Chapter 83 Disorders of Purine and Pyrimidine Metabolism
The inherited disorders of purine and pyrimidine metabolism cover a broad spectrum of illnesses with various presentations. These include hyperuricemia, acute renal failure, renal stones, gout, unexplained neurologic deficits (seizures, muscle weakness, choreoathetoid and dystonic movements), developmental disability, intellectual disability, compulsive self-injury and aggression, autistic-like behavior, unexplained anemia, failure to thrive, susceptibility to recurrent infection (immune deficiency), and deafness. When such disorders are identified, all family members should be screened.
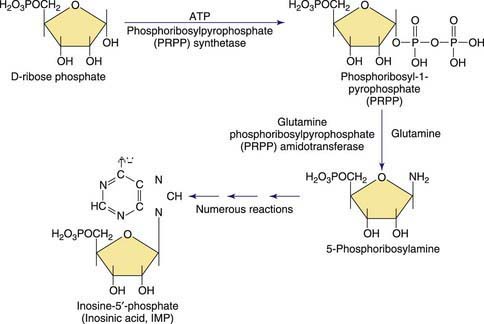
Purines are involved in all biologic processes; all cells require a balanced supply of purines for growth and survival. They provide the primary source of cellular energy through adenosine triphosphate (ATP) and, together with pyrimidines, provide the source for the RNA and DNA that stores, transcribes, and translates genetic information. Purines provide the basic coenzymes (NAD, NADH) for metabolic regulation and play a major role in signal transduction (GTP, cAMP, cGMP). Metabolically active nucleotides are formed from heterocyclic nitrogen-containing purine bases (guanine and adenine) and pyrimidine bases (cytosine, uridine, and thymine). The early steps in the biosynthesis of the purine ring are shown in Figure 83-1. Purines are primarily produced from endogenous sources and, in usual circumstances, dietary purines have a small role. The end product of purine metabolism in humans is uric acid (2,6,8-trioxypurine).
Uric acid is not a specific disease marker, so the cause of its elevation must be determined. The level of uric acid present at any time depends on the size of the purine nucleotide pool, which is derived from de novo purine synthesis, catabolism of tissue nucleic acids, and increased turnover of preformed purines. Uric acid is poorly soluble and must be excreted continuously to avoid toxic accumulations in the body. Its renal excretion involves the following components: (1) glomerular filtration, (2) reabsorption in the proximal convoluted tubule, (3) secretion near the terminus of the proximal tubule, and (4) limited reabsorption near these secretory sites. Thus, renal loss of uric acid is a result of renal tubular excretion and is a function of serum uric acid concentration and a homeostatic mechanism to avoid hyperuricemia. Because renal tubule excretion is greater in children than in adults, serum uric acid levels are a less reliable indicator of uric acid production in children than in adults, and consequently, measurement of the level in urine may be required to determine excessive production. Clearance of a smaller portion of uric acid is via the gastrointestinal tract (biliary and intestinal secretion). Owing to poor solubility of uric acid under normal circumstances, uric acid is near the maximal tolerable limits, and small alterations in production or solubility or changes in secretion may result in high serum levels. In renal insufficiency, urate excretion is increased by residual nephrons and by the gastrointestinal tract.
Increased production of uric acid is found in malignancy; Reye syndrome; Down syndrome; psoriasis; sickle cell anemia; cyanotic congenital heart disease; pancreatic enzyme replacement; glycogen storage disease types I, III, IV, and V; hereditary fructose intolerance; acyl coenzyme A dehydrogenase deficiency; and gout.
The metabolism of both purines and pyrimidines can be divided into 2 biosynthetic pathways and a catabolic pathway. The 1st, the de novo pathway, involves a multistep biosynthesis of phosphorylated ring structures from precursors such as CO2, glycine, and glutamine. Purine and pyrimidine nucleotides are produced from ribose-5-phosphate or carbamyl phosphate, respectively. The 2nd, a single-step salvage pathway, recovers purine and pyrimidine bases derived from either dietary intake or the catabolic pathway (Figs. 83-2 and 83-3; also see Fig. 83-1). In the de novo pathway, the nucleosides guanosine, adenosine, cytidine, uridine, and thymidine are formed by the addition of ribose-1-phosphate to the purine bases guanine or adenine, and to the pyrimidine bases cytosine, uracil, and thymine. The phosphorylation of these nucleosides produces monophosphate, diphosphate, and triphosphate nucleotides. Under usual circumstances, the salvage pathway predominates over the biosynthetic pathway. Synthesis is most active in tissues with high rates of cellular turnover, such as gut epithelium, skin, and bone marrow. The 3rd pathway is catabolism. The end product of the catabolic pathway of the purines is uric acid, whereas catabolism of pyrimidines produces citric acid cycle intermediates. Only a small fraction of the purines turned over each day are degraded and excreted.
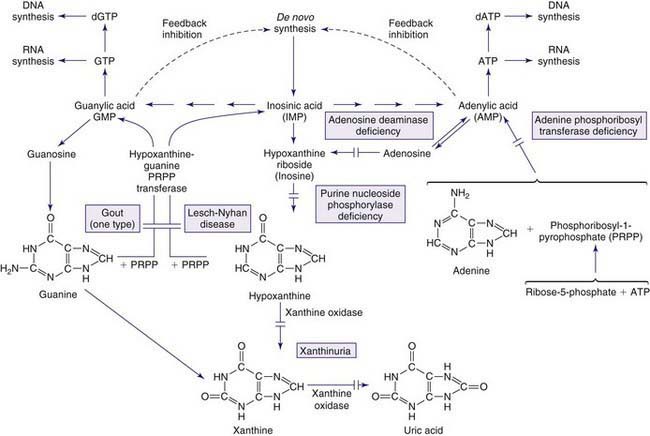
Inborn errors in the synthesis of purine nucleotides include: (1) phosphoribosylpyrophosphate synthetase superactivity, (2) adenylosuccinase deficiency, and (3) 5-amino-4-imidazolecarboxamide (AICA) riboside deficiency (AICA-ribosiduria). Disorders resulting from abnormalities in purine catabolism include: (1) muscle adenosine monophosphate (AMP) deaminase deficiency, (2) adenosine deaminase deficiency, (3) purine nucleoside phosphorylase deficiency, and (4) xanthine oxidoreductase deficiency. Disorders resulting from the purine salvage pathway include: (1) hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency, and (2) adenine phosphoribosyltransferase (APRT) deficiency.
Inborn errors of pyrimidine metabolism include disorders of pyrimidine synthesis and of pyrimidine nucleotide degradation. Disorders include: (1) hereditary orotic aciduria (uridine monophosphate synthase deficiency), (2) dihydropyrimidine dehydrogenase (DPD) deficiency, (3) dihydropyrimidinase (DPH) deficiency, (4) β-ureidopropionase deficiency, (5) UMPH-1 deficiency (previously pyrimidine 5′-nucleotidase deficiency), (6) pyrimidine nucleoside depletion and overactive cytosolic 5′-nucleotidase, (7) thymidine kinase 2 deficiency, and (8) thymidine phosphorylase deficiency.
Gout
Gout presents with hyperuricemia, uric acid nephrolithiasis, and acute inflammatory arthritis. Gouty arthritis is due to monosodium urate crystal deposits that result in inflammation in joints and surrounding tissues. The presentation is most commonly monoarticular, typically in the metatarsophalangeal joint of the big toe. Tophi, deposits of monosodium urate crystals, may occur over points of insertion of tendons at the elbows, knees, and feet or over the helix of the ears. Primary gout, ordinarily occurring in middle-aged men, results from overproduction of uric acid, decreased renal excretion of uric acid, or both. The biochemical etiology of gout is unknown for most of those affected, and it is considered to be a polygenic trait. When hyperuricemia and gout occur in childhood, it is most often secondary gout, the result of another disorder in which there is rapid tissue breakdown or cellular turnover leading to increased production or decreased excretion of uric acid. Gout occurs in any condition that leads to reduced clearance of uric acid: during therapy for malignancy or with dehydration, lactic acidosis, ketoacidosis, starvation, diuretic therapy, and renal shutdown. Excessive purine, alcohol, or carbohydrate ingestion may increase uric acid levels.
Gout is associated with hereditary disorders in three different enzyme disorders that result in hyperuricemia. These include the severe form of HPRT deficiency (Lesch-Nyhan disease) and partial HPRT deficiency, superactivity of PP-ribose-P synthetase, and glycogen storage disease type I (glucose-6-phosphatase deficiency) (Chapter 81.1). In the 1st two, the basis of hyperuricemia is purine nucleotide and uric acid overproduction, whereas in the 3rd, it is both excessive uric acid production and diminished renal excretion of urate. Glycogen storage disease types III, V, and VII are associated with exercise-induced hyperuricemia, the consequence of rapid ATP utilization and failure to regenerate it effectively during exercise (Chapter 81.1). Autosomal dominant juvenile hyperuricemia, gouty arthritis, medullary cysts, and progressive renal insufficiency are features associated with familial juvenile hyperuricemic nephropathy (FJHN) and medullary cystic kidney disease type 1 (MCKD1) and type 2 (MCKD2). MCKD1 has been mapped to chromosome 1q21. FJHN and MCKD2 have been mapped to chromosome 16p11.2. FJHN and MCKD2 are proposed to be allelic and can result from uromodulin (UMOD) mutations; the term uromodulin-associated kidney disease (UAKD) has been proposed. Unlike the three inherited purine disorders that are X-linked and the recessively inherited glycogen storage disease, these are autosomal dominant conditions. Familial juvenile gout or familial juvenile hyperuricemic nephropathy is associated with severe renal hypoexcretion of uric acid. Although it most commonly presents from puberty up to the 3rd decade, it has been reported in infancy. It is characterized by early onset, hyperuricemia, gout, familial renal disease, and low urate clearance relative to glomerular filtration rate. It occurs in both males and females and is frequently associated with a rapid decline in renal function that may lead to death unless diagnosed and treated early. Once FJHN is recognized, presymptomatic detection is of critical importance to identify asymptomatic family members with hyperuricemia and to begin treatment, when indicated, to prevent nephropathy.
Treatment of hyperuricemia involves the combination of allopurinol (a xanthine oxidase inhibitor) to decrease uric acid production, probenecid to increase uric acid clearance in those with normal renal function, alkalinization of the urine to increase the solubility of uric acid, and increased fluid intake to reduce the concentration of uric acid. A low-purine diet, weight reduction, and reduced alcohol intake are recommended.
Abnormalities in Purine Salvage
Lesch-Nyhan Disease (LND)
LND is a rare X-linked disorder of purine metabolism that results from HPRT deficiency. This enzyme is normally present in each cell in the body, but its highest concentration is in the brain, especially in the basal ganglia.
Epidemiology
The prevalence of the classic LND has been estimated at 1/100,000 to 1/380,000. The incidence of partial variants is not known. Those with classic LND rarely survive the 3rd decade because of renal or respiratory compromise. The life span may be normal for patients with partial HPRT deficiency without severe renal involvement.
Pathology
No specific brain abnormality is documented after detailed histopathology and electron microscopy of affected brain regions. Magnetic resonance imaging has documented reductions in the volume of basal ganglia nuclei. Abnormalities in neurotransmitter metabolism have been identified in 3 autopsied cases. All 3 patients had very low HPRT levels (<1% in striatal tissue and 1-2% of control in thalamus and cortex). There was a functional loss of 65-90% of the nigrostriatal and mesolimbic dopamine terminals, although the cells of origin in the substantia nigra did not show dopamine reduction. The brain regions primarily involved were the caudate nucleus, putamen, and nucleus accumbens. It is proposed that the neurochemical changes may be linked to functional abnormalities, possibly resulting from a diminution of arborization or branching of dendrites rather than cell loss. A neurotransmitter abnormality is demonstrated by changes in cerebral spinal fluid neurotransmitters and their metabolites, and confirmed by positron emission tomography scans of dopamine function. Reductions in vivo in the presynaptic dopamine transporter density have been documented in the caudate and putamen of 6 individuals.
Pathogenesis
The HPRT gene has been localized to the long arm of the X chromosome (q26-q27). The complete amino acid sequence for HPRT is known (≈44 kb; 9 exons). The disorder appears in males; occurrence in females is extremely rare and ascribed to nonrandom inactivation of the normal X chromosome. Absence of HPRT prevents the normal metabolism of hypoxanthine resulting in excessive uric acid production and manifestations of gout, necessitating specific drug treatment (allopurinol). Because of the enzyme deficiency, hypoxanthine accumulates in the cerebrospinal fluid, but uric acid does not; uric acid is not produced in the brain and does not cross the blood-brain barrier. The behavior disorder is not caused by hyperuricemia or excess hypoxanthine because patients with partial HPRT deficiency, the variants with hyperuricemia, do not self-injure and infants having isolated hyperuricemia from birth do not develop self-injurious behavior.
The mechanism whereby HPRT leads to the neurologic and behavioral symptoms is unknown. Both hypoxanthine and guanine metabolism is affected; guanosine triphosphate (GTP) and adenosine have substantial effects on neural tissues. There is a functional link between purine nucleotides and the dopamine system that involves guanine, the precursor of GTP. Dopamine binding to its receptor results in either an activation (D1 receptor) or an inhibition (D2 receptor) of adenylcyclase. Both receptor effects are mediated by G proteins (GTP-binding proteins) dependent on guanosine diphosphate (GDP) in the GDP/GTP exchange for cellular activation. Dopamine and adenosine systems are also linked through the role of adenosine as a neuroprotective agent in preventing neurotoxicity. Adenosine agonists mimic the biochemical and behavioral actions of dopamine antagonists, whereas adenosine receptor antagonists act as functional dopamine agonists. Dopamine reduction in brain is documented in HPRT-deficient strains of mutant mice.
Clinical Manifestations
LND is characterized by hyperuricemia, intellectual disability, dystonic movement disorder that may be accompanied by choreoathetosis and spasticity, dysarthric speech, and compulsive self-biting, usually beginning with the eruption of teeth. There are several clinical presentations of HPRT deficiency. HPRT levels are related to the extent of motor symptoms, to the presence or absence of self-injury, and possibly to the level of cognitive function. The majority of individuals with classic LND have low or undetectable levels of the HPRT enzyme. Partial deficiency in HPRT (Kelley-Seegmiller syndrome) with >1.5-2.0% enzyme is associated with hyperuricemia and variable neurologic dysfunction (neurologic HPRT deficiency). HPRT deficiency with levels ≥8% leads to a severe form of gout, with apparently normal cerebral functioning (HPRT-related hyperuricemia) although cognitive deficits may occur. Qualitatively similar cognitive deficit profiles have been reported in both LND and variant cases. Variants produced scores that are intermediate between those of patients with LND and normal controls on nearly every neuropsychologic measure tested.
At birth, infants with LND have no apparent neurologic dysfunction. After several months, developmental delay, intellectual disability, and neurologic signs become apparent. Before the age of 4 mo, hypotonia, recurrent vomiting, and difficulty with secretions may be noted. By about 8-12 mo, extrapyramidal signs appear, primarily dystonic movements. In some cases, spasticity may become apparent at this time or, in some instances, later in life.
Cognitive function is usually reported to be in the mild-to-moderate range of intellectual disability, although some individuals test in the low normal range. Because test scores may be influenced by difficulty in testing the subjects owing to their movement disorder and dysarthric speech, overall intelligence may be underestimated.
The age of onset of self-injury may be as early as 1 yr and, occasionally, as late as the teens. Self-injury occurs, although all sensory modalities, including pain, are intact. The self-injurious behavior usually begins with self-biting, although other patterns of self-injurious behavior emerge with time. Most characteristically, the fingers, mouth, and buccal mucosa are mutilated. Self-biting is intense and causes tissue damage and may result in the amputation of fingers and substantial loss of tissue around the lips (Fig. 83-4). Extraction of primary teeth may be required. The biting pattern can be asymmetric, with preferential mutilation of the left or right side of the body. The type of behavior is different from that seen in other intellectual disability syndromes involving self-injury; self-hitting and head banging are the most common initial presentations in other syndromes. The intensity of the self-injurious behavior generally requires that the patient be restrained. When restraints are removed, the individual with LND may appear terrified, and stereotypically place a finger in the mouth. The patient may ask for restraints to prevent elbow movement; when the restraints are placed or replaced, the patient may appear relaxed and better humored. Dysarthric speech may cause interpersonal communication problems; the higher-functioning children can express themselves fully and participate in verbal therapy.
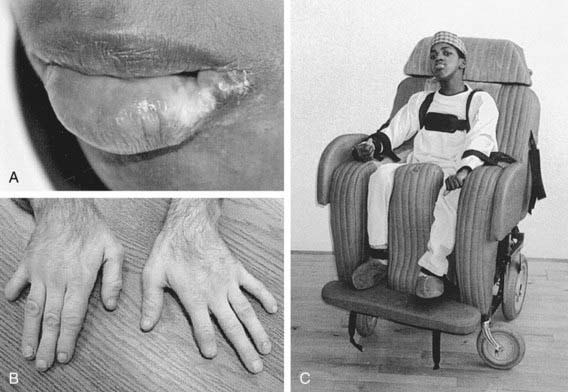
Figure 83-4 Self-injury in Lesch-Nyhan disease. Tissue damage to the lip (A) and fingers (B) was self-inflicted. Management of this problem requires covering any dangerous portions of the wheelchair in combination with protective restraints (C).
(From Visser JE, Bär PR, Jinah HA: Lesch-Nyhan disease and the basal ganglia, Brain Res Rev 32:449–475, 2000.)
The self-mutilation presents as a compulsive behavior that the child tries to control but frequently is unable to resist. Older individuals may enlist the help of others and notify them when they are comfortable enough to have restraints removed. In some instances, the behavior may lead to deliberate self-harm. Individuals with LND may also show compulsive aggression and inflict injury to others through pinching, grabbing, or hitting or by using verbal forms of aggression. Afterwards they may apologize, stating that their behavior was out of their control. Other maladaptive behaviors include head or limb banging, eye poking, and psychogenic vomiting.
Laboratory Findings
Lesch-Nyhan syndrome is caused by deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT), which catalyzes the conversion of hypoxanthine to inosine monophosphate (inosinic acid, IMP) and guanine to guanine monophosphate (guanylic acid, GMP) in the presence of phosphoribosylpyrophosphate. The urate-to-creatinine ratio, calculated from the concentration of uric acid and creatinine in the urine, provides a reliable measure of uric acid overproduction. A urate-to-creatinine ratio above 2 is characteristic for affected individuals under age 10 yr, but is not considered diagnostic. Twenty-four–hour urate excretion of more than 20 mg/kg is characteristic but not diagnostic. Hyperuricemia (serum uric acid concentration >8 mg/dL) is often present but not sensitive or specific enough for diagnostic purposes. HPRT enzyme activity in cells from any tissue (e.g., blood, cultured fibroblasts, or lymphoblasts) is performed on erythrocytes in anticoagulant but can also be performed on dried blood spots on filter paper.
Diagnosis and Differential Diagnosis
The diagnosis of Lesch-Nyhan syndrome is suspected in males with developmental delay during the 1st year of life who manifest the characteristic neurologic, cognitive, and behavioral disturbances. The presence of dystonia along with self-mutilation of the mouth and fingers suggests LND. With partial HPRT deficiency, recognition is linked to either hyperuricemia alone or hyperuricemia and a dystonic movement disorder. Serum levels of uric acid >4-5 mg uric acid/dL and a urine uric acid : creatinine ratio of 3 : 4 or more are highly suggestive of HPRT deficiency, particularly when associated with neurologic symptoms. The definitive diagnosis requires an analysis of the HPRT enzyme. This is assayed in an erythrocyte lysate. Individuals with classic LND have near 0% enzyme activity and those with partial variants show values between 1.5% and 60%. The intact cell HPRT assay in skin fibroblasts offers a good correlation between enzyme activity and the severity of the disease. Molecular techniques are used for gene sequencing and the identification of carriers.
Differential diagnosis includes other causes of infantile hypotonia and dystonia. Children with LND are often initially incorrectly diagnosed as having athetoid cerebral palsy. When a diagnosis of cerebral palsy is suspected in an infant with a normal prenatal, perinatal, and postnatal course, LND should be considered. Partial HPRT deficiency may be associated with acute renal failure in infancy; therefore, clinical awareness of partial HPRT deficiency is of particular importance.
An understanding of the molecular disorder has led to effective drug treatment for uric acid accumulation and arthritic tophi, renal stones, and neuropathy. Reduction in uric acid alone does not influence the neurologic and behavioral aspects of LND. Despite treatment from birth for uric acid elevation, behavioral and neurologic symptoms are unaffected. The most significant complications of LND are renal failure and self-mutilation.
Treatment
Medical management of this disorder focuses on the prevention of renal failure by pharmacologic treatment of hyperuricemia with high fluid intake along with alkali and allopurinol. Allopurinol treatment must be monitored because urinary oxypurine excretion is sensitive to allopurinol, resulting in an increased concentration of xanthine, which is extremely insoluble and can result in xanthinuria and xanthine urolithiasis. Self-mutilation is reduced through behavior management and the use of restraints, removal of teeth, or both. Injection of botulism toxin into the masseter muscles was useful in 1 patient. Pharmacologic approaches to decrease anxiety and spasticity with medication have mixed results. Drug therapy focuses on symptomatic management of anticipatory anxiety, mood stabilization, and reduction of self-injurious behavior. Diazepam may be helpful for anxiety symptoms, and carbamazepine or gabapentin for mood stabilization. Each of these medications may reduce self-injurious behavior by helping to reduce anxiety and stabilize mood.
Bone marrow transplantation (BMT) has been carried out in several patients, based on the possibility that the CNS damage is produced by a circulating metabolic toxin. Several infant patients have died of complications of BMT. In 1 adult case in which the transplantation was successful, there was no change in neurologic symptoms or in behavior. In this case, dopamine receptors measured by positron emission tomography before and after BMT showed no changes in receptor density after the transplantation. To date, there is no evidence that BMT is a beneficial treatment approach; it remains an experimental and potentially dangerous therapy.
Successful preimplantation genetic diagnosis and in vitro fertilization to prevent LND has been reported with the birth of an unaffected male infant.
Case reports document modest improvement in dystonic movement disorder and substantial improvement or elimination of self-injurious behavior in several cases following deep brain stimulation involving bilateral chronic stimulation of the globus pallidus internus. The safety and efficacy of this approach is the subject of ongoing investigation. In the reported cases improvement in quality of life for both patient and family are reported.
Both the motivation for self-injury and its biologic basis must be addressed in treatment programs. Yet behavioral techniques alone, using operant conditioning approaches, have not proved to be an adequate general treatment. Although behavioral procedures have had some selective success in reducing self-injury, generalization outside the experimental setting limits this approach and patients under stress may revert to their previous self-injurious behavior. Behavioral approaches may also focus on reducing the self-injurious behavior through the treatment of phobic anxiety associated with being unrestrained. The most common techniques are systematic desensitization, extinction, and differential reinforcement of other (competing) behavior. Stress management has been recommended to assist patients to develop more effective coping mechanisms. Individuals with LND do not respond to contingent electric shock or similar aversive behavioral measures. An increase in self-injury may be observed when aversive methods are utilized.
Supportive Care
Restraint (day and night) and dental procedures are common means to prevent self-injury. The time in restraints is linked to the age of onset of self-injury. Children with LND can participate in making decisions regarding restraints and the type of restraints. The time in restraints may potentially be reduced with systematic behavior treatment programs. Many patients have teeth extracted to prevent self-injury. Others use a protective mouth guard designed by a dentist. Most parents suggest that stress reduction and awareness of the patient’s needs are the most effective in reducing self-injury. Positive behavioral techniques of reinforcing appropriate behavior are rated effective by almost half of the families.
Adenine Phosphoribosyltransferase (APRT) Deficiency
APRT, a purine salvage enzyme, catalyzes the synthesis of AMP from adenine and 5-phosphoribosyl-1-pyrophosphate (PP-ribose-P). The absence of this enzyme results in the inability to utilize adenine and accumulated adenine being oxidized by xanthine dehydrogenase to 2,8-dihydroxyadenine, which is extremely insoluble. APRT deficiency is present from birth, becoming apparent as early as 5 mo and as late as the 7th decade.
Pathogenesis
The disorder is an autosomal recessive trait with considerable clinical heterogeneity. The APRT gene is located on chromosome 16q (16q24.3) and encompasses 2.8 kb of genomic DNA. There is an APRT knockout mouse model that replicates the disease process. Clinical manifestations include urinary calculus formation with crystalluria, urinary tract infections, hematuria, renal colic, dysuria, and acute renal failure. The presence of brownish spots on the infant’s diaper or of yellow-brown crystals in the urine is suggestive of the diagnosis. Laboratory findings: Urinary levels of adenine, 8-hydroxyadenine, and 2,8-dihydroxyadenine are elevated, whereas plasma uric acid is normal. The deficiency may be complete (type I) or partial (type II); the partial deficiency is reported in Japan. The diagnosis is made based on the level of residual enzyme in erythrocyte lysates. The renal calculi, composed of 2,8-dihydroxyadenine, are radiolucent, soft, and easily crushed. These stones are not distinguishable from uric acid stones by routine tests but require high-pressure liquid chromatography (HPLC), ultraviolet or infrared radiation detection, mass spectroscopy (MS), x-ray crystallography, or capillary electrophoresis for diagnosis, particularly to distinguish from stones in HPRT deficiency.
Treatment
Treatment includes high fluid intake, dietary purine restriction, and allopurinol, which inhibits the conversion of adenine to its metabolites, further 2,8-dihydroxyadenine excretion, and further stone formation. Alkalinization of the urine is to be avoided, because, unlike that of uric acid, the solubility of 2,8-dihydroxyadenine does not increase up to a pH of 9. Shock-wave lithotripsy has been reported to be successful. The prognosis depends on renal function at the time of diagnosis. Early treatment is critical in the prevention of stones because severe renal insufficiency may accompany late recognition.
Disorders Linked to Purine Nucleotide Synthesis
Phosphoribosylpyrophosphate (PRPP) Synthetase Superactivity
PRPP is a substrate involved in the synthesis of essentially all nucleotides and important in the regulation of the de novo pathways of purine and pyrimidine nucleotide synthesis. This enzyme produces PRPP from ribose-5-phosphate and ATP, as shown in Figures 83-1 and 83-2. PRPP is the 1st intermediary compound in the de novo synthesis of purine nucleotides that lead to the formation of inosine monophosphate. Superactivity of the enzyme results in an increased generation of PRPP. Because PRPP amidotransferase, the 1st enzyme of the de novo pathway, is not physiologically saturated by PRPP, the synthesis of purine nucleotides increases, and, consequently, the production of uric acid is increased. PRPP synthetase superactivity is 1 of the few hereditary disorders in which there is enhancement of the activity of an enzyme.
Pathogenesis
Phosphoribosylpyrophosphate synthetase (PRS) superactivity is inherited as an X-linked trait and presents with 2 clinical phenotypes with varying degrees of severity. Three distinct PRS cDNAs have been cloned and sequenced. Two forms are X linked to Xq22-q24 and Xp22.2-p.22.3 (escapes X inactivation), respectively, and are widely expressed; the 3rd maps to human chromosome 7 and appears to be transcribed only in the testes. Even though the defect is X linked it should be considered in a child or young adult of either sex with hyperuricemia and/or hyperuricosuria and normal HPRT activity in lysed red cells. Clinical manifestations in the more severe type in affected hemizygous males include signs of uric acid overproduction that are apparent in infancy or early childhood, neurodevelopmental retardation, and, in some cases, sensorineural deafness. Hypotonia, delays in motor milestones, ataxia, and autistic-like behavior have been described. Heterozygous female carriers may also develop gout and hearing impairment. The late juvenile to early adult onset type is found in males who show gout or uric acid urolithiasis but no neurologic signs. A mechanism for the neurologic symptoms is unknown. Laboratory findings: Blood uric acid may be 2-3 times normal values, and the urinary excretion of uric acid is increased. The diagnosis requires enzyme analysis of erythrocytes and cultured fibroblasts. This disorder must be differentiated from partial HPRT deficiency involving the salvage pathway, which also results in neurologic HPRT deficiency or hyperuricemia without neurologic features.
Treatment
Treatment is with allopurinol, which inhibits xanthine oxidase, the last enzyme of the purine catabolic pathway. Uric acid production is reduced and is replaced by hypoxanthine, which is more soluble than xanthine. The initial dose of allopurinol is 10-20 mg/kg/24 hr in children and is adjusted to maintain normal uric acid levels in plasma. Occasionally, xanthine calculi may form. Consequently, a low-purine diet (one free of organ meats, dried beans, and sardines), high fluid intake, and alkalinization of the urine to establish a urinary pH of 6.0-6.5 is necessary. These measures control the hyperuricemia and urate neuropathy but do not affect the neurologic symptoms. There is no known treatment for the neurologic complications.
Adenylosuccinate Lyase (ADSL) Deficiency
ADSL deficiency is an inherited deficiency of de novo purine synthesis in humans. Adenylosuccinase lyase is an enzyme that catalyzes 2 pathways in de novo synthesis and purine nucleotide recycling. These are the conversion of succinylaminoimidazole carboxamide ribotide (SAICAR) into aminoimidazole carboxamide ribotide (AICAR), the 8th step in the de novo synthesis of purine nucleotides, and the conversion of adenylosuccinate (S-AMP) into adenosine monophosphate (AMP); the latter is the 2nd step in the conversion of inosine monophosphate (IMP) into AMP in the purine nucleotide cycle. ADSL deficiency results in the accumulation in urine, cerebrospinal fluid, and, to a smaller extent, in plasma, of SAICA riboside (SAICAr) and succinyladenosine (S-Ado), dephosphorylated derivatives of SAICAR and S-AMP, respectively.
Pathology
CT and MRI of the brain may show hypotrophy or hypoplasia of the cerebellum, particularly the vermis. It is proposed that rather than being caused by purine nucleotide deficiency, the symptoms are due to the neurotoxic effects of accumulating succinyl purines. The ratio of S-Ado/SAICAr has been linked to phenotype severity suggesting that SAICAr is the more toxic compound and that S-Ado might be neuroprotective.
This is an autosomal recessive disorder; the gene has been mapped to chromosome 22q13.1-q13.2 and ≈20 gene mutations have been identified.
Clinical Manifestations
Clinical manifestations include varying degrees of psychomotor retardation, generally accompanied by a seizure disorder and/or autistic-like behaviors (poor eye contact and repetitive behaviors). ADSL deficiency may present with prenatal growth restriction, fetal and neonatal hypokinesia, and rapidly fatal neonatal encephalopathy. Neonatal seizures and a severe infantile epileptic encephalopathy are often the 1st manifestations of this disorder. Others demonstrate moderate to severe intellectual disability sometimes associated with growth retardation and muscle hypotonia. One reported case, a girl, tested in the mild range of intellectual disability. The form with profound intellectual disability has been designated type I, the variant case with mild intellectual disability as type II. Other patients have an intermediate clinical symptom pattern with moderately delayed psychomotor development, seizures, stereotypies, and agitation
Laboratory Studies
Laboratory investigations show the presence in urine and cerebrospinal fluid of succinylpurines, which are normally undetectable. It is based on the presence in urine and cerebrospinal fluid of SAICAr and S-Ado; both are usually undetectable. Prenatal diagnosis has been reported. Systematic screening with the Bratton-Marshall test is suggested in infants and children with unexplained psychomotor retardation and/or seizure disorders. No successful treatment has been demonstrated for this disorder.
5-Amino-4-Imidazolecarboxamide (AICA) Riboside Deficiency (AICA-Ribosiduria)
AICA riboside is the dephosphorylated product of AICAR, also termed ZMP. It, along with its diphosphates and triphosphates, accumulates in red blood cells and fibrocytes in AICA riboside deficiency. The enzyme defect is in the conversion of AICAR to FAICAR (formyl-AICAR), catalyzed by the bifunctional enzyme AICAR transformylase/IMP cyclohydrolase (ATIC). The transformylase is deficient in fibroblasts in this disorder. This is an inborn error of purine biosynthesis caused by a mutation of ATIC gene effecting AICAR transformylase activity. In a reported case, AICAR transformylase was profoundly deficient, whereas the IMP cyclohydrolase level was 40% of normal.
The disorder is described in a female infant with profound intellectual disability, epilepsy, dysmorphic features (prominent forehead and metopic suture, brachycephaly, wide mouth with thin upper lip, low-set ears, and prominent clitoris due to fused labia minora), and congenital blindness. Urinary screening with the Bratton-Marshall test resulted in the identification of this disorder. No successful treatment is described.
Disorders Resulting from Abnormalities in Purine Catabolism
Myoadenylate Deaminase Deficiency (Muscle Adenosine Monophosphate Deaminase Deficiency)
Myoadenylate deaminase is a muscle-specific isoenzyme of AMP deaminase that is active in skeletal muscle. During exercise, the deamination of AMP leads to increased levels of IMP and ammonia in proportion to the work performed by the muscle. Two forms of myoadenylate deaminase deficiency are known: an inherited (primary) form that may be asymptomatic or associated with cramps or myalgia with exercise, and a secondary form that may be associated with other neuromuscular or rheumatologic disorders.
Pathogenesis
The inherited form of the disorder is an autosomal recessive trait. AMP-D1, the gene responsible for encoding muscle AMP deaminase, is located on the short arm of chromosome 1 (1p13-21). Population studies reveal that this mutant allele is found at high frequency in white populations. The disorder may be screened for by performing the forearm ischemic exercise test. The normal elevation of venous plasma ammonia after exercise that is seen in normal subjects is absent in AMP deaminase deficiency. Clinical manifestations are, most commonly, isolated muscle weakness, fatigue, myalgias after moderate-to-vigorous exercise, or cramps. Myalgias may be associated with an increased serum creatine kinase level and detectable electromyelographic abnormalities. Muscle wasting or histologic changes on biopsy are absent. The age of onset may be as early as 8 mo of life with ≈25% of cases recognized between 2 and 12 yr of age. The enzyme defect has been identified in asymptomatic family members. Secondary forms of muscle AMP deaminase deficiency have been identified in Werdnig-Hoffmann disease, Kugelberg-Welander syndrome, polyneuropathies, and amyotrophic lateral sclerosis (Chapter 604). The metabolic disorder involves the purine nucleotide cycle. The enzymes involved in this cycle are AMP deaminase, adenylosuccinate synthetase, and adenylosuccinase (see Fig. 83-2). It is proposed that muscle dysfunction in AMP deaminase deficiency results from impaired energy production during muscle contraction. It is unclear how individuals may carry the deficit and be asymptomatic. In addition to muscle dysfunction, a mutation of liver AMP deaminase has been proposed as a cause of primary gout, leading to overproduction of uric acid.
Laboratory Findings
The final diagnosis is made by histochemical or biochemical assays of a muscle biopsy. The primary form is distinguished by the finding of enzyme levels <2% with little or no immunoprecipitable enzyme. Treatment with ribose (2-60 g/24 hr orally, in divided doses) or xylitol, which is converted to ribose, has been reported to improve endurance and muscle strength in some cases but is ineffective in others. Affected individuals are advised to exercise with caution to prevent rhabdomyolysis and myoglobinuria. Although there are no documented fully effective treatments, it has been proposed that enhancing the rate of replenishment of the ATP pool might be beneficial. Genetic approaches may be feasible in the future for inherited cases, whereas treatment of the underlying condition is essential in secondary cases.
Xanthine Oxidoreductase Deficiency, Hereditary Xanthinuria/Molybdenum Cofactor Deficiency
Xanthine oxidoreductase (XOR) is the catalytic enzyme in the final step of the purine catabolic pathway and catalyzes the oxidation of hypoxanthine to xanthine and xanthine to uric acid. Because XOR exists in 2 forms, xanthine dehydrogenase and xanthine oxidase, the deficiency is also referred to as xanthine dehydrogenase/xanthine oxidase (XDH/XO) deficiency.
Pathogenesis
The inheritance of types I and II is autosomal recessive; the human XOR gene is located on chromosome 2p22. In both forms of the deficiency, xanthine, the immediate precursor of uric acid, is less soluble than uric acid in urine and deficiency of the enzyme results in xanthinuria. Xanthine oxidoreductase deficiency may occur in an isolated form (xanthinuria type I), in a combined form involving xanthine oxidoreductase deficiency and aldehyde oxidase deficiency (xanthinuria type II), or in a combined form with deficiency of xanthine oxidoreductase, aldehyde oxidase, and sulfite oxidase (molybdenum cofactor deficiency). The isolated form results in an almost total replacement of uric acid by hypoxanthine and xanthine. Patients can oxidize allopurinol to oxypurinol via aldehyde oxidase.
Clinical Manifestations
Patients with the isolated form are usually asymptomatic or have mild symptoms; however, renal stones, often not visible on radiography, are a risk for renal damage and may appear at any age. Crystalline xanthine deposits in muscle may result in muscle pain following exertion. Rarely, xanthine stones have been reported as a result of allopurinol administration. In type II, the clinical presentation is similar to type I. Type II patients are deficient in both oxidases and cannot metabolize allopurinol. Molybdenum cofactor deficiency (failure to synthesize the molybdenum cofactor) involves all 3 molybdoenzymes and, like isolated sulfite oxidase deficiency, results in neonatal feeding problems, neonatal seizures, increased or decreased muscle tone, ocular lens dislocation, severe intellectual disability, and death in early childhood.
Laboratory Findings
The diagnosis is made by measuring plasma concentrations of uric acid; plasma uric acid is low (<1 mg/dL) and xanthine is elevated in plasma and urine. Urinary uric acid is reduced, being replaced by xanthine and hypoxanthine. In molybdenum cofactor deficiency, there is, in addition, an excessive excretion of sulfite and other sulfur-containing metabolites. Enzyme diagnostic measurement requires jejunal or liver biopsy because these are the only human tissues that contain appreciable amounts of xanthine oxidoreductase. Sulfite oxidase and the molybdenum cofactor can be measured in liver and fibroblasts. Although the isolated deficiency is generally benign, treatment with a low-purine diet and increased fluid intake are recommended. Allopurinol has been recommended for those with residual xanthine oxidoreductase activity. It completely blocks the conversion of hypoxanthine into the far less soluble xanthine. The prognosis for molybdenum cofactor deficiency is very poor.
Disorders of Pyrimidine Metabolism
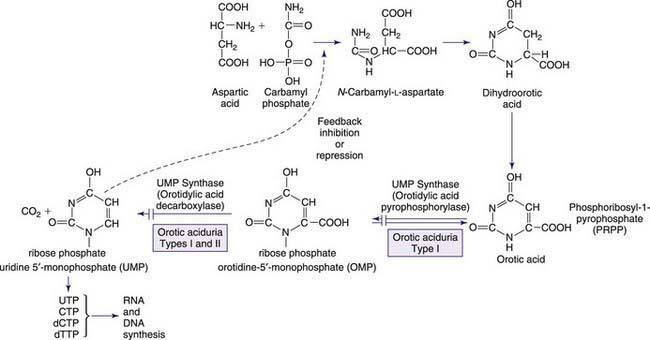
The pyrimidines are the building blocks of DNA and RNA and involved in the formation of coenzymes, as active intermediates in carbohydrate and phospholipid metabolism, glucuronidation in detoxification processes, and glycosylation of proteins and lipids. Pyrimidine synthesis differs from that of purines in that the single pyrimidine ring is 1st assembled and then linked to ribose phosphate to form uridine 5′-monophosphate (UMP). The pyrimidines uracil and thymine are degraded in 4 steps, as shown in Figure 83-3. Purine metabolism has an easily measurable end point in uric acid; however, there is no equivalent compound in pyrimidine metabolism. The 1st defect, hereditary orotic aciduria, is in the de novo synthetic pathway, whereas the other disorders involve either overactivity of or defects in the pyrimidine degradation pathway. Degradation disorders may present as anemia, neurologic disorders, or multisystem mitochondrial disorders. The 1st three steps of the degradation pathways for thymine and uracil make use of the same enzymes (DPD, DPH, and UP). These 3 steps result in the conversion into β-alanine from uracil. There is increasing evidence that pyrimidines play an important role in the regulation of the nervous system. Reduced production of the neurotransmitter function of β-alanine is hypothesized to produce clinical symptoms. These rare disorders may be overlooked because symptoms are not highly specific; they should be considered as possible causes of anemia and neurologic disease and are a contraindication for treatment of cancer patients with certain pyrimidine analogs, for example, 5-fluorouracil.
Hereditary Orotic Aciduria (Uridine Monophosphate Synthase Type 1 Deficiency)
This is a disorder of pyrimidine synthesis associated with deficient activity of the last 2 enzymes of the de novo pyrimidine synthetic pathway, orotate phosphoribosyltransferase (OPRT) and orotidine-5′-monophosphate decarboxylase (ODC). The activities of these 2 enzymes reside in separate domains to a single polypeptide coded by a single gene. This bifunctional protein, uridine 5′-monophosphate (UMP) synthase, catalyzes the 2-step conversion of orotic acid to UMP, via orotidine monophosphate (OMP). Hereditary orotic aciduria results in the excessive accumulation of orotic acid.
Pathogenesis
This disorder is an autosomal recessive disorder. Two functional domains are encoded on a single gene. The gene for UMP synthase is located on the long arm of chromosome 3 (3q13). Genetic metabolic defects that involve 4 of the 6 enzymes associated with the urea cycle may result in orotic aciduria secondary to PP-ribose-P depletion resulting from a substantial increased flux through the de novo pathway.
Clinical Manifestations
Patients with hereditary orotic aciduria (UMP synthase type 1 deficiency) have a macrocytic hypochromic megablastic anemia that is unresponsive to the usual forms of therapy (iron, folic acid, and B12) and may have leukopenia. Onset is usually in the 1st months of life. Untreated, this disorder can lead to developmental disability, intellectual disability, failure to thrive, cardiac disease, strabismus, crystalluria, and occasional ureteric obstruction. Renal function is generally normal. Heterozygotes may have mild orotic aciduria but are not otherwise affected. The clinical features may be related to pyrimidine nucleotide depletion. Metabolites derived from several pharmacologic agents (5-azauridine, allopurinol) produce secondary orotic aciduria and orotidinuria by specifically inhibiting ODC. Orotic aciduria may also occur in association with parenteral nutrition, essential amino acid deficiency, and Reye syndrome. The enzymatic defect may be demonstrated in liver, lymphoblasts, erythrocytes, leukocytes, and cultured skin fibroblasts. A carrier detection test is available, as is prenatal diagnosis. In ODC deficiency (UMP synthase type 2 deficiency), the clinical symptoms show neurologic abnormalities without megaloblastic anemia. In reported cases, both orotidinuria and orotic aciduria were found.
Treatment
The administration of uridine in doses of 50-300 mg/kg/day has been an effective treatment in most cases and led to clinical improvement and reduction in orotic acid excretion. Lifelong treatment is required. Uracil is ineffective because, unlike purines, pyrimidine salvage occurs at the nucleoside (uridine) level. The long-term prognosis in uncomplicated cases is good; congenital malformations and other associated features may adversely affect outcome.
Dihydropyrimidine Dehydrogenase (DPD) Deficiency
DPD catalyzes the initial and rate-limiting step in the degradation of the pyrimidine bases uracil and thiamine. DPD has been identified in most tissues, with the highest activity being in lymphocytes. Pathogenesis: DPD deficiency is an autosomal recessive disorder mapping to chromosome 1p22 with at least 32 polymorphisms detected. It is estimated that the frequency of the heterozygote may be as high as 3%.
Clinical Manifestations
In children these may include seizure disorder, intellectual disability, and motor delay. Less frequent are growth retardation, microcephaly, and autistic-like behavior and ocular anomalies. Others do not show developmental abnormalities but may have milder neurologic symptoms and language disorder. In most cases, there is an initial period of normal psychomotor development, followed by subsequent developmental delays. Symptoms may be linked to altered uracil, thymine, and β-alanine homeostasis. DPD is the initial and rate-limiting enzyme in the catabolism of the neoplastic drug 5-fluorouracil (5-FU), being responsible for 80% of its catabolism. Patients with a partial deficiency of this enzyme are at risk for developing a severe 5-FU–associated toxicity. In adult patients, neurotoxicity (headache, somnolence, visual illusions, and memory impairment) linked to pyrimidinemia after 5-FU treatment for cancer is reported in previously healthy individuals.
Laboratory Findings
DPD deficiency is characterized by a variable phenotype and diagnosed by the accumulation of thiamine and uracil in urine, plasma, and cerebrospinal fluid and no activity in fibroblasts. Diagnostic tests use high-pressure liquid chromatography (HPLC) or gas chromatography–mass spectroscopy (GC-MS). Alternatively, DPD deficiency may be confirmed by measuring the enzyme in cultured fibroblasts and leukoblasts. Uric acid levels have been reported to be normal. Because β-alanine is a structural analog of γ-aminobutyric acid (GABA) and glycine, it has been proposed that it may affect inhibitory neurotransmission. Prenatal diagnosis has been reported.
Dihydropyrimidinase (DPH) Deficiency (Dihydropyrimidinuria)
DPH is the 2nd enzyme in the 3-step degradation pathway of uracil and thiamine leading to increased urinary excretion. DPH deficiency is characterized by increased urinary secretion of dihydrouracil and dihydrothymine as well as uracil and thymine. There is a variable clinical phenotype.
Pathogenesis
There is an autosomal recessive form of inheritance with the gene mapped to chromosome 8q22. In 1 study, there was no significant difference in residual activity between mutations observed in symptomatic and asymptomatic individuals. Population prevalence in a Japanese sample is 0.1%
Clinical Manifestations
Clinical manifestations in 3 unrelated affected cases included seizures with dysmorphic features and developmental delay in 2 of these cases. Three unrelated infant and 2 adult asymptomatic cases were identified, however, in a screening program for pyrimidine-degradation disorders in Japan and were asymptomatic despite the accumulation of pyrimidine degradation products in body fluids.
N-Carbamyl-β-Amino Aciduria (Deficiency of β-Ureidopropionase)
The pyrimidine bases uracil and thymine are degraded via the consecutive action of 3 enzymes to β-alanine and β-aminoisobutyric acid, respectively. The 3rd enzyme in the pathway is β-ureidopropionase (UP) and its deficiency leads to N-carbamyl-β-amino aciduria. Urinary analysis in a reported case showed elevated levels of N-carbamyl-β-alanine and N-carbamyl-β-aminoisobutyric acid. The enzyme is expressed only in the liver and no activity of β-ureidopropionase is detected in a liver biopsy.
Pathogenesis
Fluorescence in situ hybridization localized the human β-ureidopropionase gene to 22q11.2.
Clinical Manifestations
Clinically observed features in a reported case included muscular hypotonia, dystonic movements, and severe developmental delay.
Laboratory Findings
Neuropathology involves both gray and white matter. UP deficiency leads to pathologic accumulation of 3-UPA in body fluids. 3-UPA acts as an endogenous neurotoxin via inhibition of mitochondrial energy metabolism, resulting in the initiation of secondary, energy-dependent excitotoxic mechanisms. There is no known treatment for UP deficiency.
Uridine Monophosphate Hydrolase 1 Deficiency (Pyrimidine 5′-Nucleotidase Deficiency)
Erythrocyte maturation is accompanied by RNA degradation and the release of mononucleotides. Pyrimidine 5′-nucleotidase is the 1st degradative enzyme of the pyrimidine salvage cycle and catalyzes the hydrolysis of pyrimidine 5′-nucleotides to the corresponding nucleosides. Enzyme deficiency results in the accumulation of high levels of cytidine and uridine nucleotides in the erythrocytes of those affected, which results, in turn, in hemolysis. Deficiency of pyrimidine 5′-nucleotidase is at least in part compensated in vivo by other nucleosidases or perhaps other nucleotide metabolic pathways.
Clinical Manifestations
In pyrimidine 5′-nucleotidase deficiency, patients, who are homozygotes, present with a defect restricted to erythrocytes that is characterized by nonspherocytic hemolytic anemia with basophilic stippling. Other characteristic features include splenomegaly, increased indirect bilirubin, and hemoglobinuria. Lead is a powerful inhibitor of pyrimidine 5′-nucleotidase and assessment of lead levels should be included whenever the hemolytic anemia, pyrimidine 5′-nucleotidase deficiency, and basophilic stippling are found together.
Diagnosis
Diagnosis requires demonstration of a complete deficiency of the major isoenzyme uridine monophosphate hydrolase-1.
Uridine Monophosphate Hydrolase 1 Deficiency and Overactive Cytosolic 5′-Nucleotidase
Pyrimidine nucleoside depletion and overactive cytosolic 5′-nucleotidase may lead to a neurodevelopmental disorder.
Pathogenesis
Four unrelated patients showed 6- to 10-fold elevation in the activity of pyrimidine 5′-nucleotidase in fibroblasts with both purine and pyrimidine substrates.
Laboratory Findings
Investigation in cultured fibroblasts derived from these patients showed normal incorporation of purine bases into nucleotides but decreased incorporation of uridine and orotic acid. Affected patients show electroencephalogram (EEG) abnormalities
Clinical Manifestations
Clinical manifestations include developmental delay, seizures, ataxia, recurrent infections, severe language deficit, hyperactivity, short attention span, and aggressive behavior appearing in the 1st few yr of life.
Metabolic testing is normal except for persistent hypouricosuria. It is proposed that increased catabolic activity and decreased pyrimidine salvage cause a deficiency of pyrimidine nucleotides.
Thymidine Phosphorylase Deficiency
Thymidine phosphorylase catalyzes the conversion of thymidine to thymine. This enzyme is also known as platelet-derived endothelial cell growth factor due to its angiogenic or “gliostatin” properties, indicating its inhibitory effects on glial cell proliferation. It has been implicated in mitochondrial nucleoside metabolism. Plasma thymidine level is increased >20-fold in patients compared to controls. Loss of function of thymidine phosphorylase causes mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), an autosomal recessive disorder with mitochondrial DNA alterations.
Pathogenesis
This is an autosomal recessive disorder. The gene encoding thymidine phosphorylase has been identified as the MNGIE gene and mapped to chromosome 22q13.32-qter.
Clinical Manifestations
Clinical manifestations of MNGIE include ptosis, progressive external ophthalmoparesis, gastrointestinal dysmotility and malabsorption, cachexia, peripheral neuropathy, skeletal muscle myopathy, and leukoencephalopathy.
Thymidine Kinase 2 (TK2) Deficiency
Thymidine kinase 2 (TK2) is a key enzyme in mitochondrial DNA (mtDNA) precursor synthesis. TK deficiencies cause tissue-specific depletion of mtDNA. TK2 normally phosphorylates deoxythymidine, deoxycytidine, and deoxyuridine.
Augoustides-Savvopoulou P, Papachristou F, Fairbanks LD, et al. Partial hypoxanthine-guanine phosphoribosyltransferase deficiency as the unsuspected cause of renal disease spanning three generations: a cautionary tale. Pediatrics. 2002;109:E17.
Cameron JS, Simmonds HA. Hereditary hyperuricemia and renal disease. Semin Nephrol. 2005;25:9-18.
Camici M, Micheli V, Ipata PL, et al. Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem Int. 2010;56:367-378.
Ceballos-Picot I, Mockel L, Potier MC, et al. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: implications for Lesch-Nyhan disease pathogenesis. Hum Mol Genet. 2009;18(13):2317-2327. Jul 1
Dabrowski E, Smathers SA, Ralstrom CS, et al. Botulinum toxin as a novel treatment for self-mutilation in Lesch-Nyhan syndrome. Dev Med Child Neurol. 2005;47:636-639.
Hartmann S, Okun JG, Schmidt C, et al. Comprehensive detection of disorders of purine and pyrimidine metabolism by HPLC with electrospray ionization tandem mass spectrometry. Clin Chem.. 2006;52:1127-1137.
Hladnik U, Nyhan WL, Bertelli M. Variable expression of HPRT deficiency in 5 members of a family with the same mutation. Arch Neurol. 2008;65:1240-1243.
Jinnah HA, De Gregorio L, Harris JC, et al. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res. 2000;463:309-326.
Jinnah HA, Visser JE, Harris JC, et al. Lesch-Nyhan Disease International Study Group. Delineation of the motor disorder of Lesch-Nyhan disease. Brain. 2006;129:1201-1217.
Loffler M, Fairbanks LD, Zameitat E, et al. Pyrimidine pathways in health and disease. Mol Med. 2005;11:430-437.
Marinaki AM, Escuredo E, Duley JA, et al. Genetic basis of hemolytic anemia caused by pyrimidine 5’ nucleotidase deficiency. Blood. 2001;97:3327-3332.
Mouchegh K, Zikánová M, Hoffmann GF, et al. Lethal fetal and early neonatal presentation of adenylosuccinate lyase deficiency: observation of 6 patients in 4 families. J Pediatr. 2007;150:57-61. e2
Nishino I, Spinazzola A, Hirano M. MNGIE: from nuclear DNA to mitochondrial DNA. Neuromuscul Disord. 2001;11:7-10.
Nyhan WL. Disorders of purine and pyrimidine metabolism. Mol Genet Metab. 2005;86:25-33.
Pralong E, Pollo C, Coubes P, et al. Electrophysiological characteristics of limbic and motor globus pallidus internus (GPI) neurons in two cases of Lesch-Nyhan syndrome. Neurophysiol Clin. 2005;35:168-173.
Pralong E, Pollo C, Villemure JG, et al. Opposite effects of internal globus pallidus stimulation on pallidal neurons activity. Mov Disord. 2007;22:1879-1884.
Race V, Marie S, Vincent MF, et al. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum Mol Genet. 2000;9:2159-2165.
Schretlen DJ, Harris JC, Park K, et al. Neurocognitive functioning in Lesch-Nyhan disease and partial hypoxanthine-guanine phosphoribosyltransferase deficiency. J Int Neuropsychol Soc. 2001;7:805-812.
Sempere A, Arias A, Farré G, et al. Study of inborn errors of metabolism in urine from patients with unexplained mental retardation. J Inherit Metab Dis. 2010;33:1-7.
Teijeira S, San Millán B, Fernández JM, et al. Myoadenylate deaminase deficiency: clinico-pathological and molecular study of a series of 27 Spanish cases. Clin Neuropathol. 2009;28(2):136-142.
Torres RJ, Prior C, Puig JG. Efficacy and safety of allopurinol in patients with hypoxanthine-guanine phosphoribosyltransferase deficiency. Metabolism. 2007;56:1179-1186.