Chapter 622 Disorders of the Retina and Vitreous
Retinopathy of Prematurity
Retinopathy of prematurity (ROP) is a complex disease of the developing retinal vasculature in premature infants. It may be acute (early stages) or chronic (late stages). Clinical manifestations range from mild, usually transient changes of the peripheral retina to severe progressive vasoproliferation, scarring, and potentially blinding retinal detachment. ROP includes all stages of the disease and its sequelae. Retrolental fibroplasia, the previous name for this disease, described only the cicatricial stages.
Pathogenesis
Beginning at 16 wk of gestation, retinal angiogenesis normally proceeds from the optic disc to the periphery, reaching the outer rim of the retina (ora serrata) nasally at about 36 wk and extending temporally by approximately 40 wk. Injury to this process results in various pathologic and clinical changes. The first observation in the acute phase is cessation of vasculogenesis. Rather than a gradual transition from vascularized to avascular retina, there is an abrupt termination of the vessels, marked by a line in the retina. The line can then grow into a ridge composed of mesenchymal and endothelial cells. Cell division and differentiation might later resume, and vascularization of the retina can proceed. Alternatively, there may be progression to an abnormal proliferation of vessels out of the plane of the retina, into the vitreous, and over the surface of the retina. Cicatrization and traction on the retina can follow, leading to retinal detachment.
The risk factors associated with ROP are not fully known, but prematurity and the associated retinal immaturity at birth represent the major factors. Oxygenation, respiratory distress, apnea, bradycardia, heart disease, infection, hypercarbia, acidosis, anemia, and the need for transfusion are thought by some to be contributory factors. Generally, the lower the gestational age, the lower the birthweight, and the sicker the infant, the greater the risk is for ROP.
The basic pathogenesis of ROP is still unknown. Exposure to the extrauterine environment including the necessarily high inspired oxygen concentrations produces cellular damage, perhaps mediated by free radicals. Later in the course of the disease, peripheral hypoxia develops and vascular endothelial growth factors (VEGFs) are produced in the nonvascularized retina. These growth factors stimulate abnormal vasculogenesis, and neovascularization can occur. Because of poor pulmonary function, a state of relative retinal hypoxia occurs. This causes upregulation of VEGF, which, in susceptible infants, can cause abnormal fibrovascular growth. This neovascularization can then lead to scarring and vision loss.
Classification
The currently used international classification of ROP describes the location, extent, and severity of the disease. To delineate location, the retina is divided into three concentric zones, centered on the optic disc. Zone I, the posterior or inner zone, extends twice the disc-macular distance, or 30 degrees in all directions from the optic disc. Zone II, the middle zone, extends from the outer edge of zone I to the ora serrata nasally and to the anatomic equator temporally. Zone III, the outer zone, is the residual crescent that extends from the outer border of zone II to the ora serrata temporally. The extent of involvement is described by the number of circumferential clock hours involved.
The phases and severity of the disease process are classified into 5 stages. Stage 1 is characterized by a demarcation line that separates vascularized from avascular retina. This line lies within the plane of the retina and appears relatively flat and white. Often noted is abnormal branching or arcading of the retinal vessels that lead into the line. Stage 2 is characterized by a ridge; the demarcation line has grown, acquiring height, width, and volume and extending up and out of the plane of the retina. Stage 3 is characterized by the presence of a ridge and by the development of extraretinal fibrovascular tissue (Fig. 622-1A). Stage 4 is characterized by subtotal retinal detachment caused by traction from the proliferating tissue in the vitreous or on the retina. Stage 4 is subdivided into 2 phases: (a) subtotal retinal detachment not involving the macula and (b) subtotal retinal detachment involving the macula. Stage 5 is total retinal detachment.
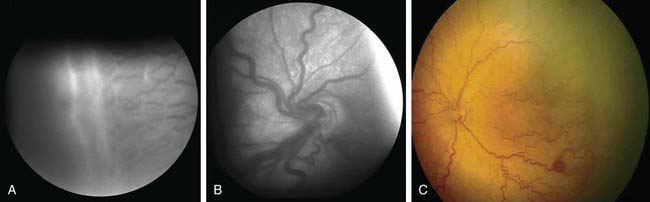
Figure 622-1 Retinopathy of prematurity (ROP). A, In stage 3, there is a ridge and extraretinal vascular tissue. B, Retinal vessels are dilated and tortuous in active ROP plus disease. C, Zone 1, stage ROP with plus disease.
When signs of posterior retinal vascular changes accompany the active stages of ROP, the term plus disease is used (see Fig. 622-1B,C). Patients reaching the point of dilatation and tortuosity of the retinal vessels also often demonstrate the associated findings of engorgement of the iris, pupillary rigidity, and vitreous haze.
Clinical Manifestations and Prognosis
In >90% of at-risk infants, the course is one of spontaneous arrest and regression, with little or no residual effects or visual disability. Less than 10% of infants progress to severe disease, with significant extraretinal vasoproliferation, cicatrization, detachment of the retina, and impairment of vision.
Some children with arrested or regressed ROP are left with demarcation lines, undervascularization of the peripheral retina, or abnormal branching, tortuosity, or straightening of the retinal vessels. Some are left with retinal pigmentary changes, dragging of the retina (dragged disc), ectopia of the macula, retinal folds, or retinal breaks. Others proceed to total retinal detachment, which commonly assumes a funnel-like configuration. The clinical picture is often that of a retrolental membrane, producing leukocoria (a white reflex in the pupil). Some patients develop cataracts, glaucoma, and signs of inflammation. The end stage is often a painful blind eye or a degenerated phthisical eye. The spectrum of ROP also includes myopia, which is often progressive and of significant degree in infancy. The incidence of anisometropia, strabismus, amblyopia, and nystagmus may also be increased.
Diagnosis
Systematic serial ophthalmologic examinations of infants at risk are recommended. In 2006 the American Academy of Pediatrics (AAP) published new screening guidelines for ROP. Infants with a birth weight of <1,500 g or gestational age of ≤32 wk and selected infants with a birth weight between 1,500 and 2,000 g or gestational age of >32 wk with an unstable clinical course, including those requiring cardiorespiratory support and who are believed by their attending pediatrician or neonatologist to be at high risk, should have retinal screening examinations. The timing of the initial screening exam is based on the infant’s age. Table 622-1 was developed from an evidence-based analysis of the Mutlicenter Trial of Cryotherapy for ROP. The examination can be stressful to fragile preterm infants, and the dilating drops can have untoward side effects. Infants must be carefully monitored during and after the examination. Some neonatologists and ophthalmologists advocate the use of topical tetracaine and/or oral sucrose to reduce the discomfort and stress to the infant. Follow-up is based on the initial findings and risk factors but is usually at 2 wk or less.
Table 622-1 TIMING OF FIRST EYE EXAMINATION BASED ON GESTATIONAL AGE AT BIRTH
| GESTATIONAL AGE AT BIRTH (wk) | AGE AT INITIAL EXAMINATION (wk) | |
|---|---|---|
| Postmenstrual | Chronologic | |
| 22 | 31 | 9 |
| 23 | 31 | 8 |
| 24 | 31 | 7 |
| 25 | 31 | 6 |
| 26 | 31 | 5 |
| 27 | 31 | 4 |
| 28 | 32 | 4 |
| 29 | 33 | 4 |
| 30 | 34 | 4 |
| 31 | 35 | 4 |
| 32 | 36 | 4 |
Treatment
In selected cases, cryotherapy or laser photocoagulation of the avascular retina reduces the more-severe complications of progressive ROP. Advances in vitreoretinal surgical techniques have led to limited success in reattaching the retina in infants with total retinal detachment (stage 5 ROP), but the visual results are often disappointing. The Early Treatment for Retinopathy of Prematurity cooperative study demonstrated the importance of plus disease and the presence of posterior retinal involvement in the determination of when to treat ROP. This study also supported the fact that laser is the treatment modality of choice. Peripheral retinal ablation should be considered for any eye with type 1 ROP. Serial examinations are indicated for any eye with type 2 ROP; treatment is considered if type 2 progresses to type 1 or if threshold ROP develops. Intravitreal bevacizumab, an inhibitor of vascular endothelial growth factor, may also have a role in treatment.
Prevention
Prevention of ROP ultimately depends on prevention of premature birth and its attendant problems. The association between ROP and oxygen saturation has been studied for decades. More-recent research has focused on keeping severely premature infants at lower oxygen saturation (85-92%) at age <34 wk and maintaining them at higher oxygen saturation (92-97%) at age >34 wk. This reduction in oxygen saturation early in the infant’s life effectively reduces the phase I hyperoxia and can stimulate the retina to develop normally. The reversal of the hypoxic phase II by elevating the oxygen saturation might ultimately decrease the incidence of severe ROP by down-regulating the secretion of VEGF. Most likely, a multicenter, prospective, randomized study will need to be performed to answer this question. Some investigators have suggested supplemental vitamin E for its antioxidant properties in infants at risk for ROP. Its efficacy has not been proved; at certain dosage levels, it can produce untoward side effects (Chapter 91.2).
Persistent Fetal Vasculature
Persistent fetal vasculature (PFV), formerly called persistent hyperplastic primary vitreous, includes a spectrum of manifestations caused by the persistence of various portions of the fetal hyaloid vascular system and associated fibrovascular tissue.
Pathogenesis
During development of the eye, the hyaloid artery extends from the optic disc to the posterior aspect of the lens; it sends branches into the vitreous and ramifies to form the posterior portion of the vascular capsule of the lens. The posterior portion of the hyaloid system normally regresses by the 7th fetal mo and the anterior portion regresses by the 8th fetal mo. Small remnants of the system, such as a tuft of tissue at the disc (Bergmeister papilla) or a tag of tissue on the posterior capsule of the lens (Mittendorf dot), are common findings in healthy persons. More-extensive remnants and associated complications constitute PFV. Two major forms are described, anterior PFV and posterior PFV. Variability is great, and mixed or intermediate forms occur.
Clinical Manifestations
The usual clinical feature of anterior PFV is the presence of a vascularized plaque of tissue on the back surface of the lens in an eye that is microphthalmic or slightly smaller than normal. The condition is usually unilateral and can occur in infants with no other abnormalities and no history of prematurity. The fibrovascular tissue tends to undergo gradual contracture. The ciliary processes become elongated, and the anterior chamber can become shallow. The lens usually is smaller than normal and may be clear but often develops cataracts and can swell or absorb fluid. Large or anomalous vessels of the iris may be present. The anterior chamber angle can have abnormalities. In time, the cornea can become cloudy.
Anterior PFV is usually noted in the 1st wk or mo of life. The most common presenting signs are leukocoria (white pupillary reflex), strabismus, and nystagmus. The course is usually progressive and the outcome poor. Major complications are spontaneous intraocular hemorrhage, swelling of the lens caused by rupture of the posterior capsule, and glaucoma. The eye might eventually deteriorate. The spectrum of posterior PFV includes fibroglial veils around the disc and macula, vitreous membranes and stalks containing hyaloid artery remnants projecting from the disc, and meridional retinal folds. Traction detachment of the retina can occur. Vision may be impaired, but the eye is usually retained.
Treatment
Surgery is performed in an effort to prevent complications, to preserve the eye and a reasonably good cosmetic appearance, and, in some cases, to salvage vision. Surgical treatment usually involves aspirating the lens and excising the abnormal tissue. If useful vision is to be attained, refractive correction and aggressive amblyopia therapy are required. In some cases, the affected eye is enucleated because distinguishing between this white mass and retinoblastoma can be difficult. Ultrasonography and CT are valuable diagnostic aids.
Retinoblastoma
Retinoblastoma (Fig. 622-2, Chapter 496) is the most common primary malignant intraocular tumor of childhood. It occurs in approximately 1/15,000 live births; 250-300 new cases are diagnosed in the United States annually. Hereditary and nonhereditary patterns of transmission occur; there is no gender or race predilection. The hereditary form is usually bilateral and multifocal, whereas the nonhereditary form is generally unilateral and unifocal. About 15% of unilateral cases are hereditary. Bilateral cases often manifest earlier than unilateral cases. Unilateral tumors are often large by the time they are discovered. The average age at diagnosis is 15 mo for bilateral cases, compared with 25 mo for unilateral cases. It is unusual for a child to present with a retinoblastoma after 3 yr of age. Rarely, the tumor is discovered at birth, during adolescence, or even in early adulthood.
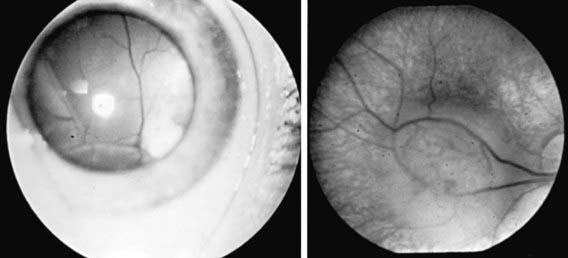
Clinical Manifestations
The clinical manifestations of retinoblastoma vary, depending on the stage at which the tumor is detected. The initial sign in the majority of patients is a white pupillary reflex (leukocoria). Leukocoria results because of the reflection of light off the white tumor. The second most common initial sign of retinoblastoma is strabismus. Less-common presenting signs include pseudohypopyon (tumor cells layered inferiorly in front of the iris) caused by tumor seeding in the anterior chamber of the eye, hyphema (blood layered in front of the iris) secondary to iris neovascularization, vitreous hemorrhage, and signs of orbital cellulitis. On examination, the tumor appears as a white mass, sometimes small and relatively flat, sometimes large and protuberant. It might appear nodular. Vitreous haze or tumor seeding may be evident.
The retinoblastoma gene is a recessive suppressor gene located on chromosome 13 at the 13q14 region. Because of the hereditary nature of retinoblastoma, family members of affected children should undergo a complete ophthalmologic examination and genetic counseling. Newborn siblings and children of affected patients should be referred to an ophthalmologist shortly after birth, when the peripheral retina can be evaluated without the need for an examination under anesthesia.
Diagnosis
Diagnosis is made by direct observation by an experienced ophthalmologist. Ancillary testing such as CT or ultrasonography can help to confirm the diagnosis and demonstrate calcification within the mass. MRI can better detect the presence of an associated pinealblastoma (trilateral retinoblastoma). A definitive diagnosis occasionally cannot be made, and removal of the eye must be considered to avoid the possibility of lethal metastasis of the tumor. Because a biopsy can lead to spread of the tumor, histologic confirmation before enucleation is not possible in most cases. Therefore, removal of a blind eye in which the diagnosis of retinoblastoma is likely may be appropriate.
Treatment
Therapy varies, depending on the size and location of the tumor as well as whether it is unilateral or bilateral. Advanced tumors may be treated by enucleation. Other treatment modalities include external beam irradiation, radiation plaque therapy, laser or cryotherapy, and chemotherapy. Since the turn of the century there has been a dramatic shift in the treatment of retinoblastomas. Chemoreduction (systemic chemotherapy) followed by local therapies (laser, cryotherapy, and brachytherapy) have markedly reduced the use of external beam radiation and is a more vision-sparing technique. Children who are irradiated during their 1st year of life are 2-8 times more likely to develop second cancers as those irradiated after 1 yr of age. Patients treated with radiation tend to develop brain tumors and sarcomas of the head and neck. Secondary cataracts can also develop from radiation.
Nonocular secondary tumors are common in patients with germinal mutations estimated to occur with an incidence of 1% per year of life. The most common secondary tumors are osteogenic sarcoma of the skull and long bones.
The prognosis for children with retinoblastoma depends on the size and extension of the tumor. When confined to the eye, most tumors can be cured. The prognosis for long-term survival is poor when the tumor has extended into the orbit or along the optic nerve.
Retinitis Pigmentosa
Retinitis pigmentosa (RP) is a progressive retinal degeneration characterized by pigmentary changes, arteriolar attenuation, usually some degree of optic atrophy, and progressive impairment of visual function. Dispersion and aggregation of the retinal pigment produce various ophthalmoscopically visible changes, ranging from granularity or mottling of the retinal pigment pattern to distinctive focal pigment aggregates with the configuration of bone spicules (Fig. 622-3). Other ocular findings include subcapsular cataract, glaucoma, and keratoconus.
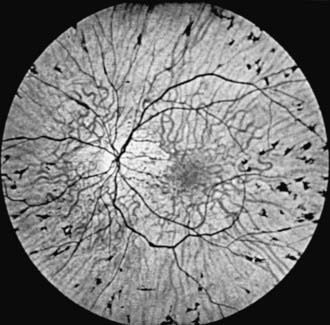
Impairment of night vision or dark adaptation is often the first clinical manifestation noted in adolescents. Progressive loss of peripheral vision, often in the form of an expanding ring scotoma or concentric contraction of the field, is usual. There may be loss of central vision. Retinal function, as measured by electroretinography (ERG), is characteristically reduced. The disorder may be autosomal recessive, autosomal dominant, or X linked. There are >45 identified genes that account for only 60% of patients. These genes are involved in the phototransduction cascade, vitamin A metabolism, cytoskeletal structure, signaling or synaptic pathways, trafficking of intracellular proteins, maintenance of cilia, pH regulation, and phagocytosis. Children with autosomal recessive RP are more likely to become symptomatic at an earlier age (median age, 10.7 yr). Those with autosomal dominant RP are more likely to present in their 20s. Only supportive treatment is available. Vitamin A palmitate and omega-3–rich fish oil might slow disease progression.
A special form of RP is Leber congenital retinal amaurosis (LCA), in which the retinal changes tend to be pleomorphic, with various degrees of pigment disorder, arteriolar attenuation, and optic atrophy. The retina can appear normal during infancy. Vision impairment, nystagmus, and poor pupillary reaction is usually evident soon after birth, and the ERG findings are abnormal early and confirm the diagnosis. LCA is caused by mutations in at least 13 genes. Type 2 LCA is seen in approximately 6% of patients and is due to a mutation in the RPE65 gene, which produces 11-cis-retinal from all-trans-retinyl esters. Gene replacement therapy (subretinal injection) currently shows early promise for children affected with LCA type 2.
Clinically similar secondary pigmentary retinal degenerations that need to be differentiated from RP occur in a wide variety of metabolic diseases, neurodegenerative processes, and multifaceted syndromes. Examples include the progressive retinal changes of the mucopolysaccharidoses (particularly Hurler, Hunter, Scheie, and Sanfilippo syndromes) and certain of the late-onset gangliosidoses (Batten-Mayou, Spielmeyer-Vogt, and Jansky-Bielschowsky diseases), the progressive retinal degeneration that is associated with progressive external ophthalmoplegia (Kearns-Sayre syndrome), and the RP-like changes in the Laurence-Moon and Bardet-Biedl syndromes. The retinal manifestations of abetalipoproteinemia (Bassen-Kornzweig syndrome) and Refsum disease are also similar to those found in RP. The diagnosis of these latter two disorders in a patient with presumed RP is important because treatment is possible. There is also an association of RP and congenital hearing loss, as in Usher syndrome (Chapters 80 and 82).
Stargardt Disease (Fundus Flavimaculatus)
Stargardt disease is an autosomal recessive retinal disorder characterized by slowly progressive bilateral macular degeneration and vision impairment. It usually appears at 8-14 yr of age and is often initially misdiagnosed as functional visual loss. The foveal reflex becomes obtunded or appears grayish, pigment spots develop in the macular area, and macular depigmentation and chorioretinal atrophy eventually occur. Macular hemorrhages can develop. Some patients also have white or yellow spots beyond the macula or pigmentary changes in the periphery; the term fundus flavimaculatus is commonly used for this condition. It is now recognized that Stargardt disease and fundus flavimaculatus represent different parts on the spectrum of the same disease. Central visual acuity is reduced, often to 20/200, but total loss of vision does not occur. ERG findings vary. The condition is not associated with central nervous system abnormalities and is to be differentiated from the macular changes of many progressive metabolic neurodegenerative diseases. The genetic mutation responsible for Stargardt macular dystrophy has been identified.
Best Vitelliform Degeneration
Best vitelliform degeneration is a macular dystrophy characterized by a distinctive yellow or orange discoid subretinal lesion in the macula, resembling the intact yolk of a fried egg. Diagnosis is usually made at 3-15 yr of age, with a mean age of presentation of 6 yr. Vision is usually normal at this stage. The condition may be progressive; the yolklike lesion can eventually degenerate (“scramble”) and result in pigmentation, chorioretinal atrophy, and vision impairment. The condition is usually bilateral. There is no association with systemic abnormalities. Inheritance is usually autosomal dominant. The vitelliform macular dystrophy gene (VMD2) has been identified and DNA testing is available. In vitelliform macular degeneration, the ERG response is normal. Electro-oculographic findings are abnormal in affected patients and carriers, and this test is useful in diagnosis and in genetic counseling.
Cherry Red Spot
Because of the special histologic features of the macula, certain pathologic processes affecting the retina produce an ophthalmoscopically visible sign referred to as a cherry red spot, a bright to dull red spot at the center of the macula surrounded and accentuated by a grayish-white or yellowish halo. The halo is a result of a loss of transparency of the retinal ganglion cell layer secondary to edema, lipid accumulation, or both. Because ganglion cells are not present in the fovea, the retina surrounding the fovea is opacified but the fovea transmits the normal underlying choroidal color (red), accounting for the presence of the cherry red spot. A cherry red spot typically occurs in certain sphingolipidoses, principally in Tay-Sachs disease (GM2 type 1), in the Sandhoff variant (GM2 type 2), and in generalized gangliosidosis (GM1 type 1). Similar but less-distinctive macular changes occur in some cases of metachromatic leukodystrophy (sulfatide lipidosis), in some forms of neuronopathic Niemann-Pick disease, and in certain mucolipidoses (Chapters 80.4 and 80.5). The cherry red spot that characteristically occurs as a result of retinal ischemia secondary to vasospasm, ocular contusion, or occlusion of the central retinal artery must be differentiated from the cherry red spot of neurodegenerative disease.
Phakomas
Phakomas (Chapter 589) are the herald lesions of the hamartomatous disorders. In Bourneville disease (tuberous sclerosis), the distinctive ocular lesion is a refractile, yellowish, multinodular cystic lesion arising from the disc or retina; the appearance of this typical lesion is often compared with that of an unripe mulberry (Fig. 622-4). Equally characteristic and more common in tuberous sclerosis are flatter, yellow to whitish retinal lesions, varying in size from minute dots to large lesions approaching the size of the disc. These lesions are benign astrocytic proliferations. Rarely, similar retinal phakomas occur in von Recklinghausen disease (neurofibromatosis). In von Hippel-Lindau disease (angiomatosis of the retina and cerebellum), the distinctive fundus lesion is a hemangioblastoma; this vascular lesion usually appears as a reddish globular mass with large paired arteries and veins passing to and from the lesion. In Sturge-Weber syndrome (encephalofacial angiomatosis), the fundus abnormality is a choroidal hemangioma; the hemangioma can impart a dark color to the affected area of the fundus, but the lesion is best seen with fluorescein angiography.
Retinoschisis
Congenital hereditary retinoschisis, also referred to as juvenile X-linked retinoschisis, is a bilateral vitreoretinal dystrophy that has a bimodal age of presentation. The first group presents with strabismus and nystagmus at a mean age of 1.5-2 yr and is the most severely affected group. The second group presents at 6-7 yr with poor vision. It is characterized by splitting of the retina into inner and outer layers. The usual ophthalmoscopic finding in affected boys is an elevation of the inner layer of the retina, most commonly in the inferotemporal quadrant of the fundus, often with round or oval holes visible in the inner layer. Schisis of the fovea is virtually pathognomonic and is found in almost 100% of patients. Ophthalmoscopically, this appears in early stages as small, fine striae in the internal limiting membrane. These striae radiate outward in a petaloid or spoke-wheel configuration. In some cases, frank retinal detachment or vitreous hemorrhage occurs.
Vision impairment varies from mild to severe; visual acuity can worsen with age, but good vision is often retained. Carrier females are asymptomatic, but linkage studies may be useful to help detect carriers.
Retinal Detachment
A retinal detachment is a separation of the outer layers of the retina from the underlying retinal pigment epithelium (RPE). During embryogenesis, the retina and RPE are initially separated. During ocular development, they join and are held in apposition to each other by various physiologic mechanisms. Pathologic events leading to a retinal detachment return the retina-RPE to its former separated state. The detachment can occur as a congenital anomaly but more commonly arises secondary to other ocular abnormalities or trauma.
Three types of detachment are described; each can occur in children. Rhegmatogenous detachments result from a break in the retina that allows fluid to enter the subretinal space. In children, these are usually a result of trauma (such as child abuse) but can occur secondary to myopia or ROP or after congenital cataract surgery. Tractional retinal detachments result when vitreoretinal membranes pull on the retina. They can occur in diabetes, sickle cell disease, and ROP. Exudative retinal detachments result when exudation exceeds absorption. This can be seen in Coats disease, retinoblastoma, and ocular inflammation.
The presenting sign of retinal detachment in an infant or child may be loss of vision, secondary strabismus or nystagmus, or leukocoria (white pupillary reflex). In addition to direct examination of the eye, special diagnostic studies such as ultrasonography and neuroimaging (CT, MRI) may be necessary to establish the cause of the detachment and the appropriate treatment. Prompt treatment is essential if vision is to be salvaged.
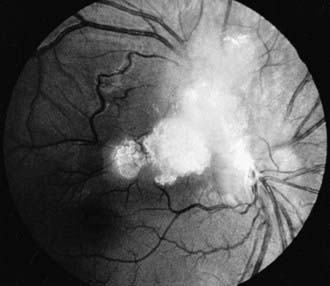
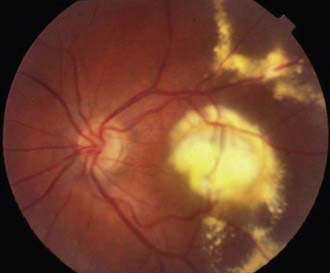
Coats Disease
Coats disease is an exudative retinopathy of unknown cause characterized by telangiectasia of retinal vessels with leakage of plasma to form intraretinal and subretinal exudates and by retinal hemorrhages and detachment (Fig. 622-5). The condition is usually unilateral. It predominantly affects boys, usually appearing in the 1st decade. The condition is nonfamilial and for the most part occurs in otherwise healthy children. The most common presenting signs are blurring of vision, leukocoria, and strabismus. Rubeosis of the iris, glaucoma, and cataract can develop. Treatment with photocoagulation or cryotherapy may be helpful.
Familial Exudative Vitreoretinopathy
Familial exudative vitreoretinopathy (FEVR) is a progressive retinal vascular disorder of unknown cause, but clinical and angiographic findings suggest an aberration of vascular development. Avascularity of the peripheral temporal retina is a significant finding in most cases, with abrupt cessation of the retinal capillary network in the region of the equator. The avascular zone often has a wedge- or V-shaped pattern in the temporal meridian. Glial proliferation or well-marked retinochoroidal atrophy may be found in the avascular zone. Excessive branching of retinal arteries and veins, dilatation of the capillaries, arteriovenous shunt formation, neovascularization, and leakage from retinal vessels of the farthest vascularized retina occur. Vitreoretinal adhesions are usually present at the peripheral margin of the vascularized retina. Traction, retinal dragging and temporal displacement of the macula, falciform retinal folds, and retinal detachment are common. Intraretinal or subretinal exudation, retinal hemorrhage, and recurrent vitreous hemorrhages can develop. Patients can also develop cataracts and glaucoma. Vision impairment of varying severity occurs. The condition is usually bilateral. FEVR is usually an autosomal dominant condition with incomplete penetrance. Asymptomatic family members often display a zone of avascular peripheral retina.
The findings in FEVR can resemble those of ROP in the cicatricial stages, but unlike ROP, the neovascularization of FEVR seems to develop years after birth and most patients with FEVR have no history of prematurity, oxygen therapy, prenatal or postnatal injury or infection, or developmental abnormalities. FEVR is also to be differentiated from Coats disease, angiomatosis of the retina, peripheral uveitis, and other disorders of the posterior segment.
Hypertensive Retinopathy
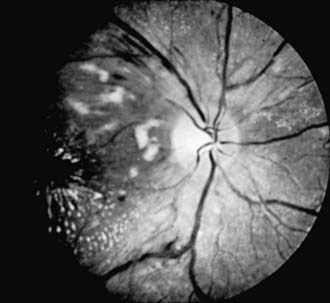
In the early stages of hypertension, no retinal changes may be observable. Generalized constriction and irregular narrowing of the arterioles are usually the first signs in the fundus. Other alterations include retinal edema, flame-shaped hemorrhages, cotton-wool spots (retinal nerve fiber layer infarcts), and papilledema (Fig. 622-6). These changes are reversible if the hypertension can be controlled in the early stages, but in long-standing hypertension, changes may be irreversible. Thickening of the vessel wall can produce a silver- or copper-wire appearance. Hypertensive retinal changes in a child should alert the physician to renal disease, pheochromocytoma, collagen disease, and cardiovascular disorders, particularly coarctation of the aorta.
Diabetic Retinopathy
The retinal changes of diabetes mellitus are classified as nonproliferative or proliferative. Nonproliferative diabetic retinopathy is characterized by retinal microaneurysms, venous dilatation, retinal hemorrhages, and exudates. The microaneurysms appear as tiny red dots. The hemorrhages may be of both the dot and blot type, representing deep intraretinal bleeding, and the splinter or flame-shaped type, involving the superficial nerve fiber layer. The exudates tend to be deep and to appear waxy. There may also be superficial nerve fiber infarcts called cytoid bodies or cotton-wool spots, as well as retinal edema. These signs may wax and wane. They are seen primarily in the posterior pole, around the disc and macula, well within the range of direct ophthalmoscopy. Involvement of the macula can lead to decreased vision.
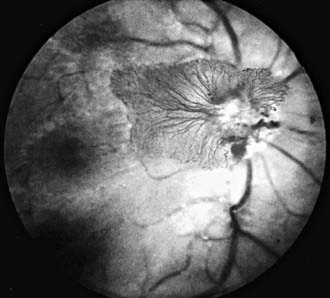
Proliferative retinopathy, the more serious form, is characterized by neovascularization and proliferation of fibrovascular tissue on the retina, extending into the vitreous. Neovascularization can occur on the optic disc (NVD), elsewhere on the retina (NVE), or on the iris and in the anterior chamber angle (NVI, or rubeosis irides) (Fig. 622-7). Traction on these new vessels leads to hemorrhage and eventually scarring. The vision-threatening complications of proliferative diabetic retinopathy are retinal and vitreous hemorrhages, cicatrization, traction, and retinal detachment. Neovascularization of the iris can lead to secondary glaucoma if not treated promptly.
Diabetic retinopathy involves the alteration and nonperfusion of retinal capillaries, retinal ischemia, and neovascularization, but its pathogenesis is not yet completely understood, either in terms of location of the primary pathogenetic mechanism (retinal vessels vs surrounding neuronal or glial tissue) or the specific biochemical factors involved. The better the degree of long-term metabolic control, the lower the risk of diabetic retinopathy.
Clinically, the prevalence and course of retinopathy relate to a patient’s age and to disease duration. Detectable microvascular changes are rare in prepubertal children, with the prevalence of retinopathy increasing significantly after puberty, especially after the age of 15 yr. The incidence of retinopathy is low during the first 5 yr of disease and increases progressively thereafter, with the incidence of proliferative retinopathy becoming substantial after 10 yr and with increased risk of visual impairment after 15 yr or more.
Ophthalmic examination guidelines have been proposed by the AAP. An initial exam is recommended at age 9 yr if the diabetes is poorly controlled. If the diabetes is well controlled, an initial exam 3 years after puberty with annual follow-up is recommended.
In addition to retinopathy, patients with juvenile-onset diabetes can develop optic neuropathy, characterized by swelling of the disc and blurring of vision. Patients with diabetes can also develop cataracts, even at an early age, sometimes with rapid progression.
Treatment
Macular edema is the leading cause of visual loss in diabetic persons. Photocoagulation may be used to decrease the risk of continued vision loss in patients with macular edema.
Proliferative retinopathy causes the most-severe vision loss and can lead to total loss of vision and even loss of the eye. Patients who have proliferative disease and who display certain high-risk characteristics should undergo panretinal photocoagulation to preserve their central vision. Neovascularization of the iris is also treated with panretinal photocoagulation to stop the development of neovascular glaucoma.
Vitrectomy and other intraocular surgery may be necessary in patients with nonresolving vitreous hemorrhage or traction retinal detachment. The value of technologic advances, such as insulin infusion pumps and pancreatic transplants, in preventing ocular complications is under investigation (Chapter 583).
Subacute Bacterial Endocarditis
At some time during the course of the disease, retinopathy is present in approximately 40% of cases of subacute bacterial endocarditis. The lesions include hemorrhages, hemorrhages with white centers (Roth spots), papilledema, and, rarely, embolic occlusion of the central retinal artery.
Blood Disorders
In primary and secondary anemias, retinopathy in the form of hemorrhages and cotton-wool patches can occur. Vision can be affected if hemorrhage occurs in the macular area. The hemorrhages may be light and feathery or dense and preretinal. In polycythemia vera, the retinal veins are dark, dilated, and tortuous. Retinal hemorrhages, retinal edema, and papilledema may be observed. In leukemia, the veins are characteristically dilated, with sausage-shaped constrictions; hemorrhages, particularly white-centered hemorrhages and exudates, are common during the acute stage. In the sickling disorders, fundus changes include vascular tortuosity, arterial and venous occlusions, salmon patches, refractile deposits, pigmented lesions, arteriolar-venous anastomoses, and neovascularization (with sea-fan formations), sometimes leading to vitreous hemorrhage and retinal detachment. Patients with HbSC and HbS-β-thalassemia hemoglobinopathies are at a higher risk for developing retinopathy than are those with HbSS disease. It is thought that the more anemic state of patients with SS disease offers protection from vascular occlusions in the retina.
Trauma-Related Retinopathy
Retinal changes can occur in patients who suffer trauma to other parts of the body. The occurrence of retinal hemorrhages in infants who have been physically abused is well documented (Fig. 622-8; Chapter 37). Retinal, subretinal, subhyaloid, and vitreous hemorrhages have been described in infants and young children with inflicted neurotrauma. Often there are no signs of direct trauma to the eye, periocular region, or head. Such cases can result from violent shaking of an infant, and permanent retinal damage can result.
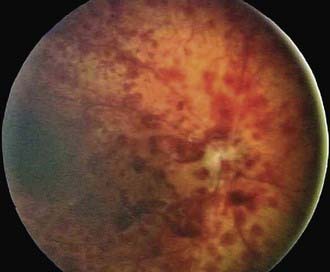
Figure 622-8 Shaken baby syndrome (inflicted neurotrauma). Retinal hemorrhages in multiple layers too numerous to count into far periphery.
In patients with severe head or chest compressive trauma, a traumatic retinal angiopathy known as Purtscher retinopathy can occur. This is characterized by retinal hemorrhage, cotton-wool spots, possible disc swelling, and decreased vision. The pathogenesis is unclear, but there is evidence of arteriolar obstruction in this condition. A Purtscher-like fundus picture can also occur in several nontraumatic settings, such as acute pancreatitis, lupus erythematosus, and childbirth.
Myelinated Nerve Fibers
Myelination of the optic nerve fibers normally terminates at the level of the disc, but in some children, ectopic myelination extends to nerve fibers of the retina. The condition is most commonly seen adjacent to the disc, although more peripheral areas of the retina may be involved. The characteristic ophthalmoscopic picture is a focal white patch with a feathered edge or brush-stroke appearance. Because the macula is generally unaffected, the visual prognosis is good. A relative or absolute visual field defect corresponding to areas of ectopic myelination is usually the only associated ocular abnormality. Extensive unilateral involvement, however, has been associated with ipsilateral myopia, amblyopia, and strabismus. If unilateral high myopia and amblyopia are present, appropriate optical correction and occlusion therapy should be instituted. For unknown reasons, the disorder is more commonly encountered in patients with craniofacial dysostosis, oxycephaly, neurofibromatosis, and Down syndrome.
Coloboma of the Fundus
The term coloboma describes a defect such as a gap, notch, fissure, or hole. The typical fundus coloboma is a result of malclosure of the embryonic fissure, which leaves a gap in the retina, RPE, and choroid, thus baring the underlying sclera. The defect may be extensive, involving the optic nerve, ciliary body, iris, and even lens, or it may be localized to 1 or more portions of the fissure. The usual appearance is of a well-circumscribed, wedge-shaped white area extending inferonasally below the disc, sometimes involving or engulfing the disc. In some cases, there is ectasia or cyst formation in the area of the defect. Less-extensive colobomatous defects might appear as only single or multiple focal punched-out chorioretinal defects or anomalous pigmentation of the fundus in the line of the embryonic fissure. Colobomas can occur in 1 or both eyes. A visual field defect usually corresponds to the chorioretinal defect. Visual acuity may be impaired, particularly if the defect involves the disc or macula.
Fundus colobomas can occur in isolation as sporadic defects or as an inherited condition. Isolated colobomatous anomalies are commonly inherited in an autosomal dominant manner with highly variable penetrance and expressivity. Family members of affected patients should receive appropriate genetic counseling. Colobomas may also be associated with such abnormalities as microphthalmia, glioneuroma of the eye, cyclopia, or encephale. They occur in children with various chromosomal disorders, including trisomies 13 and 18, triploidy, cat-eye syndrome, and 4p-. Ocular colobomas also occur in many multisystem disorders, including the CHARGE association (coloboma, heart disease, atresia choanae, retarded growth and development and/or central nervous system anomalies, genetic anomalies and/or hypogonadism, ear anomalies and/or deafness); Joubert, Aicardi, Meckel, Warburg, and Rubinstein-Taybi syndromes; linear sebaceous nevus; Goldenhar and Lenz microphthalmia syndromes; and Goltz focal dermal hypoplasia.
Abramson DH, Frank CM, Susman M, et al. Presenting signs of retinoblastoma. J Pediatr. 1998;132:505-508.
American Academy of Pediatrics. Screening examinations of premature infants for retinopathy of prematurity. Pediatrics. 2006;117:572-576.
Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231-2239.
Chang M, McLean IW, Merritt JC. Coats’ disease: a study of 62 histologically confirmed cases. J Pediatr Ophthalmol Strabismus. 1984;21:163-168.
Chiang MF, Wang L, Busuioc M, et al. Telemedical retinopathy of prematurity diagnosis. Arch Ophthamol. 2007;125:1531-1538.
Cremers FPM, Collin RWJ. Promised and challenges of genetic therapy for blindness. Lancet. 2009;374:1569-1570.
Cryotherapy for Retinopathy of Prematurity Cooperative Group. 15-year outcomes following threshold retinopathy of prematurity. Arch Ophthalmol. 2005;123:311-318.
Dass AB, Trese MT. Surgical results of persistent hyperplastic primary vitreous. Ophthalmology. 1999;106:280-284.
Early Treatment Diabetic Retinopathy Research Study Group. Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report 1. Arch Ophthalmol. 1985;103:1796-1806.
Early Treatment for Retinopathy of Prematurity Cooperative Group. Revised indications for the treatment of retinopathy of prematurity: results of the Early Treatment for Retinopathy of Prematurity randomized trial. Arch Ophthalmol. 2003;121:1684-1696.
El-Aziz MM, Barragan I, O’Driscoll CA, et al. EYS, encoding an ortholog of Drosophila, Spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genetics. 2008;40:1285-1287.
Gallie B. Canadian guidelines for retinoblastoma care. Can J Ophthalmol. 2009;44:639-642.
George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthmol. 1996;114:274-280.
Good WV. Retinopathy of prematurity and the peripheral retina. J Pediatr. 2008;153:591-592.
Hardwig P, Robertson DM. Von Hippel-Lindau disease: a familial, often lethal, multi-system phakomatosis. Ophthalmology. 1984;91:263-270.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795-1809.
Hellström A, Ley D, Hansen-Pupp I, et al. New insights into the development of retinopathy of prematurity—importance of early weight gain. Acta Paediatr. 2009;99:502-508.
Hollands H, Johnson D, Brox AC, et al. Acute-onset floaters and flashes. JAMA. 2009;302:2243-2249.
Kim JW, Abramson DH, Dunkel IJ. Current management strategies for intraocular retinoblastoma. Drugs. 2007;67(15):2173-2185.
Koenekoop RK. Successful RPE65 gene replacement and improved visual function in humans. Ophthalm Genet. 2008;29(3):89-91.
Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597-1604.
Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240-2248.
Mintz-Hittner HA, Kennedy KA, Chuang AZ. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364(7):603-614.
Miyakulo H, Hashimoto K, Miyakulo S. Retinal vascular pattern in familial exudative vitreoretinopathy. Ophthalmology. 1984;91:1524-1530.
Mohler CW, Fine SL. Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmology. 1981;88:688-692.
Noble KG, Carr RE. Leber’s congenital amaurosis: a retrospective study of 33 cases and a histopathological study of one case. Arch Ophthalmol. 1978;96:818-821.
Pawlik D, Lauterbach R, Turyk E. Fish-oil fat emulsion supplementation may reduce the risk of severe retinopathy in VLBW infants. Pediatrics. 2011;127(2):223-228.
Richter GM, Williams SL, Starren J, et al. Telemedicine for retinopathy of prematurity diagnosis: evaluation and challenges. Surv Ophthalmol. 2009;54:671-685.
Sage J. Hope in sight for retinoblastoma. Nat Med. 2007;13:30-31.
Sears JE, Pietz J, et al. A change in oxygen supplementation can decrease the incidence of ROP. Ophthalmology. 2009;116(3):513-518.
Shalev B, Farr A, Repka MX. Randomized comparison of diode laser photocoagulation versus cryotherapy for threshold retinopathy of prematurity: seven year outcome. Am J Ophthalmol. 2001;132:76-80.
Shields CL, Gorry T, Shields JA. Outcome of eyes with unilateral sporadic retinoblastoma based on the initial external findings by the family and the pediatrician. J Pediatr Ophthalmol Strabismus. 2004;41:143-149.
Shields CL, Palamar M, Sharma P, et al. Retinoblastoma regression patterns following chemoreduction and adjuvant therapy in 557 tumors. Arch Ophthalmol. 2009;127:282-290.