Chapter 63 Neurologic Emergencies and Stabilization
The care of critically ill children has advanced greatly over the past decades, and mortality rates have fallen. A remaining challenge is optimizing recovery after critical neurologic insults.
Neurocritical Care Principles
The brain has high metabolic demands, which are further increased during growth and development. Preservation of nutrient supply to the brain is the mainstay of care for children with evolving brain injuries. Intracranial dynamics describes the physics of the interactions of the contents—brain parenchyma, blood (arterial, venous, capillary) and cerebrospinal fluid (CSF)—within the cranium. Normally, brain parenchyma accounts for up to 85% of the contents of the cranial vault, and the remaining portion is divided between CSF and blood. The brain resides in a relatively rigid cranial vault, and cranial compliance decreases with age as the skull ossification centers gradually replace cartilage with bone. The intracranial pressure (ICP) is derived from the volume of its components and the bony compliance. The perfusion pressure of the brain (cerebral perfusion pressure [CPP]) is equal to the pressure of blood entering the cranium (mean arterial pressure [MAP]) minus the ICP, in most cases.
Increases in intracranial volume can result from swelling, masses, or increases in blood and CSF volumes. As these volumes increase, compensatory mechanisms decrease ICP by (1) decreasing CSF volume (CSF is displaced into the spinal canal or absorbed by arachnoid villi), (2) decreasing cerebral blood volume (venous blood return to the thorax is augmented), and/or (3) increasing cranial volume (sutures pathologically expand or bone is remodeled). Once compensatory mechanisms are exhausted (the increase in cranial volume is too large), small increases in volume lead to large increases in ICP or intracranial hypertension (Fig. 63-1). As ICP continues to increase, brain ischemia can occur as CPP falls. Further increases in ICP can ultimately displace the brain downward into the foramen magnum—a process called cerebral herniation, which can become irreversible in minutes and may lead to severe disability or death.
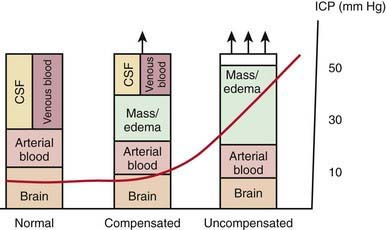
Figure 63-1 The Munro-Kellie doctrine describes intracranial dynamics in the setting of an expanding mass lesion (i.e., hemorrhage, tumor) or brain edema. In the normal state, the brain parenchyma, arterial blood, cerebrospinal fluid (CSF), and venous blood occupy the cranial vault at a low pressure, generally <10 mm Hg. With an expanding mass lesion or brain edema, initially there is a compensated state as a result of reduced CSF and venous blood volumes, and intracranial pressure (ICP) remains low. Further expansion of the lesion, however, leads to an uncompensated state when compensatory mechanisms are exhausted and intracranial hypertension results. See text for details.
Oxygen and glucose are required by brain cells for normal functioning, and these nutrients must be constantly supplied by cerebral blood flow (CBF). Normally, CBF is constant over a wide range of blood pressures (blood pressure autoregulation of CBF) via actions mainly within the cerebral arterioles. Cerebral arterioles are maximally dilated at lower blood pressures and maximally constricted at higher pressures so that CBF does not vary during normal fluctuations (Fig. 63-2). Acid-base balance of the CSF (often reflected by acute changes in PaCO2), body/brain temperature, glucose utilization, and other vasoactive mediators (i.e., adenosine, nitric oxide) can also affect the cerebral vasculature.
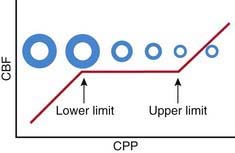
Figure 63-2 Schematic of the relationship between cerebral blood flow (CBF) and cerebral perfusion pressure (CPP). The diameter of a representative cerebral arteriole is also shown across the center of the y axis to facilitate understanding of the vascular response across CPP that underlies blood pressure autoregulation of CBF. CPP is generally defined as the mean arterial pressure (MAP) minus the intracranial pressure (ICP). At normal values for ICP, this generally represents MAP. Thus, normally, CBF is kept constant between the lower limit and upper limit of autoregulation; in normal adults, these values are ≈50 mm Hg and 150 mm Hg, respectively. In children, the upper limit of autoregulation is likely proportionally lower than the adult value relative to normal MAP for age. However, according to the work of Vavilala et al (2003), lower limit values are surprisingly similar in infants and older children. Thus, infants and young children may have less reserve for adequate CPP. See text for details.
Knowledge of these concepts is instrumental to preventing secondary brain injury. Increases in CSF pH that occur because of inadvertent hyperventilation (decreased PaCO2) can produce cerebral ischemia. Hyperthermia-mediated increases in cerebral metabolic demands may damage vulnerable brain regions after injury. Hypoglycemia can produce neuronal death when CBF fails to compensate. Prolonged seizures can lead to permanent injuries if hypoxemia occurs from loss of airway control.
Attention to detail and constant reassessment are paramount in managing children with critical neurologic insults. Among the most valuable tools for serial, objective assessments of neurologic condition is the Glasgow Coma Scale (GCS) (see Table 62-3). Originally developed to assess level of consciousness after traumatic brain injury (TBI) in adults, the GCS is also valuable in pediatrics. Modifications to the GCS have been made for nonverbal children and are available for infants and toddlers (see Table 62-3). Serial assessments of the GCS score along with a focused neurologic examination are invaluable to detection of injuries before permanent damage occurs in the vulnerable brain.
The best-studied monitoring device in clinical practice is the ICP monitor. Monitoring is accomplished by a catheter inserted either into the cerebral ventricle (externalized ventricular drain) or into brain parenchyma (parenchymal transducer). ICP-directed therapies are standard of care in TBI and are used in other conditions such as intracranial hemorrhage, Reye syndrome, and some cases of encephalopathy, meningitis, and encephalitis. Other devices being studied include catheters that measure brain tissue oxygen concentration (PbtO2), external probes that noninvasively assess brain oxygenation by absorbance of near-infrared light (near-infrared spectroscopy [NIRS]), monitors of brain electrical activity (continuous electroencephalography [EEG] or somatosensory, visual, or auditory evoked potentials), and CBF monitors (transcranial Doppler, xenon CT, perfusion MRI, or tissue probes).
Traumatic Brain Injury
Etiology
Mechanisms of TBI include motor vehicle crashes, falls, assaults, and abusive head trauma. Most TBIs in children are from closed-head injury.
Epidemiology
TBI is one of the most important pediatric public health problems, resulting in the death of about 7,000 children annually in the USA.
Pathology
Epidural, subdural, and parenchymal intracranial hemorrhages can result. Injury to gray or white matter is also commonly seen and includes focal cerebral contusions, diffuse cerebral swelling, axonal injury, and injury to the cerebellum or brainstem. Patients with severe TBI often have multiple findings; diffuse and potentially delayed cerebral swelling is common.
Pathogenesis
TBI results in primary and secondary injury. Primary injury from the impact produces irreversible tissue disruption. In contrast, 2 types of secondary injury are targets of neurointensive care. First, some of the ultimate damage seen in the injured brain evolves over hours or days, and the underlying mechanisms involved (edema, apoptosis, and secondary axotomy) are therapeutic targets. Second, the injured brain is vulnerable to additional insults because injury disrupts normal autoregulatory defense mechanisms; disruption of autoregulation of CBF can lead to ischemia from hypotension that would otherwise be tolerated by the uninjured brain.
Clinical Manifestations
The hallmark of severe TBI is coma (GCS score 3-8). Often, coma is seen immediately after the injury and is sustained. In some cases, such as with an epidural hematoma, a child may be alert presentation but may deteriorate after a period of hours. A similar picture can be seen in children with diffuse swelling, in whom a talk and die scenario has been described. Clinicians should also not be lulled into underappreciating the potential for deterioration of a child with moderate TBI (GCS score 9-12) with a significant contusion, because progressive swelling can potentially lead to devastating complications. In the comatose child with severe TBI, the second key clinical manifestation is the development of intracranial hypertension. The development of increased ICP with impending herniation may be heralded by new-onset or worsening headache, depressed level of consciousness, vital sign changes (hypertension, bradycardia, irregular respirations), and signs of 6th (lateral rectus palsy) or 3rd (anisocoria [dilated pupil], ptosis, down-and-out position of globe due to rectus muscle palsies) cranial nerve compression. Increased ICP can be appropriately managed only with continuous ICP monitoring. The development of brain swelling is progressive. Significantly raised ICP (>20 mm Hg) can occur early after severe TBI, but peak ICP generally is seen at 48-72 hr. Need for ICP-directed therapy may persist for longer than a week. A few children have coma without increased ICP, resulting from axonal injury or brainstem injury.
Laboratory Findings
Cranial CT should be obtained immediately after stabilization (Figs. 63-3 to 63-11). Generally, other laboratory findings are normal in isolated TBI, although occasionally coagulopathy or the development of the syndrome of inappropriate antidiuretic hormone secretion (SIADH) or, rarely, cerebral salt wasting is seen. In the setting of TBI with polytrauma, other injuries can result in laboratory abnormalities, and a full trauma survey is important in all patients with severe TBI (Chapter 66).
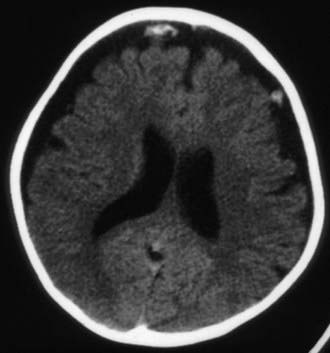
Figure 63-3 Abusive head trauma in an infant. Note the subdural fluid collections, dilated ventricles, and blood.
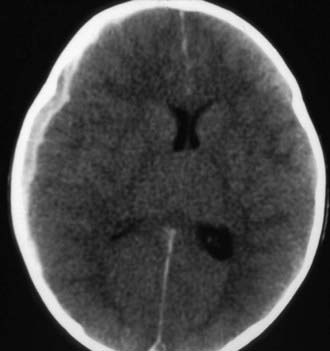
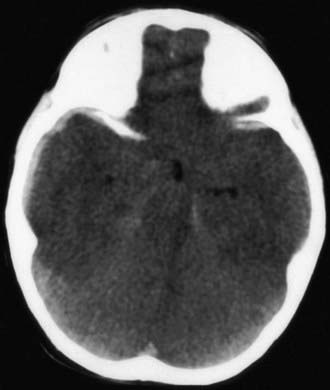
Figure 63-5 Abusive head trauma with massive cerebral edema, with loss of gray matter–white matter differentiation, loss of the ventricular system, and probable herniation of the brainstem.
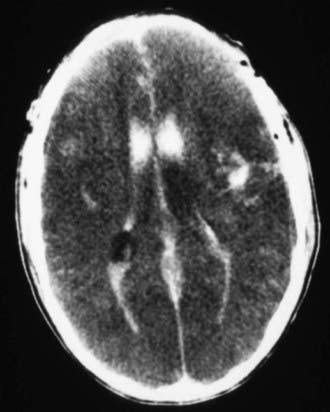
Figure 63-6 Abusive head trauma with significant intraventricular, intracerebral, and subdural hematomas, with loss of gray matter–white matter differentiation, suggestive of massive cerebral edema.
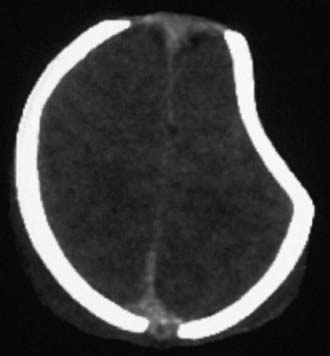
Figure 63-7 A depressed skull fracture due to traumatic delivery with forceps. Brain swelling can be seen.
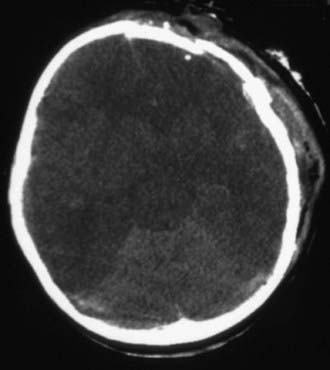
Figure 63-8 Malignant brain edema. A common pattern in severe head injury that is associated with significant secondary brain injury and a very high mortality rate. Cisterns are absent on the CT scan. This type of injury is associated with hypoxia and hypoxemia and hypotension.
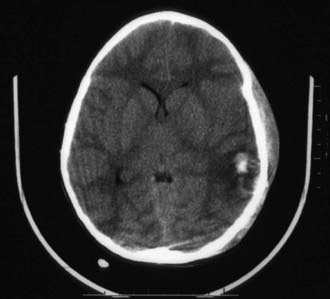
Figure 63-9 Significant closed-head injury with subgaleal hematoma, intracerebral hemorrhage, and loss of gray matter–white matter differentiation.
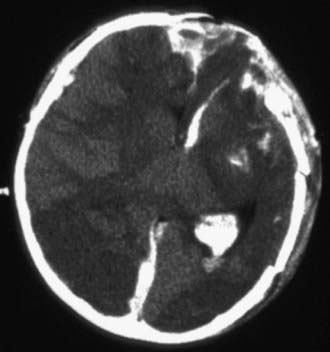
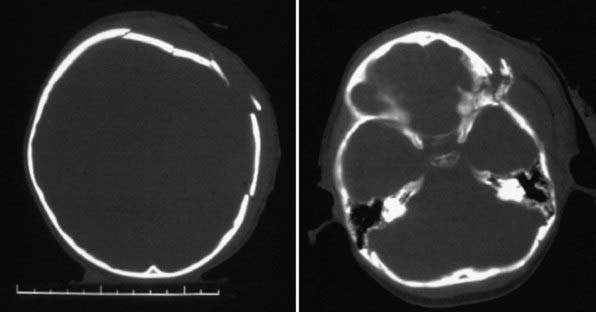
Diagnosis and Differential Diagnosis
In severe TBI, the diagnosis is generally obvious from the history and clinical presentation. Occasionally, TBI severity can be overestimated because of concurrent alcohol or drug intoxication. The diagnosis of TBI can be problematic in cases of inflicted TBI or following an anoxic event such as near drowning or smoke inhalation.
Treatment
Infants and children with severe or moderate TBI (GCS score 3-8 or 9-12, respectively) receive intensive care unit (ICU) monitoring. Evidence-based guidelines for management of severe TBI have been published (Fig. 63-12). This approach to ICP-directed therapy is also reasonable for other conditions in which ICP is monitored. Care involves a multidisciplinary team comprising pediatric caregivers from neurologic surgery, critical care medicine, surgery, and rehabilitation, and is directed at preventing secondary insults and managing raised ICP. Initial stabilization of infants and children with severe TBI includes rapid sequence tracheal intubation with spine precautions along with maintenance of normal extracerebral hemodynamics, including blood gas values (PaO2, PaCO2), MAP, and temperature. Intravenous fluid boluses may be required to treat hypotension. Euvolemia is the target, and hypotonic fluids should be rigorously avoided; normal saline is the fluid of choice. Pressors may be needed as guided by monitoring of central venous pressure (CVP), with avoidance of both fluid overload and exacerbation of brain edema. A trauma survey should be performed. Once stabilized, the patient should be taken for CT scanning to rule out the need for emergency neurosurgical intervention. If surgery is not required, an ICP monitor should be inserted to guide the treatment of intracranial hypertension.
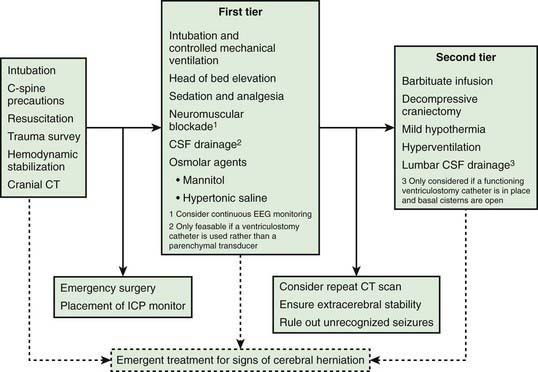
Figure 63-12 Schematic outlining the approach to management of a child with severe traumatic brain injury (TBI). It is based on the 2003 guidelines for the management of severe TBI, along with minor modifications from later literature. The intracranial pressure (ICP) and cerebral perfusion pressure (CPP) targets are discussed in the text. This schematic is specifically presented for severe TBI, for which the experience with ICP-directed therapy is greatest. Nevertheless, the general approach provided here is relevant to the management of intracranial hypertension in other conditions for which evidence-based data on ICP monitoring and ICP-directed therapy are lacking. Please see text for details.
During stabilization or at any time during the treatment course, patients can present with signs and symptoms of cerebral herniation (pupillary dilatation, systemic hypertension, bradycardia, extensor posturing). Because herniation and its devastating consequences can sometimes be reversed if promptly addressed, it should be treated as a medical emergency, with use of hyperventilation with an FIO2 of 1.0, and intubating doses of either thiopental or pentobarbital and either mannitol (0.25-1.0 g/kg, IV) or hypertonic saline (3% solution, 5-10 mL/kg IV).
ICP should be maintained <20 mm Hg; age-dependent CPP targets are ≈50 mm Hg for children 2-6 yr; 55 mm Hg for those 7-10 yr; and 65 mm Hg for those 11-16 yr. First-tier therapy includes elevation of the head of the bed, ensuring midline positioning of the head, controlled mechanical ventilation, and sedation and analgesia (i.e., benzodiazepines and narcotics). If neuromuscular blockade is needed, it may be desirable to monitor EEG continuously because status epilepticus can occur; this complication will not be recognized in a paralyzed patient and is associated with raised ICP and unfavorable outcome. If a ventricular rather than parenchymal catheter is used to monitor ICP, therapeutic CSF drainage is available and can be provided either continuously (often targeting an ICP > 5 mm Hg) or intermittently in response to ICP spikes, generally 20 mm Hg. Other first-tier therapies include the osmolar agents mannitol (0.25-1.0 g/kg IV over 20 min), given in response to ICP spikes >20 mm Hg or with a fixed (q4-6h) dosing interval, and hypertonic saline (often given as a continuous infusion of 3% saline at 0.1-1.0 mL/kg/hr). Choice of osmolar agent depends on the preference of the treating center. These two agents can be used concurrently. It is recommended to avoid serum osmolality >320 mOsm/L. A Foley urinary catheter should be placed to monitor urine output.
If ICP remains refractory to treatment, careful reassessment of the patient is needed to rule out unrecognized hypercarbia, hypoxemia, fever, hypotension, hypoglycemia, pain, and seizures. Repeat imaging should be considered to rule out a surgical lesion. Guidelines-based second-tier therapies for refractory raised ICP are available, but evidence favoring a given second-tier therapy is limited. In some centers, decompressive craniectomy is used. Others use a pentobarbital infusion, with a loading dose of 5-10 mg/kg over 30 min followed by 5 mg/kg every hour for 3 doses and then maintenance with an infusion of 1 mg/kg/hr. Careful blood pressure monitoring is required because of the possibility of drug-induced hypotension and the frequent need for support with fluids and/or pressors. Mild hypothermia (32-34°C) to control refractory ICP can be induced and maintained by means of surface cooling. Sedation and neuromuscular blockade are used to prevent shivering, and rewarming should be slow, no faster than 1°C every 4-6 hr. Hypotension should be prevented during rewarming. Refractory raised ICP can also be treated with hyperventilation (PaCO2 = 25-30 mm Hg). Other second-tier therapies (e.g., lumbar CSF drainage) are options.
Supportive Care
Euvolemia should be maintained, and isotonic fluids are recommended until resolution of intracranial hypertension. SIADH and salt wasting can develop and are important to differentiate, because management of the former is fluid restriction and that of the latter is sodium replacement. Severe hyperglycemia (blood glucose level >200 mg/dL) should be avoided and treated. The blood glucose level should be monitored frequently. Early nutrition with enteral feedings is advocated. Corticosteroids should generally not be used unless adrenal insufficiency is documented. Tracheal suctioning can exacerbate raised ICP. Timing of the use of sedation around suctioning events and/or use of tracheal or IV lidocaine can be helpful. Anticonvulsant prophylaxis with phenytoin or carbamazepine is a common treatment option.
Prognosis
Mortality rates for children with severe TBI who reach the PICU range between 10% and 30%. Ability to control ICP is related to patient survival, and the extent of cranial and systemic injuries correlates with quality of life. Motor and cognitive sequelae resulting from severe TBI generally benefit from rehabilitation to minimize long-term disabilities. Recovery from TBI may take months to achieve. Physical therapy, and in some centers methylphenidate, helps with motor and behavioral recovery.
Mild Traumatic Brain Injury
The majority (>90%) of children with blunt, closed-head trauma do not experience severe life- or brain-threatening complications. Children with mild TBI are defined as having a GCS score between 13 and 15 upon arrival at the hospital with or without the following acute symptoms: a history of loss of consciousness and antegrade or retrograde amnesia, as well as headache, vomiting, nausea, dizziness, or disorientation.
Cranial CT scanning is often considered in the evaluation of children with mild TBI, and findings are often negative. Nonetheless, the concern that the patient may have an acute intracranial hematoma that will necessitate immediate neurosurgical evacuation has resulted in identifying high-risk criteria to help determine whether the patient needs CT scanning. Although not all studies agree on all the criteria, the following are reasonable indications for CT imaging: loss of consciousness or amnesia >5 min; persistent dizziness; mental status changes; seizures; focal neurologic defects; a depressed skull fracture; signs of a basilar skull fracture; drug or alcohol use; and age <2 yr. The mechanisms of the injury are also important; they include: suspected child abuse; falls from >3 m; and high-speed injuries from projectiles, automobile, bicycle, or pedestrian-automobile crashes.
The postconcussive syndrome is an important sequela of an acute mild TBI. It often includes subjective complaints related to somatic, cognitive, or emotional symptoms. These include tiredness, headache, memory loss, dizziness, irritability, poor attention, depression, difficulty thinking (concentration), sleep problems, and personality changes. Postconcussive symptoms are more common after high-risk complications of mild TBI. The postconcussive syndrome usually resolves in 2-3 months, but subtle symptoms may last longer. Management includes avoiding excessive “brain activity” (TV, computer games, home or school work) and permitting the child to rest or sleep. In some high-risk children, symptoms may persist longer than a year after the acute injury. Children may need support in school with individualized learning programs. Parents need to know the spectrum of the postconcussive syndrome and to be assured that their children are not malingering or seeking attention.
Abusive Head Trauma
Abusive head trauma is the most common cause of death from TBI in infants (Chapter 37) (see Figs. 63-3 to 63-6). Most cases occur in the initial 2 years of life. Affected infants can be initially misdiagnosed; severe inflicted TBI can be complicated by repeated injury and/or extracerebral injuries. Delayed deterioration despite normal initial GCS score can be seen. MRI findings and serum biomarker test results indicate that these patients often exhibit more evidence of hypoxic-ischemic brain injury than is seen in non-inflicted TBI. This may result from delay in presentation, seizures, apnea, or other factors; the history is often conflicting, and time of injury may be unclear. The patients are often managed with an approach similar to that outlined previously for non-inflicted TBI, including ICP-directed therapy. Severe TBI secondary to abuse often has a poor prognosis.
Global Hypoxic-Ischemic Insults and Hypoxic-Ischemic Encephalopathy
Etiology
The leading cause of global hypoxic-ischemic insults resulting in hypoxic-ischemic encephalopathy (HIE) in infants and children is asphyxial arrest. This event can result from a variety of conditions, such as drowning, airway obstruction, trauma, hanging, infections, and perinatal asphyxia.
Epidemiology
Cardiac arrest is seen in about 8-20/100,000 children in the USA (Chapter 62). The incidence of perinatal HIE is between 1and 6/1,000 live full-term births.
Pathology
Global hypoxic-ischemic insults damage selectively vulnerable brain regions such as the hippocampus, Purkinje neurons in the cerebellum, the basal ganglia, and brainstem. Longer durations of arrest produce infarcts in watershed areas and ultimately brain death. In term newborns, hypoxic-ischemic insults can damage periventricular white matter tracks—albeit much less than in preterm babies (Chapter 93).
Pathogenesis
The pathogenesis of HIE is poorly understood; much of what is believed to occur is based on studies in experimental models. Threshold insults to the brain produced by asphyxia lead to a period of anoxic perfusion followed by cardiovascular demise. A period of “no flow” follows, with cerebral energy failure. Reperfusion may trigger secondary injury in the brain, which manifests as excitotoxicity and seizures, activation of neuronal death pathways (i.e., apoptosis and necrosis), oxidative and nitrative stress, mitochondrial damage, and inflammation.
Clinical Manifestations
After cardiac arrest, infants and children are routinely managed in the ICU, and coma or acute HIE based on GCS score and/or seizures are generally the indications for neurointensive care. In perinatal asphyxia, fetal acidosis, a 5-min Apgar score of 0 to 3, neurologic dysfunction, and/or abnormal EEG findings define the need for neuroprotective interventions.
Laboratory Findings
In the ICU, blood gas, lactate, or electrolyte abnormalities may be seen and should be serially followed. Evidence of multiorgan injury or failure, including markers of myocardial, renal, and hepatic injury/function, can be seen and should be serially evaluated. Acute echocardiographic assessment and cranial CT should be strongly considered. EEG can identify encephalopathy, seizures, and subclinical electrical status epilepticus, particularly in the child with post-resuscitation coma. If neuromuscular blockade is required, continuous EEG should be considered. MRI is useful in the subacute period to define the extent of brain injury (Fig. 63-13).
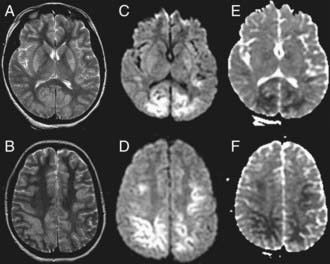
Figure 63-13 Magnetic resonance imaging of hypoxic-ischemic encephalopathy in a 1 yr old infant after asphyxial cardiac arrest from drowning. A and B, High signal intensity is seen in the basal ganglia and cortex on T2-weighted images. Brain edema as identified by restricted diffusion is noted in the deep layers of the occipital, parietal, and frontal cortex on diffusion weighted imaging (C and D) and on apparent diffusion coefficient images (E and F).
Diagnosis and Differential Diagnosis
The history is often clear with regard to the etiology of the hypoxic-ischemic insult, but if it is not, the cause of the arrest should be determined. In children, poisoning, hyperkalemia, unrecognized trauma, child abuse, myocarditis, cardiomyopathy, and prolonged QT syndrome should be considered (Chapter 430). Pertinent obstetric history should be sought in a newborn with perinatal asphyxia. HIE in children may also be secondary to other conditions, such as septic shock.
Complications
Hypoxic-ischemic insults are life-threatening conditions, and their complications include death, persistent vegetative state, severe disability, systemic inflammatory response syndrome, and the multiple organ dysfunction syndrome.
Treatment
Acute resuscitation from cardiac arrest is addressed in Chapter 62. Neurointensive care focuses on the post-resuscitation phase in the PICU. The first goal is to optimize cardiac output and cerebral perfusion. Mechanical ventilation should target normalization of PaO2 and PaCO2—avoiding inadvertent hyperoxia or hypocarbia. Systemic hemodynamics should be optimized by normalizing MAP for age; systemic perfusion and capillary refill; central venous oxygen saturation (>65%); and pH. Volume expansion with isotonic fluids should be performed to treat shock and should be further guided by urine output (>1.0 mL/kg/hr) and CVP. Inotropes, pressors, and/or vasodilators may be required to prevent re-arrest and to optimize cerebral and systemic perfusion. Hyperglycemia and hypoglycemia should be prevented or, if present, treated. Arrhythmias should be treated. If conventional hemodynamic support is inadequate, extracorporeal membrane oxygenation (ECMO) should be considered.
Mild hypothermia should be considered as a treatment option in comatose children after cardiac arrest when restoration of spontaneous circulation (ROSC) with hemodynamic stability has been achieved. Similarly, in perinatal asphyxia, fetal acidosis, an Apgar score of 0 to 3 after 5 min, neurologic dysfunction, and/or abnormal EEG findings are criteria for use of this therapy in term neonates. Exclusion criteria have included coagulopathy, bleeding, and hemodynamic instability. According to the American Heart Association (AHA) guidelines (predominantly for adults after a cardiac arrest when the initial event was associated with ventricular fibrillation), cooling should be initiated as soon as possible after ROSC but may be beneficial even if delayed (4-6 hr); it should be induced by means of surface cooling with cooling blankets, application of ice packs to the groin, axillae, and neck, use of wet towels, and fanning. Infusion of 20 mL/kg IV of ice-cold saline over 30 min can be considered in children and may reduce core temperature by ≈2°C. If hypothermia is used in children, a temperature of 32-34°C should be use for 12-72 hr, according to physician preference. The rewarming rate should be no greater than 1°C every 4-6 hr. In perinatal asphyxia, cooling should be maintained for 72 hr. Shivering should be prevented with sedation and neuromuscular blockade. Temperature should be continuously monitored. Hypothermia in children has been associated with an increased risk for neutropenia, sepsis, and, in some studies (traumatic injury), no improvement in neurologic sequelae.
Supportive Care
Optimal supportive care includes maintenance of euvolemia with isotonic fluids. Hyperglycemia, hypoglycemia, hyponatremia, hyperosmolality, and metabolic acidosis should be prevented. If hypothermia is not used, careful attention should be paid to preventing fever in the initial 72 hr. Therapies needed to treat underlying conditions should be included in the post-resuscitation treatment plan when appropriate.
Prognosis
Outcome from HIE in children depends on the location of the insult. In out-of-hospital cardiac arrest, survival to discharge is only ≈10%, and < 50% of survivors have favorable neurologic outcome. In-hospital cardiac arrests survival is ≈30%, with favorable outcome in most survivors. In perinatal asphyxia, moderate encephalopathy is associated with 10% and 30% risks of mortality and disability, respectively; severe encephalopathy is associated with 60% mortality and nearly 100% disability.
Status Epilepticus
Etiology
Status epilepticus is defined as a seizure of sufficient duration to provide an enduring epileptic focus. In more practical terms, persistent, disorganized, involuntary brain activity lasting for more than a proscribed amount of time (varying from 1 min to 30 min; usually 15-30 min) is characteristic of status epilepticus. The causes are myriad; epilepsy, abrupt discontinuation of anticonvulsive medications, febrile seizures, TBI, and encephalitis and other central nervous system infections are the leading causes in children (Chapter 586.8).
Epidemiology
The incidence of status epilepticus ranges between 10 and 60/100,000 population in various studies. Status epilepticus is most common in children <5 yr of age, with incidence in this age group >100/100,000.
Pathology and Pathogenesis
Status epilepticus can cause injury to the brain. It increases cerebral oxygen and glucose consumption markedly. In self-limited seizures, a concomitant increase in CBF to prevent energy failure is seen. As seizures persist, compensatory mechanisms fail, and relative cerebral ischemia can occur. Status epilepticus is associated with increases in brain levels of excitatory amino acids (glutamate), which bind to specific receptors (N-methyl-D-aspartate), causing greater neuronal activity and activation of intracellular pathways leading to cell death.
Clinical Manifestations
In the ICU, presentations of status epilepticus vary between easily recognizable signs and symptoms, such as tonic-clonic movements of all extremities, to a lack of physical findings in a comatose child in whom subclinical (or nonconvulsive) electrical status epilepticus is manifest only on EEG.
Laboratory Findings
Status epilepticus is not associated with significant laboratory abnormalities. Electrolyte abnormalities (i.e., hyponatremia, hypocalcemia, hypoglycemia) should be ruled out. Examination of CSF is warranted when infections are suspected. Determination of serum concentrations of chronically administered antiepileptic drugs is important to define potential etiology, adjust dosages, and ensure future compliance in these refractory cases. The definitive diagnosis of status epilepticus is made from EEG findings.
Diagnosis and Differential Diagnosis
Diagnosis is made with EEG, which shows disorganized, abnormal seizure activity during an event. The differential diagnosis for convulsive status epilepticus includes movement disorders (chorea, tics), rigors, clonus with stimulation, and decerebrate/decorticate posturing. For nonconvulsive status epilepticus, other causes of coma must be considered and eliminated by thorough testing.
Complications
Untreated status epilepticus can lead to relative cerebral ischemia and permanent brain injury. Physical injuries can occur from convulsions and should be prevented through control of the child’s environment during this critical period.
Treatment
Several antiepileptic drugs have been advocated as first-line therapies for status epilepticus, including benzodiazepines (rectal valium, IV midazolam or lorazepam), phenytoin (or fosphenytoin), and barbiturates (phenobarbital). Therapy is adjusted for both symptoms and EEG evidence of seizures. In the PICU, for refractory cases, a continuous infusion of barbiturates and/or benzodiazepines may be necessary. Specifically, continuous IV infusions of midazolam (starting at 0.1 mg/kg/hr) or pentobarbital (loading dose of 2-10 mg/kg and a continuous infusion starting at 1 mg/kg/hr) should be considered. Bolus doses in the lower portion of this range may minimize untoward cardiovascular side effects. For refractory cases that progress to this level of therapeutic intensity, respiratory support and hemodynamic monitoring and/or support should already be in place. As therapy escalates, continuous EEG monitoring should be considered to help titrate therapy. For refractory status epilepticus, newer therapies include mapping of the seizure focus followed by neurosurgical resection, IV lidocaine, or levetiracetam.
Supportive Care
Effective cardiopulmonary resuscitation is critical to maximize outcome, because airway compromise can be common during the ictal event or may result from the drugs used to treat the seizure. Tracheal intubation should be strongly considered if the child becomes obtunded, loses airway reflexes, or has respiratory insufficiency. Neuromuscular blockade to facilitate intubation will mask epileptic movements but not the abnormal brain activity, and thus, treatment for the underlying ictus should be continued. Hypotension and decreased cardiac output may also be seen in severe cases or with high-dose anticonvulsant drug therapy; administration of fluids or inotropic drugs or use of hemodynamic monitoring may be indicated. In cases of prolonged refractory status epilepticus, meticulous ICU care, including pulmonary toilet, optimal nutrition, and infection surveillance and treatment, is needed to minimize morbidity.
Stroke and Intracerebral Hemmorrhage
Etiology
The predominant causes of ischemic stroke in children are sickle cell disease and heart disease (either acquired or congenital), which are responsible for ≈50% of strokes after the neonatal period (Chapter 594). A variety of other conditions, including carotid or vertebral arterial dissection, infectious (meningitis, sinusitis), hematologic (prothrombotic states, polycythemia, chronic anemias), traumatic, and autoimmune (systemic lupus erythematosus, inflammatory bowel) disorders, and vasculitis, are risk factors. Intracerebral hemorrhage results from abnormal vascular development and subsequent rupture of cerebral vessels, with arteriovenous malformations, hemangiomas, or aneurysms. Cerebral venous sinus thrombosis is often caused by severe dehydration and hypercoagulable states.
Epidemiology
In the USA, there is an overall incidence of 2.3/100,000 (1.2/100,000 for ischemic stroke and 1.1/100,000 for intracerebral hemorrhage) in children.
Pathology and Pathogenesis
Ischemic strokes in children are generally not the result of atherosclerotic plaque migration, as they are in adults. Instead, damage to the intima of cerebral arteries can form a thrombotic nidus. In sickle cell disease, chronic turbulent blood flow likely leads to vascular damage. In intracerebral hemorrhage, blood vessel wall integrity is compromised, leading to extravasation of blood into the parenchyma or dural spaces. The usual pathology in children with heart disease is embolism from diseased valves (or intracardiac devices) and right-to-left shunts that leads to cerebrovascular occlusion.
Clinical Manifestations
The predominant presentation of children with stroke consists of the abrupt onset of focal neurologic deficits, and that of children with intracerebral hemorrhage is coma (if large portions of the cortex or brainstem are involved). In sickle cell disease, strokes are often unrecognized until imaging studies are obtained.
Laboratory Findings
In large hemorrhagic infarctions, release of tissue factor from the brain can lead to a prolonged prothrombin time (PT), which can exacerbate the injury. Conversely, in children with hypercoagulable phenotypes, abnormalities in factor V Leiden, protein S, protein C, or other factors may be present. Homocystinuria is another cause of hypercoagulable state in children.
Diagnosis and Differential
Detailed history and physical examination can often localize the location of lesions. However, CT or MRI studies of the brain are required. Differential diagnoses can include complex migraines, seizures, and other organic brain syndromes.
Complications
The major complications of stroke are hemorrhagic transformation of thrombotic lesions and vasospasm after aneurysmal subarachnoid hemorrhage (SAH). The incidence of hemorrhagic transformation in children has not been defined, but the incidence in adults is ≈3%. The incidence of vasospasm in children is unclear; case reports suggest that vasospasm can occur for up to 14 days after SAH.
Treatment
Close monitoring in a PICU or intermediate care unit is appropriate for most acute cases of stroke or intracerebral hemorrhage. The child with an evolving stroke may have progressive neurologic deterioration, particularly if the stroke affects motor control of the airway or is associated with cerebral edema. The only approved acute therapy for ischemic stroke is recombinant tissue plasminogen activator (rTPA) in adults with known thrombosis of a major cerebral artery by imaging—with the limitation that therapy be initiated within 3 hr if rTPA is given IV or within 6 hr if it is given intra-arterially into the occlusion. Recombinant TPA has been reported as effective in individual cases or small series in children, but dosing of this potentially dangerous agent has not been studied. Moreover, risk factors for hemorrhagic transformation in adults (extent of parenchymal hypoattenuation on baseline CT scan, a history of heart failure, increasing age, and baseline systolic blood pressure) may not be applicable to children. A focus at this time is determining which children would benefit from anticoagulation therapy (standard heparin, low-molecular-weight heparin, warfarin, aspirin) after stroke. Pediatric guidelines-based recommendations germane to the neurocritical care aspects of stroke include (1) consideration of ICP monitoring for spontaneous intracerebral hemorrhage (classification of recommendation: I, level of evidence: C), (2) providing red blood cell exchange/transfusion therapy for children with sickle cell disease and acute stroke (class IIa, level C), (3) anticoagulation and/or thrombolytics in the acute period if ICP management is not warranted (class IIa, level C and class IIb, level C, respectively), continuous EEG monitoring for children with tracheal intubation (class IIb, level C), and consideration of thrombolytics for children with cerebral sinus venous thrombosis (class IIb, level C).
In SAH, there are no pediatric guidelines. Adult guidelines recommend surgical clipping or endovascular coiling, along with medical management of vasospasm that includes induced hypertension and hypervolemic therapy, administration of calcium channel blockers, particularly nimodipine, and ICP monitoring in some cases. Collaboration between pediatric neurosurgery and ICU teams is essential.
Supportive Care
Controlling airway and respiration and avoiding secondary injury are essential. Hypotension should be treated. Hypertension, however, is a more difficult problem. Adult guidelines for acute ischemic stroke recommend antihypertensive therapies only if thrombolytic treatments are used, because a 25% risk of hemorrhagic transformation has been reported in adults with systolic blood pressure >165 mm Hg with use of rTPA. The conundrum faced by the clinician is whether the cause of the hypertension is related to a Cushing response due to raised ICP or is independent of ICP. Treatment of the former should target a reduction in ICP, whereas that of the latter should target a safe reduction in MAP—maintaining adequate CPP in the injured brain while preventing cerebral edema formation if blood pressure has exceeded the upper limit of the autoregulation. This problem is compounded in children by the fact that the upper limit of blood pressure autoregulation has not been defined. It is probably less than the adult value of MAP, 150 mm Hg, and is age-dependent. When this upper limit is exceeded, CBF rises proportionately with increases in MAP, leading to the propensity for cerebral edema formation. In intracerebral hemorrhage, the 2007 AHA Guidelines recommend aggressive reduction of blood pressure in patients with systolic blood pressure >200 mm Hg. What that threshold represents in children across the age spectrum is unclear. If a reduction of MAP is desired in a child with brain injury, it is best accomplished by means of continuous infusions of medications that have minimal effects on cerebral vasomotor tone (i.e., β-blockers such as esmolol or mixed α/β-blockers such as labetalol). These agents minimize the vasodilation and increases in cerebral blood volume that may exacerbate ICP. If such agents are contraindicated (i.e., by bradycardia or reactive airway disease), continuous infusion of a calcium channel blocker (nicardipine or diltiazem) can be effective.
Adelson PD, Bratton SL, Carney NA, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Pediatr Crit Care Med. 2003;4:S1-S75.
Atabaki SM, Stiell IG, Bazarian JJ, et al. A clinical decision rule for cranial computed tomography in minor pediatric head trauma. Arch Pediatr Adolesc Med. 2008;162:439-445.
Bernard S. Hypothermia after cardiac arrest: expanding the therapeutic scope. Crit Care Med. 2009:S227-S233.
Bishop NB. Traumatic brain injury: a primer for primary care physicians. Curr Probl Pediatr Adolesc Health Care. 2006;36:313-340.
Bulger EM, May S, Brasel KJ, et al. Out-of-hospital hypertonic resuscitation following severe traumatic brain injury. JAMA. 2010;304(13):1455-1464.
Chambers IR, Stobbart L, Jones PA, et al. Age-related differences in intracranial pressure and cerebral perfusion pressure in the first 6 hours of monitoring after children’s head injury: association with outcome. Childs Nerv Syst. 2005;21:195-199.
Deverber G, Kirkham F. Guidelines for the treatment and prevention of stroke in children. Lancet Neurol. 2008;7:983-985.
Dunning J, Daly JP, Lomas JP, et al. Children’s head injury algorithm for the prediction of important clinical events study group: Derivation of the children’s head injury algorithm for the prediction of important clinical events decision rule for head injury in children. Arch Dis Child. 2006;91:885-891.
Hammell CL, Henning JD. Prehospital management of severe traumatic brain injury. BMJ. 2009;338:1262-1266.
Hutchinson JS, Ward RE, Lacroix J, et al. Hypothermia therapy after traumatic brain injury in children. N Engl J Med. 2008;358:2447-2456.
Klein GW, Hojsak JM, Schmeidler J, Rapaport R. Hyperglycemia and outcome in the pediatric intensive care unit. J Pediatr. 2008;153:379-384.
MRC CRASH Trial CollaboratorsPerel P, Arango M, Clayton T, et al. Predicting outcome after traumatic brain injury: practical prognostic models based on large cohort of international patients. BMJ. 2009;336:425-429.
Mullen MT, McKinney JS, Kasner SE. Blood pressure management in acute stroke. J Hum Hypertens. 2009;23:559-569.
Polderman KH. Induced hypothermia and fever control for prevention and treatment of neurological injuries. Lancet. 2009;371:1955-1969.
Riviello JJJr, Ashwal S, Hirtz D, et al. Practice parameter: diagnostic assessment of the child with status epilepticus (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2006;67:1542-1550.
Roach ES, Golomb MR, Adams R, et al. Management of stroke in infants and children: a scientific statement from a special writing group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008;39:2644-2691.
SAFE Study Investigators, Australian and New Zealand Intensive Care Society Clinical Trials Group, Australian Red Cross Blood Service; George Institute for International HealthMyburgh J, Cooper DJ, Finfer S, et al. Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;357:874-884.
Sagalyn E, Band RA, Gaieski DF, et al. Therapeutic hypothermia after cardiac arrest in clinical practice: review and compilation of recent experiences. Crit Care Med. 2009;37:S223-S226.
Shankaran S. Neonatal encephalopathy: treatment with hypothermia. J Neurotrauma. 2009;26:437-443.
Smits M, Dippel DWJ, Steyerberg EW, et al. Predicting intracranial traumatic finding on computed tomography in patients with minor head injury: the CHIP prediction rule. Ann Intern Med. 2007;146:397-405.
Stein SC, Fabbri A, Servadei F, et al. A critical comparison of clinical decision instruments for computed tomographic scanning in mild closed traumatic brain injury in adolescents and adults. Ann Emerg Med. 2009;53:180-188.
Willmot C, Ponsford J. Efficacy of methylphenidate in the rehabilitation of attention following traumatic brain injury: a randomized, crossover, double blind, placebo controlled inpatient trial. J Neurosurg Psychiatry. 2009;80:552-557.
Yeates KO, Taylor HG, Rusin J, et al. Longitudinal trajectories of postconcussive symptoms in children with mild traumatic brain injuries and their relationship to acute clinical status. Pediatrics. 2009;123:735-743.
Young GB. Neurologic prognosis after cardiac arrest. N Engl J Med. 2009;361:605-611.
63.1 Brain Death
Brain death is the irreversible cessation of all functions of the entire brain, including the brainstem. It is also known as the determination of death using neurologic criteria. Although brain death is legally accepted in the USA as the equivalent of death due to the irreversible cessation of circulatory and respiratory functions, it remains a concept that is sometimes difficult to understand and is not universally accepted.
Epidemiology
In children brain death most commonly develops following TBI (including brain injury due to nonaccidental trauma) or asphyxial injury. Pathogenesis is multifactorial, with the end result being the irreversible loss of brain and brainstem function.
Clinical Manifestations
Brain death is primarily a clinical diagnosis. Although tests such as EEG and studies to assess CBF are sometimes used to confirm the diagnosis, repeated clinical examination is the standard for diagnosis.
Guidelines for the determination of brain death in children were published in 1987 by a Special Task Force to the American Academy of Pediatrics, and they have not been revised. Practice parameters for determining brain death in adults were published by the American Academy of Neurology in 1995. Despite these published guidelines, there exists substantial variability in institutional policies, the practice of performing brain death examinations, and documentation.
The 3 key components of clinical brain death diagnosis are demonstrations of irreversible coma/unresponsiveness, absence of brainstem reflexes, and apnea.
Before a determination of brain death may be made, it is of utmost importance that the cause of the coma be determined through the use of historical, radiologic, and laboratory data to rule out a reversible condition. Potentially reversible causes of coma include metabolic disorders, toxins, sedative drugs, paralytic agents, hypothermia, hypoxia, hypotension/shock, hypoglycemia/hyperglycemia, hyponatremia/hypernatremia, hypercalcemia, hypermagnesemia, nonconvulsive status epilepticus, hypothyroidism, hypocortisolism, hypercarbia, liver or renal failure, sepsis, meningitis, encephalitis, SAH, and surgically remediable brainstem lesions.
The state of coma requires that the patient be unresponsive, even to noxious stimuli. Any purposeful motor response, such as localization, does not constitute coma. Likewise, any posturing (decerebrate or decorticate) is not consistent with coma, and therefore not consistent with brain death. The presence of spinal cord reflexes— even complex reflexes—does not preclude the diagnosis of brain death. Some complex spinal movements may be particularly pronounced following removal of the ventilator, and the family of a patient whose ventilator is being removed should be alerted to this possibility beforehand. The best-known series of movements is the Lazarus sign, which consists of extension of the upper extremities followed by flexion of the arms with the hands reaching to midsternal level. Flexion of the body at the waist may occur. A variety of other movements has been reported.
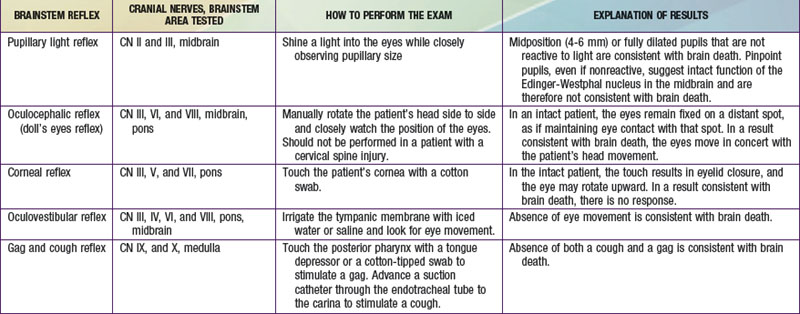
Brainstem reflexes must be absent. A listing of the brainstem reflexes to be tested, the brainstem location of each reflex, and the result of each test that is consistent with a diagnosis of brain death can be found in Table 63-1.
Apnea is the absence of respiratory effort in response to an adequate stimulus. A PCO2 value >60 mm Hg is usually considered an adequate stimulus as long as the patient’s previous PCO2 value was normal (≈40 mm Hg). If the patient had an elevated baseline PCO2 for some reason, then a PCO2 value 20 mm Hg above baseline is considered sufficient. There are case reports of children who demonstrated spontaneous breathing efforts at higher PCO2 (70-110 mm Hg); nevertheless, the generally accepted threshold is still 60 mm Hg. Apnea is clinically confirmed through the apnea test. Because the apnea test has the potential to destabilize the patient, this test is performed only if the first 2 criteria for brain death (irreversible coma and absence of brainstem reflexes) are already confirmed.
The apnea test assesses the function of the medulla in driving ventilation. It is performed by first ensuring the adequacy of hemodynamics and temperature and the absence of apnea-producing drug effects or significant metabolic derangements. The test is performed by first preoxygenating the patient with 100% oxygen for approximately 10 min and adjusting ventilation to achieve a PCO2 of about 40 mm Hg. A baseline blood gas result documents the starting values. During the test, oxygenation must be maintained on continuous positive airway pressure CPAP and 100% oxygen by means of the ventilator circuit or a resuscitation bag such as a Mapleson device, which has an adjustable CPAP valve; alternatively the patient may be disconnected from the ventilator, and 100% oxygen may be delivered via a large-bore catheter advanced down the endotracheal tube. Throughout the test, the child is assessed for breathing efforts through observation and auscultation. A blood gas sample is obtained approximately 10 min into the test and every 5 min thereafter until the target PCO2 is surpassed; ventilatory support is resumed at that time. If at any point during the test the patient becomes hypoxic or hypotensive, the test is aborted and ventilatory support is resumed. Absence of respiratory efforts with a PCO2 > 60 mm Hg or more that 20 mm Hg above an elevated baseline value is consistent with brain death.
Observation Periods
To establish the diagnosis of brain death, the findings must remain consistent over a period of observation. The recommended length of this period of observation varies by age. For children aged 7 days to 2 mo, 2 examinations separated by at least 48 hr are recommended. For children aged 2 mo to 1 yr, 2 examinations separated by at least 24 hr are recommended. For children older than 1 yr the recommendation is a 12-hr observation period between exams. If the cause of the coma is hypoxic-ischemic brain injury and the first exam is performed shortly after the insult, a period of at least 24 hr is recommended before the second exam. A second exam is not needed if a nuclear medicine cerebral flow scan demonstrates absence of CBF.
Confirmatory Tests
Brain death is primarily a clinical diagnosis, but sometimes confirmatory testing is necessary or desired. According to the 1987 recommendations, confirmatory testing should be performed on all children <1 yr of age. Confirmatory testing is also used in situations in which a portion of the clinical exam is impossible to perform or the results are suspected to be unreliable. The 2 most commonly used confirmatory tests are EEG and studies to confirm the absence of CBF, such as nuclear medicine cerebral flow scans.
A valid EEG to confirm suspected brain death must be performed according to accepted technical requirements, under conditions of normothermia, and in the absence of drug levels sufficient to suppress the EEG response. An EEG that demonstrates electrocerebral silence over a 30-min recording time under these conditions supports the diagnosis of brain death. There have been reports of children who meet the clinical criteria for brain death but continue to have some electrical activity on EEG. Advantages of this study are its wide availability and low risk. Disadvantages include potential confounders, such as artifact in the tracing and the presence of suppressing levels of drugs such as barbiturates.
A nuclear medicine cerebral flow scan consists of intravenous injection of a radiopharmaceutical agent followed by imaging of the brain to look for cerebral uptake. Like EEG, nuclear medicine scans are widely available and low risk. Unlike EEG, these studies are not affected by drug levels. A study that shows absence of uptake in the brain demonstrates absence of CBF and is confirmatory of brain death. In infants with open sutures and fontanels, the scan may show residual flow. This finding does not contradict a clinical diagnosis of brain death. Four-vessel intracranial contrast angiography has previously been used as the definitive confirmatory test, but practical technical difficulties and risks have led to the use of nuclear medicine scans.
Documentation
Documentation is an important aspect of diagnosing brain death. Complete documentation should include statements of the following:
Supportive Care
Following a diagnosis of brain death, supportive care may continue for hours to days as the family makes decisions about potential organ donation and comes to terms with the diagnosis. A diagnosis of brain death may not be accepted by the family for personal, religious, or cultural reasons. It is important for care providers to be patient and supportive of the family dealing with this difficult situation.
Objections to the Idea of Brain Death
Although the concept of brain death is widely accepted and very useful in facilitating organ transplantation, it is not accepted by all. Several countries do not recognize brain death, and many individuals, both medical personnel and laypeople, object to the idea of brain death.
It has been pointed out that some patients who meet brain death criteria continue to show evidence of integrative functioning, such as control over free-water homeostasis (absence of diabetes insipidus), presence of some electrical activity on EEG, control of temperature regulation, capacity for growth and wound healing, and variability of heart rate and blood pressure in response to stimulus. Along with scientific arguments, there are also philosophical arguments about what constitutes death and whether a person who lacks function of the brain, but not of the body, is truly dead.
Ashwal S, Serna-Fonseca T. Brain death in infants and children. Crit Care Nurse. 2006;26:117-128.
Greer DM, Varelas PN, Haque S, et al. Variability of brain death determination guidelines in leading US neurologic institutions. Neurology. 2008;70:284-289.
Mathur M, Petersen L, Stadtler M, et al. Variability in pediatric brain death determination and documentation in Southern California. Pediatrics. 2008;121:988-993.
Miller FG, Truog RD. The incoherence of determining death by neurologic criteria: a commentary on Controversies in the Determination of Death, a white paper by the President’s Council on Bioethics. Kennedy Inst Ethics J. 2009;19:185-193.
Morenski JD, Oro JJ, Tobias JD, et al. Determination of death by neurologic criteria. J Intensive Care Med. 2003;18:211-221.
1995 Practice parameters for determining brain death in adults (summary statement): the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1995;45:1012-1014.
1987 Report of Special Task Force. Guidelines for the determination of brain death in children, American Academy of Pediatrics Task Force on Brain Death in Children. Pediatrics. 1987;80:298-300.
Saposnik G, Basile VS, Young GB. Movements in brain death: a systematic review. Can J Neurol Sci. 2009;36:154-160.
Shemie SD, Pollack MM, Morioka M, et al. Diagnosis of brain death in children. Lancet Neurol. 2007;6:87-92.