Inflammatory Mediators and Modulators of Pain
Introduction
A long-standing interest for pain scientists has been the identification of chemical mediators released into injured or diseased tissues that are responsible for the abnormal pain states associated with these disorders. For some time, attention was focused on a small number of molecules such as prostaglandins and bradykinin. These factors were known to be produced as a result of tissue damage or inflammation and were thought to be responsible for activation and sensitization of peripheral pain-signaling sensory neurons; that is, they were seen as the principal peripheral pain mediators. During the past decade or so, evidence has emerged for many novel pain mediators. The old ones have not disappeared, although their roles have been redefined in some cases. Prostaglandin E2 (PGE2), for instance, is now recognized as playing a prominent role in central nervous system (CNS) as well as peripheral tissues. The newly identified mediators include a variety of factors produced and released from non-neuronal cells, often immune and glial cells. There is now a rapidly expanding evidence base that these are important mediators of persistent pain states and can act at a number of loci.
This chapter focuses on the cellular characteristics of nociceptive afferent neurons, their ion channels, and their signal transduction pathways and discusses the ways in which inflammatory mediators impinge on these basic properties. In particular, we first review the cellular mechanisms of activation and sensitization of nociceptors. Then we discuss the roles and actions of particular immune cells and specific pain mediators, starting with a group of small molecules often rapidly released into damaged tissue. We conclude with a review of the actions of another group of peripheral pain mediators and modulators: the pro-inflammatory cytokines, some chemokines, and some neurotrophic factors, which in addition to their traditionally recognized roles, are all capable of changing the response properties of pain-signaling neurons. The topic of neuro-immune interactions within the CNS is considered in Chapter 4.
Overview Of Inflammatory Mediator Actions
A large number of endogenously generated factors produce pain when injected into peripheral tissue. Many of these substances can also sensitize nociceptors. That is, they reduce the threshold for activation of nociceptors by one or more stimulus modalities and/or increase the responsiveness of nociceptors to suprathreshold stimulation. This process of sensitization is recognized as being of critical importance in many chronic pain states; it is precisely this aberrant excitability of nociceptors that causes a large part of the sensory abnormality. Some features of the sensitization process are described in Chapter 1. Here we first review the cellular mechanisms by which sensitization occurs.
Receptors and Effectors
Sensory nerves express a variety of receptors for inflammatory mediators. Different classes of nociceptors express distinct patterns of receptors. The receptors fall into three main classes: G protein–coupled receptors (GPCRs), ligand-gated ion channels, and the cytokine receptors or receptor tyrosine kinases (Fig. 3-1).
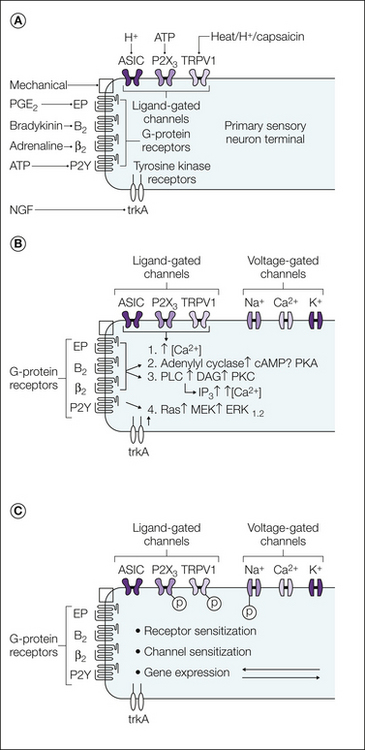
Figure 3-1 Peripheral sensitization of nociceptive neurons.
A, Some of the different stimuli (and the receptors that they act on) that can lead to activation and sensitization of the peripheral terminals of nociceptive neurons. B and C show the main effector mechanisms and second-messenger cascades underlying sensitization, respectively. ASIC, acid-sensing ion channel; DAG, diacylglycerol; ERK, extracellular signal–regulated kinase; IP3, inositol triphosphate; MEK, mitogen-activated protein/ERK kinase; NGF, nerve growth factor; PGE2, prostaglandin E2; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; TRPV1, transient receptor potential vanilloid 1. Many changes are effected by phosphorylation of receptors or channels (P).
G Protein–Coupled Receptors
Many mediators produced during inflammation, such as bradykinin, serotonin, prostaglandins, and chemokines, act via GPCRs. These receptors elicit a specific biochemical response that depends on the type of G protein that is activated. Activation of Gs stimulates adenylate cyclase to raise the level of cyclic adenosine monophosphate (cAMP) and activate protein kinase A (PKA) in the neuron, whereas Gi inhibits the activity of adenylate cyclase to lower cAMP levels. Although many cAMP effects are mediated by PKA, other mechanisms may be operative. For example, cAMP can activate Epac (exchange protein directly activated by cAMP), a guanine nucleotide exchange factor, which leads to activation of the ε isoform of protein kinase C (PKC-ε). Stimulation of Gq/11 activates phospholipases, notably phospholipase C (PLC), which generates inositol triphosphate (IP3) and diacylglycerol (DAG) from the membrane lipid precursor phosphatidylinositol 4,5-bisphosphate (PIP2). Gq activation can also stimulate PLA2, which cleaves membrane phospholipids at the sn-2 position to produce the prostaglandin precursor arachidonic acid. G-protein control of cellular function can also involve direct action of βγ subunits on ion channels and enzymes, such as PLC (see Smrcka 2008, Zylbergold et al 2010).
Ion Channels
Some inflammatory mediators act by directly gating the ion channels expressed by sensory neurons. Notable examples in this class are adenosine triphosphate (ATP; acting via P2X channels), protons (acting via acid-sensing ion channels [ASICs] and transient receptor potential vanilloid 1 [TRPV1]), and the lipid activators of TRPV1. All these ion channels are cation selective and are permeable to either sodium ions or both monovalent and divalent cations. In all cases the ion flow evoked by channel opening depolarizes the sensory neurons and leads to neuronal firing.
Receptor Tyrosine Kinases
The third general type of receptor includes cytokine receptors activated by mediators such as interleukin-1 (IL-1) or tumor necrosis factor-α (TNF-α) and the receptor tyrosine kinases for neurotrophic factors, such as the receptors for nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), glial cell line–derived neurotrophic factor (GDNF), and artemin. Both classes of receptors have monomers derived from a single transmembrane segment with a large extracellular ligand-binding domain. The cytosolic domain of receptor tyrosine kinases contains an intrinsic protein tyrosine kinase catalytic site, whereas the cytosolic domain of cytokine receptors is generally associated with a separate protein kinase that is recruited to the complex either directly or via adapter proteins. The functional receptors are either dimers or trimers, which either exist normally or are formed by cross-linking of adjacent monomers by the ligand. In either case, ligand binding activates kinase pathways that affect gene transcription and can also elicit acute effects on neuronal function.
Nitric Oxide and Cyclic Guanosine Monophosphate
In addition to receptor-mediated signaling, cells also signal via nitric oxide (NO). NO is an important intercellular mediator and is produced by many cells that have close physical association with neurons both in the periphery and within the spinal cord. NO is formed from L-arginine following activation of the enzyme nitric oxide synthase (NOS) by calcium and other co-factors, including calmodulin. Three forms of NOS have been identified: neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS), each with a distinct physiological role. nNOS and eNOS are both Ca2+/calmodulin dependent and are present in both the spinal cord and brain, whereas iNOS is functionally Ca2+ independent and normally expressed in macrophages, inflammatory cells, and glia (for review see Benarroch 2011). NO diffuses to its site of action, where it stimulates guanylate cyclase to produce cyclic guanosine monophosphate (cGMP). In turn, cGMP modifies intracellular processes, including activation of protein kinases, ion channels, and phosphodiesterases. NO can also act in other ways, for example, by activating cyclooxygenase (COX) enzymes and by S-nitrosylation of proteins (Tegeder et al 2011).
Intracellular Signaling Pathways
Sensory nerves are activated and sensitized by inflammatory mediators in several ways (see Fig. 3-1B). Some mediators directly activate cation channels and thus depolarize neurons toward the voltage for initiation of an action potential. Other receptors activate intracellular pathways and influence neuronal sensitivity and excitability indirectly. These mechanisms include GPCR-mediated production of the second-messenger molecules NO, COX, and lipoxygenase products of arachidonic acid. Phosphorylation or dephosphorylation of membrane proteins often regulates the transduction and transmission of sensory signals (Fig. 3-1C), and this can occur via PKA-, PKC-, mitogen-activated protein kinase (MAPK)-, or phosphatidylinositol-3′-kinase/Akt-mediated phosphorylation or by dephosphorylation via protein phosphatases such as calcineurin. In addition to phosphorylation, some of the mediators that act on nociceptors can stimulate biochemical processes such as methylation and lipid modification of proteins, and these pathways may be important in nociceptive neurons.
In general, the effect of sensitization is to increase the probability that a given stimulus (ligand or voltage) will activate the target receptor or ion channel or increase the probability that the neuron will be excited. Protein phosphorylation is a well-known mechanism for controlling the activity of ion channels. For example, activity of the heat-sensitive ion channel TRPV1 is modified by both PKC- and PKA-mediated phosphorylation (Bhave et al 2003, Mohaptra and Nau 2005), and the level of membrane expression is regulated by src-mediated phosphorylation (Zhang et al 2005b). Control of transduction channel activity can also be regulated by hydrolysis of PIP2 and removal of the tonic inhibition caused by PIP2 binding to the ion channel (see, e.g., Dai et al 2007). Ion channels that control the excitability and firing frequency of sensory neurons are also substrates for regulation by PIP2 (Suh and Hille 2008) and phosphorylation (Gold 1999, Baker 2005, Beyak and Vanner 2005, Stamboulian et al 2010, Emery et al 2011).
Neuronal sensitization can occur through changes in the level of protein expression, either by transcriptional control altering the production of proteins or by changing the trafficking such that an altered amount of the protein is functionally expressed. Transcriptional control is an important long-term mechanism underlying the effects of neurotrophin receptor activation. In some cases, sensitization has been associated with the de novo expression of molecules important for nociception in neurons that do not normally express the protein (Hudson et al 2001, Vellani et al 2004).
Specific Pain Mediators
There is a considerable body of evidence that kinins contribute to the pathophysiological processes accompanying both acute and chronic inflammation. Bradykinin and the related peptide kallidin (Lys0-bradykinin) are formed from kininogen precursor proteins following the activation of plasma or tissue kallikrein enzymes during inflammation, tissue damage, or anoxia. The activity of these kinins is terminated by several degradative enzymes. Kininase I liberates the biologically active metabolites des-Arg9-bradykinin and des-Arg10-kallidin, whereas kininase II and endopeptidases form inactive metabolites (Calixto et al 2000, Marceau and Regoli 2004). The biologically active kinins activate two distinct types of G protein–linked receptors. Bradykinin and kallidin act preferentially at the B2 receptor, whereas des-Arg9-bradykinin and des-Arg10-kallidin act with much higher affinity at the B1 receptor than at the B2 receptor.
B2 receptors are expressed constitutively on a wide range of cell types, including nociceptive sensory nerves, and administration of bradykinin evokes pain and sensitizes polymodal nociceptors (see Mizumura et al 2009). Bradykinin acts directly on sensory nerves and can also act indirectly by evoking the release of other inflammatory mediators from non-neuronal cells. There is good pharmacological evidence that the acute and some of the long-term effects of bradykinin are mediated via the B2 receptor. For example, peptide and non-peptide B2 receptor antagonists have analgesic and anti-hyperalgesic actions in animal models of inflammatory pain (Dray and Perkins 1993; Perkins and Kelly 1993, 1994; Asano et al 1997; Burgess et al 2000; Cuhna et al 2007; Valenti et al 2010), as well as in some neuropathic pain models (Werner et al 2007, Luiz et al 2010). Interestingly, thermal hypersensitivity is still evoked by complete Freund’s adjuvant (CFA)-induced inflammation in mice lacking the B2 receptor (Boyce et al 1996, Rupniak et al 1997, Ferreira et al 2001), but carrageenan-evoked thermal hypersensitivity is reduced (Boyce et al 1996, Rupniak et al 1997).
In contrast to B2 receptors, B1 receptors are not normally expressed at significant levels in normal tissue, except in some vascular beds, but their expression is induced by tissue injury and infection. This up-regulation of B1 receptors requires de novo protein synthesis (Regoli et al 1978, Bouthillier et al 1987, DeBlois et al 1991), and there is evidence that the induction is stimulated by the release of cytokines such as IL-1β and TNF-α from immunocompetent cells in the damaged tissue (Calixto et al 2004, Cuhna et al 2007). Some effects of B1 agonists are mediated via non-neuronal cells, where activation of the B1 receptor evokes the release of PGE2 and PGI2, NO, and various cytokines (Leeb-Lundberg et al 2005, Kuhr et al 2010). There is also immunocytochemical and autoradiographic evidence that the B1 receptor is expressed in a subset of sensory neurons (Wotherspoon and Winter 2000, Ma 2001, Petcu et al 2008) and that the level of expression is increased during inflammation (Fox et al 2003). The mechanisms regulating expression of the B1 receptor in sensory neurons are not well understood but are likely to involve cytokines, as found in other cell types, and neurotrophins. Functional expression of sensory neuron B1 receptors is up-regulated by exposure to the neurotrophins GDNF and neurturin. Under such conditions, B1 receptor activation evokes sustained enhancement of the heat-gated current mediated by TRPV1 (Vellani et al 2004).
There is good pharmacological evidence that B1 receptors have an important role in the hyperalgesia associated with persistent inflammation. Although B1 agonists do not normally affect nociceptive thresholds in animals, they evoke hyperalgesia following inflammation (Davis and Perkins 1994, Perkins and Kelly 1994, Fox et al 2003). Furthermore, peptide B1 antagonists such as des-Arg10-HOE140 and des-Arg8Leu9-bradykinin (Perkins and Kelly 1993, Perkins et al 1993, Campos and Calixto 1995, Rupniak et al 1997, Fox et al 2003), as well as non-peptide B1 antagonists (Fox et al 2005, Hawkinson et al 2007), inhibit thermal or mechanical hyperalgesia in models of joint, paw, or tail inflammation. These data are consistent with the finding that mice lacking the B1 receptor show reduced thermal (Ferreira et al 2001) and mechanical (Fox et al 2005) hyperalgesia after CFA treatment.
The relative importance of the changes in subtypes of bradykinin receptors is variable and depends on the inflammatory condition, with evidence of a shift toward a dominant role of B1 receptors in chronic conditions in which B1 receptor expression is up-regulated (see, e.g., Cuhna et al 2007). Although many studies have focused on the peripheral role of kinin receptors, there is also evidence from studies involving selective antagonists and knockout mice that B1 and B2 receptors expressed in the spinal cord influence spinal processing of nociceptive signals in inflammatory conditions (Pesquero et al 2000; Ferriera et al 2001, 2002).
Bradykinin Receptor Signaling
B1 and B2 receptors couple through Gqα to stimulate PLC, which results in phosphoinositide hydrolysis, DAG production, and mobilization of intracellular Ca2+ from intracellular stores. They can also act through Giα to inhibit adenylate cyclase and stimulate the MAPK pathways (Leeb-Lundberg et al 2005, Cheng and Ji 2008). A significant body of evidence supports the idea that bradykinin activates sensory neurons via a DAG–PKC pathway. Bradykinin causes the translocation of a specific PKC isoform, PKC-ε, from the cytoplasm to the plasma membrane of dorsal root ganglion (DRG) neurons (Cesare et al 1999), and the excitatory effects of bradykinin are inhibited by the PKC inhibitor staurosporine (Burgess et al 1989), which also attenuates the responses of skin afferents (Dray et al 1992). Furthermore, the bradykinin responses of many, but not all, neurons are reduced or abolished when PKC activity is down-regulated by prolonged exposure to phorbol esters (Rang and Ritchie 1988, Burgess et al 1989).
PKC activators depolarize sensory neurons by opening a cation-permeable ion channel (Burgess et al 1989, McGehee and Oxford 1991), and several pieces of information indicate that bradykinin exerts its effects, in part, by sensitizing or opening the heat-sensitive TRPV1 ion channel. Bradykinin activates ion channels in DRG neurons with properties similar to those of TRPV1 channels (Premkumar and Ahern 2000); this agonistic effect requires the presence of PKC-ε and is blocked by PKC inhibitors (Cesare et al 1999, Premkumar and Ahern 2000). Bradykinin also increases the capsaicin sensitivity of TRPV1 and reduces the temperature threshold for activation from approximately 42°C toward or below normal body temperature via a PKC mechanism (Vellani et al 2001, Sugiura et al 2002).
Activation of TRPV1 cannot explain all the excitatory effects of bradykinin inasmuch as activation of vagal and visceral afferents by bradykinin is retained in TRPV1 knockout mice (Kollarik and Undem 2004, Rong et al 2004) and bradykinin can stimulate DRG neurons from TRPV1−/− mice (Katanosaka et al 2008). Bradykinin can also act via PLC to activate TRPA1 (Bandell et al 2004), and bradykinin-evoked responses were significantly attenuated in sensory neurons from both TRPV1 and TRPA1 knockout mice (Bautista et al 2006). One possibility is that TRPV1 and TRPA1 act in concert. In this scenario (Bautista et al 2006), activation of PLC evokes TRPV1 gating and calcium influx. Because TRPA1 is often co-expressed with TRPV1 and because TRPA1 can be activated by increases in the intracellular calcium concentration (Doerner et al 2007, Zuborg et al 2007), a small calcium influx through TRPV1 may activate TRPA1.
Failure to inhibit bradykinin responses in all sensory neurons with staurosporine or prolonged exposure to phorbol esters (Burgess et al 1989, Rang and Ritchie 1988) suggests that excitation can be mediated by a PKC-independent mechanism. Other evidence points to different phospholipase-linked mechanisms resulting in activation of TRPV1. One proposal is that binding of PIP2 to TRPV1 inhibits channel activity (Prescott and Julius 2003) and its hydrolysis by B2 receptor–mediated activation of PLC potentiates channel opening by removing this tonic inhibition (Chuang et al 2001). However, the inhibitory influence of PIP2 on TRPV1 has been challenged, and there is evidence that PIP2 binding potentiates rather than inhibits TRPV1 (Klein et al 2008, Yao and Qin 2009, Sowa et al 2010). Phosphoinositide binding may have both inhibitory and potentiating effects on TRPV1, depending on the level of stimulation (Lukacs et al 2007).
B2 receptor activation also stimulates the 12-lipoxygenase pathway and leads to the production of endogenous TRPV1 agonists (e.g., 12-hydroperoxyarachidonate [HPETE] and leukotriene B4 [LTB4]. Bradykinin-evoked activation of TRPV1-like currents, neuronal firing, and behavioral responses are blocked by lipoxygenase inhibitors, consistent with a contribution of this pathway (Shin et al 2002, Carr et al 2003, Calixto et al 2004, Wu and Pan 2007). Other data point to a role of COX products since the COX inhibitor flurbiprofen inhibits the heat sensitization induced by bradykinin in a skin–nerve preparation (Petho et al 2001).
Two other ionic mechanisms have recently been proposed for bradykinin-evoked activation of DRG neurons. Depolarization resulting from inhibition of M-type potassium currents and activation of a calcium-activated chloride current, encoded by TMEM16A, have been proposed as important PLC-linked mechanisms for the excitatory actions of bradykinin (Liu et al 2010).
Arachidonic Acid Metabolites
The enzymatic breakdown of arachidonic acid yields a variety of bioactive lipid molecules that have diverse physiological roles, including important actions in inflammation and pain. These molecules are not stored but are synthesized de novo from membrane lipids. The first step is release of arachidonic acid from phospholipids by the action of PLA2 enzymes. Arachidonic acid is then metabolized to prostaglandins via the COX enzymes; to leukotrienes, 5-HPETE, and 5-hydroxyeicosatetraenoic acid (HETE) via 5-lipoxygenase; to 12-HPETE and 12-HETE via 12-lipoxygenase; to lipoxins via 15-lipoxygenase; and to epoxyeicosatetraenoic acids via the action of cytochrome P450.
Prostaglandins
Non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit COX enzymes, are the most widely used and effective drugs for the clinical treatment of inflammatory pain and hyperalgesia. NSAIDs have no obvious effect on normal pain thresholds but attenuate the abnormal pain responses in inflammatory conditions. Two COX enzymes, COX-1 and COX-2, are responsible for the first steps in prostaglandin synthesis. These enzymes have two catalytic enzymatic activities: a COX activity responsible for the production of PGG2 from arachidonic acid and a peroxidase activity that reduces PGG2 to form PGH2, the first steps in prostanoid biosynthesis.
In general, COX-1 is considered to have a “housekeeping” role in almost all tissues mediating physiological responses. In contrast, COX-2 is not constitutively expressed (except in the kidney, vas deferens, and importantly, the brain) but is induced in inflammatory conditions. In the periphery, COX-2 expression is induced in cells involved in inflammation (macrophages, monocytes, and synoviocytes) and is primarily responsible for synthesis of the prostaglandins involved in acute and chronic inflammatory states. COX-2 expression is induced in peripheral tissues in animal models of arthritis, and up-regulated expression is seen in human rheumatoid arthritic joints, although relatively little expression has been noted in human osteoarthritic joints. Both COX-1 and COX-2 are expressed constitutively in DRG neurons and in the spinal cord. Normally, COX-1 is expressed in small and medium-sized DRG neurons and in neurons and astrocytes in the spinal cord. Enzyme expression in both neuronal and non-neuronal cells in the spinal cord is up-regulated after peripheral inflammation and nerve injury (see Samad et al 2002, Svensson and Yaksh 2002), and intraspinal release of PGE2 is enhanced during peripheral inflammation (Yang et al 1996, Ebersberger et al 1999).
The important roles of spinal cord COX enzymes are not discussed in detail here but are covered in Chapter 28. The available information indicates that COX inhibition at both peripheral and central sites can contribute to the anti-hyperalgesic effects, with the predominant clinical effect being mediated centrally. Certainly, prostaglandins produced in the periphery after tissue injury can sensitize peripheral nerves and induce hyperalgesia in animal models of inflammation, thus suggesting that a component of hyperalgesia could be due to a peripheral action. However, the finding that intrathecal administration of COX-2–selective inhibitors suppresses experimentally induced inflammatory hyperalgesia also argues for a central site of action (Samad et al 2001). The observations that COX-2 inhibitors have clinical efficacy similar to that of non-selective NSAIDs and that COX-2 inhibitors exert a rapid effect after surgery also argue that they act in these conditions at central sites where COX-2 is constitutively expressed.
PGH2 is metabolized by different prostaglandin synthetases to a range of prostaglandins. Prostaglandins such as PGE2, PGD2, and PGI2 are produced during inflammation and act with some specificity on different prostanoid receptors, termed EP, DP, and IP, respectively. Each of the prostanoid receptors has distinct coupling to G proteins, and the pattern of coupling determines the biochemical consequence of receptor activation. Four major types of EP receptors (EP1–4) have been described, and splice variants of the EP3 subclass have also been identified, which probably explains the multiplicity of transduction pathways that have been associated with this receptor. In situ hybridization studies have shown the presence of mRNA for IP, EP1, EP3, and EP4 receptors in DRG neurons. About half the neurons express EP3 receptor mRNA; 40%, IP mRNA; 30%, EP1 mRNA; and 20%, EP4 mRNA, with some degree of co-expression (Sugimoto et al 1994, Oida et al 1995). Of these, EP1, EP4, IP, and some splice variants of EP3 receptors (EP3B and EP3C) couple positively via Gs to stimulate adenylate cyclase and raise cAMP levels.
A major peripheral effect of PGE2 and PGI2 is to sensitize afferent neurons to noxious chemical, thermal, and mechanical stimuli (see, for example, Mizumura et al 1987, Schaible and Schmidt 1988, Birrell et al 1991). In contrast, PGD2 shows little or no such activity (Rueff and Dray 1992). The importance of these receptor subtypes in the periphery is confirmed by the findings that EP3−/− and IP−/− mice show reduced hyperalgesia after lipopolysaccharide (LPS) administration (Ueno et al 2001). In contrast, intrathecal administration of PGE2 induced normal mechanical allodynia in wild-type and EP3−/− mice but not in EP1−/− mice, thus illustrating that the EP1 receptor plays a role in prostaglandin-induced spinal sensitization (Minami et al 2001).
Lipoxygenase Products
The potential role of lipoxygenase products in inflammatory pain is less clear, and although the levels are increased in inflammatory conditions, evidence of a direct role in nociception is lacking. The major effect of these lipids is to recruit immune cells and alter microvascular permeability. Intradermal injection of LTB4 or 8R,15S-diHETE decreases mechanical and thermal thresholds in rats (Levine et al 1984, 1985, 1986a; Martin et al 1987; Martin 1990) and humans (Bisgaard and Kristensen 1985), and LTB4 sensitizes dental afferents (Madison et al 1992). The sensitizing actions of LTB4 require the presence of polymorphonuclear (PMN) leukocytes and are thus likely to be indirect (Levine et al 1984, 1985). 8R,15S-diHETE reduces the thermal and mechanical thresholds of C fibers (Taiwo et al 1989, White et al 1990) and excites some C-fiber neuromas (Devor et al 1992). A role of LTB4 in experimental antigen (ovalbumin)-induced mechanical hyperalgesia has been shown by using the LTB4 antagonist CP10596 (Cunha et al 2003). More recently, the cysteinyl-leukotriene receptor CysLT2 was found to be expressed in about 40% of rat DRG neurons, preferentially in small-diameter neurons. Intraplantar administration of the CysLT2 agonist LTC4 strongly enhanced the nocifensive response evoked by the P2X3 agonist αβ-me-ATP but was without effect on thermal sensitivity, thus suggesting a lack of effect on TRPV1 channels (Okubo et al 2010).
One probable action for some lipoxygenase products is to activate TRPV1 channels inasmuch as 12S-HPETE, 15S-HPETE, 5S-HETE, and LTB4 all open TRPV1 channels in DRG neurons (Hwang et al 2000). The behavioral effects of 8R,15S-diHETE noted earlier are unlikely to be due to such an action since this lipid shows very weak agonist effects on TRPV1.
Other Fatty Acid Metabolites
In addition to the leukotrienes, lipoxygenases can also convert eicosapentaenoic and docosahexaenoic acids into active signaling molecules. Formation of some of these metabolites requires the sequential action of COX-2 or cytochrome P450 followed by lipoxygenase-mediated oxidation (Bannenberg and Serhan 2010). The resulting molecules have been named resolvins because of the roles that they are thought to play in the resolution phase of inflammation, and they have attracted interest for their analgesic potential (Ji et al 2011). The resolvins RvE1 and RvD1 potently reduce thermal and mechanical hypersensitivity in inflammatory pain models. Resolvins produce these effects by stimulating Gi/o-coupled GPCRs located both on DRG neurons and in the spinal cord, thereby effectively inhibiting the activity of the sensory neuron ion channels TRPA1 and TRPV1, as well as C-fiber evoked long-term potentiation in the spinal cord (Xu et al 2010, Park et al 2011).
Linoleic acid is converted into several hydroxyl and carbonyl derivatives (9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE) by both lipoxygenase pathways and non-enzymatic lipid peroxidation reactions. In experimental situations, formation of these mediators is increased by depolarization of the spinal cord with a high-K+ solution (Patwardhan et al 2009). Extended exposure to heat also significantly increases the tissue concentration of these oxidized linoleic acid metabolites in mouse skin biopsy samples. Application of 9-HODE to cultured trigeminal neurons stimulates TRPV1, and administration in vivo evokes nocifensive behavior and thermal hypersensitivity, which is absent in Trpv1−/− mice, thereby demonstrating that TRPV1 mediates the nociceptive effect of 9-HODE (Patwardhan et al 2010). Thus, oxidized linoleic acid metabolites, such as the endocannabinoid anandamide and several lipoxygenase products formed from arachidonic acid, can act as direct TRPV1 agonists (Zygmunt et al 1999, Hwang et al 2000).
During conditions characterized by oxidative stress, such as inflammation or reperfusion after ischemia, a range of lipid peroxidation products are formed in reactions between free radicals and membrane lipids. Many of the lipids formed are well-known reactive, electrophilic molecules that bind covalently to proteins such as hydroxynenonal, cyclopentenone prostaglandins, isoprostanes, and related species. The covalent modification of redox-sensitive transcription factors initiates specific signaling cascades that may act to modify or protect against oxidative conditions, but the electrophilic lipids also stimulate nociceptive sensory neurons directly by activating TRPA1 (Trevisani et al 2007, Andersson et al 2008).
Protease-Activated Receptors
Four types of G protein–coupled protease-activated receptors (PARs) have been identified (PAR1–4). These receptors are activated by a unique mechanism whereby extracellular, soluble, or surface-associated proteases cleave at specific residues in the extracellular N-terminal domain of the G protein to expose a novel N-terminal sequence that acts as a tethered ligand and activates the receptor by binding to other regions of the protein. These agonist effects can be mimicked by short synthetic peptides based on the sequence of the tethered ligands of the different PARs. PAR1, PAR3, and PAR4 are activated by thrombin produced during the blood-clotting cascade, whereas PAR2 activation is triggered by tryptase, which is known to be released from mast cells in inflammatory conditions, as well as by the blood-clotting factors VIIa and Xa and the cysteine protease cathepsin S (Soh et al 2010, Cattaruzza et al 2011). In this way PARs are activated as a result of tissue damage and inflammation. Because activation involves an irreversible enzymatic cleavage, restoration of PAR sensitivity requires internalization of the receptors and insertion of new receptor into the plasma membrane.
Protease-Activated Receptor Signaling
Activation of PARs can trigger a variety of intracellular signaling pathways. PAR1 and PAR2 couple to either Gq/11α, G12/13α, or Giα; PAR3 signals through Gq/11α activation; and PAR 4 through either Gq/11α or G12/13α (Russo et al 2009, Soh et al 2010). In this way, activation of PAR1 and PAR2 may stimulate PLC-β to activate the DAG–PKC and IP3–Ca2+ pathways (Gq11α), Rho and Rho-kinase (G12/13α), and the MAPK cascade and inhibit adenylate cyclase (Giα).
Protease-Activated Receptor Expression
PARs were initially detected in platelets, endothelial cells, and fibroblasts, but they are also expressed in the nervous system. All four PARs are expressed on peripheral sensory neurons. Expression of PAR2 is almost exclusively restricted to small-diameter unmyelinated neurons in rat and mouse DRG neurons, a majority of which are also positive for calcitonin gene–related peptide (CGRP) expression (Zhu et al 2005, Vellani et al 2010).
Studies using PAR subtype–selective peptide agonists and knockout mice suggest that the hyperalgesic effect of PAR activation is mediated primarily through PAR2, although in vitro, PAR1 and PAR4 receptor activation can sensitize TRPV1-mediated heat responses (Vellani et al 2010). Intraplantar injection of PAR2 synthetic agonists, as well as tryptase, evokes prolonged thermal and mechanical hyperalgesia and c-fos expression in laminae I and II in the spinal cord (Kawabata et al 2001, 2002; Vergnolle et al 2001). This hyperalgesia occurs with low concentrations of agonists that do not cause overt inflammation, and it is not seen in mice lacking the neurokinin 1 (NK1; substance P) receptor or in the presence of centrally acting NK1 receptor antagonists. Mast cells are known to be closely associated with sensory nerves in normal as well as inflammatory conditions (Stead et al 1997), and the hyperalgesia evoked by the mast cell–degranulating agent 48/80 is significantly reduced in PAR2−/− mice (Vergnolle et al 2001). These findings suggest a direct action of PAR2 activation on sensory nerve function. Such a direct action has been demonstrated in isolated DRG neurons, where activation of PAR2 sensitizes TRPV1 and TRPA1 to agonist stimulation. The sensitizing effect of PAR2 activation on TRPV1 appears to be mediated by PKC since it is inhibited by PKC inhibitors and a PKC-ε translocation inhibitor (Amadesi et al 2004, Dai et al 2004). In contrast, PAR2-mediated sensitization of TRPA1 is independent of PKC and instead depends on activation of PLC and subsequent reduction of PIP2 levels (Dai et al 2007). In vivo, administration of a selective PAR2 agonist enhances the nocifensive responses evoked by the TRPA1 agonists AITC and cinnamaldehyde in the rat (Dai et al 2007). An important role for TRPV1 in vivo is also shown by the finding that the thermal hyperalgesia, mechanical allodynia, and spinal cord c-fos expression evoked by the intraplantar injection of a PAR2 agonist peptide are significantly attenuated in TRPV1−/− mice (Amadesi et al 2004, Dai et al 2004).
Activation of PAR1 may have complex effects on nociception. Sub-inflammatory doses of PAR1 agonists have been reported to increase nociceptive thresholds and significantly reduce the inflammatory hyperalgesia induced by carrageenan (Asfaha et al 2002). However, higher doses of PAR1 agonists are pro-nociceptive, and it is possible that stimulation of PAR1 on different neuronal populations (small TRPV1-containing nociceptors and larger non-nociceptive neurons) can explain these apparently contradictory observations (Vellani et al 2010). The pro-nociceptive effect of PAR1 stimulation appears to depend on PKC-ε and sensitization of TRPV1.
Serotonin
Serotonin is one of many mediators released from platelets (rats and humans) and mast cells (rats) in injured and inflamed tissues. In humans, intradermal dialysis of 5-hydroxytryptamine (5-HT) evokes burning pain (Lischetski et al 2001), and intramuscular injection of 5-HT elicits pain and sensitization to pressure stimuli (Ernberg et al 2000, Ernberg et al 2006). In situ hybridization studies have shown that DRG neurons normally express mRNA for 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2B, 5-HT3B, and 5-HT4 receptors (Nicholson et al 2003), with other evidence for the expression of 5-HT7 receptors (Amaya-Castellanos et al 2011). Expression of some of these receptor subtypes (5-HT2A, 5-HT3, 5-HT4, and 5-HT7) is increased with inflammation (Wu et al 2001, Liu et al 2005).
Some of the excitatory actions of serotonin have been ascribed to activation of the 5-HT3 receptor/ion channel. 5-HT3 receptor agonists enhance the excitability of unmyelinated C-fibers (Moalem et al 2005, Lang et al 2006), and relatively selective 5-HT3 antagonists reduce the pain evoked by peripheral administration of serotonin or carrageenan in rats (Eschalier et al 1985, Richardson et al 1985, Sufka et al 1992).
Serotonin can also activate and sensitize nociceptors by actions on G protein–coupled 5-HT receptors. 5-HT2A receptors are expressed mainly in small-diameter (Aq- and C-fiber) peptidergic and non-peptidergic sensory neurons, and there is significant overlap with TRPV1 expression (Okamoto et al 2002, van Steenwinckel et al 2009). 5-HT2A receptors play a significant role in inflammatory thermal hypersensitivity. Intraplantar administration of 5-HT2A agonists into rats produces thermal hyperalgesia (Abbott et al 1996, Tokunaga et al 1998), and activation of peripheral 5-HT2A receptors induces Fos expression in dorsal horn neurons, indicative of sensory neuron excitation (Doi-Saika et al 1997). Conversely, peripheral administration of 5-HT2A receptor antagonists reduces the thermal hyperalgesia induced by either CFA or carrageenan (Okamoto et al 2002, Wei et al 2005, Huang et al 2009). In addition to the strong evidence for a role of 5-HT2A receptors, there is also pharmacological evidence that 5-HT2B receptors play a role in inflammatory mechanical hypersensitivity but not in thermal hyperalgesia (Lin et al 2011). The cellular mechanisms responsible for these effects are unclear. 5-HT2 receptors are usually linked to the PLC pathway, and the sensitization mechanism or mechanisms may be attributable to PKC-mediated modulation of ion channels.
Relatively few data are available on the roles of peripheral 5-HT4 and 5-HT7 receptors in inflammatory conditions, although some pharmacological evidence indicates that these receptor subtypes have roles in the longer-term (days) mechanical allodynia following intraplantar administration of formalin (Godinez-Chapiro et al 2011). These receptors are positively coupled to adenylate cyclase, and receptor activation stimulates cAMP production. An increase in cAMP can result in a PKA-mediated modification of ion channel function, notably, increased activity of tetrodotoxin (TTX)-resistant sodium channels (Cardenas et al 2001, Scroggs 2011).
Nitric Oxide
Although the actions of NO on nociceptive processes are primarily spinal and evident after intrathecal administration of drugs, there is controversial evidence of a peripheral action of NO. The cellular source of NO is unclear, and both neuronal and non-neuronal sources are likely. NO is produced in the periphery during inflammation (see Toriyabe et al 2004). nNOS appears to be responsible for synthesis in the early phase of inflammation and nNOS and iNOS at later phases (Omote et al 2001). Experimentally, intradermal and intravascular injection of NO evokes a concentration-dependent pain in human volunteers (Holthusen and Arndt 1994, 1995), whereas topical administration of NO donors is antinociceptive. The site of action appears to be important. Studies in rats have shown that intradermal administration of the NO precursor L-arginine or an NO donor (3-[4-morphinolinyl]-sydnonimine hydrochloride [SIN-1]) evokes mechanical hypersensitivity. In contrast, subcutaneous injection of these agents had little effect on baseline mechanical thresholds but reversed PGE2-induced hypersensitivity (Vivancos et al 2003) via an NO/cGMP pathway (Sachs et al 2004). A pro-nociceptive action of NO in inflammatory conditions is supported by the finding that local administration of the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) reduces both mechanical and thermal hyperalgesia, as well as the inflammation induced by carrageenan (Lawand et al 1997, Nakamura et al 1996). Similarly, co-injection of another NOS inhibitor, NG-methyl-L-arginine (L-NMA), inhibited PGE2-induced mechanical hyperalgesia, whereas intradermal injection of the NOS substrate L-arginine or the NO donor SIN-1 evoked mechanical hyperalgesia (Aley et al 1998). In peripheral nerves the NO-sensitive (soluble) guanylate cyclase is expressed by non-neuronal cells and not by sensory neurons (Schmidtko et al 2007), so the sensory neuron effects of activating the NO/cGMP pathway are likely to be indirect. NO can also nitrosylate ion channels, and this may be a more important mechanism for any direct pro- or antinociceptive NO effects. NO can stimulate DRG neurons by activation of both TRPA1 and TRPV1, and studies of genetically modified mice show that the thermal hyperalgesia elicited by injection of an NO donor is largely dependent on TRPV1 expression. In addition, both TRPA1 and TRPV1 appear to play roles in the acute nociceptive behavioral response to NO donor injection after pre-activation of the PLC/PKA pathways (Miyamoto et al 2009). Conversely NO activates ATP-sensitive K+ channels (Kawano et al 2009) and inhibits voltage-gated sodium channels (Renganathan et al 2002) in DRG neurons; both actions will inhibit neuronal firing and could contribute to antinociception. Many of the peripheral effects of NO or NOS inhibition are likely to involve other cells and mediators, including alterations in cytokine levels (Chen et al 2010b).
ATP and Adenosine
There has been considerable debate about the role of ATP in activation of peripheral nerves, especially in inflammatory conditions. ATP is released from damaged cells, and ATP levels are elevated in damaged and inflamed tissues (Gordon 1986, Cook and McCleskey 2002). It has also been proposed that ATP has a role in the genesis of pain associated with malignancy inasmuch as ATP levels at tumor sites are higher than those in normal tissues (Pellegatti et al 2008). In humans, application of ATP to the skin evokes the sensation of pain (Bleehen and Keele 1977, Coutts et al 1981), which is enhanced after ultraviolet irradiation (Hamilton et al 2000), and intracutaneous administration of ATP excites human C fibers (Hilliges et al 2002). Similar pain behavior has been noted in animals, with nocifensive behavior being evoked by intraplantar administration of ATP (Bland-Ward and Humphrey 1997, Hamilton et al 1999, Jarvis et al 2001), and this is augmented by treatment with PGE1 and the inflammatory agent carrageenan (Sawynok and Reid 1997, Hamilton et al 1999). These behavioral responses are probably mediated by Aδ and C fibers because these fibers are excited by ATP both in vivo (Dowd et al 1998) and in isolated nerve preparations (Hamilton et al 2001) and the pain response evoked by ATP in human skin is markedly reduced after the topical application of capsaicin to functionally desensitize the TRPV1-expressing fibers (Hamilton et al 2000).
The receptors responsible for this excitation are likely to contain the P2X3 receptor subtype (i.e., P2X3 homomeric or P2X2/3 heteromeric receptors) because sensory fibers are excited by the P2X3 agonist α,β-me-ATP (see Irnich et al 2002). Furthermore, P2X3 receptor expression is restricted to small-diameter sensory afferents (Vulchanova et al 1997, Bradbury et al 1998), and their expression is up-regulated in experimental inflammatory conditions (Xu and Huang 2002, Shinoda et al 2005). One mechanism for this up-regulation is an increased supply of the growth factors NGF and GDNF in sensory nerves during inflammation since administration of both these growth factors (by intrathecal administration) increased P2X3 receptor immunoreactivity in rat DRG neurons (Ramer et al 2001). Similarly, P2X3 receptor expression is elevated following injection of NGF into skeletal muscle (Liu et al 2011). Inflammatory mediators may also increase ATP sensitivity via PKA- and PKC-mediated phosphorylation of P2X3-containing receptors (Paukert et al 2001, Fabbretti et al 2006).
A role of P2X3 receptors in inflammatory pain is supported by the finding that intrathecal delivery of antisense oligonucleotides or small interfering RNA (siRNA) directed against P2X3 mRNA, which reduces P2X3 protein expression by about 50%, partially reverses inflammatory thermal and mechanical hyperalgesia (Barclay et al 2002, Honore et al 2002, Dorn et al 2004). In addition, reversal of inflammatory thermal and mechanical hyperalgesia, as well as thermal and mechanical hyperalgesia after nerve injury, is seen after the administration of selective antagonists (A317491 and AF-353) (Jarvis et al 2002, Oliveira et al 2009, Ford 2012). AF-353 is also effective in models of bone cancer pain, where it reversed mechanical hypersensitivity and improved weight bearing on the affected limb (Kaan et al 2010). The marked effects of antisense oligonucleotide treatment and pharmacological antagonism contrast with the relatively mild phenotypic changes seen in P2X3-null mice (Cockayne et al 2000, Souslova et al 2000), which display a modest reduction in the behavioral response to intraplantar administration of formalin. The paradoxical finding that P2X3-null mice show increased thermal hyperalgesia after injection of CFA suggests that some adaptive processes occur when the P2X3 receptor is ablated.
P2Y Receptors
ATP can also stimulate sensory neurons by activating G protein–coupled P2Y receptors. Of the known P2Y receptors, mRNA for the Gq/11α-linked receptors P2Y1, P2Y2, P2Y4, and P2Y6 is expressed in sensory ganglia. P2Y1 and P2Y2 receptors, which are expressed by sensory neurons (Molliver et al 2002, Kobayashi et al 2006), have received the most attention. Expression of P2Y2 is increased during inflammation induced by CFA, whereas P2Y1, P2Y4, and P2Y6 are reduced (Malin et al 2008). Both P2Y1 and P2Y2 receptors are Gq11 linked and signal via IP3–DAG pathways, which is consistent with the finding that activation of either receptor subtype evokes a rise in intracellular calcium levels and an increase in excitability in DRG neurons that is blocked by PLC and PKC inhibition (Usachev et al 2002, Malin and Molliver 2010, Yousuf et al 2011). On the other hand, stimulation of the Gi/o-coupled receptors P2Y12–14 reduced the excitation of DRG neurons in a pertussis toxin–sensitive fashion. In vivo, peripheral administration of P2Y13 and P2Y14 agonists reduced the inflammatory hyperalgesia induced by CFA (Malin and Molliver 2010). P2Y receptor activation in DRG neurons also activates the transcription factor cAMP response element–binding protein (CREB), which is likely to lead to longer-term changes in the cell phenotype (Molliver et al 2002). P2Y receptor activation by the P2Y2/P2Y4 agonist uridine triphosphate (UTP) evokes sustained action potential firing in capsaicin-sensitive C fibers and some Aδ fibers (Stucky et al 2004). This effect is probably mediated through P2Y2 receptors since these receptors appear to be expressed at very low levels by sensory neurons (Sanada et al 2002).
The mechanisms underlying P2Y receptor–mediated excitation involve sensitization of TRPV1 and modulation of ion channels that regulate the firing frequency of action potentials. P2Y2 receptor activation potentiates the capsaicin-evoked TRPV1 currents and [Ca2+]i responses in isolated sensory neurons, and this potentiation is lost in P2Y2-null mice (Moriyama et al 2003, Malin et al 2008). P2Y1 receptor activation also lowers the heat activation threshold for TRPV1 in rat DRG neurons (Tominaga et al 2001) and increases sensitivity to the TRPV1 agonist capsaicin (Yousuf et al 2011).
P2Y1/2 receptor activation can also increase the excitability of DRG neurons by inhibiting Kv7 potassium channels (Yousuf et al 2011), which is also a mechanism described for bradykinin sensitization. Furthermore, P2Y2 activation sensitizes mechanotransduction channels (Lechner and Lewin 2009) and purinergic P2X2 and P2X3 channels (Chen et al 2010a) and may underlie the ATP-induced potentiation of TTX-resistant sodium channel (Nav1.8) currents (Baker 2005).
Adenosine
It is almost certain that some of the effects of ATP in vivo are mediated by adenosine diphosphate (ADP) (Bleehen and Keele 1977, Coutts et al 1981), AMP, and adenosine (Bleehen and Keele 1977) formed by rapid sequential ectonucleotidase cleavage of ATP. All these agents produce pain when applied to human skin. However, the underlying mechanisms probably differ because the nocifensive response to ADP seen in animal studies differs from that evoked by ATP (Bland-Ward and Humphrey 1997).
During inflammation, adenosine is released from a variety of cell types (endothelial cells, mast cells, neutrophils, and fibroblasts), in addition to release from neurons. The effects of adenosine are complex, with evidence of both pro-nociceptive and analgesic effects (see Sawynok and Liu 2003) mediated through various receptor subtypes (A1, A2A, A2B, and A3) at peripheral and spinal sites. Although some of the effects are probably directly on nerves, others are more likely to be mediated via activation of adenosine receptors on other cell types, such as mast cells. Nevertheless, there is clear evidence that adenosine can activate sensory nerves since intravenous administration of adenosine produces pain in human volunteers (Sylven 1989) and application of adenosine sensitizes cat myelinated and unmyelinated vagal afferents (Cherniak et al 1987). Isolated segments of human nerve are also depolarized by ATP; the pharmacological properties are consistent with an effect mediated by adenosine acting on Gs-coupled A2B receptors (Irnich et al 2002). In other experiments, A1 agonists have been reported to activate C fibers in the rat (Esquisatto et al 2001, Sawynok et al 2000), and stimulation of A1 receptors induces an inward current and action potential firing in guinea pig jugular and spinal esophageal TRPV1-positive nociceptors (Ru et al 2011). In contrast to the predominating pro-nociceptive peripheral effects produced by adenosine, intrathecal administration of adenosine has well-recognized analgesic effects mediated by A1 receptor activation (Sawynok and Liu 2003). Accordingly, intrathecal administration of the ectonucleotidase prostatic acid phosphatase (PAP) has been shown to produce long-lasting antinociception and anti-hyperalgesia mediated by hydrolysis of extracellular AMP to adenosine, which in turn stimulates adenosine A1 receptors (Zylka et al 2008).
Low pH
The pH of the extracellular environment is known to fall in a number of pathophysiological conditions, such as hypoxia and anoxia, as well as with inflammation and tumors. Acidic conditions can have direct effects on sensory nerves. Low-pH solutions evoke prolonged activation of sensory nerves and produce a sharp stinging pain in humans (Lindahl 1962, Steen and Reeh 1993, Jones et al 2004). Several mechanisms are thought to underlie the neuronal excitation observed. One key effect of acid solutions is activation and sensitization of the thermosensitive ion channel TRPV1 (Tominaga et al 1998, McLatchie and Bevan 2001, Leffler et al 2006). A second mechanism is direct activation of ASICs (see Deval et al 2010), notably ASIC3, which is expressed in the sensory innervation of the heart and activated by modest reductions in extracellular pH (to about pH 7). ASIC3 has been proposed to be the sensor in cardiac nociceptors that triggers cardiac pain in response to myocardial acidity (Sutherland et al 2001) and may play a role in sensing acidic conditions in other tissues such as skin (Deval et al 2008) and skeletal muscle (Sluka et al 2003). Finally, low pH can augment or stimulate neuronal firing by inhibiting K+ channel activity (Baumann et al 2004).
Immune Cells and Pain
It is now well established that the immune system, as well as the factors that it produces, can alter sensory processing and play a pivotal role in the development and maintenance of persistent pain (Marchand et al 2005, Ren and Dubner 2010). For example, not only are cytokines and chemokines an important means of communication between immune cells, but such factors can also act as pain mediators and have a direct sensitizing action on nociceptors. The importance of the immune system is not restricted to inflammatory pain states but extends to neuropathic conditions since nerve injury evokes a profound immune response. Many of the pain mediators discussed below are closely linked to this system through either their release by or their action on different immune cells. We discuss the role of particular immune cells in different pain states below and summarize these actions in Table 3-1.
Table 3-1
Contribution of Peripheral Immune Cells to Animal Models of Persistent Pain
CELL TYPE* |
INFLAMMATORY PAIN† |
NEUROPATHIC PAIN‡ |
| Macrophage | ↑ Infiltration (joint, muscle) ↓ Mechanical, spontaneous |
↑↑ Infiltration (nerve) ↓/↔ Mechanical, ↓/↔thermal, ↓ spontaneous |
| Dendritic cell/Langerhans cell | − | ↑ Infiltration/activation (skin, nerve) |
| Mast cell | ↑ Degranulation (skin) ↓ Mechanical, thermal, spontaneous, visceral |
↑ Degranulation (skin, nerve) ↓ Mechanical, thermal |
| Neutrophils | ↑↑ Infiltration (skin, joint) ↓ Mechanical, thermal |
↑ Infiltration (nerve) ↓ Thermal |
| T cells | ↑ Infiltration (joint) ↓ Mechanical |
↑ Infiltration (nerve) ↓ Mechanical, thermal |
| Natural killer cells | − | ↑ Infiltration (nerve) |
| B cells | − | ↑ Infiltration (nerve) ↔ Mechanical |
This table highlights the involvement of immune cells in both inflammatory and neuropathic pain by using data from animal models. Following the injection of an inflammogen or damage to a peripheral nerve (either traumatic or drug induced), various immune cells infiltrate the relevant peripheral tissue and/or alter their response state. In addition, via genetic, chemical, or pharmacological approaches, certain immune cell populations can be depleted, their infiltration suppressed, or their activation prevented, thereby leading to the attenuation of persistent pain. The data in this table is a summary of the studies discussed in this section.
*Although microglia are important in the development and/or maintenance of persistent pain, they are central nervous system immune cells and therefore have not been mentioned in this table. Work regarding these cells is discussed in Chapter 4.
†Inflammatory models include complete Freund’s adjuvant, carrageenan, zymosan, nerve growth factor, lipopolysaccharide, formalin, collagen- or antigen-induced arthritis, and acetic acid.
‡Neuropathic pain models include partial sciatic nerve ligation, chronic constriction injury, spinal nerve ligation, spared nerve injury, vincristine, paclitaxel, and streptozocin.
Mast Cells
Mast cells are found in areas of the body that interact with the external environment, such as the skin and mucosal layers, and these cells are normally situated in close proximity to blood vessels and nerves. Mast cell granules contain numerous chemicals, including histamine, and they can also synthesize and release many cytokines and chemokines (Metcalfe et al 1997).
Mast cells can be degranulated by the compound 48/80, which when applied to human skin causes thermal hyperalgesia, thus indicating that chemicals in the granules of mast cells are pro-algesic (Drummond 2004). Chronic treatment with this compound prevents re-granulation of these cells, and in this state some common models of inflammatory pain, including those precipitated by injection of acetic acid or zymosan and the second phase of the formalin test, show reduced pain-like behavior (Ribeiro et al 2000, Parada et al 2001). Treatment with 48/80 to deplete mast cell granules also reduces both the thermal and mechanical hyperalgesia produced by CFA (Woolf et al 1996). One of the mechanisms by which NGF induces thermal hyperalgesia (see below) is thought to be mediated via its action on mast cells (Lewin et al 1994). Thus, NGF was not able to sensitize nociceptors to thermal stimuli in mice deficient in these cells (Rueff and Mendell 1996), and these mice do not fully develop pain-like symptoms in a model of cystitis, which also seems to be strongly dependent on the release of mast cell mediators (Rudick et al 2008).
Mast cells are also present in the sciatic nerve. Following partial sciatic nerve ligation (PSNL, a model of neuropathic pain), very few intact mast cells remain at the site of injury or directly distal to it, thus suggesting that the majority have released the contents of their granules. Stabilization of these cells increases the presence of intact mast cells and reduces the development of both mechanical and thermal hyperalgesia (Theodosiou et al 1999, Zuo et al 2003). Although the chemicals released by mast cells may act directly to sensitize nociceptors, such agents may also act to recruit and activate other immune cells within the injured nerve. Histamine is one of the mediators released by mast cells. However, the analgesic effect of antihistamine treatment is modest, and in some neuropathic pain models such agents have limited effects on mechanical pain–related hypersensitivity. Stabilization of mast cells with cromoglycate can reduce neuropathic hypersensitivity. Some of this action is likely to be indirect since such treatment also reduced both neutrophil and macrophage infiltration into the injured nerve (Zuo et al 2003). Mast cells can produce NGF (Leon et al 1994), and this might also contribute to the pro-algesic action of these cells.
Neutrophils
Neutrophils are PMN granulocytes and make up around 60% of the circulating white blood cells, which puts them in an ideal position to react, in large numbers, to pathogens or tissue injury. Rodent models of inflammatory pain are commonly induced by the local injection of an antigen, such as zymosan, LPS, or carrageenan, and subsequent activation of the innate and adaptive immune system (Cunha et al 2008a, 2008b; Guerrero et al 2008; Ting et al 2008). Accumulation of neutrophils occurs in all these models and can be reduced by blocking receptors that mediate the rolling, attachment, and transmigration of these cells from blood into tissue. Complement component 5a (C5a), a complement activation product, is a potent chemotactic factor for neutrophils (Shin et al 1968). Following injection of zymosan into the paw, pharmacological inhibition of the C5a receptor attenuated both mechanical hypersensitivity and neutrophil influx (Ting et al 2008). The chemokine receptors CXCR1 and CXCR2 are both important in neutrophil migration and activation in numerous inflammatory states (Bizzarri et al 2006). Dual inhibition of these receptors was able to significantly reduce the accumulation of neutrophils and abnormal sensory behavior induced by zymosan, carrageenan, and LPS (Cunha et al 2008a). More recently, specific antagonism of the CXCR2 receptor via the small molecule SB225002 reduced both pain-related hypersensitivity and neutrophil accumulation in the carrageenan model (Manjavachi et al 2010). Other factors with strong chemotactic effects on neutrophils include the lipoxygenase product LTB4 (Ford-Hutchinson et al 1980). Both pharmacological and genetic inhibition of the action of LTB4 reduced the hypersensitivity produced by joint inflammation (Guerrero et al 2008). In agreement with these data, chemical depletion of neutrophils decreased their accumulation in skin after both zymosan and carrageenan treatment and prevented full development of the abnormal sensory behavior in these models (Ting et al 2008). Although recruitment of these cells is important, blockade of the C5a receptor in the LPS and carrageenan models did not affect neutrophil recruitment but did attenuate pain-like behavior, thus suggesting that certain molecules such as C5a may, in some instances of inflammation, be more important for activation than for direct recruitment of these cells (Ting et al 2008). In naïve animals, intradermal injection of neutrophil chemotactic factors such as LTB4, N-formylmethionyl-leucyl-phenylalanine (fMLP), C5a, and chemokine C-X-C motif ligand 1 (CXCL1) induces pain-related hypersensitivity (Levine et al 1985, 1986a; Cunha et al 2008a). Interestingly the prominent pro-algesic properties of NGF are also reported to depend on neutrophil recruitment (Bennett et al 1998b). Recently, IL-17 has been shown to be a pro-nociceptive cytokine, particularly in the setting of antigen-induced arthritis, where neutralization of its effect reduced pain-related hypersensitivity and neutrophil recruitment in a TNF-α–dependent manner (Pinto et al 2010). In addition, intraplantar injection of this cytokine produces both thermal and mechanical hypersensitivity associated with the accumulation of neutrophils in the dermis (Kim and Moalem-Taylor 2011b, McNamee et al 2011). However, it must be stated that neutrophil attraction alone may not be sufficient to cause pain-like behavior because the activation status of these cells is also likely to be important. The chemotactic factor glycogen results in neutrophil recruitment but does not cause any significant pain-like hypersensitivity (Levine et al 1985). Nevertheless, systemic depletion of neutrophils significantly reduced the pain-like behavior elicited by LTB4, C5a, fMLP, and NGF administration, thus suggesting that activated neutrophils are crucial in the pro-algesic properties of these and other factors (Levine et al 1985, 1986a; Bennett et al 1998b; Ting et al 2008). In vitro experiments have shown that in a co-cultured system, dissociated DRG neurons increase their excitability following neutrophil activation, which suggests that neutrophils do release factors that can act directly on nociceptive neurons (Shaw et al 2008). Clinically, it seems that neutrophils play an important role in inflammatory diseases; in particular, they are present in the joint fluid and synovial membrane of patients with rheumatoid arthritis (RA) (Wright et al 2010). Interestingly, therapies used to treat RA, such as antibodies against TNF-α, reduce pain scores in these patients and decrease the influx of neutrophils into the joint (den Broeder et al 2003).
Neutrophils are normally completely absent from the naïve sciatic nerve. However, in animals in which the nerve has been injured to induce neuropathic pain–like behavior, substantial neutrophil infiltration takes place (Perkins and Tracey 2000, Zuo et al 2003, Kim and Moalem-Taylor 2011a). In addition, cytokine recruitment of neutrophils into the non-injured nerve can recapitulate this pain-like behavior (Kim and Moalem-Taylor 2011b). Some of the strongest evidence for a role of these cells in the development of neuropathic pain–like behavior comes from depletion studies. Systemic depletion of neutrophils before injury reduced the development of thermal hypersensitivity (Perkins and Tracey 2000). However, an attempt to deplete neutrophils 8 days after injury had no effect on pain behavior (Perkins and Tracey 2000), a finding suggestive of an important role in the initiation rather than the maintenance of neuropathic pain. Neutrophils can release numerous chemokines (Scapini et al 2000), and it is likely that their algogenic effects, like those of mast cells, may partly be due to the subsequent recruitment and activation of other immune cells such as macrophages.
Macrophages
Macrophages are leukocytes and represent a heterogeneous group of cells resident in the majority of tissues. They are continually being replenished from a circulating peripheral blood mononuclear cell population, which itself originates from bone marrow. These cells have homeostatic actions in their tissue of residence, such as clearing cell debris, as well as repairing and remodeling tissue following damage and inflammation. Macrophages derive from monocytes, which also generate a range of other specialized cells contributing to innate immunity, including microglia in the CNS, alveolar macrophages in the lung, Langerhans cells in the skin, osteoclasts in bone, Kupffer cells in the liver, and histocytes in connective tissue, as well as resident cells in the spleen, gastrointestinal tract, and the peritoneum (Gordon and Taylor 2005). Following tissue damage or infection, the macrophage population is augmented by blood-derived monocytes. The resident as well as the infiltrating macrophages react to endogenous danger signals released by necrotic cells or exogenous signals such as factors produced by microorganisms and appropriately release cytokines to orchestrate the innate and adaptive immune response. A strong body of evidence suggests a role of macrophages in the development of both inflammatory and neuropathic pain.
Intraperitoneal injection of acetic acid or zymosan is used as a model of visceral pain and induces overt pain-like behavior in rodents in the form of a writhing response. This behavior can be exacerbated by increasing the macrophage population (Ribeiro et al 2000). Inhibiting the production of inflammatory mediators by macrophages through treatment with either anti-inflammatory cytokines or pentoxifylline (which reduces activation of these cells via a poorly defined mechanism) has been shown to reduce inflammatory pain (Vale et al 2003, 2004). Mice deficient in the purinergic receptor P2X4 demonstrate reduced mechanical hyperalgesia following either CFA or carrageenan application. This effect is attributed to a reduction in the release of PGE2 from tissue-resident macrophages, which would normally occur in a P2X4-dependent manner, and in agreement, injection of ATP-stimulated macrophages from wild-type mice into P2X4-deficient mice was able to induce mechanical hyperalgesia (Ulmann et al 2010).
Macrophages have an important role in the development and maintenance of neuropathic pain. Traumatic injury to a peripheral nerve results in degeneration of axons separated from their cell bodies and breakdown of the associated myelin sheath in a process termed wallerian degeneration. Macrophages have an important role in phagocytosing and clearing myelin debris; because such debris is inhibitory to axon regeneration, clearance is vital for effective nerve repair. Chronic constriction injury (CCI) of the sciatic nerve in mice results in an increase in macrophage infiltration over a 28-day period that is strongly associated with neuropathic pain–like behavior (Myers et al 1996). Naturally occurring mutant mice that exhibit slow wallerian degeneration display delayed macrophage recruitment and reduced cytokine production in injured nerves (Sommer and Schafers 1998). Consistent with this attenuated inflammatory response, such mice also show delayed onset/reduced mechanical and thermal pain–related hypersensitivity (Myers et al 1996, Ramer et al 1997). Systemic depletion of macrophages also reduced both thermal and mechanical hyperalgesia in the PSNL model of neuropathic pain (Liu et al 2000, Barclay et al 2007).
Another means of inhibiting the pro-algesic actions of macrophages is to reduce their recruitment from the circulation to the injured nerve. An important molecule for macrophage chemotaxis is CCL3, blockade of which reduces macrophage infiltration, as well as thermal and mechanical pain–related hypersensitivity (Kiguchi et al 2010). The toll-like receptors (TLRs) are pattern recognition receptors that respond to structural motifs on pathogens and the products of tissue injury. They have an important role in macrophage recruitment and activation. Mice lacking TLR2 demonstrate absent macrophage recruitment and reduced neuropathic pain–like behavior (Shi et al 2011).
Another option for modulating the functional properties of these cells is to alter their functional status and thereby reduce the production of pro-inflammatory cytokines (Kiguchi et al 2010). This can be achieved by treatment with anti-inflammatory cytokines such as IL-10 (Wagner et al 1998). Trying to change the phenotype of macrophages from a pro- to an anti-inflammatory state may be a better therapeutic option than trying to globally inhibit their recruitment to or function within the injured nerve because they are essential for effective nerve repair (Barrette et al 2008).
Dendritic Cells
Dendritic cells (DCs) are closely related to macrophages; they are primarily antigen-presenting cells but also have phagocytic capabilities and can release cytokines and chemokines. Some of the pro-nociceptive effects of IL-17 may be mediated by these cells (Ruts et al 2010, Kim and Moalem-Taylor 2011b). In the epidermis these cells are referred to as Langerhans cells (LCs). Following traumatic nerve injury, epidermal nerve fiber density is decreased. However, spared fibers that intermingle with degenerating axons share innervation territories, and these spared axons have an important role in the generation of neuropathic pain. The endings of these spared axons show increased association with LCs after nerve injury (Lin et al 2001, Lindenlaub and Sommer 2002). In chemotherapy-induced neuropathic pain–like states these LCs express OX-6, a marker of activation associated with pro-inflammatory cytokine production (Siau et al 2006). This same phenomenon has been seen in skin biopsy samples taken from patients with complex regional pain syndrome (Calder et al 1998), and in diabetic patients with small-fiber neuropathy, the number of LCs is increased in the skin in comparison to control samples (Casanova-Molla et al 2012). DCs also infiltrate the injured sciatic nerve distal to and at the site of injury. This infiltration is delayed and occurs 1 week after the initial injury when a robust thermal and mechanical pain–related hypersensitivity takes place (Kim and Moalem-Taylor 2011a). Although these cells respond to a number of different types of tissue injury, there is as yet no direct evidence that they contribute to persistent pain states.
T Cells
T cells are lymphocytes activated by the presentation of antigens. They mediate cellular immunity either by directing the immune response via the release of cytokines to activate innate immune cells or through the destruction of infected cells. More than most, T cells represent a heterologous group of immune cells loosely divided into CD4+ (helper) and CD8+ (cytotoxic) with type 1 and type 2 subsets. Th1 cells release pro-inflammatory cytokines that activate neutrophils, macrophages, and natural killer (NK) cells, whereas Th2 cells release anti-inflammatory cytokines that activate humoral immunity and strongly deactivate macrophages (O’Garra and Arai 2000).
T cells have a pivotal role in autoimmune diseases. RA represents one such disease that is commonly associated with persistent pain, T-cell infiltration, and cytokine production (Toh and Miossec 2007). Abatacept is a fusion protein that prevents the activation of T cells and decreases pain in patients with RA. In models of neuropathic pain induced by either CCI or PSNL, the number of T cells is significantly increased in the injured nerve in comparison to controls (Cui et al 2000, Kim and Moalem-Taylor 2011a). This increase is associated with both thermal and mechanical pain–related hypersensitivity (Cui et al., 2000). Neuropathic pain–related behavior is reduced in T cell–deficient mice or in mice that lack the ability to produce mature T cells (Moalem et al 2004, Kleinschnitz et al 2006, Cao and Deleo 2008, Costigan et al 2009). Passive transfer of Th1 cells into mature T cell–deficient mice is able to restore full neuropathic pain–like behavior, and this pain behavior can be attenuated by Th2 cells in immune-competent mice (Moalem et al 2004). Guillain-Barré syndrome is an autoimmune disorder affecting the peripheral nervous system. The syndrome is a form of peripheral neuropathy and is commonly associated with abnormal pain (Ruts et al 2010). Rats with experimental autoimmune neuritis model this syndrome, and these animals display robust neuropathic pain–like behavior and significant expression of T cells in the affected nerves (Moalem-Taylor et al 2007, Ruts et al 2010).
Other Immune Cells
Many other cells have immune functions and orchestrate both innate and adaptive immunity. NK cells are lymphocytes and constitute up to 20% of the mononuclear cells found in the blood and spleen. They target and kill infected or “stressed” cells, thereby playing a major role in tumor rejection, and can release many pro-inflammatory cytokines. NK cell activity is not altered in the CCI model of neuropathic pain (Tsai and Won 2001), and although their numbers do increase in some nerve injuries, this is not associated with the development of hyperalgesia (Cui et al 2000). B lymphocytes mediate the humoral arm of the adaptive immune response and produce specific antibodies against presented antigens. A recent study looking at immune cell infiltration into the sciatic nerve found a significant increase in the number of B cells 3–14 days after injury, particularly at the site of damage (Kim and Moalem-Taylo, 2011a). However, there does not seem to be any direct evidence linking B cells to the development or maintenance of persistent pain. In fact, full neuropathic pain–like behavior develops in B cell–deficient mice following nerve injury (Costigan et al 2009). Eosinophils and basophils are, like neutrophils, PMN cells that play a role in both parasitic infection and the body’s response to allergens. There is little evidence linking these cells to pain modulation. Both these cell types can release a variety of pro-algesic factors, and further study is required to elucidate any possible role in pain pathophysiology.
Production of Immune Mediators by Non-immune Cells
Many immune cells in their quiescent state do not appear to interact with nociceptive systems. However, following tissue injury some cells undergo a profound phenotypic change that results in the release of cytokines and chemokines. These factors can recruit more immune cells and may also act as pain mediators. Non-immune cells can play an important role in initiating this process. An example is keratinocytes found within the epidermis, an important innervation target in which the naked endings of nociceptors terminate. Following injury or disease, keratinocytes can release an array of cytokines, chemokines, and growth factors (Pastore et al 2006, Li et al 2011), which can have sensitizing actions on nociceptors, as well as endogenous opioids, which can have an analgesic action (Khodorova et al 2003). A further example is Schwann cells, which in normal nerves are intimately associated with axons: myelinating Schwann cells wrap around peripheral axons to form the myelin sheaths that facilitate axonal conduction. There are also non-myelinating Schwann cells, which in nerve fibers ensheathe small-diameter unmyelinated axons, usually in groups called Remak bundles (Griffin and Thompson 2008). In the process of wallerian degeneration these cells de-differentiate and proliferate. They produce a variety of pro-algesic factors such as NGF (Bandtlow et al 1987, Heumann et al 1987, Matsuoka et al 1991); cytokines such as TNF-α, IL-1β, and IL-6 (Bolin et al 1995, Shamash et al 2002, Wagner and Myers 1996); and chemokines such as CCL2 (Fu et al 2010). Such factors may act directly by sensitizing the remaining intact axons within injured nerves (Sorkin et al 1997) and may also have a role in the recruitment of immune cells (Tofaris et al 2002, Perrin et al 2005), thereby contributing to the development of neuropathic pain.
Clearly, then, there is a large body of evidence showing that immune cells are important contributors to the development of multiple types of persistent pain. Immunosuppressive strategies, however, are in general not useful in treating pain because, of course, many aspects of inflammation are of use in promoting tissue repair. A more productive strategy is likely to be the identification of mediators produced by immune cells that lead to activation and sensitization of nociceptors. Below we consider the evidence for such specific mediators.
Immune Cell and Neurotrophic Factors as Pain Mediators and Modulators
One well-recognized consequence of inflammation is the production of various prostanoids, but the limited efficacy of NSAIDs that target COX enzymes—and therefore prostanoid production—strongly suggests a role for other inflammatory mediators. The inflammatory process, triggered by the recruitment of immunocompetent cells and the generation of free radicals, leads to the release of several other algogenic mediators. NGF is one such mediator, and the biology of this factor is discussed at some length below since anti-NGF therapies are now being tested in the clinic. TNF-α and IL-1β are two inflammatory cytokines that also contribute to inflammatory pain. Administration of small doses of TNF-α or IL-1β to adult animals and humans can produce pain and hyperalgesia that start within minutes in some cases and typically persist for several hours (see Watkins and Maier 2003, McMahon and Cafferty 2004, Sommer and Kress 2004). Both these factors are capable of activating and sensitizing peripheral nociceptive neurons and thereby contribute to ongoing pain and hyperalgesia. There is evidence demonstrating that neutralization or block of IL-1β and TNF-α is also effective in preventing abnormal pain behavior in some inflammatory pain models (see Sommer and Kress 2004). Antibodies against TNF-α and IL-1β are now in clinical use for the treatment of inflammatory arthritis and are proving very successful both in treating disease symptoms, including pain, and in modifying the course of the disease (Fleischmann et al 2004, Iannone et al 2007, Laas et al 2009). The analgesic effects of blocking TNF-α are also seen in patients with osteoarthritis (OA) of the hand, for which they have been shown to significantly reduce spontaneous as well as pressure-evoked pain (Fioravanti et al 2009).
Sensory neurons are known to express receptor components capable of transducing extracellular TNF-α (Pollock et al 2002) and IL-1β (Gardiner et al 2002). Intraneural injection of either TNF-α or IL-1β can induce both thermal and mechanical hyperalgesia (Zelenka et al 2005), and blocking either of these factors peripherally following nerve injury attenuates such pain behavior (Lindenlaub et al 2000, Schafers et al 2003, Kiguchi et al 2010). For TNF-α these effects seem to be mediated via TNFR1 and not TNFR2 (Sommer et al 1998). In addition, intraplantar injection of TNF-α (Cunha et al 1992, Perkins et al 1994) or IL-1β (Safieh-Garabedian et al 1995, Amaya et al 2006) can induce hypersensitivity to both thermal and mechanical stimuli. These effects can be mediated directly on nociceptors; both TNF-α and IL-1β have been shown to increase the excitability of nociceptive neurons by enhancing TTX-resistant sodium channel currents via the activation of intracellular cascades involving p38 MAPK (Jin and Gereau 2006, Binshtok et al 2008). TNF-α can also enhance the sensitivity of TRPV1 to contribute to thermal hypersensitivity (Nicol et al 1997, Jin and Gereau 2006). Intriguingly, trimers of TNF-α have been reported to insert into membranes and form functional voltage-dependent sodium channels (Kagan et al 1992), which may allow generalized sensitization of sensory neurons in the absence of functional TNF-α receptors. In addition to these direct actions on sensory neurons, it is clear that a large proportion of the algogenic effect of TNF-α and IL-1β is mediated via NGF. Neutralizing antisera or other molecules blocking NGF prevent the pain produced by these inflammatory cytokines. Mast cells also express trkA (Horigome et al 1993) and in response to NGF proliferate, degranulate, and release inflammatory mediators, including TNF-α (Woolf et al 1996). Because mast cells also release NGF, there is the possibility of a vicious circle of events promoting pain.
Leukemia inhibitory factor (LIF) and IL-6 both belong to a family of neuropoietic cytokines defined by their binding to the common receptor gp130. Other members include IL-11, ciliary-derived neurotrophic factor, oncostatin M, and cardiotrophin-1. LIF signals via a receptor complex of the LIF receptor-β and gp130 and is retrogradely transported by a population of small-diameter DRG cells (Thompson et al 1997). Levels of LIF are normally very low. However, following nerve injury, LIF expression increases at the site of injury (Banner and Patterson 1994). Nerve injury also results in a large increase in expression of the neuropeptide galanin within sensory neurons. Evidence from both animals deficient in LIF (Sun and Zigmond 1996) and the administration of exogenous LIF (Thompson et al 1998) indicates that this cytokine is responsible for up-regulation of galanin. LIF may also be implicated in the sprouting of post-ganglionic sympathetic neurons that occurs around DRG cell bodies following nerve injury (Thompson and Majithia 1998).
The actions of LIF are not restricted to nerve injury but also extend to inflammatory conditions (Banner et al 1998). LIF levels increase during inflammation. LIF knockout mice have an enhanced inflammatory reaction. Conversely, administration of exogenous LIF can attenuate both the hyperalgesia and the increased NGF expression that normally occur during inflammation. Confusingly, the effects of exogenous LIF may be dose dependent in that another study has found that administration of this factor to naïve animals may itself produce hyperalgesia (Thompson et al 1996). Endogenous LIF, however, appears to have an interesting role as a mediator that suppresses the inflammatory reaction possibly at an early stage by negatively regulating the expression of IL-1β and NGF.
IL-6 can exert its biological effect through the binding of either a membrane-bound IL-6 receptor or a soluble receptor subunit, both of which need to form a complex with gp130 for signal transduction. Sensory neurons, which constitutively express gp130 (Gardiner et al 2002), lack the IL-6 membrane receptor, and it is therefore likely that direct actions of IL-6 on these cells involve it and the soluble form of the receptor binding to a cell. Intraplantar injection of IL-6 induces a dose-dependent mechanical hyperalgesia (Cunha et al 1992). Mice deficient in IL-6 show both reduced thermal and mechanical hyperalgesia following inflammation or nerve injury (Xu XJ et al 1997, Ramer et al 1998). By measuring release of CGRP, it seems that IL-6, in combination with its soluble receptor, can sensitize nociceptors in the skin to thermal stimuli (Obreja et al 2002), and electrophysiological experiments have also reported that this cytokine can sensitize joint afferents to mechanical stimulation (Brenn et al 2007). The effect of IL-6 on pain behavior could be direct since IL-6 can elicit calcium transients in around 33% of DRG neurons in vitro (von Banchet et al 2005). This evidence is suggestive of a role of IL-6 as a peripheral pain mediator, and this notion has been highlighted recently by a study using an antigen-induced arthritis model. Here, local neutralization of the IL-6/soluble IL-6 receptor complex with soluble gp130 in the knee joint significantly attenuated mechanical hyperalgesia to a greater extent than did repeated systemic delivery (Boetteger et al 2010). Like IL-1β and TNF-α, therapies that block IL-6 are used clinically and have been shown to reduce pain scores in RA patients (Smolen et al 2008, Burmester et al 2011).
These same immune-related factors and others can also act as pain mediators in the CNS, and such actions are discussed in Chapter 4.
Chemokines
Chemokines are chemotactic cytokines and have a key role in immune cell recruitment. They are small molecules (8–10 kDa) and are structurally related with four conserved cysteine residues. They signal via GPCRs, and a level of complexity is added by the fact that multiple chemokines may signal via one receptor. Like a number of cytokines, there is good preclinical evidence to suggest that some chemokines, particularly CCL2 and CX3CL1, are able to modulate pain processing at the level of the spinal cord. However, there is also evidence suggesting that this family of chemotactic cytokines can act as peripheral pain mediators. For example, inflammation or tissue injury can up-regulate numerous chemokine ligands, and the application of such factors can induce pain-related behavior (as highlighted in Table 3-2 and in Fig. 3-2A). These actions can be achieved either through direct actions on nociceptive neurons or through the recruitment of immune cells (as shown in Fig. 3-2B) and the subsequent release of other algogenic factors.
Table 3-2
Chemokines as Peripheral Pain Mediators
GENE NAME |
FC IN RAT |
FC IN HUMAN |
| CXCL5 | 51.3 (20.5–128.2)* | 82.5 (45.4–150.0)* |
| iNOS | 34.3 (3.1–385.1)‡ | −1.1 (−2.2–1.8) |
| IL-24 | 32.7 (8.3–128.8)* | 63.7 (44.5–91.3)* |
| CXCL2 | 24.6 (3.1–198.4)* | 12.0 (8.0–18.0)* |
| CCL4 | 15.4 (6.6–35.8)* | 2.5 (1.4–4.5)‡ |
| IL-6 | 14.8 (4.1–53.9)* | 54.7 (30.3–99.0)* |
| CCL2 | 14.6 (5.3–40.6)* | 5.1 (3.8–7.0)* |
| CCL7 | 14.2 (6.2–32.6)* | 13.8 (4.2–44.8)* |
| CXCL7 | 14.0 (2.8–70.5)§ | 4.0 (1.8–8.6) |
| CCL11 | 11.6 (5.9–22.9)* | 4.2 (1.1–16.6) |
| IL-10 | 10.7 (5.4–21.2)* | 8.0 (4.1–158)* |
| IL-3 | 9.0 (3.4–23.3)§ | ND |
| G-CSF | 7.1 (−1.2–62.2) | 25.0 (10.7–58.5)* |
| IL-19 | 6.2 (3.0–12.6)§ | ND |
| CCL3 | 6.0 (2.9–18.9)§ | 16.6 (10.7–25.7)* |
| CXCL4 | 6.0 (3.9–9.2)* | 2.1 (−1.1–5.0) |
| KGF | 5.8 (3.2–10.5)* | 4.3 (2.9–6.5)* |
| CXCL1 | 5.4 (1.9–15.5)§ | 18.9 (12.6–28.4)* |
| IL-1β | 5.0 (1.3–18.7)‡ | 10.3 (5.9–17.8)* |
| COX-2 | 4.6 (2.0–10.6)§ | 5.3 (3.0–9.5)* |
Numerous chemokines can be up-regulated by tissue injury or inflammation, and one such example is ultraviolet B (UVB) irradiation. In both human and rat skin the transcriptional expression of various chemokines is increased at the peak of UVB-induced hyperalgesia when compared with control skin.
§P < 0.01; mean FC (±1 SD range).
From Dawes JM, Calvo M, Perkins JR, et al 2011 CXCL5 mediates UVB irradiation–induced pain. Science Translational Medicine 3(90):90ra60, Table 1.
Figure 3-2 Chemokines as peripheral pain mediators.
A, When injected into naïve rats, the chemokine, CXCL5, was able to recapitulate ultraviolet B–induced mechanical hypersensitivity. B, In addition, the increase in mechanical sensitivity was associated with the infiltration of numerous neutrophils and macrophages into the chemokine-treated skin. (From Dawes JM, Calvo M, Perkins JR, et al 2011 CXCL5 mediates UVB irradiation–induced pain. Science Translational Medicine 3(90):90ra60, Fig. 3A–C.)
A number of chemokines have been shown to modulate peripheral pain pathways. This was first shown with the intraplantar injection of exogenous human CXCL8, also known as IL-8, which induced dose-dependent mechanical hyperalgesia in rats (Cunha et al 1991). Interestingly, there is no direct rodent ortholog of this chemokine, but CXCL1 seems to elicit similar effects when given to naïve rats, and antiserum against it attenuates carrageenan-induced mechanical hyperalgesia (Lorenzetti et al 2002, Qin et al 2005). In terms of the influence that chemokines have in persistent pain states, the majority of work suggests a prominent role for the CCL2/CCR2 axis. CCL2 is up-regulated in peripheral tissues in neuropathic (Perrin et al 2005, Fu et al 2010) and inflammatory pain states such as intraplantar CFA injection (Jeon et al 2008). Indeed, up-regulation in a number of different pain models was recently emphasized in a meta-analysis of micro-array studies (LaCroix-Fralish et al 2011). When injected into the paw, CCL2 is able to elicit both thermal and mechanical hyperalgesia (Abbadie et al 2003, Qin et al 2005, Bogen et al 2009). In addition to these findings, ablation of CCR2, the predominant receptor for CCL2, prevents the development of mechanical hyperalgesia following nerve injury (Abbadie et al 2003). CCL2 therefore acts as a peripheral pain mediator in the setting of nerve injury and/or inflammation. Another closely related chemokine, CCL3, can produce pain-related hypersensitivity when applied peripherally (Zhang et al 2005a, Eijkelkamp et al 2010) and is able to recapitulate neuropathic pain–like behavior when given intraneurally (Kiguchi et al 2010). One possible mechanism by which chemokines may influence the perception of nociceptive input is via direct interaction with sensory afferents. A number of chemokines can directly act on DRG neurons, as seen with calcium imaging (Oh et al 2001). These same chemokines were also able to induce pain-related behavior when injected into the paw. Subsequent to this work it was shown that CCL3 was capable of sensitizing TRPV1 on DRG neurons and that sodium currents in cultured sensory neurons could be enhanced by incubation with CXCL1 (Wang et al 2008). In addition, CCL3 might also be able to desensitize opioid receptors on sensory neurons, thereby preventing the analgesic effects of endogenous opioids released following tissue injury (Zhang et al 2004). The idea that chemokines can act directly on sensory neurons requires that appropriate receptors be expressed by these cells. Following nerve injury the chemokine receptors CCR2, CCR5, and CXCR4 can be expressed by DRG neurons, and cells in this condition have been shown to increase their responsiveness to a number of chemokine ligands, including CCL2, CCL5, CXCL10, and CXCL12 (White et al 2005; Sun et al 2006; Bhangoo et al 2007, 2009; Jung et al 2008). One study using in vivo electrophysiological techniques showed that after a chronic compression injury of the spinal nerve, DRG neurons were depolarized by CCL2 and lowered their threshold for activation with this ligand (Sun et al 2006). Although this effect was clear in DRG neurons from injured animals, some responses were also measured in neurons from uninjured ganglia (Sun et al 2006). Via an immunohistochemical approach in naïve rats, expression of both CCR1 and CXCR2 has been found in a high proportion of sensory neurons from naïve animals (Zhang et al 2005a, Wang et al 2008). In addition, Oh and colleagues (2001) detected mRNA for CXCR4, CX3CR1, CCR4, and CCR5 in DRGs, as well as staining for CXCR4 and CCR4 in vitro. Therefore, a range of chemokines released by either resident or infiltrating cells into damaged tissue could act directly on nociceptor nerve terminals to enhance pain perception.
Chemokines may also act indirectly to induce pain-related hypersensitivity. For example, the pro-algesic actions of both CXCL8 and CXCL1 have been attributed to their ability to induce the release of sympathetic amines from resident cells (Cunha et al 1991, 1992, 2005; Ben-Baruch et al 1995; Lorenzetti et al 2002), which can act to directly sensitize nociceptors. The majority of these indirect effects, however, are most likely to involve the recruitment of immune cells, cells that are known to infiltrate areas of damaged tissue. CXCL1-induced hyperalgesia is associated with neutrophil recruitment into the treated peripheral tissue (Cunha et al 2008a, 2008b). This chemokine attracts neutrophils predominantly through activation of the CXCR2 receptor. Systemic treatment with a CXCR1/2 inhibitor or specific antagonism of CXCR2 is able to attenuate the mechanical hyperalgesia induced by peripheral injection of carrageenan, zymosan, LPS, and CFA and that caused by nerve injury (Cunha et al 2008a, Manjavachi et al 2010). These analgesic effects are associated with reduced neutrophil infiltration. The ultraviolet B (UVB) model of inflammatory pain is associated with both neutrophil and macrophage infiltration and up-regulation of numerous chemokines at the peak of both thermal and mechanical hyperalgesia (Dawes et al 2011). Neutralization of one of the most overexpressed chemokines, CXCL5, which also acts via the CXCR2 receptor, was able to reduce the UVB-induced mechanical hyperalgesia and infiltration of immune cells. In addition, the pro-algesic properties of this chemokine in naïve animals involved the recruitment of both neutrophils and macrophages (Dawes et al 2011).
A number of chemokines are up-regulated in injured peripheral nerves. One of these, CCL2, is particularly pivotal in the recruitment of macrophages (Toews et al 1998)—cells that seem crucial for the full development of neuropathic pain (see the previous section on immune cells; Liu et al 2000, Barclay et al 2007). With the use of bi-transgenic reporter mice in a focal demyelination model of neuropathic pain it has been suggested that a large proportion of the pro-nociceptive effects of CCL2 occur in peripheral nerves because of its action on CCR2-expressing leukocytes (Jung et al 2009). In this same model, disruption of this interaction with a CCR2 antagonist significantly attenuates neuropathic pain–like behavior (Bhangoo et al 2007). CCL3 is also up-regulated in injured nerves (Perrin et al 2005, Kiguchi et al 2010). Local neutralization of CCL3 was able to attenuate both thermal and mechanical hyperalgesia following nerve damage, and this was associated with a reduction in the level of macrophages (Kiuchi et al 2010).
Resolution of Inflammation
The inflammatory cascade is under complex regulatory control, and regulatory factors include anti-inflammatory cytokines (IL-4, IL-10, IL-13, transforming growth factor [TGF-β]), promoters of resolution (lipoxins, neuroprotectins, maresins, resolvins), endocannabinoids, and inhibitors of pro-inflammatory signaling pathways (inhibitor of the nuclear factor NF-κB, complement inhibitors, IL-1R antagonist, co-stimulatory molecules, and MAPK phosphatases). These systems may be exploited to terminate inflammation, and a number of these mediators have been shown to have analgesic actions. An example is the resolvins, which are endogenous lipid mediators derived from ω-3 fatty acids. These factors promote resolution of inflammation through inhibition of leukocyte recruitment and can directly modulate sensory transduction and synaptic plasticity within the dorsal horn (Serhan et al 2002, Xu et al 2010, Park et al 2011). They have been shown to have potent analgesic actions in inflammatory pain states (Xu et al 2010, Park et al 2011).
Neurotrophic Factors
In 1996 a study was published on the genetic basis of the congenital insensitivity to pain observed in a single family (Indo et al 1996). A mutation was identified in the gene encoding a tyrosine kinase receptor known as trkA. This protein is the high-affinity receptor for a single trophic factor, NGF, and the mutation disrupts the normal signaling of NGF. This single example provides a startling example of the importance of trophic factors in general and NGF in particular for normal nociceptive functioning. Several clinical trials have indicated the analgesic efficacy of blocking NGF, and we now have a good understanding of the mechanisms by which NGF interacts with pain-signaling systems, which we review here.
Neurotrophic factors can be defined as factors that regulate the long-term survival, growth, or differentiated function of discrete populations of nerve cells. There are many neurotrophic factors, and multiple factors can affect a single population of neurons. However, trophic factors fall into a smaller number of families, with members being related by high levels of structural homology or by the common or related receptors that they use in exerting biological actions. The number of factors identified as affecting sensory processing is limited. Most data relate to just two families of factors: the neurotrophin family and the GDNF-related family. Here we primarily consider one member of the neurotrophin family—NGF. Other members of the neurotrophin family are BDNF, neurotrophin-3, and neurotrophin-4/5 (Gotz et al 1994, Lindsay 1996). In general, these members share around 50% of their amino acid sequence. At physiological concentrations, the neurotrophins exist as homodimers. The neurotrophins are initially expressed as pre-pro-precursors, which when processed, yield highly basic mature proteins of around 13 kDa (120 amino acids). These pre-pro-precursors themselves might be biologically active, and currently there is much discussion whether some of the pro-forms of neurotrophins are secreted and act on specific receptors (Teng et al 2010). Binding studies have demonstrated the presence of both high- and low-affinity binding sites for NGF on responsive cell lines (Bothwell 1995). Two different classes of neurotrophin receptor have now been characterized (for reviews see Barbacid 1995, Chao and Hempstead 1995). The first to be cloned was the p75 or low-affinity NGF receptor LNGFR, which binds all the neurotrophins more or less equally with relatively low affinity (Chao et al 1986). Additionally, there is a family of high-affinity receptors, trks, that are tyrosine kinase receptors (Kaplan et al 1991). The p75 receptor is thought to play several roles and may serve as the preferred receptor for pro-NGF. It can also interact with the trk receptors and modulate the specificity and sensitivity of neurotrophin interaction. There are three known members of the trk family of receptors, trkA, trkB, and trkC, and all show different specificities for the neurotrophins. NGF is the preferred ligand for trkA, BDNF and neurotrophin-4/5 are the preferred ligands for trkB, and neurotrophin-3 is the preferred ligand for trkC (Ip et al 1993).
A great deal of the information about signal transduction following trk activation comes from the study of events following activation of trk by NGF in PC12 cells. After NGF binding, receptor dimerization occurs, which is critical for receptor activation (Clary et al 1994). The tyrosine kinase domain of the receptor is activated and a number of substrates are phosphorylated; autophosphorylation of the receptor also occurs. There is now a large body of evidence demonstrating that neurotrophin receptors are expressed in specific populations of DRG cells. With double-labeling techniques it has been possible to relate receptor expression to different functional classes of DRG cells. Multiple approaches have demonstrated that approximately 40% of DRG cells express the NGF receptor trkA (Verge et al 1989, 1992; McMahon et al 1994; Averill et al 1995; Kashiba et al 1995; Molliver et al 1995; Wetmore and Olson 1995), and cells that express trkA are principally of small cell diameter. TrkA is expressed principally in the peptidergic population of small-diameter DRG cells, whereas very few non-peptidergic (isolectin B4-binding) small-diameter DRG cells express trkA (Averill et al 1995, Molliver et al 1995). Some of the myelinated DRG cells (i.e., those that express neurofilament 200) do express trkA (around 20%). TrkA-immunoreactive terminals within the spinal cord are present within laminae I and IIouter. TrkA is therefore expressed in small-diameter DRG cells that express CGRP and project to the superficial laminae of the spinal cord. These are all characteristic of nociceptive afferents. Thus, about half of the nociceptors in adult animals express both p75 and trkA and are therefore likely to be sensitive to NGF. The other half of the nociceptor population does not express any trk receptor, nor p75. Rather, they express receptors for members of the GDNF family of receptors. Interestingly, these two populations of C fibers have different central terminations, even though it is not yet clear whether they have distinct functional roles (although the receptors that they express do indicate that they can be activated by different putative pain mediators).
During development it appears that functionally distinct groups of sensory neurons depend on different neurotrophins for survival. Animals with a gene deletion of either NGF or trkA are born with DRGs lacking virtually all small-diameter primary sensory neurons, including the peptidergic neurons expressing CGRP and substance P (Crowley et al 1994). These animals are, as expected, profoundly hypo-algesic. In utero deprivation of NGF, achieved by antibody treatment, produces similar effects (Johnson et al 1980, Ruit et al 1992), and this phenotype is equivalent to that seen in patients with loss-of-function mutations in trkA, which also leads to loss of peripheral pain-signaling neurons.
The developmental dependence of nociceptors on NGF for survival is lost in the postnatal period, some time before the second week of life in the rat and presumably in the first few years of life in humans. However, NGF continues to exert profound effects on adult nociceptors. Adult DRG neurons can be cultured in the absence of added trophic factors (Lindsay 1988). If NGF is then added to these cultures, extensive neurite outgrowth of trkA-positive cells is promoted. NGF in these cultures also regulates expression of the neuropeptides substance P and CGRP (Lindsay and Harmar 1989). In addition, NGF regulates the chemical sensitivity of cultured sensory neurons. For example, the sensitivity of cultured sensory neurons to the potent algogen capsaicin is increased by NGF, as is their sensitivity to protons and to γ-aminobutyric acid (GABA) (Winter et al 1988, Bevan and Winter 1995). Expression of bradykinin binding sites in cultured sensory neurons has also been shown to be regulated by NGF (Petersen et al 1998), apparently in a p75 receptor–dependent manner. This marked regulation of the chemosensitivity of cultured sensory neurons by NGF is interesting in relation to the association between NGF and inflammatory pain, and the in vivo effects of NGF are discussed here.
The effects of NGF extend from the peripheral to the central terminals of sensory neurons, and many are mediated via altered gene expression in neurons expressing trkA. These effects are summarized in Figure 3-3.
Figure 3-3 Summary of the biological effects of exogenous nerve growth factor (NGF) on pain-signaling systems in normal animals (A) and in animals with nerve injury (B).
In normal animals, NGF causes peripheral sensitization of some nociceptors, in part directly as a result of NGF binding to receptors on nociceptors and in part indirectly by the release of algogens from other cell types. NGF also regulates the expression of many genes in trkA-expressing neurons, ranging from transmitters and modulators to ion channels and receptors. In the spinal cord, NGF produces central sensitization via altered expression of putative neuromodulators, particularly brain-derived neurotrophic factor (BDNF). CGRP, calcitonin gene–related peptide; TRPV1, transient receptor potential vanilloid 1.
Administration of small doses of NGF to adult animals and humans can produce pain and hyperalgesia. In rodents, thermal hyperalgesia is present within 30 minutes of systemic NGF administration and both thermal and mechanical hyperalgesia after a couple of hours (Lewin et al 1993). Subcutaneous injections of NGF also produce both thermal and mechanical hyperalgesia at the injection site. In humans, intravenous injections of very low doses of NGF produce widespread aching pain in deep tissues and hyperalgesia at the injection site (Petty et al 1994). Detailed quantitative sensory testing in human volunteers has demonstrated long-lasting mechanical and thermal hypersensitivity following the intradermal injection of NGF (Rukweid et al 2010). The rapid onset of some of these effects and their localization to the injection site strongly suggest that they arise, at least in part, from a local effect on the peripheral terminals of nociceptors. This has been substantiated by the observation that acute administration of NGF can sensitize nociceptive afferents to thermal and chemical stimuli (Rueff and Mendell 1996). Cutaneous nociceptors chronically exposed to elevated NGF levels (in an NGF-overexpressing mouse) show marked heat sensitization (Stucky et al 1998). Recordings of primary afferents innervating porcine skin following NGF application have demonstrated reduced activity-dependent slowing in mechanically insensitive afferents and increased ongoing activity, thus emphasizing the potential effects of NGF on axonal excitability (Obreja et al 2011).
NGF produces sensitization of nociceptors by several mechanisms. Some of these mechanisms are direct (that is, they follow activation of trkA on nociceptors), and some are indirect and mediated by NGF inducing the release of other algogens from a variety of peripheral cell types. The direct mechanisms involve both altered gene expression and post-translational regulation of receptors and ion channels. There are now multiple examples of post-translational changes induced by NGF that involve phosphorylation of receptors and ion channels, although other actions are possible, such as altered trafficking of receptors. The heat sensitization of nociceptors induced by NGF is prominent and rapid. Phosphorylation of particular residues on TRPV1 receptors appears to account for most of the effect. However, the intracellular cascades responsible are disputed, with published data supporting a critical role for PKA, PKC, MAPK ERK1/2, or a mechanism involving inhibition of PIP2 (see Bonnington and McNaughton 2003). NGF has also been shown to enhance mechanically activated currents in cultured sensory neurons (Di Castro et al 2006). The post-translational modification of some ion channels, particularly TTX-resistant sodium channels, by NGF may likewise contribute to the sensitization of nociceptors by this agent (see Zhang et al 2002 and references therein).
Because cellular elements other than nociceptors in peripheral tissues express trkA, some of the sensitization of nociceptors by NGF may arise indirectly, and some of these elements have already been discussed. Mast cells contain a number of inflammatory mediators known to excite primary afferents, including histamine and serotonin (Leon et al 1994), and some types of mast cells express trkA receptors (Horigome et al 1994). NGF can produce mast cell degranulation (Mazurek et al 1986, Horigome et al 1994) and increase the proliferation of mast cells resident in tissue. In peritoneal mast cell cultures, NGF induces the expression of a number of cytokines (Bullock and Johnson 1996). Mast cell degranulators and serotonin antagonists have both been demonstrated to partially prevent the thermal but not the mechanical hyperalgesia (Lewin et al 1994, Woolf et al 1996) that occurs in response to NGF. These degranulators can significantly reduce hyperalgesia (both thermal and mechanical) and the up-regulation of NGF expression induced by CFA (Woolf et al 1996).
In skin, NGF may also affect keratinocytes, some of which express p75 receptors. NGF increases the proliferation of keratinocytes in culture (Paus et al 1994, Fantini et al 1995), and this process may contribute to tissue remodeling after inflammation. In addition, NGF may also target eosinophils and convert circulating eosinophils into tissue-type eosinophils (Hamada et al 1996), and it has been reported to increase B- and T-cell proliferation (Otten et al 1989), thus suggesting a role for NGF as an immunoregulatory factor.
There may be an interaction between NGF and sympathetic neurons during inflammation. Sympathetic efferents also possess the trkA receptor (Smeyne et al 1994). Surgical or chemical sympathectomy can reduce the short-latency thermal and mechanical hyperalgesia evoked by NGF (Andreev et al 1995, Woolf et al 1996). Production of eicosanoids by sympathetic efferents within the skin has been suggested to contribute to hyperalgesia in some inflammatory conditions (Levine et al 1986b). However, a role for eicosanoids in NGF-induced hyperalgesia is unlikely since it is unaffected by treatment with the NSAID indomethacin (Amann et al 1996).
Effects of NGF on Gene Expression and the Phenotypic Properties of Sensory Neurons
Not all the algogenic and hyperalgesic effects of NGF can readily be explained by peripheral processes. Some aspects of NGF actions are centrally mediated via altered gene expression in nociceptors. There appear to be a group of peptides that are constitutively expressed in trkA cells and whose expression is controlled mainly by NGF, with an increase following NGF supplementation and a decrease following NGF depletion (resulting, for instance, from peripheral axotomy). CGRP and substance P belong to this group (Goedert et al 1981, Otten et al 1984, Verge et al 1995). Based on NGF’s ability to reverse some axotomy-induced increases in peptide, it would appear that there is also a group of peptides whose production is partly inhibited by neurotrophins; vasoactive intestinal peptide, cholecystokinin, neuropeptide Y, and galanin belong to this group. NGF also represses expression of the transcription factor ATF3 (Averill et al 2004). In addition to an effect on substance P and CGRP, NGF has been shown to produce a dramatic up-regulation of BDNF mRNA and protein in trkA-expressing DRG cells (Apfel et al 1996, Michael et al 1997). This is interesting since there is good evidence to suggest that BDNF may serve as a central regulator of excitability (Pezet et al 2002, Coull et al 2005).
NGF also regulates the expression of some of the receptors expressed by nociceptors. Capsaicin sensitivity is increased in vivo by NGF. NGF-sensitive nociceptors (i.e., those expressing trkA) have the highest levels of TRPV1 (Tominaga et al 1998). Because TRPV1 is sensitive to heat and also to protons, regulation by NGF is likely to have consequences for the responsiveness of nociceptors to noxious stimuli. NGF can also positively regulate the expression of other ligand-gated ion channels in nociceptors, including several ASICs (Mamet et al 2003) and the ATP receptor P2X3. In addition, NGF may alter the excitability of sensory neurons by changing the expression of several voltage-gated ion channels, for instance, the sodium channel Nav1.8 (Black and Waxman 1996, Boucher et al 2000). Because some forms of nociceptor sensitization appear to be mediated through this channel (Gold and Levine 1996), this provides another potential mechanism by which NGF might regulate pain signaling.
Actions on Spinal Processing of Nociceptive Information: Central Sensitization
NGF given systemically fails to penetrate into the spinal cord. There is also little trkA expression in the spinal cord, with the receptor largely being restricted to the terminals of primary afferent nociceptors (Averill et al 1995). One might therefore imagine that exogenously administered NGF would have little effect on spinal nociceptive processes. However, several of the biological effects of NGF described earlier are capable of leading to secondary effects on the spinal cord. First, activation and sensitization of primary afferent nociceptors may lead to sufficient afferent activity to trigger central changes. Second, the altered chemistry of afferent neurons produced by NGF may lead to increased neurotransmitter or neuromodulator release from nociceptive afferent terminals (Malcangio et al 1997). Release of some sensory neuropeptides can contribute to the induction of central sensitization. One might therefore expect that peripheral NGF treatment could lead to the induction of central sensitization.
It has been shown that several hours after systemic NGF treatment, C-fiber stimulation produces greater than normal amounts of central sensitization, seen as wind-up of ventral root reflexes (Thompson et al 1995). A fibers also develop the novel ability to produce wind-up. Such changes in the central processing of nociceptive information may occur during inflammation secondary to expression of substance P within A fibers (Neumann et al 1996). There is considerable evidence that the ability of NGF to up-regulate BDNF expression in some nociceptors may prove to be the most significant mechanism by which NGF regulates the sensitivity of spinal processing of noxious stimuli.
Functional Role of NGF
Because mice with NGF or trkA deletions rarely survive past the first postnatal week, most of what we know about endogenous NGF function in the adult has been determined by the use of blocking agents. A number of studies have used autoimmune models of NGF deprivation or infusions of NGF antisera to study the effects of NGF in normal adult animals. Local infusion of trkA–IgG (an NGF-neutralizing reagent; Shelton et al 1995) into the rat hindpaw leads to thermal hypo-algesia and a decrease in CGRP content in DRG neurons projecting to the infused area (McMahon et al 1995). These changes take several days to develop. In addition, there is a decrease in the thermal and chemical sensitivity of nociceptors projecting to the area and a reduction in epidermal innervation density (Bennett et al 1998a). These results provide strong evidence that NGF continues to play an important role in regulating the function of small, peptidergic sensory neurons in the adult.
By far the most extensively studied area of endogenous NGF function in the adult is in models of relatively persistent inflammatory pain (lasting at least several days). NGF is found in many cell types in tissues subjected to an inflammatory insult, and much evidence now supports the hypothesis that up-regulation of NGF levels is a common component of the inflammatory response that relates to hyperalgesia. Elevated NGF levels have been found in a variety of inflammatory states in humans, including in the bladder of patients with cystitis (Lowe et al 1997), and increased levels are present in the synovial fluid of patients with arthritis (Aloe et al 1992) and in the cerebrospinal fluid of fibromyalgia patients (Sarchielli et al 2007). In animal studies, NGF is found in the exudate produced during blistering of the skin (Weskamp and Otten 1987) and is elevated in skin after inflammation induced by CFA (Donnerer et al 1992, Woolf et al 1994), IL-1β (Safieh-Garabedian et al 1995), ultraviolet light (Gillardon et al 1995), or TNF-α (Woolf et al 1997).
There is now widespread agreement that blocking NGF can impede many of the effects of inflammation on sensory nerve function. For instance, intraplantar injection of carrageenan produces an acute inflammatory reaction, which has previously been widely used to study the analgesic effects of NSAIDs. When the trkA–IgG molecule was co-administered with carrageenan, it could almost completely prevent development of the thermal hyperalgesia that normally occurs (McMahon et al 1995; Fig. 3-4A). The properties of nociceptive afferents innervating carrageenan-inflamed skin have also been studied. Following carrageenan inflammation, there was a marked increase in spontaneous activity in these afferents and increased thermal and chemical sensitivity (Fig. 3-4B). This probably represents the multiple peripheral actions of NGF described earlier. Administration of the trkA–IgG molecule could largely prevent these changes (Bennett et al 1996), and similar results have now been found in a number of different inflammatory models (see Pezet and McMahon 2006).
Figure 3-4 The role of nerve growth factor (NGF) in inflammation as revealed by use of the NGF-sequestering protein trkA–IgG.
A, The thermal hyperalgesia that develops in rats in the hours following intraplantar carrageenan. The ordinate plots the ratio of the withdrawal times to radiant noxious heat applied to the inflamed paw and the non-inflamed contralateral paw. Most of the expected thermal hyperalgesia fails to develop in animals inflamed and concurrently treated with trkA–IgG. B, Effects of carrageenan inflammation on the properties of primary afferent nociceptors. Recordings were made from an isolated skin–nerve preparation a few hours after the inflammatory stimulus was given in vivo, and afferents were tested for their responses to a ramp increase in skin temperature. In inflamed skin, some nociceptors develop spontaneous activity and show thermal sensitization. In animals inflamed with carrageenan and concurrently treated with trkA–IgG, the thermal sensitization of nociceptors is completely blocked. The inset on the right shows the average stimulus–response functions for nociceptors in these groups. (After McMahon SB, Bennett DL, Priestley JV, et al 1995 The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nature Medicine 1:774–780; and Koltzenburg M, Bennett DL, Shelton DL, et al 1999 Neutralization of endogenous NGF prevents the sensitization of nociceptors supplying inflamed skin. European Journal of Neuroscience 11:1698–1704.)
The increased NGF levels observed after inflammatory stimuli result from increased synthesis and release of NGF from a variety of cell types, including keratinocytes, smooth muscle cells, and Schwann cell (Heumann et al 1987, Raychaudhuri et al 1998, Freund et al 2002). IL-1β and TNF-α have been shown to drive changes in NGF expression during inflammation in vivo, and the hyperalgesia produced by these cytokines can be prevented by NGF antagonism.
Role of NGF in Clinical Pain States
Findings from the sequestration of endogenous NGF and the administration of exogenous NGF suggest that this factor is important in modulating the sensitivity of the sensory nervous system to noxious stimuli. The evidence that NGF levels increase during inflammation, which is derived from studies using NGF antagonism, makes a strong case for NGF being a critical mediator of inflammatory pain. NGF clearly has a powerful neuroprotective effect on small-diameter sensory neurons, and NGF levels have been shown to change in a number of models of nerve injury. The idea that NGF does act as a mediator in persistent pain states has recently been tested in a series of clinical trials with encouraging results. The persistent pain associated with OA has a strong peripheral drive and represents an ideal platform to test whether NGF may act as a peripheral pain mediator in this context. In a 16-week study of patients with OA of the knee, tanezumab, a humanized monoclonal antibody directed against NGF, given at 8-week intervals dose-dependently reduced the pain associated with walking (Lane et al 2010). This effect lasted for the whole study period and was maximal with the highest dose, which on average reduced pain scores by about 75% (Fig. 3-5). The analgesic effects of blocking NGF are not limited to OA. Tanezumab also significantly reduced pain scores in patients with lower back pain and pain arising from inflammation of the bladder (Evans et al 2011, Katz et al 2011). In addition, in patients with lower back pain, tanezumab outperformed the NSAID naproxen (Katz et al 2011). These findings represent clear evidence of the important role that a signal mediator can play in patients with persistent pain. However The development of tanezumab and other anti-NGF antibodies for widespread use was halted because of adverse events observed in a phase II trial involving OA. Here, worsening OA developed in 16 tanezumab-treated patients and joint replacement therapy was required. The mechanisms behind these osteonecrotic events are unclear but potentially suggest a modulatory role of NGF on joint homeostasis. Alternatively, some level of hyponociception caused by excessive NGF neutralization may have resulted in accelerated progression of OA because of the overuse of damaged joints. However, it seems that in more than half the cases, the bone necrosis occurred in previously unaffected joints. Many other trials in which NGF was neutralized have not observed such severe adverse events, and it has been reported that repeated doses of tanezumab induce a favorable side effect profile (Schnitzer et al 2011). The block on NGF clinical trials was lifted in early 2012, and it is likely that analgesic efficacy will now be examined in multiple clinical trials.
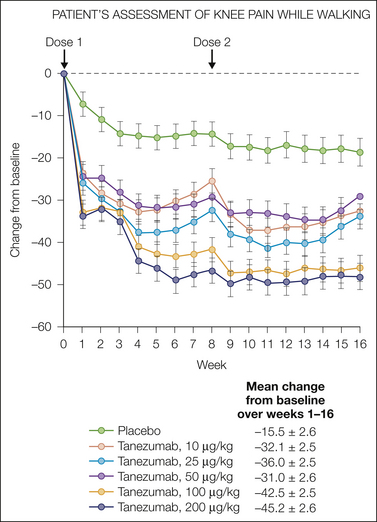
Figure 3-5 Neutralizing nerve growth factor in patients with osteoarthritis significantly reduces pain scores.
At baseline, visual analog scale scores for patients were obtained to assess the level of pain experienced while walking. Patients then received either placebo or varying doses of tanezumab (10, 25, 50, 100, 200 μg/kg) at the start of the study and again after 8 weeks. A decrease in the score represents a reduction in pain scores. (Data from Lane NE, Schnitzer TJ, Birbara CA, et al 2010 Tanezumab for the treatment of pain from osteoarthritis of the knee. New England Journal of Medicine 363:1521–1531.)
Other Neurotrophic Factors As Peripheral Pain Mediators
In addition to NGF, preclinical data also suggest that other neurotrophic factors can act as peripheral pain mediators. The neurotrophins NT3 and BDNF both induce thermal hypersensitivity in rats when injected into the hindpaw, and following nerve injury, neutralization of BDNF in the periphery can reduce the increase in thermal hypersensitivity (Theodosiou et al 1999). In the adult system the non-peptidergic nociceptive fibers lack trkA but instead express c-Ret, the prominent receptor for GDNF (Snider and McMahon 1998). When injected into the paw of naïve animals, GDNF is reported to lower thermal pain–related thresholds (Malin et al 2006). However, this factor might not act as a pro-algesic mediator in persistent pain states since its application to nerve-injured rats is analgesic (Boucher et al 2000). Artemin, a member of the GDNF family of ligands, is also able elicit thermal hypersensitivity when given intradermally. In inflammation induced by CFA injection, artemin is greatly up-regulated at a transcriptional level in the skin in the first 24 hours (Malin et al 2007). It has also been observed that in genetically modified mice that overexpress artemin in the skin, sensitivity to both thermal and cold stimuli is increased (Elitt et al 2006).
Conclusion
The number of inflammatory pain mediators and modulators has grown steadily and now includes not only a variety of small molecules such as bradykinin, prostanoids, ATP, protons, and NO but also numerous cytokines, chemokines, and growth factors. The number of sources of such mediators has also increased and includes several or many immune cells, glial cells, and neurons. Finally, it has become clear that these mediators have diverse mechanisms and sites of action, including activation and sensitization of nociceptive terminals, regulation of primary nociceptive phenotype, and in the spinal cord, presynaptic control of nociceptor transmitter release and post-synaptic control of neuronal excitability. One of the most critical issues, of course, is to identify the relative importance of all these different mediators and mechanisms in particular pain states. This may seem a difficult job given the known interaction of many inflammatory mediators. However, the success of one series of new agents, TNF-α function–blocking molecules, as both disease-modifying and pain-relieving agents in several autoimmune disorders, including RA, and also the promise shown by anti-NGF antibodies give hope that this increased understanding of basic mechanisms will translate into effective new therapies for painful disorders.
The references for this chapter can be found at www.expertconsult.com.
References
Abbadie C., Lindia J.A., Cumiskey A.M., et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7947–7952.
Abbott F.V., Hong Y., Blier P. Activation of 5-HT2A receptors potentiates pain produced by inflammatory mediators. Neuropharmacology. 1996;35:99–110.
Aley K.O., McCarter G., Levine J.D. Nitric oxide signaling in pain and nociceptor sensitization in the rat. Journal of Neuroscience. 1998;18:7008–7014.
Aloe L., Tuveri M.A., Carcassi U., et al. Nerve growth factor in the synovial fluid of patients with chronic arthritis. Arthritis and Rheumatism. 1992;35:351–355.
Amadesi S., Nie J., Vergnolle N., et al. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. Journal of Neuroscience. 2004;24:4300–4312.
Amaya F., Wang H., Costigan M., et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. Journal of Neuroscience. 2006;26:12852–12860.
Amaya-Castellanos E., Pineda-Farias J.B., Castaneda-Corral G., et al. Blockade of 5-HT7 receptors reduces tactile allodynia in the rat. Pharmacology, Biochemistry, and Behavior. 2011;99:591–597.
Amann R., Schuligoi R., Herzeg G., et al. Intraplantar injection of nerve growth factor into the rat hind paw: local edema and effects on thermal nociceptive threshold. Pain. 1996;64:323–329.
Andersson D.A., Gentry C., Moss S., et al. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. Journal of Neuroscience. 2008;28:2485–2494.
Andreev N.Y., Dimitrieva N., Koltzenburg M., et al. Peripheral administration of nerve growth factor in the adult rat produces a thermal hyperalgesia that requires the presence of sympathetic post-ganglionic neurons. Pain. 1995;63:109–115.
Apfel S.C., Wright D.E., Wiideman A.M., et al. Nerve growth factor regulates the expression of brain-derived neurotrophic factor mRNA in the peripheral nervous system. Molecular and Cellular Neurosciences. 1996;7:134–142.
Asano M., Hatori C., Inamura N., et al. Effects of a nonpeptide bradykinin B2 receptor antagonist, FR167344, on different in vivo animal models of inflammation. British Journal of Pharmacology. 1997;122:1436–1440.
Asfaha S., Brussee V., Chapman K., et al. Proteinase-activated receptor-1 agonists attenuate nociception in response to noxious stimuli. British Journal of Pharmacology. 2002;135:1101–1106.
Averill S., McMahon S.B., Clary D.O., et al. Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. European Journal of Neuroscience. 1995;7:1484–1494.
Baker M.D. Protein kinase C mediates up-regulation of tetrodotoxin-resistant, persistent Na+ current in rat and mouse sensory neurones. Journal of Physiology. 2005;567:851–867.
Bandell M., Story G.M., Hwang S.W., et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857.
Bandtlow C.E., Heumann R., Schwab M.E., et al. Cellular localization of nerve growth factor synthesis by in situ hybridization. EMBO Journal. 1987;6:891–899.
Bannenberg G., Serhan C.N. Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochimica et Biophysica Acta. 2010;1801:1260–1273.
Banner L.R., Patterson P.H. Major changes in the expression of the mRNAs for cholinergic differentiation factor leukemia inhibitory factor and its receptor after injury to adult peripheral-nerves and ganglia. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:7109–7113.
Banner L.R., Patterson P.H., Allchorne A., et al. Leukemia inhibitory factor is an anti-inflammatory and analgesic cytokine. Journal of Neuroscience. 1998;18:5456–5462.
Barbacid M. Neurotrophic factors and their receptors. Current Opinion in Cell Biology. 1995;7:148–155.
Barclay J., Clark A.K., Ganju P., et al. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain. 2007;130:225–234.
Barclay J., Patel S., Dorn G., et al. Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. Journal of Neuroscience. 2002;22:8139–8147.
Barrette B., Hebert M.A., Filali M., et al. Requirement of myeloid cells for axon regeneration. Journal of Neuroscience. 2008;28:9363–9376.
Baumann T.K., Chaudhary P., Martenson M.E. Background potassium channel block and TRPV1 activation contribute to proton depolarization of sensory neurons from humans with neuropathic pain. European Journal of Neuroscience. 2004;19:1343–1351.
Bautista D.M., Jordt S.E., Nikai T., et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282.
Benarroch E.E. Nitric oxide: a pleiotropic signal in the nervous system. Neurology. 2011;77:1568–1576.
Ben-Baruch A., Michiel D.F., Oppenheim J.J. Signals and receptors involved in recruitment of inflammatory cells. Journal of Biological Chemistry. 270, 1995. 11703–11706, 1995
Bennett D.L.H., Koltzenburg M., Priestley J.V., et al. Endogenous nerve growth factor regulates the sensitivity of nociceptors in the adult rat. European Journal of Neuroscience. 1998;10:1282–1291.
Bennett D.L.H., McMahon S.B., Shelton D., et al. NGF sequestration using a trkA–IgG fusion molecule prevents primary afferent sensitisation to carrageenan inflammation. 8th World Congress on Pain. 1996;35:120.
Bennett G., Al-Rashed S., Hoult J.R., et al. Nerve growth factor induced hyperalgesia in the rat hind paw is dependent on circulating neutrophils. Pain. 1998;77:315–322.
Bevan S., Winter J. Nerve growth factor (NGF) differentially regulates the chemosensitivity of adult rat cultured sensory neurons. Journal of Neuroscience. 1995;15:4918–4926.
Beyak M.J., Vanner S. Inflammation-induced hyperexcitability of nociceptive gastrointestinal DRG neurones: the role of voltage-gated ion channels. Neurogastroenterology and Motility. 2005;17:175–186.
Bhangoo S., Ren D., Miller R.J., et al. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Molecular Pain. 2007;3:38.
Bhangoo S.K., Ripsch M.S., Buchanan D.J., et al. Increased chemokine signaling in a model of HIV1-associated peripheral neuropathy. Molecular Pain. 2009;5:48.
Bhave G., Hu H.J., Glauner K.S., et al. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12480–12485.
Binshtok A.M., Wang H., Zimmermann K., et al. Nociceptors are interleukin-1beta sensors. Journal of Neuroscience. 2008;28:14062–14073.
Birrell G.J., McQueen D.S., Iggo A., et al. PGI2-induced activation and sensitization of articular mechanonociceptors. Neuroscience Letters. 1991;124:5–8.
Bisgaard H., Kristensen J.K. Leukotriene B4 produces hyperalgesia in humans. Prostaglandins. 1985;30:791–797.
Black J.A., Waxman S.G. Sodium channel expression: a dynamic process in neurons and nonneuronal cells. Developmental Neuroscience. 1996;18:139–152.
Bland-Ward P.A., Humphrey P.P. Acute nociception mediated by hindpaw P2X receptor activation in the rat. British Journal of Pharmacology. 1997;122:365–371.
Bleehen T., Keele C.A. Observations on the algogenic actions of adenosine compounds on the human blister base preparation. Pain. 1977;3:367–377.
Bonnington J.K., McNaughton P.A. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. Journal of Physiology (London). 2003;551:433–446.
Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annual Review of Neuroscience. 1995;18:223–253.
Boucher T.J., Okuse K., Bennett D.L., et al. Potent analgesic effects of GDNF in neuropathic pain states. Science. 2000;290:124–127.
Bouthillier J., DeBois D., Marceau F. Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vitro and in vivo. British Journal of Pharmacology. 1987;92:257–264.
Bizzarri C., Beccari A.R., Bertini R., et al. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacology & Therapeutics. 2006;112:139–149.
Boettger M.K., Leuchtweis J., Kummel D., et al. Differential effects of locally and systemically administered soluble glycoprotein 130 on pain and inflammation in experimental arthritis. Arthritis Research & Therapy. 2010;12:R140.
Bogen O., Dina O.A., Gear R.W., et al. Dependence of monocyte chemoattractant protein 1 induced hyperalgesia on the isolectin B4–binding protein versican. Neuroscience. 2009;159:780–786.
Bolin L.M., Verity A.N., Silver J.E., et al. Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. Journal of Neurochemistry. 1995;64:850–858.
Boyce S., Rupniak N.M., Carlson E.J., et al. Nociception and inflammatory hyperalgesia in B2 bradykinin receptor knockout mice. Immunopharmacology. 1996;33:333–335.
Bradbury E.J., Burnstock G., McMahon S.B. The expression of P2X3 purinoreceptors in sensory neurons: effects of axotomy and glial-derived neurotrophic factor. Molecular and Cellular Neurosciences. 1998;12:256–268.
Brenn D., Richter F., Schaible H.G. Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: an inflammatory mechanism of joint pain. Arthritis and Rheumatism. 2007;56:351–359.
Bullock E.D., Johnson E.M., Jr. Nerve growth factor induces the expression of certain cytokine genes and bcl-2 in mast cells. Potential role in survival promotion. Journal of Biological Chemistry. 1996;271:27500–27508.
Burgess G.M., Mullaney J., McNeil M., et al. Second messengers involved in the action of bradykinin on cultured sensory neurones. Journal of Neuroscience. 1989;9:3314–3325.
Burgess G.M., Perkins M.N., Rang H.P., et al. Bradyzide, a potent non-peptide B2 bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. British Journal of Pharmacology. 2000;129:77–86.
Burmester G.R., Feist E., Kellner H., et al. Effectiveness and safety of the interleukin 6-receptor antagonist tocilizumab after 4 and 24 weeks in patients with active rheumatoid arthritis: the first phase IIIb real-life study (TAMARA). Annals of the Rheumatic Diseases. 2011;70:755–759.
Calder J.S., Holten I., McAllister R.M. Evidence for immune system involvement in reflex sympathetic dystrophy. Journal of Hand Surgery, (Edinburgh, Scotland). 1998;23:147–150.
Calixto J.B., Cabrini D.A., Ferreira J., et al. Kinins in pain and inflammation. Pain. 2000;87:1–5.
Calixto J.B., Medeiros R., Fernandes E.S., et al. Kinin B1 receptors: key G-protein–coupled receptors and their role in inflammatory and painful processes. British Journal of Pharmacology. 2004;143:803–818.
Campos M.M., Calixto J.B. Involvement of B1 and B2 receptors in bradykinin-induced rat paw oedema. British Journal of Pharmacology. 1995;114:1005–1013.
Cao L., Deleo J.A. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection–induced neuropathic pain. European Journal of Immunology. 2008;38:448–458.
Cardenas L.M., Cardenas C.G., Scroggs R.S. 5HT increases excitability of nociceptor-like rat dorsal root ganglion neurons via cAMP-coupled TTX-resistant Na+ channels. Journal of Neurophysiology. 2001;86:241–248.
Carr M.J., Kollarik M., Meeker S.N., et al. A role for TRPV1 in bradykinin-induced excitation of vagal airway afferent nerve terminals. Journal of Pharmacology and Experimental Therapeutics. 2003;304:1275–1279.
Casanova-Molla J., Morales M., Planas-Rigol E., et al. Epidermal Langerhans cells in small fiber neuropathies. Pain. 2012;153:982–989.
Cattaruzza F., Lyo V., Jones E., et al. Cathepsin S is activated during colitis and causes visceral hyperalgesia by a PAR2-dependent mechanism in mice. Gastroenterology. 2011;141:1864–1874.
Cesare P., Dekker L.V., Sardini A., et al. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624.
Chao M.V., Bothwell M.A., Ross A.H., et al. Gene transfer and molecular cloning of the human NGF receptor. Science. 1986;232:518–521.
Chao M.V., Hempstead B.L. p75 and trk: a two-receptor system. Trends in Neurosciences. 1995;18:321–326.
Chen X., Molliver D.C., Gebhart G.F. The P2Y2 receptor sensitizes mouse bladder sensory neurons and facilitates purinergic currents. Journal of Neuroscience. 2010;30:2365–2372.
Chen Y., Boettger M.K., Reif A., et al. Nitric oxide synthase modulates CFA-induced thermal hyperalgesia through cytokine regulation in mice. Molecular Pain. 2010;6:13.
Cheng J.K., Ji R.R. Intracellular signaling in primary sensory neurons and persistent pain. Neurochemistry Research. 2008;33:1970–1978.
Cherniak N.S., Runold M., Prabhakar N.R., et al. Effect of adenosine on vagal sensory pulmonary afferents. Federation Proceedings. 1987;46:825.
Chuang H.H., Prescott E.D., Kong H., et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962.
Clary D.O., Weskamp G., Austin L.R., et al. TrkA cross-linking mimics neuronal responses to nerve growth factor. Molecular Biology of the Cell. 1994;5:549–563.
Cockayne D.A., Hamilton S.G., Zhu Q.M., et al. Urinary bladder hyporeflexia and reduced pain related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015.
Cook S.P., McCleskey E.W. Cell damage excites nociceptors through release of cytosolic ATP. Pain. 2002;95:41–47.
Costigan M., Moss A., Latremoliere A., et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain–like hypersensitivity. Journal of Neuroscience. 2009;29:14415–14422.
Coutts A.A., Jorizzo J.L., Eady R.A., et al. Adenosine triphosphate–evoked vascular changes in human skin: mechanism of action. European Journal of Pharmacology. 1981;76:391–401.
Cui J.G., Holmin S., Mathiesen T., et al. Possible role of inflammatory mediators in tactile hypersensitivity in rat models of mononeuropathy. Pain. 2000;88:239–248.
Cunha F.Q., Lorenzetti B.B., Poole S., et al. Interleukin-8 as a mediator of sympathetic pain. British Journal of Pharmacology. 1991;104:765–767.
Cunha F.Q., Poole S., Lorenzetti B.B., et al. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. British Journal of Pharmacology. 1992;107:660–664.
Cunha J.M., Sachs D., Canetti C.A., et al. The critical role of leukotriene B4 in antigen-induced mechanical hyperalgesia in immunised rats. British Journal of Pharmacology. 2003;139:1135–1145.
Cunha T.M., Barsante M.M., Guerrero A.T., et al. Treatment with DF 2162, a non-competitive allosteric inhibitor of CXCR1/2, diminishes neutrophil influx and inflammatory hypernociception in mice. British Journal of Pharmacology. 2008;154:460–470.
Cunha T.M., Verri W.A., Jr., Fukada S.Y., et al. TNF-alpha and IL-1beta mediate inflammatory hypernociception in mice triggered by B1 but not B2 kinin receptor. European Journal of Pharmacology. 2007;573:221–229.
Cunha T.M., Verri W.A., Jr., Schivo I.R., et al. Crucial role of neutrophils in the development of mechanical inflammatory hypernociception. Journal of Leukocyte Biology. 2008;83:824–832.
Cunha T.M., Verri W.A., Jr., Silva J.S., et al. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1755–1760.
Dai Y., Moriyama T., Higashi T., et al. Proteinase activated receptor 2–mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. Journal of Neuroscience. 2004;24:4293–4299.
Dai Y., Wang S., Tominaga M., et al. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. Journal of Clinical Investigation. 2007;117:1979–1987.
Davis A.J., Perkins M.N. Induction of B1 receptors in vivo in a model of persistent inflammatory mechanical hyperalgesia in the rat. Neuropharmacology. 1994;33:127–133.
Dawes J.M., Calvo M., Perkins J.R., et al. CXCL5 mediates UVB irradiation–induced pain. Science Translational Medicine. 3(90), 2011. 90ra60
DeBlois D., Bouthillier J., Marceau F. Effect of glucocorticoids, monoamines and growth factors on the spontaneously developing responses of the rabbit isolated aorta to des-Arg9-bradykinin. British Journal of Pharmacology. 1988;93:969–977.
den Broeder A.A., Wanten G.J., Oyen W.J., et al. Neutrophil migration and production of reactive oxygen species during treatment with a fully human anti–tumor necrosis factor-alpha monoclonal antibody in patients with rheumatoid arthritis. Journal of Rheumatology. 2003;30:232–237.
Deval E., Gasull X., Noel J., et al. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacology & Therapeutics. 2010;128:549–558.
Deval E., Noel J., Lay N., et al. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO Journal. 2008;27:3047–3055.
Devor M., White D.M., Goetzl E.J., et al. Eicosanoids, but not tachykinins, excite C-fiber endings in rat sciatic nerve-end neuromas. Neuroreport. 1992;3:21–24.
Di Castro A., Drew L.J., Wood J.N., et al. Modulation of sensory neuron mechanotransduction by PKC- and nerve growth factor–dependent pathways. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4699–4704.
Doerner J.F., Gisselmann G., Hatt H., et al. Transient receptor potential channel A1 is directly gated by calcium ions. Journal of Biological Chemistry. 2007;282:13180–13189.
Donnerer J., Schuligoi R., Stein C. Increased content and transport of substance P and calcitonin gene–related peptide in sensory nerves innervating inflamed tissue: evidence for a regulatory function of nerve growth factor in vivo. Neuroscience. 1992;49:693–698.
Dorn G., Patel S., Wotherspoon G., et al. siRNA relieves chronic neuropathic pain. Nucleic Acids Research. 2004;32:e49.
Dowd E., McQueen D.S., Chessell I.P., et al. P2X receptor–mediated excitation of nociceptive afferents in the normal and arthritic rat knee joint. British Journal of Pharmacology. 1998;125:341–346.
Dray A., Perkins M. Bradykinin and inflammatory pain. Trends in Neurosciences. 1993;16:99–104.
Dray A., Patel I.A., Perkins M.N., et al. Bradykinin-induced activation of nociceptors: receptor and mechanistic studies on the neonatal rat spinal cord–tail preparation in vitro. British Journal of Pharmacology. 1992;107:1129–1134.
Drummond P.D. The effect of cutaneous mast cell degranulation on sensitivity to heat. Inflammation Research. 2004;53:309–315.
Ebersberger A., Grubb B.D., Willingale H.L., et al. The intraspinal release of prostaglandin E2 in a model of acute arthritis is accompanied by an up-regulation of cyclo-oxygenase-2 in the spinal cord. Neuroscience. 1999;93:775–781.
Eijkelkamp N., Heijnen C.J., Willemen H.L., et al. GRK2: a novel cell-specific regulator of severity and duration of inflammatory pain. Journal of Neuroscience. 2010;30:2138–2149.
Elitt C.M., McIlwrath S.L., Lawson J.J., et al. Artemin overexpression in skin enhances expression of TRPV1 and TRPA1 in cutaneous sensory neurons and leads to behavioral sensitivity to heat and cold. Journal of Neuroscience. 2006;26:8578–8587.
Emery E.C., Young G.T., Berrocoso E.M., et al. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. 2011;333:1462–1466.
Ernberg M., Hedenberg-Magnusson B., Kurita H., et al. Effects of local serotonin administration on pain and microcirculation in the human masseter muscle. Journal of Orofacial Pain. 2006;20:241–248.
Ernberg M., Lundeberg T., Kopp S. Pain and allodynia/hyperalgesia induced by intramuscular injection of serotonin in patients with fibromyalgia and healthy individuals. Pain. 2000;85:31–39.
Esquisatto L.C., Costa S.K., Camargo E.A., et al. The plasma protein extravasation induced by adenosine and its analogues in the rat dorsal skin: evidence for the involvement of capsaicin sensitive primary afferent neurones and mast cells. British Journal of Pharmacology. 2001;134:108–115.
Evans R.J., Moldwin R.M., Cossons N., et al. Proof of concept trial of tanezumab for the treatment of symptoms associated with interstitial cystitis. Journal of Urology. 2011;185:1716–1721.
Fabbretti E., D’Arco M., Fabbro A., et al. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene–related peptide. Journal of Neuroscience. 2006;26:6163–6171.
Fantini F., Giannetti A., Benassi L., et al. Nerve growth factor receptor and neurochemical markers in human oral mucosa: an immunohistochemical study. Dermatology. 1995;190:186–191.
Ferreira J., Campos M.M., Araujo R., et al. The use of kinin B1 and B2 receptor knockout mice and selective antagonists to characterize the nociceptive responses caused by kinins at the spinal level. Neuropharmacology. 2002;43:1188–1197.
Ferreira J., Campos M.M., Pesquero J.B., et al. Evidence for the participation of kinins in Freund’s adjuvant–induced inflammatory and nociceptive responses in kinin B1 and B2 receptor knockout mice. Neuropharmacology. 2001;41:1006–1012.
Fioravanti A., Fabbroni M., Cerase A., et al. Treatment of erosive osteoarthritis of the hands by intra-articular infliximab injections: a pilot study. Rheumatology International. 2009;29:961–965.
Fleischmann R., Stern R., Iqbal I. Anakinra: an inhibitor of IL-1 for the treatment of rheumatoid arthritis. Expert Opinion on Biological Therapy. 2004;4:1333–1344.
Ford A.P. In pursuit of P2X3 antagonists: novel therapeutics for chronic pain and afferent sensitization. Purinergic Signal. 2012;8:3–26.
Ford-Hutchinson A.W., Bray M.A., Doig M.V., et al. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:264–265.
Fox A., Wotherspoon G., McNair K., et al. Regulation and function of spinal and peripheral neuronal B1 bradykinin receptors in inflammatory mechanical hyperalgesia. Pain. 2003;104:683–691.
Fox A., Kaur S., Li B., et al. Antihyperalgesic activity of a novel nonpeptide bradykinin B1 receptor antagonist in transgenic mice expressing the human B1 receptor. British Journal of Pharmacology. 2005;144:889–899.
Fu E.S., Zhang Y.P., Sagen J., et al. Transgenic inhibition of glial NF-kappa B reduces pain behavior and inflammation after peripheral nerve injury. Pain. 2010;148:509–518.
Gardiner N.J., Cafferty W.B., Slack S.E., et al. Expression of gp130 and leukaemia inhibitory factor receptor subunits in adult rat sensory neurones: regulation by nerve injury. Journal of Neurochemistry. 2002;83:100–109.
Gillardon F., Eschenfelder C., Rush R.A., et al. Increase in neuronal Jun immunoreactivity and epidermal NGF levels following UV exposure of rat skin. Neuroreport. 1995;6:1322–1324.
Godinez-Chaparro B., Barragan-Iglesias P., Castaneda-Corral G., et al. Role of peripheral 5-HT(4), 5-HT(6), and 5-HT(7) receptors in development and maintenance of secondary mechanical allodynia and hyperalgesia. Pain. 2011;152:687–697.
Goedert M., Stoeckel K., Otten U. Biological importance of the retrograde axonal transport of nerve growth factor in sensory neurons. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:5895–5898.
Gold M.S. Tetrodotoxin-resistant Na+ currents and inflammatory hyperalgesia. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7645–7649.
Gold M.S., Levine J.D. DAMGO inhibits prostaglandin E2–induced potentiation of a TTX-resistant Na+ current in rat sensory neurons in vitro. Neuroscience Letters. 1996;212:83–86.
Gordon J.L. Extracellular ATP: effects, sources and fate. Biochemical Journal. 1986;233:309–319.
Gordon S., Taylor P.R. Monocyte and macrophage heterogeneity. Nature Reviews. Immunology. 2005;5:953–964.
Gotz R., Koster R., Winkler C., et al. Neurotrophin-6 is a new member of the nerve growth factor family. Nature. 1994;372:266–269.
Griffin J.W., Thompson W.J. Biology and pathology of nonmyelinating Schwann cells. Glia. 2008;56:1518–1531.
Guerrero A.T., Verri W.A., Jr., Cunha T.M., et al. Involvement of LTB4 in zymosan-induced joint nociception in mice: participation of neutrophils and PGE2. Journal of Leukocyte Biology. 2008;83:122–130.
Hamada A., Watanabe N., Ohtomo H., et al. Nerve growth factor enhances survival and cytotoxic activity of human eosinophils. British Journal of Haematology. 1996;93:299–302.
Hamilton S.G., McMahon S.B., Lewin G.R. Selective activation of nociceptors by P2X receptor agonists in normal and inflamed rat skin. Journal of Physiology. 2001;534:437–445.
Hamilton S.G., Wade A., McMahon S.B. The effects of inflammation and inflammatory mediators on nociceptive behaviour induced by ATP analogues in the rat. British Journal of Pharmacology. 1999;126:326–332.
Hamilton S.G., Warburton J., Bhattacharjee A., et al. ATP in human skin elicits a dose-related pain response which is potentiated under conditions of hyperalgesia. Brain. 2000;123:1238–1246.
Hawkinson J.E., Szoke B.G., Garofalo A.W., et al. Pharmacological, pharmacokinetic, and primate analgesic efficacy profile of the novel bradykinin B1 receptor antagonist ELN441958. Journal of Pharmacology and Experimental Therapeutics. 2007;322:619–630.
Heumann R., Korsching S., Bandtlow C., et al. Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection. Journal of Cell Biology. 1987;104:1623–1631.
Hilliges M., Weidner C., Schmelz M., et al. ATP responses in human C nociceptors. Pain. 2002;98:59–68.
Holthusen H., Arndt J.O. Nitric oxide evokes pain in humans on intracutaneous injection. Neuroscience Letters. 1994;165:71–74.
Honore P., Kage K., Mikusa J., et al. Analgesic profile of intrathecal P2X3 antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain. 2002;99:11–19.
Horigome K., Bullock E.D., Johnson E.M., Jr. Effects of nerve growth factor on rat peritoneal mast cells. Survival promotion and immediate—early gene induction. Journal of Biological Chemistry. 1994;269:2695–2702.
Horigome K., Pryor J.C., Bullock E.D., et al. Mediator release from mast cells by nerve growth factor. Neurotrophin specificity and receptor mediation. Journal of Biological Chemistry. 1993;268:14881–14887.
Huang J., Cai Q., Chen Y., et al. Treatment with ketanserin produces opioid-mediated hypoalgesia in the late phase of carrageenan-induced inflammatory hyperalgesia in rats. Brain Research. 2009;1303:39–47.
Hudson L.J., Bevan S., Wotherspoon G., et al. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. European Journal of Neuroscience. 2001;13:2105–2114.
Hwang S.W., Cho H., Kwak J., et al. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6155–6160.
Iannone F., Trotta F., Montecucco C., et al. Etanercept maintains the clinical benefit achieved by infliximab in patients with rheumatoid arthritis who discontinued infliximab because of side effects. Annals of the Rheumatic Diseases. 2007;66:249–252.
Ip N.Y., Stitt T.N., Tapley P., et al. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149.
Irnich D., Tracey D.J., Polten J., et al. ATP stimulates peripheral axons in human, rat and mouse-differential involvement of A2B adenosine and P2X purinergic receptors. Neuroscience. 2002;110:123–129.
Jarvis M.F., Burgard E.C., McGaraughty S., et al. A-317491, a novel potent and selective nonnucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:17179–17184.
Jarvis M.F., Wismer C.T., Schweitzer E., et al. Modulation of BzATP and formalin induced nociception: attenuation by the P2X receptor antagonist, TNP-ATP and enhancement by the P2X3 allosteric modulator, cibacron blue. British Journal of Pharmacology. 2001;132:259–269.
Jeon S.M., Lee K.M., Park E.S., et al. Monocyte chemoattractant protein-1 immunoreactivity in sensory ganglia and hindpaw after adjuvant injection. Neuroreport. 2008;19:183–186.
Ji R.R., Xu Z.Z., Strichartz G., et al. Emerging roles of resolvins in the resolution of inflammation and pain. Trends in Neurosciences. 2011;34:599–609.
Jin X., Gereau R.W. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. Journal of Neuroscience. 2006;26:246–255.
Johnson E.M., Jr., Gorin P.D., Brandeis L.D., et al. Dorsal root ganglion neurons are destroyed by exposure in utero to maternal antibody to nerve growth factor. Science. 1980;210:916–918.
Jones N., Slater R., Cadiou H., et al. Acid-induced pain and its modulation in humans. Journal of Neuroscience. 2004;24:10974–10979.
Jung H., Bhangoo S., Banisadr G., et al. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. Journal of Neuroscience. 2009;29:8051–8062.
Jung H., Toth P.T., White F.A., et al. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. Journal of Neurochemistry. 2008;104:254–263.
Kaan T.K., Yip P.K., Patel S., et al. Systemic blockade of P2X3 and P2X2/3 receptors attenuates bone cancer pain behaviour in rats. Brain. 2010;133:2549–2564.
Kagan B.L., Baldwin R.L., Munoz D., et al. Formation of ion-permeable channels by tumor necrosis factor-α. Science. 1992;255:1427–1430.
Kaplan D.R., Hempstead B.L., Martin-Zanca D., et al. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558.
Kashiba H., Noguchi K., Ueda Y., et al. Coexpression of trk family members and low-affinity neurotrophin receptors in rat dorsal root ganglion neurons. Molecular Brain Research. 1995;30:158–164.
Katanosaka K., Banik R.K., Giron R., et al. Contribution of TRPV1 to the bradykinin-evoked nociceptive behavior and excitation of cutaneous sensory neurons. Neuroscience Research. 2008;62:168–175.
Katz N., Borenstein D.G., Birbara C., et al. Efficacy and safety of tanezumab in the treatment of chronic low back pain. Pain. 2011;152:2248–2258.
Kawabata A., Kawao N., Kuroda R., et al. Peripheral PAR-2 triggers thermal hyperalgesia and nociceptive responses in rats. Neuroreport. 2001;12:715–719.
Kawabata A., Kawao N., Kuroda R., et al. Specific expression of spinal Fos after PAR-2 stimulation in mast cell-depleted rats. Neuroreport. 2002;13:511–514.
Kawano T., Zoga V., Kimura M., et al. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: action by direct S-nitrosylation. Molecular Pain. 2009;5:12.
Khodorova A., Navarro B., Jouaville L.S., et al. Endothelin-B receptor activation triggers an endogenous analgesic cascade at sites of peripheral injury. Nature Medicine. 2003;9:1055–1061.
Kiguchi N., Maeda T., Kobayashi Y., et al. Macrophage inflammatory protein-1alpha mediates the development of neuropathic pain following peripheral nerve injury through interleukin-1beta up-regulation. Pain. 2010;149:305–315.
Kim C.F., Moalem-Taylor G. Detailed characterization of neuro-immune responses following neuropathic injury in mice. Brain Research. 2011;1405:95–108.
Kim C.F., Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. Journal of Pain. 2011;12:370–383.
Klein R.M., Ufret-Vincenty C.A., Hua L., et al. Determinants of molecular specificity in phosphoinositide regulation. Phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) is the endogenous lipid regulating TRPV1. Journal of Biological Chemistry. 2008;283:26208–26216.
Kleinschnitz C., Hofstetter H.H., Meuth S.G., et al. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Experimental Neurology. 2006;200:480–485.
Kobayashi K., Fukuoka T., Yamanaka H., et al. Neurons and glial cells differentially express P2Y receptor mRNAs in the rat dorsal root ganglion and spinal cord. Journal of Comparative Neurology. 2006;498:443–454.
Kollarik M., Undem B.J. Activation of bronchopulmonary vagal afferent nerves with bradykinin, acid and vanilloid receptor agonists in wild-type and TRPV1−/− mice. Journal of Physiology. 2004;555:115–123.
Kuhr F., Lowry J., Zhang Y., et al. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides. 2010;44:145–154.
LaCroix-Fralish M.L., Austin J.S., Zheng F.Y., et al. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152:1888–1898.
Laas K., Peltomaa R., Puolakka K., et al. Early improvement of health-related quality of life during treatment with etanercept and adalimumab in patients with rheumatoid arthritis in routine practice. Clinical and Experimental Rheumatology. 2009;27:315–320.
Lane N.E., Schnitzer T.J., Birbara C.A., et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. New England Journal of Medicine. 2010;363:1521–1531.
Lang P.M., Moalem-Taylor G., Tracey D.J., et al. Activity-dependent modulation of axonal excitability in unmyelinated peripheral rat nerve fibers by the 5-HT(3) serotonin receptor. Journal of Neurophysiology. 2006;96:2963–2971.
Lawand N.B., Willis W.D., Westlund K.N. Blockade of joint inflammation and secondary hyperalgesia by L-NAME, a nitric oxide synthase inhibitor. Neuroreport. 1997;8:895–899.
Lechner S.G., Lewin G.R. Peripheral sensitisation of nociceptors via G-protein–dependent potentiation of mechanotransduction currents. Journal of Physiology. 2009;587:3493–3503.
Leeb-Lundberg L.M., Marceau F., Muller-Esterl W., et al. International Union of Pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacological Reviews. 2005;57:27–77.
Leffler A., Monter B., Koltzenburg M. The role of the capsaicin receptor TRPV1 and acid-sensing ion channels (ASICS) in proton sensitivity of subpopulations of primary nociceptive neurons in rats and mice. Neuroscience. 2006;139:699–709.
Leon A., Buriani A., Dal T.R., et al. Mast cells synthesize, store, and release nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3739–3743.
Levine J.D., Gooding J., Donatoni P., et al. The role of the polymorphonuclear leukocyte in hyperalgesia. Journal of Neuroscience. 1985;5:3025–3029.
Levine J.D., Lam D., Taiwo Y.O., et al. Hyperalgesic properties of 15-lipoxygenase products of arachidonic acid. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:5331–5334.
Levine J.D., Lau W., Kwiat G., et al. Leukotriene B4 produces hyperalgesia that is dependent on polymorphonuclear leukocytes. Science. 1984;225:743–745.
Levine J.D., Taiwo Y.O., Collins S.D., et al. Noradrenaline hyperalgesia is mediated through interaction with sympathetic postganglionic neurone terminals rather than activation of primary afferent nociceptors. Nature. 1986;323:158–160.
Lewin G.R., Rueff A., Mendell L.M. Peripheral and central mechanisms of NGF-induced hyperalgesia. European Journal of Neuroscience. 1994;6:1903–1912.
Li T., Qi J., Cowley E.A. Activation of the EP prostanoid receptor induces prostaglandin E and pro-inflammatory cytokine production in human airway epithelial cells. Pulmonary Pharmacology & Therapeutics. 2011;24:42–48.
Lin S.Y., Chang W.J., Lin C.S., et al. Serotonin receptor 5-HT2B mediates serotonin-induced mechanical hyperalgesia. Journal of Neuroscience. 2011;31:1410–1418.
Lin Y.W., Tseng T.J., Lin W.M., et al. Cutaneous nerve terminal degeneration in painful mononeuropathy. Experimental Neurology. 2001;170:290–296.
Lindahl O. Pain: a chemical explanation. Acta Rheumatologica Scandinavica. 1962;8:161–169.
Lindenlaub T., Sommer C. Epidermal innervation density after partial sciatic nerve lesion and pain-related behavior in the rat. Acta Neuropathologica. 2002;104:137–143.
Lindenlaub T., Teuteberg P., Hartung T., et al. Effects of neutralizing antibodies to TNF-alpha on pain-related behavior and nerve regeneration in mice with chronic constriction injury. Brain Research. 2000;866:15–22.
Lindsay R.M. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. Journal of Neuroscience. 1988;8:2394–2405.
Lindsay R.M. Role of neurotrophins and trk receptors in the development and maintenance of sensory neurons: an overview. Philosophical Transactions of the Royal Society of London. Series B. Biological Sciences. 1996;351:365–373.
Lindsay R.M., Harmar A.J. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364.
Liu B., Linley J.E., Du X., et al. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl− channels. Journal of Clinical Investigation. 2010;120:1240–1252.
Liu J., Li J.D., Lu J., et al. Contribution of nerve growth factor to upregulation of P2X expression in DRG neurons of rats with femoral artery occlusion. American Journal of Physiology. Heart and Circulatory Physiology. 2011;301:H1070–H1079.
Liu T., Van R.N., Tracey D.J. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain. 2000;86:25–32.
Liu X.Y., Wu S.X., Wang Y.Y., et al. Changes of 5-HT receptor subtype mRNAs in rat dorsal root ganglion by bee venom–induced inflammatory pain. Neuroscience Letters. 2005;375:42–46.
Lorenzetti B.B., Veiga F.H., Canetti C.A., et al. Cytokine-induced neutrophil chemoattractant 1 (CINC-1) mediates the sympathetic component of inflammatory mechanical hypersensitivity in rats. European Cytokine Network. 2002;13:456–461.
Lowe E.M., Anand P., Terenghi G., et al. Increased nerve growth factor levels in the urinary bladder of women with idiopathic sensory urgency and interstitial cystitis. British Journal of Urology. 1997;79:572–577.
Luiz A.P., Schroeder S.D., Chichorro J.G., et al. Kinin B(1) and B(2) receptors contribute to orofacial heat hyperalgesia induced by infraorbital nerve constriction injury in mice and rats. Neuropeptides. 2010;44:87–92.
Lukacs V., Thyagarajan B., Varnai P., et al. Dual regulation of TRPV1 by phosphoinositides. Journal of Neuroscience. 2007;27:7070–7080.
Ma Q.P. The expression of bradykinin B1 receptors on primary sensory neurones that give rise to small caliber sciatic nerve fibres in rats. Neuroscience. 2001;107:665–673.
Madison S., Whitsel E.A., Suarez-Roca H., et al. Sensitizing effects of leukotriene B4 on intradental primary afferents. Pain. 1992;49:99–104.
Malcangio M., Garrett N.E., Cruwys S., et al. Nerve growth factor– and neurotrophin-3–induced changes in nociceptive threshold and the release of substance P from the rat isolated spinal cord. Journal of Neuroscience. 1997;17:8459–8467.
Malin S.A., Davis B.M., Koerber H.R., et al. Thermal nociception and TRPV1 function are attenuated in mice lacking the nucleotide receptor P2Y2. Pain. 2008;138:484–496.
Malin S.A., Molliver D.C. Gi- and Gq-coupled ADP (P2Y) receptors act in opposition to modulate nociceptive signaling and inflammatory pain behavior. Molecular Pain. 2010;6:21.
Malin S.A., Molliver D.C., Koerber H.R., et al. Glial cell line–derived neurotrophic factor family members sensitize nociceptors in vitro and produce thermal hyperalgesia in vivo. Journal of Neuroscience. 2006;26:8588–8599.
Manjavachi M.N., Quintao N.L., Campos M.M., et al. The effects of the selective and non-peptide CXCR2 receptor antagonist SB225002 on acute and long-lasting models of nociception in mice. European Journal of Pain. 2010;14:23–31.
Marceau F., Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nature Reviews. Drug Discovery. 2004;3:845–852.
Marchand F., Perretti M., McMahon S.B. Role of the immune system in chronic pain. Nature Reviews. Neuroscience. 2005;6:521–532.
Martin H.A. Leukotriene B4 induced decrease in mechanical and thermal thresholds of C-fiber mechanonociceptors in rat hairy skin. Brain Research. 1990;509:273–279.
Martin H.A., Basbaum A.I., Kwiat G.C., et al. Leukotriene and prostaglandin sensitization of cutaneous high threshold C- and A-™ mechanoreceptors in the hairy skin of rat hindlimbs. Neuroscience. 1987;22:651–659.
Matsuoka I., Meyer M., Thoenen H. Cell-type–specific regulation of nerve growth factor (NGF) synthesis in non-neuronal cells: comparison of Schwann cells with other cell types. Journal of Neuroscience. 1991;11:3165–3177.
Mazurek N., Weskamp G., Erne P., et al. Nerve growth factor induces mast cell degranulation without changing intracellular calcium levels. FEBS Letters. 1986;198:315–320.
McGehee D.S., Oxford G.S. Bradykinin modulates the electrophysiology of cultured rat sensory neurons through a pertussis toxin–insensitive G protein. Molecular Cellular Neuroscience. 1991;2:21–30.
McLatchie L.M., Bevan S. The effects of pH on the interaction between capsaicin and the vanilloid receptor in rat dorsal root ganglia neurons. British Journal of Pharmacology. 2001;132:899–908.
McMahon S.B., Armanini M.P., Ling L.H., et al. Expression and coexpression of Trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron. 1994;12:1161–1171.
McMahon S.B., Bennett D.L., Priestley J.V., et al. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nature Medicine. 1995;1:774–780.
McMahon S.B., Cafferty W.B.J., Neurotrophic influences on neuropathic pain. Pathological pain: from molecular to clinical aspects. Novartis Foundation Symposium, 261. Chichester, UK: Wiley; 2004:68–102.
McNamee K.E., Alzabin S., Hughes J.P., et al. IL-17 induces hyperalgesia via TNF-dependent neutrophil infiltration. Pain. 2011;152:1838–1845.
Metcalfe D.D., Baram D., Mekori Y.A. Mast cells. Physiological Reviews. 1997;77:1033–1079.
Michael G.J., Averill S., Nitkunan A., et al. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. Journal of Neuroscience. 1997;17:8476–8490.
Minami T., Nakano H., Kobayashi T., et al. Characterization of EP receptor subtypes responsible for prostaglandin E2–induced pain responses by use of EP1 and EP3 receptor knockout mice. British Journal of Pharmacology. 2001;133:438–444.
Miyamoto T., Dubin A.E., Petrus M.J., et al. TRPV1 and TRPA1 mediate peripheral nitric oxide–induced nociception in mice. PLoS One. 2009;4:e7596.
Mizumura K., Sato J., Kumazawa T. Effects of prostaglandins and other putative chemical intermediaries on the activity of canine testicular polymodal receptors studied in vitro. Pflügers Archiv. 1987;408:565–572.
Mizumura K., Sugiura T., Katanosaka K., et al. Excitation and sensitization of nociceptors by bradykinin: what do we know? Experimental Brain Research. 2009;196:53–65.
Moalem G., Grafe P., Tracey D.J. Chemical mediators enhance the excitability of unmyelinated sensory axons in normal and injured peripheral nerve of the rat. Neuroscience. 2005;134:1399–1411.
Moalem G., Xu K., Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129:767–777.
Moalem-Taylor G., Allbutt H.N., Iordanova M.D., et al. Pain hypersensitivity in rats with experimental autoimmune neuritis, an animal model of human inflammatory demyelinating neuropathy. Brain, Behavior, and Immunity. 2007;21:699–710.
Mohapatra D.P., Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. Journal of Biological Chemistry. 2005;280:13424–13432.
Molliver D.C., Cook S.P., Carlsten J.A., et al. ATP and UTP excite sensory neurons and induce CREB phosphorylation through the metabotropic receptor, P2Y2. European Journal of Neuroscience. 2002;16:1850–1860.
Molliver D.C., Radeke M.J., Feinstein S.C., et al. Presence or absence of trkA protein distinguishes subsets of small sensory neurons with unique cytochemical characteristics and dorsal horn projections. Journal of Comparative Neurology. 1995;361:404–416.
Moriyama T., Iida T., Kobayashi K., et al. Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1–mediated thermal hypersensitivity. Journal of Neuroscience. 2003;23:6058–6062.
Myers R.R., Heckman H.M., Rodriguez M. Reduced hyperalgesia in nerve-injured WLD mice: relationship to nerve fiber phagocytosis, axonal degeneration, and regeneration in normal mice. Experimental Neurology. 1996;141:94–101.
Nakamura A., Fujita M., Shiomi H. Involvement of endogenous nitric oxide in the mechanism of bradykinin-induced peripheral hyperalgesia. British Journal of Pharmacology. 1996;117:407–412.
Neumann S., Doubell T.P., Leslie T., et al. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature. 1996;384:360–364.
Nicholson R., Small J., Dixon A.K., et al. Serotonin receptor mRNA expression in rat dorsal root ganglion neurons. Neuroscience Letters. 2003;337:119–122.
Nicol G.D., Lopshire J.C., Pafford C.M. Tumor necrosis factor enhances the capsaicin sensitivity of rat sensory neurons. Journal of Neuroscience. 1997;17:975–982.
Obreja O., Ringkamp M., Turnquist B., et al. Nerve growth factor selectively decreases activity-dependent conduction slowing in mechano-insensitive C-nociceptors. Pain. 2011;152:2138–2146.
Obreja O., Schmelz M., Poole S., et al. Interleukin-6 in combination with its soluble IL-6 receptor sensitises rat skin nociceptors to heat, in vivo. Pain. 2002;96:57–62.
O’Garra A., Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends in Cell Biology. 2000;10:542–550.
Oh S.B., Tran P.B., Gillard S.E., et al. Chemokines and glycoprotein 120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. Journal of Neuroscience. 2001;21:5027–5035.
Oida H., Namba T., Sugimoto Y., et al. In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. British Journal of Pharmacology. 1995;116:2828–2837.
Okamoto K., Imbe H., Morikawa Y., et al. 5-HT2A receptor subtype in the peripheral branch of sensory fibers is involved in the potentiation of inflammatory pain in rats. Pain. 2002;99:133–143.
Okubo M., Yamanaka H., Kobayashi K., et al. Expression of leukotriene receptors in the rat dorsal root ganglion and the effects on pain behaviors. Molecular Pain. 2010;6:57.
Oliveira M.C., Pelegrini-da-Silva A., Tambeli C.H., et al. Peripheral mechanisms underlying the essential role of P2X3,2/3 receptors in the development of inflammatory hyperalgesia. Pain. 2009;141:127–134.
Omote K., Hazama K., Kawamata T., et al. Peripheral nitric oxide in carrageenan-induced inflammation. Brain Research. 2001;912:171–175.
Otten U., Baumann J.B., Girard J. Nerve growth factor induces plasma extravasation in rat skin. European Journal of Pharmacology. 1984;106:199–201.
Otten U., Ehrhard P., Peck R. Nerve growth factor induces growth and differentiation of human B lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:10059–10063.
Parada C.A., Tambeli C.H., Cunha F.Q., et al. The major role of peripheral release of histamine and 5-hydroxytryptamine in formalin-induced nociception. Neuroscience. 2001;102:937–944.
Park C.K., Xu Z.Z., Liu T., et al. Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin D1, D2, and E1. Journal of Neuroscience. 2011;31:18433–18438.
Pastore S., Mascia F., Girolomoni G. The contribution of keratinocytes to the pathogenesis of atopic dermatitis. European Journal of Dermatology. 2006;16:125–131.
Patwardhan A.M., Scotland P.E., Akopian A.N., et al. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18820–18824.
Paukert M., Osteroth R., Geisler H.S., et al. Inflammatory mediators potentiate ATP-gated channels through the P2X3 subunit. Journal of Biological Chemistry. 2001;276:21077–21082.
Paus R., Luftl M., Czarnetzki B.M. Nerve growth factor modulates keratinocyte proliferation in murine skin organ culture. British Journal of Dermatology. 1994;130:174–180.
Pellegatti P., Raffaghello L., Bianchi G., et al. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One. 2008;3:e2599.
Perkins M.N., Campbell E., Dray A. Antinociceptive activity of the B1 and B2 receptor antagonists desArg9Leu8Bk and HOE 140, in two models of persistent hyperalgesia in the rat. Pain. 1993;53:191–197.
Perkins M.N., Kelly D. Induction of bradykinin B1 receptors in vivo in a model of ultra-violet irradiation–induced thermal hyperalgesia in the rat. British Journal of Pharmacology. 1993;110:1441–1444.
Perkins M.N., Kelly D. Interleukin-1®–induced desArg9bradykinin-mediated thermal hyperalgesia in the rat. Neuropharmacology. 1994;33:657–660.
Perkins M.N., Kelly D., Davis A.J. Bradykinin B1 and B2 receptor mechanisms and cytokine-induced hyperalgesia in the rat. Canadian Journal of Physiology and Pharmacology. 1995;73:832–836.
Perkins N.M., Tracey D.J. Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience. 2000;101:745–757.
Perrin F.E., Lacroix S., Aviles-Trigueros M., et al. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in wallerian degeneration. Brain. 2005;128:854–866.
Pesquero J.B., Araujo R.C., Heppenstall P.A., et al. Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:8140–8145.
Petcu M., Dias J.P., Ongali B., et al. Role of kinin B1 and B2 receptors in a rat model of neuropathic pain. International Immunopharmacology. 2008;8:188–196.
Petersen M., Segond V.B., Heppelmann B., et al. Nerve growth factor regulates the expression of bradykinin binding sites on adult sensory neurons via the neurotrophin receptor p75. Neuroscience. 1998;83:161–168.
Petho G., Derow A., Reeh P.W. Bradykinin-induced nociceptor sensitization to heat is mediated by cyclooxygenase products in isolated rat skin. European Journal of Neuroscience. 2001;14:210–218.
Petty B.G., Cornblath D.R., Adornato B.T., et al. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Annals of Neurology. 1994;36:244–246.
Pezet S., Malcangio M., McMahon S.B. BDNF: a neuromodulator in nociceptive pathways? Brain Research Reviews. 2002;40:240–249.
Pezet S., McMahon S.B. Neurotrophins: mediators and modulators of pain. Annual Review of Neuroscience. 2006;29:507–538.
Pinto L.G., Cunha T.M., Vieira S.M., et al. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain. 2010;148:247–256.
Pollock J., McFarlane S.M., Connell M.C., et al. TNF-α receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology. 2002;42:93–106.
Premkumar L.S., Ahern G.P. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990.
Prescott E.D., Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284–1288.
Qin X., Wan Y., Wang X. CCL2 and CXCL1 trigger calcitonin gene–related peptide release by exciting primary nociceptive neurons. Journal of Neuroscience Research. 2005;82:51–62.
Ramer M.S., Bradbury E.J., McMahon S.B. Nerve growth factor induces P2X3 expression in sensory neurons. Journal of Neurochemistry. 2001;77:864–875.
Ramer M.S., French G.D., Bisby M.A. Wallerian degeneration is required for both neuropathic pain and sympathetic sprouting into the DRG. Pain. 1997;72:71–78.
Ramer M.S., Murphy P.G., Richardson P.M., et al. Spinal nerve lesion–induced mechanoallodynia and adrenergic sprouting in sensory ganglia are attenuated in interleukin-6 knockout mice. Pain. 1998;78:115–121.
Rang H.P., Ritchie J.M. Depolarization of nonmyelinated fibers of the rat vagus nerve produced by activation of protein kinase C. Journal of Neuroscience. 1988;8:2606–2617.
Regoli D., Marceau F., Barabe J. De novo formation of vascular receptors for bradykinin. Canadian Journal of Physiology and Pharmacology. 1978;56:674–677.
Ren K., Dubner R. Interactions between the immune and nervous systems in pain. Nature Medicine. 2010;16:1267–1276.
Renganathan M., Cummins T.R., Waxman S.G. Nitric oxide blocks fast, slow, and persistent Na+ channels in C-type DRG neurons by S-nitrosylation. Journal of Neurophysiology. 2002;87:761–775.
Ribeiro R.A., Vale M.L., Thomazzi S.M., et al. Involvement of resident macrophages and mast cells in the writhing nociceptive response induced by zymosan and acetic acid in mice. European Journal of Pharmacology. 2000;387:111–118.
Rong W., Hillsley K., Davis J.B., et al. Jejunal afferent nerve sensitivity in wild-type and TRPV1 knockout mice. Journal of Physiology. 2004;560:867–881.
Ru F., Surdenikova L., Brozmanova M., et al. Adenosine-induced activation of esophageal nociceptors. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2011;300:G485–G493.
Rudick C.N., Bryce P.J., Guichelaar L.A., et al. Mast cell–derived histamine mediates cystitis pain. PLoS One. 2008;3:e2096.
Rueff A., Dray A. 5-hydroxytryptamine–induced sensitization and activation of peripheral fibers in the neonatal rat are mediated via different 5-hydroxytryptamine receptors. Neuroscience. 1992;50:899–905.
Rueff A., Mendell L.M. Nerve growth factor NT-5 induce increased thermal sensitivity of cutaneous nociceptors in vitro. Journal of Neurophysiology. 1996;76:3593–3596.
Ruit K.G., Elliott J.L., Osborne P.A., et al. Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron. 1992;8:573–587.
Rukwied R., Mayer A., Kluschina O., et al. NGF induces non-inflammatory localized and lasting mechanical and thermal hypersensitivity in human skin. Pain. 2010;148:407–413.
Rupniak N.M., Boyce S., Webb J.K., et al. Effects of the bradykinin B1 receptor antagonist des-Arg9[Leu8]bradykinin and genetic disruption of the B2 receptor on nociception in rats and mice. Pain. 1997;71:89–97.
Russo A., Soh U.J., Trejo J. Proteases display biased agonism at protease-activated receptors: location matters!. Molecular Interventions. 2009;9:87–96.
Ruts L., Drenthen J., Jongen J.L., et al. Pain in Guillain-Barré syndrome: a long-term follow-up study. Neurology. 2010;75:1439–1447.
Sachs D., Cunha F.Q., Ferreira S.H. Peripheral analgesic blockade of hypernociception: activation of arginine/NO/cGMP/protein kinase G/ATP-sensitive K+ channel pathway. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3680–3685.
Safieh-Garabedian B., Poole S., Allchorne A., et al. Contribution of interleukin-1 beta to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. British Journal of Pharmacology. 1995;115:1265–1275.
Samad T.A., Moore K.A., Sapirstein A., et al. Interleukin-1®–mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475.
Samad T.A., Sapirstein A., Woolf C.J. Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends in Molecular Medicine. 2002;8:390–396.
Sanada M., Yasuda H., Omatsu-Kanbe M., et al. Increase in intracellular Ca2+ and calcitonin gene–related peptide release through metabotropic P2Y receptors in rat dorsal root ganglion neurons. Neuroscience. 2002;111:413–422.
Sawynok J., Liu X.J. Adenosine in the spinal cord and periphery: release and regulation of pain. Progress in Neurobiology. 2003;69:313–340.
Sawynok J., Reid A. Peripheral adenosine 5′-triphosphate enhances nociception in the formalin test via activation of a purinergic p2X receptor. European Journal of Pharmacology. 1997;330:115–121.
Sawynok J., Reid A., Liu X.J. Involvement of mast cells, sensory afferents and sympathetic mechanisms in paw oedema induced by adenosine A1 and A2B/3 receptor agonists. European Journal of Pharmacology. 2000;395:47–50.
Scapini P., Lapinet-Vera J.A., Gasperini S., et al. The neutrophil as a cellular source of chemokines. Immunological Reviews. 2000;177:195–203.
Schafers M., Svensson C.I., Sommer C., et al. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. Journal of Neuroscience. 2003;23:2517–2521.
Schaible H.-G., Schmidt R.F. Excitation and sensitization of fine articular afferents from cat’s knee joint by prostaglandin E2. Journal of Physiology. 1988;403:91–104.
Schmidtko A., Gao W., Konig P., et al. cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. Journal of Neuroscience. 2008;28:8568–8576.
Schnitzer T.J., Lane N.E., Birbara C., et al. Long-term open-label study of tanezumab for moderate to severe osteoarthritic knee pain. Osteoarthritis and Cartilage. 2011;19:639–646.
Scroggs R.S. Up-regulation of low-threshold tetrodotoxin-resistant Na+ current via activation of a cyclic AMP/protein kinase A pathway in nociceptor-like rat dorsal root ganglion cells. Neuroscience. 2011;186:13–20.
Serhan C.N., Hong S., Gronert K., et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. Journal of Experimental Medicine. 2002;196:1025–1037.
Shamash S., Reichert F., Rotshenker S. The cytokine network of wallerian degeneration: tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. Journal of Neuroscience. 2002;22:3052–3060.
Shaw S.K., Owolabi S.A., Bagley J., et al. Activated polymorphonuclear cells promote injury and excitability of dorsal root ganglia neurons. Experimental Neurology. 2008;210:286–294.
Shi X.Q., Zekki H., Zhang J. The role of TLR2 in nerve injury–induced neuropathic pain is essentially mediated through macrophages in peripheral inflammatory response. Glia. 2011;59:231–241.
Shin J., Cho H., Hwang S.W., et al. Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10150–10155.
Shin H.S., Snyderman R., Friedman E., et al. Chemotactic and anaphylatoxic fragment cleaved from the fifth component of guinea pig complement. Science. 1968;162:361–363.
Shinoda M., Ozaki N., Asai H., et al. Changes in P2X3 receptor expression in the trigeminal ganglion following monoarthritis of the temporomandibular joint in rats. Pain. 2005;116:42–51.
Siau C., Xiao W., Bennett G.J. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: loss of epidermal innervation and activation of Langerhans cells. Experimental Neurology. 2006;201:507–514.
Sluka K.A., Price M.P., Breese N.M., et al. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain. 2003;106:229–239.
Smeyne R.J., Klein R., Schnapp A., et al. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249.
Smolen J.S., Beaulieu A., Rubbert-Roth A., et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–997.
Smrcka A.V. G protein betagamma subunits: central mediators of G protein–coupled receptor signaling. Cellular and Molecular Life Sciences. 2008;65:2191–2214.
Snider W.D., McMahon S.B. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–632.
Soh U.J., Dores M.R., Chen B., et al. Signal transduction by protease-activated receptors. British Journal of Pharmacology. 2010;160:191–203.
Sommer C., Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neuroscience Letters. 2004;361:184–187.
Sommer C., Schafers M. Painful mononeuropathy in C57BL/Wld mice with delayed wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Research. 1998;784:154–162.
Sommer C., Schmidt C., George A. Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Experimental Neurology. 1998;151:138–142.
Sorkin L.S., Xiao W.H., Wagner R., et al. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255–262.
Souslova V., Cesare P., Ding Y., et al. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–1017.
Sowa N.A., Street S.E., Vihko P., et al. Prostatic acid phosphatase reduces thermal sensitivity and chronic pain sensitization by depleting phosphatidylinositol 4,5-bisphosphate. Journal of Neuroscience. 2010;30:10282–10293.
Stamboulian S., Choi J.S., Ahn H.S., et al. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. Journal of Neuroscience. 2010;30:1637–1647.
Stead R.H., Tomioka M., Quinonez G., et al. Intestinal mucosal mast cells in normal and nematode-infected rat intestines are in intimate contact with peptidergic nerves. Proceedings of the National Academy of Sciences of the United States of America. 1997;84:2975–2979.
Steen K.H., Reeh P.W. Sustained graded pain and hyperalgesia from harmless experimental tissue acidosis in human skin. Neuroscience Letters. 1993;154:113–116.
Stucky C.L., Dechiara T.M., Lindsay R.M., et al. Neurotrophin 4 is required for the survival of a subclass of hair follicle receptors. Journal of Neuroscience. 1998;18:7040–7046.
Stucky C.L., Medler K.A., Molliver D.C. The P2Y agonist UTP activates cutaneous afferent fibers. Pain. 2004;109:36–44.
Sufka K.J., Schomburg F.M., Giordano J. Receptor mediation of 5-HT–induced inflammation and nociception in rats. Pharmacology, Biochemistry, and Behavior. 1992;41:53–56.
Sugimoto Y., Shigemoto R., Namba T., et al. Distribution of the messenger RNA for the prostaglandin E receptor subtype EP3 in the mouse nervous system. Neuroscience. 1994;62:919–928.
Sugiura T., Tominaga M., Katsuya H., et al. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. Journal of Neurophysiology. 2002;88:544–548.
Suh B.C., Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annual Review of Biophysics. 2008;37:175–195.
Sun J.H., Yang B., Donnelly D.F., et al. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. Journal of Neurophysiology. 2006;96:2189–2199.
Sun Y., Zigmond R.E. Leukaemia inhibitory factor induced in the sciatic nerve after axotomy is involved in the induction of galanin in sensory neurons. European Journal of Neuroscience. 1996;8:2213–2220.
Sutherland S.P., Benson C.J., Adelman J.P., et al. Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:711–716.
Svensson C.I., Yaksh T.L. The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Annual Review of Pharmacology and Toxicology. 2002;42:553–583.
Sylven C. Angina pectoris. Clinical characteristics, neurophysiological and molecular mechanisms. Pain. 1989;36:145–167.
Taiwo Y.O., Bjerknes L.K., Goetzl E.J., et al. Mediation of primary afferent hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32:577–580.
Tegeder I., Scheving R., Wittig I., et al. SNO-ing at the nociceptive synapse? Pharmacological Reviews. 2011;63:366–389.
Teng K.K., Felice S., Kim T., et al. Understanding proneurotrophin actions: recent advances and challenges. Developmental Neurobiology. 2010;70:350–359.
Theodosiou M., Rush R.A., Zhou X.F., et al. Hyperalgesia due to nerve damage: role of nerve growth factor. Pain. 1999;81:245–255.
Thompson S.W.N., Dray A., McCarson K.E., et al. Nerve growth factor induces mechanical allodynia associated with novel A fibre–evoked spinal reflex activity and enhanced neurokinin-1 receptor activation in the rat. Pain. 1995;62:219–231.
Thompson S.W., Dray A., Urban L. Leukemia inhibitory factor induces mechanical allodynia but not thermal hyperalgesia in the juvenile rat. Neuroscience. 1996;71:1091–1094.
Thompson S.W., Majithia A.A. Leukemia inhibitory factor induces sympathetic sprouting in intact dorsal root ganglia in the adult rat in vivo. Journal of Physiology. 1998;506:809–816.
Thompson S.W., Priestley J.V., Southall A. gp130 cytokines, leukemia inhibitory factor and interleukin-6, induce neuropeptide expression in intact adult rat sensory neurons in vivo: time-course, specificity and comparison with sciatic nerve axotomy. Neuroscience. 1998;84:1247–1255.
Thompson S.W., Vernallis A.B., Heath J.K., et al. Leukaemia inhibitory factor is retrogradely transported by a distinct population of adult rat sensory neurons: co-localization with trkA and other neurochemical markers. European Journal of Neuroscience. 1997;9:1244–1251.
Ting E., Guerrero A.T., Cunha T.M., et al. Role of complement C5a in mechanical inflammatory hypernociception: potential use of C5a receptor antagonists to control inflammatory pain. British Journal of Pharmacology. 2008;153:1043–1053.
Toews A.D., Barrett C., Morell P. Monocyte chemoattractant protein 1 is responsible for macrophage recruitment following injury to sciatic nerve. Journal of Neuroscience Research. 1998;53:260–267.
Tofaris G.K., Patterson P.H., Jessen K.R., et al. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. Journal of Neuroscience. 2002;22:6696–6703.
Toh M.L., Miossec P. The role of T cells in rheumatoid arthritis: new subsets and new targets. Current Opinion in Rheumatology. 2007;19:284–288.
Tokunaga A., Saika M., Senba E. 5-HT2A receptor subtype is involved in the thermal hyperalgesic mechanism of serotonin in the periphery. Pain. 1998;76:349–355.
Tominaga M., Caterina M.J., Malmberg A.B., et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543.
Tominaga M., Wada M., Masu M. Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6951–6956.
Toriyabe M., Omote K., Kawamata T., et al. Contribution of interaction between nitric oxide and cyclooxygenases to the production of prostaglandins in carrageenan-induced inflammation. Anesthesiology. 2004;101:983–990.
Trevisani M., Siemens J., Materazzi S., et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13519–13524.
Tsai Y.C., Won S.J. Effects of tramadol on T lymphocyte proliferation and natural killer cell activity in rats with sciatic constriction injury. Pain. 2001;92:63–69.
Ueno A., Matsumoto H., Naraba H., et al. Major roles of prostanoid receptors IP and EP3 in endotoxin-induced enhancement of pain perception. Biochemical Pharmacology. 2001;62:157–160.
Ulmann L., Hirbec H., Rassendren F. P2X4 receptors mediate PGE2 release by tissue-resident macrophages and initiate inflammatory pain. EMBO Journal. 2010;29:2290–2300.
Usachev Y.M., DeMarco S.J., Campbell C., et al. Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron. 2002;33:113–122.
Vale M.L., Benevides V.M., Sachs D., et al. Antihyperalgesic effect of pentoxifylline on experimental inflammatory pain. British Journal of Pharmacology. 2004;143:833–844.
Vale M.L., Marques J.B., Moreira C.A., et al. Antinociceptive effects of interleukin-4, -10, and -13 on the writhing response in mice and zymosan-induced knee joint incapacitation in rats. Journal of Pharmacology and Experimental Therapeutics. 2003;304:102–108.
Valenti C., Giuliani S., Cialdai C., et al. Anti-inflammatory synergy of MEN16132, a kinin B(2) receptor antagonist, and dexamethasone in carrageenan-induced knee joint arthritis in rats. British Journal of Pharmacology. 2010;161:1616–1627.
Van Steenwinckel J., Noghero A., Thibault K., et al. The 5-HT2A receptor is mainly expressed in nociceptive sensory neurons in rat lumbar dorsal root ganglia. Neuroscience. 2009;161:838–846.
Vellani V., Kinsey A.M., Prandini M., et al. Protease activated receptors 1 and 4 sensitize TRPV1 in nociceptive neurones. Molecular Pain. 2010;6:61.
Vellani V., Mapplebeck S., Moriondo A., et al. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. Journal of Physiology. 2001;534:813–825.
Vellani V., Zachrisson O., McNaughton P.A. Functional bradykinin B1 receptors are expressed in nociceptive neurones and are upregulated by the neurotrophin GDNF. Journal of Physiology. 2004;560:391–401.
Verge V.M., Merlio J.P., Grondin J., et al. Colocalization of NGF binding sites, trk mRNA, and low-affinity NGF receptor mRNA in primary sensory neurons: responses to injury and infusion of NGF. Journal of Neuroscience. 1992;12:4011–4022.
Verge V.M., Richardson P.M., Benoit R., et al. Histochemical characterization of sensory neurons with high-affinity receptors for nerve growth factor. Journal of Neurocytology. 1989;18:583–591.
Verge V.M., Richardson P.M., Wiesenfeld-Hallin Z., et al. Differential influence of nerve growth factor on neuropeptide expression in vitro: a novel role in peptide suppression in adult sensory neurons. Journal of Neuroscience. 1995;15:2081–2096.
Vergnolle N., Bunnett N.W., Sharkey K.A., et al. Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nature Medicine. 2001;7:821–826.
Vivancos G.G., Parada C.A., Ferreira S.H. Opposite nociceptive effects of the arginine/NO/cGMP pathway stimulation in dermal and subcutaneous tissues. British Journal of Pharmacology. 2003;138:1351–1357.
Vulchanova L., Riedl M.S., Shuster S.J., et al. Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology. 1997;36:1229–1242.
von Banchet G.S., Kiehl M., Schaible H.G. Acute and long-term effects of IL-6 on cultured dorsal root ganglion neurones from adult rat. Journal of Neurochemistry. 2005;94:238–248.
Wagner R., Janjigian M., Myers R.R. Anti-inflammatory interleukin-10 therapy in CCI neuropathy decreases thermal hyperalgesia, macrophage recruitment, and endoneurial TNF-alpha expression. Pain. 1998;74:35–42.
Wagner R., Myers R.R. Schwann cells produce tumor necrosis factor alpha: expression in injured and non-injured nerves. Neuroscience. 1996;73:625–629.
Wang J.G., Strong J.A., Xie W., et al. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Molecular Pain. 2008;4:38.
Watkins L.R., Maier S.F. Glia: a novel drug discovery target for clinical pain. Nature Reviews. Drug Discovery. 2003;2:973–985.
Wei H., Chen Y., Hong Y. The contribution of peripheral 5-hydroxytryptamine2A receptor to carrageenan-evoked hyperalgesia, inflammation and spinal Fos protein expression in the rat. Neuroscience. 2005;132:1073–1082.
Werner M.F., Kassuya C.A., Ferreira J., et al. Peripheral kinin B(1) and B(2) receptor–operated mechanisms are implicated in neuropathic nociception induced by spinal nerve ligation in rats. Neuropharmacology. 2007;53:48–57.
Weskamp G., Otten U. An enzyme-linked immunoassay for nerve growth factor (NGF): a tool for studying regulatory mechanisms involved in NGF production in brain and in peripheral tissues. Journal of Neurochemistry. 1987;48:1779–1786.
Wetmore C., Olson L. Neuronal and nonneuronal expression of neurotrophins and their receptors in sensory and sympathetic ganglia suggest new intercellular trophic interactions. Journal of Comparative Neurology. 1995;353:143–159.
White D.M., Basbaum A.I., Goetzl E.J., et al. The 15-lipoxygenase product, 8R,15S-diHETE, stereospecifically sensitizes C-fiber mechanoheat nociceptors in hairy skin of rat. Journal of Neurophysiology. 1990;63:966–970.
White F.A., Sun J., Waters S.M., et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:14092–14097.
Winter J., Forbes C.A., Sternberg J., et al. Nerve growth factor (NGF) regulates adult rat cultured dorsal root ganglion neuron responses to the excitotoxin capsaicin. Neuron. 1988;1:973–981.
Woolf C.J., Allchorne A., Safieh-Garabedian B., et al. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor-α. British Journal of Pharmacology. 1997;121:417–424.
Woolf C.J., Ma Q.P., Allchorne A., et al. Peripheral cell types contributing to the hyperalgesic action of nerve growth factor in inflammation. Journal of Neuroscience. 1996;16:2716–2723.
Woolf C.J., Safieh-Garabedian B., Ma Q.P., et al. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331.
Wotherspoon G., Winter J. Bradykinin B1 receptor is constitutively expressed in the rat sensory nervous system. Neuroscience Letters. 2000;294:175–178.
Wright H.L., Moots R.J., Bucknall R.C., et al. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford). 2010;49:1618–1631.
Wu S., Zhu M., Wang W., et al. Changes of the expression of 5-HT receptor subtype mRNAs in rat dorsal root ganglion by complete Freund’s adjuvant–induced inflammation. Neuroscience Letters. 2001;307:183–186.
Wu Z.Z., Pan H.L. Role of TRPV1 and intracellular Ca2+ in excitation of cardiac sensory neurons by bradykinin. American Journal of Physiology. Regulatory. Integrative and Comparative Physiology. 2007;293:R276–R283.
Xu G.Y., Huang L.Y. Peripheral inflammation sensitizes P2X receptor–mediated responses in rat dorsal root ganglion neurons. Journal of Neuroscience. 2002;22:93–102.
Xu X.J., Hao J.X., Andell-Jonsson S., et al. Nociceptive responses in interleukin-6–deficient mice to peripheral inflammation and peripheral nerve section. Cytokine. 1997;9:1028–1033.
Xu Z.Z., Zhang L., Liu T., et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nature Medicine. 2010;16:592–597.
Yao J., Qin F. Interaction with phosphoinositides confers adaptation onto the TRPV1 pain receptor. PLoS Biology. 2009;7:e46.
Yang L.C., Marsala M., Yaksh T.L. Characterization of time course of spinal amino acids, citrulline and PGE2 release after carrageenan/kaolin-induced knee joint inflammation: a chronic microdialysis study. Pain. 1996;67:345–354.
Yousuf A., Klinger F., Schicker K., et al. Nucleotides control the excitability of sensory neurons via two P2Y receptors and a bifurcated signaling cascade. Pain. 2011;152:1899–1908.
Zelenka M., Schafers M., Sommer C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. 2005;116:257–263.
Zhang N., Inan S., Cowan A., et al. A pro-inflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4536–4541.
Zhang N., Rogers T.J., Caterina M., et al. Pro-inflammatory chemokines, such as C-C chemokine ligand 3, desensitize mu-opioid receptors on dorsal root ganglia neurons. Journal of Immunology. 2004;173:594–599.
Zhang Y.H., Vasko M.R., Nicol G.D. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. Journal of Physiology. 2002;544:385–402.
Zhang X., Huang J., McNaughton P.A. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO Journal. 2005;24:4211–4223.
Zhu W.J., Yamanaka H., Obata K., et al. Expression of mRNA for four subtypes of the proteinase-activated receptor in rat dorsal root ganglia. Brain Research. 2005;1041:205–211.
Zuo Y., Perkins N.M., Tracey D.J., et al. Inflammation and hyperalgesia induced by nerve injury in the rat: a key role of mast cells. Pain. 2003;105:467–479.
Zurborg S., Yurgionas B., Jira J.A., et al. Direct activation of the ion channel TRPA1 by Ca2+. Nature Neuroscience. 2007;10:277–279.
Zygmunt P.M., Petersson J., Andersson D.A., et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457.
Zylbergold P., Ramakrishnan N., Hebert T. The role of G proteins in assembly and function of Kir3 inwardly rectifying potassium channels. Channels (Austin). 2010;4:411–421.
Zylka M.J., Sowa N.A., Taylor-Blake B., et al. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron. 2008;60:111–122.
Andersson D.A., Gentry C., Moss S., et al. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. Journal of Neuroscience. 2008;28:2485–2494.
Baker M.D. Protein kinase C mediates up-regulation of tetrodotoxin-resistant, persistent Na+ current in rat and mouse sensory neurones. Journal of Physiology. 2005;567:851–867.
Barclay J., Clark A.K., Ganju P., et al. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain. 2007;130:225–234.
Binshtok A.M., Wang H., Zimmermann K., et al. Nociceptors are interleukin-1beta sensors. Journal of Neuroscience. 2008;28:14062–14073.
Bizzarri C., Beccari A.R., Bertini R., et al. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacology & Therapeutics. 2006;112:139–149.
Bradbury E.J., Burnstock G., McMahon S.B. The expression of P2X3 purinoreceptors in sensory neurons: effects of axotomy and glial-derived neurotrophic factor. Molecular and Cellular Neurosciences. 1998;12:256–268.
Dai Y., Wang S., Tominaga M., et al. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. Journal of Clinical Investigation. 2007;117:1979–1987.
Dawes J.M., Calvo M., Perkins J.R., et al. CXCL5 mediates UVB irradiation–induced pain. Science Translational Medicine. 3(90), 2011. 90ra60
Ford A.P. In pursuit of P2X3 antagonists: novel therapeutics for chronic pain and afferent sensitization. Purinergic Signal. 2012;8:3–26.
Gold M.S. Tetrodotoxin-resistant Na+ currents and inflammatory hyperalgesia. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7645–7649.
Ji R.R., Xu Z.Z., Strichartz G., et al. Emerging roles of resolvins in the resolution of inflammation and pain. Trends in Neurosciences. 2011;34:599–609.
Kaan T.K., Yip P.K., Patel S., et al. Systemic blockade of P2X3 and P2X2/3 receptors attenuates bone cancer pain behaviour in rats. Brain. 2010;133:2549–2564.
Lane N.E., Schnitzer T.J., Birbara C.A., et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. New England Journal of Medicine. 2010;363:1521–1531.
Levine J.D., Lam D., Taiwo Y.O., et al. Hyperalgesic properties of 15-lipoxygenase products of arachidonic acid. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:5331–5334.
Lewin G.R., Rueff A., Mendell L.M. Peripheral and central mechanisms of NGF-induced hyperalgesia. European Journal of Neuroscience. 1994;6:1903–1912.
Marceau F., Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nature Reviews. Drug Discovery. 2004;3:845–852.
Marchand F., Perretti M., McMahon S.B. Role of the immune system in chronic pain. Nature Reviews. Neuroscience. 2005;6:521–532.
McMahon S.B., Bennett D.L., Priestley J.V., et al. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nature Medicine. 1995;1:774–780.
Obreja O., Ringkamp M., Turnquist B., et al. Nerve growth factor selectively decreases activity-dependent conduction slowing in mechano-insensitive C-nociceptors. Pain. 2011;152:2138–2146.
Oh S.B., Tran P.B., Gillard S.E., et al. Chemokines and glycoprotein 120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. Journal of Neuroscience. 2001;21:5027–5035.
Pezet S., McMahon S.B. Neurotrophins: mediators and modulators of pain. Annual Review of Neuroscience. 2006;29:507–538.
Ren K., Dubner R. Interactions between the immune and nervous systems in pain. Nature Medicine. 2010;16:1267–1276.
Safieh-Garabedian B., Poole S., Allchorne A., et al. Contribution of interleukin-1 beta to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. British Journal of Pharmacology. 1995;115:1265–1275.
Serhan C.N., Hong S., Gronert K., et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. Journal of Experimental Medicine. 2002;196:1025–1037.
Smolen J.S., Beaulieu A., Rubbert-Roth A., et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–997.
Sommer C., Schafers M. Painful mononeuropathy in C57BL/Wld mice with delayed wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Research. 1998;784:154–162.
Sorkin L.S., Xiao W.H., Wagner R., et al. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255–262.
Teng K.K., Felice S., Kim T., et al. Understanding proneurotrophin actions: recent advances and challenges. Developmental Neurobiology. 2010;70:350–359.
Tominaga M., Caterina M.J., Malmberg A.B., et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543.
Ulmann L., Hirbec H., Rassendren F. P2X4 receptors mediate PGE2 release by tissue-resident macrophages and initiate inflammatory pain. EMBO Journal. 2010;29:2290–2300.
White F.A., Sun J., Waters S.M., et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:14092–14097.
Xu Z.Z., Zhang L., Liu T., et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nature Medicine. 2010;16:592–597.
Zygmunt P.M., Petersson J., Andersson D.A., et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457.