Chapter 425 Cyanotic Congenital Heart Disease
Lesions Associated with Increased Pulmonary Blood Flow
425.1 D-Transposition of the Great Arteries
Transposition of the great vessels, a common cyanotic congenital anomaly, accounts for ≈5% of all congenital heart disease. In this anomaly, the systemic veins return normally to the right atrium and the pulmonary veins return to the left atrium. The connections between the atria and ventricles are also normal (atrioventricular concordance). The aorta arises from the right ventricle and the pulmonary artery from the left ventricle (Fig. 425-1). In normally related great vessels, the aorta is posterior and to the right of the pulmonary artery; in d-transposition of the great arteries (d-TGA), the aorta is anterior and to the right of the pulmonary artery (the d indicates a dextropositioned aorta, transposition indicates that it arises from the anterior right ventricle). Desaturated blood returning from the body to the right side of the heart goes inappropriately out the aorta and back to the body again, whereas oxygenated pulmonary venous blood returning to the left side of the heart is returned directly to the lungs. Thus, the systemic and pulmonary circulations exist as two parallel circuits. Survival in the immediate newborn period is provided by the foramen ovale and the ductus arteriosus, which permit some mixture of oxygenated and deoxygenated blood. About 50% of patients with d-TGA also have a ventricular septal defect (VSD), which usually provides for better mixing. The clinical findings and hemodynamics vary in relation to the presence or absence of associated defects (e.g. VSD or pulmonary stenosis). d-TGA is more common in infants of diabetic mothers and in males (3 : 1). d-TGA, especially when accompanied by other cardiac defects such as pulmonic stenosis or right aortic arch, can be associated with deletion of chromosome 22q11 (DiGeorge syndrome [Chapter 418]). Before the modern era of corrective or palliative surgery, mortality was >90% in the 1st yr of life.
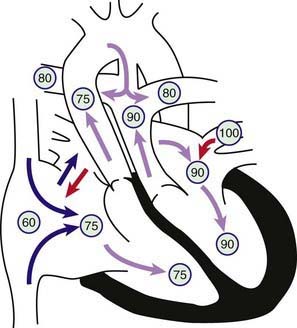
Figure 425-1 Physiology of d-transposition of the great arteries (d-TGA). Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. Desaturated blood enters the right atrium, flows through the tricuspid valve into the right ventricle, and is ejected into the transposed aorta with resultant severe aortic desaturation. Fully saturated pulmonary venous blood flows into the left atrium, across the mitral valve into the left ventricle, and across the transposed pulmonary artery into the lungs. Pulmonary arterial oxygen saturation is thus increased. This lesion would not be compatible with life were it not for the ability of blood to shunt via two fetal pathways: the patent foramen ovale (PFO) and patent ductus arteriosus (PDA). Blood may shunt left to right or bidirectionally at the PFO. Because systemic vascular resistance tends to be higher than pulmonary vascular resistance, blood tends to shunt across the PDA mostly from the aorta to the pulmonary artery. As pulmonary resistance drops in the 1st few weeks of life, pulmonary blood flow will gradually increase in patients with d-TGA.
425.2 D-Transposition of the Great Arteries with Intact Ventricular Septum
D-TGA with an intact ventricular septum is also referred to as simple TGA or isolated TGA. Before birth, oxygenation of the fetus is only slightly abnormal, but after birth, once the ductus arteriosus begins to close, the minimal mixing of systemic and pulmonary blood via the patent foramen ovale is usually insufficient and severe hypoxemia ensues, generally within the 1st few days of life.
Clinical Manifestations
Cyanosis and tachypnea are most often recognized within the 1st hrs or days of life. Untreated, the vast majority of these infants would not survive the neonatal period. Hypoxemia is usually moderate to severe, depending on the degree of atrial level shunting and whether the ductus is partially open or totally closed. This condition is a medical emergency, and only early diagnosis and appropriate intervention can avert the development of prolonged severe hypoxemia and acidosis, which lead to death. Physical findings, other than cyanosis, may be remarkably nonspecific. The precordial impulse may be normal, or a parasternal heave may be present. The 2nd heart sound is usually single and loud, although it may be split. Murmurs may be absent, or a soft systolic ejection murmur may be noted at the midleft sternal border.
Diagnosis
The electrocardiogram is usually normal, showing the expected neonatal right-sided dominant pattern. Roentgenograms of the chest may show mild cardiomegaly, a narrow mediastinum (the classic “egg-shaped heart”), and normal to increased pulmonary blood flow. In the early newborn period, the chest roentgenogram is generally normal. As pulmonary vascular resistance drops during the 1st several weeks of life, evidence of increased pulmonary blood flow becomes apparent. Arterial PO2 is low and does not rise appreciably after the patient breathes 100% oxygen (hyperoxia test), although this test may not be totally reliable. Echocardiography is diagnostic and confirms the transposed ventricular-arterial connections (Fig. 425-2). The size of the interatrial communication and the ductus arteriosus can be visualized and the degree of mixing assessed by pulsed and color Doppler examination. The presence of any associated lesion, such as left ventricular outflow tract obstruction or a VSD, can also be assessed. The origins of the coronary arteries can be imaged, although echocardiography is generally not as accurate as catheterization for this purpose. Cardiac catheterization may be performed in patients for whom noninvasive imaging is diagnostically inconclusive, where an unusual coronary artery anomaly is suspected, or in patients who require emergency balloon atrial septostomy. Catheterization will show right ventricular pressure to be systemic because this ventricle is supporting the systemic circulation. The blood in the left ventricle and pulmonary artery has a higher oxygen saturation than that in the aorta. Depending on the age at catheterization, left ventricular and pulmonary arterial pressure can vary from systemic level to <50% of systemic-level pressure. Right ventriculography demonstrates the anterior and rightward aorta originating from the right ventricle, as well as the intact ventricular septum. Left ventriculography shows that the pulmonary artery arises exclusively from the left ventricle.
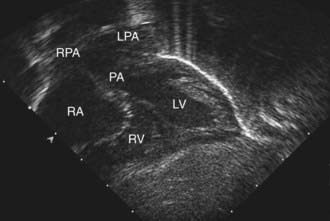
Figure 425-2 Subcostal four-chamber two-dimensional echocardiographic demonstration of d-transposition of the great arteries. The pulmonary artery (PA) can be seen arising directly from the left ventricle (LV). The immediate bifurcation of this great vessel into the branch pulmonary arteries differentiates it from the aorta, which branches more distally from the heart. LPA, left pulmonary artery; RA, right atrium; RPA, right pulmonary artery; RV, right ventricle.
Anomalous coronary arteries are noted in 10-15% of patients and defined by an aortic root injection or by selective coronary arteriography.
Treatment
When transposition is suspected, an infusion of prostaglandin E1 should be initiated immediately to maintain patency of the ductus arteriosus and improve oxygenation (dosage, 0.01-0.20 µg/kg/min). Because of the risk of apnea associated with prostaglandin infusion, an individual skilled in neonatal endotracheal intubation should be available. Hypothermia intensifies the metabolic acidosis resulting from hypoxemia, and thus the patient should be kept warm. Prompt correction of acidosis and hypoglycemia is essential.
Infants who remain severely hypoxic or acidotic despite prostaglandin infusion should undergo Rashkind balloon atrial septostomy (Fig. 425-3). A Rashkind atrial septostomy is also usually performed in all patients in whom any significant delay in surgery is necessary. If surgery is planned during the 1st 2 wk of life, and the patient is stable, catheterization and atrial septostomy may be avoided.
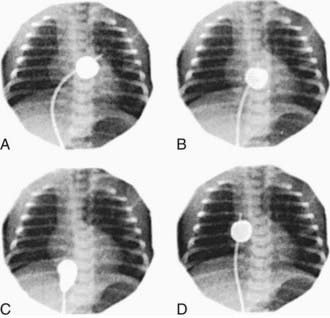
Figure 425-3 Rashkind balloon atrial septostomy. Four frames from a continuous cineangiogram show the creation of an atrial septal defect in a hypoxemic newborn infant with transposition of the great arteries and an intact ventricular septum. A, Balloon inflated in the left atrium. B, The catheter is jerked suddenly so that the balloon ruptures the foramen ovale. C, Balloon in the inferior vena cava. D, Catheter advanced to the right atrium to deflate the balloon. The time from A to C is <1 sec.
A successful Rashkind atrial septostomy should result in a rise in PaO2 to 35-50 mm Hg and elimination of any pressure gradient across the atrial septum. Some patients with TGA and VSD (Chapter 425.3) may require balloon atrial septostomy because of poor mixing, even though the VSD is large. Others may benefit from decompression of the left atrium to alleviate the symptoms of increased pulmonary blood flow and left-sided heart failure.
The arterial switch (Jatene) procedure is the surgical treatment of choice for neonates with d-TGA and an intact ventricular septum and is usually performed within the 1st 2 wk of life. The reason for this time frame is that as pulmonary vascular resistance declines after birth, pressure in the left ventricle (connected to the pulmonary vascular bed) also declines. This drop in pressure results in a decrease in left ventricular mass over the 1st few weeks of life. If the arterial switch operation is attempted after left ventricular pressure (and mass) has declined too far, the left ventricle will be unable to generate adequate pressure to pump blood to the high pressure systemic circulation. The arterial switch operation involves dividing the aorta and pulmonary artery just above the sinuses and re-anastomosing them in their correct anatomic positions. The coronary arteries are removed from the old aortic root along with a button of aortic wall and reimplanted in the old pulmonary root (the “neoaorta”). By using a button of great vessel tissue, the surgeon avoids having to suture directly onto the coronary artery (Fig. 425-4); this is the major innovation that has allowed the arterial switch to replace previous atrial switch operations for d-TGA. Rarely, a two-stage arterial switch procedure, with initial placement of a pulmonary artery band, may be used in patients presenting late who already have had a reduction in left ventricular muscle mass and pressure.
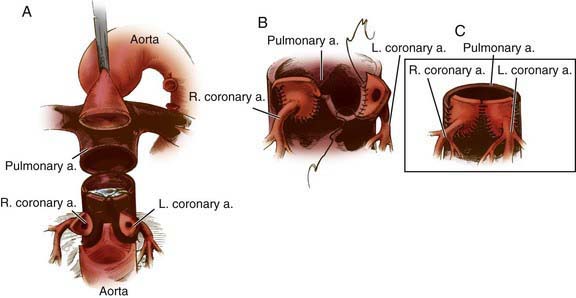
Figure 425-4 Method for translocating the coronary arteries in the arterial switch (Jantene) procedure. A, The aorta (anterior) and the pulmonary artery (posterior) have been transected to allow visualization of the left and right coronary arteries. The coronaries have been excised from their respective sinuses, including a large flap (button) of arterial wall. Equivalent segments of the wall of the pulmonary artery (which will become the neoaorta) are also removed. B, The aortocoronary buttons are sutured into the proximal portion of the neoaorta. With this technique all sutures are placed in the button of aortic wall rather than directly on the coronary arteries. C, Completed anastomosis of the left and right coronary arteries to the neoaorta.
(Adapted from Castañeda AR, Jonas RA, Mayer JE Jr, et al: Cardiac surgery of the neonate and infant, Philadelphia, 1994, WB Saunders.)
In experienced centers, the arterial switch procedure has a survival rate of 95% for uncomplicated d-TGA. It restores the normal physiologic relationships of systemic and pulmonary arterial blood flow and eliminates the long-term complications of the previously used atrial switch procedure.
Previous operations for d-TGA consisted of some form of atrial switch procedure (Mustard or Senning operation). These procedures produced excellent early survival (≈85-90%), but had significant long-term morbidities. Atrial switch procedures reverse blood flow at the atrial level by the creation of an inter-atrial baffle that directs systemic venous blood returning from the vena cavae to the left atrium, where it will enter the left ventricle and then, via the pulmonary artery, the lungs. The same baffle also permits oxygenated pulmonary venous blood to cross over to the right atrium, right ventricle, and aorta. Atrial switch procedures involve significant atrial surgery and have been associated with the late development of atrial conduction disturbances, sick sinus syndrome with bradyarrhythmia and tachyarrhythmia, atrial flutter, sudden death, superior or inferior vena cava syndrome, edema, ascites, and protein-losing enteropathy. The atrial switch procedure also leaves the right ventricle as the systemic pumping chamber and these “systemic” right ventricles often begin to fail in young adulthood. Atrial switch operations are currently reserved for patients whose anatomy is such that they are not candidates for the arterial switch procedure.
425.3 Transposition of the Great Arteries with Ventricular Septal Defect
If the VSD associated with d-TGA is small, the clinical manifestations, laboratory findings, and treatment are similar to those described previously for transposition with an intact ventricular septum. A harsh systolic murmur is audible at the lower left sternal border, resulting from flow through the defect. Many of these small defects eventually close spontaneously and may not be addressed at the time of surgery.
When the VSD is large and not restrictive to ventricular ejection, significant mixing of oxygenated and deoxygenated blood usually occurs and clinical manifestations of cardiac failure are seen. The degree of cyanosis may be subtle and sometimes may not be recognized until an oxygen saturation measurement is performed. The murmur is holosystolic and generally indistinguishable from that produced by a large VSD in patients with normally related great arteries. The heart is usually significantly enlarged.
Cardiomegaly, a narrow mediastinal waist, and increased pulmonary vascularity are demonstrated on the chest roentgenogram. The electrocardiogram shows prominent P waves and isolated right ventricular hypertrophy or biventricular hypertrophy. Occasionally, dominance of the left ventricle is present. Usually, the QRS axis is to the right, but it can be normal or even to the left. The diagnosis is confirmed by echocardiography, and the extent of pulmonary blood flow can also be assessed by the degree of enlargement of the left atrium and ventricle. In equivocal cases, the diagnosis can be confirmed by cardiac catheterization. Right and left ventriculography indicate the presence of arterial transposition and demonstrate the site and size of the VSD. Systolic pressure is equal in the 2 ventricles, the aorta, and the pulmonary artery. Left atrial pressure may be much higher than right atrial pressure, a finding indicative of a restrictive communication at the atrial level. At the time of cardiac catheterization, Rashkind balloon atrial septostomy may be performed to decompress the left atrium, even when adequate mixing is occurring at the ventricular level.
Surgical treatment is advised soon after diagnosis, because heart failure and failure to thrive are difficult to manage and pulmonary vascular disease can develop unusually rapidly in these patients. Preoperative management with diuretics lessens the symptoms of heart failure and stabilizes the patient prior to surgery.
Patients with d-TGA and a VSD without pulmonic stenosis can be treated with an arterial switch procedure combined with VSD closure. In these patients, the arterial switch operation can be safely performed after the 1st 2 wk of life because the VSD results in equal pressure in both ventricles and prevents regression of left ventricular muscle mass. At major centers, however, there is no reason to delay repair, as results are excellent whether the surgery is performed in the neonatal period or later.
425.4 L-Transposition of the Great Arteries (Corrected Transposition)
In l-transposition (l-TGA), the atrioventricular relationships are discordant: the right atrium is connected to the left ventricle and the left atrium to the right ventricle (also known as ventricular inversion). The great arteries are also transposed, with the aorta arising from the right ventricle and the pulmonary artery from the left. In contrast to d-TGA, the aorta arises to the left of the pulmonary artery (hence the designation l for levo-transposition). The aorta may be anterior to the pulmonary artery, although often they are nearly side by side.
The physiology of l-TGA is quite different from that of d-TGA. Desaturated systemic venous blood returns via the vena cavae to a normal right atrium, from which it passes through a bicuspid atrioventricular (mitral) valve into a right-sided ventricle that has the architecture and smooth wall morphologic features of the normal left ventricle (Fig. 425-5). Because transposition is also present, however, the desaturated blood ejected from this left ventricle enters the transposed pulmonary artery and flows into the lungs, as it would in the normal circulation. Oxygenated pulmonary venous blood returns to a normal left atrium, passes through a tricuspid atrioventricular valve into a left-sided ventricle, which has the trabeculated morphologic features of a normal right ventricle, and is then ejected into the transposed aorta. The double inversion of the atrioventricular and ventriculoarterial relationships result in desaturated right atrial blood appropriately flowing to the lungs and oxygenated pulmonary venous blood appropriately flowing to the aorta. The circulation is thus physiologically “corrected.” Without other defects, the hemodynamics would be nearly normal. In most patients, however, associated anomalies coexist: VSD, Ebstein-like abnormalities of the left-sided atrioventricular (tricuspid) valve, pulmonary valvular or subvalvular stenosis (or both), and atrioventricular conduction disturbances (complete heart block, accessory pathways such as Wolff-Parkinson-White syndrome).
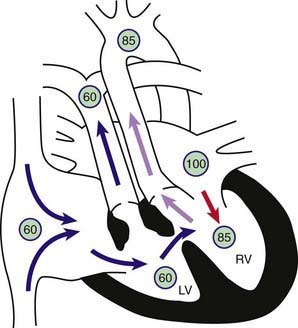
Figure 425-5 Physiology of l- or corrected transposition of the great arteries (l-TGA) with a ventricular septal defect and pulmonic stenosis (VSD + PS). Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. Blood from the right atrium flows through the mitral valve into the “inverted” left ventricle. The left ventricle is, however, attached to the transposed pulmonary artery. Therefore, despite the anomalies, desaturated blood still winds up in the pulmonary circulation. Saturated blood returns to the left atrium, traverses the tricuspid valve into the “inverted” right ventricle, and is pumped into the transposed aorta. This circulation would be totally “corrected” were it not for the frequent association of other congenital anomalies, in this case, VSD + PS. Because of the stenotic pulmonary valve, some left ventricular blood flow crosses the VSD and into the right ventricle and the ascending aorta, and systemic desaturation results.
Clinical Manifestations
Symptoms and signs are widely variable and are determined by the associated lesions. If pulmonary outflow is unobstructed, the clinical signs are similar to those of an isolated VSD. If l-TGA is associated with pulmonary stenosis and a VSD, the clinical signs are more similar to those of tetralogy of Fallot.
Diagnosis
The chest roentgenogram may suggest the abnormal position of the great arteries; the ascending aorta occupies the upper left border of the cardiac silhouette and has a straight profile. The electrocardiogram, in addition to any atrioventricular conduction disturbances, may show abnormal P waves; absent Q waves in V6; abnormal Q waves in leads III, aVR, aVF, and V1; and upright T waves across the precordium. The echocardiogram is diagnostic. The characteristic echocardiographic features of the right ventricle (moderator band, coarser trabeculations, tricuspid valve that sits more inferiorly compared to the bicuspid mitral valve, and a smooth muscular conus or infundibulum separating the atrioventricular valve from the semilunar valve) allow the echocardiographer to determine the presence of atrioventricular discordance (right atrium connected to left ventricle; left atrium to right ventricle).
Surgical treatment of the associated anomalies, most often the VSD, is complicated by the position of the bundle of His, which can be injured at the time of surgery and result in heart block. Identification of the usual course of the bundle in corrected transposition (running superior to the defect) has been accomplished by mapping of the conduction system so that the surgeon can avoid the bundle of His during repair. Even without surgical injury, patients with l-TGA are at risk for heart block as they grow older.
Because simple surgical correction leaves the right ventricle as the systemic pumping chamber, and hence vulnerable to late ventricular failure, surgeons have become more aggressive about trying operations that utilize the left ventricle as the systemic pumping chamber. This is accomplished by performing an atrial switch operation, to reroute the systemic and pulmonary venous returns, in combination with an arterial switch operation to reroute the ventricular outflows (double switch procedure). The long-term benefit of this approach in preserving systemic ventricular function is still under investigation.
Beca J, Gunn J, Coleman L, et al. Pre-operative brain injury in newborn infants with transposition of the great arteries occurs at rates similar to other complex congenital heart disease and is not related to balloon atrial septostomy. J Am Coll Cardiol. 2009;53:1807-1811.
Bellinger DC, Newburger JW, Wypij D, et al. Behaviour at eight years in children with surgically corrected transposition: The Boston Circulatory Arrest Trial. Cardiol Young. 2009;19:86-97.
Bellinger DC, Wypij D, duDuplessis AJ, et al. Neurodevelopmental status at eight years in children with dextro-transposition of the great arteries: The Boston Circulatory Arrest Trial. J Thorac Cardiovasc Surg. 2003;126:1385-1396.
Blume ED, Wernovsky G. Long-term results of arterial switch repair of transposition of the great vessels. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 1998;1:129-138.
Dearani JA, Danielson GK, Puga FJ, et al. Late results of the Rastelli operation for transposition of the great arteries. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2001;4:3-15.
Dunbar-Masterson C, Wypij D, Bellinger DC, et al. General health status of children with D-transposition of the great arteries after the arterial switch operation. Circulation. 2001;104(Suppl 1):I138-I142.
Hayes CJ, Gersony WM. Arrhythmias after the Mustard operation for transposition of the great arteries: a long-term study. J Am Coll Cardiol. 1986;7:133-137.
Khairy P, Van Hare GF. Catheter ablation in transposition of the great arteries with Mustard or Senning baffles. Heart Rhythm. 2009;6:283-289.
Ly M, Belli E, Leobon B, et al. Results of the double switch operation for congenitally corrected transposition of the great arteries. Eur J Cardiothorac Surg. 2009;35:879-883.
Neufeld RE, Clark BG, Robertson CM, et al. Five-year neurocognitive and health outcomes after the neonatal arterial switch operation. J Thorac Cardiovasc Surg. 2008;136:1413-1421.
Pedra SR, Pedra CA, Abizaid AA, et al. Intracoronary ultrasound assessment late after the arterial switch operation for transposition of the great arteries. J Am Coll Cardiol. 2005;45:2061-2068.
425.5 Double-Outlet Right Ventricle without Pulmonary Stenosis
In double-outlet right ventricle without pulmonary stenosis, both the aorta and the pulmonary artery arise from the right ventricle (Chapter 424.5). The only outlet from the left ventricle is through a VSD. In the absence of obstruction to pulmonary blood flow, clinical manifestations are similar to those of an uncomplicated VSD with a large left-to-right shunt, although mild systemic desaturation may be present because of mixing of oxygenated and deoxygenated blood in the right ventricle. The electrocardiogram usually shows biventricular hypertrophy. Echocardiography is diagnostic and shows the right ventricular origin of both great arteries, their anteroposterior relationship, as well as the relationship of the VSD to each of the great arteries. Surgical correction is dependent on these relationships. If the VSD is subaortic, it is accomplished by creation of an intracardiac tunnel. Blood is then ejected from the left ventricle via the VSD into the aorta. If the VSD is subpulmonic, an arterial switch may be performed in combination with an intracardiac tunnel. If pulmonary blood flow is excessive enough to cause congestive heart failure, pulmonary arterial banding may be required in infancy, followed by surgical correction when the child is bigger. When associated pulmonary stenosis is present, cyanosis is more marked, pulmonary blood flow is decreased, and clinical presentation may be similar to that of tetralogy of Fallot (Chapter 425.5).
425.6 Double-Outlet Right Ventricle with Malposition of the Great Arteries (Taussig-Bing Anomaly)
In double-outlet right ventricle with malposed great arteries, the VSD is usually directly subpulmonary and the aorta distant from the left ventricle. Sometimes both the pulmonary and aortic valves may be located close to the VSD (doubly committed VSD) and sometimes neither is (doubly uncommitted VSD). The term malposition is used instead of transposition since both great arteries arise from the right ventricle. Aortic obstructive lesions are common, including valvular and subvalvular aortic stenosis, coarctation of the aorta, and interruption of the aortic arch. Because pulmonary blood flow is unobstructed, patients experience cardiac failure early in infancy and are at risk for the development of pulmonary vascular disease and cyanosis. If aortic obstructive lesions are a component, patients can present with poor systemic output and cardiovascular collapse, particularly after the ductus begins to close. Cardiomegaly is usual, and a parasternal systolic ejection murmur is audible, sometimes preceded by an ejection click and loud closure of the pulmonary valve. The electrocardiogram shows right axis deviation and right, left, or biventricular hypertrophy. The roentgenogram shows cardiomegaly and prominence of the pulmonary vasculature. The anatomic features of the anomaly and associated abnormalities are usually demonstrated by echocardiography, augmented if necessary by either cardiac catheterization, MRI, or CT. Palliation may be achieved by pulmonary arterial banding in infancy and surgical correction at a later age, which may be accomplished by an arterial switch procedure (Chapter 425.2) combined with an intracardiac baffle, or some modification of the Rastelli procedure (Chapter 425.8).
425.7 Total Anomalous Pulmonary Venous Return
Pathophysiology
Abnormal development of the pulmonary veins may result in either partial or complete anomalous drainage into the systemic venous circulation. Partial anomalous pulmonary venous return is usually an acyanotic lesion (Chapter 420.4). Total anomalous pulmonary venous return (TAPVR) is associated with total mixing of systemic venous and pulmonary venous blood flow within the heart and thus produces cyanosis.
In TAPVR, the heart has no direct pulmonary venous connection into the left atrium. The pulmonary veins may drain above the diaphragm into the right atrium directly, into the coronary sinus, or into the superior vena cava via a “vertical vein,” or they may drain below the diaphragm and join into a “descending vein” that enters into the inferior vena cava or one of its major tributaries, often via the ductus venosus. This latter form of anomalous venous drainage is most commonly associated with obstruction to venous flow, usually as the ductus venosus closes soon after birth, although supracardiac anomalous veins may also become obstructed. Occasionally, the drainage may be mixed, with some veins draining above and others below the diaphragm.
All forms of TAPVR involve mixing of oxygenated and deoxygenated blood before or at the level of the right atrium (total mixing lesion). This mixed right atrial blood either passes into the right ventricle and pulmonary artery or passes through an atrial septal defect (ASD) or patent foramen ovale into the left atrium, which will be the only source of systemic blood flow. The right atrium and ventricle and the pulmonary artery are generally enlarged, whereas the left atrium and ventricle may be normal or small. The clinical manifestations of TAPVR depend on the presence or absence of obstruction of the venous channels (Table 425-1). If pulmonary venous return is obstructed, severe pulmonary congestion and pulmonary hypertension develop; rapid deterioration occurs without surgical intervention. Obstructed TAPVR is a pediatric cardiac surgical emergency because prostaglandin therapy is usually not effective.
Table 425-1 TOTAL ANOMALOUS PULMONARY VENOUS RETURN
| SITE OF CONNECTION (% OF CASES) | % WITH SIGNIFICANT OBSTRUCTION |
|---|---|
| Supracardiac (50) | |
| Left superior vena cava (40) | 40 |
| Right superior vena cava (10) | 75 |
| Cardiac (25) | |
| Coronary sinus (20) | 10 |
| Right atrium (5) | 5 |
| Infracardiac (20) | 95-100 |
| Mixed (5) | |
Clinical Manifestations
Two major clinical patterns of TAPVR are seen, depending on the presence or absence of obstruction. Those neonates with severe obstruction to pulmonary venous return, most prevalent in the infracardiac group (see Table 425-1), present with severe cyanosis and respiratory distress. Murmurs may not be present. These infants are severely ill and fail to respond to mechanical ventilation. Rapid diagnosis and surgical correction are necessary for survival. In contrast, those with mild or no obstruction to pulmonary venous return are usually characterized by the development of heart failure as the pulmonary vascular resistance falls, with mild to moderate degrees of desaturation. Systolic murmurs may be audible along the left sternal border, and a gallop rhythm may be present. Some infants may have mild obstruction in the neonatal period and develop worsening obstruction as time passes.
Diagnosis
The electrocardiogram demonstrates right ventricular hypertrophy (usually a qR pattern in V3R and V1, and the P waves are frequently tall and spiked). In neonates with marked pulmonary venous obstruction, the chest roentgenogram demonstrates a very dramatic perihilar pattern of pulmonary edema and a small heart. This appearance can sometimes be confused with primary pulmonary disease and the differential diagnosis includes persistent pulmonary hypertension of the newborn, respiratory distress syndrome, pneumonia (bacterial, meconium aspiration), pulmonary lymphangiectasia, and other heart defects (hypoplastic left heart syndrome). In older children, if the anomalous pulmonary veins enter the innominate vein and persistent left superior vena cava (Fig. 425-6), a large supracardiac shadow can be seen, which together with the normal cardiac shadow forms a “snowman” appearance. In most cases without obstruction, the heart is enlarged, the pulmonary artery and right ventricle are prominent, and pulmonary vascularity is increased.
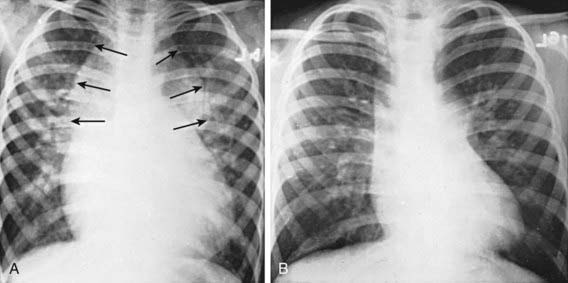
Figure 425-6 Chest x-ray of total anomalous pulmonary venous return to the left superior vena cava. A, Preoperative image. Arrows point to the supracardiac shadow, which produces the snowman or figure 8 configuration. Cardiomegaly and increased pulmonary vascularity are evident. B, Postoperative image showing a decrease in the size of the heart and the supracardiac shadow.
The echocardiogram demonstrates a large right ventricle and usually identifies the pattern of abnormal pulmonary venous connections (Fig. 425-7). The demonstration of any vein with Doppler flow away from the heart is pathognomonic of TAPVR since normal venous flow is usually towards the heart. Shunting occurs from right to left at the atrial level. The size of the left atrium and left ventricle can be measured and the presence of any associated cardiac defects determined.
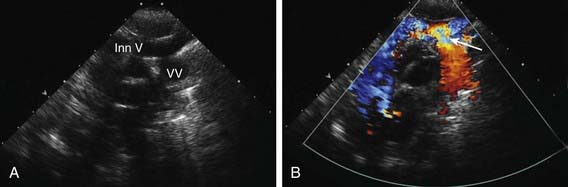
Figure 425-7 Suprasternal two-dimensional echocardiographic views demonstrating supracardiac total anomalous pulmonary venous return (type I). A, The large vertical ascending vein can be seen entering the innominate vein. There is a moderate narrowing where the anomalous vein enters the upper body venous system. B, Color Doppler examination shows a venous flow signal (red color) indicating that blood is moving away toward the transducer and thus from the heart (all venous flow should normally return toward the heart), diagnostic of anomalous pulmonary venous return. The turbulent acceleration of flow can be seen (arrow) where the vertical vein enters the innominate. Inn V, innominate vein; VV, vertical vein.
Echocardiography should be adequate to demonstrate TAPVR in most cases, however, if there is question about the drainage of one or more pulmonary veins, cardiac catheterization, MRI, or CT is performed. Catheterization shows that the oxygen saturation of blood in both atria, both ventricles, and the aorta is similar, indicative of a total mixing lesion. An increase in systemic venous saturation occurs at the site of entry of the abnormal pulmonary venous channel, either above or below the diaphragm. In older patients, pulmonary arterial and right ventricular pressure may be only moderately elevated, but in infants with pulmonary venous obstruction, pulmonary hypertension is usual. Selective pulmonary arteriography shows the anatomy of the pulmonary veins and their point of entry into the systemic venous circulation.
Treatment
Surgical correction of TAPVR is indicated during infancy, with emergent repair performed for those patients with venous obstruction. If surgery cannot be performed urgently, extracorporeal membrane oxygenation (ECMO) may be required to maintain oxygenation. Surgically, the pulmonary venous confluence is anastomosed directly to the left atrium, the ASD is closed, and any connection to the systemic venous circuit is interrupted. Early results are generally good, even for critically ill neonates. The postoperative period may be complicated by pulmonary vascular hypertensive crises. In some patients, especially those in whom the diagnosis was delayed or the obstruction was severe, recurrent stenosis and development of pulmonary veno-occlusive disease may occur. Attempts have been made to treat recurrent stenosis with surgery, balloon angioplasty, stents, and antiproliferative chemotherapy. To date, the long-term prognosis in these patients is very guarded and in those with veno-occlusive disease heart-lung transplantation may be the only option (Chapter 437.2).
Holt DB, Moller JH, Larson S, et al. Primary pulmonary vein stenosis. Am J Cardiol. 2007;99:568-572.
Huhta J, Gutgesell HP, Nihill MR. Cross sectional echocardiographic diagnosis of total anomalous pulmonary venous connection. Br Heart J. 1985;53:525-534.
Kirshbom PM, Flynn TB, Clancy RR, et al. Late neurodevelopmental outcome after repair of total anomalous pulmonary venous connection. J Thorac Cardiovasc Surg. 2005;129:1091-1097.
Lakshminrusimha S, Wynn RJ, Youssfi M, et al. Use of CT angiography in the diagnosis of total anomalous venous return. J Perinatol. 2009;29:458-461.
Morales DL, Braud BE, Booth JH, et al. Heterotaxy patients with total anomalous pulmonary venous return: improving surgical results. Ann Thorac Surg. 2006;82:1621-1627.
425.8 Truncus Arteriosus
Pathophysiology
In truncus arteriosus, a single arterial trunk (truncus arteriosus) arises from the heart and supplies the systemic, pulmonary, and coronary circulations. A VSD is always present, with the truncus overriding the defect and receiving blood from both the right and left ventricles (Fig. 425-8). The number of truncal valve cusps varies from 2 to as many as 6 and the valve may be stenotic, regurgitant, or both. The pulmonary arteries can arise together from the posterior left side of the persistent truncus arteriosus and then divide into left and right pulmonary arteries (type I). In types II and III truncus arteriosus, no main pulmonary artery is present, and the right and left pulmonary arteries arise from separate orifices on the posterior (type II) or lateral (type III) aspects of the truncus arteriosus. Type IV truncus is a term no longer used, since in this case there is no identifiable connection between the heart and pulmonary arteries, and pulmonary blood flow is derived from major aortopulmonary collateral arteries (MAPCAs) arising from the transverse or descending aorta; this is essentially a form of pulmonary atresia (Chapter 424.2).
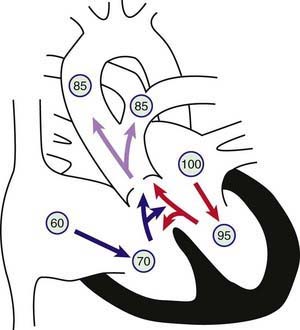
Figure 425-8 Physiology of truncus arteriosus. Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. Desaturated blood enters the right atrium, flows through the tricuspid valve into the right ventricle, and is ejected into the truncus. Saturated blood returning from the left atrium enters the left ventricle and is also ejected into the truncus. The common aortopulmonary trunk gives rise to the ascending aorta and to the main or branch pulmonary arteries. Oxygen saturation in the aorta and pulmonary arteries is usually the same (definition of a total mixing lesion). As pulmonary vascular resistance decreases in the 1st few weeks of life, pulmonary blood flow increases dramatically and mild cyanosis and congestive heart failure result.
Both ventricles are at systemic pressure and both eject blood into the truncus. When pulmonary vascular resistance is relatively high immediately after birth, pulmonary blood flow may be normal; as pulmonary resistance drops in the 1st mo of life, blood flow to the lungs is greatly increased and heart failure ensues. Truncus arteriosus is a total mixing lesion with complete admixture of pulmonary and systemic venous return. Because of the large volume of pulmonary blood flow, clinical cyanosis is usually mild. If the lesion is left untreated, pulmonary resistance eventually increases, pulmonary blood flow decreases, and cyanosis becomes more prominent (Eisenmenger physiology; Chapter 427.2).
Clinical Manifestations
The clinical signs of truncus arteriosus vary with age and depend on the level of pulmonary vascular resistance. In the immediate newborn period, signs of heart failure are usually absent; a murmur and minimal cyanosis may be the only initial findings. Over the next 1-2 mo of life, pulmonary blood flow begins to become torrential and the clinical picture is dominated by heart failure, with still mild cyanosis. Runoff of blood from the truncus to the pulmonary circulation may result in a wide pulse pressure and bounding pulses. These findings will be further exaggerated if truncal valve insufficiency is present. The heart is usually enlarged, and the precordium is hyperdynamic. The 2nd heart sound is loud and single. A systolic ejection murmur, sometimes accompanied by a thrill, is generally audible along the left sternal border. The murmur is frequently preceded by an early systolic ejection click due to the abnormal truncal valve. In the presence of truncal valve insufficiency, a high-pitched early diastolic decrescendo murmur is heard at the mid-left sternal border. An apical mid-diastolic rumbling murmur caused by increased flow through the mitral valve is often audible with the bell of the stethoscope, especially as heart failure develops. Truncus arteriosus is a conotruncal malformation and may be associated with DiGeorge syndrome, linked to a deletion of a large region of chromosome 22q11 (Chapter 418).
Diagnosis
The electrocardiogram shows right, left, or combined ventricular hypertrophy. The chest roentgenogram also shows considerable variation. Cardiac enlargement will develop over the 1st several weeks of life, and is due to prominence of both ventricles. The truncus may produce a prominent shadow that follows the normal course of the ascending aorta and aortic knob; the aortic arch is right-sided in 50% of patients. Sometimes a high bulge left of the aortic knob is produced by the main or left pulmonary artery. Pulmonary vascularity is increased after the 1st few weeks of life. Echocardiography is diagnostic and demonstrates the large truncal artery overriding the VSD and the pattern of origin of the branch pulmonary arteries (Fig. 425-9). Associated anomalies such as an interrupted aortic arch may be noted. Pulsed and color Doppler studies are used to evaluate truncal valve regurgitation. If required, cardiac catheterization shows a left-to-right shunt at the ventricular level, with right-to-left shunting into the truncus. Systolic pressure in both ventricles and the truncus is similar. Angiography reveals the large truncus arteriosus and more defines the origin of the pulmonary arteries.
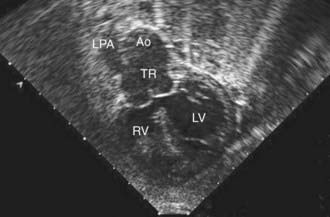
Figure 425-9 Subcostal two-dimensional echocardiographic demonstration of truncus arteriosus. The large truncal valve can be seen overriding the ventricular septal defect. In this case, only the left pulmonary artery (LPA) arises from the truncus (TR). The pulmonary arteries are discontinuous and the right pulmonary artery arises from the descending aorta via the ductus arteriosus (not shown). Ao, aorta; LV, left ventricle; RV, right ventricle.
Prognosis and Complications
Surgical results have been excellent, and many patients with repaired truncus are now entering adulthood. The need to replace the right ventricular to pulmonary artery conduit as the child grows means that these patients will need to undergo multiple operations by the time they reach adulthood. When truncus arteriosus is associated with DiGeorge syndrome, the associated endocrine, immunologic, craniofacial, and airway abnormalities may complicate recovery.
Treatment
In the 1st few weeks of life, many of these infants can be managed with anticongestive medications; as pulmonary vascular resistance falls, heart failure symptoms worsen and surgery is indicated, usually within the 1st few months. Delay of surgery much beyond this time period may increase the likelihood of pulmonary vascular disease; many centers now perform routine neonatal repair at the time of diagnosis. At surgery, the VSD is closed, the pulmonary arteries are separated from the truncus, and continuity is established between the right ventricle and the pulmonary arteries with a homograft conduit. Immediate surgical results are excellent, but these conduits will develop either regurgitation or stenosis over time, and must be replaced, often several times, as the child grows.
Colon M, Anderson RH, Weinberg P, et al. Anatomy, morphogenesis, diagnosis, management, and outcomes for neonates with common arterial trunk. Cardiol Young. 2008;18(Suppl 3):52-62.
Goldmuntz E, Driscoll DA, Emanuel BS, et al. Evaluation of potential modifiers of the cardiac phenotype in the 22q11.2 deletion syndrome. Birth Defects Res A Clin Mol Teratol. 2009;85:125-129.
Reddy VM, Hanley F. Late results of repair of truncus arteriosus. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 1998;1:139-146.
Swanson TM, Selamet Tierney ES, Tworetzky W, et al. Truncus arteriosus: diagnostic accuracy, outcomes, and impact of prenatal diagnosis. Pediatr Cardiol. 2009;30:256-261.
Thompson LD, McElhinney DB, Reddy M, et al. Neonatal repair of truncus arteriosus: Continuing improvement in outcomes. Ann Thorac Surg. 2001;72:391-395.
Wilson DI, Burn J, Scambler P, et al. DiGeorge syndrome: part of CATCH 22. J Med Genet. 1993;30:852-856.
425.9 Single Ventricle (Double-Inlet Ventricle, Univentricular Heart)
Pathophysiology
With a single ventricle, both atria empty through a common atrioventricular valve or via 2 separate valves into a single ventricular chamber, with total mixing of systemic and pulmonary venous return. This chamber may have left, right, or indeterminate ventricular anatomic characteristics. The aorta and pulmonary artery both arise from this single chamber, although 1 of the great vessels may originate from a rudimentary outflow chamber. The aorta may be posterior, anterior (malposition), or side by side with the pulmonary artery and either to the right or to the left. Pulmonary stenosis or atresia is common.
Clinical Manifestations
The clinical picture is variable and depends on the associated intracardiac anomalies. If pulmonary outflow is obstructed, the findings are usually similar to those of tetralogy of Fallot: marked cyanosis without heart failure. If pulmonary outflow is unobstructed, the findings are similar to those of transposition with VSD: minimal cyanosis with increasing heart failure.
In patients with pulmonary stenosis, cyanosis is present in early infancy. Cardiomegaly is mild or moderate, a left parasternal lift is palpable, and a systolic thrill is common. The systolic ejection murmur is usually loud; an ejection click may be audible, and the 2nd heart sound is single and loud. In patients with unobstructed pulmonary flow, as pulmonary vascular resistance drops, torrential pulmonary blood flow develops, and these patients present with tachypnea, dyspnea, failure to thrive, and recurrent pulmonary infections. Cyanosis is only mild or moderate. Cardiomegaly is generally marked, and a left parasternal lift is palpable. A systolic ejection murmur is present but is not usually loud or harsh, and the 2nd heart sound is loud and closely split. A 3rd heart sound is common and may be followed by a short mid-diastolic rumbling murmur caused by increased flow through the atrioventricular valves. The eventual development of pulmonary vascular disease reduces pulmonary blood flow so that the cyanosis increases and signs of cardiac failure appear to improve (Eisenmenger physiology; Chapter 427.2).
Diagnosis
Findings on the electrocardiogram are nonspecific. P waves are normal, spiked, or bifid. The precordial lead pattern suggests right ventricular hypertrophy, combined ventricular hypertrophy, or sometimes left ventricular dominance. The initial QRS forces are usually to the left and anterior. Roentgenographic examination confirms the degree of cardiomegaly. If present, a rudimentary outflow chamber may produce a bulge on the upper left border of the cardiac silhouette in the posteroanterior projection. In the absence of pulmonary stenosis, pulmonary vasculature is increased, whereas in the presence of pulmonary stenosis, pulmonary vasculature is diminished. Echocardiography will confirm the absence or near absence of the ventricular septum and can usually determine whether the single ventricle has right, left, or mixed morphologic features. The presence of a rudimentary outflow chamber under one of the great vessels can be identified, and pulsed Doppler can be used to determine whether flow through this communication (known as a bulboventricular foramen) is obstructed.
If cardiac catheterization is performed, the pressure in the single ventricular chamber is at systemic level; however, a gradient may be demonstrated across the entrance to a rudimentary outflow chamber. Pressure measurements and angiography demonstrate whether pulmonary stenosis is present.
Prognosis and Complications
Unoperated, some patients succumb during infancy from heart failure. Others may survive to adolescence and early adult life but finally succumb to the effects of chronic hypoxemia or, in the absence of pulmonary stenosis, to the effects of pulmonary vascular disease. Patients with moderate pulmonary stenosis have the best prognosis because pulmonary blood flow, though restricted, is still adequate. Surgical palliation, eventually leading to Fontan-type circulatory physiology (Chapter 424.4), has very good short- and intermediate-term results.
Treatment
If pulmonary stenosis is severe, a Blalock-Taussig aortopulmonary shunt is performed to provide a reliable source of pulmonary blood flow (Chapter 424.1). If pulmonary blood flow is unrestricted, pulmonary arterial banding is used to control heart failure and prevent progressive pulmonary vascular disease. The bidirectional Glenn shunt is usually performed at between 2 and 6 mo of age, followed by a modified Fontan operation (cavopulmonary isolation procedure, Chapter 424.4) at 2-3 yr of age. If subaortic stenosis is present because of a restrictive connection to a rudimentary outflow chamber, (restrictive bulboventricular foramen) surgical relief can be provided by anastomosing the proximal pulmonary artery to the side of the ascending aorta (Damus-Stansyl-Kaye operation).
425.10 Hypoplastic Left Heart Syndrome
Pathophysiology
The term hypoplastic left heart is used to describe a related group of anomalies that include underdevelopment of the left side of the heart (atresia of the aortic or mitral orifice) and hypoplasia of the ascending aorta. The left ventricle may be moderately hypoplastic, very small and nonfunctional, or totally atretic; in the immediate neonatal period the right ventricle maintains both the pulmonary circulation and the systemic circulation via the ductus arteriosus (Fig. 425-10). Pulmonary venous blood passes through an atrial septal defect or dilated foramen ovale from the left to the right side of the heart, where it mixes with systemic venous blood (total mixing lesion). When the ventricular septum is intact, which is usually the case, all the right ventricular blood is ejected into the main pulmonary artery; the descending aorta is supplied via the ductus arteriosus, and flow from the ductus also fills the ascending aorta and coronary arteries in a retrograde fashion. The major hemodynamic abnormalities are inadequate maintenance of the systemic circulation and, depending on the size of the atrial-level communication, either pulmonary venous hypertension (restrictive foramen ovale) or pulmonary overcirculation (moderate or large ASD).
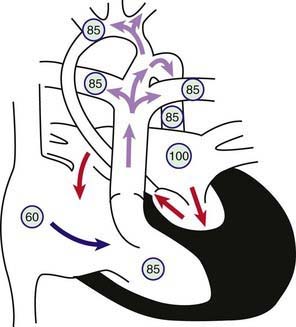
Figure 425-10 Physiology of hypoplastic left heart syndrome (HLHS). Circled numbers represent oxygen saturation values. HLHS is not a single lesion but a constellation of different degrees of hypoplasia of the left-sided heart structures. This drawing shows a patent mitral valve, a small left ventricular cavity, and a diminutive ascending aorta. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. Desaturated blood enters the right atrium, flows through the tricuspid valve into the right ventricle, and is ejected into the pulmonary artery. Because of the markedly decreased left ventricular compliance, most of the pulmonary venous blood returning to the left atrium shunts left to right at the atrial level. A small amount of left atrial blood will cross the mitral valve and be ejected into the tiny ascending aorta. The right ventricular oxygen saturation represents a mixing of desaturated systemic venous blood and saturated pulmonary venous blood. Pulmonary artery blood flows into the pulmonary arteries as well as right to left across the patent ductus arteriosus (PDA) into the aorta. Ductal blood flows prograde to the descending aorta as well as retrograde to the ascending aorta, where it supplies the head and neck vessels in addition to the coronary arteries (which arise off the small ascending aorta). Closure of the PDA results in profound hypoxia and circulatory collapse.
Clinical Manifestations
Although cyanosis may not always be obvious in the 1st 48 hr of life, a grayish-blue color of the skin is soon apparent and denotes a mix of cyanosis and poor perfusion. The condition is diagnosed in most infants in the 1st few hours or days of life. Once the ductus arteriosus begins to closes, signs of poor systemic perfusion and shock predominate. All of the peripheral pulses may be weak or absent. A palpable right ventricular parasternal lift may be present along with a nondescript systolic murmur.
This lesion may be isolated or associated in 5-15% of patients with known genetic syndromes, such as Turner syndrome, trisomy 13 or 18, Jacobsen syndrome (11q deletion), Holt-Oram syndrome, and Rubinstein-Taybi syndrome. In these circumstances, noncardiac manifestations of the syndrome may be evident and influence the clinical outcomes.
Diagnosis
On the chest roentgenogram, the heart is variable in size in the 1st days of life, but cardiomegaly develops rapidly and is associated with increased pulmonary vascularity. The initial electrocardiogram may show only the normal neonatal pattern of right ventricular dominance, but later, P waves become prominent and right ventricular hypertrophy is usual with reduced left ventricular forces. The echocardiogram is diagnostic and demonstrates absence or hypoplasia of the mitral valve and aortic root, a variably small left atrium and left ventricle, and a large right atrium and right ventricle (Fig. 425-11). The size of the atrial communication, by which pulmonary venous blood leaves the left atrium, can be assessed directly and by pulsed and color flow Doppler studies. The small ascending aorta and transverse aortic arch are identified and a discrete coarctation of the aorta in the juxtaductal area may be present, although in the presence of a large ductus, it may be difficult to identify. Doppler echocardiography demonstrates the absence of anterograde flow in the ascending aorta, which is supplied by retrograde flow via the ductus arteriosus. The diagnosis of hypoplastic left heart syndrome can usually be made without need for cardiac catheterization. If catheterization is necessary, the hypoplastic ascending aorta is demonstrated by angiography.
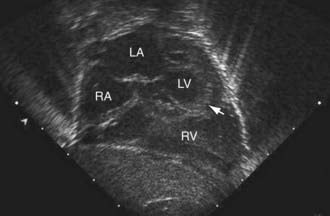
Figure 425-11 Subcostal two-dimensional echocardiographic diagnosis of hypoplastic left heart syndrome. The small left ventricular chamber can be seen, the apex of which (arrowhead) does not form the apex of the heart. The atrial septum can be seen bowing from the left to the right, indicating that the communication between the two atria is pressure restrictive. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
Prognosis and Complications
Untreated patients most often succumb during the 1st months of life, usually during the 1st or 2nd wk. Occasionally, unoperated patients may live for months or rarely years. Up to 30% of infants with hypoplastic left heart syndrome have evidence of either a major or minor central nervous system abnormality. Other dysmorphic features may be found in up to 40% of patients. Thus, careful preoperative evaluation (genetic, neurologic, ophthalmologic) should be performed in patients being considered for surgical therapy.
Long-term follow up after the Norwood procedure demonstrates reduced neurodevelopmental outcomes and poor exercise tolerance. Whether the poor neurodevelopmental outcome is due to prenatal associated central nervous system injury or malformation, the alterations of cerebral hemodynamics during bypass surgery, or poor postoperative perfusion is unknown.
Treatment
Surgical therapy for hypoplastic left heart syndrome has been associated with improving survival rates, reported as high as 95% for the 1st-stage palliation in experienced centers. The 1st-stage repair is designed to construct a reliable source of systemic blood flow arising from the single right ventricle using a combination of aortic and pulmonary arterial tissue, and to limit pulmonary blood flow to avoid heart failure and prevent the development of pulmonary vascular disease. The 2 surgical procedures most commonly utilized are the Norwood procedure (Fig. 425-12) or the Sano procedure. Primary heart transplantation, previously advocated by a few centers, is much less common due to the substantially improved survival rates with standard surgery and the limited supply of donor organs in this age group.
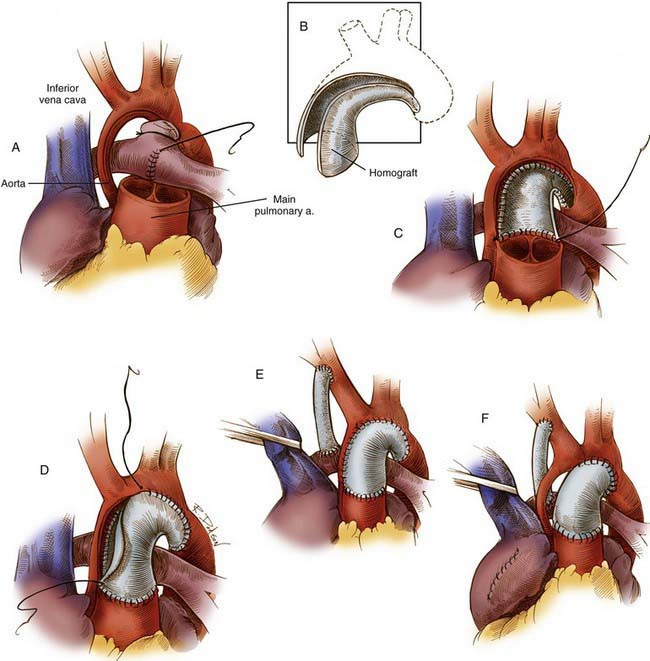
Figure 425-12 The Norwood procedure, one of the two current techniques for 1st-stage palliation of hypoplastic left heart syndrome. A, Incisions used for the procedure incorporate a cuff of arterial wall allograft. The distal divided main pulmonary artery may be closed by direct suture or with a patch. B, Dimensions of the cuff of the arterial wall allograft. C, The arterial wall allograft is used to supplement the anastomosis between the proximal divided main pulmonary artery and the ascending aorta, aortic arch, and proximal descending aorta. D and E, The procedure is completed by an atrial septectomy and a 3.5-mm modified right Blalock shunt. F, When the ascending aorta is particularly small, an alternative procedure involves placement of a complete tube of arterial allograft. The tiny ascending aorta may be left in situ, as indicated, or implanted into the side of the neoaorta.
(From Castañeda AR, Jonas RA, Mayer JE Jr, et al: Single-ventricle tricuspid atresia. In Cardiac surgery of the neonate and infant, Philadelphia, 1994, WB Saunders.)
If a Norwood or Sano procedure is to be performed, preoperative medical management includes correction of acidosis and hypoglycemia, maintenance of ductus arteriosus patency with prostaglandin E1 (0.01-0.20 µg/kg/min) to support systemic blood flow, and prevention of hypothermia. Preoperative management should avoid excessive pulmonary blood flow; either through management of ventilator settings, increasing the concentration of inspired CO2, or decreasing the concentration of inspired O2. Balloon dilatation of the atrial septum may be indicated.
The Norwood procedure is usually performed in 3 stages. Stage I (see Fig. 425-12) includes an atrial septectomy and transection and ligation of the distal main pulmonary artery; the proximal pulmonary artery is then connected to the transversely opened hypoplastic aortic arch to form a neoaorta, extending through the coarcted segment of the juxtaductal aortic arch. A synthetic aortopulmonary (Blalock-Taussig) shunt connects the aorta to the main pulmonary artery to provide controlled pulmonary blood flow. In the Sano modification, a right ventricle to pulmonary artery conduit is used instead of an aortopulmonary shunt to provide pulmonary blood flow, temporarily creating a double-outlet right ventricle. The operative risk for these 1st-stage procedures has improved dramatically in the past 2 decades and the best reported results demonstrate a 90-95% survival rate.
Stage II consists of a Glenn anastomosis to connect the superior vena cava to the pulmonary arteries (Chapter 425.4), at between 2 and 6 mo of age. Stage III, usually performed at 2-3 yr of age, consists of a modified Fontan procedure (cavopulmonary isolation) to connect the inferior vena cava to the pulmonary arteries via either an intra-atrial or external baffle. After stage III, all systemic venous return enters the pulmonary circulation directly. Pulmonary venous flow enters the left atrium and is directed across the atrial septum to the tricuspid valve and subsequently to the right (now the systemic) ventricle. Blood leaves the right ventricle via the neoaorta, which supplies the systemic circulation. The old aortic root now attached to the neoaorta provides coronary blood flow. The risks associated with stages II and III are even less than those of stage I; interstage mortality (usually between stages I and II) has been reduced with the use of home monitoring programs. The short- and long-term benefits of using the Norwood vs the Sano procedure remain to be demonstrated.
An alternative therapy is cardiac transplantation, either in the immediate neonatal period, thereby obviating stage I of the Norwood procedure, or after a successful stage I Norwood procedure is performed as a bridge to transplantation. After transplantation, patients usually have normal cardiac function and no symptoms of heart failure; however, these patients have the chronic risk of organ rejection and lifelong immunosuppressive therapy (Chapter 437.1). The combination of donor shortage and improved results with standard surgical procedures has caused most centers to stop recommending transplantation except when associated lesions make the Norwood operation an exceptionally high-risk procedure, or for patients who develop poor ventricular function at some time after the standard surgical approach.
Prevention
Serial fetal echocardiographic studies demonstrate that in some fetuses, hypoplastic left heart syndrome may be a progressive lesion, beginning with simple valvar aortic stenosis in midgestation. The decreased flow through the stenotic aortic valve reduces flow through the left ventricle during development, resulting in gradual ventricular chamber hypoplasia. The potential for preventing this hypoplasia has been demonstrated by performing in utero aortic balloon valvuloplasty in midgestation fetuses (Fig. 425-13). Early results are encouraging, although even if the aortic valve is successfully opened, adequate ventricular growth occurs in only about 30% of patients. At present, this procedure is regarded as experimental.
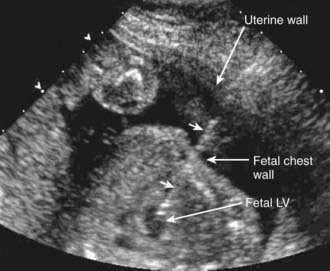
Figure 425-13 Fetal treatment of critical aortic stenosis to prevent development of hypoplastic left heart syndrome. Fetal ultrasound showing insertion of a needle (arrowheads) via the maternal abdominal wall, through the uterus and the fetal chest wall, and into the fetal left ventricle (LV). A balloon catheter is next inserted via the needle into the left ventricular chamber and across the stenotic aortic valve. The balloon is inflated to dilate the valve, the catheter and needle are removed.
(Courtesy of Dr. Stanton Perry, Stanford University, Stanford, CA.)
Barron DJ, Kilby MD, Davies B, et al. Hypoplastic left heart syndrome. Lancet. 2009;374:551-564.
Chiavarelli M, Gundry SR, Razzouk AJ, et al. Cardiac transplantation for infants with hypoplastic left heart syndrome. JAMA. 1993;270:2944-2947.
Connor JA, Arons RR, Figueroa M, et al. Clinical outcomes and secondary diagnoses for infants born with hypoplastic left heart syndrome. Pediatrics. 2004;114:e160-e165.
Daebritz SH, Nollert GD, Zurakowski D, et al. Results of Norwood stage I operation: comparison of hypoplastic left heart syndrome with other malformations. J Thorac Cardiovasc Surg. 2000;119:358-367.
Forbess JM, Visconti KJ, Bellinger DC, et al. Neurodevelopmental outcomes in children after the Fontan operation. Circulation. 2001;104(Suppl 1):I127-I132.
Hinton RB, Martin LJ, Rame-Gowda S, et al. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J Am Coll Cardiol. 2009;53:1065-1071.
Jenkins PC, Chinnock RE, Jenkins KJ, et al. Decreased exercise performance with age in children with hypoplastic left heart syndrome. J Pediatr. 2008;152:507-512.
Marshall AC, Tworetzky W, Bergersen L, et al. Aortic valvuloplasty in the fetus: technical characteristics of successful balloon dilation. J Pediatr. 2005;147:535-539.
Odegard KC, Zurakowski D, DiNardo JA, et al. Prospective longitudinal study of coagulation profiles in children with hypoplastic left heart syndrome from stage I through Fontan completion. J Thorac Cardiovasc Surg. 2009;137:934-941.
Ohye RG, Sleeper LA, Mahony L, et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010;362(21):1980-1992.
Phelps HM, Mahle WT, Kim D, et al. Postoperative cerebral oxygenation in hypoplastic left heart syndrome after the Norwood procedure. Ann Thorac Surg. 2009;87:1490-1494.
Sano S, Ishino K, Kado H, et al. Outcome of right ventricle-to-pulmonary artery shunt in first-stage palliation of hypoplastic left heart syndrome: a multi-institutional study. Ann Thorac Surg. 2004;78:1951-1958.
Sarajuuri A, Jokinen E, Puosi R, et al. Neurodevelopment in children with hypoplastic left heart syndrome. J Pediatr. 2010;157:414-420.
Tabbutt S, Nord AS, Jarvik GP, et al. Neurodevelopmental outcomes after staged palliation for hypoplastic left heart syndrome. Pediatrics. 2008;121:476-483.
Theilen U, Shekerdemian L. The intensive care of infants with hypoplastic left heart syndrome. Arch Dis Fetal Neonatal Ed. 2005;90:F97-F102.
Tworetzky W, Wilkins-Haug L, Jennings RW, et al. Balloon dilation of severe aortic stenosis in the fetus: potential for prevention of hypoplastic left heart syndrome: candidate selection, technique, and results of successful intervention. Circulation. 2004;110:2125-2131.
Wernovsky G. The paradigm shift toward surgical intervention for neonates with hypoplastic left heart syndrome. Arch Pediatr Adolesc Med. 2008;162:849-854.
425.11 Abnormal Positions of the Heart and the Heterotaxy Syndromes (Asplenia, Polysplenia)
Classification and diagnosis of abnormal cardiac position are best performed via a segmental approach, with the position of the viscera and atria defined first, and then the ventricles, followed by the great vessels. Determination of visceroatrial situs can be made by roentgenographic demonstration of the position of the abdominal organs and the tracheal bifurcation for recognition of the right and left bronchi and by echocardiography. The atrial situs is usually similar to the situs of the viscera and lungs. In situs solitus, the viscera are in their normal positions (stomach and spleen on the left, liver on the right), the 3-lobed right lung is on the right, and the 2-lobed left lung on the left; the right atrium is on the right, and the left atrium is on the left. When the abdominal organs and lung lobation are reversed, an arrangement known as situs inversus occurs, the left atrium is on the right and the right atrium on the left. If the visceroatrial situs cannot be readily determined, a condition known as situs indeterminus or heterotaxia exists. The 2 major variations are (1) asplenia syndrome (right isomerism or bilateral right-sidedness), which is associated with a centrally located liver, absent spleen, and 2 morphologic right lungs; and (2) polysplenia syndrome (left isomerism or bilateral left-sidedness), which is associated with multiple small spleens, absence of the intrahepatic portion of the inferior vena cava, and 2 morphologic left lungs. The heterotaxia syndromes are usually associated with severe congenital heart lesions: ASD, VSD, atrioventricular septal defect, hypoplasia of 1 of the ventricles, pulmonary stenosis or atresia, and anomalous systemic venous or pulmonary venous return (Table 425-2).
Table 425-2 COMPARISON OF CARDIOSPLENIC HETEROTAXY SYNDROMES
| FEATURE | ASPLENIA (RIGHT ISOMERISM) | POLYSPLENIA (LEFT ISOMERISM) |
|---|---|---|
| Spleen | Absent | Multiple |
| Sidedness (isomerism) | Bilateral right | Bilateral left |
| Lungs | Bilateral trilobar with eparterial bronchi | Bilateral bilobar with hyparterial bronchi |
| Sex | Male (65%) | Female ≥ male |
| Right-sided stomach | Yes | Less common |
| Symmetric liver | Yes | Yes |
| Partial intestinal rotation | Yes | Yes |
| Dextrocardia (%) | 30-40 | 30-40 |
| Pulmonary blood flow | Decreased (usually) | Increased (usually) |
| Severe cyanosis | Yes | No |
| Transposition of great arteries (%) | 60-75 | 15 |
| Total anomalous pulmonary venous return (%) | 70-80 | Rare |
| Common atrioventricular valve (%) | 80-90 | 20-40 |
| Single ventricle (%) | 40-50 | 10-15 |
| Absent inferior vena cava with azygos continuation | No | Characteristic |
| Bilateral superior vena cava | Yes | Yes |
| Other common defects | PA, PS | Partial anomalous pulmonary venous return, ventricular septal defect, double-outlet right ventricle |
| Risk of sepsis | Yes | No |
| Howell-Jolly and Heinz bodies, pitted erythrocytes | Yes | No |
| Absent gallbladder; biliary atresia | No | Yes |
PA, pulmonary atresia; PS, pulmonary stenosis.
The next segment is localization of the ventricles, which depends on the direction of development of the embryonic cardiac loop. Initial protrusion of the loop to the right (d-loop) carries the future right ventricle anteriorly and to the right, whereas the left ventricle remains posterior and on the left. With situs solitus, a d-loop yields normal atrioventricular connections (right atrium connecting to the right ventricle, left atrium to the left ventricle). Protrusion of the loop to the left (l-loop) carries the future right ventricle to the left and the left ventricle to the right. In this case, in the presence of situs solitus, the right atrium connects with the left ventricle and the left atrium with the right ventricle (ventricular inversion).
The final segment is that of the great vessels. With each type of cardiac loop, the ventricular-arterial relationships may be regarded as either normal (right ventricle to the pulmonary artery, left ventricle to the aorta) or transposed (right ventricle to the aorta, left ventricle to the pulmonary artery). A further classification can be based on the position of the aorta (normally to the right and posterior) relative to the pulmonary artery. In transposition, the aorta is usually anterior and either to the right of the pulmonary artery (d-transposition) or to the left (l-transposition). These segmental relationships can usually be determined by echocardiographic studies demonstrating both atrioventricular and ventriculoarterial relationships. The clinical manifestations of these syndromes of abnormal cardiac position are determined primarily by their associated cardiovascular anomalies.
Dextrocardia occurs when the heart is in the right side of the chest; levocardia (the normal situation) is present when the heart is in the left side of the chest. Dextrocardia without associated situs inversus and levocardia in the presence of situs inversus are most often complicated by other severe cardiac malformations. Surveys of older children and adults indicate that dextrocardia with situs inversus and normally related great arteries (“mirror-image” dextrocardia) is often associated with a functionally normal heart, although congenital heart disease of a less severe nature is common.
Anatomic or functional abnormalities of the lungs, diaphragm, and thoracic cage may result in displacement of the heart to the right (dextroposition). In this case, however, the cardiac apex is pointed normally to the left. This anatomic position is less often associated with congenital heart lesions, although hypoplasia of a lung may be accompanied by anomalous pulmonary venous return from that lung (scimitar syndrome [Chapter 420.4]).
The electrocardiogram is difficult to interpret in the presence of lesions with discordant atrial, ventricular, and great vessel anatomy. Diagnosis usually requires detailed echocardiographic and sometimes MRI, CT, or cardiac catheterization studies. The prognosis and treatment of patients with one of the cardiac positional anomalies are determined by the underlying defects and are covered in their respective chapters. Asplenia increases the risk of serious infections such as bacterial sepsis and thus requires daily antibiotic prophylaxis. Patients with polysplenia frequently have poor splenic function and may also require prophylaxis against pneumococcal sepsis.
Britz-Cunningham SH, Shah MM, Zuppan CW, et al. Mutation of the connexin 43 gap-junction gene in patients with heart malformations and defects of laterality. N Engl J Med. 1995;332:1323-1329.
Cohen MS, Anderson RH, Cohen MI, et al. Controversies, genetics, diagnostic assessment, and outcomes relating to the heterotaxy syndrome. Cardiol Young. 2007;17(Suppl 2):29-43.
Ferdman B, States L, Gaynor JW, et al. Abnormalities of intestinal rotation in patients with congenital heart disease and the heterotaxy syndrome. Congenit Heart Dis. 2007;2:12-18.
Foerster SR, Gauvreau K, McElhinney DB, et al. Importance of totally anomalous pulmonary venous connection and postoperative pulmonary vein stenosis in outcomes of heterotaxy syndrome. Pediatr Cardiol. 2008;29:536-544.