Chapter 124 Disorders of Phagocyte Function
Neutrophils are particularly important in protecting the skin, the lining of the respiratory and gastrointestinal tracts, and other mucous membranes as part of the 1st line of defense against microbial invasion. During the critical 2-4 hr after microbial invasion, these phagocytes arrive at the site of inflammation to contain the infection and prevent hematogenous dissemination.
Immunologic evaluation of patients with suspected immunodeficiency (Chapter 116) should focus on disorders of phagocyte function (Table 124-1) in patients with recurrent or unusual bacterial infections (Fig. 124-1).
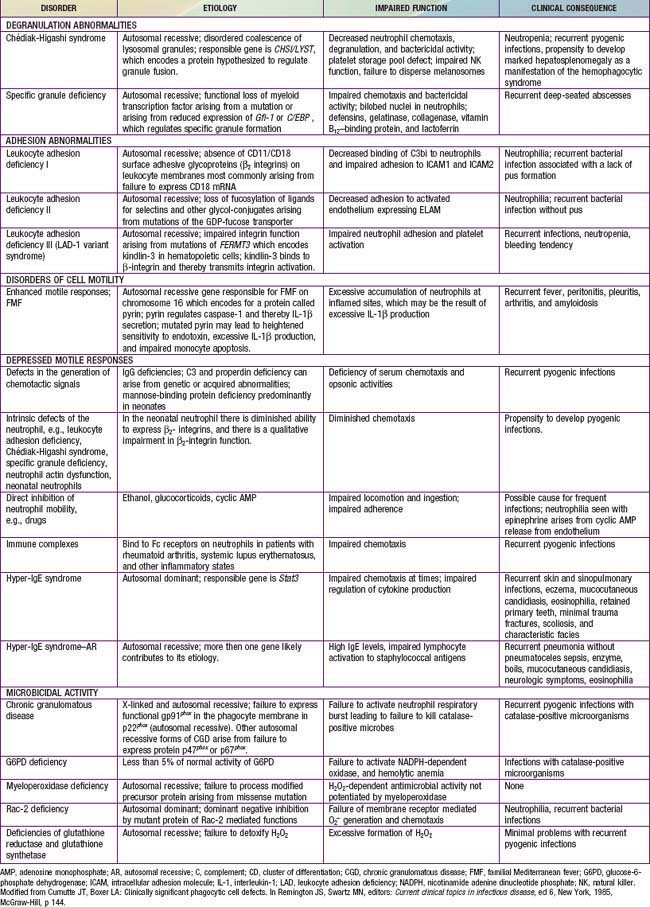
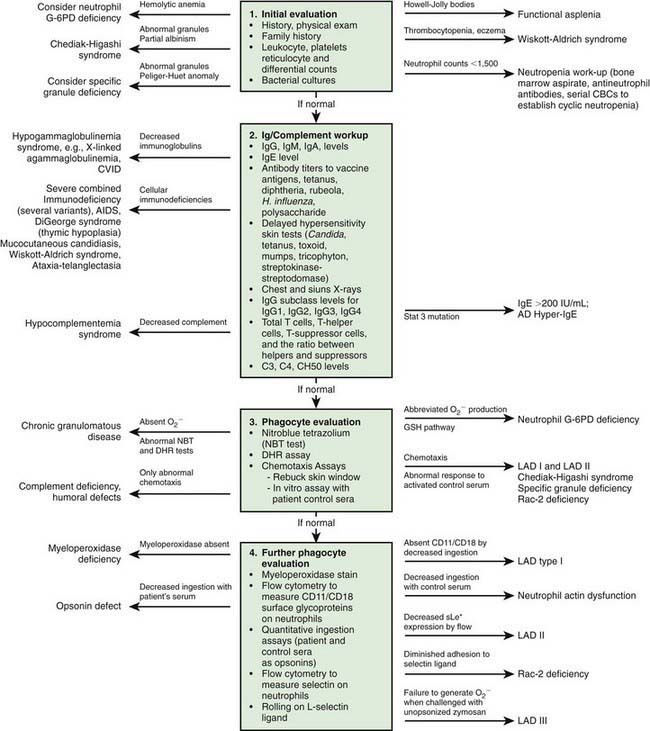
Figure 124-1 Algorithm for clinical evaluation of patients with recurrent infections. AD, autosomal dominant; AR, autosomal recessive; C, complement; CBC, complete blood count; CD, cluster of differentiation; CVID, common variable immunodeficiency; DHR, dihydrorhodamine; G-6PD, glucose-6-phosphate dehydrogenase; Ig, immunoglobulin; LAD, leukocyte adhesion deficiency; NK, natural killer; X, X-linked.
(Modified from Curnutte JT, Boxer LA: Clinically significant phagocyte cell defects. In Remington JS, Swartz MN, editors: Current clinical topics in infectious diseases, ed 6, New York, 1985, McGraw-Hill, p 144.)
Chemotaxis, the direct migration of cells into sites of infection, involves a complex series of events (Chapter 121). Studies of defective in vitro chemotaxis of neutrophils obtained from children having various clinical conditions have not established whether frequent infections arise from a primary chemotactic abnormality or occur as secondary medical complications of the underlying disorder. For example, variable but at times severe abnormalities in neutrophil motility accompany the hyper-IgE syndrome, which is characterized by markedly elevated levels of IgE, chronic dermatitis, and recurrent sinopulmonary infections, as well as coarse facial features, retention of primary teeth, and a propensity for recurrent bone fractures (Chapter 123).
Leukocyte Adhesion Deficiency
Leukocyte adhesion deficiency 1 (LAD-1), 2 (LAD-2), and 3 (LAD-3) are rare autosomal recessive disorders of leukocyte function. LAD-1 affects about 1 per 10 million individuals and is characterized by recurrent bacterial and fungal infections and depressed inflammatory responses despite striking blood neutrophilia.
Genetics and Pathogenesis
LAD-1 results from mutations of the gene on chromosome 21q22.3 encoding CD18, the 95-kD β2 leukocyte integrin subunit. Normal neutrophils express 4 heterodimeric adhesion molecules: LFA-1 (CD11a/CD18), Mac-1 (CD11b/CD18, also known as CR3 or iC3b receptor), p150,95 (CD11c/CD18), and αd β2 (CD11d/CD18). These 4 transmembrane adhesion molecules are composed of unique α1 subunits of 185 kD, 190 kD, 150 kD, and 160 kD, respectively, encoded on chromosome 16, and share a common β2 subunit. This group of leukocyte integrins is responsible for the tight adhesion of neutrophils to the endothelial cell surface, egress from the circulation, and adhesion to iC3b-coated microorganisms, which promotes phagocytosis and particulate activation of the phagocyte NADPH oxidase.
Mutations in the CD18 gene either impair gene expression or affect the structure of the synthesized CD18 peptide, leading to functionally abnormal CD11/CD18. Some mutations of CD11/CD18 allow a low level of assembly and activity of integrin molecules. These children retain some neutrophil integrin adhesion function and have a moderate phenotype. Failure of neutrophils to bear the β2-integrins leads to inability to migrate to sites of inflammation outside the blood vessel lumen because of their inability to adhere firmly to surfaces and undergo transendothelial migration. Failure of the CD11/CD18–deficient neutrophils to undergo transendothelial migration occurs because the β2-integrins bind to intercellular adhesion molecules 1 (ICAM-1) and 2 (ICAM-2) expressed on inflamed endothelial cells (Chapter 121). Neutrophils that do arrive at inflammatory sites by CD11/CD18–independent processes fail to recognize microorganisms opsonized with complement fragment iC3b, an important stable opsonin formed by the cleavage of C3b. Hence, other neutrophil functions such as degranulation and oxidative metabolism normally triggered by iC3b binding are also markedly compromised in LAD-1 neutrophils, resulting in impaired phagocytic function and high risk for serious and recurrent bacterial infections.
Monocyte function is also impaired, with poor fibrinogen-binding function, an activity that is promoted by the CD11/CD18 complex. Consequently, such cells are unable to participate effectively in wound healing.
Children with LAD-2 share the clinical features of LAD-1 but have normal CD11/CD18 integrins. Features unique to LAD-2 include neurologic defects, cranial facial dysmorphism, and absence of the erythrocyte ABO blood group antigen (Bombay phenotype). LAD-2 (also known as congenital disorder of glycosylation IIc) derives from mutations in the gene encoding a specific GDP-L-fucose transporter of the Golgi apparatus. This abnormality prevents the incorporation of fucose into various cell surface glycoproteins, which are expressed on cell surface membranes. These include the erythrocyte carbohydrate blood group markers and neutrophil the carbohydrate structure sialyl Lewis X. Absence of this selectin ligand renders the cells incapable of rolling adhesion to activated endothelial cells, an initial step necessary for subsequent integrin-mediated activation, spreading, and transendothelial migration. Infections in LAD-2 are milder than that in LAD-1.
LAD-3 is characterized by a Glanzmann thrombasthenia-like bleeding disorder, delayed separation of the umbilical cord as well as serious skin and soft tissue infections similar to that seen in LAD-1, and failure of leukocytes to undergo β2 and β1 integrin mediated adhesion and migration. Mutations in KINDLIN3 affect integrin activation.
Clinical Manifestations
Patients with the severe clinical form of LAD-1 express <0.3% of the normal amount of the β2-integrin molecules, whereas patients with the moderate phenotype may express 2-7% of the normal amount. Children with severe disease present in infancy with recurrent, indolent bacterial infections of the skin, mouth, respiratory tract, lower intestinal tract, and genital mucosa. They may have a history of delayed separation of the umbilical cord, usually with associated infection (omphalitis) of the cord stump. However, 10% of healthy infants can have cord separation at age 3 wk or later, so this sign alone should not be sufficient to raise suspicion of LAD-1. Skin infection may progress to large chronic ulcers with polymicrobial infection, including anaerobic organisms. The ulcers heal slowly, need months of antibiotic treatment, and often require plastic surgical grafting. Severe gingivitis can lead to early loss of primary and secondary teeth.
The pathogens infecting patients with LAD-1 are similar to those affecting patients with severe neutropenia (Chapter 125) and include Staphylococcus aureus and enteric gram-negative organisms such as Escherichia coli. These patients are also susceptible to opportunistic infection by fungi such as Candida and Aspergillus. Typical signs of inflammation such as swelling, erythema, and warmth may be absent. Pus does not form, and few neutrophils are identified microscopically in biopsy specimens of infected tissues. Despite the paucity of neutrophils within the affected tissue, the circulating neutrophil count during infection typically exceeds 30,000/µL and can surpass 100,000/µL. During intervals between infections, the peripheral blood neutrophil count may chronically exceed 12,000/µL. LAD-1 genotypes producing moderate amounts of functional integrins at the surface of the neutrophil significantly reduce the severity and frequency of infections compared to children with the severe form.
Similar clinical syndromes have been reported in patients with endothelial cell E-selectin deficiency, which manifests with delayed separation of the umbilical cord and omphalitis, and in a child with an autosomal dominant mutation of Rac2 (a Rho GTPase needed to regulate actin organization and superoxide production). Rac2 deficiency is characterized by delayed separation of the umbilical cord, leukocytosis, and absence of pus at sites of infection.
Laboratory Findings
The diagnosis of LAD-1 is established most readily by flow cytometric measurements of surface CD11b/CD18 in stimulated and unstimulated neutrophils. Assessments of neutrophil and monocyte adherence, aggregation, chemotaxis, and iC3b-mediated phagocytosis generally demonstrate striking abnormalities that directly correspond to the molecular deficiency. Delayed-type hypersensitivity reactions are normal, and most individuals have normal specific antibody synthesis. However, some patients have impaired T lymphocyte–dependent antibody responses that can be demonstrated by suboptimal responses to repeat vaccination with tetanus toxoid, diphtheria toxoid, and poliovirus. The diagnosis of LAD-2 is established by flow cytometric measurement of sialyl Lewis X (CD15) on neutrophils.
Treatment
Treatment of LAD-1 depends on the phenotype as determined by the level of expression of functional CD11/CD18 integrins. Early allogeneic hematopoietic stem cell transplantation (HSCT) is the treatment of choice for severe LAD-1 (and LAD-3) associated with complete absence of the CD11/CD18 integrins. Other treatment is largely supportive. Patients can be maintained on prophylactic trimethoprim-sulfamethoxazole and should have close surveillance for early identification of infections and initiation of empirical treatment with broad-spectrum antibiotics. Specific determination of the etiologic agent by culture or biopsy is important because of the prolonged antibiotic treatment required in the absence of neutrophil function.
Some LAD-2 patients have responded to fucose supplementation, which induced a rapid reduction in the circulating leukocyte count and appearance of the sialyl Lewis X molecules accompanied by marked improvement in leukocyte adhesion.
Prognosis
The severity of infectious complications correlates with the degree of β2-integrin deficiency. Patients with severe deficiency may die in infancy, and those surviving infancy have a susceptibility to severe life-threatening systemic infections. Patients with moderate deficiency have infrequent life-threatening infections and relatively long survival.
Chédiak-Higashi Syndrome
Chédiak-Higashi syndrome (CHS) is a rare autosomal recessive disorder characterized by increased susceptibility to infection due to defective degranulation of neutrophils, a mild bleeding diathesis, partial oculocutaneous albinism, progressive peripheral neuropathy, and a tendency to develop a life-threatening form of hemophagocytic lymphohistiocytosis (Chapter 501). CHS is a disorder of generalized cellular dysfunction caused by a fundamental defect in granule morphogenesis that results in abnormally large granules in multiple tissues. Pigmentary dilution involving the hair, skin, and ocular fundi results from pathologic aggregation of melanosomes. Neurologic deficits are associated with a failure of decussation of the optic and auditory nerves. Patients exhibit an increased susceptibility to infection that can be explained in part by defects in neutrophil chemotaxis, degranulation, and bactericidal activity. Giant granules in the neutrophils probably interfere with the cells’ ability to traverse the narrow passages between endothelial cells into tissue.
Genetics and Pathogenesis
LYST (for lysosomal traffic regulator), the gene mutated in CHS, is located at chromosome 1q2-q44. The LYST/CHS protein is thought to regulate vesicle transport by mediating protein-protein interaction and protein-membrane associations. Loss of function may lead to indiscriminate interactions with lysosomal surface proteins, yielding giant granules through uncontrolled fusion of lysosomes with each other.
Almost all cells of patients with CHS show some oversized and dysmorphic lysosomes, storage granules, or related vesicular structures. Melanosomes are oversized, and delivery to the keratinocytes and hair follicles is compromised because of the failure to disperse the giant melanosomes properly, resulting in hair shafts devoid of pigment granules. This abnormality in melanosomes leads to the macroscopic impression of hair and skin that is lighter than expected from parental coloration. The same abnormality in melanocytes leads to the partial ocular albinism associated with light sensitivity.
Beginning early in neutrophil development, spontaneous fusion of giant primary granules with each other or with cytoplasmic membrane components results in huge secondary lysosomes with reduced contents of hydrolytic enzymes, including proteinases, elastase, and cathepsin G. This deficiency of proteolytic enzymes may be responsible for the impaired killing of microorganisms by CHS neutrophils. The cell membranes of CHS leukocytes are more fluid than normal, possibly leading to defective regulation of membrane activation. Changes in membrane fluidity may also affect cell function by altering expression of membrane receptors, which may in turn promote fusion of neutrophil azurophilic granules with each other.
Clinical Manifestations
Patients with CHS have light skin and silvery hair and frequently complain of solar sensitivity and photophobia that is associated with rotary nystagmus. Other signs and symptoms vary considerably, but frequent infections and neuropathy are common. The infections involve mucous membranes, skin, and respiratory tract. Affected children are susceptible to gram-positive bacteria, gram-negative bacteria, and fungi, with S. aureus being the most common offending organism. The neuropathy may be sensory or motor in type, and ataxia may be a prominent feature. Neuropathy often begins in the teenage years and becomes the most prominent problem.
Patients with CHS have prolonged bleeding times with normal platelet counts, resulting from impaired platelet aggregation associated with a deficiency of the dense granules containing adenosine diphosphate and serotonin. Natural killer cell function of large granular lymphocytes is also impaired.
The most life-threatening complication of CHS is the development of an accelerated phase characterized by pancytopenia, high fever, and lymphohistiocytic infiltration of liver, spleen, and lymph nodes. The onset of the accelerated phase, which can occur at any age, may be related to the inability of these patients to contain and control Epstein-Barr virus infection, as well as other viruses, leading to features of hemophagocytic lymphohistiocytosis (HLH). The accelerated phase is associated with secondary bacterial and viral infections and usually results in death.
Laboratory Findings
The diagnosis of CHS is established by finding large inclusions in all nucleated blood cells. These can be seen on Wright-stained blood films and are accentuated by a peroxidase stain. Because of impaired egress from the bone marrow, cells containing the large inclusions may be missed on peripheral blood smear but readily identified on bone marrow examination.
Treatment
High-dose ascorbic acid (200 mg/day for infants, 2,000 mg/day for adults) improves the clinical status of some children in the stable phase. Although controversy surrounds the efficacy of ascorbic acid, given the safety of the vitamin, it is reasonable to administer ascorbic acid to all patients.
The only curative therapy to prevent the accelerated phase is HSCT. Normal stem cells reconstitute hematopoietic and immunologic function, correct the natural killer cell deficiency, and prevent conversion to the accelerated phase, but can not correct or prevent the peripheral neuropathy. If the patient is in the accelerated phase with active HLH, HSCT often fails to prevent death.
Myeloperoxidase Deficiency
Myeloperoxidase (MPO) deficiency is an autosomal recessive disorder of oxidative metabolism and is one of the most common inherited disorders of phagocytes, occurring at a frequency approaching 1 per 2,000 individuals. MPO is a green heme protein located in the azurophilic lysosomes of neutrophils and monocytes and is the basis for the greenish tinge to pus accumulated at a site of infection. Most individuals with the trait do not have an increased rate of infection or other clinical manifestations of disease.
Genetics and Pathogenesis
Mutations in the MPO gene causing this defect provide insight into the post-translational processing of this granule protein. MPO mRNA is transcribed exclusively during the promyelocytic stage of granulopoiesis. MPO deficiency is caused by a missense mutation in the MPO gene that results in an MPO precursor that does not incorporate heme. Although this mutation is the most common cause of MPO deficiency, many patients are compound heterozygotes with 1 allele bearing the common mutation and the other possessing a mutation not yet identified. A partial deficiency results if only 1 allele is normal.
Partial or complete MPO deficiency leads to diminished production of hypochlorous acid (HOCl) and HOCl-derived chloramines. The deficiency in HOCl leads to early depression of gram-positive and gram-negative bacterial rates of killing in vitro that normalizes after 1 hr incubation. These data indicate that deficient cells use an MPO-independent microbicidal system that is slower to kill pathogens than the MPO-H2O2-halide system used by normal neutrophils.
Clinical Manifestations
MPO deficiency is usually clinically silent. Rarely, patients may have disseminated candidiasis, usually in conjunction with diabetes mellitus. Acquired partial MPO deficiency can develop in acute myelogenous leukemia and in myelodysplastic syndromes.
Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is characterized by neutrophils and monocytes capable of normal chemotaxis, ingestion, and degranulation, but unable to kill catalase-positive microorganisms because of a defect in the generation of microbicidal oxygen metabolites. CGD is a rare disease with an incidence of 4 to 5 per million individuals, caused by 4 genes, 1 X-linked and 3 autosomal recessive in inheritance.
Genetics and Pathogenesis
Activation of the phagocyte NADPH oxidase requires stimulation of the neutrophils and involves assembly from cytoplasmic and integral membrane subunits (see Fig. 121-3). Oxidase activation initiates with phosphorylation of a cationic cytoplasmic protein, p47phox (47-kd phagocyte oxidase protein). Phosphorylated p47phox, together with 2 other cytoplasmic components of the oxidase, p67phox and the low molecular weight GTPase Rac-2, translocates to the membrane where they combine with the cytoplasmic domains of the transmembrane flavocytochrome b558 to form the active oxidase complex (see Fig. 121-3). The flavocytochrome is a heterodimer composed of p22phox and highly glycosylated gp91phox. Current models predict that 3 transmembrane domains within the N-terminus of the flavocytochrome contain the histidines that coordinate heme binding. The p22phox component is required for stability of gp91phox and provides a docking site for the cytoplasmic subunits. The gp91phox glycoprotein catalyzes electron transport through its NADPH-binding, flavin-binding, and heme-binding domains. The cytoplasmic p47phox, p67phox, and Rac-2 components appear to serve as regulatory elements for activation of cytochrome b558. An addition protein, p40phox, stabilizes the preactivation cytoplasmic complex of p67phox and p47phox and protects p67phox from degradation. Defects in any of these NADPH oxidase components can lead to CGD.
Approximately 65% of patients with CGD are males who inherit their disorder as a result of mutations in CYBB, an X-chromosome gene encoding gp91phox. About 35% of patients inherit CGD in an autosomal recessive fashion resulting from mutations in the NCF1 gene on chromosome 7, encoding p47phox. Defects in the genes encoding p67phox (NCF2 on chromosome 1) and p22phox (CYBA on chromosome 16) are inherited in an autosomal recessive manner and account for about 5% of cases of CGD.
The metabolic deficiency of the CGD neutrophil predisposes the host to infection. The CGD phagocytic vacuoles lack microbicidal reactive oxygen species and remain acidic, so bacteria are not killed or digested properly (Fig. 124-2). Hematoxylin-eosin–stained sections from patients’ tissues show multiple granulomas that give CGD its descriptive name; the macrophages may contain a golden pigment that reflects an abnormal accumulation of ingested material and contributes to granuloma formation. The metabolic impairment in the CGD neutrophil leads to delayed neutrophil apoptosis and subsequent clearance of degenerating neutrophils by macrophages, which in turn leads to ongoing local tissue damage from release of proteases and other granule proteins.
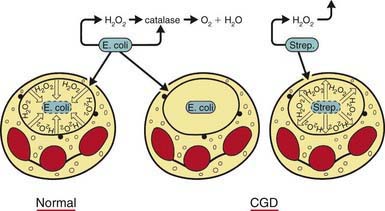
Figure 124-2 The pathogenesis of chronic granulomatous disease (CGD). The manner in which the metabolic deficiency of the CGD neutrophil predisposes the host to infection is shown schematically. Normal neutrophils stimulate hydrogen peroxide in the phagosome containing ingested Escherichia coli. Myeloperoxidase is delivered to the phagosome by degranulation, as indicated by the closed circles. In this setting, hydrogen peroxide acts as a substrate for myeloperoxidase to oxidize halide to hypochlorous acid and chloramines that kill the microbes. The quantity of hydrogen peroxide produced by the normal neutrophil is sufficient to exceed the capacity of catalase, a hydrogen peroxide-catabolizing enzyme of many aerobic microorganisms, including Staphylococcus aureus, most gram-negative enteric bacteria, Candida albicans, and Aspergillus. When organisms such as E. coli gain entry into CGD neutrophils, they are not exposed to hydrogen peroxide because the neutrophils do not produce it, and the hydrogen peroxide generated by microorganisms themselves is destroyed by their own catalase. When CGD neutrophils ingest streptococci, which lack catalase, the organisms generate enough hydrogen peroxide to result in a microbicidal effect. As indicated (middle), catalase-positive microbes such as E. coli can survive within the phagosome of the CGD neutrophil.
(Modified from Boxer LA: Quantitative abnormalities of granulocytes. In Beutler E, Lichtman MA, Coller BS, et al, editors: Williams hematology, ed 6, New York, 2001, McGraw-Hill, p 845.)
Clinical Manifestations
Although the clinical presentation is variable, several features suggest the diagnosis of CGD. Any patient with recurrent pneumonia, lymphadenitis, hepatic or other abscesses, osteomyelitis at multiple sites, a family history of recurrent infections, or any infection with an unusual catalase-positive organism requires evaluation. Residual NADPH oxidase may attenuate CGD.
The onset of clinical signs and symptoms may occur from early infancy to young adulthood. The attack rate and severity of infections are exceedingly variable. The most common pathogen is S. aureus, although any catalase-positive microorganism may be involved. Other organisms frequently causing infections include Serratia marcescens, Burkholderia cepacia, Aspergillus, Candida albicans, Nocardia, and Salmonella. There may also be increased susceptibility to mycobacterium including the BCG vaccine. Pneumonia, lymphadenitis, osteomyelitis, and skin infections are the most common illnesses encountered. Bacteremia or fungemia occur but are much less common than focal infections. Patients may suffer from the sequelae of chronic infection, including anemia of chronic disease, poor growth, lymphadenopathy, hepatosplenomegaly, chronic purulent dermatitis, restrictive lung disease, gingivitis, hydronephrosis, and pyloric outlet narrowing. Perirectal abscesses and recurrent skin infections, including folliculitis, cutaneous granulomas, and discoid lupus erythematosus also suggest the possibility of CGD. Granuloma formation and inflammatory processes are a hallmark of CGD and may be the presenting symptoms that prompt testing for CGD if they cause pyloric outlet obstruction, bladder outlet or ureter obstruction, or rectal fistulae and granulomatous colitis simulating Crohn disease.
Laboratory Findings
The diagnosis is most often made by performing flow cytometry using dihydrorhodamine 123 (DHR) to measure oxidant production through its increased fluorescence when oxidized by H2O2. The nitroblue tetrazolium (NBT) dye test is frequently cited in the literature but is now only rarely used clinically.
A few individuals have been described with apparent CGD due to severe glucose-6-phosphate dehydrogenase (G6PD) deficiency, leading to insufficient NADPH substrate for the phagocyte oxidase. The erythrocytes of these patients also lack the enzyme, leading to chronic hemolysis.
Treatment
HSCT is the only known cure for CGD. For supportive care, patients with CGD should be given daily oral trimethoprim-sulfamethoxazole and an antifungal drug, such as itraconazole (see later), for prophylaxis of infections. Cultures must be obtained as soon as infection is suspected. Most abscesses require surgical drainage for therapeutic and diagnostic purposes. Prolonged use of antibiotics is often required. Aspergillus or Candida infection requires treatment with intravenous antifungal drugs. Granulocyte transfusions may be necessary if antibiotics are ineffective. If fever occurs without an obvious focus, it is advisable to consider the use of radiographs of the chest and skeleton as well as CT scans of the liver to determine if pneumonia, osteomyelitis, or liver abscesses are present. The cause of fever cannot always be established, and empirical treatment with broad-spectrum parenteral antibiotics is often required. The erythrocyte sedimentation rate (ESR) may be used to help determine the duration of antibiotic treatment.
Corticosteroids may also be useful for the treatment of children with antral and urethral obstruction or severe granulomatous colitis. Granulomas may be sensitive to low doses of prednisone (0.5 mg/kg/day); treatment should be tapered over several weeks. Inhibitors of tumor necrosis factor alpha pathways, such as infliximab, should be avoided if possible due to the very high risk of invasive fungal infection.
Interferon-γ (IFN-γ) 50 µg/m2 3 times/wk reduces the number of hospitalizations and serious infections. The mechanism of action of IFN-γ therapy in CGD is unknown. Itraconazole (200 mg/day for patients >50 kg and 100 mg/day for patients <50 kg and ≤5 yr of age) administered prophylactically reduces the frequency of fungal infections.
Genetic Counseling
Identifying a patient’s specific genetic subgroup is useful primarily for genetic counseling and prenatal diagnosis. In cases of suspected X-linked CGD, further analysis is not necessary if the fetus is initially demonstrated to be a 46,XX female. Fetal blood sampling and oxidase function analysis of fetal neutrophils can be used for prenatal diagnosis of CGD. DNA analysis of amniotic fluid cells or chorionic villus biopsy is an option for early prenatal diagnosis in families in which the specific mutation is known.
Prognosis
The overall mortality rate for CGD is about 2 patient deaths/yr/100 cases, with the highest mortality among young children. The development of effective infection prophylactic regimens, close surveillance for signs of infections, and aggressive surgical and medical interventions has improved the prognosis.
Beauté J, Obenga G, Le Mignot L, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease. Pediatr Infect Dis J. 2011;30(1):57-62.
Bender JM, Rand TH, Ampofo K, et al. Family clusters of variant X-linked chronic granulomatous disease. Pediatr Infect Dis J. 2009;28:529-533.
Dale DC, Boxer L, Liles W. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935.
Dinauer MC, Newburger PE. The phagocyte system and disorders of granulopoiesis and granulocyte function. In: Orkin SH, Ginsburg D, Nathan DG, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders/Elsevier; 2009:1109-1217.
Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am. 2007;28:277-291.
Hansson M, Olsson I, Naufeef W. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys. 2006;445:214.
Kuhns DB, Alvord G, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600-2610.
Lee PPW, Chan KW, Jiang L, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease. Pediatr Infect Dis J. 2008;27:224-230.
Malech HL, Hickstein DD. Genetics, biology and clinical management of myeloid cell primary immune deficiencies: chronic granulomatous disease and leukocyte adhesion deficiency. Curr Opin Hematol. 2007;14:29.
Nussbaum C, Moser M, Sperandio M. Leukocyte adhesion deficiency-III: when leukocytes cannot stop. Pediatr Res. 2010;67:339.
Qasim W, Cavazzana-Calvo M, Davies EG, et al. Allogeneic hematopoietic stem-cell transplantation for leukocyte adhesion deficiency. Pediatrics. 2009;123:836-840.
Reichenbach J, Van de Velde H, De Rycke M, et al. First successful bone marrow transplantation for X-linked chronic granulomatous disease by using preimplantation female gender typing and HLA matching. Pediatrics. 2008;122:e778-e782.
Seger RA. Modern management of chronic granulomatous disease. Brit J Haematol. 2008;140:255.
Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198-203.
Svensson L, Howarth K, McDowall A, et al. Leukocyte adhesion deficiency-III is caused by mutations on KINDLIN3 affecting integrin activation. Nat Med. 2009;15:306-312.