Chapter 125 Leukopenia
Marked developmental changes in normal values for the total white blood cell (WBC) count occur during childhood (Chapter 708). The mean WBC count at birth is high, followed by a rapid fall beginning at 12 hr until the end of the 1st wk. Thereafter, values are stable until 1 yr of age. A slow, steady decline in the WBC count continues throughout childhood until reaching the adult value during adolescence. Leukopenia in adolescents and adults is defined as a total WBC count <4,000/µL. Evaluation of patients with leukopenia, neutropenia, or lymphopenia begins with a thorough history, physical examination, family history, and screening laboratory tests (Table 125-1).
Table 125-1 DIAGNOSTIC APPROACH FOR PATIENTS WITH LEUKOPENIA
| EVALUATION | ASSOCIATED CLINICAL DIAGNOSES |
|---|---|
| INITIAL EVALUATION | |
| • History of acute or chronic leukopenia | |
| • General medical history | Congenital syndromes (Shwachman-Diamond, Wiskott-Aldrich, Fanconi anemia, dyskeratosis congenita, glycogen storage disease type Ib, disorders of vesicular transport) |
| • Physical examination: stomatitis, gingivitis, dental defects, congenital anomalies | |
| • Spleen size | Hypersplenism |
| • History of drug exposure | Drug-associated neutropenia |
| • Complete blood count with differential and reticulocyte counts | Neutropenia, aplastic anemia, autoimmune cytopenias |
| IF ANC <1,000/µL | |
| EVALUATION OF ACUTE ONSET NEUTROPENIA | |
| • Repeat blood counts in 3-4 weeks | Transient myelosuppression (e.g., viral) |
| • Serology and cultures for infectious agents | Active or chronic infection with viruses (e.g., EBV, CMV), bacteria, mycobacteria, rickettsia |
| • Discontinue drug(s) associated with neutropenia | Drug-associated neutropenia |
| • Test for antineutrophil antibodies | Autoimmune neutropenia |
| • Measure quantitative immunoglobulins (G, A, and M), lymphocyte subsets | Neutropenia associated with disorders of immune function |
| IF ANC <500/µL ON 3 SEPARATE TESTS | |
| • Bone marrow aspiration and biopsy, with cytogenetics | Severe congenital neutropenia, Shwachman-Diamond syndrome, myelokathexis; chronic benign or idiopathic neutropenia |
| • Serial CBCs (3/week for 6 weeks) | Cyclic neutropenia |
| • Exocrine pancreatic function | Shwachman-Diamond syndrome |
| • Skeletal radiographs | Shwachman-Diamond syndrome, cartilage-hair hypoplasia, Fanconi anemia |
| IF ABSOLUTE LYMPHOCYTE COUNT <1000/µL | |
| • Repeat blood counts in 3-4 weeks | Transient leukopenia (e.g., viral) |
| IF ALC <1000/µL ON 3 SEPARATE TESTS | |
| • HIV-1 antibody test | HIV-1 infection, AIDS |
| • Quantitative immunoglobulins (G, A, and M), lymphocyte subsets | Congenital or acquired disorders of immune function |
| IF THERE IS PANCYTOPENIA | |
| • Bone marrow aspiration and biopsy | Bone marrow replacement by malignancy, fibrosis, granulomata, storage cells |
| • Bone marrow cytogenetics | Myelodysplasia, leukemia |
| • Vitamin B12 and folate levels | Vitamin deficiencies |
ANC, absolute neutrophil count; CBC, complete blood count; CMV, cytomegalovirus; EBV, Epstein-Barr virus.
Neutropenia
Neutropenia is an absolute neutrophil count (ANC), calculated as the WBC count × % of neutrophils and bands, more than 2 standard deviations below the normal mean. Normal neutrophil counts must be stratified for age and race. For whites over the age of 12 mo, the lower limit of normal for the neutrophil count is 1,500/µL, and for blacks over 12 mo old, the lower limit of normal is 1,200/µL. The relatively lower limit in blacks probably reflects a relative decrease in neutrophils in the storage compartment of the bone marrow. Neutropenia may be characterized as mild neutropenia, with an ANC of 1,000-1,500/µL; moderate neutropenia, with an ANC of 500-1,000/µL; or severe neutropenia, with an ANC <500/µL. This stratification aids in predicting the risk of pyogenic infection; only patients with severe neutropenia have significantly increased susceptibility to life-threatening infections.
Etiology
Acute neutropenia evolving over a few days often occurs when neutrophil use is rapid and production is compromised. Chronic neutropenia lasting months or years can arise from reduced production, increased destruction, or excessive splenic sequestration of neutrophils. Neutropenia may be classified by whether it arises secondary to factors extrinsic to marrow myeloid cells (Table 125-2), which is common; as an acquired disorder of myeloid progenitor cells (Table 125-3), which is less common; or, more rarely, as an intrinsic defect affecting proliferation and maturation of myeloid progenitor cells (Table 125-4).
Table 125-2 CAUSES OF NEUTROPENIA EXTRINSIC TO MARROW MYELOID CELLS
| CAUSE | ETIOLOGIC FACTORS/AGENTS | ASSOCIATED FINDINGS |
|---|---|---|
| Infection | Viruses, bacteria, protozoa, rickettsia, fungi | Redistribution from circulating to marginating pools, impaired production, accelerated destruction |
| Drug-induced | Phenothiazines, sulfonamides, anticonvulsants, penicillins, aminopyrine | Hypersensitivity reaction (fever, lymphadenopathy, rash, hepatitis, nephritis, pneumonitis, aplastic anemia), antineutrophil antibodies |
| Immune neutropenia | Alloimmune, autoimmune | Variable arrest from metamyelocyte to segmented neutrophils in bone marrow |
| Reticuloendothelial sequestration | Hypersplenism | Anemia, thrombocytopenia, neutropenia |
| Bone marrow replacement | Malignancy (lymphoma, metastatic solid tumor, etc.) | Presence of immature myeloid and erythroid precursors in peripheral blood |
| Cancer chemotherapy or radiation therapy to bone marrow | Suppression of myeloid cell production | Bone marrow hypoplasia, anemia, thrombocytosis |
Table 125-3 ACQUIRED DISORDERS OF MYELOID CELLS
| CAUSE | ETIOLOGIC FACTORS/AGENTS | ASSOCIATED FINDINGS |
|---|---|---|
| Aplastic anemia | Stem cell destruction and depletion | Pancytopenia |
| Vitamin B12 or folate deficiency | Malnutrition; congenital deficiency of B12 absorption, transport, and storage; vitamin avoidance | Megaloblastic anemia, hypersegmented neutrophils |
| Acute leukemia, chronic myelogenous leukemia | Bone marrow replacement with malignant cells | Pancytopenia, leukocytosis |
| Myelodysplasia | Dysplastic maturation of stem cells | Bone marrow hypoplasia with megaloblastoid red cell precursors, thrombocytopenia |
| Prematurity with birthweight <2 kg | Impaired regulation of myeloid proliferation and reduced size of postmitotic pool | Maternal preeclampsia |
| Chronic idiopathic neutropenia | Impaired myeloid proliferation and/or maturation | None |
| Paroxysmal nocturnal hemoglobinuria | Acquired stem cell defect secondary to mutation of PIG-A gene | Pancytopenia, thrombosis |
Table 125-4 INTRINSIC DISORDERS OF MYELOID PRECURSOR CELLS
| SYNDROME | INHERITANCE (GENE) | CLINICAL FEATURES (INCLUDING STATIC NEUTROPENIA UNLESS OTHERWISE NOTED) |
|---|---|---|
| PRIMARY DISORDERS OF MYELOPOIESIS | ||
| Cyclic neutropenia | AD (ELA2) | Periodic oscillation (21-day cycles) in ANC |
| Severe congenital neutropenia | AD (ELA2, GFI1, others) | Risk of MDS and AML |
| X-linked (WAS) | Neutropenic variant of Wiskott-Aldrich syndrome | |
| Kostmann syndrome | AR (HAX1) | Neurological abnormalities, risk of MDS and AML |
| DISORDERS OF RIBOSOMAL FUNCTION | ||
| Shwachman-Diamond syndrome | AR (SBDS) | Pancreatic insufficiency, variable neutropenia, other cytopenias, metaphysical dysostosis |
| Dyskeratosis congenita | Telomerase defects: XL (DKC1), AD (TERC), AR (TERT) | Nail dystrophy, leukoplakia, reticulated hyperpigmentation of the skin; 30-60% develop bone marrow failure |
| DISORDERS OF GRANULE SORTING | ||
| Chédiak-Higashi syndrome | AR (LYST) | Partial albinism, giant granules in myeloid cells, platelet storage pool defect, impaired natural killer cell function, hemophagocytic lymphohistiocytosis |
| Griscelli syndrome, type II | AR (RAB27a) | Partial albinism, impaired natural killer cell function, hemophagocytic lymphohistiocytosis |
| Cohen syndrome | AR (COH1) | Partial albinism |
| Hermansky-Pudlak syndrome, type II | AR (AP3P1) | Cyclic neutropenia, partial albinism |
| p14 deficiency | probable AR (MAPBPIP) | Partial albinism, decreased B and T cells |
| DISORDERS OF METABOLISM | ||
| Glycogen storage disease, type 1b | AR (G6PT1) | Hepatic enlargement, growth retardation, impaired neutrophil motility |
| G6Pase, catalytic subunit 3, deficiency | AR (G6PC3) | Structural heart defects, urogenital abnormalities, venous angiectasia |
| Barth syndrome | XL (TAZ1) | Episodic neutropenia, dilated cardiomyopathy, methylglutaconic aciduria |
| Pearson’s syndrome | Mitochondrial (DNA deletions) | Episodic neutropenia, pancytopenia; defects in exocrine pancreas, liver, and kidneys |
| NEUTROPENIA IN DISORDERS OF IMMUNE FUNCTION | ||
| Common variable immunodeficiency | Familial, sporadic (TNFRSF13B) | Hypogammaglobulinemia, other immune system defects |
| IgA deficiency | Unknown (Unknown or TNFRSF13B) | Decreased IgA |
| Severe combined immunodeficiency | AR, XL (multiple loci) | Absent humoral and cellular immune function |
| Hyper-IgM syndrome | XL (HIGM1) | Absent IgG, elevated IgM, autoimmune cytopenia |
| WHIM syndrome | AD (CXCR4) | Warts, hypogammaglobulinemia, infections, myelokathexis |
| Cartilage-hair hyperplasia | AR (RMKP) | Lymphopenia, short-limbed dwarfism, metaphysical chondrodysplasia, fine sparse hair |
| Schimke immuno-osseous dysplasia | probable AR (SMARCAL1) | Lymphopenia, pancytopenia, spondyloepiphyseal dysplasia, growth retardation, renal failure |
AD, autosomal dominant; AML, acute myelogenous leukemia; ANC, absolute neutrophil count; AR, autosomal recessive; MDS, myelodysplasia; XL, X-linked.
Infectious Causes
Transient neutropenia often accompanies or follows viral infections (Table 125-5). Neutropenia associated with common childhood viral disease occurs during the 1st 1-2 days of illness and may persist for 3-8 days. It usually corresponds to a period of acute viremia and is related to virus-induced redistribution of neutrophils from the circulating to the marginating pool. Neutrophil sequestration possibly occurs after virus-induced tissue damage. Moderate to severe neutropenia may also be associated with a wide variety of other infectious causes. Bacterial sepsis is a particularly serious cause of neutropenia and neonates are particularly vulnerable to developing neutropenia because of a deficient pool of reserve neutrophils in the bone marrow.
Table 125-5 INFECTIONS THAT ARE ASSOCIATED WITH NEUTROPENIA
VIRAL
Respiratory syncytial virus
Dengue fever
Colorado tick fever
Mumps
Viral hepatitis
Infectious mononucleosis (Epstein-Barr virus)
Influenza
Measles
Rubella
Varicella
Cytomegalovirus
Human immunodeficiency virus
Sandfly fever
BACTERIAL
Pertussis
Typhoid fever
Paratyphoid fever
Tuberculosis (disseminated)
Brucellosis
Tularemia
Gram-negative sepsis
Psittacosis
FUNGAL
Histoplasmosis (disseminated)
PROTOZOA
Malaria
Leishmaniasis (kala-azar)
RICKETTISIAL
Rocky Mountain spotted fever
Typhus fever
Rickettsialpox
From Boxer LA, Blackwood RA: Leukocyte disorders: quantitative and qualitative disorders of the neutrophil, part 1, Pediatr Rev 17:19–28, 1996.
Chronic neutropenia often accompanies infection with Epstein-Barr virus, cytomegalovirus, or HIV. The neutropenia associated with AIDS probably arises from a combination of impaired neutrophil production, the accelerated destruction of neutrophils mediated by antineutrophil antibodies, and sometimes effects of antiretroviral or other drugs.
Drug-Induced Neutropenia
Drugs constitute one of the most common causes of neutropenia (Table 125-6). The incidence of drug-induced neutropenia increases dramatically with age; only 10% of cases occur among children and young adults, and the majority of cases among adults over age 65 yr. Drug-induced neutropenia has several underlying mechanisms (immune-mediated, toxic, idiosyncratic, hypersensitivity reactions) that are distinct from the severe neutropenia that predictably occurs after administration of cytoreductive cancer drugs or radiotherapy.
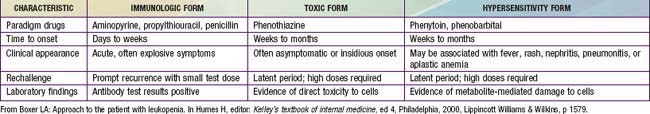
Drug-induced neutropenia due to immune mechanisms usually develops abruptly, is accompanied by fever, and lasts for about 1 wk after discontinuation of the drug. The process likely arises from effects of drugs such as propylthiouracil or penicillin that act as haptens to stimulate antibody formation, or drugs such as quinine that induce immune complex formation. Other drugs, including the antipsychotic drugs such as the phenothiazines, can cause neutropenia when given in toxic amounts, but some individuals, such as those with pre-existing neutropenia, may be susceptible to levels at the high end of the usual therapeutic range. Late-onset neutropenia can occur after rituximab therapy. Idiosyncratic reactions, for example to chloramphenicol, are unpredictable with regard to dose or duration of use. Hypersensitivity reactions are rare and may involve arene oxide metabolites of aromatic anticonvulsants. Fever, rash, lymphadenopathy, hepatitis, nephritis, pneumonitis, and aplastic anemia are often associated with hypersensitivity-induced neutropenia. Acute hypersensitivity reactions such as those caused by phenytoin or phenobarbital may last for only a few days if the offending drug is discontinued. Chronic hypersensitivity may last for months to years. Drug-induced neutropenia may occasionally be asymptomatic despite severely reduced numbers of neutrophils and is noted only because of regular monitoring of WBC counts during drug therapy.
Neutropenia commonly and predictably follows the use of anticancer drugs or radiation therapy, especially radiation therapy directed at the pelvis or vertebrae, secondary to cytotoxic effects on rapidly replicating myeloid precursors. A decline in the WBC count typically occurs 7-10 days after administration of the anticancer drug and may persist for 1-2 wk. The neutropenia accompanying malignancy or following cancer chemotherapy is frequently associated with compromised cellular immunity, thereby predisposing patients to a much greater risk of infection (Chapter 171) than found in disorders associated with isolated neutropenia.
Bone Marrow Replacement
Various acquired bone marrow disorders lead to neutropenia, usually accompanied by anemia and thrombocytopenia. Hematologic malignancies, including leukemia and lymphoma, and metastatic solid tumors (e.g., neuroblastoma, rhabdomyosarcoma, and Ewing sarcoma) suppress myelopoiesis by infiltrating the bone marrow. Neutropenia may also accompany myelodysplastic disorders or preleukemic syndromes, which are characterized by peripheral cytopenias and macrocytic blood cells associated with impaired survival of myeloid precursors. Aplastic anemia arising from damage (generally immune-mediated) and depletion of stem cells also causes neutropenia in the setting of pancytopenia.
Reticuloendothelial Sequestration
Splenic enlargement resulting from intrinsic splenic disease (e.g., storage disease), portal hypertension, or systemic causes of splenic hyperplasia (e.g., inflammation, neoplasia) can lead to neutropenia. The neutropenia is usually mild to moderate and accompanied by corresponding degrees of thrombocytopenia and anemia, and may be corrected by successfully treating the underlying disease. The reduced neutrophil survival corresponds to the size of the spleen, and the extent of the neutropenia is inversely proportional to bone marrow compensatory mechanisms. In selected cases, splenectomy may be necessary to restore the neutrophil count to normal but results in increased risk of infections by encapsulated bacterial organisms.
Immune Neutropenia
Immune neutropenias are usually associated with the presence of circulating antineutrophil antibodies, which may mediate neutrophil destruction by complement-mediated lysis or splenic phagocytosis of opsonized neutrophils, or by accelerated apoptosis of mature neutrophils or myeloid precursors.
Alloimmune Neonatal Neutropenia
This form of neonatal neutropenia occurs after transplacental transfer of maternal alloantibodies directed against antigens on the infant’s neutrophils, analogous to Rh hemolytic disease. Prenatal sensitization induces maternal IgG antibodies to neutrophil antigens on fetal cells. The antibodies are usually complement activating and are frequently directed to neutrophil-specific antigens. Symptomatic infants may present with delayed separation of the umbilical cord, mild skin infections, fever, and pneumonia within the 1st 2 wk of life. The neutropenia is often severe and associated with fever and infections due to the usual microbes that cause neonatal disease. By 7 wk of age, the neutrophil count usually returns to normal, reflecting the decay of maternal antibodies in the infant’s circulation. Treatment consists of supportive care and appropriate antibiotics for clinical infections.
Neonatal Passive Autoimmune Neutropenia
Mothers with autoimmune disease may give birth to infants who develop transient neutropenia. The duration of the neutropenia depends on the time required for the infant to clear the maternally transferred circulating IgG antibody. It persists in most cases for a few weeks to a few months. Neonates almost always remain asymptomatic.
Autoimmune Neutropenia
Autoimmune neutropenia is analogous to autoimmune hemolytic anemia and thrombocytopenia. Antibodies causing neutropenia have been detected in patients who have no other signs of autoimmune disease, in patients who have additional antibodies against red blood cells and/or platelets, and in patients who have a connective tissue disorder. Autoimmune neutropenia is distinguished from other forms of neutropenia only by the demonstration of antineutrophil antibodies and the appearance of myeloid hyperplasia on bone marrow examination. Antineutrophil antibody assays are quite prone to false-negative and false-positive results. Autoimmune neutropenia may occur in children with congenital or acquired immune deficiencies, including common variable immunodeficiency, dysgammaglobulinemias, or in the settings of systemic lupus erythematosus or autoimmune lymphoproliferative syndrome.
Treatment with recombinant human granulocyte colony–stimulating factor (rhG-CSF; filgrastim [Neupogen]) is generally effective at raising the ANC and preventing infection. Often, very low doses (<1-2 µg/kg/day) are effective, and administration of “standard” doses can lead to severe bone pain due to marrow expansion.
Autoimmune Neutropenia of Infancy (ANI)
This benign condition is diagnosed more frequently as techniques for detection of antineutrophil antibodies have become more available. The exact incidence of ANI remains unknown, but because of its benign nature, the disorder may be more common than currently appreciated. In 1 study, ANI occurred with an annual incidence of approximately 1/100,000 among children between infancy and 10 yr. All patients recognized as having ANI have severe neutropenia on presentation, with an ANC usually <500/µL, but the total WBC count is generally within normal limits. Monocytosis or eosinophilia may occur but does not affect the low rate of infection. The age at diagnosis is usually between 5 and 15 mo, with a female to male ratio of 6:4. None of the affected children has evidence of other autoimmune diseases. Children with ANI present with minor infections such as otitis media, gingivitis, respiratory tract infections, gastroenteritis, and cellulitis. The diagnosis often is considered only after the blood count reveals neutropenia. Occasionally, children may present with more severe infections, including pneumonia, sepsis, or abscesses. Longitudinal studies of infants with ANI demonstrate a median duration of disease of ≈7-24 mo. The diagnosis is established by the presence of antineutrophil antibodies in serum.
Treatment is not generally necessary, but rhG-CSF may be useful in providing temporary remission in infants with severe infections or requiring surgical intervention.
Ineffective Myelopoiesis
Extrinsic Disorders
Ineffective myelopoiesis may result from congenital or acquired vitamin B12 or folic acid deficiency. Megaloblastic pancytopenia also can result from extended use of antibiotics such as trimethoprim-sulfamethoxazole, which inhibit folic acid metabolism, and from the use of phenytoin, which may impair folate absorption in the small intestine. Neutropenia also occurs with starvation and marasmus in infants, with anorexia nervosa, and occasionally among patients receiving prolonged parenteral feedings without vitamin supplementation.
Intrinsic Disorders of Myeloid Precursors
The isolated disorders of proliferation and maturation of myeloid precursor cells are rare. Table 125-4 presents a classification based on genetics and molecular mechanisms; selected disorders are discussed in the next sections.
Primary Disorders of Granulocytopoiesis
Cyclic neutropenia, an autosomal dominant disorder, is characterized by regular, periodic oscillation in the number of peripheral neutrophils from normal to neutropenic values with a mean oscillatory period of 21 ± 3 days. During the neutropenic phase, most patients suffer from fever, stomatitis or pharyngitis, and occasionally lymph node enlargement. Serious infections may occur at the ANC nadirs, including pneumonia, periodontitis, and recurrent ulcerations of the oral, vaginal, and rectal mucosa, leading to life-threatening clostridial sepsis. Cyclic neutropenia arises from a regulatory abnormality involving early hematopoietic precursor cells and is associated with mutations in the neutrophil elastase gene, ELA2, that lead to accelerated apoptosis due to abnormal protein folding. Many patients experience abatement of symptoms with age. The cycles tend to become less noticeable in older patients, and the hematologic picture often begins to resemble that of chronic neutropenia. Treatment with rhG-CSF elevates the neutrophil counts and improves outcome.
Severe congenital neutropenia is characterized by an arrest in myeloid maturation at the promyelocyte stage in the bone marrow, resulting in consistent ANCs <200/µL. This disorder occurs sporadically or with autosomal dominant or recessive inheritance. The dominant form is caused most often by mutations in ELA2, while the recessive form (Kostmann disease) arises from mutations in HAX1, which protects cells against apoptosis. Patients typically show monocytosis and eosinophilia and suffer from recurrent, severe pyogenic infections, especially of the skin, mouth, and rectum. Anemia of chronic inflammation is often present. Approximately 20% of patients develop acute myelogenous leukemia or myelodysplasia associated with monosomy 7 and sometimes preceded by acquisition of mutations in the gene encoding the G-CSF receptor. Before the advent of treatment with rhG-CSF, most of these patients died of fatal infections before reaching adolescence.
Disorders of Ribosomal Function
Shwachman-Diamond syndrome is an autosomal recessive disorder characterized by pancreatic insufficiency and neutropenia. Shwachman-Diamond syndrome is caused by pro-apoptotic mutations of the SBDS gene, which encodes a protein that may play a role in ribosome biogenesis or RNA processing. The initial symptoms are usually diarrhea and failure to thrive because of malabsorption, which develops in almost all infants by 4 mo of age. Some patients have respiratory problems with pneumonia and frequent otitis media, as well as eczema. Virtually all patients with Shwachman-Diamond syndrome have neutropenia, with the ANC periodically <1,000/µL associated with hypoplastic myelopoiesis. Some children have been reported to have a chemotactic defect that may contribute to the increased susceptibility to pyogenic infection. The illness may progress to bone marrow hypoplasia with moderate thrombocytopenia and anemia. Myelodysplasia and acute myelogenous leukemia associated with monosomy 7 have also been reported in this syndrome. The neutropenia responds to treatment with rhG-CSF.
Granule Sorting Disorders
This constellation of very rare autosomal recessive disorders combine neutropenia with partial oculocutaneous albinism, immunodeficiencies, and other features, all derived from defects in formation or trafficking of lysosome-related organelles (see Table 125-4). Treatment usually includes hematopoietic stem cell transplantation.
Chédiak-Higashi syndrome, best known for the characteristic giant cytoplasmic granules in neutrophils, monocytes, and lymphocytes, is a disorder of subcellular vesicular dysfunction due to mutations in the LYST gene, with resultant fusion of cytoplasmic granules in all granule-bearing cells. Patients have increased susceptibility to infections, mild bleeding diathesis, progressive peripheral neuropathy, and predisposition to life-threatening hemophagocytic syndrome. The only curative treatment remains allogeneic stem cell transplantation.
Griscelli syndrome type II also features neutropenia, albinism, and a high risk of hemophagocytic syndrome, but peripheral blood granulocytes do not show giant granules. The autosomal recessive disorder is caused by mutations in RAB27a, which encodes a small GTPase that regulates granule secretory pathways.
Disorders of Metabolism
Recurrent infections with neutropenia are a distinctive feature of glycogen storage disease (GSD) type Ib. Both classic von Gierke glycogen storage disease (GSDIa) and GSDIb cause massive enlargement of liver and severe growth retardation (Chapter 81.1). In contrast to GSDIa, glucose-6-phosphatase (G6Pase) enzyme activity is normal, but mutations in the G6P transporter 1, G6PT1, inhibit glucose transport in GSDIb, resulting in both defective neutrophil motility and increased apoptosis associated with neutropenia and recurrent bacterial infections. Treatment with rhG-CSF can correct the neutropenia.
Metabolically related but phenotypically distinct mutations in G6PC3, encoding glucose-6-phosphatase, catalytic subunit 3, result in neutropenia with heart defects, urogenital abnormalities, and venous angiectasia but not symptomatic disruption of glucose transport.
Neutropenia in Disorders of Immune Function
Congenital immunologic disorders that have severe neutropenia as a clinical feature include common variable immunodeficiency, the severe combined immunodeficiencies, hyper-IgM syndrome, WHIM syndrome, and a number of even rarer immunodeficiency syndromes (see Table 125-4).
Unclassified Disorders
Acquired idiopathic chronic neutropenia is characterized by onset of neutropenia after 2 yr of age. Patients with an ANC persistently <500/µL are afflicted with recurrent pyogenic infections involving the skin, mucous membranes, lungs, and lymph nodes. Bone marrow examination reveals variable patterns of myeloid formation with arrest generally occurring between the myelocyte and band forms (see Table 125-3). Often there is overlap with the diagnoses of chronic benign or autoimmune neutropenias.
Chronic benign neutropenia of childhood represents a common group of disorders characterized by mild to moderate neutropenia that does not lead to an increased risk of pyogenic infections. Spontaneous remissions are often reported, although these may represent misdiagnosis of autoimmune neutropenia of infancy, in which remissions occur commonly during childhood. Chronic benign neutropenia may be inherited in either a dominant or recessive form. An autosomal recessive form of benign neutropenia is encountered in Yemenite Jews. Because of the relatively low risk of serious infection, patients should be not subjected to the potentially toxic therapy.
Clinical Manifestations of Neutropenia
Individuals with neutrophil counts <500/µL are at substantial risk for developing infections, primarily from their endogenous flora as well as from nosocomial organisms. Some patients with isolated chronic neutropenia with an ANC <200/µL may not experience many serious infections, probably because the remainder of the immune system remains intact. In contrast, children whose neutropenia is secondary to acquired disorders of production such as with cytotoxic therapy, immunosuppressive drugs, or radiation therapy are likely to develop serious bacterial infections because many arms of the immune system are markedly compromised.
Neutropenia associated with leukopenia, that is, additional monocytopenia or lymphocytopenia, is more highly associated with serious infection than neutropenia alone. The integrity of skin and mucous membranes, the vascular supply to tissues, and nutritional status also influence the risk of infection.
The most common clinical presentation of profound neutropenia includes fever >38°C, aphthous stomatitis and gingivitis, cellulitis, furunculosis, perirectal inflammation, colitis, sinusitis, and otitis media are also frequent accompaniments of profound neutropenia in children. Other clinical manifestations of profound neutropenia include hepatic abscesses, recurrent pneumonias, and septicemia. Isolated neutropenia does not heighten a patient’s susceptibility to parasitic or viral infections or to bacterial meningitis.
The most common pathogens causing infections in neutropenic patients are Staphylococcus aureus and gram-negative bacteria. The usual signs and symptoms of local infection and inflammation such as exudate, fluctuance, and regional lymphadenopathy are generally diminished in the absence of neutrophils because of the inability to form pus. Patients with complete agranulocytosis still experience fever and feel pain at sites of inflammation.
Laboratory Findings
Isolated absolute neutropenia has a limited number of causes (see Tables 125-1 through 125-4). The duration and severity of the neutropenia greatly influence the extent of laboratory evaluation. Patients with chronic neutropenia since infancy and a history of recurrent fevers and chronic gingivitis should have WBC counts and differential counts determined 3 times weekly for 6 wk to evaluate the periodicity suggestive of cyclic neutropenia. Bone marrow aspiration and biopsy should be performed on selected patients to assess cellularity. Additional marrow studies such as cytogenetic analysis and special stains for detecting leukemia and other malignant disorders should be obtained for patients with suspected intrinsic defects in the myeloid progenitors and for patients with suspected malignancy. Selection of further laboratory tests is determined by the duration and severity of the neutropenia and the associated findings on physical examination (see Table 125-1).
Treatment
The management of acquired transient neutropenia associated with malignancies, myelosuppressive chemotherapy, or immunosuppressive chemotherapy differs from that of congenital or chronic forms of neutropenia. In the former situation, infections sometimes are heralded only by fever, and sepsis is a major cause of death. Early recognition and treatment of infections may be lifesaving (Chapter 171).
Therapy of severe chronic neutropenia is dictated by the clinical manifestations. Patients with benign neutropenia and no evidence of repeated bacterial infections or chronic gingivitis require no specific therapy. Superficial infections in children with mild to moderate neutropenia may be treated with appropriate oral antibiotics. In patients who have invasive or life-threatening infections, broad-spectrum intravenous antibiotics should be started promptly.
Subcutaneously administered rhG-CSF can provide effective treatment of severe chronic neutropenia including severe congenital neutropenia, chronic symptomatic idiopathic neutropenia, and cyclic neutropenia. Doses ranging from 2 to over 100 µg/kg/day lead to dramatic increases in neutrophil counts, resulting in marked attenuation of infection and inflammation. rhG-CSF, often at very low doses, may also benefit patients who have immune or drug-induced neutropenias. The long-term effects of rhG-CSF therapy are unknown but include a propensity for the development of moderate splenomegaly, thrombocytopenia, and, occasionally, vasculitis. Autoimmune neutropenia may be responsive to intermittent corticosteroids, especially if it is part of an underlying disease process such as systemic lupus erythematosus.
Patients with severe congenital neutropenia or Shwachman-Diamond syndrome who develop myelodysplasia or acute myelogenous leukemia respond only to allogeneic stem cell transplantation. Chemotherapy is ineffective. Hematopoietic stem cell transplantation is also the treatment of choice for aplastic anemia or hemophagocytic lymphohistiocytosis complicating syndromes discussed earlier.
Lymphopenia
Lymphopenia by itself usually causes no symptoms and is often detected in the evaluation of other illnesses, particularly recurrent viral, fungal, and parasitic infections. Lymphocyte subpopulations can be measured by multiparameter flow cytometry, which uses the pattern of antigen expression to classify and characterize these cells.
Inherited Causes of Lymphocytopenia
Inherited immunodeficiency disorders may have a quantitative or qualitative stem cell abnormality resulting in ineffective lymphocytopoiesis (Table 125-7). Other disorders such as Wiskott-Aldrich syndrome may have associated lymphocytopenia arising from accelerated destruction of T cells. A similar mechanism is present in patients with adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency. Lymphocyte counts may also be decreased in some forms of inherited bone marrow failure, such as reticular dysgenesis, severe congenital neutropenia secondary to GFI1 mutation, or dyskeratosis congenita.
Table 125-7 CAUSES OF LYMPHOCYTOPENIA
ACQUIRED CAUSES
Infectious Diseases
AIDS
Viral hepatitis
Influenza
Tuberculosis
Typhoid fever
Sepsis
Iatrogenic
Immunosuppressive therapy
Corticosteroids
High-dose PUVA therapy
Cytotoxic chemotherapy
Radiation
Thoracic duct drainage
Systemic and Other Diseases
Systemic lupus erythematosus
Myasthenia gravis
Hodgkin disease
Protein-losing enteropathy
Renal failure
Sarcoidosis
Thermal injury
Aplastic anemia
Dietary Deficiency
Dietary deficiency associated with ethanol abuse
INHERITED CAUSES
Aplasia of lymphopoietic stem cells
Severe combined immunodeficiency
Ataxia-telangiectasia
Wiskott-Aldrich syndrome
Immunodeficiency with thymoma
Cartilage-hair hypoplasia
Idiopathic CD4 T lymphocytopenia
ADA, adenosine deaminase; IL-2, interleukin 2; PNP, purine nucleoside phosphorylase; PUVA, psoralen and ultraviolet A irradiation.
From Boxer LA: Approach to the patient with leukopenia. In Humes HD, editor: Kelley’s textbook of internal medicine, ed 4, Philadelphia, 2000, Lippincott Williams & Wilkins, p 1580.
Acquired Lymphocytopenia
AIDS is the most common infectious disease associated with lymphocytopenia, which results from destruction of CD4 T cells infected with HIV-1 or HIV-2. Other viral and bacterial diseases may be associated with lymphocytopenia. In some instances of acute viremia with other viral infections, lymphocytes may undergo accelerated destruction from intracellular viral replication, become trapped in the spleen or nodes, or migrate to the respiratory tract.
Systemic autoimmune diseases such as systemic lupus erythematosus are associated with lymphocytopenia. Other conditions such as protein-losing enteropathy and aberrant or surgical drainage of the thoracic duct are associated with lymphocyte depletion. Iatrogenic lymphocytopenia may be caused by cytotoxic chemotherapy, radiation therapy, and administration of antilymphocyte globulin. Long-term treatment of psoriasis with psoralen and ultraviolet irradiation may also destroy T lymphocytes. Corticosteroids can cause lymphopenia through increased cell destruction.
Andersohn F, Konzen C, Garbe E. Systematic review: agranulocytosis induced by nonchemotherapy drugs. Ann Intern Med. 2007;146:657-665.
Berliner N. Lessons from congenital neutropenia: 50 years of progress in understanding myelopoiesis. Blood. 2008;111:5427-5432.
Boxer LA, Newburger PE. A molecular classification of congenital neutropenia syndromes. Pediatr Blood Cancer. 2007;49:609-614.
Bruin M, Dassen A, Pajkrt D, et al. Primary autoimmune neutropenia in children: a study of neutrophil antibodies and clinical course. Vox Sang. 2005;88:52-59.
Donini M, Fontana S, Savoldi S, et al. G-CSF treatment of severe congenital neutropenia reverses neutropenia but does not correct the underlying functional deficiency of the neutrophil in defending against microorganisms. Blood. 2007;109:4716-4723.
Hsieh MM, Everhart JE, Byrd-Holt DD, et al. Prevalence of neutropenia in the U.S. population: age, sex, smoking status, and ethnic differences. Ann Intern Med. 2007;146:486-492.
Klein C, Grudzien M, Appaswamy G, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007;39:86-92.
Latger-Cannard V, Bensoussan D, Bordigoni P. The WHIM syndrome shows a peculiar dysgranulopoiesis: myelokathexis. Br J Haematol. 2006;132:669.
Melis D, Fulceri R, Parenti G, et al. Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature. Eur J Pediatr. 2005;164:501-508.
Pannicke U, Hönig M, Hess I, et al. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet. 2009;41:101-105.
Patel S, de la Fuente J, Atra A, et al. Where have all the neutrophils gone? Arch Dis Child Educ Pract Ed. 2009;94:74-77.
Rosenberg PS, Alter BP, Bolyard AA, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107:4628-4635.
Shimamura A. Shwachman-Diamond syndrome. Semin Hematol. 2006;43:178-188.
Townshend J, Clark J, Cant A, et al. Congenital neutropenia. Arch Dis Child Educ Pract Ed. 2008;93:14-18.