowed from Chen ZL. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209-233.)
A few additional pathways may play significant roles in regeneration. cAMP also directly upregulates interleukin-6 (IL-6) and inhibits myelin-associated Rho activation.23 Moreover, Raivich and coworkers discovered that c-Jun knockout mice show impaired induction of several molecules involved in regeneration, including CD44, galanin, and integrin α7β1.24
Environmental Factors in Peripheral Axon Regeneration
Many argue that environmental differences can explain why PNS regeneration succeeds while CNS regeneration does not. CNS axon regeneration is made especially difficult by myelin-associated inhibitory molecules and the formation of a glial scar.23 The PNS response to injury prevents accumulation of myelin-associated inhibitory proteins.25 Following an injury in the PNS, Schwann cells and macrophages rapidly remove myelin debris and Schwann cells dedifferentiate and down-regulate myelin proteins.25 Moreover, Nogo-A, a strong inhibitor in the CNS, is not normally expressed in the PNS.26 A glial scar does not form in the PNS after injury because astrocytes, the glial cells that form glial scars, are absent in the PNS.
Successful regeneration may also depend on axon guidance cues, including extracellular matrix (ECM) proteins and neuronal adhesion molecules. One ECM protein, laminin, has attracted significant attention in recent years, for it has been shown that laminin plays an important role in neurite outgrowth in vitro. Laminins are heterotrimeric glycoproteins composed of a α-, β-, and γ-chains and are major components of the basal lamina. Substantial evidence shows that laminin plays an important role in neurite outgrowth in vitro (for review, see Luckenbill-Edds 199727). Of the 15 known laminin isoforms, laminin-2 and laminin-8 are expressed in the endoneurium of peripheral nerves28 and are up-regulated after peripheral nerve injury.29-31 Because laminin can directly promote neurite extension32 and Schwann cells are dramatically affected by laminin knockout mice,33 it is believed that laminin effects axonal regeneration by directly serving as a substrate for elongation or by indirectly supporting Schwann cell function, or both. Although the exact mechanism by which laminin supports regeneration is not completely understood, in vitro studies have shown that laminin plays a critical role in axon establishment through the PI/AKT signaling pathway.34-36 Laminin binding to receptors (e.g., integrins, dystroglycan) triggers phosphorylation and activation of PI (phosphoinositide) 3-kinase. PI 3-kinase activation triggers the activation of AKT.34-36 Active AKT then phosphorylates and inhibits glycogen synthase kinase-3 beta (GSK-3β) activity, triggering cytoskeleton-binding proteins to organize cytoskeleton elongation.34-36
Other Factors Affecting Peripheral Nerve Regeneration
Several neurotrophic factors, cytokines, and transcription factors may play a significant role in creating a permissive environment for regeneration. Neurotrophic factors prevent neuronal death and thus make axon regeneration possible. One major group of neurotrophic factors is the neurotrophins, four structurally related proteins: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4). Neurotrophin signaling is mediated by two types of receptors: three high-affinity Trk receptor kinases (each neurotrophin-specific) and a low-affinity p75NTR (which binds all neurotrophins with similar affinity).
NGF, BDNF, and NT-3 are all believed to be involved in peripheral nerve regeneration. NGF is the first neuronal growth factor to be discovered, is specific for a subset of primary sensory neurons and for sympathetic neurons, and is involved in nerve cell survival and maintenance in the normal state. Because NGF messenger RNA expression is greatly increased following peripheral nerve injury, it appears that NGF is an important component of the nerve repair process. Although NGF has been shown to bind to both low-affinity (p75) and high-affinity (trkA) receptors, studies have shown that only neurons that express the high-affinity receptor (TrkA) are affected by NGF. Because motor neurons do not express TrkA, regeneration in motor nerves is likely not affected by NGF,37,38 although some studies contest this.39
In addition to NGF, BDNF and NT-3 are being explored as potential mediators of peripheral nerve regeneration. Both BDNF and NT-3 (as well as NT-3 and NT-4/5) undergo receptor-mediated retrograde transport to neurons to prevent neuronal death,40 promote survival and differentiation of motor neurons in vitro,41 and regulate the function of developing neuromuscular synapses.42 Taken together, these qualities suggest that BDNF and NT-3 could potentially be used to promote regeneration. Two studies recently reported that BDNF promotes functional recovery after peripheral nerve injury,43,44 but others present seemingly conflicting results.45,46 Moreover, NT-3, which is down-regulated following peripheral nerve injury, was recently shown to enhance peripheral nerve regeneration through fibronection mats.47
Cytokines are also thought to be involved in peripheral nerve regeneration. Cytokines are similar to neurotrophins, except cytokines are inducible, whereas neurotrophins are constitutively expressed. Arguably the most widely studied cytokine is ciliary neurotrophic factor (CNTF). CNTF has a broad range of functions in the nervous system and has a trophic effect of denervated muscle. The neuronal specificity of CNTF overlaps considerably with that of the neurotrophins; however, its cellular localization, receptor structure, and signaling pathway are distinct.48-50 In normal peripheral nerves, CNTF expression is abundant and localized in the cytoplasm of myelinating Schwann cells.51,52 After axotomy, CNTF promotes survival of motor neurons in vitro and in neonatal animals, and it is hypothesized that CNTF acts as an “injury factor,” being released by glial cells in response to injury.53 Moreover, CNTF has been shown to protect denervated muscle from atrophy.54 A second cytokine involved in peripheral nerve regeneration is interleukin-6 (IL-6). IL-6 acts as both a proinflammatory and anti-inflammatory cytokine (cellular communication protein). IL-6 mRNA is induced in the DRG sensory neurons after injury and at degeneration sites during WD after sciatic nerve crush. In IL-6 knockout mice, the adult mouse shows sensory defects and delayed regeneration of sensory axons after crush injury. Another cytokine, leukemia inhibitory factor (LIF), is induced after sciatic nerve crush. LIF can be retrogradely transported to DRG sensory neurons to induce gene expression changes to promote regeneration, and LIF knockout mice show impaired peripheral nerve regeneration. In addition to the effects mentioned earlier, elevated cAMP levels following nerve injury causes IL-6 and LIF to activate the transcription factor STAT-3 (signal transducers and activators of transcription 3),55 which results in the transcription of regeneration-related genes like growth associated protein-43 (GAP-43).56 STAT-3 conditional knockout mice show that STAT-3 is required for motor neuron survival. Additional neurotrophic factors are thought to be linked to peripheral nerve regeneration, but a complete discussion of each is outside the scope of this chapter (for a review, see Terenghi 199957).
Remyelination
Functional recovery is only possible if, in addition to regeneration, Schwann cells remyelinate the regrown axons. At any time, Schwann cells can only be in one state: dedifferentiated–proliferating or differentiated–myelinating. Schwann cells destined to myelinate differentiate to promyelinating Schwann cells and extend cytoplasmic bundles to form a 1 to 1 ratio with individual axons (radial sorting) and enwrap them in myelin sheath.58,59
The process by which multipotent neural crest cells differentiate into mature, myelinating Schwann cells is not well understood; however, it is known that the establishment of axonal contact triggers Schwann cell development and the absence of contact leads to degeneration. β-Neuregulin, a protein product of neuregulin-1 (see section on Neurotrophic Factors and Peripheral Remyelination), is thought to play a pivotal role in this contact-mediated effect. Moreover, it has been discovered that the rate of differentiation is positively regulated by fibroblast growth factor and negatively by endothelin. When Schwann cells receive promyelinogenic signals from the axon, they produce large amounts of plasma membrane and up-regulate several proteins that give them the ability to myelinate, including MAG, periaxin, P0, myelin basic protein (MBP), and PMP22. Two transcription factors, Oct-6 and Krox-20, have been shown to promote and mediate the phenotypic transition from promyelinating to myelinating. Oct-6, which is expressed in Schwann cell progenitors but not in mature myelinating Schwann cells, is a strong repressor of the end-stage myelin-specific genes P0 and MBP. Krox-20, which is expressed only in mature myelinating Schwann cells, promotes the Schwann cells’ withdrawal from the cell cycle.
As previously mentioned, Schwann cells undergo phenotypic changes following nerve injury, and these changes play pivotal roles in degeneration–demyelination as well as regeneration–remyelination. Stoll and Muller showed that the loss of Schwann cell–axon contact following nerve injury leads to Schwann cell dedifferentiation followed by a series of proliferations.60 Once the Schwann cell recontacts a regenerated axon, Schwann cell redifferentiation is triggered. As in normal development, Oct-6 expression is followed by Krox-20 expression and the reinduction of myelin-specific genes.61-63
In recent years, secondary to research into demyelinating diseases, it has been discovered that the Schwann cell phenotype is also largely influenced by ECM proteins, neurotrophic factors, and hormones.
Extracellular Matrix Proteins and Peripheral Remyelination
Bunge and associates have provided substantial evidence that Schwann cells require the formation of a basal lamina to properly ensheath and myelinate axons.64,65 The basal lamina is a layer of ECM that envelops the Schwann cell–neurite units. Laminins, as discussed earlier, are major components of the basal lamina that are especially important to myelination. In fact, recent studies have shown that signals from laminins, and not the assembly of a continuous basal lamina per se, are required for myelination.66 Yang and colleagues67 and Yu and coworkers68 showed that disruption of laminins in the PNS results in aberrant Schwann cell differentation, a lack of radial sorting of axons, and severe hypomyelination. Yu and coworkers disrupted the laminin-γ1 gene, which is one of the most abundant laminin chains and is present in all known PNS isoforms.68 The results indicated that Schwann cells lacking laminin do not extend processes required for axonal sorting and mediating axon–Schwann cell interaction.68 In a related study, Yang and colleagues found that combined laminin-2/laminin-8 deficiencies caused nearly complete amyelination.67 A close analysis of the data revealed that laminin-2 and laminin-8 have a dominant role in defasciculation, and that laminin-8 promotes myelination without basal lamina.67
Laminin’s ability to significantly affect myelination depends on its ability to bind to the appropriate receptor. Therefore, laminin receptors are now being explored as possible means of promoting myelination. Laminin receptors include integrin receptors and other plasma membrane molecules, such as dystroglycan. Integrins are cell surface receptors that interact with ECM and mediate various intracellular signals. Structurally, integrins are obligate heterodimers with two distinct chains, composed of α- and β-subunits. Two laminin-binding integrins expressed by Schwann cells are α6β1- and α6β4-integrins. Dystroglycan, on the other hand, is one of the dystrophin-associated glycoproteins that, in skeletal muscle, acts as a transmembrane linkage between the ECM and the cytoskeleton. Lefort and associates69 and Masaki and colleagues70 showed that remyelinating Schwann cells up-regulate expression of β1-integrin and dystroglycan.69,70 Later studies revealed that β1-integrin is necessary for proper sorting of axons,71 whereas dystroglycan maintains the myelin sheath and regulates myelin thickness.72 Curiosity over how basal lamina components on the Schwann cell outside surface transduce a signal to influence axon sorting (a process affecting the inner and lateral surface) led to the discovery that laminin signaling activates the Rho family GTPase Rac1 in Schwann cells, which leads to radial sorting and subsequent myelination of axons.73,74
In addition to laminins and their receptors, extracellular proteases (e.g., plasmin) also regulate remyelination. Plasmin is an important blood enzyme that degrades fibrin. Studies have revealed that fibrinogen infiltrates the nerve following nerve injury. Fibrinogen is converted to fibrin, which accumulates and prevents two crucial steps to regeneration: migration and remyelination of Schwann cells.75,76 In response to fibrin accumulation, Schwann cells produce tissue plasminogen activator (tPA),77 which activates the fibronolytic cascade, paving the way for Schwann cell remyelination.
Neurotrophic Factors and Peripheral Remyelination
Originally, attention was drawn to neurotrophic factors for their role in neuron survival and regeneration. However, recent studies have revealed that neurotrophic factors also significantly affect Schwann cell differentiation and myelination.
As discussed earlier, one major group of neurotrophic factors, the neurotrophins (e.g., NGF, BDNF, and NT-3), are thought to play a significant role in remyelination following peripheral nerve injury. One study revealed that the addition of endogenous BDNF or depletion of exogenous NT-3 enhances myelination, suggesting that BDNF is a positive modulator and NT-3 is a negative modulator of peripheral nerve regeneration.78 Another study showed that inhibition of TrkB (specific for BDNF) did not prevent myelination, whereas blockage of p75NTR showed inhibitory effects on myelination.79 This result implies that p75NTR is necessary for proper myelination. Not surprisingly, the down-regulation of NT-3 and the up-regulation of both BDNF and p75NTR promotes remyelination following nerve injury. Further analysis of neurotrophin expression and transport may help direct the innovation of a therapeutic intervention. For instance, one interesting study recently revealed that sensory neurons of the DRG are a major source of BDNF, and DRGs transport and secrete endogenous BDNF along the surface of axons in the anterograde fashion.80 Understanding the mechanism of expression is a key step to successfully harnessing the intrinsic ability to promote myelination.
Beyond the neurotrophins, a number of other neurotrophic factors affect Schwann cell differentiation, but a review of each is outside the scope of this paper. Neuregulin-1 (NGR-1) and transforming growth factor-β (TGF-β) are the two major neurotrophic factors under active investigation. Neuregulins are a family of four structurally related proteins that are part of the epidermal growth factor (EGF) family of proteins. The NRG-1 family includes more than 15 EGF-like ligands that interact with ErbB receptor tyrosine kinase to control many aspects of neural development. During development, NRG-1 isoforms positively regulate Schwann cells at multiple stages of cell lineage.81 NRG-1 type III isoform is the principal regulator of early Schwann cell lineage (including precursors), and a threshold level of NRG-1 type III triggers Schwann cell myelination.82 Additionally, NRG-1 type III regulates myelin sheath thickness to match axon caliber.82 Carroll and associates revealed that neuregulin and ErbB expression increased after axotomy of the sciatic nerve.83 Nevertheless, a recent study showed that disruption of ErbB2 in mature Schwann cells does not impair regeneration after injury, diminishing the enthusiasm for the potential use of NRG-1 to promote remyelination.84 Notably, this study emphasizes that the developmental process of Schwann cell differentiation is not identical to Schwann cell differentiation after injury.
During development, TGF-β maintains the nonmyelinating, proliferating state of Schwann cells and regulates Schwann cell survival.81 Scherer and colleagues demonstrated that following sciatic nerve injury, TGF-β1 mRNA in the distal nerve stump increases, whereas TGF-β3 mRNA falls (TGF-β2 is not detected).85 This shows that Schwann cells alter expression of TGF-β1 and TGF-β2 in response to nerve injury.85 Studies have demonstrated that, as in development, TGF-β blocks Schwann cells differentiation–myelination after injury. TGF-β exerts its effects by preventing the expression of Ski, a proto-oncogene that coordinates the transition from Schwann cell proliferation to differentiation.86 Ski expression is decreased 4 days after nerve injury, but returns to preinjury levels following complete regeneration and remyelination. It would be interesting to explore the manipulation of Ski expression to promote remyelination after peripheral nerve injury.
Hormones and Peripheral Remyelination
Beyond ECM proteins and neurotrophic factors, studies have shown that myelination and remyelination are affected by a number of hormones, including progesterone, thyroid hormone (triiodothyronine, T3), and erythropoietin. Progesterone, a steroid hormone secreted by the adrenal glands, is involved in the female menstrual cycle, embryogenesis, and pregnancy (supports gestation). Koening and coworkers found that blockage of the effects of progesterone decreases the thickness of myelin sheaths of remyelinating axons after injury, whereas administration of exogenous progesterone to the injury site promotes myelin sheath formation.87 The thryoid hormone T3, a tyrosine-based hormone produced by the thyroid gland, plays an integral role in regulating metabolism and homeostasis. Voinesco and associates discovered that administration of T3 to an injured rat sciatic nerve increased nerve regeneration and increased the number, diameter, and myelin thickness of remyelinated axons.88 Erythropoietin (EPO), a glycoprotein hormone, regulates red blood cell production and is expressed in PNS axons and Schwann cells.89 Recent studies suggest that EPO may facilitate peripheral nerve regeneration by increasing Schwann cell proliferation and decreasing tumor necrosis factor-alpha (TNF-α) mediated injury and death.90,91
Chronic Nerve Compression Injury and Healing
CNC injuries, such as carpal tunnel syndrome, cubital tunnel syndrome, and spinal nerve root stenosis, affect millions of individuals. In contrast to acute nerve injuries, CNC injuries develop over the course of weeks to months. Until recently, they were considered variants of acute nerve injuries that developed over time and had a similar pathogenesis. Much of the earlier literature described compression neuropathies as the by-product of mild WD that occurred at the site of injury. Recent research using both in vivo and in vitro modeling systems has called that notion into question. Active research into chronic nerve compression injuries has revealed important details about their unique pathogenesis and corresponding repair mechanisms. Chapter 46 provides additional information on the pathophysiology of nerve compression, whereas this section focuses on Schwann cell changes.
Chronic nerve compression injury induces both Schwann cell proliferation and apoptosis, with minimal morphometric evidence of early axonal pathology.92 Design-based stereologic techniques revealed that Schwann cell number increased sixfold relative to the normal site of compression at 1 month and then slowly declined toward control levels.92 Assays of apoptosis revealed extensive Schwann cell apoptosis at 2 weeks after compression, demonstrating a concurrent Schwann cell proliferation and apoptosis that was confirmed with counts of bromodeoxyuridine-labeled Schwann cells.92 Electron microscopic analysis confirmed that these dramatic changes in Schwann cells occurred in the absence of axon degeneration and axonal swelling and before there was any detectable alterations in nerve conduction velocity.92 Although WD is known to trigger Schwann cell proliferation, alternative hypotheses exist such as mechanical stimuli providing Schwann cell mitogenic signals.92 An in vitro model to deliver shear stress in the form of laminar fluid flow to pure populations of Schwann cells confirmed that mechanical stimuli do, indeed, induce Schwann cell proliferation.92 These findings suggest that CNC injury induces Schwann cell turnover with minimal axonal injury and support the idea that mechanical stimuli have a direct mitogenic effect on Schwann cells.92
As Schwann cells undergo marked cellular turnover in the early stages of CNC injury, it is possible that demyelination and remyelination occur at the same time.93 Through nerve teasing techniques and unbiased stereology, myelination in nerves 1 month and 8 months after compression was recently assessed.93 Evaluations of myelin thickness and axonal diameter using design-based, unbiased stereology revealed a dramatic decrease in the average internodal length (IL) and an increase in the proportion of thinly myelinated axons, indicative of remyelination.93 The mean IL was reduced after 1 month of chronic nerve injury with no further decrease in IL at 8 months.93 There was limited change in average axonal diameter at both 1 and 8 months.93 Measures of myelin thickness revealed a sixfold increase in the number of axons with very thin (<5-µm thickness) myelin sheaths, as well as a proportional decrease in the number of axons with the thick myelin sheaths characteristic of normal nerve.93 These results confirm that an early consequence of CNC is local demyelination and remyelination, which may be the primary cause of alterations in nerve function during the early postcompression period.93
In addition to stimulating Schwann cell proliferation, in vitro mechanical loading alters the phenotype of many different cell types.94 After exposure to sustained shear stress, Schwann cells decreased their mRNA expression and protein levels of MAG and MBP. The mRNA expression of MAG and MBP was down-regulated 21% and 18%, respectively, whereas the Western blot showed down-regulation in MAG and MBP protein expression by 29% and 35%, respectively.94 The data suggests that down-regulation of myelin-associated proteins may be a direct response to physical stimulus that is not secondary to axonal injury.94 Immunohistochemistry data analysis revealed significant axonal sprouting at early time points, when MAG and MBP is down-regulated.94 Thus, the data suggest that down-regulation of MAG and MBP may create an environment permissive to axonal sprouting.94
Quantititative microscopic techniques helped to define the nature of this sprouting response and explore whether down-regulation of MAG by proliferating Schwann cells induces local sprouting.95 In the outer regions of CNC nerves, axonal sprouting was observed without evidence of WD.95 Immunolabeling and Western blot analysis revealed a local down-regulation of MAG protein within the site of injury, and local delivery of purified MAG protein abrogates the axonal sprouting response.95 These data demonstrate that CNC injury triggers axonal sprouting and suggest that a local down-regulation of MAG within the peripheral nerve secondary to CNC injury is the critical signal for sprouting response.95
Within the region of CNC injury, studies have shown that, in addition to Schwann cell changes, vascular permeability and neural vascularity increase.96 Vascular endothelial growth factor (VEGF) is a potent mitogen for endothelial cells and promotes blood vessel sprouting and vascular permeabilization.96 VEGF mRNA and protein expression increased within Schwann cells as early as 2 weeks after compression and peaked by 1 month, with a subsequent marked increase in the number of blood vessels.96
In addition to changes that result from Schwann cell phenotypic changes, CNC injuries vary markedly from acute injuries in terms of macrophage recruitment.97 In acute injuries, there is an immediate recruitment of macrophages, with a peak as early as 24 to 96 hours after injury.97 In CNC injuries, macrophage recruitment occurs slowly.97 Moreover, at 1 month after compression, macrophages are mainly localized in the outer third cross-section, to diffuse out later.97
After injury, hematogenously recruited macrophages express inducible nitric oxide synthase (iNOS), which generates localized increases in nitric oxide (NO).97 NO is a free-radical gas that acts as a free messenger and plays an integral role in regeneration by inhibiting neurotoxins.97 The majority of the iNOS expression is in the perineurium not the periphery, suggesting that iNOS up-regulation is due to ischemia regulated at the blood–nerve barrier.97 Macrophages also secrete Schwann cell mitogenic factors.97 In macrophage-depleting experiments, it was clearly shown that the hematogenously derived macrophages that are recruited to the region of CNC injury are integral to the alteration of the blood–nerve barrier, but had no detectable influence on Schwann cell proliferation after CNC injury.97
Summary
Clinicians should understand the basic science behind peripheral nerve anatomy, injury, and healing. It is equally important to recognize that recent research has revealed that the pathogenesis and recovery of acute and chronic nerve injuries differ in numerous and significant ways. A strong basic science background may improve the functional recovery, which is rarely complete today.
REFERENCES
1. Mackinnon SE, Dellon DA. Surgery of the peripheral nerve. New York: George Thieme Medical Publishers; 1992.
2. Bell MA, Waddell AG. A morphometric study of intrafascicular vessels of mammalian sciatic nerve. Muscle Nerve. 1984;7:524–534.
3. Lundborg G. The intrinsic vascularization of human peripheral nerves: structural and functional aspects. J Hand Surg [Am]. 1979;4:34–41.
4. Seddon H. Three types of nerve injury. Brain. 1943;66:237–288.
5. Sunderland S. A classification of peripheral nerve injuries producing loss of function. Brain. 1951;74:491–516.
6. Millesi H. Chirurgie der peripheren nerven. Munchen, Urban and Schwarzenberg. 1992.
7. Lee SK, Wolfe SW. Peripheral nerve injury and repair. J Am Acad Orthop Surg. 2000;8:243–252.
8. Burnett MG, Zager EL. Pathophysiology of peripheral nerve injury: a brief review. Neurosurg Focus. 2004;16:E1.
9. Creange A, Lefaucheur JP, Authier FJ, Gherardi RK. Cytokines and peripheral neuropathies. Rev Neurol (Paris). 1998;154:208–216.
10. Leskovar A, Moriarty LJ, Turkek JJ, et al. The macrophage in acute neural injury: changes in cell numbers over time and levels of cytokine production in mammalian central and peripheral nervous systems. J Exp Biol. 2000;203:1783–1795.
11. Sunderland S. The anatomy and physiology of nerve injury. Muscle Nerve. 1990;13:771–784.
12. Sunderland S, Bradley KC. Endoneurial tube shrinkage in the distal segment of a severed nerve. J Comp Neurol. 1950;93:411–420.
13. Magill CK, Tong A, Kawamura D, et al. Reinnervation of the tibialis anterior following sciatic nerve crush injury: a confocal microscopic study in transgenic mice. Exp Neurol. 2007;207:64–74.
14. Seckel BR. Enhancement of peripheral nerve regeneration. Muscle Nerve. 1990;13:785–800.
15. Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91.
16. Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793.
17. Shen YJ, DeBellard ME, Salzer JL, et al. Myelin-associated glycoprotein in myelin and expressed by Schwann cells inhibits axonal regeneration and branching. Mol Cell Neurosci. 1998;12:79–91.
18. Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893.
19. Qiu J, Cai D, Dai H, et al. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903.
20. Qiu J, Cai D, Filbin MT. A role for cAMP in regeneration during development and after injury. Prog Brain Res. 2002;137:381–387.
21. Cai D, Deng K, Mellado W, et al. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron. 2002;35:711–719.
22. Gao Y, Deng K, Hou J, et al. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–621.
23. Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nature Rev. 2003;4:703–713.
24. Raivich G, Bohatschek M, Da Costa C, et al. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:57–67.
25. Schafer M, Fruttiger M, Montag D, et al. Disruption of the gene for the myelin-associated glycoprotein improves axonal regrowth along myelin in C57BL/Wlds mice. Neuron. 1996;16:1107–1113.
26. Pot C, Simonen M, Weinmann O, et al. Nogo-A expressed in Schwann cells impairs axonal regeneration after peripheral nerve injury. J Cell Biol. 2002;159:29–35.
27. Luckenbill-Edds L. Laminin and the mechanism of neuronal outgrowth. Brain Res Rev. 1997;23(1-2):1–27.
28. Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521.
29. Doyu M, Sobue G, Ken E, et al. Laminin A, B1, and B2 chain gene expression in transected and regenerating nerves: regulation by axonal signals. J Neurochem. 1993;60:543–551.
30. Kuecherer-Ehret A, Graeber MB, Edgar D, et al. Immunoelectron microscopic localization of laminin in normal and regenerating mouse sciatic nerve. J Neurocytol. 1990;19:101–109.
31. Wallquist W, Patarroyo M, Thams S, et al. Laminin chains in rat and human peripheral nerve: distribution and regulation during development and after axonal injury. J Comparative Neurol. 2002;454:284–293.
32. Menager C, Arimura N, Fukata Y, Kaibuchi K. PIP3 is involved in neuronal polarization and axon formation. J Neurochem. 2004;89:109–118.
33. Chen ZL, Strickland S. Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J Cell Biol. 2003;163:889–899.
34. Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135.
35. Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75.
36. Yoshimura T, Kawano Y, Arimura N, et al. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149.
37. He C, Chen Z, Chen Z. Enhancement of motor nerve regeneration by nerve growth factor. Microsurgery. 1992;13:151–154.
38. Whitworth IH, Brown RA, Dore CJ, et al. Nerve growth factor enhances nerve regeneration through fibronectin grafts. J Hand Surg (Edinburgh). 1996;21:514–522.
39. Ang LC, Bhaumick B, Juurlink BH. Neurite promoting activity of insulin, insulin-like growth factor I and nerve growth factor on spinal motoneurons is astrocyte dependent. Brain Res. 1993;74:83–88.
40. Sendtner M, Holtmann B, Kolbeck R, et al. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1992;360:757–759.
41. Henderson CE, Camu W, Mettling C, et al. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature. 1993;363:266–270.
42. Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353.
43. Lewin SL, Utley DS, Cheng ET, et al. Simultaneous treatment with BDNF and CNTF after peripheral nerve transection and repair enhances rate of functional recovery compared with BDNF treatment alone. Laryngoscope. 1997;107:992–999.
44. Utley DS, Lewin SL, Cheng ET, et al. Brain-derived neurotrophic factor and collagen tubulization enhance functional recovery after peripheral nerve transection and repair. Archiv Otolaryngol–Head Neck Surg. 1996;122:407–413.
45. Ohnishi A, Yamamoto T, Her QY, et al. The effect of brain-derived neurotrophic factor on elongation of axons after transection with suture-morphometric evaluation. Journal of University of Occupational and Environmental Health, Japan. 1997;19:23–28.
46. Shirley DM, Williams SA, Santos PM. Brain-derived neurotrophic factor and peripheral nerve regeneration: a functional evaluation. Laryngoscope. 1996;106:629–632.
47. Sterne GD, Brown RA, Green CJ, Terenghi G. Neurotrophin-3 delivered locally via fibronectin mats enhances peripheral nerve regeneration. Europ J Neurosci. 1997;9:1388–1396.
48. Ip NY, Nye SH, Boulton TG, et al. CNTF and LIF act on neuronal cells via shared signaling pathways that involve the IL-6 signal transducing receptor component gp130. Cell. 1992;69:1121–1132.
49. Ip NY, Yancopoulos GD. Ciliary neurotrophic factor and its receptor complex. Prog Growth Factor Res. 1992;4:139–155.
50. Richardson PM. Ciliary neurotrophic factor: a review. Pharmacology & therapeutics. 1994;63:187–198.
51. Friedman B, Scherer SS, Rudge JS, et al. Regulation of ciliary neurotrophic factor expression in myelin-related Schwann cells in vivo. Neuron. 1992;9:295–305.
52. Rende M, Muir D, Ruoslahti E, et al. Immunolocalization of ciliary neuronotrophic factor in adult rat sciatic nerve. Glia. 1992;5:25–32.
53. Adler R. Ciliary neurotrophic factor as an injury factor. Curr Opinion Neurobiol. 1993;3:785–789.
54. Helgren ME, Squinto SP, Davis HL, et al. Trophic effect of ciliary neurotrophic factor on denervated skeletal muscle. Cell. 1994;76:493–504.
55. Qiu J, Cafferty WB, McMahon SB, Thompson SW. Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J Neurosci. 2005;25:1645–1653.
56. Cafferty WB, Gardiner NJ, Das P, et al. Conditioning injury-induced spinal axon regeneration fails in interleukin-6 knock-out mice. J Neurosci. 2004;24:4432–4443.
57. Terenghi G. Peripheral nerve regeneration and neurotrophic factors. J Anatomy. 1999;194(Pt 1):1–14.
58. Webster HD. Development of periphery nerve fibers. Dyck IJ, Thomas PK, Low PA, Poduslo JF, eds. Peripheral Neuropathy.Vol. 1. Philadelphia, WB: Saunders; 1993:243–266.
59. Mirsky R, Jessen KR. The neurobiology of Schwann cells. Brain Pathol. 1999;9:293–311.
60. Stoll G, Muller HW. Nerve injury, axonal degeneration and neural regeneration: basic insights. Brain Pathol (Zurich). 1999;9:313–325.
61. LeBlanc AC, Poduslo JF. Axonal modulation of myelin gene expression in the peripheral nerve. J Neurosci Res. 1990;26:317–326.
62. Scherer SS, Wang DY, Kuhn R, et al. Axons regulate Schwann cell expression of the POU transcription factor SCIP. J Neurosci. 1994;14:1930–1942.
63. Zorick TS, Syroid DE, Arroyo E, et al. The transcription factors SCIP and Krox-20 mark distinct stages and cell fates in Schwann cell differentiation. Mol Cell Neurosci. 1996;8:129–145.
64. Bunge MB, Clark MB, Dean AC, et al. Schwann cell function depends upon axonal signals and basal lamina components. Annals N Y Acad Sci. 1990;580:281–287.
65. Bunge RP, Bunge MB, Eldridge CF. Linkage between axonal ensheathment and basal lamina production by Schwann cells. Annu Rev Neurosci. 1986;9:305–328.
66. Podratz JL, Rodriguez E, Windebank AJ. Role of the extracellular matrix in myelination of peripheral nerve. Glia. 2001;35:35–40.
67. Yang D, Bierman J, Tarumi YS, et al. Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J Cell Biol. 2005;168:655–666.
68. Yu WM, Feltri ML, Wrabetz L, et al. Schwann cell-specific ablation of laminin gamma1 causes apoptosis and prevents proliferation. J Neurosci. 2005;25:4463–4472.
69. Lefcort F, Venstrom K, McDonald JA, Reichardt LF. Regulation of expression of fibronectin and its receptor, alpha 5 beta 1, during development and regeneration of peripheral nerve. Development (Cambridge). 1992;116:767–782.
70. Masaki T, Matsumura K, Saito F, et al. Expression of dystroglycan and laminin-2 in peripheral nerve under axonal degeneration and regeneration. Acta Neuropathol. 2000;99:289–295.
71. Feltri ML, Graus Porta D, Previtali SC, et al. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199–209.
72. Saito F, Moore SA, Barresi R, et al. Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron. 2003;38:747–758.
73. Benninger Y, Thurnherr T, Pereira JA, et al. Essential and distinct roles for cdc42 and rac1 in the regulation of Schwann cell biology during peripheral nervous system development. J Cell Biol. 2007;177:1051–1061.
74. Nodari A, Zambroni D, Quattrini A, et al. Beta1 integrin activates Rac1 in Schwann cells to generate radial lamellae during axonal sorting and myelination. J Cell Biol. 2007;177:1063–1075.
75. Akassoglou K, Akpinar P, Murray S, Strickland S. Fibrin is a regulator of Schwann cell migration after sciatic nerve injury in mice. Neurosci Lett. 2003;338:185–188.
76. Akassoglou K, Strickland S. Nervous system pathology: the fibrin perspective. Biol Chem. 2002;383:37–45.
77. Akassoglou K, Kombrinck KW, Degen JL, Strickland S. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol. 2000;149:1157–1166.
78. Chan JR, Cosgaya JM, Wu YJ, Shooter EM. Neurotrophins are key mediators of the myelination program in the peripheral nervous system. Proc Nat Acad Sci USA. 2001;98:14661–14668.
79. Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science. 2002;298:1245–1248.
80. Ng BK, Chen L, Mandemakers W, et al. Anterograde transport and secretion of brain-derived neurotrophic factor along sensory axons promote Schwann cell myelination. J Neurosci. 2007;27:7597–7603.
81. Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nature Rev. 2005;6:671–682.
82. Nave KA, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opinion Neurobiol. 2006;16:492–500.
83. Carroll SL, Miller ML, Frohnert PW, et al. Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J Neurosci. 1997;17:1642–1659.
84. Atanasoski S, Scherer SS, Sirkowski E, et al. ErbB2 signaling in Schwann cells is mostly dispensable for maintenance of myelinated peripheral nerves and proliferation of adult Schwann cells after injury. J Neurosci. 2006;26:2124–2131.
85. Scherer SS, Kamholz J, Jakowlew SB. Axons modulate the expression of transforming growth factor-betas in Schwann cells. Glia. 1993;8:265–276.
86. Atanasoski S, Notterpek L, Lee HY, et al. The protooncogene Ski controls Schwann cell proliferation and myelination. Neuron. 2004;43:499–511.
87. Koenig HL, Schumacher M, Ferzaz B, et al. Progesterone synthesis and myelin formation by Schwann cells. Science. 1995;268:1500–1503.
88. Voinesco F, Glauser L, Kraftsik R, Barakat-Walter I. Local administration of thyroid hormones in silicone chamber increases regeneration of rat transected sciatic nerve. Exp Neurol. 1998;150:69–81.
89. Campana WM, Myers RR. Erythropoietin and erythropoietin receptors in the peripheral nervous system: changes after nerve injury. Faseb J. 2001;15:1804–1806.
90. Campana WM, Li X, Shubayev VI, Angert M, et al. Erythropoietin reduces Schwann cell TNF-alpha, Wallerian degeneration and pain-related behaviors after peripheral nerve injury. Eur J Neurosci. 2006;23:617–626.
91. Li X, Gonias SL, Campana WM. Schwann cells express erythropoietin receptor and represent a major target for Epo in peripheral nerve injury. Glia. 2005;51:254–265.
92. Gupta R, Steward O. Chronic nerve compression induces concurrent apoptosis and proliferation of Schwann cells. J Comp Neurol. 2003;461:174–186.
93. Gupta R, Rowshan K, Chao T, et al. Chronic nerve compression induces local demyelination and remyelination in a rat model of carpal tunnel syndrome. Exp Neurol. 2004;187:500–508.
94. Gupta R, Truong L, Bear D, et al. Shear stress alters the expression of myelin-associated glycoprotein (MAG) and myelin basic protein (MBP) in Schwann cells. J Orthop Res. 2005;23:1232–1239.
95. Gupta R, Rummler LS, Palispis W, et al. Local down-regulation of myelin-associated glycoprotein permits axonal sprouting with chronic nerve compression injury. Exp Neurol. 2006;200:418–429.
96. Gupta R, Gray M, Chao T, Bear D, et al. Schwann cells upregulate vascular endothelial growth factor secondary to chronic nerve compression injury. Muscle Nerve. 2005;31:452–460.
97. Gupta R, Lin YM, Bui P, et al. Macrophage recruitment follows the pattern of inducible nitric oxide synthase expression in a model for carpal tunnel syndrome. J Neurotrauma. 2003;20:671–680.