Nerve Response to Injury and Repair

▪ Peripheral nerve end-organs require innervation to stay viable. The loss of the trophic stimulus of the nerve axon dooms the end-organ to atrophy and eventual death.
▪ Denervated muscles can be made to function by external electrical stimulation, and this can prevent some of the changes caused by denervation.
▪ Nerve injuries can result from many different mechanisms. They can result from compression by internal forces (e.g., tumors, fracture, callus) or by external forces (tourniquet or “crutch palsy”), or from ischemia, traction, x-radiation, inadvertent injection injury, or electrical injury.
▪ Regeneration of the peripheral nerve after injury is influenced by mechanical forces, delay to repair, the patient’s age, and the level of the injury.
▪ The nerve axon has the greatest likelihood of accurate regeneration when minimum scar is interposed between the severed nerve ends and if there is accurate alignment of the fascicles.
▪ The most commonly used technique for peripheral nerve microcoaptation is the epineurial repair technique.
▪ Electrophysiologic studies supported the use of grafts in lieu of coaptation under tension and showed excellent recovery even in the face of two suture lines.
There are many diverse causes of peripheral nerve dysfunction, and most treatments are outside the scope of the hand surgeon and therapist. Although some of the sequelae of nerve dysfunction can be treated surgically (e.g., tendon transfer or decompression), essentially only traumatic peripheral nerve injuries—transection, crush, compression, and stretch—can be repaired or reconstructed surgically. Because each particular type of nerve injury carries with it a different prognosis, it is important to fully appreciate the effects of each kind of nerve injury and the extent to which they affect the nerve cell, axon, and target organ. With this knowledge, the surgeon and therapist can offer the patient reasonable advice regarding prognosis and the expected results of surgical reconstruction.1 For a complete discussion of the basic science of nerve injury and repair, refer to Chapter 42.
As long as a nerve cell body remains alive, it has an unlimited potential for regeneration. This is not the case for the sensory or motor end-organ. Peripheral nerve end-organs require innervation to stay viable. The loss of the trophic stimulus of the nerve axon dooms the end-organ to atrophy and eventual death. However, this deterioration does differ in time for each kind of end-organ, and the understanding of the pathophysiology of end-organ degeneration will help determine the time constraints for surgical reconstruction.
The response of the sensory end-organ to denervation varies along the continuum from atrophy to frank degeneration and disappearance. This response not only is time dependent but also depends on the life cycle of the receptor in question. Muscle spindles, which undergo no further division after differentiation, respond to denervation by atrophy. Other cell types that are characterized by a short life cycle and high turnover—such as the taste bud—respond to denervation by degeneration and complete disappearance within 1 to 2 weeks.2 The integrity of sensory receptors apparently depends on an intact nerve fiber, although there need not necessarily be efferent impulses. Spinal ganglia, severed of their connections with the spinal cord, are able to maintain sensory structures as long as the axon is intact.3
The three most common sensory end-organs—the Meissner corpuscle, the Merkel cell–neurite complex, and the Pacinian corpuscle—have similar responses to denervation. As a result of Wallerian degeneration, the axon terminal progressively degenerates over time and is absent by 9 months. The lamellar components of the Meissner corpuscles and the Pacinian corpuscles become atrophic but never completely disappear, as do the supporting structures of the Merkel cell–neurite complex.4-8 The Merkel cells seem to reduce in number, become atrophic, and possibly even become differentiated into transitional cells or keratinocytes after denervation.9 If the sensory receptor does proceed to complete degeneration after long-term denervation, it is lost to the receptor pool because the adult mammal appears to have lost the ability to form new sensory receptors de novo.10,11 The muscle spindle, innervated with both motor and sensory fibers, responds in a similar fashion. When the γ-innervation is severed, the polar regions become atrophic, while the central sensory component (nuclear bag or chain regions) remains intact. When the dorsal roots, which supply the sensory portions, are injured, the central regions decrease in size, lose their equatorial collections of nuclei, and are eventually replaced by striated muscle.2
After the denervation of skeletal muscle, many changes are apparent. Clinically, the muscle ceases to function and there is gross muscle atrophy. Muscle atrophy after nerve injury results in a decrease of total muscle weight, a loss of total protein, and an overall reduction in muscle cross-sectional area.12 There is not a decrease in the number of individual muscle fibers, even though there is a relative increase in the amount of connective tissue stroma.13 Rates of atrophy can vary widely, with reports of as much as a 40% decrease in muscle mass within 1 to 3 weeks of denervation.3 This does not represent an irreversible degradation, however, and anecdotal reports have documented successful restoration of function after 22 years of denervation.14 Realistically, however, functional reinnervation is unlikely after 2 years of denervation. Denervated muscles can be made to function by external electrical stimulation, and this can prevent some of the changes caused by denervation.15-19 However, the role functional electrical stimulation plays in the reconstruction and rehabilitation of the peripheral nerve injury remains to be seen.
Whenever a patient has sustained a peripheral nerve injury, the treating physician and therapist must fully understand the nature of the injury and the mechanism by which the nerve was injured. Each different type of injury carries with it a different prognosis, and at the time of acute injury, the clinical findings of numbness, paralysis, paresthesia, or pain shed little light on the pathophysiology and ultimate prognosis. By knowing the mechanism of the injury and the pathologic changes produced by that injury, the clinician may better predict the outcome and be better able to plan the care and ultimate reconstruction of the patient’s nerve injury.
Nerve injuries can result from many different mechanisms. They can result from compression by internal forces (e.g., tumors, fracture, callus) or by external forces (tourniquet or “crutch palsy”), or from ischemia, traction, x-radiation, inadvertent injection injury, or electrical injury. Although each mechanism represents a different type of injury, all of the injuries result from mechanical deformation, ischemia-induced metabolic failure, or both.
Mechanical deformation lesions encompass the entire spectrum of nerve injury, from first-degree neuropraxic injuries to fifth-degree injuries. In general, the effects of mechanical deformation depend on the rate of application of the deforming forces, the area of the nerve over which they are applied, and the magnitude of the forces. The regional anatomy of the nerve and its adjacent structures, as well as the nerve’s proximity to underlying bone and unyielding fascial bands, must be considered. Internal nerve anatomy also is important.
Peripheral nerves organized as a single fascicle are much more vulnerable to injury than nerves consisting of many fascicles surrounded by a larger amount of protective connective tissue. The mechanisms of injury creating mechanical deformation include acute and chronic compression, crush, and stretch. All of these lesions involve some degree of vascular injury. Occlusion of the supporting vasculature accompanies any deformation and may remain after the release of the deforming force.20 Recovery may depend on the degree of resultant ischemia that persists. When a peripheral nerve is subjected to a severe, abruptly applied deforming force, three grades of injury may result. There may be a rapidly reversible physiologic block, a local demyelinating block, or Wallerian degeneration.21 Although it is not completely understood which mechanism—mechanical deformation or ischemia—causes the injury, each factor has some role, and they are probably additive.22
Acute compression injuries are especially amenable to experimental examination and can be reproducibly created by the use of a pneumatic tourniquet.23 At low pressures (up to 30 mm Hg), impaired venular flow is observed20 and endoneurial fluid pressures are seen to be up to three times normal.24 Physiologically, decreased nerve conduction velocities are noted.25 At pressures of 60 mm Hg, nerve fiber viability is endangered by the creation of a local metabolic block, secondary to ischemia. In addition, at this level of compression, mechanical deformation within the nerve fiber is seen. At higher levels of compression (90 mm Hg and higher), there is even greater mechanical deformation of the axon and the supporting structures (Schwann cells), as well as collapse of the intraneural microcirculation, which is likely to persist upon the release of the deforming force.24,26
Application of a tourniquet at pressures above systolic but at levels insufficient to cause cellular damage results in progressive centripetal sensory loss and paralysis within 30 to 40 minutes.27 In these first-degree neuropraxic injuries, the earliest pathophysiologic change is the inability of the nerve to transmit repeated impulses. Although the exact defect is unknown, ischemia from compression surely creates anoxic block of ionic and axonal transport. In addition, compression at high levels causes narrowing of the involved axons, increased endoneurial fluid pressure, and subsequent intraneural edema.28,29 The classic examples of acute neuropraxic compression neuropathy include the transient conduction block, typified by paresthesias, that results from local pressure on a peripheral nerve. This causes the familiar sensation that one’s leg or arm “goes to sleep.” The conduction block rapidly recovers when the pressure is relieved or posture altered. When a motor nerve is involved, the sensation is one of “pseudocramps.”30 With extended tourniquet application at suprasystolic pressures, motor deficits and mild sensory losses occur. There is a higher degree of impairment in faster-conducting larger axons (primarily motor, proprioceptive, and light-touch fibers), whereas the smaller and nonmyelinated axons (pain, temperature, and autonomic function) are spared.31 Higher levels of compression yield a longer-lasting conduction block caused by focal demyelination without disruption of axonal continuity. This local conduction block is caused by mechanical deformation (nodal intussusception).21,32 Higher levels of pressure on a nerve create further mechanical deformation, which results in the shearing of the mesaxonal Schwann cell from the adaxonal portion of the cell. This damages the myelin, which then degenerates and leaves an area of exposed axon. Conduction is not restored until remyelination is complete,21,32,33 and function returns in most cases by 3 to 6 weeks.33 Because axonal continuity is maintained, there is little, if any, target-organ degeneration. Because of the differential susceptibility of axons, complete paralysis (large fibers) can occur without loss of cutaneous sensibility (smaller myelinated and unmyelinated fibers).34
Long periods of compression or high levels of pressure over a relatively small area of a peripheral nerve can produce a crushing injury that may be second, third, or even fourth degree. These are lesions-in-continuity, and the prognosis for recovery depends on the magnitude of the intraneural disruption. When the crush injury is axonotmetic, the axonal basement membrane remains intact, and regeneration progresses with an exact target-organ match and good recovery. Although recovery can be delayed, most patients have achieved 50% return by 4 months.23 When the crush injury is neurotmetic (third or fourth degree), function is mildly or moderately reduced by the failure of some axons to achieve a proper end-organ match. Axonal admixture as well as increased amounts of intraneural scar prevents complete return of function, and budding axons can become lost in the interposed scar and develop a neuroma-in-continuity.
Injury created by low-grade chronic compression differs from the acute compression injury in all aspects of etiology, histology, and clinical presentation. The susceptibility of peripheral nerves to chronic compression is a function of internal anatomy. Proximal nerves, which contain many fascicles and an abundant amount of supportive connective tissue, are much less vulnerable to compression than distal nerves. Within each nerve, peripheral fascicles are more affected than central fascicles, and within each fascicle, peripheral axons are more likely to be injured than the central ones.35 There is also a greater susceptibility to compression if the patient is afflicted with malnutrition, alcoholism, diabetes, or renal failure.
Histologic assessment of chronic nerve compression shows myelin sheath asymmetry, epineurial fibrosis, perineurial thickening, and, in severe stages, endoneurial fibrosis. Larger fibers appear to “drop out.” There is some Wallerian degeneration, and simultaneous regeneration of axons is seen.35 In contradistinction to the acute compressive neuropathy that creates nodal intussusception, the terminal loops of the inner lamellae of the thinned myelin near the entrapment become detached from the axon at the node and retract. An abundance of myelin appears at the opposite end of the internode, which produces bulbous paranodal swelling. The detachment and subsequent myelin retraction leave multiple consecutive internodes demyelinated. This partially accounts for the reduced conduction velocity seen in nerves injured by chronic compression. The conduction block seen in acute compression injuries is rarely seen in these chronic lesions.36
Chronic compression also affects the vascular supply of the involved segment. In pressures as low as 30 mm Hg, impaired venular flow is observed, ultimately leading to congestion and anoxia. This induced ischemia leads to further vascular dilation, nerve swelling, and more compression, beginning a vicious cycle.37 The rapid reversibility of some chronic compression injuries supports the vascular etiology.38
Axoplasmic flow is decreased in chronic compressive injuries, and even when the force is insufficient to create a demyelinating lesion, there is a profound effect on the function of the nerve. Distal to the compressive force, the nerve becomes more sensitive to low levels of pressure. Multiple subclinical levels of compression along any given nerve can lead to symptoms of compressive neuropathy. This double-crush syndrome implies that serial constraints of axoplasmic flow are additive in nature.39,40
Histologic changes are observed in nerves subject to chronic compression. Epineurial fibrosis and perineurial thickening are noted, and there is a decrease in the number of large axons in the periphery of the affected fascicles. There is proteinaceous intraneural edema from the loss of the blood–nerve barrier, within which fibroblasts proliferate and ultimately render the nerve segment permanently scarred and potentially anoxic.41 Late chronic compression injuries are more likely to require extensive external neurolysis (and sometimes even internal neurolysis) than early lesions because of the extensive nature of the intraneural scar. Patients with chronic compression injuries often report the sensation of pain, but this can arise along any area of the affected nerve’s course and is often a misleading diagnostic sign. A more useful diagnostic sign that correlates with the site of compression is the Tinel’s sign.42 At surgical exploration, the affected nerve often is seen to be swollen, edematous, and hyperemic proximal to the area of compression. The nerve underlying the compression is often pale and narrow.43 At the time of surgical decompression, almost all patients obtain relief of their pain, although improvement in conduction delay takes weeks or months to return.41
Peripheral nerves must incorporate within their structure the ability to accommodate changes in joint position. To do this, nerves have the inherent ability to stretch, recoil, and glide within their beds on their loose mesoneurial attachments.44-47 Under conditions of minimal tension, the nerve fibers assume an undulating course within the fascicle. As longitudinal tension is applied to the nerve, the nerve slides within the bed and begins to take up the load and become stressed. The epineurium—an elastic structure—begins to elongate by “stretch” and the fascicles within completely straighten out. Upon release of the longitudinal tension, the nerve recoils and resumes its natural resting length.35,44,48,49 As long as the nerve remains free to glide within its bed, significant stretch can be tolerated without injury. Normal excursions of nerves vary greatly, from a maximum of 15.3 mm (average for the brachial plexus) to a minimum of 1.15 mm (average for a digital nerve).45-47
When a nerve is injured (through compression, scar, entrapment, or adhesions) and is subsequently anchored by scar tissue to its bed, it can be subjected to “overstretch” traction injury during normal physiologic demands. There are also certain traumatic states that can exceed the normal nerve excursion or limits of elasticity and therein create injury.47 A nerve achieves its strength through the perineurium. This layer has three orientations of collagen fibers in its outer sheath: circumferential, longitudinal, and oblique.49,50 As longitudinal tension is applied the perineurium lengthens, but at the expense of the cross-sectional area. This creates an increase in the intrafascicular pressure along the entire length of the nerve.47-51 As long as the perineurium remains intact, the nerve maintains its elastic characteristics, but intraneural damage occurs far below the point of mechanical failure.51 The elastic limit, or allowable stretch limit, is about 20%. At that level there can be one or many areas of intraneural tearing, with axonal and fascicular disruption and areas of hemorrhage. Fibroblastic proliferation follows, which ultimately leads to intraneural scarring.44,52,53
Although the true incidence of nerve stretch injuries is unknown, these injuries are commonly associated with traumatic events, such as fractures, dislocations, obstetric trauma, and occasionally inadvertent retraction during surgery. Of nerve injuries associated with fractures, 95% occur in the upper extremities, and of the five most commonly injured nerves, 58% are radial, 18% ulnar, 16% peroneal, 6% median, and 2% sciatic.54 In a prospective study of 648 nerve traction lesions in which the nerve was seen to be in continuity at the time of surgery, Omer55 reported that 70% achieved spontaneous recovery. In low-velocity gunshot wounds, 69% recovered after 3 to 8 months. Patients with high-velocity gunshot wounds also recovered from nerve lesions 69% of the time, but recovery required up to 9 months. In patients with nerve injuries associated with fractures and dislocations, 83% recovered spontaneously in 1 to 4 months, and in patients with nerve traction (stretch) injuries, 86% recovered in 3 to 6 months.55
Not all traction injuries are traumatic. Some are the result of attempts to overcome an excessive nerve gap during reconstruction. This may be by stretch on the nerve stumps to achieve coaptation or by positioning the joints in flexion and creating the traction by the progressive postoperative extension of the joints. The progressive extension may lead to an ischemic injury or even exceed the elastic limits of the nerve.56 It has been shown that some length can be gained with slow stretch over time.51 This finding has been exploited using tissue expansion techniques that allow progressive nerve lengthening before nerve reconstruction to overcome nerve gaps. When caring for a patient with a major traction injury, we must be aware that the injury can affect the entire length of the nerve. The most clinically significant injury may be remote from the actual site of extremity injury, and this may test even the most astute diagnostician.
The peripheral nerve requires a continuous and adequate supply of oxygen for aerobic metabolism to drive the normal functions of axoplasmic transport; maintain cell integrity; and remain primed for the generation, maintenance, and restoration of the membrane potentials necessary for conduction of impulses. To accomplish this goal, the nerve has an elaborate dynamic plexus of blood vessels composed of two integrated but functionally independent systems.57 The peripheral nerve can survive relatively long anoxic periods with a rapid recovery of function,58-60 but longer periods of acute ischemia or chronic hypoxia may produce irreversible injury. Muscle weakness, pain, paresthesias, hypersensitivity, and sensory deficits all are symptoms of ischemic nerve lesions.13,61,62
Ischemic injury may result from three different pathologic processes. There may be large-vessel occlusion, arteriolar angiopathy, or nutrient capillary disease. Large-vessel occlusion caused by conditions such as trauma or embolism is amenable to medical management or direct surgical reconstruction. Arteriolar and capillary disease is indirectly approached by attempts at improving the nerve environment (e.g., flap reconstruction of the nerve bed) or by release of the offending perineurial and intraneural scar (or both).63,64 Some of the pathologic states that affect the arteriolar (50 to 400 mm in diameter) vessels are necrotizing angiopathic disorders such as polyarteritis nodosa, rheumatoid arthritis, Churg-Strauss syndrome, Wegener’s granulomatosis, and thromboangiitis obliterans (Buerger’s disease). All of these disease states affect the epineurial arterioles and result in patchy occlusion and ischemic nerve damage.63,65-67
The length of time the ischemic insult persists is the most important determinant of anoxic damage. Within the first 10 minutes of nerve ischemia, there is a rapid decrease in the membrane resting potential and electrical resistance. By 15 minutes, the action potential decreases and there is a further decrease in the resting potential, which blocks conduction.68 There is a complete loss in conductivity by 30 to 40 minutes.21 Reoxygenation brings recovery within 1 to 2 minutes, and recovery is usually complete by 10 minutes. This implies that the pathologic insult of ischemia is a metabolic phenomenon and not a morphologic one. In chronically ischemic nerves there is segmental demyelination,66,69,70 and irreversible axonal infarction may occur.71,72 If regeneration occurs there is a favorable prognosis, because the intraneural destruction leaves the axonal basement membrane intact. This ensures directed axonal regrowth and proper nerve end-organ connectivity.73
The question of tolerance to ischemia becomes very important when the issue of replantation or free-tissue transfer is raised. A normal nerve apparently can tolerate up to 8 hours of warm ischemia (room temperature) and suffer little morphologic damage. Nutrient blood flow is rapidly restored upon revascularization. After 8 hours there is a breakdown of the blood–nerve barrier, and the resultant influx of proteinaceous fluid negatively affects nerve regeneration by ultimately stimulating fibroblastic proliferation and subsequent intraneural scarring.60
For more proximal amputations in which there is a significant amount of muscle involved, the tolerance to ischemia of the nerve is not of clinical importance, because the target organs suffer irreversible damage before the nerves. However, injured nerve fibers are more susceptible to induced ischemia than are normal nerves. This may be because there is a reduction in axoplasmic flow in the injured nerve (especially if severed).74
An electrical injury to the upper extremity can run the gamut from minor to life-threatening and is associated with a significant percentage of resultant amputations (32.5%).75 The severity of these injuries depends on the current pathway and the relevant features of voltage level, tissue resistance, and current duration. The neurologic defects following electrical injury are usually immediate in onset and, for reasons incompletely understood, more commonly involve motor nerves. Most injured nerves show some recovery over time, but complete resolution of significant injuries is rare.76
The major pathologic change in the electrically injured nerve is one of coagulation necrosis resulting from the generation of heat energy. The electrical current follows the path of least resistance, and resistance to flow increases in various tissues in the following order: nerve, blood vessel, muscle, skin, tendon, fat, and bone.77,78 The electrical injury therefore preferentially follows the neurovascular bundles and creates deep-tissue destruction along these pathways. Flash thermal burns at the entrance and exit sites accompany these injuries. In 22% of reported cases of electrical burns, direct nerve destruction was the initial result of the injury.75 In nondestructive lesions, electrical injuries are found to cause an increase in threshold stimulus and a loss of amplitude of the response to supramaximal stimulation. Although some of these changes were reversible, the electrical injury left a persistent increase in latency and decreased conduction velocity.79 Severe injuries can lead to patchy necrosis of the entire nerve as well as central necrosis. Hemorrhage and subarachnoid bleeding also are common.75,78,80-82
If the nerve is not destroyed, total demyelination in a multifocal distribution is seen, and blood vessels in the vasa nervorum sustain significant damage, thus creating a late ischemic injury. Complicating electrical injury are violent tetanic contractures that can result in hemorrhage, muscle rupture, and broken bones. Late changes principally result from chronic ischemic changes associated with vascular damage and progressive perineurial fibrosis. Unlike the previously mentioned vascular pathology, this chronic ischemic injury may respond to neurolysis and revascularization of the nerve (e.g., muscle flap, omental transfer).77,82
As radiation treatment becomes more precise and refined, its use as a treatment modality in the therapy for cancer is increasing. With it come increasing survival rates for patients with cancer and also greater opportunity to observe the late effects of radiation injury on surrounding soft tissues and nerves. In the past, orthovoltage radiation (less than 1 million volts) was the standard, and this technique had very shallow tissue penetration. There were marked skin changes that limited the total dose of radiation before significant nerve damage could result. Today, megavoltage radiation (1 million to 35 million volts) has allowed increased penetrance to deeper tissue planes with minimal apparent skin damage, and the result is a much higher dose of radiation to the adjacent structures within the field.83
Radiation injury to the peripheral nerve is poorly understood. Injury results from direct cell injury and indirectly from damage caused to the supportive vascular and connective tissues. These injuries are synergistic.84 Fortunately for neural tissue, there is relative stability in cellular population (i.e., there is little mitotic activity, and therefore little biologically significant damage occurs at the genetic encoding level) until attempts at neural regeneration.85,86 Cellular damage is more pronounced if the radiation follows a nerve injury that stimulates the cellular supportive proliferation.87
Radiation injury is permanent and does not seem to abate with time. Histologically, there is axonal dropout and patchy loss of myelin within the radiated segment. Attempts at Schwann cell proliferation yield a decreased total number of cells that produce abnormally thin myelin sheaths. An increased nerve cross-sectional area implies that there is persistent intraneural edema associated with abnormal endoneurial vessel permeability. Late examination reveals marked intraneural and perineurial fibrosis with apparent fibrous replacement of fascicles and a marked amount of thick pale scar.83,84,87,88
Because of the common use of radiation in the treatment of cancers of the breast, brachial plexus radiation injury has been the most extensively studied type of radiation injury. The incidence of brachial plexopathy varies greatly, depending on the mode of delivery and the total dosage. In one series, using 4-MV radiation, 15% of patients developed neurologic symptoms after 5775 rads (5.775 Gy); after 6300 rads (6.3 Gy) the percentage increased to 73%.83 After radiation given by a 15-MV betatron, 22% of patients receiving between 400 and 5000 rads (0.4 and 5 Gy) developed an actinic plexopathy, increasing to 47% after 550 to 6600 rads (0.55 to 6.6 Gy).89 The latent period between radiation and the onset of symptoms of plexopathy has ranged from as short as 5 months to as long as 20 years, with a mean latency of 4.25 years.88,90 Pain is by far the most common presentation of brachial plexopathy, with as many as 80% of patients reporting some amount. Fifty percent of patients describe their pain as severe. Sixty-six percent of patients presented with muscle weakness and atrophy, and this sign was often associated with marked upper extremity lymphedema. Most of the patients with sensory and motor deficits presented with median and ulnar involvement.88,90
The typical patient who presents to the hand surgeon with upper extremity pain and who has had radiation for the treatment of a carcinoma presents a diagnostic dilemma. The problem is in distinguishing an actinic plexopathy from local recurrence or metastatic involvement of the nerves by tumor. Both present with like signs and symptoms and have the same mean onset. Both progress steadily over years, but progression of the plexopathy without the development of other metastatic sites is the best presumptive evidence for a radiation-induced lesion. In all surgical cases, absence of metastatic disease must be confirmed by liberal biopsy.90-93
The strongest indication for surgical intervention in radiation-induced nerve injury is intractable pain, but surgery should not be approached in a cavalier fashion. Downgrading of function is a likely outcome, because the compromised nerves will not tolerate much surgical manipulation. The goals of surgical intervention should be to gently excise the strangulating fibrotic scar and to improve the vascularity of the involved nerves by transposition to an improved bed or by flap reconstruction.
The peripheral nerve is vulnerable to direct injection in many circumstances, and this can result in permanent damage to the nerve. It is common to inject various substances in the immediate vicinity of nerves when administering local anesthetic agents for regional anesthesia or injecting steroid preparations for the local treatment of inflammatory conditions. In addition, the intramuscular injection of materials such as antibiotics can result in inadvertent injection into an underlying peripheral nerve. Most of these injuries could be avoided by knowledge of the surface and underlying anatomy.94 When a nerve injection injury occurs, the patient can experience severe pain at the site of injection that radiates to the distribution of the nerve. This is often associated with a neurologic deficit—sensory, motor, or both.95-98
Injection injury was thought to be caused by mechanical needle injury, allergic neuritis, ischemia, and the development of circumferential scar, as well as the intraneural deposition of neurotoxic substances. Several studies have shown that only the intraneural injection of neurotoxic substances causes significant nerve fiber injury. Only with diazepam, chlordiazepoxide, chlorpromazine, and benzylpenicillin was extrafascicular injection associated with nerve injury. The most severe injuries were related to the intrafascicular injection of meperidine, diazepam, chlorpromazine, hydrocortisone, triamcinolone hexacetonide, procaine, and tetracaine. Less severe, but still significant, injuries were produced by gentamicin, cephalothin, methylprednisolone, triamcinolone acetonide, lidocaine (worse if with epinephrine), and bupivacaine hydrochloride with epinephrine.97-101 Several of these drugs contain similar buffers, and these may be the offending agents.102,103
Acutely, axonal dropout and Wallerian degeneration are seen. Alterations of the blood–nerve barrier change the normal endoneurial environment and may lead to late changes caused by the attendant swelling, ischemia, and intraneural scar. By 8 weeks after injury, there is severe intraneural fibrosis associated with minimal external scar.96,99
When a peripheral nerve injection injury occurs, observation is indicated for the first 3 months, with electrophysiologic studies obtained about 6 weeks after the injury.97 Early surgical exploration with irrigation of the offending agent or external neurolysis is not recommended. Because the damage is intraneural, extraneural manipulations do not address the pathology. If there is no clinical recovery by 4 months, exploration is indicated. An internal neurolysis procedure should be done to decompress the scarred fascicles, and several months should be allowed to elapse to see what functional recovery follows. If little improvement is gained, one should perform resection of the neuroma incontinuity, followed by reconstruction.97,104,105
Nerve laceration is likely to be one of the most common injuries that the peripheral nerve surgeon treats. Lacerations are either complete or partial, and all are fifth-degree neurotmetic lesions. A sharp instrument causes most such injuries, but some are associated with sharp bone fragments in a closed fracture. All are associated with a clearly defined neural motor or sensory deficit that will not improve without surgical intervention. Approximately 20% of all nerve lacerations that appear to be complete are in fact only partial lacerations, with contusion and stretch being responsible for the neuropraxic or axonotmetic deficit of the remaining intact fascicles.105 Nerve lacerations are essentially low-velocity crush injuries isolated to very small areas of the involved nerve. Even under ideal conditions, a divided nerve will have some component of crush injury in the proximal and distal stumps, which must be treated by careful debridement at the time of surgical reconstruction.106
Peripheral nerve reconstruction is built on a foundation of principles that, when followed, facilitate the natural regenerative process of the nerve. Regeneration of the peripheral nerve after injury is influenced by mechanical forces, delay to repair, the patient’s age, and the level of the injury.
Mechanical interference to nerve regeneration is in the form of scar tissue and inappropriate topographic orientation. The nerve axon has the greatest likelihood of accurate regeneration when minimum scar is interposed between the severed nerve ends and if there is accurate alignment of the fascicles. This increases the probability that the regenerating axons will enter their native nerve fibers and achieve appropriate connectivity.107
Delay before reconstruction is another important factor. The capacity for a nerve cell body to regenerate is essentially unlimited as long as cell death does not occur. It is the passage of time, however, that eliminates the possibility for regeneration by time-related changes in the distal nerve segment (scar and progressive decrease in diameter) and in the target organs (atrophy and degeneration).13
There is little question that there is a consistently better functional return in the young after nerve injury.108 Rates of nerve regeneration are age-related. They decline with increasing age and may be related to the decreasing rates of slow-axonal transport with age.109 In addition, trophic mechanisms seem to function over greater distances in the young, and this may facilitate more accurate end-organ connectivity.107 The differences in the quality of return of sensibility between the young and old may be attributable to the diminution in receptor populations that occurs naturally in the old (at least for populations of Meissner corpuscles).110 Finally, the consistently better functional outcomes in the young may result from greater central plasticity. This is the cortical ability to relearn or reorganize spatially disrupted input and thereby overcome the inexact peripheral connectivity.111
The injury level (proximal versus distal) is one of the greatest factors in determining the prognosis for successful outcome of regeneration. Proximal injuries generally carry a worse prognosis than distal injuries. In a proximal injury, regenerative demand on the cell body is greater because the axon must regenerate for a greater distance. Also, in proximal injuries there is a greater likelihood of neuronal death.36,112 Second, if the injury is proximal, there is a greater distance to the target organ, and in the time the axon takes to regenerate over the great distance to the end-organ, considerable atrophy or degeneration may have occurred.105,112,113 Intraneural topography also plays an important role. The more proximal the nerve, the greater the fascicular heterogeneity. In higher level injuries, there is more axonal mixing during regeneration; this makes appropriate target-organ connectivity difficult if not impossible.13
The cause of the nerve injury is important, and the associated structures—skin, bone, joint, and vascular system—must be stabilized before any definitive reconstruction. Devitalized tissues must be debrided, bones must be stabilized, and blood vessels (when injured) must be repaired. Severe injuries may result in multilevel nerve lesions from traction, compression, or ischemia. As an injury increases in severity, there is more scarring around the nerve and its bed, thereby decreasing the quality of regeneration. In addition, a patient in an unstable condition may require delay in the repair. Nerve repair or reconstruction should not be performed in the absence of good skin cover, skeletal stability (ideally with supple joints), and without correction of vascular insufficiency. Infection should be aggressively treated, and adjunctive procedures such as flap coverage should be performed if they will facilitate earlier nerve reconstruction.
Reinnervation of a target organ involves much more than just axonal regeneration and connectivity. After an axon reaches the target organ, neuromuscular junctions and the axon terminus of sensory receptors must be reestablished, and this does not occur if there is an axon target-organ mismatch.
Because of the randomness of regeneration for all of the higher level injuries, there are five potential outcomes of regeneration. An axon may achieve exact reinnervation by establishing continuity with its native target organ. If the end-organ is not irreversibly damaged, return of function can be essentially normal. If the end-organ has degenerated, there will be no useful return of function. The wrong receptor may be reinnervated within the proper territory, resulting in improper input, or the axon may achieve connectivity with the appropriate receptor in the wrong territory and create false localization. Finally, the axon may be frustrated and not achieve end-organ connectivity, thereby rendering the results of regeneration fruitless.
When a regenerating motor axon reaches a denervated muscle, reinnervation generally occurs at old motor end plates.3,114,115 The axon then sprouts and reinnervates contiguous muscle fibers, creating histochemically uniform giant motor units.116 Recovery of motor function does not immediately occur upon reestablishment of the neuromuscular junctions. There is an 18-day delay before nerve stimulation will produce contraction, and another 5 days elapse before functional reflex activity can occur.117 Recovery of functional activity best correlates with the return of the γ-efferent control of the intrafusal fibers.118 Without adequate γ-return, the muscle function is downgraded clinically by imprecision of motion despite good return.119
The recovery of sensibility occurs in a repeatable orderly sequence that correlates with the morphology of the reinnervated receptor populations. The perception of pain and temperature precede the return of touch, and the touch submodalities recover in the following sequence: 30-Hz frequencies, moving touch, constant touch, and finally 256-Hz stimuli.10
Over the past 20 years, the therapeutic approach toward the patient with nerve injury has significantly changed, facilitated by new technologies of intraoperative electrodiagnosis, by better instruments and magnification, and by better understanding of peripheral nerve structure and function. Atraumatic nerve handling and suture techniques have improved the potential for nerve reconstruction. Awareness of fascicular anatomy has made the results of nerve repair more precise, and the appreciation of the untoward effects of tension at the repair site has made nerve interposition grafting commonplace. After reconstruction, knowledge of the patterns of sensory recovery has led to elaborate schemes of sensory reeducation that enable more functional use.
There are no absolute rules regarding the timing of nerve repair, and the decisions should be made after careful consideration of the nature of the injury, the condition of the patient, and the status of the associated injuries. Nerve injuries can be divided into two broad categories. First are those injuries in which there is a suspected transection. These must be handled with primary reconstruction if all conditions permit. Second are those injuries in which the nerve is expected to be in continuity or in which there are multilevel lesions or marked contusion. These injuries require secondary reconstruction. By definition, primary repair is that which is done within 48 hours of injury. Early secondary repairs are those performed within the first 6 weeks, and late secondary repairs are performed after 3 months.105,106,120,121
If the limits of the nerve injury can be delineated, there is a definite advantage to performing repair or reconstruction primarily. If the repair is performed within 4 days, electrical stimulation can be used to identify distal fascicles and nerve stump retraction is limited. If the repair is delayed beyond 4 days, Wallerian degeneration has progressed and electrical stimulation is not possible. If the wound is contaminated or if there is a soft-tissue deficit or fracture, nerve repair must be delayed until a clean stable wound can be obtained. With adequate debridement, soft-tissue reconstruction, and fracture stabilization, the nerve repair can be done simultaneously. If a primary nerve repair fails because it is done under poor conditions, the patient is likely to obtain a worse result than if repair had been delayed until a secondary repair could be done under good conditions.
There are times when the surgeon should delay repair or reconstruction after an injury. These are when there is an expected first-, second-, or third-degree injury. No surgical reconstruction can provide a better result than an intact fascicle that has healed on its own, and time should be allowed to elapse for the resolution of low-grade lesions. It is appropriate to allow 8 to 16 weeks for the resolution of nontransecting blunt or stretch injuries.
The greatest advantage of primary nerve reconstruction is the saving of time.121 The great disadvantage is the inability to detect the precise extent of the nerve injury, especially if there is an undetected traction or multilevel injury. If repair is done under the latter circumstances (undetected injury), some function can return with time, but the surgeon is then faced with the decision to reoperate in hopes of achieving greater return or to wait for improvement that is not likely to come. A great advantage of all secondary repairs is that the surgeon and the operating team can perform the surgery electively, without fatigue, and under proper operating room conditions, making it perhaps easier to achieve a more meticulous microcoaptation.
Exploration after an injury always should be done under tourniquet control if possible. This allows for more accurate dissection and identification of the lesion. Nerve microcoaptation does not require a bloodless field as long as the structures are properly positioned and tagged. In fact, it is sometimes preferable to deflate the tourniquet before the microsurgical repair to obtain homeostasis, because this can be difficult to accomplish after a nerve repair without jeopardizing the coaptation.
Nerve lesions should be approached with wide exposure, proximal and distal. The nerve is dissected from the uninjured to the injured areas of the nerve, with attempt made to preserve all vascular attachments. For lacerations, high magnification is used to determine the fascicular pattern of the nerve; this allows the orientation of both the proximal and distal ends of the nerve to be appreciated. High magnification also should be used to perform the fascicular dissection for lesions that are in continuity. As the lesion is reached, fascicles that do not contribute to the neuroma can be carefully freed from the offending scar and involved fascicles. Electrical stimulation or intraoperative electrodiagnosis can be used to determine which of the fascicles retain sufficient conduction and can be spared. The fascicles that contribute to the neuroma then can be resected and repaired or reconstructed with interposition grafts.
The most commonly used technique for peripheral nerve microcoaptation is the epineurial repair technique (Fig. 43-1).122 This technique is applied to the completely transected nerve, and its advantages are simplicity, rapid execution time, and minimal requirement for magnification.
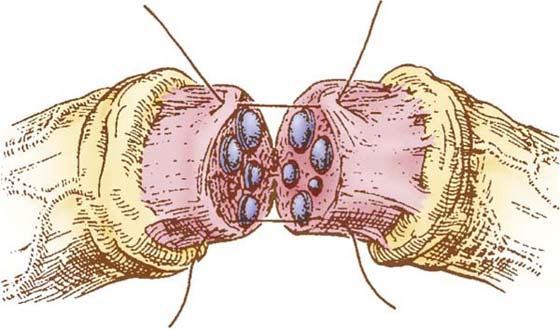
Figure 43-1 Epineurial repair. Diagrammatic representation of the microcoaptation. The outer epineurium is resected proximally and distally. After careful alignment of the nerve stumps, sutures are placed in the inner epineurium. Two or three tension-relieving sutures can be used if necessary, using 8-0 nylon, and the coaptation is completed using 10-0 nylon. (From Terzis JK, Smith KL. The Peripheral Nerve: Structure, Function and Reconstruction. New York: Raven Press, 1990.)
The cut nerve end is debrided carefully and serially sectioned until the axoplasmic outflow mushrooms under positive intrafascicular pressure and the fascicular pattern is identified and is relatively free of scar.123 The same is done for the distal stump, and then, with the nerves lying without tension next to one another, the magnification is increased to 25× and 10-0 nylon sutures are placed in the epineurium, as one carefully realigns the fascicular bundles to achieve exact coaptation. A minimum number of sutures are used to complete the repair. Usually, 8 or 10 sutures are necessary for a large nerve and as few as 2 sutures for a small one. If the nerve gap is so large that the first 10-0 suture cannot hold the nerve ends in apposition, one or two guide sutures of 8-0 can be used.
The leading causes of failure of the epineurial repair are gapping, overriding, buckling, and straddling of the fascicle ends. Even slight tension can create a significant intraneural gap, which is quickly filled with scar tissue, making regeneration difficult at best.124-126
The second common technique of peripheral nerve reconstruction is the perineurial repair (Fig. 43-2). A great deal of discussion focuses on whether an epineurial repair or a perineurial repair (fascicular repair) is the preferred method of peripheral nerve reconstruction, but numerous articles report no difference in the regeneration and functional recovery associated with either technique.127-130 When significant debridement is required before microcoaptation, fascicular matching is improved with this technique; it is also used extensively when interposition grafts are indicated.
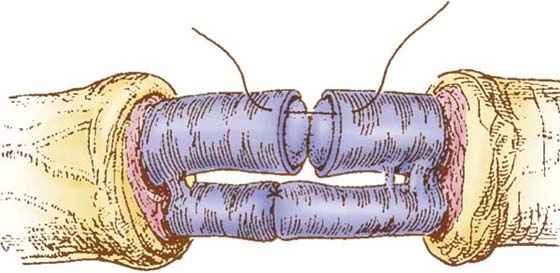
Figure 43-2 Perineurial (fascicular) repair. Diagrammatic representation of the microcoaptation. The outer and inner epineurium is dissected from the proximal and distal stumps. Fascicles and fascicular groups are aligned, and the coaptations are performed with the minimum number of 10-0 nylon sutures necessary (as few as two). Note the care taken to place the sutures within the inner epineurium or perineurium and not to violate the endoneurium. (From Terzis JK, Smith KL. The Peripheral Nerve: Structure, Function and Reconstruction. New York: Raven Press, 1990.)
Under high magnification, the nerve ends are prepared and the epineurium is dissected away. Fascicles are separated, and coaptation is performed between matching fascicles, with sutures placed into the inner epineurium. Sutures are carefully placed so as not to enter the endoneurium. The great advantage of the perineurial technique is the accurate coaptation of similar-size fascicles. The greatest disadvantage is the stimulation of greater amounts of intraneural scar by increased dissection and foreign material (suture).130-133
After any nerve injury in which there is a gap between the nerve endings, either after trauma or as required by debridement, the surgeon faces a dilemma. Should the nerve gap be bridged by extensive mobilization and stretch (with the nerve repair under tension); should the defect be bridged with a nerve graft, forcing the axons to cross two coaptations134; or should the limb be postured in flexion to bring the nerve ends together and then slowly returned to full extension in hopes of stretching the nerves out to length?
Tension at the site of coaptation invites the proliferation of scar tissue at the repair. As the scar matures, it tends to constrict the regenerating axons within.105 If the joints are flexed, mobilization can create a second traction lesion in the already injured nerve. If the regenerating axons manage to cross the suture line and grow down the nerve fiber, they are unlikely to achieve functional reinnervation in a great number of cases. Studies have shown that repeated stretching of a sutured nerve fails to result in its elongatation, and instead, repetitive traction injuries result.56,135
Comparing the results of nerve grafting with those of coaptation under tension, Millesi determined that there was little interposed scar at the coaptation if the nerve was repaired without tension, with or without a graft. Electrophysiologic studies supported the use of grafts in lieu of coaptation under tension and showed excellent recovery even in the face of two suture lines.43,136,137 Nerve grafting is approached surgically like any nerve exploration. The nerve ends are exposed and prepared, and a suitable donor nerve (commonly the sural nerve) is harvested and cut to length. Depending on the caliber of the injured nerve, the number of interposition grafts is chosen to match the cross-sectional area. Fascicular matching is conducted, and the perineurial repairs are done.
Autogenous nerve grafting is no longer the only approach available to reconstruct the nerve gap. The nerve gap can be mechanically shortened by nerve-lengthening techniques (tissue expansion or nerve distraction)138 or bridged by tubes of biologic or nonbiologic materials.
To avoid donor-site morbidity from autogenous nerve harvest, interest has been rekindled in various tubulization techniques and in homografting (transplant). Like an autogenous nerve graft, the nonneural tube offers a conduit for the budding axons, and growing nerves can be directed across a nerve gap to achieve connectivity with the distal nerve stump. Examples of conduits currently in use are those constructed from polyglycolic acid, autogenous vein, and amnion.139,140 Results of reconstruction with conduits of short nerve gaps (less than 3 cm) in digital nerves show functional results equal to tensionless coaptations, thereby making this technique quite advantageous in view of the elimination of donor-site morbidity.141 However, the maximum gap that can presently be bridged with a conduit seems to be 3 cm, and reconstruction with conduits requires 2 to 3 weeks of orthotic positioning in the postoperative period.
Promising results have also been obtained with tissue-expansion techniques. Slow expansion of Wallerian-degenerated nerve results in accelerated Schwann cell proliferation and increased vascularity that seem to facilitate nerve regeneration.142 Promising results have also been obtained with tissue-expansion techniques. Slow expansion of Wallerian-degenerated nerve results in accelerated Schwann cell proliferation and increased vascularity that seem to facilitate nerve regeneration.142 Permanent elongation of 30% could be achieved with expansion techniques yielding results of repair equal to tensionless primary nerve coaptation. However, the proximal segment tolerated expansion better than the distal segment.143
With successful repair, the progressing Tinel’s sign can follow regeneration as the budding axons travel along the nerve. It is important to follow the punctum maximum, because this delineates the most distal extent of the majority of axons. If the Tinel’s sign fails to progress and remains at the site of coaptation, this is an indication for reexploration and repair. If Tinel’s sign stalls distally to the repair, one must assume there is a second lesion previously unknown.
After the nerve repair, it is important that the nerve coaptation be protected for 7 to 10 days by immobilization. Then, during the period of nerve regeneration, therapy should concentrate on keeping the affected areas supple, mobile, and ready to accept the growing axons. As soon as sensory reinnervation is seen, sensory reeducation programs should begin.
Over the last decades, peripheral nerve reconstructive surgery has evolved from rather crude reapproximation of severed nerve ends performed without much regard for tension or topographic orientation, to technically precise methods of surgery for which better understanding of the intraneural anatomy has allowed better fascicular alignment and has led to improved results. These still fall below the ideal of axon-to-axon realignment, however. Perhaps in the future, new understanding of tropic and trophic manipulations will allow us to achieve even better results and achieve exact target-organ reinnervation, and techniques of tensionless sutureless coaptation, and scar manipulation will serve to eliminate fibrotic interference with regeneration. Today, we can appreciate that the nerve cell has an essentially unlimited regenerative capability. It is the job of the surgeon to perform as precise a repair after a nerve injury as possible, and it is the job of the therapist to assist the patient in the maintenance of the end-organs by protective orthotic positioning, range-of-motion therapy, massage, and modalities as indicated, to achieve the best possible functional results after nerve injuries and repair.
1. Mohammad J, Shenaq J, Rabinovsky E, Shenaq S. Modulation of peripheral nerve regeneration: a tissue-engineering approach. The role of amnion tube conduit across a 1-centimeter nerve gap. Plast Reconstr Surg. 2000;105:660–666.
2. Trojaborg W. Rate of recovery in motor and sensory fibers of the radial nerve: clinical and electrophysiological aspects. J Neurol Neurosurg Psychiatry. 1970;33:625–638.
3. Rudge P. Tourniquet paralysis with prolonged conduction block. An electro-physiological study. J Bone Joint Surg Br. 1974;56-B:716–720.
4. Brown MC, Holland RL, Hopkins WG. Motor nerve sprouting. Annu Rev Neurosci. 1981;4:17–42.
5. Denny-Brown D, Hoherty MM. Effects of transient stretching of peripheral nerve. Arch Neurol Psychiatr. 1945;54:116.
6. Eversman WW, Ritsick JA. Intraoperative changes in motor nerve conduction latency in carpal tunnel syndrome. J Hand Surg. 1978;3:77–81.
7. Kutz JE, Shealy G, Lubbers L. Interfascicular nerve repair. Orthop Clin North Am. 1981;12:277–286.
8. Panse F. Electrical lesions of the nervous system. Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology.Vol 7. New York: Elsevier; 1970.
9. English KB. The ultrastructure of cutaneous type I mechanoreceptors (Haarscheiben) in cats following denervation. J Comp Neurol. 1977;172:137–163.
10. Dellon AL, Witebsky FG, Terrill RE. The denervated Meissner corpuscle: a sequential histologic study after nerve division in the Rhesus monkey. Plast Reconstr Surg. 1975;46:182–193.
11. Onne L. Recovery of sensibility and sudomotor activity in the hand after nerve suture. Acta Chir Scand Suppl. 1962;300:1–69.
12. Dellon AL. Evaluation of Sensibility and Reeducation of Sensation in the Hand. Baltimore: Williams & Wilkins; 1981.
13. Sunderland S. Nerves and Nerve Injuries. Edinburgh: E & S Livingstone; 1973.
14. Dyck PJ, Conn DL, Okazaki H. Necrotizing angiopathic neuropathy: three-dimensional morphology of fiber degeneration related to sites of occluded vessels. Mayo Clin Proc. 1972;47:461–475.
15. Lomo T, Westgaard RH. Control of ACh sensitivity in rat muscle fibers. Cold Spring Harb Symp Quant Biol. 1975;40:263–274.
16. Lomo T, Westgaard RH. Further studies on the control of ACh sensitivity by muscle activity in the rat. J Physiol. 1975;252:603–626.
17. Love S. An experimental study of peripheral nerve regeneration after x-irradiation. Brain. 1983;106:39–54.
18. Ridley A. Silver staining of the innervation of Meissner corpuscles in peripheral neuropathy. Brain. 1968;91:539–552.
19. Woltman HW, Wilder RM. Diabetes mellitus: pathologic changes in the spinal cord and peripheral nerves. Arch Intern Med. 1927;44:576.
20. Rydevik B, Lundborg G, Nordborg C. Intraneural tissue reactions induced by internal neurolysis. An experimental study on the blood-nerve barrier, connective tissues and nerve fibres of rabbit tibial nerve. Scand J Plast Reconstr Surg. 1976;10:3–8.
21. Gilliatt RW, Wilson TG. Schemic sensory loss in patients with peripheral nerve lesions. J Neurol Neurosurg Psychiatry. 1954;17:104–114.
22. Bora FW, Osterman AL. Compression neuropathy. Clin Orthop. 1982;163:20–32.
23. Rydevik B, Lundborg G, Bagge U. Effects of graded compression on intraneural blood flow. An in vivo study on rabbit tibial nerve. J Hand Surg Am. 1981;6:3–12.
24. Mackinnon SE, Dellon AL. Experimental study of chronic nerve compression. Clinical implications. Hand Clin. 1986;2:639–650.
25. Hess K, Eames RA, Darveniza P, Gilliatt RW. Acute ischemic neuropathy in the rabbit. J Neurol Sci. 1979;44:19–43.
26. Schut L. Nerve injuries in children. Surg Clin North Am. 1972;52:1307–1312.
27. Lieberman AR. The axon reaction: a review of the principle features of perikaryal response to axonal injury. Int Rev Neurobiol. 1971;14:49–124.
28. Karnash LJ. Sciatic causalgia due to nerve trunk ischemia. J Nerv Ment Dis. 1936;84:283.
29. Skoulis TG, Lovice D, von Fricken K, Terzis JK. Nerve expansion. The optimal answer for the short nerve gap. Behavioral analysis. Clin Orthop. 1995;314:84–94.
30. Ochs S. Fast axoplasmic transport in the fibers of chromatolysed neurons. J Physiol (Lond). 1976;255:249–261.
31. Stoll BA, Andrews JT. Radiation-induced neuropathy. Br Med J. 1966;1:834–837.
32. Ochoa JL, Torebjörk HE. Paresthesia from ectopic impulse generation in human sensory nerves. Brain. 1980;103:835–855.
33. Truge K. Management of established Volkmann’s contracture. In: Green DP, ed. Operative Hand Surgery. New York: Churchill Livingstone; 1982.
34. Frischbier HJ, Lohbeck HD. Strahlenchaden nach elektronentherapie beim mammacarcinom. Strahlentherapie. 1970;139:684–694. Cited in reference 104
35. Sunderland S. The nerve lesion in the carpal tunnel syndrome. J Neurol Neurosurg Psychiatry. 1976;39:615–626.
36. Bunge MB. Initial endocytosis of peroxidase or ferritin by growth cones of cultured nerve cells. J Neurocytol. 1977;6:407–439.
37. Sunderland S. The pros and cons of funicular nerve repair—Founder’s Lecture–American Society for Surgery of the Hand. J Hand Surg. 1979;4:201–211.
38. Forman DS, Berenberg RA. Regeneration of motor axons in the rat sciatic nerve studied by labelling with axonally transported radioactive proteins. Brain Res. 1978;156:213–225.
39. Selander D, Brattsand R, Lundborg G, et al. Local anesthetics: importance of mode of application, concentration, and adrenaline for the appearance of nerve lesions, An experimental study of axonal degeneration and barrier damage after intrafascicular injection or topical application of bupivacaine (Marcain). Acta Anaesthesiol Scand. 1979;23:127–136.
40. Urbaniak JR. Fascicular nerve suture. Clin Orthop Relat Res. 1982;163:57–64.
41. Grafstein B. The nerve cell body’s response to axotomy. Exp Neurol. 1975;48:32–51.
42. Mackinnon SE, Hudson AR, Gentili F, et al. Peripheral nerve injection injury with steroid agents. Plast Reconstr Surg. 1962;69:482–490.
43. Mira JC. Quantitative studies of the regeneration of rat myelinated fibres: variations in the number and size of regenerating nerve fibres after repeated localized freezings. J Anat (Lond). 1979;129:77–93.
44. Hargens AR, Romine JS, Sipe JC, et al. Peripheral nerve conduction block by high muscle compartment pressure. J Bone Joint Surg. 1979;61:192–200.
45. McLellan DL, Swash M. Longitudinal sliding of the median nerve during movements of the upper limb. J Neurol Neurosurg Psychiatry. 1976;39:566–570.
46. Miller RG. Acute vs. chronic compression neuropathy. Muscle Nerve. 1984;7:427–430.
47. Yates SK, Hurst LM, Brown WF. The pathogenesis of pneumatic tourniquet paralysis in man. J Neurol Psychiatr. 1981;44:759–767.
48. Millesi H. Nerve grafting. In: Terzis JK, ed. Microreconstruction of Nerve Injuries. Philadelphia: WB Saunders; 1987.
49. Thulin CA. Electrophysiological studies of peripheral nerve regeneration with special reference to small diameter (gamma) fibers. Exp Neurol. 1960;2:598–612.
50. Thomas PK. The deposition of collagen in relation to Schwann cell basement membrane during peripheral nerve regeneration. J Cell Biol. 1964;23:375–382.
51. Terzis JK. Microreconstruction of Nerve Injuries. Philadelphia: WB Saunders; 1987.
52. DiVincenti FC, Moncrief JA, Pruitt BA. Electrical injuries: a review of 65 cases. J Trauma. 1977;9:497–507.
53. Ochoa J, Fowler TJ, Gilliatt RW. Anatomical changes in the peripheral nerves compressed by a pneumatic tourniquet. J Anat. 1972;113:433–455.
54. Goodman HV, Gilliatt RW. The effect of treatment on median nerve conduction in patients with the carpal tunnel syndrome. Ann Phys Med. 1961;6:137–155.
55. Omer GE. Nerve response to injury and repair. In: Hunter JM, ed., et al. Rehabilitation of the Hand. St Louis: Mosby; 1984.
56. Highet WB, Saunders FK. The effect of stretching nerves after suture. Br J Surg. 1943;30:355–369.
57. Lundborg G, Schildt B. Microvascular permeability in irradiated rabbits. Acta Radiol Ther Phys Biol. 1971;10:311–320.
58. Davis HL, Kiernan JA. Effect of nerve abstract on atrophy of denervated or immobilized muscles. Exp Neurol. 1981;72:582–591.
59. Lundborg G. Intraneural microvascular pathophysiology as related to ischemia and nerve injury. In: Daniel RK, Terzis JK, eds. Reconstructive Microsurgery. Boston: Little, Brown; 1977.
60. Lundborg G. The intrinsic vascularization of human peripheral nerves: structure and functional aspects. J Hand Surg. 1979;4:34–41.
61. Kline DG. Timing for exploration of nerve lesions and evaluation of the neuroma-in-continuity. Clin Orthop Relat Res. 1982;163:42–49.
62. Ugland OM. Electrical injuries to peripheral nerves in animals. A preliminary report. Acta Chir Scand. 1966;131:432–437.
63. Dykes RW. Central consequences of peripheral nerve injuries. Ann Plast Surg. 1984;13:412–422.
64. Schnapp B, Mugnaini E. Membrane architecture of myelinated fibers seen by freeze-fracture. In: Waxman SG, ed. Physiology and Pathology of Axons. New York: Raven Press; 1974.
65. Barker NW. Lesions of peripheral nerves in thrombangiitis obliterans. Arch Intern Med. 1938;62:271.
66. Dyck PJ, Karnes J, Lais A, et al. Pathologic alterations of the peripheral nervous system of humans. In: Dyck PV, ed., et al. Peripheral Neuropathy. Philadelphia: WB Saunders; 1984.
67. Rosenthal J. Trophic interactions of neurons. In: Bookhart JM, Mountcastle VB, eds. Handbook of Physiology. Bethesda, Md: American Physiological Society; 1977.
68. McDonald WI. Physiological consequence of demyelination. In: Sumner AJ, ed. The Physiology of Peripheral Nerve Disease. Philadelphia: WB Saunders; 1980.
69. Edshage S. Peripheral nerve suture: a technique for improved intraneural topography. Acta Chir Scand Suppl. 1964;331:1.
70. Korthals JK, Korthals MA, Wisniewski HM. Peripheral nerve ischemia. Part 2: accumulation of organelles. Ann Neurol. 1978;4:487–498.
71. Highet WB, Holmes W. Traction injuries to the lateral popliteal nerve and traction injuries to peripheral nerves after suture. Br J Surg. 1943;30:212–233.
72. Kurosumi K, Kurosumi U, Inoue K. Morphological and morphometric studies with the electron microscope on the Merkel cells and associated nerve terminals in normal and denervated skin. Arch Histol Jpn. 1979;42:243–261.
73. Woltman HW, Wilder RM. Diabetes mellitus: pathologic changes in the spinal cord and peripheral nerves. Arch Int Med. 1929;44:574–603.
74. Goodall RJ. Nerve injuries in fresh fractures. Texas State J Med. 1956;52:93–95.
75. Ducker TB, Kempe LG, Hayes GJ. The metabolic background for peripheral nerve surgery. J Neurosurg. 1969;30:270–280.
76. Purves D, Sakmann B. The effect of contractile activity on fibrillation and extrajunctional acetylcholine sensitivity in rat muscle maintained in organ culture. J Physiol. 1974;237:157–182.
77. Powell HC, Myers RR. Pathology of the peripheral myelinated axon. In: Adachi M, Hirano A, Aronson SM, eds. The Pathology of the Myelinated Axon. New York: Igaku-Shoin; 1985.
78. Simpson JA. Nerve injuries, general aspects. In: Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology, Diseases of Nerves. New York: Elsevier; 1970.
79. Upton ARM, McComas AJ. The double crush syndrome in nerve entrapment syndromes. Lancet. 1973;2:359–362.
80. Letourneau PC. Cell-to-substratum adhesion and guidance of axonal elongation. Dev Biol. 1975;44:92–101.
81. Pockett S, Slack JR. Source of stimulus for nerve terminal sprouting in partially denervated muscle. Neuroscience. 1982;7:3173–3176.
82. Souttar HS. Nerve injuries in children. Br Med J. 1945;2:349–350.
83. Strauch B. Use of nerve conduits in peripheral nerve repair. Hand Clin. 2000;16:123–130.
84. Lundborg G, Gelberman RH, Minteer-Convery M, et al. Median nerve compression in the carpal tunnel–functional response to experimentally induced controlled pressure. J Hand Surg. 1982;7:252–259.
85. Nobel W. Peroneal palsy due to hematoma in the common peroneal nerve sheath after distal torsional fractures and inversion ankle sprains. J Bone Joint Surg Am. 1966;48:1484–1495.
86. Silversides J. The neurologic sequelae of electrical injury. Can Med Assoc J. 1964;91:195–204.
87. Lubinska L. Early course of wallerian degeneration in myelinated nerve fibres of the rat phrenic nerve. Brain Res. 1979;130:47–63.
88. Castaldo JE, Ochoa JL. Mechanical injury of peripheral nerves, fine structure and dysfunction. Clin Plast Surg. 1984;11:9–16.
89. Gentili F, Hudson AR, Hunter D. Clinical and experimental aspects of injection injuries of peripheral nerves. Can J Neurol Sci. 1980;7:143–151.
90. Thomas PK. The connective tissue of peripheral nerve: an electron microscopic study. J Anat. 1963;97:35–44.
91. Korthals JK, Gieron MA, Maki T, Belsole RJ. Peripheral demyelination after ransient ischemia. Neurology (NY). 1984;34:168.
92. Lewis T, Pickering GW, Rothschild P. Centripetal paralysis arising out of arrested blood flow to the limb, including notes on a form of tingling. Heart. 1931;16:1–32.
93. Thomas PK, Landon DN, King RHM. Normal structure of the peripheral nerve. In: Adams JH, Corcellis J, Duchen LW, eds. Greenfield’s Neuropathology. New York: John Wiley & Sons; 1984.
94. Lindsay WK, Walker FG, Farmer AW. Traumatic peripheral nerve injuries in children. Results of repair. Plast Reconstr Surg Transplant Bull. 1962;30:462–468.
95. Gentili F, Hudson A, Kline DG, Hunter D. Peripheral nerve injection injury: an experimental study. Neurosurgery. 1979;4:244–253.
96. Gentili F, Hudson AR, Hunter D, Kline DG. Nerve injection injury with local anesthetic agents: a light and electron microscopic, fluorescent microscopic, and horseradish peroxidase study. Neurosurgery. 1980;6:263–272.
97. Jewett DL. Functional blockade of impulse tissues by acute nerve compression. In: Jewett DL, McCarroll HR, eds. Nerve Repair and Regeneration. St Louis: Mosby; 1980.
98. Margiotta MS, Usal H, Karp NS, et al. A nerve distraction model in the rat. Ann Plast Surg. 1998;40:486–489.
99. Gentili F, Hudson AR, Kline D, Hunter D. Early changes following injection injury of peripheral nerves. Can J Surg. 1980;23:177–182.
100. Gilliatt RW. Acute compression block. In: Sumner AJ, ed. The Physiology of Peripheral Nerve Disease. Philadelphia: WB Saunders; 1980.
101. Selzer ME. Regeneration of the peripheral nerve. In: Sumner AJ, ed. The Physiology of Peripheral Nerve Disease. Philadelphia: WB Saunders; 1980.
102. Chiu DT. Autogenous venous nerve conduits: a review. Hand Clinics. 1999;15:667–671.
103. Clodius L, Uhlschmidt G, Hess K. Irradiation plexitis of the brachial plexus. In: Terzis JK, ed. Microreconstruction of Nerve Injuries. Philadelphia: WB Saunders; 1987.
104. Clark WK. Peripheral nerve injection injury with steroid agents. Discussion of Mackinnon SE, et al Plast Reconstr Surg. 1982;69:490.
105. Daniel RK, Terzis JK. Reconstructive Microsurgery. Boston: Little, Brown; 1977.
106. Kline DG, Hackett ER. Reappraisal of timing for exploration of civilian peripheral nerve injuries. Surgery. 1975;78:54–65.
107. Shack RB, Lynch JB. Radiation dermatitis. Clin Plast Surg. 1987;14:391–401.
108. Birch R, Achan P. Peripheral nerve repairs and their results in children. Hand Clin. 2000;16:579–595.
109. Black MM, Lasek RJ. Slowing of the rate of axonal regeneration during growth and maturation. Exp Neurol. 1979;63:108–119.
110. Roberts JT. The effect of occlusive arterial diseases of the extremities on the blood supply of nerves: experimental and clinical studies on the role of the vasa nervorum. Am Heart J. 1948;35:369–392.
111. Eames RA, Lange LS. Clinical and pathological study of ischemic neuropathy. J Neurol Neurosurg Psychiatry. 1967;30:215–226.
112. Brown WF, Ferguson GG, Jones MW, Yates SK. The location of conduction abnormalities in human entrapment neuropathies. Can J Neurol Sci. 1976;3:111–122.
113. Gutman E, Young JZ. The reinnervation of muscle after various periods of atrophy. J Anat (Lond). 1944;18:15–43.
114. Brown PW. Factors influencing the success of the surgical repair of peripheral nerves. Surg Clin North Am. 1972;52:1137–1155.
115. Ponten B, Erickson U, Johansson S, Olding L. New observations on tissue changes along the pathway of the current in an electrical injury. Scand J Plast Reconstr Surg. 1970;4:75–82.
116. Mira JC. Degeneration and regeneration of peripheral nerves: ultrastructural and electrophysiological observation, quantitative aspects and muscle changes during reinnervation. Intern J Microsurg. 1981;3:102–120.
117. Haftek J. Stretch injury of peripheral nerve, acute effects of stretching on rabbit peripheral nerve. J Bone Joint Surg Br. 1970;52:354–365.
118. Tinel J. The sign of “tingling” in lesions of the peripheral nerves. J Presse Med. 1915;23:388. (Translated in Arch Neurol. 1971;24:574.)
119. Hudson AR. Nerve injection injuries. In: Terzis JK, ed. Microreconstruction of Nerve Injuries. Philadelphia: WB Saunders; 1987.
120. Kline DG, Hudson AR, Bratton BR. Experimental study of fascicular nerve repair with and without epineurial closure. J Neurosurg. 1981;54:513–520.
121. Millesi H. The nerve gap: theory and clinical practice. Hand Clin. 1986;2:651–663.
122. Terzis JK, Strauch B. Microsurgery of the peripheral nerve: a physiological approach. Clin Orthop Relat Res. 1978;133:39–48.
123. Bristow WR. Injuries of peripheral nerves in two world wars. Br J Surg. 1947;34:333–348.
124. Bora FW, Pleasure DE, Didizian NA. A study of nerve regeneration and neuroma formation after nerve suture by various techniques. J Hand Surg. 1976;1:138–143.
125. Engh CA, Schofield BH. A review of the central response to peripheral nerve injury and its significance in nerve regeneration. J Neurosurg. 1972;37:195–203.
126. Sunderland S. Nerves and Nerve Injuries. Edinburgh: E & S Livingstone; 1968.
127. Cabaud HE, Rodkey WG, McCarroll HR Jr, et al. Epineurial and perineurial fascicular nerve repairs: a critical comparison. J Hand Surg. 1976;1:131–137.
128. Chino N, Awad EA, Kottke FJ. Pathology of propylene glycol administered by perineural and intramuscular injection in rats. Arch Phys Med Rehabil. 1974;55:33–38.
129. Kori SH, Foley KM, Posner JB. Brachial plexus lesions in patients with cancer: 100 cases. Neurology. 1981;31:45–50.
130. Palmer P. Ultrastructural alterations of Merkel cells following denervation. Anat Rec. 1965;151:396–397.
131. Langworthy OR. Histological changes in nerve cells following injury. Bull Johns Hopkins Hosp. 1930;47:11.
132. Sunderland S, Bradley KC. Denervation atrophy of the distal stump of a severed nerve. J Comp Neurol. 1950;93:401–409.
133. Weiss P. Nerve patterns. The mechanics of nerve growth. Growth. 1941;5(Suppl):163–203.
134. Terzis JK, Smith KL. The Peripheral Nerve: Structure, Function and Reconstruction. New York: Raven Press; 1990.
135. Horch K. Guidance of regrowing sensory axons after cutaneous nerve lesions in the rat. J Neurophysiol. 1979;42:1437–1449.
136. Millesi H. Reappraisal of nerve repair. Surg Clin North Am. 1981;61:321–340.
137. Maruyama Y, Mylrea MM, Logothetis J. Neuropathy following irradiation. An unusual late complication of radiotherapy. AJR Am J Roentgenol. 1967;101:216–219.
138. Maruhashi J, Wright EB. Effect of oxygen lack in the single isolated mammalian (rat) nerve fiber. J Neurophysiol. 1967;30:434–452.
139. Clark WK. Surgery for injection injuries of peripheral nerves. Surg Clin North Am. 1972;52:1325–1328.
140. Sunderland S. Factors influencing the course of regeneration and the quality of the recovery after nerve suture. Brain. 1952;75:19–54.
141. Watson WE. An autoradiographic study of the incorporation of nucleic acid precursors by neurons and glia during nerve regeneration. J Physiol. 1965;180:741–753.
142. Omer GE. Injuries to nerves of the upper extremity. J Bone Joint Surg Am. 1974;56:1615–1624.
143. Solem L, Fischer RP, Strate RG. The natural history of electrical injury. J Trauma. 1977;17:487–492.