Cancer Pain
Causes, Consequences, and Therapeutic Opportunities
The Clinical Problem
In 2010 cancer will be diagnosed in more than 12 million people worldwide (excluding non-melanoma skin cancer), and 8 million individuals will die of cancer (Boyle and Levin 2008). Cancer incidence rates are stable or slightly falling in developed countries, whereas in developing countries, these rates are increasing because smoking, obesity, and increased life expectancy have led to a rapid rise in the incidence of cancer (Boyle and Levin 2008, Khan et al 2010, Jemal et al 2011). Additionally, as detection and treatment of most cancers have dramatically improved, survival rates have increased so that even patients with metastatic cancer are surviving years to decades beyond their initial diagnosis (Jemal et al 2010).
Cancer-associated pain is one of the most common initial symptoms that results in the diagnosis of cancer. Although pain can be present at any time during the course of the disease, it generally increases with disease progression such that 75–90% of patients with metastatic or advanced-stage cancer will experience significant cancer pain (van den Beuken-van Everdingen et al 2007, Costantini et al 2009). Figure 72-1 outlines some of the mechanisms that drive cancer pain, including cancer therapy, factors released from tumors that sensitize or excite primary afferent neurons, tumor-induced nerve injury, and tumor-induced nerve sprouting and neuroma formation (Mantyh 2006, Gordon-Williams and Dickenson 2007, Jimenez-Andrade et al 2010a, Mantyh et al 2010).
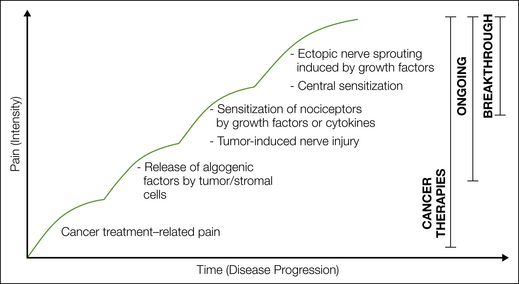
Figure 72-1 An evolving set of mechanisms drives cancer pain.
Cancer pain may initially be directly related to treatment of the cancer (i.e., diagnostic or therapeutic surgical procedures such as biopsy, resection) or be a side effect or toxicity related to the therapies used to treat cancer. Early ongoing pain is driven primarily by factors released from cancer cells and their associated stromal cells. Cancer pain becomes more intense as nociceptors are injured by the growing tumor and become sensitized by release of factors such as nerve growth factor, which can be released by cancer and stromal cells. With progression of cancer, the severity of the pain tends to increase as spontaneous and movement-evoked pain/breakthrough pain occurs. This breakthrough pain may in part be due to tumor- and stromal-induced pathological sprouting of nociceptors and the formation of neuroma-like structures. Breakthrough pain also appears to be due in part to changes in the central nervous system, which include neurochemical and cellular reorganization of the spinal cord, as well as enhanced synaptic transmission mediated through Aδ and C fibers in the substantia gelatinosa of the spinal cord.
In patients with cancer, undergoing surgery or receiving the full dose of radiation therapy or chemotherapy is one of the most significant factors in determining patient survival (Mantyh 2006). However, peripheral nerve neurotoxicity and the accompanying pain are major side effects of radiation therapy and many of the most commonly used antineoplastic agents, including the taxanes (e.g., paclitaxel, docetaxel), vinca alkaloids (e.g., vincristine, vinblastine), platinum-based compounds (e.g., cisplatin and oxaliplatin), and proteasome inhibitors (e.g., bortezomib, disulfiram) (Quasthoff and Hartung 2002, Cata et al 2006, Mantyh 2006, Mielke et al 2006, Bennett 2010). If the chemotherapy-induced peripheral neuropathy becomes severe enough, the oncologist or patient may reduce or cease chemotherapy treatment, which decreases the survival rate of the patient and the likelihood that the patient will be disease free.
In cases in which the tumor is inoperable or relapse occurs, cancer and its associated stromal cells can induce significant pain. This pain can arise from the original site of the cancer (i.e., pancreatic cancer, head and neck cancer, osteosarcoma) (Dreghorn et al 1990, Zhu et al 1999, Lam and Schmidt 2011) or from distant sites (such as bone, liver, and lung), where common cancers such as breast, prostate, kidney, and lung cancer avidly metastasize (Coleman 2006). The original quality of the tumor-induced pain is usually described as dull in character, constant, and gradually increasing in intensity with time (Dy et al 2008) (see Fig. 72-1). If the disease continues, a second type of cancer pain known as “breakthrough” or severe “incident pain” can emerge (Mercadante 1997). Incident or breakthrough pain, which is defined as a transitory flare of extreme pain superimposed on an otherwise stable pain pattern in patients treated with opioids (Casuccio et al 2009), can occur spontaneously or with movement or weight-bearing of a tumor-bearing organ or tissue (Mercadante et al 2004). Since breakthrough pain is frequently acute and unpredictable in onset, this pain can be severe, debilitating, and difficult to fully control (Coleman 1997, Mercadante 1997).
Currently, tumor-induced pain is largely managed with an “analgesic ladder” that was originally promulgated by the World Health Organization in 1986 (see Chapter 75). This ladder begins with a non-steroidal anti-inflammatory drug (NSAID); if the pain worsens, an NSAID plus a mild opiate; and finally, when the pain becomes severe, an NSAID plus a strong opiate. In addition to this three-step ladder, other adjuvant therapies, including radiation therapy, radioisotopes, nerve blocks, nerve lesions, antiepileptics (e.g., gabapentin, carbamazepine), antidepressants (amitriptyline, imipramine), and steroids, are commonly used to control cancer pain (Desandre and Quest 2009). It should be stressed that most cancer pain can be controlled if it is closely monitored and these therapies are used in a timely and proactive manner. However, the abovementioned therapies all have significant unwanted side effects (Montagnini and Zaleon 2009), and closely monitoring and fully controlling the cancer pain (especially if breakthrough pain is present) can be very time-consuming for the patient, caregiver, and physician (Lossignol and Dumitrescu 2010). Developing new analgesic therapies that are efficacious and have fewer side effects than current analgesics do and incorporating these advances into mainstream cancer therapy will significantly improve the quality of life and functional status of both the patient and the caregiver.
Development of a Murine Model of Bone Cancer
Given the enormous consequences in terms of human suffering that cancer pain can cause, it was surprising to us when we first began exploring the mechanisms driving cancer pain that no well-established animal model was available for studying any form of cancer pain. However, there were two commonly used in vivo models to study tumor-induced bone destruction. In the first model, tumor cells are injected into the left ventricle of the heart and then spread to multiple sites, including the bone marrow, where they multiply, grow, and destroy the surrounding bone (Arguello et al 1988, Yoneda et al 1994). Although this model replicates the observation that most tumor cells metastasize to multiple sites, including bone, a major problem with the model is animal-to-animal variability in the sites, size, and extent of the metastasis. Since the tumors frequently metastasize to vital organs such as the lung or liver, the general health of the animal is also variable, which makes behavioral assessment difficult. Additionally, because the tumors frequently metastasize to bone in the vertebral column, tumor growth in the vertebrae can result in collapse of the vertebral column and compression of the spinal cord with resultant spinal dysfunction and paralysis. Given these problems, development of a model of bone cancer pain using intracardiac injection proved difficult at best.
The second major model used to study tumor-induced bone destruction involved the direct injection of lytic sarcoma cells into the intramedullary space of the mouse tibia or femur. The major problem with this model was that since the injection site could not be plugged with conventional sealing agents (because it is a wet, bony surface), the tumor cells rapidly escape and grow avidly in nearby skin and joints, thereby resulting in large extraskeletal tumor masses that not only interfered with behavioral analysis but also destroyed nerves passing though these sites and generated a neuropathic pain state. We chose to adapt and modify this model by plugging the injection hole with a dental amalgam that tightly binds and seals the injection hole in the proximal head of the femur. Plugging of the injection site allowed us to contain the tumor cells within the intramedullary space and prevented invasion of tumor cells into the surrounding soft tissue (Fig. 72-2) (Honore et al 2000). This advance, along with techniques with which we could simultaneously measure bone cancer–induced pain behavior, tumor growth, and tumor-induced bone remodeling has provided us with the first preclinical cancer pain model, which we then used to define the mechanisms that generate and maintain bone cancer pain.
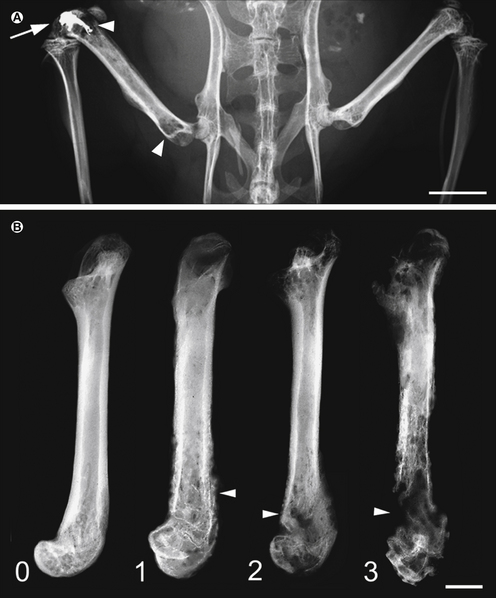
Figure 72-2 Progressive destruction of mineralized bone in mice with bone cancer.
A, Low-power anteroposterior radiograph of the mouse pelvis and hindlimbs after unilateral injection of sarcoma cells into the distal part of the femur and closure of the injection site with an amalgam plug (arrow), which prevents the tumor cells from growing outside the bone; arrowheads indicate areas of significant tumor-induced bone destruction of mineralized bone. B, Radiographs of murine femora show the progressive loss of mineralized bone caused by tumor growth. These images are representative of the stages of bone destruction in the murine femur. At week 1, there is minor loss of bone near the distal head (arrowhead); at week 2, substantial loss of mineralized bone at both the proximal and distal (arrowhead) heads; and at week 3, loss of mineralized bone throughout the entire femur and fracture of the distal head (arrowhead). Scale bar = 2 mm. (Modified from Schwei MJ, Honore P, Rogers SD, et al 1999 Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. Journal of Neuroscience 19:10886–10897. Copyright 1999 by the Society for Neurosciences).
After injection and confinement of primarily osteolytic 2472 murine osteosarcoma tumor cells to the intramedullary space of the mouse femur, the tumor cells grow in a highly reproducible fashion as they proliferate and replace the hematopoietic cells in the bone marrow (Schwei et al 1999, Sabino et al 2002). Eventually, the entire marrow space is homogeneously filled with tumor cells and tumor-associated inflammatory/immune cells. In terms of bone remodeling, injection of osteosarcoma cells into the femur induces a dramatic proliferation and hypertrophy of osteoclasts at the tumor–bone interface, with significant bone destruction in both the proximal and distal heads of the femur (see Fig. 72-2). In the osteosarcoma model, ongoing pain and movement-evoked pain-related behavior increased in severity with time, and this pain-related behavior correlated with the tumor growth and progressive tumor-induced bone destruction, which mirrors what occurs in patients with primary or metastatic bone cancer. Although sarcoma cells constituted the tumor used in the first bone cancer pain model that we developed, we have since developed other bone cancer pain models using prostate, breast, melanoma, colon, and lung tumors, all of which have provided insight into the similarities and differences by which different tumors drive bone cancer pain (Sabino et al 2003).
The Tumor Environment and Cancer Pain
A tumor is composed of not only cancer cells but also tumor-associated stromal cells. In most tumors, stromal cells far outnumber cancer cells and include endothelial cells, fibroblasts, and a host of inflammatory and immune cells, including macrophages, mast cells, neutrophils, and T lymphocytes (Joyce and Pollard 2009) (Fig. 72-3). Both cancer cells and their associated stromal cells secrete a wide variety of factors (Mantyh 2006, Joyce and Pollard 2009), many of which have been shown to sensitize or directly excite primary afferent neurons (Julius and Basbaum 2001).
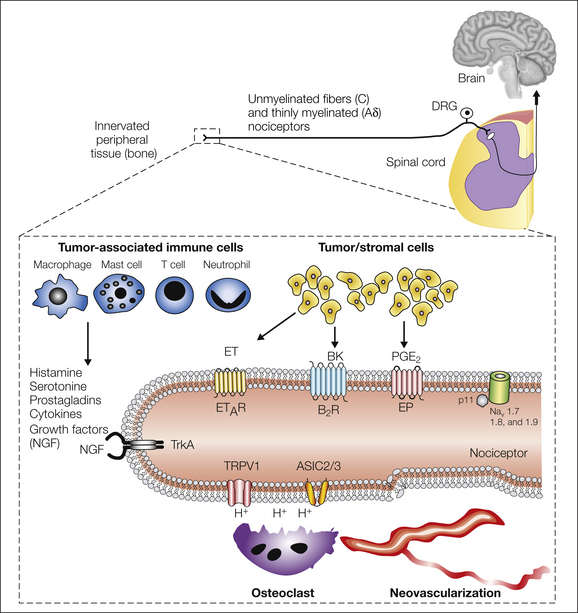
Figure 72-3 Primary afferent sensory nerve fibers and the generation and maintenance of cancer pain.
Primary afferent neurons innervating the body have their cell bodies in the dorsal root ganglia (DRG) and transmit sensory information from the periphery to the spinal cord and brain. Unmyelinated C fibers and thinly myelinated Aδ fibers contain small-diameter cell bodies that project centrally to the superficial spinal cord. These fibers are involved in detecting multiple noxious stimuli (chemical, thermal, and mechanical). Box: Nociceptors use several different types of receptor to detect and transmit signals about noxious stimuli produced by cancer and stromal cells (yellow), tumor-associated immune cells (blue), or other aspects of the tumor microenvironment. Multiple factors may contribute to the pain associated with cancer. Transient receptor potential vanilloid 1 (TRPV1) and acid-sensing ion channels (ASICs) detect the extracellular protons produced by tumor-induced tissue damage or abnormal osteoclast-mediated bone resorption. Several mechanosensitive ion channels may be involved in detecting the high-threshold mechanical stimuli that occur when distal aspects of the sensory nerve fiber are distended by mechanical pressure as a result of the growing tumor or as a result of destabilization or fracture of bone. Tumor cells and associated inflammatory (immune) cells produce a variety of chemical mediators, including prostaglandins (PGE2), nerve growth factor (NGF), endothelins (ET), bradykinin, (BK) ,and extracellular adenosine triphosphate. Several of these pro-inflammatory mediators have receptors on peripheral terminals and can directly activate or sensitize nociceptors. NGF, together with its cognate receptor TrkA, may serve as a master regulator of bone cancer pain by modulating the sensitivity and increasing the expression of several receptors and ion channels that contribute to the increased excitability of nociceptors in the vicinity of the tumor.
Tumor-Induced Acidosis
The finding that a subpopulation of sensory neurons express transient receptor potential vanilloid 1 (TRPV1, which is also known as the capsaicin receptor) and the acid-sensing ion channel 3 (ASIC3) and that both these channels respond to acidosis is of significant interest to researchers studying cancer pain (Joyce and Pollard 2009) because cancer cells in general have a lower pH (6.8) than normal cells do (pH 7.2) (Griffiths 1991). Importantly, many tumors that metastasize to bone induce a marked proliferation and hypertrophy of osteoclasts. Osteoclasts avidly resorb bone by generating a pH of 2–4 in their resorption bay, which drives the excessive bone resorption that can ultimately lead to fracture of the tumor-bearing bone (Clohisy et al 2000). To test whether TRPV1 channels were expressed by sensory nerve fibers that innervate tumor-bearing tissue and whether TRPV1 contributed to cancer pain, an in vivo model of bone cancer pain was explored (Ghilardi et al 2005). In these studies it was shown that a subpopulation of nerve fibers that innervate the tumor-bearing bone express TRPV1, that acute or chronic administration of a TRPV1 antagonist attenuates bone cancer pain, and that disruption of the TRPV1 gene results in attenuation of bone cancer pain (Ghilardi et al 2005). Furthermore, administration of a TRPV1 antagonist to TRPV1 null animals with bone cancer resulted in no further reduction in the pain that was already present in the TRPV1 null mice (Ghilardi et al 2005). To date, the results of human clinical trials with TRPV1 or ASIC3 channel antagonists have not been reported in subjects with cancer pain. However, as discussed later, understanding the role that TRPV1 plays in driving bone cancer pain has provided insight into why therapies that inhibit osteoclasts, including bisphosphonates and denosumab (an anti-RANKL fully humanized monoclonal antibody), are efficacious in reducing bone cancer pain (von Moos et al 2008, Stopeck et al 2010, Henry et al 2011).
The skeleton is the most common site for distant metastasis from prostate, breast, thyroid, lung, and renal carcinoma (Coleman 2006). Once tumor cells have metastasized to bone, a cycle of tumor growth, bone destruction, and the formation of woven bone begins and results in significant pain, skeletal fractures, and hypercalcemia (Coleman 2006). Cancer cells themselves do not destroy bone but rather they and their associated stromal cells express the receptor activator of nuclear factor κB ligand (RANKL), which binds to the receptor RANK expressed by osteoclasts. Activation of the RANKL/RANK pathway promotes the proliferation and hypertrophy of these bone-destroying osteoclasts (Clohisy and Mantyh 2004). Osteoclasts resorb bone by forming a highly acidic resorption “bay” or “pit” between the osteoclast and bone, which can stimulate TRPV1 or ASIC3 channels and drive bone cancer pain (Clohisy and Mantyh 2004). In the past decade multiple studies have shown that two therapies that reduce osteoclast function also significantly reduce bone cancer pain (Honore et al 2000, Lipton 2008, von Moos et al 2008, Stopeck et al 2010, Henry et al 2011).
The first and most widely used therapy is the class of compounds known as bisphosphonates, which avidly bind to bone. Once the bisphosphonate has bound to bone, osteoclasts that are resorbing bone generally need to actively take up the breakdown products of bone at the apical (bone facing) surface and transfer these products via transcytosis for release at the distal surface of the osteoclast so that they can be disposed by exocytosis (Stenbeck 2002). However, if a bisphosphonate is tightly bound to the bone that is being resorbed, the bisphosphonate will also be taken up by endocytosis (Rogers et al 2000). Once internalized, the bisphosphonate interferes either with adenosine triphosphate energy metabolism (non–nitrogen-containing bisphosphonates) or with the mevalonate pathway (nitrogen-containing bisphosphonates), which results first in osteoclast dysfunction and ultimately in osteoclast apoptosis (Rogers et al 2000, Clezardin et al 2005). Because a significant population of nerve fibers that innervate bone express TRPV1 (Ghilardi et al 2005), one way that bisphosphonates appear to relieve bone pain is by decreasing osteoclast-induced acidosis, which in turn will decrease activation of the ion-sensing TRPV1 or ASIC3 receptors that are expressed by sensory nerve fibers.
Another method that is highly effective in reducing tumor-induced osteoclast bone resorption in both animals and humans is interference in the binding of RANKL to RANK, which is required for osteoclast proliferation and maturation (Lipton and Jun 2008). Within 2 days of administration of therapies that interfere with binding of RANKL to RANK (such as osteoprotegerin or denosumab), there is an almost complete loss of activated osteoclasts, a marked reduction in plasma markers of bone resorption, and significant attenuation of bone cancer pain in a mouse model of bone cancer pain (Honore et al 2000).
Tumor-Induced Mechanical Instability of Bone
Therapies that inhibit osteoclast-induced bone resorption also maintain the mechanical strength of bone even though tumor cells are present in the bone. Thus, in addition to acidosis, excessive tumor-induced osteoclast bone resorption destroys bone and leads to mechanical instability and fracture of bone, which causes mechanical distortion of the nerve fibers innervating the bone (Yates and Smith 1994, Jimenez-Andrade et al 2007). Thus, following significant weakening or fracture secondary to tumor-induced bone remodeling, significant movement-evoked pain can occur, presumably because of mechanical distortion of the mechanosensitive sensory nerve fibers that innervate the bone. Clearly, pain associated with the fracture is attenuated if the bone is stabilized and repositioned into its normal orientation (Rubert and Malawer 2000). Both osteolytic and osteoblastic tumors induce loss of the mechanical strength and stability of mineralized bone (Arrington et al 2006), so with significant bone remodeling, normally innocuous mechanical stress can now result in distortion and activation of the mechanosensitive nerve fibers that innervate the bone. Since bisphosphonates and anti-RANKL therapies reduce tumor-induced osteoclast bone remodeling, preserve the mechanical strength of bone, reduce bone fractures, and decrease osteoclast-induced acidosis in both animals and humans, these therapies are highly useful in managing pain resulting from metastasis of cancer to bone.
Factors Released by Cancer/Stromal Cells That Drive Cancer Pain
One area that has significantly contributed to our understanding of what drives cancer pain is work examining the factors released by tumor/stromal cells that drive cancer pain and influence disease progression. These factors include bradykinin, cannabinoids, endothelins, interleukin-6 (IL-6), granulocyte-macrophage colony-stimulating factor (GM-SCF), nerve growth factor (NGF), proteases, and tumor necrosis factor-α (TNF-α). Because there are several recent studies in this area, we will briefly summarize some of the most striking research on this topic.
Cannabinoids
Recent studies in animals have revealed that the cannabinoid system plays an important role in modulating cancer pain. It has been reported that tumor-induced mechanical hyperalgesia in the hindpaw is associated with decreased levels of anandamide (endogenous agonist of the cannabinoid CB1 and CB2 receptors) and increased degradation of anandamide in the hindpaw skin ipsilateral to the tumor-bearing paw (Khasabova et al 2008). Furthermore, injection into the hindpaw of anandamide or an inhibitor of the enzyme that degrades anandamide reduced the tumor-induced hyperalgesia. A recent report also demonstrated that acute and sustained administration of a cannabinoid CB2 agonist attenuates both spontaneous and evoked pain behavior in a bone cancer pain model (Lozano-Ondoua et al 2010). Although these studies suggest that the cannabinoid system plays a role in driving cancer pain, future clinical studies are warranted to evaluate the analgesic as well as the potential central nervous system (CNS) side effects of these drugs.
Colony-Stimulating Factors
Human studies have shown that several non-hematopoietic tumors secrete colony-stimulating factors that act on their receptors expressed on myeloid cells, tumor cells, and nerve fibers. Recently, Schweizerhof and co-authors (2009) reported that levels of granulocyte colony-stimulating factor (G-CSF) and GM-CSF in the lysates of bone marrow from tumor-bearing mice were significantly increased in comparison to the levels in naïve mice. Additionally, GM-CSF sensitized the nerves to mechanical stimuli, potentiated the release of calcitonin gene–related peptide (CGRP), and caused sprouting of sensory nerve endings in the skin (Schweizerhof et al 2009). The actions of these colony-stimulating factors are mediated by activation of their receptors (G-CSFR and GM-CSFRα, expressed in peripheral nerves innervating the tumor-bearing tissue) since the administration of neutralizing antisera against these receptors and the sensory nerve–specific knockdown of GM-CSF receptors reduced the tumor-induced pain behavior (Schweizerhof et al 2009). Based on these studies, colony-stimulating factors may be a potential target therapeutic to be exploited in the cancer pain field.
Endothelins
Endothelin antagonists are another group of pharmacological agents that offer promise in managing cancer pain and reducing progression of the disease. Endothelins (ET-1, -2, and -3) are a family of vasoactive peptides that are expressed at high levels by several types of tumors, including those that arise from the prostate (Nelson et al 1995, 1996). Clinical studies have shown a correlation between the severity of the pain and plasma levels of endothelins in prostate cancer patients (Nelson et al 1995). Electrophysiological studies have shown that ET-1 may directly sensitize or excite C-fiber nociceptors innervating the tumor-bearing tissue through activation of endothelin A receptors (ETAs) (Hamamoto et al 2008), which are expressed by a subset of small unmyelinated primary afferent neurons (Pomonis et al 2001). Furthermore, direct application of endothelin to peripheral nerves induces the activation of primary afferent fibers and pain-related behavior (Davar et al 1998, Hamamoto et al 2008). These results suggest that endothelins play a critical role in tumor-induced thermal and mechanical hyperalgesia. Currently, several ongoing human clinical trials are examining the effects that ETA antagonists have on cancer pain and disease progression.
Cytokines
Tumors release a variety of chemical agents that sensitize peripheral afferent neurons, including cytokines. One of the most studied cytokines in the pain field is the pro-inflammatory and pro-algesic cytokine TNF-α. This cytokine is produced by inflammatory/immune cells, Schwann cells, and some tumor cells (Beutler 1999). It has been reported that TNF-α levels are significantly increased in tumor micro-perfusates and tumor site homogenates in comparison to the levels in naïve mice or the contralateral hindlimb (Wacnik et al 2005). Furthermore, injection of TNF-α into tumor-bearing mice results in heat and mechanical hyperalgesia, which is blocked by administering the TNF-α antagonist etanercept (Constantin et al 2008).
Another cytokine that has been suggested to be involved in driving cancer pain is the pleiotropic cytokine IL-6. Several studies have shown that levels of this cytokine are up-regulated under various pathological conditions (Poole et al 1995, Smith et al 2001, Nishimoto and Kishimoto 2006, Rose-John et al 2006) and that direct injection (intradermally, intramuscularly, intrathecally) of IL-6 results in mechanical and thermal hyperalgesia (Poole et al 1995, DeLeo et al 1996, Dina et al 2008). IL-6 is produced by different inflammatory/immune cells and by some tumor cells (Nishimoto and Kishimoto 2006). The actions of IL-6 are mediated by binding to its specific receptor, IL-6R, which exists in both transmembrane and soluble form (Rose-John and Heinrich 1994, Rose-John et al 2006). Binding of IL-6 triggers an association of IL-6R with the transducer glycoprotein gp130 (Taga et al 1989). Recently, it has been reported that nociceptor-specific depletion of gp130 results in a significant reduction in the heat hyperalgesia induced by the subcutaneous injection of LL2 carcinoma cells into the hindpaw without affecting tumor growth (Andratsch et al 2009). These results suggest that blocking TNF-α or IL-6/gp130 may be a viable therapeutic opportunity for the treatment of cancer pain.
Tyrosine Receptor Kinases
One important concept that has emerged over the past decade is that in addition to NGF being able to directly activate TrkA-expressing sensory neurons, NGF activation of TrkA appears to play a key role in the sensitization of nociceptors (see for review see Pezet and McMahon 2006; also see Chapter 3). Thus, in addition to inducing rapid phosphorylation and sensitization of TRPV1, retrograde transport of the NGF/TrkA complex to the neuronal cell body of nociceptors induces increased synthesis of the neurotransmitters substance P and CGRP and increased expression of receptors (bradykinin receptor), channels (P2X3, TRPV1, ASIC3, and sodium channels), transcription factors (activated transcription factor 3 [ATF3]), and structural molecules (neurofilaments and the sodium channel–anchoring molecule p11) (for review see Pezet and McMahon 2006). Additionally, NGF appears to modulate the trafficking and insertion of sodium channels such as Nav1.8 (Gould et al 2000) and TRPV1 (Ji et al 2002) in sensory neurons, as well as modulate the expression profile of supporting cells in the dorsal root ganglion (DRG) and peripheral nerve, such as non-myelinating Schwann cells and macrophages (Heumann et al 1987a, 1987b; Obata et al 2002).
In light of the potential role that NGF may play in driving bone cancer pain, therapies that block NGF or TrkA have been examined in breast, prostate, and sarcoma models of bone cancer pain. Interestingly, even though prostate cancer cells did not express detectable levels of mRNA coding for NGF (Halvorson et al 2005), in all three models of bone cancer pain, not only was administration of anti-NGF therapy (using an antibody that sequesters extracellular NGF) highly efficacious in reducing both early- and late-stage bone cancer pain–related behavior, but this reduction in pain-related behavior was also greater than that achieved with the acute administration of 10 or 30 mg/kg of morphine sulfate (Halvorson et al 2005, Sevcik et al 2005). If cancer cells do not have to express NGF for anti-NGF to have an analgesic effect, what cells might be synthesizing and releasing NGF? Previous studies have shown that many tumor-associated stromal cells, including macrophages, T lymphocytes, mast cells, and endothelial cells, are capable of expressing and releasing NGF (Vega et al 2003, Pezet and McMahon 2006). Thus, therapies that target NGF or its cognate receptor TrkA may be efficacious in attenuating the pain in other cancers such as ovarian, pancreatic, and head and neck carcinoma, in which a large number of tumor-associated stromal cells express and release NGF.
Therapies targeting ETA receptors, NGF, and TNF-α are currently in human clinical trials in patients with cancer pain (www.clinicaltrials.gov). Whether therapies targeting these and the other aforementioned factors will reach clinical trials and be successful in humans in relieving cancer pain will depend largely on issues related to safety, efficacy, and effects on tumor growth and metastasis. Choosing which type of cancer pain to target in clinical trials is currently a hit-or-miss proposition because there are relatively few models of cancer pain and different types of tumors can have very unique characteristics, as shown by chemotherapeutic agents, which can be highly effective against one cancer and completely ineffective against another. Developing a better understanding of what common and what unique factors drive different cancer pain would be of enormous benefit because it would greatly aid in defining which type of cancer pain will be most likely to respond to targeted analgesic therapies in clinical trials.
Tumor-Induced Nerve Injury and Neuropathic Pain
In both the sarcoma and prostate bone cancer pain models and a model of pancreatic cancer pain, as tumor cells invade normal tissue, the tumor appears to first come into contact, injure, and then destroy the very distal processes of sensory fibers (Peters et al 2005). Thus, although sensory fibers appear to have normal morphology at the leading edge of the tumor, with time the sensory nerve fibers begin to display a discontinuous and fragmented appearance, thus suggesting that following initial activation by the tumor cells, the distal processes of sensory fibers are ultimately injured and destroyed as the invading tumor cells first proliferate and then undergo necrosis as they outgrow the neovascularization that supports them (Peters et al 2005). This initial tumor-induced activation and then injury to sensory nerve fibers are accompanied by an increase in ongoing and movement-evoked pain behavior. Interestingly, there are several changes in the DRG, including hypertrophy of the satellite cells surrounding sensory neuron cell bodies, up-regulation of ATF3, and macrophage infiltration of the DRG, findings that have also been described in other models of peripheral nerve injury and in other non-cancerous neuropathic pain states (Peters et al 2005). These data, as well as the fact that a component of bone cancer pain is attenuated by gabapentin (which is approved for the treatment of neuropathic pain), suggests that a component of cancer pain is neuropathic in origin (Peters et al 2005).
Tumor-Induced Nerve Sprouting and Neuroma Formation
Although tumor-induced injury has been observed in both animals and humans with cancer, an intriguing but largely unexplored mechanism by which cancer pain may be generated is by active and pathological tumor-induced sprouting and neuroma formation. Previous studies in humans and experimental animals have shown that inappropriate sprouting and/or neuroma formation and can lead to a change in the phenotype of sensory and sympathetic nerve fibers, including up-regulation and inappropriate insertion of sodium channels into the distal tips of injured sensory neurons (Devor et al 1993, England et al 1996, Black et al 2008). These newly formed sensory nerve fibers (which sprout in response to peripheral nerve injury) exhibit both spontaneous and movement-evoked ectopic discharges accompanied by a pain that is both severe and difficult to manage medically (Lindqvist et al 2000, Devor 2001, Black et al 2008).
In a mouse model of pancreatic cancer pain (Lindsay et al 2005) and in breast (Bloom et al 2011), prostate (Jimenez-Andrade et al 2010a), and sarcoma (Mantyh et al 2010) models of bone cancer there was first tumor-induced nerve injury and then subsequent sprouting and formation of neuroma-like structures by sensory (Fig. 72-4) and sympathetic nerve fibers. To address what might be driving this ectopic sprouting and neuroma formation, anti-NGF therapy was given. It was found that sustained administration of anti-NGF therapy largely blocked the pathological sprouting of sensory and sympathetic nerve fibers and the formation of neuroma-like structures and significantly inhibited the development of cancer pain in this model (Mantyh et al 2010). Interestingly, injection of canine prostate cancer cells (which do not express detectable levels of mRNA encoding NGF) into the bone of nude mice induces sprouting of CGRP-expressing and NF200-expressing sensory nerve fibers and tyrosine hydroxylase (TH)-positive sympathetic nerve fibers, and nearly all of these sprouting nerve fibers co-express TrkA (Jimenez-Andrade et al 2010a). What is in some ways impressive about these results is the extent of the sprouting; even in bone marrow, which normally receives very modest innervation by sensory nerve fibers, prostate cancer–associated stromal cells can induce up to a 10–70-fold increase in the density of TrkA-positive nerve fibers (Jimenez-Andrade et al 2010a).
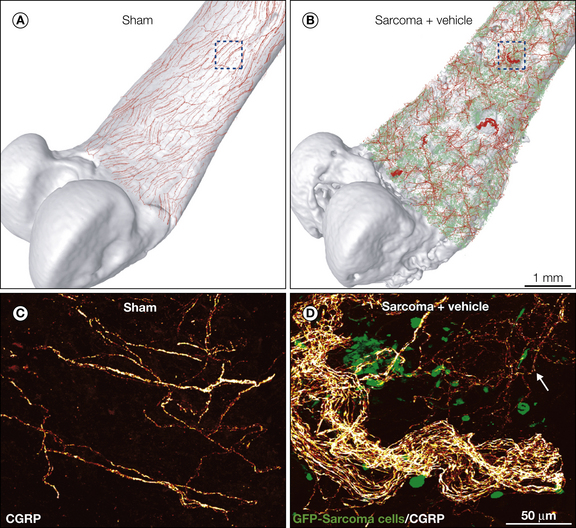
Figure 72-4 Cancer and its associated stromal cells can induce nerve sprouting and neuroma formation in the tumor-bearing organ. Confocal images of the periosteum of the mouse bone were acquired from whole-mount preparations, tiled, and overlaid (to scale) on a three-dimensional micro–computed tomography rendering of a sham femur (A) or sarcoma femur (B), respectively, with use of the Amira software. Note that bone injected with green fluorescent protein (GFP)–cancer cells (in green, B) exhibits significant cortical bone deterioration and pathological reorganization of calcitonin gene–related peptide (CGRP) nerve fibers (in red) when compared with the sham bone (A). The boxed areas in A and B correspond to the confocal images in C and D, respectively. High-power confocal images of non-decalcified, whole-mount preparations of the femoral periosteum from sham (C) or sarcoma mice (D) show CGRP-positive nerve fibers and GFP-positive sarcoma cancer cells. Similar nerve sprouting and formation of neuroma-like structures are also observed in breast and prostate cancer invading bone.
Data from these experiments suggested that a significant proportion of this ectopic sprouting and neuroma formation is driven by NGF. Although it was originally assumed that the majority of the NGF was derived from tumor cells, another study using a tumor cell that did not express NGF showed highly exuberant sprouting, thus suggesting that NGF released from tumor-associated stromal cells could drive this ectopic reorganization of sensory and sympathetic nerve fibers. Interestingly, in both the pancreatic (Lindsay et al 2005) and bone (Jimenez-Andrade et al 2010a, Mantyh et al 2010, Bloom et al 2011) models of cancer pain, sprouting and nerve degeneration were not mutually exclusive, but rather over a period of weeks to months nerve fibers were first injured, then sprouting occurred, and then the nerve fibers were reinjured when the tumor became necrotic because of gradual loss of the vascular supply needed to maintain tumor viability. Since damage to even the distal ends of peripheral nerves can induce neuropathic pain, this process of tumor-induced sprouting and destruction of these newly sprouted sensory and sympathetic fibers has the potential to contribute to both movement-evoked and spontaneous breakthrough cancer pain.
These studies suggest that tyrosine kinase activators, in this case NGF activating TrkA, can induce a remarkable and active reorganization of sensory and sympathetic nerve fibers that may contribute to ongoing and breakthrough pain (Jimenez-Andrade et al 2010a, Bloom et al 2011). Clearly, other tyrosine kinase activators that induce sensitization in tumor tissues in a manner similar to NGF, such as artemin (Elitt et al 2006), G-CSF (Schweizerhof et al 2009), and GM-CSF (Schweizerhof et al 2009), which have been shown to be involved in tumor-induced sensitization, may also play a significant role in promoting tumor-induced sprouting and neuroma formation. Whether similar spouting/neuroma formation occurs in painful cancers such as ovarian, renal, head, and neck cancer has yet to be explored. Previous studies have demonstrated that tyrosine kinase receptor activation can induce sprouting that is both rapid and profuse (Diamond et al 1992). Importantly, tumor cells are constantly proliferating, metastasizing, undergoing necrosis, and then regrowing at new sites. Thus, even if therapies that block NGF or TrkA are given after tumor-induced sprouting and/or neuroma formation has occurred, these therapies will block the newly forming nerve sprouting and neuroma formation (Fig. 72-5). These results emphasize the evolving nature of cancer pain and suggest that the earlier and more effective the analgesic therapy, the greater the likelihood of being able to effectively control both early- and late-stage cancer pain.
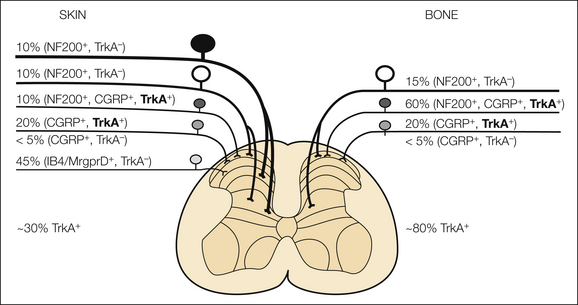
Figure 72-5 The effectiveness of analgesic therapies for different types of cancer may depend on the specific population of nerve fibers that innervate the tumor-bearing organ. This schematic illustrates the percentages and types of sensory nerve fibers that innervate the skin versus bone. The skin is innervated by thickly myelinated Aβ fibers (neurofilament 200 positive [NF200+], TrkA−), thinly myelinated Aδ fibers (NF200+,Trk− and NF200+, calcitonin gene–related peptide positive [CGRP+], TrkA+), unmyelinated peptide-rich C fibers (CGRP+, TrkA+) and unmyelinated peptide-poor C fibers (isolectin B4 positive [IB4+]), and Mas-related G protein–coupled receptor member D (Mrgprd+, TrkA−). In contrast, the bone appears to be predominantly innervated by thinly myelinated Aδ fibers (NF200+, TrkA− and NF200+, CGRP+, TrkA+) and peptide-rich C fibers (CGRP+ and TrkA+). In skin and bone there is also a small proportion (<5% of the total) of unmyelinated C fibers that are CGRP+ and TrkA−. The percentages and types of sensory nerve fibers innervating the skin were estimated with data from previous studies (Bennett et al 1996, Lu et al 2001, Ambalavanar et al 2005, Zylka et al 2005, Nakajima et al 2008, Sugiura et al 2008, Jimenez-Andrade et al 2010b). Note that although approximately 30% of the sensory nerve fibers that innervate skin are TrkA+, more than 80% of the sensory nerve fibers that innervate bone are TrkA+. The fact that a greater percentage of the sensory nerve fibers that innervate bone versus skin are TrkA+ may in part explain why therapies that block nerve growth factor or TrkA show greater efficacy in relieving skeletal than in relieving skin pain. TrkA+ subpopulations of neurons are shown in bold for emphasis.
Central SensiTIzation in Cancer Pain
The majority of what we know about the mechanisms that generate cancer pain has focused on changes in primary afferent sensory nerve fibers and sympathetic fibers that innervate the tumor-bearing organ. However, several studies have demonstrated that animals with cancer pain also exhibit significant pathological changes in the CNS that contribute to the generation and maintenance of cancer pain (for review see Gordon-Williams and Dickenson 2007). Thus, it has been reported that in mice with bone cancer pain there are simultaneous changes in segments of the spinal cord that receive input from nerve fibers innervating the tumor-bearing tissue. These changes include simultaneous alterations in dynorphin, astrocytes (Fig. 72-6), microglia, c-Fos expression, and substance P internalization (Schwei et al 1999). Other reports have demonstrated that in bone cancer models, pain-related behavior is accompanied by increased expression of NR2B, an N-methyl-D-aspartate (NMDA) receptor subunit, and IL-1β released from glial cells and thought to facilitate pain by enhancing phosphorylation of the NR1 subunit of the NMDA receptor (Zhang et al 2008, Gu et al 2010). These latter results suggest that chemical mediators released from glial cells may control the amplitude of synaptic responses in animals bearing bone cancer by changing expression levels of NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and their phosphorylation (Zhang et al 2008, Gu et al 2010).
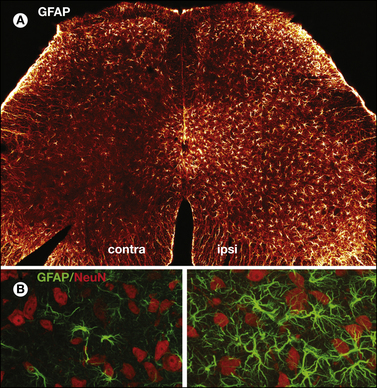
Figure 72-6 Cancer-induced reorganization of the central nervous system.
Confocal images show the increase in the astrocyte marker glial fibrillary acidic protein (GFAP) in a mouse with bone cancer pain in the right femur. Coronal sections of the L4 spinal cord were taken 21 days following the injection of osteolytic sarcoma cells into the intramedullary space of the femur. GFAP is bright orange in A and green in B and C, and NeuN staining (which labels neurons) is red. A low-power image (A) shows that up-regulation of GFAP is almost exclusively ipsilateral to the femur with the intraosseous tumor. A higher magnification of GFAP contralateral (B) and ipsilateral (C) to the femur with cancer shows that on the ipsilateral side there is marked hypertrophy of astrocytes characterized by an increase in both the size of the astrocyte cell bodies and the extent of arborization of their distal processes. Additionally, this increase in GFAP (green) is observed without a detectable loss of neurons because NeuN (red) labeling remains unchanged. These images, from 60 μm-thick tissue, are projected from 6 optical sections acquired at 4-μm intervals with a 20× lens (scale bar = 200 μm) (A) and from 12 optical sections acquired at 0.8-μm intervals with a 60× lens (scale bar = 30 μm) (B and C). (Modified from Schwei MJ, Honore P, Rogers SD, et al 1999 Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. Journal of Neuroscience 19:10886–10897. Copyright 1999 by the Society for Neuroscience.)
The possibility that cancer pain also involves changes in the CNS is supported by a recent study using patch-clamp recordings from spinal cord slices with an attached dorsal root. It was shown that tumor-bearing mice exhibit unique plastic changes in spinal excitatory synaptic transmission mediated through Aδ and C afferent fibers (Yanagisawa et al 2010). In vivo population studies in a rodent model of breast cancer–induced bone cancer pain revealed that in normal animals, the proportion of wide–dynamic range (WDR) to nociceptive-specific neurons in this lamina is 26% WDR to 74% nociceptive specific whereas on establishment of cancer pain, this ratio shifts to 47% WDR to 53% nociceptive specific. This phenotypic shift of the superficial dorsal horn population is accompanied by WDR hyperexcitability to mechanical, thermal, and electrical stimuli in the superficial and deep dorsal horn (Urch et al 2003), which correlates with the development of behavioral signs of pain and further suggests that an ongoing state of central sensitization occurs with bone cancer pain.
Other data suggest that with cancer pain, central sensitization is not confined to the spinal cord, but rather sites in the brain stem that are involved in descending inhibition and facilitation also show clear changes, thus implying that descending controls likewise play a role in the maintenance of cancer-induced bone pain (Gordon-Williams and Dickenson 2007). Together, these studies suggest that cancer pain not only sensitizes primary afferent neurons but also induces significant reorganization within the CNS. Combining preclinical cancer pain studies with brain-imaging studies in human cancer pain patients has the potential not only to provide better understanding of how cancer pain is generated and maintained but also to identify the changes in processing and perception (i.e., discriminative versus affective component) of cancer pain that occur in specific areas of the CNS.
Conclusion
The mechanisms that drive cancer pain clearly evolve and change with disease progression. Cancer cells and their associated stromal cells can generate ongoing and breakthrough pain. This pain appears to be driven in an additive fashion, first by tumor and stromal cells releasing factors that sensitize and activate nociceptors, then by injury to sensory nerve fibers, and finally by releasing growth factors that drive ectopic sprouting of nerve fibers and neuroma formation, all of which can contribute to central sensitization. Developing therapies that target the different mechanisms that contribute to cancer pain has the potential to reduce both ongoing and breakthrough pain. Moving forward, it will be important to develop novel targeted therapies to treat cancer pain and to ensure that relief of cancer pain becomes an integral part of mainstream cancer therapy that is available to all cancer patients in both the developing and developed world.
The references for this chapter can be found at www.expertconsult.com.
References
Ambalavanar R., Moritani M., Dessem D. Trigeminal P2X3 receptor expression differs from dorsal root ganglion and is modulated by deep tissue inflammation. Pain. 2005;117:280–291.
Andratsch M., Mair N., Constantin C.E., et al. A key role for gp130 expressed on peripheral sensory nerves in pathological pain. Journal of Neuroscience. 2009;29:13473–13483.
Arguello F., Baggs R.B., Frantz C.N. A murine model of experimental metastasis to bone and bone marrow. Cancer Research. 1988;48:6876–6881.
Arrington S.A., Schoonmaker J.E., Damron T.A., et al. Temporal changes in bone mass and mechanical properties in a murine model of tumor osteolysis. Bone. 2006;38:359–367.
Bennett D.L., Dmietrieva N., Priestley J.V., et al. TrkA, CGRP and IB4 expression in retrogradely labelled cutaneous and visceral primary sensory neurones in the rat. Neuroscience Letters. 1996;206:33–36.
Bennett G.J. Pathophysiology and animal models of cancer-related painful peripheral neuropathy. Oncologist. 2010;15(Suppl 2):9–12.
Beutler B.A. The role of tumor necrosis factor in health and disease. Journal of Rheumatology. 1999;57(Suppl):16–21.
Black J.A., Nikolajsen L., Kroner K., et al. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Annals of Neurology. 2008;64:644–653.
Bloom A.P., Jimenez-Andrade J.M., Taylor R.N., et al. Breast cancer–induced bone remodeling, skeletal pain and sprouting of sensory nerve fibers. Journal of Pain. 2011;12:698–711.
Boyle P., Levin B., eds. World cancer report. International Agency for Research on Cancer. Lyon: France, 2008.
Casuccio A., Mercadante S., Fulfaro F. Treatment strategies for cancer patients with breakthrough pain. Expert Opinion on Pharmacotherapy. 2009;10:947–953.
Cata J.P., Weng H.R., Lee B.N., et al. Clinical and experimental findings in humans and animals with chemotherapy-induced peripheral neuropathy. Minerva Anestesiologica. 2006;72:151–169.
Clezardin P., Ebetino F.H., Fournier P.G. Bisphosphonates and cancer-induced bone disease: beyond their antiresorptive activity. Cancer Research. 2005;65:4971–4974.
Clohisy D.R., Mantyh P.W. Bone cancer pain and the role of RANKL/OPG. Journal of Musculoskeletal & Neuronal Interactions. 2004;4:293–300.
Clohisy D.R., Perkins S.L., Ramnaraine M.L. Review of cellular mechanisms of tumor osteolysis. Clinical Orthopaedics and Related Research. 2000;373:104–114.
Coleman R.E. Skeletal complications of malignancy. Cancer. 1997;80:1588–1594.
Coleman R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clinical Cancer Research. 2006;12:6243s–6249s.
Constantin C.E., Mair N., Sailer C.A., et al. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. Journal of Neuroscience. 2008;28:5072–5081.
Costantini M., Ripamonti C., Beccaro M., et al. Prevalence, distress, management, and relief of pain during the last 3 months of cancer patients’ life. Results of an Italian mortality follow-back survey. Annals of Oncology. 2009;20:729–735.
Davar G., Hans G., Fareed M.U., et al. Behavioral signs of acute pain produced by application of endothelin-1 to rat sciatic nerve. Neuroreport. 1998;9:2279–2283.
DeLeo J.A., Colburn R.W., Nichols M., et al. Interleukin-6–mediated hyperalgesia/allodynia and increased spinal IL-6 expression in a rat mononeuropathy model. Journal of Interferon & Cytokine Research. 1996;16:695–700.
Desandre P.L., Quest T.E. Management of cancer-related pain. Emergency Medicine Clinics of North America. 2009;27:179–194.
Devor M. Neuropathic pain: what do we do with all these theories? Acta Anaesthesiologica Scandinavica. 2001;45:1121–1127.
Devor M., Govrin-Lippmann R., Angelides K. Na+ channel immunolocalization in peripheral mammalian axons and changes following nerve injury and neuroma formation. Journal of Neuroscience. 1993;13:1976–1992.
Diamond J., Foerster A., Holmes M., et al. Sensory nerves in adult rats regenerate and restore sensory function to the skin independently of endogenous NGF. Journal of Neuroscience. 1992;12:1467–1476.
Dina O.A., Green P.G., Levine J.D. Role of interleukin-6 in chronic muscle hyperalgesic priming. Neuroscience. 2008;152:521–525.
Dreghorn C.R., Newman R.J., Hardy G.J., et al. Primary tumors of the axial skeleton. Experience of the Leeds Regional Bone Tumor Registry. Spine. 1990;15:137–140.
Dy S.M., Asch S.M., Naeim A., et al. Evidence-based standards for cancer pain management. Journal of Clinical Oncology. 2008;26:3879–3885.
Elitt C.M., McIlwrath S.L., Lawson J.J., et al. Artemin overexpression in skin enhances expression of TRPV1 and TRPA1 in cutaneous sensory neurons and leads to behavioral sensitivity to heat and cold. Journal of Neuroscience. 2006;26:8578–8787.
England J.D., Happel L.T., Kline D.G., et al. Sodium channel accumulation in humans with painful neuromas. Neurology. 1996;47:272–276.
Ghilardi J.R., Rohrich H., Lindsay T.H., et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. Journal of Neuroscience. 2005;25:3126–3131.
Gordon-Williams R.M., Dickenson A.H. Central neuronal mechanisms in cancer-induced bone pain. Current Opinion in Supportive and Palliative Care. 2007;1:6–10.
Gould H.J., 3rd., Gould T.N., England J.D., et al. A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Research. 2000;854:19–29.
Griffiths J.R. Are cancer cells acidic? British Journal of Cancer. 1991;64:425–427.
Gu X., Zhang J., Ma Z., et al. The role of N-methyl-D-aspartate receptor subunit NR2B in spinal cord in cancer pain. European Journal of Pain. 2010;14:496–502.
Halvorson K.G., Kubota K., Sevcik M.A., et al. A blocking antibody to nerve growth factor attenuates skeletal pain induced by prostate tumor cells growing in bone. Cancer Research. 2005;65:9426–9435.
Hamamoto D.T., Khasabov S.G., Cain D.M., et al. Tumor-evoked sensitization of C nociceptors: a role for endothelin. Journal of Neurophysiology. 2008;100:2300–2311.
Henry D.H., Costa L., Goldwasser F., et al. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. Journal of Clinical Oncology. 2011;29:1125–1132.
Heumann R., Korsching S., Bandtlow C., et al. Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection. Journal of Cell Biology. 1987;104:1623–1631.
Heumann R., Lindholm D., Bandtlow C., et al. Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration, and regeneration: role of macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:8735–8739.
Honore P., Luger N.M., Sabino M.A., et al. Osteoprotegerin blocks bone cancer–induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nature Medicine. 2000;6:521–528.
Jemal A., Bray F., Center M.M., et al. Global cancer statistics, CA. A Cancer Journal for Clinicians. 2011;61:69–90.
Jemal A., Siegel R., Xu J., et al. Cancer statistics, 2010. CA. A Cancer Journal for Clinicians. 2010;60:277–300.
Ji R.R., Samad T.A., Jin S.X., et al. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68.
Jimenez-Andrade J.M., Bloom A.P., Stake J.I., et al. Pathological sprouting of adult nociceptors in chronic prostate cancer–induced bone pain. Journal of Neuroscience. 2010;30:14649–14656.
Jimenez-Andrade J.M., Mantyh W.G., Bloom A.P., et al. A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bone. 2010;46:306–313.
Jimenez-Andrade J.M., Martin C.D., Koewler N.J., et al. Nerve growth factor sequestering therapy attenuates non-malignant skeletal pain following fracture. Pain. 2007;133:183–196.
Joyce J.A., Pollard J.W. Microenvironmental regulation of metastasis. Nature Reviews. Cancer. 2009;9:239–252.
Julius D., Basbaum A.I. Molecular mechanisms of nociception. Nature. 2001;413:203–210.
Khan N., Afaq F., Mukhtar H. Lifestyle as risk factor for cancer: evidence from human studies. Cancer Letters. 2010;293:133–143.
Khasabova I.A., Khasabov S.G., Harding-Rose C., et al. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. Journal of Neuroscience. 2008;28:11141–11152.
Lam D.K., Schmidt B.L. Orofacial pain onset predicts transition to head and neck cancer. Pain. 2011;152:1206–1209.
Lindqvist A., Rivero-Melian C., Turan I., et al. Neuropeptide- and tyrosine hydroxylase–immunoreactive nerve fibers in painful Morton’s neuromas. Muscle & Nerve. 2000;23:1214–1218.
Lindsay T.H., Jonas B.M., Sevcik M.A., et al. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. Pain. 2005;119:233–246.
Lipton A. Emerging role of bisphosphonates in the clinic—antitumor activity and prevention of metastasis to bone. Cancer Treatment Reviews. 2008;34(Suppl 1):S25–S30.
Lipton A., Jun S. RANKL inhibition in the treatment of bone metastases. Current Opinion in Supportive and Palliative Care. 2008;2:197–203.
Lossignol D.A., Dumitrescu C. Breakthrough pain: progress in management. Current Opinion in Oncology. 2010;22:302–306.
Lozano-Ondoua A.N., Wright C., Vardanyan A., et al. A cannabinoid 2 receptor agonist attenuates bone cancer–induced pain and bone loss. Life Sciences. 2010;86:646–653.
Lu J., Zhou X.F., Rush R.A. Small primary sensory neurons innervating epidermis and viscera display differential phenotype in the adult rat. Neuroscience Research. 2001;41:355–363.
Mantyh P.W. Cancer pain and its impact on diagnosis, survival and quality of life. Nature Reviews. Neuroscience. 2006;7:797–809.
Mantyh W.G., Jimenez-Andrade J.M., Stake J.I., et al. Blockade of nerve sprouting and neuroma formation markedly attenuates the development of late stage cancer pain. Neuroscience. 2010;171:588–598.
Mercadante S. Malignant bone pain: pathophysiology and treatment. Pain. 1997;69:1–18.
Mercadante S., Villari P., Ferrera P., et al. Optimization of opioid therapy for preventing incident pain associated with bone metastases. Journal of Pain and Symptom Management. 2004;28:505–510.
Mielke S., Sparreboom A., Mross K. Peripheral neuropathy: a persisting challenge in paclitaxel-based regimes. European Journal of Cancer. 2006;42:24–30.
Montagnini M.L., Zaleon C.R. Pharmacological management of cancer pain. Journal of Opioid Management. 2009;5:89–96.
Nakajima T., Ohtori S., Yamamoto S., et al. Differences in innervation and innervated neurons between hip and inguinal skin. Clinical Orthopaedics and Related Research. 2008;466:2527–2532.
Nelson J.B., Chan-Tack K., Hedican S.P., et al. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Research. 1996;56:663–668.
Nelson J.B., Hedican S.P., George D.J., et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nature Medicine. 1995;1:944–949.
Nishimoto N., Kishimoto T. Interleukin 6: from bench to bedside. Nature Clinical Practice. Rheumatology. 2006;2:619–626.
Obata K., Tsujino H., Yamanaka H., et al. Expression of neurotrophic factors in the dorsal root ganglion in a rat model of lumbar disc herniation. Pain. 2002;99:121–132.
Peters C.M., Ghilardi J.R., Keyser C.P., et al. Tumor-induced injury of primary afferent sensory nerve fibers in bone cancer pain. Experimental Neurology. 2005;193:85–100.
Pezet S., McMahon S.B. Neurotrophins: mediators and modulators of pain. Annual Review of Neuroscience. 2006;29:507–538.
Pomonis J.D., Rogers S.D., Peters C.M., et al. Localization of endothelin receptors in peripheral nerve indicates roles for non-neuronal cells in nociceptive signalling. Society for Neuroscience. 2001. Program Number 57.6
Poole S., Cunha F.Q., Selkirk S., et al. Cytokine-mediated inflammatory hyperalgesia limited by interleukin-10. British Journal of Pharmacology. 1995;115:684–688.
Quasthoff S., Hartung H.P. Chemotherapy-induced peripheral neuropathy. Journal of Neurology. 2002;249:9–17.
Rogers M.J., Gordon S., Benford H.L., et al. Cellular and molecular mechanisms of action of bisphosphonates. Cancer. 2000;88:2961–2978.
Rose-John S., Heinrich P.C. Soluble receptors for cytokines and growth factors: generation and biological function. Biochemical Journal. 1994;300:281–290.
Rose-John S., Scheller J., Elson G., et al. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. Journal of Leukocyte Biology. 2006;80:227–236.
Rubert C.H.R., Malawer M. Orthopedic management of skeletal metastases. In: Body J.-J., ed. Tumor bone disease and osteoporosis in cancer patients. New York: Marcel Dekker; 2000:305–356.
Sabino M., Luger N., Mach D., et al. Different tumors in bone each give rise to a distinct pattern of skeletal destruction, bone cancer–related pain behaviors and neurochemical changes in the central nervous system. International Journal of Cancer. 2003;104:550–558.
Sabino M.A., Ghilardi J.R., Jongen J.L., et al. Simultaneous reduction in cancer pain, bone destruction, and tumor growth by selective inhibition of cyclooxygenase-2. Cancer Research. 2002;62:7343–7349.
Schwei M.J., Honore P., Rogers S.D., et al. Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. Journal of Neuroscience. 1999;19:10886–10897.
Schweizerhof M., Stosser S., Kurejova M., et al. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nature Medicine. 2009;15:802–807.
Sevcik M.A., Ghilardi J.R., Peters C.M., et al. Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain. 2005;115:128–141.
Smith P.C., Hobisch A., Lin D.L., et al. Interleukin-6 and prostate cancer progression. Cytokine & Growth Factor Reviews. 2001;12:33–40.
Stenbeck G. Formation and function of the ruffled border in osteoclasts. Seminars in Cell & Developmental Biology. 2002;13:285–292.
Stopeck A.T., Lipton A., Body J.J., et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. Journal of Clinical Oncology. 2010;28:5132–5139.
Sugiura A., Ohtori S., Yamashita M., et al. Existence of nerve growth factor receptors, tyrosine kinase A and p75 neurotrophin receptors in intervertebral discs and on dorsal root ganglion neurons innervating intervertebral discs in rats. Spine. 2008;33:2047–2051.
Taga T., Hibi M., Hirata Y., et al. Interleukin-6 receptor and a unique mechanism of its signal transduction. Cold Spring Harbor Symposia on Quantitative Biology. 1989;54(Pt 2):713–722.
Urch C.E., Donovan-Rodriguez T., Dickenson A.H. Alterations in dorsal horn neurones in a rat model of cancer-induced bone pain. Pain. 2003;106:347–356.
van den Beuken-van Everdingen M.H., de Rijke J.M., Kessels A.G., et al. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Annals of Oncology. 2007;18:1437–1449.
Vega J.A., Garcia-Suarez O., Hannestad J., et al. Neurotrophins and the immune system. Journal of Anatomy. 2003;203:1–19.
von Moos R., Strasser F., Gillessen S., et al. Metastatic bone pain: treatment options with an emphasis on bisphosphonates. Supportive Care in Cancer. 2008;16:1105–1115.
Wacnik P.W., Eikmeier L.J., Simone D.A., et al. Nociceptive characteristics of tumor necrosis factor-alpha in naive and tumor-bearing mice. Neuroscience. 2005;132:479–491.
Yanagisawa Y., Furue H., Kawamata T., et al. Bone cancer induces a unique central sensitization through synaptic changes in a wide area of the spinal cord. Molecular Pain. 2010;6:38.
Yates D., Smith M. Orthopaedic pain after trauma. In: Wall P.D., Melzack R., eds. Textbook of pain. Edinburgh: Churchill Livingstone; 1994:409–421.
Yoneda T., Sasaki A., Mundy G.R. Osteolytic bone metastasis in breast cancer. Breast Cancer Research and Treatment. 1994;32:73–84.
Zhang R.X., Liu B., Li A., et al. Interleukin 1beta facilitates bone cancer pain in rats by enhancing NMDA receptor NR-1 subunit phosphorylation. Neuroscience. 2008;154:1533–1538.
Zhu Z.W., Friess H., diMola F.F., et al. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. Journal of Clinical Oncology. 1999;17:2419–2428.
Zylka M.J., Rice F.L., Anderson D.J. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45:17–25.