Chapter 93 Nervous System Disorders
Central nervous system (CNS) disorders are important causes of neonatal mortality and both short- and long-term morbidity. The CNS can be damaged as a result of hypoxia, asphyxia, hemorrhage, trauma, hypoglycemia, or direct cytotoxicity. The etiology of CNS damage is often multifactorial and includes acute perinatal complications, postnatal hemodynamic instability, and developmental abnormalities that may be genetic and/or environmental. Predisposing factors for brain injury include chronic and acute maternal illness resulting in uteroplacental dysfunction, intrauterine infection, macrosomia/dystocia, malpresentation, prematurity, and intrauterine growth restriction. Acute and often unavoidable emergencies during the delivery process frequently result in mechanical and/or hypoxic-ischemic brain injury.
93.1 The Cranium
Erythema, abrasions, ecchymoses, and subcutaneous fat necrosis of facial or scalp soft tissues may be noted after a normal delivery or after forceps or vacuum-assisted deliveries. Their location depends on the area of contact with the pelvic bones or of application of the forceps. Traumatic hemorrhage may involve any layer of the scalp as well as intracranial contents (Fig. 93-1).
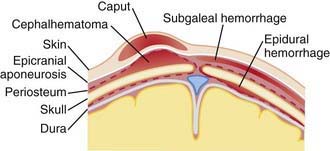
Figure 93-1 Sites of extracranial (and extradural) hemorrhages in the newborn. Schematic diagram of important tissue planes from skin to dura.
(From Volpe JJ: Neurology of the newborn, ed 4, Philadelphia, 2001, WB Saunders.)
Caput succedaneum is a diffuse, sometimes ecchymotic, edematous swelling of the soft tissues of the scalp involving the area presenting during vertex delivery (see Fig. 93-1). It may extend across the midline and across suture lines. The edema disappears within the 1st few days of life. Molding of the head and overriding of the parietal bones are frequently associated with caput succedaneum and become more evident after the caput has receded; they disappear during the 1st weeks of life. Rarely, a hemorrhagic caput may result in shock and require blood transfusion. Analogous swelling, discoloration, and distortion of the face are seen in face presentations. No specific treatment is needed, but if extensive ecchymoses are present, hyperbilirubinemia may develop.
Cephalohematoma (Fig. 93-2) is a subperiosteal hemorrhage, hence always limited to the surface of one cranial bone. Cephalohematomas occur in 1-2% of live births. No discoloration of the overlying scalp occurs, and swelling is not usually visible for several hours after birth because subperiosteal bleeding is a slow process. The lesion becomes a firm tense mass with a palpable rim localized over one area of the skull. Most cephalohematomas are resorbed within 2 wk-3 mo, depending on their size. They may begin to calcify by the end of the 2nd week. A few remain for years as bony protuberances and are detectable on radiographs as widening of the diploic space; cystlike defects may persist for months or years. An underlying skull fracture, usually linear and not depressed, may be associated with 10-25% of cases. A sensation of central depression suggesting but not indicative of an underlying fracture or bony defect is usually encountered on palpation of the organized rim of a cephalohematoma. Cephalohematomas require no treatment, although phototherapy may be necessary to treat hyperbilirubinemia. Infection of the hematoma is a very rare complication.
A subgaleal hemorrhage is a collection of blood beneath the aponeurosis that covers the scalp the entire length of the occipitofrontalis muscle. Bleeding can be very extensive into this large potential space and may even dissect into the subcutaneous tissues of the neck. There is often an association with vacuum-assisted delivery. The mechanism of injury is most likely secondary to a linear skull fracture, suture diastasis or fragmentation of the superior margin of the parietal bone, and/or rupture of the emissary vein. Extensive subgaleal bleeding is occasionally secondary to a hereditary coagulopathy (hemophilia). A subgaleal hemorrhage manifests as a firm fluctuant mass that increases in size after birth. Many patients have a consumptive coagulopathy owing to massive blood loss. Patients should be monitored for hypotension and the development of hyperbilirubinemia. These lesions typically resolve over 2-3 weeks.
Fractures of the skull may occur as a result of pressure from forceps or from the maternal symphysis pubis, sacral promontory, or ischial spines. Linear fractures, the most common, cause no symptoms and require no treatment. Depressed fractures are usually indentations of the calvaria similar to the dents in a ping-pong ball; they are generally a complication of forceps delivery or fetal compression. Affected infants may be asymptomatic unless they have associated intracranial injury; it is advisable to elevate severe depressions to prevent cortical injury from sustained pressure. Fracture of the occipital bone with separation of the basal and squamous portions almost invariably causes fatal hemorrhage because of disruption of the underlying vascular sinuses. Such fractures may result during breech deliveries from traction on the hyperextended spine of the infant while the head is fixed in the maternal pelvis.
Subconjunctival and retinal hemorrhages are frequent; petechiae of the skin of the head and neck are also common. All are probably secondary to a sudden increase in intrathoracic pressure during passage of the chest through the birth canal. Parents should be assured that these hemorrhages are temporary and the result of normal events of delivery. The lesions resolve rapidly within the 1st 2 wk of life.
93.2 Traumatic, Epidural, Subdural, and Subarachnoid Hemorrhage
Traumatic epidural, subdural, or subarachnoid hemorrhage is especially likely when the fetal head is large in proportion to the size of the mother’s pelvic outlet, with prolonged labor, in breech or precipitous deliveries, or as a result of mechanical assistance with delivery. Massive subdural hemorrhage, often associated with tears in the tentorium cerebelli or, less frequently, in the falx cerebri, is rare but is encountered more often in full-term than in premature infants. Patients with massive hemorrhage caused by tears of the tentorium or falx cerebri rapidly deteriorate and may die soon after birth. The majority of subdural and epidural hemorrhages resolve without intervention; consultation with a neurosurgeon is recommended. The diagnosis of subdural hemorrhage may be delayed until the chronic subdural fluid volume expands and produces megalocephaly, frontal bossing, a bulging fontanel, anemia, and, sometimes, seizures. CT scan and MRI are useful imaging techniques to confirm these diagnoses. Symptomatic subdural hemorrhage in large term infants should be treated by removal of the subdural fluid collection with a needle placed through the lateral margin of the anterior fontanelle. In addition to birth trauma, child abuse must be suspected in all infants with subdural effusion after the immediate neonatal period.
Subarachnoid hemorrhage (SAH) is rare and typically is clinically silent. The anastomoses between the penetrating leptomeningeal arteries or the bridging veins are the most likely source of the bleeding. The majority of affected infants have no clinical symptoms, but the SAH may be detected because of an elevated number of red blood cells in a lumbar puncture sample. Some infants experience benign seizures, which tend to occur on the 2nd day of life. Rarely, an infant has a life-threatening catastrophic hemorrhage and dies. There are usually no neurologic abnormalities during the acute episode or on follow-up. Significant neurologic findings should suggest an arteriovenous malformation; this lesion can easily be detected on CT or MRI; ultrasonography is a less sensitive tool.
93.3 Intracranial-Intraventricular Hemorrhage and Periventricular Leukomalacia
Etiology
Intracranial hemorrhage usually develops spontaneously; less commonly, it may be due to trauma or asphyxia, and rarely, it occurs from a primary hemorrhagic disturbance or congenital vascular anomaly. Intracranial hemorrhage often involves the ventricles (intraventricular hemorrhage [IVH]) of premature infants delivered spontaneously without apparent trauma. Primary hemorrhagic disturbances and vascular malformations are rare and usually give rise to subarachnoid or intracerebral hemorrhage. In utero hemorrhage associated with maternal idiopathic or, more often, fetal alloimmune thrombocytopenia may occur as severe cerebral hemorrhage or a porencephalic cyst after resolution of a fetal cortical hemorrhage. Intracranial bleeding may be associated with disseminated intravascular coagulopathy, isoimmune thrombocytopenia, and neonatal vitamin K deficiency, especially in infants born to mothers receiving phenobarbital or phenytoin.
Epidemiology
The overall incidence of IVH has decreased over the past decades as a result of improved perinatal care and increased use of antenatal corticosteroids, surfactant to treat respiratory distress syndrome (RDS), and, possibly, prophylactic indomethacin; however, it continues to be an important cause of morbidity in preterm infants. Approximately 30% of premature infants <1,500 g have IVH. The risk is inversely related to gestational age and birthweight, with the smallest and most immature infants being at the highest risk; 7% of infants 1,001-1,500 g have a severe IVH (grade III or IV), compared with 14% of infants 751-1,000 g and 24% of infants ≤750 g. In 3% of infants <1,000 g, periventricular leukomalacia (PVL) develops.
Pathogenesis
The major neuropathologic lesions associated with VLBW infants are IVH and PVL. IVH in premature infants occurs in the gelatinous subependymal germinal matrix. This periventricular area is the site of origin for embryonal neurons and fetal glial cells, which migrate outwardly to the cortex. Immature blood vessels in this highly vascular region of the developing brain combined with poor tissue vascular support predispose premature infants to hemorrhage. The germinal matrix involutes as the infant approaches full-term gestation and the tissue’s vascular integrity improves; therefore IVH is much less common in the term infant. Periventricular hemorrhagic infarction often develops after a grade IV IVH owing to venous congestion. Predisposing factors for IVH include prematurity, respiratory distress syndrome, hypoxic-ischemic or hypotensive injury, reperfusion injury of damaged vessels, increased or decreased cerebral blood flow, reduced vascular integrity, increased venous pressure, pneumothorax, thrombocytopenia, hypervolemia, and hypertension.
Understanding of the pathogenesis of PVL is evolving, and it appears to involve both intrauterine and postnatal events. A complex interaction exists between the development of the cerebral vasculature and the regulation of cerebral blood flow (both of which are gestational age dependent), disturbances in the oligodendrocyte precursors required for myelination, and maternal/fetal infection and/or inflammation. Similar factors (hypoxia-ischemia), venous obstruction from an IVH, or undetected fetal stress may result in decreased perfusion to the brain, leading in turn to periventricular hemorrhage and necrosis. PVL is characterized by focal necrotic lesions in the periventricular white matter and/or more diffuse white matter damage. The risk for PVL increases in infants with severe IVH and/or ventriculomegaly. The corticospinal tracts descend through the periventricular white matter, hence the association between cerebral white matter injury/PVL and motor abnormalities, including cerebral palsy.
Clinical Manifestations
The majority of patients with IVH, including some with moderate to severe hemorrhages, have no clinical symptoms. Some premature infants in whom severe IVH develops may have acute deterioration on the 2nd or 3rd day of life. Hypotension, apnea, pallor, or cyanosis; poor suck; abnormal eye signs; a high-pitched, shrill cry; convulsions, or decreased muscle tone; metabolic acidosis; shock; and a decreased hematocrit or failure of the hematocrit to increase after transfusion may be the 1st clinical indications. IVH may rarely manifest at birth; 50% of cases are diagnosed within the 1st day of life, and up to 75% within the 1st 3 days. A small percentage of infants have late hemorrhage, between days 14 and 30. IVH as a primary event is rare after the 1st month of life.
PVL is usually clinically asymptomatic until the neurologic sequelae of white matter damage become apparent in later infancy as spastic motor deficits. PVL may be present at birth but usually occurs later as an early echodense phase (3-10 days of life), followed by the typical echolucent (cystic) phase (14-20 days of life).
The severity of hemorrhage may be defined on CT scans by the location and degree of ventricular dilatation. In a grade I hemorrhage, bleeding is isolated to the subependymal area. In Grade II hemorrhage, there is bleeding within the ventricle but without evidence of ventricular dilatation. Grade III hemorrhage consists of IVH with ventricular dilatation. In Grade IV hemorrhage, there is intraventricular and parenchymal hemorrhage. Another grading system describes 3 levels of increasing severity of IVH detected on ultrasound: In grade I, bleeding is confined to the germinal matrix–subependymal region or to <10% of the ventricle (≈35% of IVH cases); grade II is defined as intraventricular bleeding with 10-50% filling of the ventricle (≈40% of IVH cases) and in grade III, more than 50% of the ventricle is involved, with dilated ventricles (Fig. 93-3). Ventriculomegaly is defined as mild (0.5-1 cm), moderate (1.0-1.5 cm), or severe (>1.5 cm).
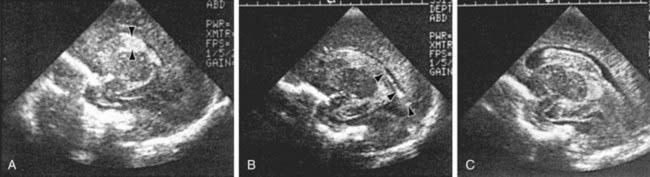
Figure 93-3 Grading the severity of germinal matrix intraventricular hemorrhage with parasagittal ultrasound scans. A, Grade I: Note the echogenic blood in the germinal matrix (arrowheads) just anterior to the anterior tip of the choroid plexus, which (normally) is also echogenic. B, Grade II: Note the echogenic blood (arrowheads) filling <50% of the ventricular area. C, Grade III: Note the large blood clot nearly completely filling and distending the entire lateral ventricle.
(From Intracranial hemorrhage: germinal matrix-intraventricular hemorrhage of the premature infant. In Volpe JJ: Neurology of the newborn, ed 4, Philadelphia, 2001, WB Saunders.)
Diagnosis
Intracranial hemorrhage is suspected on the basis of the history, clinical manifestations, and knowledge of the birthweight-specific risks for IVH. The associated clinical signs of IVH are typically nonspecific or absent; therefore, it is recommended that premature infants <32 wk of gestation be evaluated with routine real-time cranial ultrasonography through the anterior fontanel to screen for IVH. Infants <1,000 g are at highest risk and should undergo cranial ultrasonography within the 1st 3-7 days of age, when approximately 75% of lesions will be detectable. Ultrasonography is the preferred imaging technique for screening because it is noninvasive, portable, reproducible, and sensitive and specific for detection of IVH. All at-risk infants should undergo follow-up ultrasonography at 36-40 wk of postmenstrual age to evaluate adequately for PVL, because cystic changes related to perinatal injury may not be visible for at least 2-4 wk. In one study, 29% of LBW infants who later experienced cerebral palsy did not have radiographic evidence of PVL until after 28 days of age. Ultrasonography also detects the precystic and cystic symmetric lesions of PVL and the asymmetric intraparenchymal echogenic lesions of cortical hemorrhagic infarction. Furthermore, the delayed development of cortical atrophy, porencephaly, and the severity, progression, or regression of posthemorrhagic hydrocephalus can be determined by serial ultrasonographic examinations.
Approximately 3-5% of VLBW infants have posthemorrhagic hydrocephalus and require ventriculoperitoneal shunt insertion; if the initial ultrasonography findings are abnormal, additional interval ultrasonographic studies are indicated to monitor for the development of hydrocephalus.
IVH represents only one facet of brain injury in the term or preterm infant. MRI is a more sensitive tool for evaluation of extensive periventricular injury and may be more predictive of adverse long-term outcome. CT or, more reliably, diffusion-weighted MRI is indicated for term infants in whom brain injury or stroke is suspected, because ultrasonography may not reveal edema or intraparenchymal hemorrhage and infarction.
Prognosis
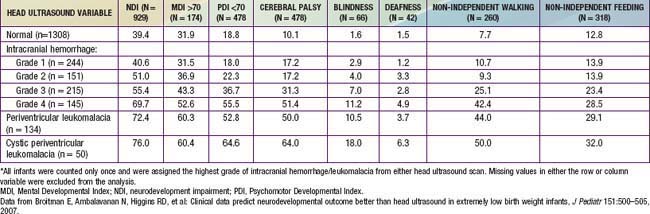
The degree of IVH and the presence of PVL are strongly linked to neurodevelopmental impairment. For infants with birthweight <1,000 g, the incidences of severe neurologic impairment (defined as mental developmental index <70, psychomotor development index <70, cerebral palsy, blindness, or deafness) are about 50%, 55%, and 70% for infants with grade II, grade III, and grade IV IVH, respectively (Table 93-1). In contrast, the rate of neurodevelopmental impairment is approximately 40% in infants without IVH and those with grade I IVH. PVL, cystic PVL, and progressive hydrocephalus requiring shunt insertion are each independently associated with a poorer prognosis.
Table 93-1 PERCENTAGE OF INFANTS WITH EACH NEUROLOGIC OUTCOME AT 18 TO 22 MONTHS CORRECTED AGE BY HEAD ULTRASOUND FINDINGS*
Most infants with IVH and acute ventricular distention do not have posthemorrhagic hydrocephalus (PHH). Ten percent to 15% of LBW neonates with IVH demonstrate hydrocephalus, which may initially be present without clinical signs, such as an enlarging head circumference, lethargy, a bulging fontanel or widely split sutures, apnea, and bradycardia. In infants in whom symptomatic hydrocephalus develops, clinical signs may be delayed 2-4 wk despite progressive ventricular distention with compression and thinning of the cerebral cortex. Many infants with PHH have spontaneous regression; 3-5% of VLBW infants with PHH require shunt insertion. Infants with PHH requiring shunt insertion have lower cognitive and psychomotor performance at 18-22 mo.
Prevention
Improved perinatal care is imperative to minimize traumatic brain injury and decrease the risk of preterm delivery. The incidence of traumatic intracranial hemorrhage may be reduced by judicious management of cephalopelvic disproportion and operative (forceps, vacuum) delivery. Fetal or neonatal hemorrhage caused by maternal idiopathic thrombocytopenic purpura or alloimmune thrombocytopenia may be reduced by maternal treatment with steroids, intravenous immunoglobulin, fetal platelet transfusion, or cesarean section. Tenacious care of the LBW infant’s respiratory status and fluid and electrolyte management—including avoidance of acidosis, hypocarbia, hypoxia, hypotension, wide fluctuations in neonatal blood pressure or PCO2, and pneumothorax—are important factors that may affect the risk for development of IVH and PVL.
A single course of antenatal corticosteroids is recommended in pregnancies 24-34 wk of gestation that are at risk for preterm delivery. Antenatal steroids decrease the risk of death, grade III and IV IVH, and PVL in the neonate. The prophylactic administration of low-dose indomethacin (0.1 mg/kg/day for 3 days) to VLBW preterm infants reduces the incidence of severe IVH.
Treatment
Although no treatment is available for IVH, it may be associated with other complications that require therapy. Seizures should be treated with anticonvulsant drugs. Anemia and coagulopathy require transfusion with packed red blood cells or fresh frozen plasma. Shock and acidosis are treated with the judicious and slow administration of sodium bicarbonate and fluid resuscitation.
Insertion of a ventriculoperitoneal shunt is the preferred method to treat progressive and symptomatic posthemorrhagic hydrocephalus; some infants require temporary cerebrospinal fluid diversion before a permanent shunt can be safety inserted. Diuretics and acetazolamide are not effective. Serial lumbar punctures, ventricular taps or reservoirs, and externalized ventricular drains are potential temporizing interventions; they have an associated risk of infection and of “puncture porencephaly” owing to injury to the surrounding parenchyma. A ventriculosubgaleal shunt inserted from the ventricle into a surgically created subgaleal pocket provides a closed system for constant ventricular decompression without these additional risk factors. Decompression is regulated by the pressure gradient between the ventricle and the subgaleal pocket.
Accardo J, Kammann H, Hoon AHJr. Neuroimaging in cerebral palsy. J Pediatr. 2004;145:S19-S27.
Adams-Chapman I, Hansen NI, Stoll BJ, et al. Neurodevelopmental outcome of extremely low birth weight infants with posthemorrhagic hydrocephalus requiring shunt insertion. Pediatrics. 2008;121:167-177.
Armstrong-Wells J, Johnston SC, Wu YW, et al. Prevalence and predictors of perinatal hemorrhagic stroke: results from the Kaiser pediatric stroke study. Pediatrics. 2009;123:823-828.
Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res. 2010;67:1-8.
Bassan H, Limperopoulos C, Visconti K, et al. Neurodevelopmental outcome in survivors of periventricular hemorrhagic infarction. Pediatrics. 2007;120:785-792.
Broitman E, Ambalavanan N, Higgins RD, et al. Clinical data predict neurodevelopmental outcome better than head ultrasound in extremely low birth weight infants. J Pediatr. 2007;151:500-505.
Brouwer A, Groenendaal F, Van Haastert IL, et al. Neurodevelopmental outcome of preterm infants with severe intraventricular hemorrhage and therapy for post-hemorrhagic ventricular dilatation. J Pediatr. 2008;152:648-654.
Fowlie PW, Davis PG: Prophylactic intravenous indomethacin for preventing mortality and morbidity in preterm infants, Cochrane Database of Systematic Reviews (3):CD000174, 2002.
Heuchan AM, Evans N, Henderson DJ, et al. Perinatal risk factors for major intraventricular haemorrhage in the Australian and New Zealand neonatal network, 1995–1997. Arch Dis Child. 2002;86:F86-F90.
Maunu J, Ekholm E, Parkkola R, et al. Antenatal Doppler measurements and early brain injury in very low birthweight infants. J Pediatr. 2007;150:51-56.
Roze E, Van Braeckel KNJA, van der Veere CH, et al. Functional outcome at school age of preterm infants with periventricular hemorrhagic infarction. Pediatrics. 2009;123:1493-1500.
Saigal S, Stoskopf BL, Streiner DL, et al. Physical growth and current health status of infants who were of extremely low birth weight and controls at adolescence. Pediatrics. 2001;108:407-415.
Schmidt B, Davis P, Moddemann PD, et al. Long-term effects of indomethacin prophylaxis in extremely-low-birth-weight infants. N Engl J Med. 2001;344:1966-1972.
Soul JS, Eichenwald E, Walter G, et al. CSF removal in infantile posthemorrhagic hydrocephalus results in significant improvements in cerebral hemodynamics. Pediatr Res. 2004;55:872-876.
Vincer MJ, Allen AC, Joseph KS, et al. Increasing prevalence of cerebral palsy among very preterm infants: a population-based study. Pediatrics. 2006;118:e1621-e1626.
Whitelaw A: Repeated lumbar or ventricular punctures in newborns with intraventricular hemorrhage, Cochrane Database Syst Rev (1):CD000216, 2002.
93.4 Brain Injury from Inflammation, Infection, and Medications
Severe IVH and PVL are the most commonly associated risk factors for adverse outcome in the VLBW infant. Other factors are also involved in the etiology of perinatal brain injury. Cytokines and prenatal or postnatal infection or inflammation may contribute to brain injury. A systemic inflammatory response syndrome in the mother, fetus, or infant may induce the production of various inflammatory mediators that are directly cytotoxic or cause decreased CNS perfusion (Fig. 93-4). Preterm infants with evidence (often subclinical) of intrauterine or postnatal infection or maternal chorioamnionitis are more likely than uninfected infants to have adverse neurodevelopmental outcome including cerebral palsy.
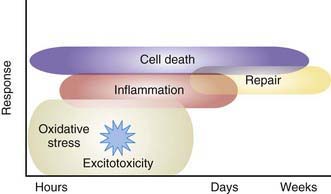
Figure 93-4 Mechanisms of brain injury in the term neonate. Oxidative stress and excitotoxicity, through downstream intracellular signaling, produce both inflammation and repair. Cell death begins immediately and continues during a period of days to weeks. The cell-death phenotype changes from an early necrotic morphology to a pathology resembling apoptosis. This evolution is called the necrosis-apoptosis continuum.
(From Ferriero DM: Neonatal brain injury, N Engl J Med 351:1985–1995, 2004.
In utero infections may involve the developing CNS and directly impair cell growth or produce cell neurosis, resulting in microcephaly, developmental delay, mental retardation, or cerebral palsy. These specific congenital or perinatal acquired infections include those due to cytomegalovirus (Chapter 247), toxoplasmosis (Chapter 282), herpes simplex (Chapter 244), syphilis (Chapter 210), rubella (Chapter 239), and human immunodeficiency virus (Chapter 268). Postnatal acquired bacterial meningitis in the 1st year, but even more so in the 1st month of life, is another major risk factor for CNS injury and associated adverse neurodevelopmental outcome (Chapter 595).
Long-term adverse neurodevelopmental outcomes are also associated with high-dose postnatal corticosteroid use in VLBW infants. Early postnatal exposure to dexamethasone, within the 1st wk of life, is associated with metabolic derangements, poor growth, increased risk for sepsis, and an increased risk of spontaneous bowel perforation. Infants exposed to postnatal steroids after the 1st wk of life have an increased risk of cerebral palsy and developmental delay. The risk may be increased with prolonged steroid use (>6 wk). At 8 yr of age, dexamethasone-treated children are smaller, have smaller head circumferences, poorer motor skills and coordination, more difficulty with visual motor integration, and lower full-scale verbal IQ and performance IQ scores. The American Academy of Pediatrics (AAP) recommends that postnatal corticosteroid use in VLBW infants be limited to exceptional clinical circumstances and that parents of infants in whom corticosteroids are used be informed of the potential adverse side effects, including increased risk for developmental delay, cerebral palsy, and impaired growth.
Necrotizing enterocolitis (NEC) affects approximately 9-14% of LBW infants and is associated with significant morbidity and mortality (Chapter 96.2). Patients with NEC requiring surgery are more likely to have Mental Developmental Index (MDI) scores <70, Psychomotor Developmental Index (PDI) scores <70, and evidence of overall neurodevelopmental impairment. Infants with severe NEC are reported to have a higher incidence of PVL, postnatal infections, and poor growth.
AAP Committee of the Fetus and Newborn. Postnatal corticosteroids to treat or prevent chronic lung disease in preterm infants. Pediatrics. 2002;109:330-338.
Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985-1995.
Miller SP, Ramaswamy V, Michelson D, et al. Patterns of brain injury in term neonatal encephalopathy. J Pediatr. 2005;146:453-460.
Pierrat V, Haiuari N, Liska A, et al. Prevalence, causes, and outcome at 2 years of age of newborn encephalopathy: population based study. Arch Dis Child. 2005;90:F257-F261.
Stark AR, Carlo WA, Tyson JE, et al. Adverse effects of early dexamethasone treatment in extremely low birth weight infants. N Engl J Med. 2001;344:95-101.
Wood NS, Costeloe K, Gibson AT, et al. The EPICure Study: Associations and antecedents of neurological and developmental disability at 30 months of age following extremely preterm birth. Arch Dis Child Fetal Neonatal Ed. 2005;90:F134-F140.
Wu YW, Colford JMJr. Chorioamnionitis as a risk factor for cerebral palsy: a meta-analysis. JAMA. 2000;284:1417-1424.
Yang SH, Choi SJ, Roh CR, et al. Multiple courses of antenatal corticosteroid therapy in patients with preterm premature rupture of membranes. J Perinat Med. 2004;32:42-48.
Yeh TF, Lin YJ, Hung C, et al. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. N Engl J Med. 2004;350:1304-1313.
93.5 Hypoxic-Ischemic Encephalopathy
Anoxia is a term used to indicate the consequences of complete lack of oxygen as a result of a number of primary causes. Hypoxemia refers to decreased arterial concentration of oxygen. Hypoxia refers to a decreased oxygenation to cells or organs. Ischemia refers to blood flow to cells or organs that is insufficient to maintain their normal function. Hypoxic-ischemic encephalopathy (HIE) is an important cause of permanent damage to CNS tissues that may result in neonatal death or manifest later as cerebral palsy or developmental delay. About 20-30% of infants with HIE die in the neonatal period, and ≈ 33-50% of survivors are left with permanent neurodevelopmental abnormalities (cerebral palsy, mental retardation). The greatest risk of adverse outcome is seen in infants with severe fetal acidosis (pH <6.7) (90% death/impairment) and a base deficit >25 mmol/L (72% mortality). Multiorgan failure and insult can occur (Table 93-2).
Table 93-2 MULTIORGAN SYSTEMIC EFFECTS OF ASPHYXIA
| SYSTEM | EFFECT(S) |
|---|---|
| Central nervous system | Hypoxic-ischemic encephalopathy, infarction, intracranial hemorrhage, seizures, cerebral edema, hypotonia, hypertonia |
| Cardiovascular | Myocardial ischemia, poor contractility, cardiac stunning, tricuspid insufficiency, hypotension |
| Pulmonary | Pulmonary hypertension, pulmonary hemorrhage, respiratory distress syndrome |
| Renal | Acute tubular or cortical necrosis |
| Adrenal | Adrenal hemorrhage |
| Gastrointestinal | Perforation, ulceration with hemorrhage, necrosis |
| Metabolic | Inappropriate secretion of antidiuretic hormone, hyponatremia, hypoglycemia, hypocalcemia, myoglobinuria |
| Integument | Subcutaneous fat necrosis |
| Hematology | Disseminated intravascular coagulation |
Etiology
Most neonatal encephalopathic or seizure disorders, in the absence of major congenital malformations or syndromes, appear to be due to perinatal events. Brain MRI or autopsy findings in full-term neonates with encephalopathy demonstrate that 80% have acute injuries, <1% have prenatal injuries, and 3% have non–hypoxic-ischemic diagnoses. Fetal hypoxia may be caused by various disorders in the mother, including (1) inadequate oxygenation of maternal blood from hypoventilation during anesthesia, cyanotic heart disease, respiratory failure, or carbon monoxide poisoning; (2) low maternal blood pressure from acute blood loss, spinal anesthesia, or compression of the vena cava and aorta by the gravid uterus; (3) inadequate relaxation of the uterus to permit placental filling as a result of uterine tetany caused by the administration of excessive oxytocin; (4) premature separation of the placenta; (5) impedance to the circulation of blood through the umbilical cord as a result of compression or knotting of the cord; and (6) placental insufficiency from toxemia or postmaturity.
Placental insufficiency often remains undetected on clinical assessment. Intrauterine growth restriction may develop in chronically hypoxic fetuses without the traditional signs of fetal distress. Doppler umbilical waveform velocimetry (demonstrating increased fetal vascular resistance) and cordocentesis (demonstrating fetal hypoxia and lactic acidosis) identify a chronically hypoxic infant (Chapter 90). Uterine contractions may further reduce umbilical oxygenation, depressing the fetal cardiovascular system and CNS and resulting in low Apgar scores and respiratory depression at birth.
After birth, hypoxia may be caused by (1) failure of oxygenation as a result of severe forms of cyanotic congenital heart disease or severe pulmonary disease; (2) severe anemia (severe hemorrhage, hemolytic disease); (3) or shock severe enough to interfere with the transport of oxygen to vital organs from overwhelming sepsis, massive blood loss, and intracranial or adrenal hemorrhage.
Pathophysiology and Pathology
The topography of injury typically correlates with areas of decreased cerebral blood flow. After an episode of hypoxia and ischemia, anaerobic metabolism occurs and generates increased amounts of lactate and inorganic phosphates. Excitatory and toxic amino acids, particularly glutamate, accumulate in the damaged tissue. Increased amounts of intracellular sodium and calcium may result in tissue swelling and cerebral edema. There is also increased production of free radicals and nitric oxide in these tissues. The initial circulatory response of the fetus is increased shunting through the ductus venosus, ductus arteriosus, and foramen ovale, with transient maintenance of perfusion of the brain, heart, and adrenals in preference to the lungs, liver, kidneys, and intestine.
The pathology of hypoxia-ischemia depends on the affected organ and the severity of the injury. Early congestion, fluid leak from increased capillary permeability, and endothelial cell swelling may then lead to signs of coagulation necrosis and cell death. Congestion and petechiae are seen in the pericardium, pleura, thymus, heart, adrenals, and meninges. Prolonged intrauterine hypoxia may result in inadequate perfusion of the periventricular white matter, resulting, in turn, in PVL. Pulmonary arteriole smooth muscle hyperplasia may develop, which predisposes the infant to pulmonary hypertension (Chapter 95.7). If fetal distress produces gasping, the amniotic fluid contents (meconium, squames, lanugo) are aspirated into the trachea or lungs.
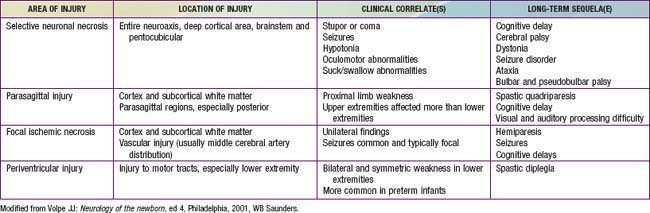
The combination of chronic fetal hypoxia and acute hypoxic-ischemic injury around the time of birth results in gestational age–specific neuropathology (Table 93-3). Term infants demonstrate neuronal necrosis of the cortex (later, cortical atrophy) and parasagittal ischemic injury. Preterm infants demonstrate PVL (later, spastic diplegia), status marmoratus of the basal ganglia, and IVH. Term more often than preterm infants have focal or multifocal cortical infarcts that manifest clinically as focal seizures and hemiplegia.
Clinical Manifestations
Intrauterine growth restriction with increased vascular resistance may be the 1st indication of fetal hypoxia. During labor, the fetal heart rate slows and beat-to-beat variability declines. Continuous heart rate recording may reveal a variable or late deceleration pattern (see Fig. 90-4). Particularly in infants near term, these signs should lead to the administration of high concentrations of oxygen to the mother and consideration of immediate delivery to avoid fetal death and CNS damage.
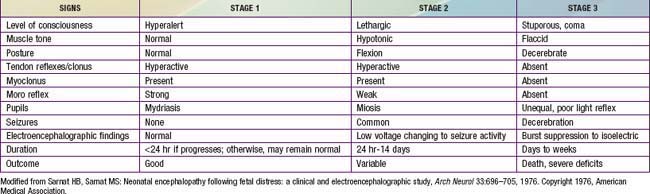
At delivery, the presence of meconium-stained amniotic fluid is evidence that fetal distress has occurred. At birth, affected infants may be depressed and may fail to breathe spontaneously. During the ensuing hours, they may remain hypotonic or change from a hypotonic to a hypertonic state, or their tone may appear normal (Tables 93-4 and 93-5). Pallor, cyanosis, apnea, a slow heart rate, and unresponsiveness to stimulation are also signs of HIE. Cerebral edema may develop during the next 24 hr and result in profound brainstem depression. During this time, seizure activity may occur; it may be severe and refractory to the usual doses of anticonvulsants. Though most often a result of the HIE, seizures in asphyxiated newborns may also be due to hypocalcemia, hypoglycemia, or infection.
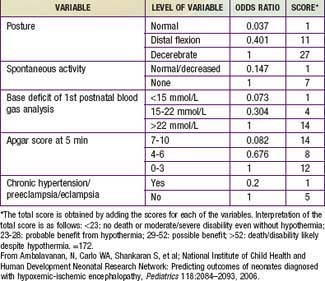
Table 93-4 PREDICTOR VARIABLES, ODDS RATIOS, AND SCORES ASSIGNED TO EACH VARIABLE FOR DEATH/DISABILITY SCORING IN INFANTS WITH HYPOXIC-ISCHEMIC ENCEPHALOPATHY
In addition to CNS dysfunction, heart failure and cardiogenic shock, persistent pulmonary hypertension, respiratory distress syndrome, gastrointestinal perforation, hematuria, and acute tubular necrosis are associated with perinatal asphyxia secondary to inadequate perfusion (see Table 93-2).
The severity of neonatal encephalopathy depends on the duration and timing of injury. Symptoms develop over a series of days, making it important to perform serial neurologic examinations (see Tables 93-4 and 93-5). During the initial hours after an insult, infants have a depressed level of consciousness. Periodic breathing with apnea or bradycardia is present, but cranial nerve functions are often spared with intact pupillary responses and spontaneous eye movement. Seizures are common with extensive injury. Hypotonia is also common as an early manifestation.
Diagnosis
Diffusion-weighted MRI is the preferred imaging modality in neonates with HIE because of its increased sensitivity and specificity early in the process and its ability to outline the topography of the lesion (Figs. 93-5 to 93-8). CT scans are helpful in identifying focal hemorrhagic lesions, diffuse cortical injury, and damage to the basal ganglia; CT has limited ability to identify cortical injury during the 1st few days of life. Ultrasonography has limited utility in evaluation of hypoxic injury in the term infant; it is the preferred modality in evaluation of the preterm infant.
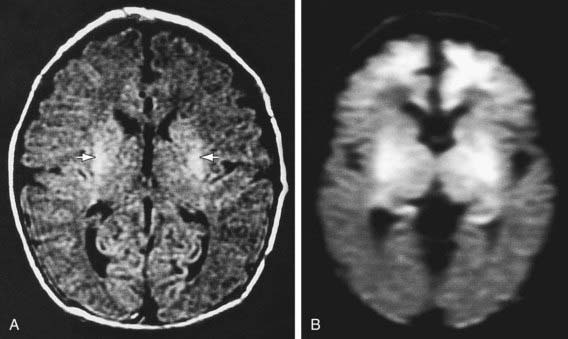
Figure 93-5 MR images of selective neuronal injury. The infant experienced intrapartum asphyxia and had seizures on the 1st postnatal day. MRI was performed on the 5th postnatal day. A, An axial, fluid-attenuated inversion recovery image shows increased signal in the putamen bilaterally (arrows) but no definite abnormality in the cerebral cortex. B, By contrast, a diffusion-weighted image shows striking increased signal intensity (i.e., decreased diffusion) in the frontal cortex (in addition to a more pronounced basal ganglia abnormality).
(From Volpe JJ, editor: Neurology of the newborn, ed 5, Philadelphia, 2008, Saunders/Elsevier, p 420.)
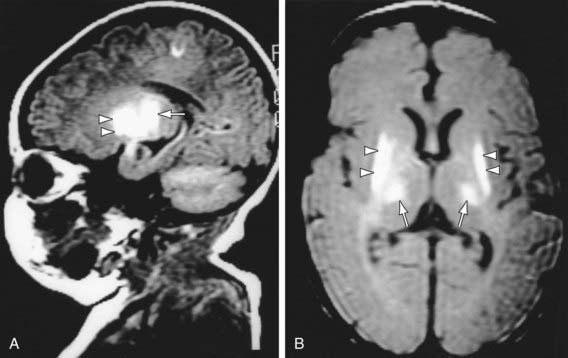
Figure 93-6 MR images of hypoxic-ischemic injury to basal ganglia and thalamus. MRI was performed in a 5-day-old infant who experienced severe perinatal asphyxia. A, Note, in this parasagittal T1-weighted image, the markedly increased signal intensity in the basal ganglia, especially the putamen (arrowheads) and the thalamus (arrow). B, An axial proton density image also demonstrates the injury well in the same distribution.
(From Volpe JJ, editor: Neurology of the newborn, ed 5, Philadelphia, 2008, Saunders/Elsevier, p 420.)
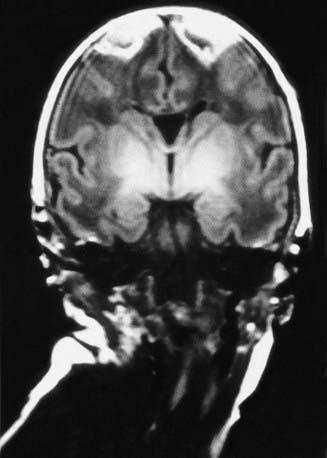
Figure 93-7 MR image of a parasagittal cerebral injury. A coronal T1-weighted image, obtained on the 5th postnatal day in an asphyxiated term infant, shows striking triangular lesions in the parasagittal areas bilaterally; increased signal intensity is also apparent in the basal ganglia and thalamus bilaterally.
(From Volpe JJ, editor: Neurology of the newborn, ed 5, Philadelphia, 2008, Saunders/Elsevier, p 421.)
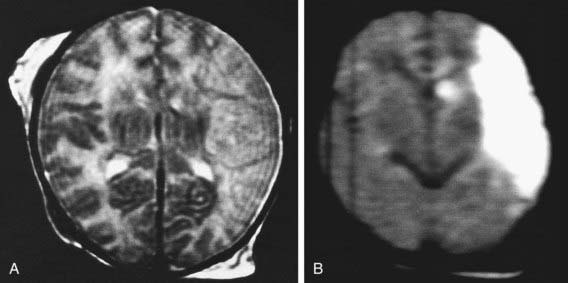
Figure 93-8 MR images of focal ischemic cerebral injury. MRI was performed on the 3rd postnatal day. A, An axial T2-weighted image shows a lesion in the distribution of the main branch of the left middle cerebral artery. B, A diffusion-weighted image demonstrates the lesion more strikingly.
(From Volpe JJ, editor: Neurology of the newborn, ed 5, Philadelphia, 2008, Saunders/Elsevier, p 422.)
Amplitude-integrated electroencephalography (aEEG) may help to determine which infants are at highest risk for long-term brain injury. A single-channel tracing is generated from 2 electrodes placed in the biparietal area. A filter is used to filter and attenuate the signal between 2 Hz and 15 Hz. This technique is simple to perform and correlates with standard EEG. It has good reliability, a positive predictive value of 85%, and a negative predictive value of 91-96% for infants who will have adverse neurodevelopmental outcome. The technique provides information quickly within the window during which intervention is most likely to be useful. Also, aEEG is able to detect seizure activity, which is common in patients with HIE. Continuous aEEG monitoring detects subclinical seizure activity during the subacute phase.
Treatment
Selective cerebral or whole body (systemic) therapeutic hypothermia reduces mortality or major neurodevelopmental impairment in term and near-term infants with HIE. Hypothermia decreases the rate of apoptosis and suppresses production of mediators known to be neurotoxic, including extracellular glutamate, free radicals, nitric oxide, and lactate. The neuroprotective effects are thought to be secondary to downregulation of the secondary mediators of injury resulting from cerebral edema, accumulation of cytokines, and seizures. Animal data suggest that the intervention is most effective when implemented within 6 hr of the event.
Several clinical trials and a meta-analysis demonstrate that either isolated cerebral cooling or systemic hypothermia to a core temperature of 33.5°C within the 1st 6 hr after birth reduces mortality and major neurodevelopmental impairment at 18 mo of age. Systemic hypothermia may result in more uniform cooling of the brain and deeper CNS structures. Infants treated with systemic hypothermia have a lower incidence of cortical neuronal injury on MRI.
Phenobarbital, the drug of choice for seizures, is given with an intravenous loading dose (20 mg/kg); additional doses of 5-10 mg/kg (up to 40-50 mg/kg total) may be needed. Phenytoin (20 mg/kg loading dose) or lorazepam (0.1 mg/kg) may be needed for refractory seizures. Phenobarbital levels should be monitored 24 hr after the loading dose has been given and maintenance therapy (5 mg/kg/24hr) is begun. Therapeutic phenobarbital levels are 20-40 µg/mL. There is some clinical evidence that high-dose prophylactic phenobarbital may decrease neurodevelopmental impairment in infants with HIE.
Additional therapy for infants with HIE includes supportive care directed at management of organ system dysfunction. Hyperthermia has been found to be associated with impaired neurodevelopment, so it is important to prevent hyperthermia before initiation of hypothermia. Careful attention to ventilatory status and adequate oxygenation, blood pressure, hemodynamic status, acid-base balance, and possible infection is important. Secondary hypoxia or hypotension due to complications of HIE must be prevented. Aggressive treatment of seizures is critical and may necessitate continuous EEG monitoring.
Prognosis
The outcome of HIE, which correlates with the timing and severity of the insult, ranges from complete recovery to death. The prognosis varies depending on the severity of the insult and the treatment. Infants with initial cord or initial blood pH <6.7 have a 90% risk for death or severe neurodevelopmental impairment at 18 mo of age. In addition, infants with Apgar scores of 0-3 at 5 min, high base deficit (>20-25 mmol/L), decerebrate posture, and lack of spontaneous activity are also at increased risk for death or impairment. These predictor variables can be combined to determine a score that helps with prognosis (see Table 93-4). Infants with the highest risk are likely to die or have severe disability despite aggressive treatment including hypothermia. Those with intermediate scores are likely to benefit from treatment. In general, severe encephalopathy, characterized by flaccid coma, apnea, absence of oculocephalic reflexes, and refractory seizures, is associated with a poor prognosis (see Table 93-5). A low Apgar score at 20 min, absence of spontaneous respirations at 20 min of age, and persistence of abnormal neurologic signs at 2 wk of age also predict death or severe cognitive and motor deficits. The combined use of early EEG and MRI is useful in predicting outcome in term infants with HIE. Normal MRI and EEG findings are associated with a good recovery, whereas severe MRI and EEG abnormalities predict a poor outcome. Microcephaly and poor head growth during the 1st year of life also correlate with injury to the basal ganglia and white matter and adverse developmental outcome at 12 mo. All survivors of moderate to severe encephalopathy require comprehensive high-risk medical and developmental follow-up. Early identification of neurodevelopmental problems allows prompt referral for developmental, rehabilitative, neurologic care, and early intervention services so that the best possible outcome can be achieved.
Brain death after neonatal HIE is diagnosed from the clinical findings of coma unresponsive to pain, auditory, or visual stimulation; apnea with PCO2 rising from 40 to >60 mm Hg without ventilatory support; and absence of brainstem reflexes (pupillary, oculocephalic, oculovestibular, corneal, gag, sucking) (Chapter 63.1). These findings must occur in the absence of hypothermia, hypotension, and elevations of depressant drugs (phenobarbital). An absence of cerebral blood flow on radionuclide scans and of electrical activity on EEG (electrocerebral silence) is inconsistently observed in clinically brain-dead neonatal infants. Persistence of the clinical criteria for 2 days in term infants and 3 days in preterm infants predicts brain death in most asphyxiated newborns. Nonetheless, no universal agreement has been reached regarding the definition of neonatal brain death. Consideration of withdrawal of life support should include discussions with the family, the health care team, and, if there is disagreement, an ethics committee. The best interest of the infant involves judgments about the benefits and harm of continuing therapy or avoiding ongoing futile therapy.
Ambalavanan N, Carlo WA, Shankaran S, et al. Predicting outcomes of neonates diagnosed with hypoxic-ischemic encephalopathy. Pediatrics. 2006;118:2084-2093.
Azzopardi DV, Strohm B, Edwards AD, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361:1349-1358.
Bonifacio SI, Glass HC, Vanderpluym J, et al. Perinatal events and early magnetic resonance imaging in therapeutic hypothermia. J Pediatr. 2011;158:360-365.
Chau V, Poskitt KJ, Sargent MA, et al. Comparison of computer tomography and magnetic resonance imaging scans on the third day of life in term newborns with neonatal encephalopathy. Pediatrics. 2009;123:319-326.
Davis AS, Hintz SR, Van Meurs KP, et al. Seizures in extremely low birth weight infants are associated with adverse outcome. J Pediatr. 2010;157:720-725.
De Vries LS, Hellstrom-Westas. Role of cerebral function monitoring in the newborn. Arch Dis Child Fetal Neonatal Ed. 2005;90:F201-F207.
Edwards AD. Hypothermic neural rescue: work continues. J Pediatr. 2010;157(3):351-352.
Glass HC, Glidden D, Jeremy RJ, et al. Clinical neonatal seizures are independently associated with outcome in infants at risk for hypoxic-ischemic brain injury. J Pediatr. 2009;155:318-323.
Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling and mild systemic hypothermia after neonatal encephalopathy: multicenter randomized trial. Lancet. 2005;365:663-670.
Gray J, Geva A, Zheng Z, et al. CoolSim: Using industrial modeling techniques to examine the impact of selective head cooling in a model of perinatal regionalization. Pediatrics. 2008;121:28-36.
Groenendaal F, De Vooght KMD, van Bel F. Blood gas values during hypothermia in asphyxiated term neonates. Pediatrics. 2009;123:170-172.
Gunn AJ, Wyatt JS, Whitelaw A, et al. Therapeutic hypothermia changes the prognostic value of clinical evaluation of neonatal encephalopathy. J Pediatr. 2008;152:55-58.
Hall RT, Hall FK, Daily DK. High-dose phenobarbital therapy in term newborn infants with severe perinatal asphyxia: a randomized, prospective study with three-year follow-up. J Pediatr. 1998;132:345-348.
Hellstrom-Westas L, Rosen I, Svenningsen NW. Predictive value of early continuous amplitude integrated EEG recordings on outcome after severe birth asphyxia in full term infants. Arch Dis Child Fetal Neonatal Ed. 1995;72:34F-38F.
Jacobs SE, Hunt R, Tarnow-Mordi WO, et al: Cooling for newborns with hypoxic ischaemic encephalopathy, Cochrane Database Syst Rev (17):CD003311, 2007.
Laptook AR, Shankaran S, Ambalavanan N, et al. Outcome of term infants using Apgar scores at 10 minutes following hypoxic-ischemic encephalopathy. Pediatrics. 2009;124:1619-1626.
Levene MI. Cool treatment for birth asphyxia, but what’s next? Arch Dis Child Fetal Neonatal Ed. 2010;95(6):F154-F156.
Logitharajah P, Rutherford MA, Cowan FM. Hypoxic-ischemic encephalopathy in preterm infants: antecedent factors, brain imaging, and outcome. Pediatr Res. 2009;66:222-229.
Murray DM, Boylan GB, Ryan CA, et al. Early EEG findings in hypoxic-ischemic encephalopathy predict outcomes at 2 years. Pediatrics. 2009;124:e459-e467.
Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574-1584.
Shankaran S, Pappas A, Laptook AR, et al. Outcomes of safety and effectiveness in a multicenter randomized, controlled trial of whole-body hypothermia for neonatal hypoxic-ischemic encephalopathy. Pediatrics. 2008;122:e791-e798.
Toet MC, Hellstrom-Westas L, Broenendaal F, et al. Amplitude integrated EEG 3 and 6 hours after birth in full term neonates with hypoxic-ischaemic encephalopathy. Arch Dis Child Fetal Neonatal Ed. 1999;81:19F-23F.
93.6 Spine and Spinal Cord
Injury to the spine/spinal cord during birth is rare but can be devastating. Strong traction exerted when the spine is hyperextended or when the direction of pull is lateral, or forceful longitudinal traction on the trunk while the head is still firmly engaged in the pelvis, especially when combined with flexion and torsion of the vertical axis, may produce fracture and separation of the vertebrae. Such injuries are most likely to occur when difficulty is encountered in delivering the shoulders in cephalic presentations and the head in breech presentations. The injury occurs most commonly at the level of the 4th cervical vertebra with cephalic presentations and the lower cervical–upper thoracic vertebrae with breech presentations. Transection of the cord may occur with or without vertebral fractures; hemorrhage and edema may produce neurologic signs that are indistinguishable from those of transection except that they may not be permanent. Areflexia, loss of sensation, and complete paralysis of voluntary motion occur below the level of injury, although the persistence of a withdrawal reflex mediated through spinal centers distal to the area of injury is frequently misinterpreted as representing voluntary motion. If the injury is severe, the infant, who from birth may be in poor condition because of respiratory depression, shock, or hypothermia, may deteriorate rapidly to death within several hours before any neurologic signs are obvious. Alternatively, the course may be protracted, with symptoms and signs appearing at birth or later in the 1st wk; immobility, flaccidity, and associated brachial plexus injuries may not be recognized for several days. Constipation may also be present. Some infants survive for prolonged periods, their initial flaccidity, immobility, and areflexia being replaced after several weeks or months by rigid flexion of the extremities, increased muscle tone, and spasms. Apnea on day 1 and poor motor recovery by 3 mo are poor prognostic signs.
The differential diagnosis of spine/spinal cord injury includes amyotonia congenita and myelodysplasia associated with spina bifida occulta. Ultrasonography or, more often, MRI confirms the diagnosis. Treatment of the survivors is supportive, including home ventilation; patients often remain permanently disabled. When a fracture or dislocation is causing spinal compression, the prognosis is related to the time elapsed before the compression is relieved.
MacKinnon JA, Perlman M, Kirpalani H, et al. Spinal cord injury at birth: diagnostic and prognostic data in 22 patients. J Pediatr. 1993;122:431-437.
Mills JF, Dargaville PA, Coleman LT, et al. Upper cervical spinal cord injury in neonates: the use of magnetic resonance imaging. J Pediatr. 2001;138:105-108.
93.7 Peripheral Nerve Injuries
Brachial Palsy
Brachial plexus injury is a common problem, with an incidence of 0.6-4.6/1,000 live births. Injury to the brachial plexus may cause paralysis of the upper part of the arm with or without paralysis of the forearm or hand or, more commonly, paralysis of the entire arm. These injuries occur in macrosomic infants and when lateral traction is exerted on the head and neck during delivery of the shoulder in a vertex presentation, when the arms are extended over the head in a breech presentation, or when excessive traction is placed on the shoulders. Approximately 45% of brachial plexus injuries are associated with shoulder dystocia. In Erb-Duchenne paralysis, the injury is limited to the 5th and 6th cervical nerves. The infant loses the power to abduct the arm from the shoulder, rotate the arm externally, and supinate the forearm. The characteristic position consists of adduction and internal rotation of the arm with pronation of the forearm. Power to extend the forearm is retained, but the biceps reflex is absent; the Moro reflex is absent on the affected side (Fig. 93-9). The outer aspect of the arm may have some sensory impairment. Power in the forearm and hand grasps is preserved unless the lower part of the plexus is also injured; the presence of hand grasp is a favorable prognostic sign. When the injury includes the phrenic nerve, alteration in diaphragmatic excursion may be observed with ultrasonography or fluoroscopy. Klumpke paralysis is a rare form of brachial palsy, in which injury to the 7th and 8th cervical nerves and the 1st thoracic nerve produces a paralyzed hand and ipsilateral ptosis and miosis (Horner syndrome) if the sympathetic fibers of the 1st thoracic root are also injured. Mild cases may not be detected immediately after birth. Differentiation must be made from cerebral injury; from fracture, dislocation, or epiphyseal separation of the humerus; and from fracture of the clavicle. MRI demonstrates nerve root rupture or avulsion.

Full recovery occurs in most patients; prognosis depends on whether the nerve was merely injured or was lacerated. If the paralysis was due to edema and hemorrhage about the nerve fibers, function should return within a few months; if it was due to laceration, permanent damage may result. Involvement of the deltoid is usually the most serious problem and may result in shoulder drop secondary to muscle atrophy. In general, paralysis of the upper part of the arm has a better prognosis than paralysis of the lower part.
Treatment consists of initial conservative management with monthly follow-up and a decision for surgical intervention by three months if function has not improved. Partial immobilization and appropriate positioning are used to prevent the development of contractures. In upper arm paralysis, the arm should be abducted 90 degrees with external rotation at the shoulder, full supination of the forearm, and slight extension at the wrist with the palm turned toward the face. This position may be achieved with a brace or splint during the 1st 1-2 wk. Immobilization should be intermittent throughout the day while the infant is asleep and between feedings. In lower arm or hand paralysis, the wrist should be splinted in a neutral position, and padding placed in the fist. When the entire arm is paralyzed, the same treatment principles should be followed. Gentle massage and range-of-motion exercises may be started by 7-10 days of age. Infants should be closely monitored with active and passive corrective exercises. If the paralysis persists without improvement for 3 months, neuroplasty, neurolysis, end-to-end anastomosis, and nerve grafting offer hope for partial recovery.
The type of treatment and the prognosis depend on the mechanism of injury and the number of nerve roots involved. The mildest injury to a peripheral nerve (neurapraxia) is due to edema and heals spontaneously within a few weeks. Axonotmesis is more severe and is due to nerve fiber disruption with an intact myelin sheath; function usually returns in a few months. Total disruption of nerves (neurotmesis) or root avulsion is the most severe, especially if it involves C5-T1; microsurgical repair may be indicated. Fortunately, most (75%) injuries are at the root level C5-C6, involve neurapraxia and axonotmesis, and should heal spontaneously. Botulism toxin may be used to treat biceps-triceps co-contractions.
Phrenic Nerve Paralysis
Phrenic nerve injury (3rd, 4th, 5th cervical nerves) with diaphragmatic paralysis must be considered when cyanosis and irregular and labored respirations develop. Such injuries, usually unilateral, are associated with ipsilateral upper brachial palsy. Because breathing is thoracic in type, the abdomen does not bulge with inspiration. Breath sounds are diminished on the affected side. The thrust of the diaphragm, which may often be felt just under the costal margin on the normal side, is absent on the affected side. The diagnosis is established by ultrasonographic or fluoroscopic examination, which reveals elevation of the diaphragm on the paralyzed side and seesaw movements of the 2 sides of the diaphragm during respiration.
No specific treatment is available; infants should be placed on the involved side and given oxygen if necessary. Initially, intravenous feedings may be needed; later, progressive gavage or oral feeding may be started, depending on the infant’s condition. Pulmonary infections are a serious complication. Recovery usually occurs spontaneously by 1-3 mo; rarely, surgical plication of the diaphragm may be indicated.
Facial Nerve Palsy
Facial palsy is usually a peripheral paralysis that results from pressure over the facial nerve in utero, from efforts during labor, or from forceps use during delivery. Rarely, it may result from nuclear agenesis of the facial nerve. Peripheral paralysis is flaccid and, when complete, involves the entire side of the face, including the forehead. When the infant cries, movement occurs only on the nonparalyzed side of the face, and the mouth is drawn to that side. On the affected side the forehead is smooth, the eye cannot be closed, the nasolabial fold is absent, and the corner of the mouth droops. The forehead wrinkles on the affected side with central paralysis because only the lower 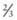 of the face is involved. The infant also usually has other manifestations of intracranial injury, most commonly 6th nerve palsy. The prognosis depends on whether the nerve was injured by pressure or the nerve fibers were torn. Improvement occurs within a few weeks in the former instance. Care of the exposed eye is essential. Neuroplasty may be indicated when the paralysis is persistent. Facial palsy may be confused with absence of the depressor muscles of the mouth, which is a benign problem.
of the face is involved. The infant also usually has other manifestations of intracranial injury, most commonly 6th nerve palsy. The prognosis depends on whether the nerve was injured by pressure or the nerve fibers were torn. Improvement occurs within a few weeks in the former instance. Care of the exposed eye is essential. Neuroplasty may be indicated when the paralysis is persistent. Facial palsy may be confused with absence of the depressor muscles of the mouth, which is a benign problem.
Other peripheral nerves are seldom injured in utero or at birth except when they are involved in fractures or hemorrhage.
Brown T, Cupido C, Scarfone H, et al. Developmental apraxia arising from neonatal brachial plexus palsy. Neurology. 2000;55:24-30.
Hoeksma AF, Wolf H, Oei SL. Obstetrical brachial plexus injuries: incidence, natural course and shoulder contracture. Clin Rehabil. 2000;14:523-526.
Noetzel MJ, Wolpaw JR. Emerging concepts in the pathophysiology of recovery from neonatal brachial plexus injury. Neurology. 2000;55:5-6.
Strombeck C, Krumlinde-Sundholm L, Forrsberg H. Functional outcome at 5 years in children with obstetrical brachial plexus palsy with and without microsurgical reconstruction. Dev Med Child Neurol. 2000;42:148-157.