Chapter 648 Photosensitivity
Photosensitivity denotes a qualitatively or quantitatively abnormal cutaneous reaction to sunlight or artificial light.
Acute Sunburn Reaction
The most common photosensitive reaction seen in children is acute sunburn. Sunburn is caused mainly by ultraviolet (UV) B radiation (290-320 nm wavelength). Sunlight contains many times more UVA (320-400 nm) than UVB radiation, but UVA must be encountered in much larger quantities than UVB radiation to produce sunburn.
Pathophysiology and Clinical Manifestations
Transmitted radiation <300 nm is largely absorbed in the epidermis, whereas that >300 nm is mostly transmitted to the dermis after variable epidermal melanin absorption. Children vary in susceptibility to UV radiation, depending on their skin type (amount of pigment) (Table 648-1). Immediate pigment darkening is due to UVA radiation–induced photo-oxidative darkening of existing melanin and its transfer from melanocytes to keratinocytes. This effect generally lasts for a few hours and is not photoprotective. UVB-induced effects appear 6-12 hr after initial exposure and reach a peak in 24 hr. Effects include redness, tenderness, edema, and blistering (Fig. 648-1). Reactive oxidation species generated by UVB induce keratinocyte membrane damage and are involved in the pathogenesis of sunburn. A portion of the vasodilatation seen in UVB-induced erythema is mediated by prostaglandins E2 and F2. Delayed melanogenesis as a result of UVB radiation begins in 2-3 days and lasts several days to a few weeks. Manufacture of new melanin in melanocytes, transfer of melanin from melanocytes to keratinocytes, increase in size and arborization of melanocytes, and activation of quiescent melanocytes produce delayed melanogenesis. This effect reduces skin sensitivity to development of UV-induced erythema. The amount of protection afforded depends on the skin type of the patient. Additional effects and possible complications of sun exposure include increased thickness of the stratum corneum, recurrence or exacerbation of herpes simplex labialis, lupus erythematosus, and many other conditions (Table 648-2).
Table 648-1 SUN-REACTIVE SKIN TYPES
| TYPE AND DEMOGRAPHICS | SUNBURN, TANNING HISTORY |
|---|---|
| I Red hair, freckles, Celtic origin | Always burns easily, no tanning |
| II Fair skin, fair-haired, blue-eyed, white | Usually burns, minimal tanning |
| III Darker-skinned white | Sometimes burns, gradual light brown tan |
| IV Mediterranean background | Minimal to no burning, always tans |
| V Middle Eastern white, Mexican | Rarely burns, tans profusely dark brown |
| VI Black | Never burns, pigmented black |
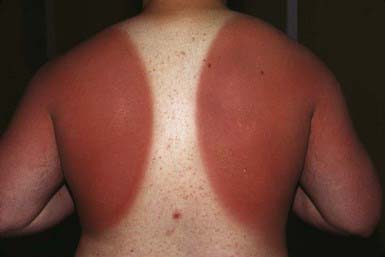
Table 648-2 CUTANEOUS REACTIONS TO SUNLIGHT
Treatment
Acute severe sunburn should be managed with cool compresses. Topical corticosteroids and oral prostaglandin inhibitors such as ibuprofen and indomethacin may decrease erythema and pain but must be administered preradiation or early in the course of the sunburn. Once peak erythema has been reached, little help is afforded by these medications. Proprietary preparations containing topical anesthetics are relatively ineffective and potentially hazardous because of their propensity to cause contact dermatitis. A bland emollient is effective in the desquamative phase.
Prognosis and Prevention of Sequelae
The long-term sequelae of chronic and intense sun exposure are not often seen in children, but most individuals receive >50% of their lifetime UV dose by age 20 yr. Therefore, pediatricians have a pivotal role in educating patients and their parents about the harmful effects, potential malignancy risks, and irreversible skin damage that result from unduly prolonged exposure to the sun and tanning lights. Premature aging, senile elastosis, actinic keratoses, squamous and basal cell carcinomas, and melanomas all occur with greater frequency in sun-damaged skin. In particular, blistering sunburns in childhood and adolescence significantly increase the risk for development of malignant melanoma. Sun protection is best achieved by sun avoidance, which includes minimizing time in the midday sun (10 AM to 3 PM), staying in the shade, and wearing protective clothing including wide-brimmed hats. Protection is enhanced by a wide variety of sunscreen agents. Physical opaque sunscreens (zinc oxide, titanium dioxide) block UV light, whereas chemical sunscreens (para-aminobenzoic acid [PABA], PABA esters, salicylates, benzophenones, dibenzoylmethanes [avobenzone], cinnamates, terephthalylidene dicamphor sulfonic acid [ecamsule]) absorb damaging radiation. The benzophenones and dibenzoylmethanes provide protection in both the UVA and UVB ranges. Stabilizers such as octocrylene and diethyl 2,6-naphthalate increase the time of function of the dibenzoylmethanes. Ecamsule and drometrizole trisiloxane are UVA sunscreens. Vitamins C and E added to sunscreen may also be beneficial in reduction of the formation of reactive oxygen species. Children with skin types I to III (see Table 648-1) require sunscreens with a sun protection factor (SPF) of at least 15, although the higher SPF, the more protection. SPF is defined as the minimal dose of sunlight required to produce cutaneous erythema after application of a sunscreen, divided by the dose required with no use of sunscreen. SPF applies only to UVB protection. UVA rating of sunscreens should be available in the near future.
Photosensitive Reactions
Photosensitizers in combination with a particular wavelength of light cause dermatitis that can be classified as a phototoxic or a photoallergic reaction. Contact of the skin with the photosensitizer may occur externally, internally by enteral or parenteral administration, or through host synthesis of photosensitizers in response to an administered drug.
Photoallergic reactions occur in only a small percentage of persons exposed to photosensitizers and light and require a time interval for sensitization to take place. Thereafter, dermatitis appears within ≈ 24 hr of re-exposure to the photosensitizer and light. Photoallergic dermatitis is a T cell–mediated delayed hypersensitivity reaction in which the drug, acting as a hapten, may combine with a skin protein to form the antigenic substance. Photoallergic reactions vary in morphology and may occur on partially covered and on light-exposed skin. Some of the important classes of drugs and chemicals responsible for photosensitivity reactions are listed in Table 648-2.
Phototoxic reactions occur in all individuals who accumulate adequate amounts of a photosensitizing drug or chemical within the skin. Prior sensitization is not required. Dermatitis develops within hours after exposure to radiation in the range of 285-450 nm. The eruption is confined to light-exposed areas and often resembles exaggerated sunburn, but it may be urticarial or bullous. It results in postinflammatory hyperpigmentation. All the drugs that cause photoallergic reactions may also cause a phototoxic dermatitis if given in sufficiently high doses. Several additional drugs and contactants cause phototoxic reactions, notably the plant-derived furocoumarins (see Table 648-2). Differentiation from contact dermatitis due to poison ivy or poison oak may be difficult, but itching is prominent in contact dermatitis. In phytophotodermatitis, burning is prominent and is confined to sun-exposed areas, sparing the upper eyelids, beneath the nose and chin, and the retroauricular areas. Postinflammatory hyperpigmentation develops rapidly and is usually the presenting sign.
Although photodermatitis caused by drugs or chemicals may be diagnosed by photopatch testing, facilities for this diagnostic procedure are not widely available. A high index of suspicion combined with an appreciation of the distribution pattern of the eruption and a history of application or ingestion of a known photosensitizing agent is all that is required to make a diagnosis. Discontinuation of the offending medication or avoidance of sun exposure, oral administration of an antihistamine, and application of a topical corticosteroid to alleviate pruritus are appropriate therapeutic measures. Severe reactions may necessitate systemic corticosteroid therapy for a brief time.
Porphyrias (Chapter 85)
Porphyrias are acquired or inborn disorders due to abnormalities of specific enzyme mutations in the heme biosynthetic pathway. Two in particular occur in children and have photosensitivity as a consistent feature. Signs and symptoms may be negligible during the winter, when sun exposure is minimal.
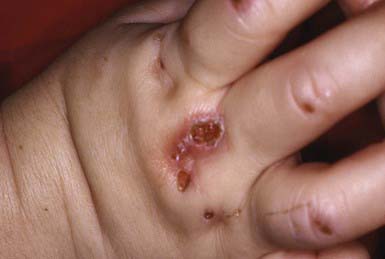
Congenital erythropoietic porphyria (Günther disease) is a rare autosomal recessive disorder. It manifests in the first few months of life as exquisite sensitivity to light, which may induce repeated severe bullous eruptions that result in mutilating scars (Fig. 648-2). Hyperpigmentation, hyperkeratosis, vesiculation, and fragility of skin develop in light-exposed areas. Hirsutism in areas of mild involvement, scarring alopecia in severely affected areas, pink to red urine, brown teeth, hemolytic anemia, splenomegaly, and increased amounts of uroporphyrin I in urine, plasma, and erythrocytes and of coproporphyrin I in feces are additional characteristic manifestations. Urine from affected patients fluoresces reddish pink under a Wood light. Total protection from sunlight is the treatment of choice.
Erythropoietic protoporphyria, an autosomal dominant trait, becomes apparent in early childhood and manifests as pain, tingling, and a burning sensation within ≈ 30 min of sun exposure, followed by erythema, edema, urticaria, and, rarely, vesicles on light-exposed areas. Nail changes consist of opacification of the nail plate, onycholysis, pain, and tenderness. Mild systemic symptoms such as malaise, chills, and fever may accompany the acute skin reaction. Recurrent sun exposure produces a chronic eczematous dermatitis with thickened, lichenified skin, especially over the finger joints (Fig. 648-3A), and persistent violaceous erythema, ulcers, and pitted or linear, crusted atrophic scars on the face (Fig. 648-3B) and rims of the ears. Pigmentation, hypertrichosis, skin fragility, and mutilation are uncommon. Liver disease is uncommon and generally mild, with only 3% of patients demonstrating severe disease.
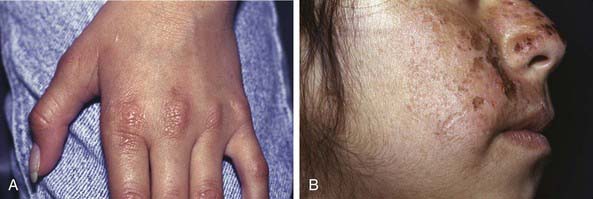
Figure 648-3 Erythropoietic porphyria. A, Erythematous thickening over the metacarpal phalangeal joints. B, Linear crusts and scarring.
The wavelengths of light mainly responsible for eliciting cutaneous reactions in porphyria are in the region of 400 nm. Window glass, which transmits wavelengths >320 nm, is not protective, and artificial lights of a certain wavelength may be pathogenic. Patients must avoid direct sunlight, wear protective clothing, and use a sunscreen agent that effectively blocks wavelengths in the region of 400 nm. Administration of beta-carotene (Solatene) 120-180 mg/day to achieve a level of 11-15 µmol/L is often effective in reducing symptoms.
Colloid Milium
Colloid milium is a rare, asymptomatic disorder that occurs on the face (nose, upper lip, upper cheeks) and may extend to the dorsum of the hands and the neck as a profuse eruption of tiny, ivory to yellow, firm, grouped papules. Lesions appear before puberty on otherwise normal skin, unlike the adult variant that develops on sun-damaged skin. Onset may follow an acute sunburn or long-term sun exposure. Most cases reach maximal severity within ≈ 3 yr and remain unchanged thereafter, although the condition may remit spontaneously after puberty. Histopathologic changes include well-circumscribed accumulations of fissured eosinophilic material, primarily in the upper dermis in contact with the epidermis. Treatment options include dermabrasion and ablative lasers.
Hydroa Vacciniforme
A vesiculobullous disorder, hydroa vacciniforme is more common in boys than in girls, begins in early childhood, but may remit at puberty. The peak incidence is in the spring and summer. Erythematous, pruritic macules develop symmetrically within hours of sun exposure over the ears, nose, lips, cheeks, and dorsal surfaces of the hands and forearms. Lesions progress to stinging tender papules and hemorrhagic vesicles and bullae. Severe lesions of hydroa vacciniforme resemble the vesicles of chickenpox. They become umbilicated, ulcerated, and crusted and heal with pitted scars and telangiectasias. Fever and malaise are noted occasionally during the acute phase. Histopathologically, lesions show intraepidermal multilocular vesicles, leading to focal epidermal and dermal necrosis. Noted early is a dermal perivascular mononuclear cell infiltrate, which later surrounds areas of necrosis. This eruption should be distinguished from erythropoietic protoporphyria, which rarely shows vesicles. Pathogenesis of hydroa vacciniforme is related to latent Epstein-Barr virus infections. Typical lesions have been reproduced with repeated doses of UVA or UVB light. A mid-potency topical corticosteroid may be useful for the inflammatory phase of the eruption. Sun avoidance and broad-spectrum sunscreens may also be helpful, as may low-dose courses of narrow-band UVB (NB-UVB) therapy or psoralen with UVA (PUVA) therapy (hardening).
Actinic Prurigo
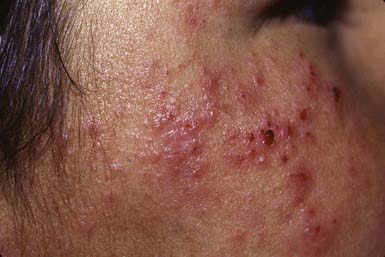
A chronic familial photodermatitis, actinic prurigo is inherited as an autosomal dominant trait among the Native Americans of North and South America. HLA DRB1*0407 (60-70%) and HLA DRB1*0401 (20%) are strongly associated with actinic prurigo. The first episode generally occurs in early childhood, several hours to 2 days after intense sun exposure. Most patients are female and are sensitive to UVA radiation. Lesions are intensely pruritic, erythematous papules on the face (Fig. 648-4), lower lip, distal extremities, and, in severe cases, buttocks. Facial lesions may heal with minute pitted or linear scarring. Lesions often become chronic, without periods of total clearing, merging into eczematous plaques that lichenify and may become secondarily infected. Associated features that distinguish this disorder from other photoeruptions and atopic dermatitis include cheilitis, conjunctivitis, and traumatic alopecia of the outer half of the eyebrows. Actinic prurigo is a chronic condition that generally persists into adult life, although it may improve spontaneously in the late teenage years. Sun avoidance, protective clothing, and broad-spectrum sunscreens may be helpful in preventing the eruption. Mid- to high-potency topical corticosteroids and antihistamines palliate the pruritus and inflammation. Thalidomide 50-100 mg/day is very effective, but its use is limited by toxicity.
Solar Urticaria
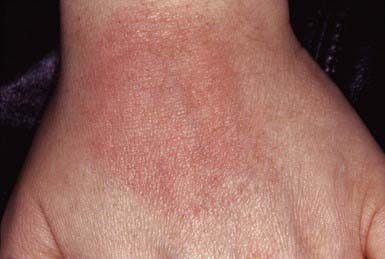
Solar urticaria is a rare disorder induced by UV or visible irradiation. The disorder is mediated by immunoglobulin (Ig) E antibodies to either an abnormal chromophore (type I) or a normal chromophore (type II), leading to mast cell degranulation and histamine release. This reaction occurs within 5-10 min of sun exposure, fades within 1-2 hr, and is characterized by widespread severe wheal formation (Fig. 648-5), which may lead to faintness, headache, nausea, syncope, or bronchospasm. H1-blocking antihistamines may be useful to prevent or abate the eruption.
Polymorphous Light Eruption
Polymorphous light eruption develops most commonly in females younger than 30 yr. The first eruption typically appears after prolonged sun exposure during the spring or summer. Onset of the eruption is delayed by hours to days after sun exposure and lasts for days to sometimes weeks. Areas of involvement tend to be symmetric and are characteristic for a given patient, including some but not all of the exposed or lightly covered skin on the face, neck, upper chest, and distal extremities. Lesions have various morphologies but most commonly are pruritic, 2- to 5-mm, grouped erythematous papules or papulovesicles or edematous plaques that are > 5 cm in diameter. Most cases involve sensitivity to UVA radiation, although some are UVB induced. Therapeutic approaches include sun avoidance, protective clothing, broad-spectrum sunscreens, mid- to high-potency topical or systemic corticosteroids, or prophylactic NB-UVB or PUVA phototherapy (hardening).
Cockayne Syndrome
Onset of Cockayne syndrome, an autosomal recessive disorder, is characterized by the appearance, at ≈ 1 yr of age, of facial erythema in a butterfly distribution after sun exposure, followed by loss of adipose tissue and development of thin, atrophic, hyperpigmented skin, particularly over the face. Associated features include dwarfism; mental retardation; large, protuberant ears; long limbs; disproportionately large hands and feet, which are sometimes cool and cyanotic; pinched nose; carious teeth; unsteady gait with tremor; limitation of joint mobility; progressive deafness; cataracts; retinal degeneration; optic atrophy; decreased sweating and tearing; and premature graying of the hair. Diffuse extensive demyelination of the peripheral and central nervous systems ensues, and patients generally die of atheromatous vascular disease before the third decade. There are 2 types of Cockayne syndrome. Type I (CSA gene) is less severe than type II (CSB gene). Xeroderma pigmentosum–Cockayne syndrome (XP-CS) demonstrates complementation with xeroderma pigmentosa groups B, D, or G. Patients with XP-CS are phenotypically more like patients with Cockayne syndrome. Photosensitivity is due to deficient rates of repair of UV-induced damage, specifically within actively transcribing regions of DNA (transcription-coupled DNA repair). The syndrome is distinguished from progeria (Chapter 84) by the presence in Cockayne syndrome of photosensitivity and the ocular abnormalities.
Xeroderma Pigmentosum
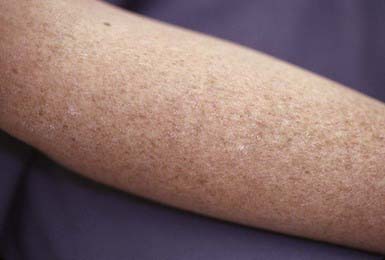
Xeroderma pigmentosum is a rare autosomal recessive disorder that results from a defect in nucleotide excision repair. Seven complementation groups have been recognized, on the basis of each group’s separate defect in ability to repair damaged DNA. Xeroderma pigmentosum variant is caused by mutations in the DNA polymerase ETA gene (POLH), which lead to a defect in the conversion of newly synthesized DNA after UV radiation. The wavelength of light that induces the DNA damage ranges from 280 to 340 nm. Skin changes are first noted during infancy or early childhood in sun-exposed areas such as the face, neck, hands, and arms; lesions may occur, however, at other sites, including the scalp. The skin lesions consist of erythema, scaling, bullae, crusting, ephelides, telangiectasia, keratoses (Fig. 648-6), basal and squamous cell carcinomas, and malignant melanomas. Ocular manifestations include photophobia, lacrimation, blepharitis, symblepharon, keratitis, corneal opacities, tumors of the lids, and possible eventual blindness. Neurologic abnormalities such as mental deterioration and sensorineural deafness may develop in ≈ 20% of patients.
This disease is a serious mutilating disorder, and the life span of an affected patient is often brief. Affected families should have genetic counseling. Xeroderma pigmentosum is detectable in cells cultured from amniotic fluid. Affected children should be totally protected from sun exposure; protective clothing, eyeglasses, and opaque broad-spectrum sunscreens should be used even for mildly affected children. Light from unshielded fluorescent bulbs and sunlight passing through glass windows are also harmful. Early detection and removal of malignancies is mandatory.
Rothmund-Thomson Syndrome
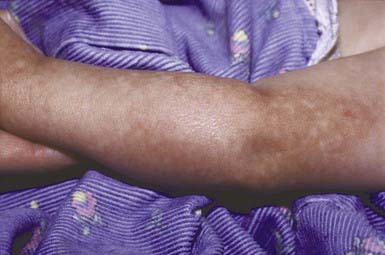
Rothmund-Thomson syndrome is also known as poikiloderma congenitale because of the striking skin changes (Fig. 648-7). It is inherited as an autosomal recessive trait. Mutations in the RECQL4 gene are found in approximately 65% of the patients. The other mutations causing Rothmund-Thomson syndrome are unknown. Skin changes are noted as early as 3 mo of age. Plaques of erythema and edema appear on the cheeks, forehead, ears, neck, dorsal portions of the hands, extensor surfaces of the arms, and buttocks and are replaced gradually by reticulated, atrophic, hyperpigmented, telangiectatic plaques. Light sensitivity is present in many cases, and exposure to the sun may provoke formation of bullae. Areas of involvement, however, are not strictly photodistributed. Short stature; small hands and feet; sparse eyebrows, eyelashes, and pubic and axillary hair, and sparse, fine, prematurely gray scalp hair or alopecia; bony defects; and hypogenitalism are common. Cataracts may also occur at an early age. Most patients have normal mental development. Keratoses and later squamous cell carcinomas may develop on exposed skin. The most worrisome association is that with osteosarcoma, which occurs only in those patients with Rothmund-Thomson syndrome and RECQL4 mutations.
Bloom Syndrome
The defect in Bloom syndrome (BLM/RECQL3 gene) is inherited in an autosomal recessive manner. Patients are sensitive to UV radiation, and their rate of chromosomal breaks and sister chromatid exchanges is markedly increased. Erythema and telangiectasia develop during infancy in a butterfly distribution on the face after exposure to sunlight. A bullous eruption on the lips and telangiectatic erythema on the hands and forearms may develop. Café-au-lait spots and hypopigmented macules may be present. Prenatal and postnatal short stature and a distinctive facies consisting of a prominent nose and ears and a small, narrow face are generally found. Intellect is average to low average. Immunodeficiency is seen in all patients, manifesting as recurrent ear and pulmonary infections. Gastrointestinal malabsorption is common. Affected children have an unusual tendency to experience both solid tumors and lymphoreticular malignancies.
Hartnup Disease (Chapter 79.5)
Hartnup disease is a rare inborn error of metabolism with autosomal recessive inheritance. Neutral amino acids, including tryptophan, are not transported across the brush border epithelium of the intestine and kidneys, resulting in deficiency of synthesis of nicotinamide and causing a photo-induced pellagra-like syndrome. The urine contains increased amounts of monoamine monocarboxylic amino acids. Cutaneous signs, which precede neurologic manifestations, initially develop during the early months of life, consisting of an eczematous, occasionally vesiculobullous eruption noted on the face and extremities in a glove-and-stocking photodistribution. Hyperpigmentation and hyperkeratosis may supervene and are intensified by further exposure to sunlight. Episodic flares may be precipitated by febrile illness, sun exposure, emotional stress, and poor nutrition. In most cases, mental development is normal, but some patients display emotional instability and episodic cerebellar ataxia. Neurologic symptoms are fully reversible. Administration of nicotinamide and protection from sunlight results in improvement of both cutaneous and neurologic manifestations.
Balk SJ, Geller AC. Teenagers and artificial tanning. Pediatrics. 2008;121:1040-1042.
Bissonnette R. Update on sunscreens. Skin Therapy Lett. 2008;13:5-7.
Botto NC, Warshaw EM. Solar urticaria. J Am Acad Dermatol. 2008;59:909-920.
Honigsmann H. Polymorphous light eruption. Photodermatol Photoimmunol Photomed. 2008;24:155-161.
Iwatsuki K, Satoh M, Yamamoto T, et al. Pathogenic link between hydroa vacciniforme and Epstein-Barr virus associated hematologic disorders. Arch Dermatol. 2006;142:587-595.
Lautenschlager S, Wulf HC, Pittelkow MR. Photoprotection. Lancet. 2007;370:528-537.
2007 A new sunscreen agent. Med Lett Drugs Ther. 2007;49:41-43.
Pourabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
Ross G, Foley P, Baker C. Actinic prurigo. Photodermatol Photoimmunol Photomed. 2008;24:272-275.
Sassa S. Modern diagnosis and management of the porphyries. Br J Haematol. 2006;135:281-292.