chapter 117 Anomalies of the Upper Urinary Tract
Congenital anomalies of the upper urinary tract comprise a group of abnormalities, ranging from complete absence to aberrant location, orientation, and shape of the kidney as well as aberrations of the collecting system and blood supply. These diverse entities are among the most common malformations in newborns. Historically, developmental biology of the urinary tract was based on observations in animal models and human fetuses. The mechanism and timing of various congenital anomalies were extrapolated from the normal process of organogenesis. Technical advances in molecular genetics have provided the opportunity to hypothesize complex mechanisms of normal and abnormal development.
Anomalies of Number
Bilateral Renal Agenesis
Of all the anomalies of the upper urinary tract, bilateral renal agenesis (BRA) has the most profound effect on the fetus. Fortunately, it occurs infrequently when compared with other renal abnormalities. Although BRA was first recognized in 1671 by Wolfstrigel, it was not until Potter’s extensive description of the constellation of associated defects that the full extent of the syndrome could be appreciated and easily recognized (Potter, 1946a, 1946b, 1952). Subsequently, many investigators have attempted to explain the syndrome by employing a single unifying etiology (Fitch and Lachance, 1972). However, we are learning that there is probably no single etiology as we begin to unravel the myriad of complex molecular events that are required for normal renal development.
The incidence of BRA is rare, with only about 500 cases reported in the literature. Potter (1965) estimated that BRA occurs once in 4800 births, but in British Columbia the incidence is 1 in 10,000 births (Wilson and Baird, 1985). Davidson and Ross (1954) noted a 0.28% incidence in autopsies of infants and children, whereas Stroup and colleagues (1990) detected an incidence of 3.5 per 100,000 in the Centers for Disease Control Birth Defects Monitoring Program. A recent study using prenatal ultrasonography in 8500 pregnancies in Poland reported an incidence of 0.25% (Forys et al, 2003). There is significant male predominance, with almost 75% of individuals being male. Increasing maternal age appears to be a risk factor (Bianca et al, 2003), but specific complications of pregnancy or any maternal disease do not appear to consistently influence the incidence of BRA (Davidson and Ross, 1954; Ruhland et al, 1998). The anomaly was reported in three infants of an insulin-dependent diabetic mother (Novak and Robinson, 1994). It has been observed in several sets of siblings (Rizza and Downing, 1971; Dicker et al, 1984) and in monozygotic twins (Thomas and Smith, 1974; Cilento et al, 1994). In four pairs of monozygotic twins, one sibling was anephric while the other had normal kidneys (Kohler, 1972; Mauer et al, 1974; Cilento et al, 1994; Klinger et al, 1997). It has been suggested that an autosomal-recessive inheritance pattern exists (Dicker et al, 1984). There is a genetic predisposition to this syndrome with a high level of penetrance (Stella, 1998). When siblings and parents of an index child with BRA were screened, 4.5% had unilateral renal agenesis (Roodhooft et al, 1984) and 3.5% had BRA (McPherson et al, 1987). This is 1000 times higher than in the general population (Stroup et al, 1990). Other investigators have suggested that this is an autosomal-dominant trait with variable penetrance (Kovacs et al, 1991; Murugasu et al, 1991; Moerman et al, 1994; Stella, 1998). Recently, McPherson (2007) evaluated renal anomalies in families of individuals with congenital solitary kidneys that included renal agenesis or a very poorly functioning kidney due to dysplasia/hypoplasia. The empiric risk of 7% for offspring, 4% for parents, and 2.5% for siblings may be an underestimation, because not all relatives underwent ultrasound screening. The incidence of BRA in offspring of congenital solitary kidney probands was ≈1%, which is significantly greater than the risk found in the general population but less than that for families with a history of BRA. Ultrasound screening has been recommended for parents and siblings of infants born with either unilateral or bilateral renal agenesis or dysgenesis (Roodhooft, 1984). McPherson (2007) has recommended prenatal ultrasound examination when either parent has a congenital solitary kidney. Ultrasound screening is also recommended for first-degree relatives of persons with congenital solitary kidney.
Syndromic Associations
BRA has been detected in higher-than-expected proportions in esophageal atresia (Saing et al, 1998) and several syndromes, including cryptophthalmos or Frazer syndrome (Fryns et al, 1997), Klinefelter syndrome (Barroeta et al, 2004), and Kallmann syndrome (Colquhoun-Kerr et al, 1999).
Renal Embryology
The intermediate kidney, or mesonephros, develops and then regresses except for the mesonephric tubules (Constantini and Shakya, 2006; Schedl, 2007; Uetani and Bouchard, 2009). In the male, these are the efferent ductules that serve as a link between the gonad and the mesonephric or wolffian duct (WD) structures (the body and tail of the epididymis and vas deferens). In the female, the mesonephric tubules link the ovary through the fimbriated end of the fallopian tube to the reproductive tract. The WD elongates caudally and fuses with the anterior cloaca. The definitive kidney differentiates from the metanephric blastema, which is a specialized region of the intermediate mesoderm termed the metanephric mesenchyme (MM).
This process requires the reciprocal induction between the metanephric blastema and the ureteral bud (UB). The metanephric blastema sends signals to the WD to initiate UB formation between the fifth and seventh weeks of gestation. As a result, the ureter is induced from the caudal end of the WD. The UB evaginates and invades the metanephric blastema and branches repeatedly in a characteristic pattern to form the collecting duct system. The ureteral tips induce nephron differentiation in the adjacent mesenchyme, forming the mature metanephros (Airik, 2007; Uetani and Bouchard, 2009). The absence of a nephrogenic ridge on the dorsolateral aspect of the coelomic cavity or the failure of a UB to develop from the WD will result in renal agenesis. Therefore in order for BRA to occur, there must be an alteration in normal molecular development or a mutation that causes renal or ureteral maldevelopment on both sides of the midline (see Molecular Mechanisms of Mammalian Kidney Organogenesis).
Relationship of the Wolffian Duct to Müllerian Duct Formation
A review of the relationship of the WD to müllerian duct (MD) development is necessary to understand the genitourinary phenotype of individuals with renal anomalies or more specifically, renal agenesis (Kobayashi and Behringer, 2003). The cellular mechanisms involved in MD formation have been partially unraveled only recently. Gene fate mapping and lineage tracing experiments show that the WD does not contribute cells to the MD, and the MDs are derived from the coelomic epithelium (Guioli et al, 2007; Orvis and Behringer, 2007). A three-phase model of MD development has recently been proposed (Guioli et al, 2007; Orvis and Behringer, 2007). In the first phase, cells of the coelomic epithelium in the cervical region of the intermediate mesoderm are specified to become MD cells and have been noted to express Lim1 (Kobayashi et al, 2005; Orvis and Behringer, 2007; Masse et al, 2009) (Fig. 117–1). After the process of specification is complete, the second phase is heralded by Wnt4 expression from the mesonephros or coelomic epithelium, which induces these cells that are destined to become the MDs to invaginate (Kobayashi et al, 2004, 2005; Orvis and Behringer, 2007). The second phase of MD development is WD independent and ends when the MD extends caudally and contacts the WD (Carroll, 2005; Kobayashi et al, 2005; Orvis and Behringer, 2007). The third phase involves elongation of the MDs posteriorly until they are joined at the urogenital sinus. This process is WD dependent, requiring the MD epithelium at its posterior end to be in close physical contact with the WD epithelium, while the MDs are separated from the coelomic epithelium by only a basement membrane (Orvis and Behringer, 2007). This intimate relationship between the WD and MD is emphasized in experiments that interrupt the formation of the WD at a specific point and show that the MD could not grow beyond that point to complete its formation (Gruenwald, 1941). Lim1 in the WD is critical for WD maintainance. Loss of Lim1 in the WD by inactivation leads to WD loss. Because the MD is dependent upon the WD in this third phase, the MDs are again incompletely formed (Kobayashi et al, 2005). This third phase is also dependent upon the Pax2 gene; mice mutants for this gene show cellular invagination but no elongation because the WDs have degenerated (Torres et al, 1995; Miyamoto et al, 1997). Studies have also shown that the WD not only acts as a physical guide but also plays a role in MD elongation through paracrine signaling. More specifically, Wnt9b is expressed by the WD epithelium, and gene inactivation results in incomplete formation of the MDs (Carroll et al, 2005). Loss of Wnt9b expression did not affect the WD, per se, or the first two phases of MD development but did affect the caudal extension and elongation of the MDs, suggesting that WD signaling by Wnt9b is one of the critical factors in directing MD formation (Carroll et al, 2005). For a clinical correlation, see Anomalies in the Female (Unilateral Renal Agenesis section).
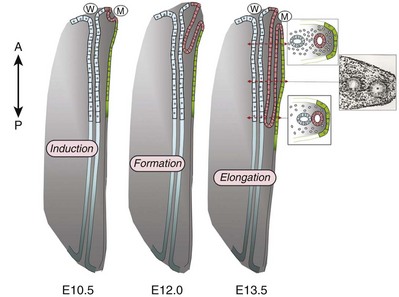
Figure 117–1 Model for müllerian duct development. At E11.75, after a subset of coelomic epithelium cells (represented in green) are specified, they invaginate in the intermediate mesoderm. Then, the invaginating cells form the müllerian duct (M, represented in pink). Anteriorly, the funnel is opened in the abdominal cavity, and caudally, the growing tip extends to and contacts the wolffian ducts (W, represented in blue) at E12.0. A phase of elongation allows the müllerian duct to elongate posteriorly in very close contact with the wolffian duct. As soon as the müllerian duct growing tip has deposited cells and elongated caudally, the physical contact between the ducts is lost by the appearance of mesenchymal cells around the müllerian duct epithelium. At E13.5, the two müllerian ducts reach the urogenital sinus and fuse together. Developmental stages indicated in this figure correspond to mouse stages.
(From Masse J, Watrin T, Laurent A, et al. The developing female genital tract: from genetics to epigenetics. Int J Dev Biol 2009;53:411–24.)
Molecular Mechanisms of Mammalian Kidney Organogenesis
Several genes play a critical role in WD development, including Pax2/8, Gata3, and Lim1. Many of the same genes affecting renal development will also affect internal duct development. If there is gene inactivation of Pax2/8, Gata3, or Lim1, there will be no formation of the kidneys (BRA), ureters, or genital tract (Uetani and Bouchard, 2009). The pathway for mammalian kidney development is regulated by reciprocal epithelial-mesenchymal inductive signaling between the UB epithelium and the MM (Yu et al, 2004). Ureteral bud formation and the induction of its branching require glial cell line–derived neurotrophic factor (GDNF), a secreted growth factor expressed in the MM (Michos et al, 2007). The GDNF ligand activates the RET receptor, which is expressed in the WD epithelium and then around the UB tips as branching proceeds. Gdnf expression and localization are positively regulated by Eya1 and Pax2 (Michos, 2009). GDNF activation of RET requires the glial cell line–derived neurotrophic factor family receptor α1 (GFRα1) and is essential for induction of UB formation and initiation of outgrowth and branching (Chi, 2009). Most genes that are thought to be essential for UB formation are also regulators of Gdnf or Ret expression. Studies of murine kidney development show that Gdnf −/− mice have renal agenesis, while Ret−/− mice have renal agenesis or dysplastic kidneys (Pichel et al, 1996; Schuchardi et al, 1996, Glassberg, 2002).
Skinner and colleagues (2008) examined the association between abnormal kidney development and mutations of RET, GDNF, and GFRα1 in 29 stillborn fetuses with BRA or unilateral renal agenesis (URA). Mutations in RET were found in 7 of 19 fetuses with BRA and 2 of 10 fetuses with URA. A mutation in GDNF was found in 1 fetus with URA who also had mutations in RET. No GFRα1 mutations were observed. These data suggest that congenital renal agenesis results from RET mutations that prevent or impede the embryonic development of RET-dependent structures.
After the GDNF/GFRα1 complex binds to RET, the Wnt11 gene is activated in the epithelial tips of the UB and is associated with UB branching (Majumdar et al, 2003). Wnt11 is a member of the Wnt gene family, which is composed of structurally related genes encoding secreted signaling proteins. These proteins are likely involved in several processes, including regulation of cell fate and patterning during embryogenesis. WNT11 signaling is required for the propagation of mesenchymal GDNF signaling, which establishes the autoregulatory epithelial-mesenchymal GDNF/WNT11 feedback signaling loop that controls the progression of metanephric branching morphogenesis after initiation of UB outgrowth (Majumdar et al, 2003; Michos et al, 2007).
Bone morphogenetic protein-4 (BMP4), a member of the transforming growth factor-α family, is expressed in the periureteral mesenchyme and is essential for morphogenesis (Glassberg, 2002; Miyazaki et al, 2000, 2003; Michos et al, 2007). In wild-type mouse embryos, BMP4 is expressed by the mesenchyme surrounding the WD and UB. Mesenchymal cells that express BMP4 inhibit UB formation, in part by inhibiting Wnt11 expression. BMP4 migrates to ectopic sites, thereby preventing ectopic UB formation. The BMP4 mesenchymal cells act in a similar fashion during UB branching by inhibiting side branching and permitting stems to lengthen. Mesenchymal cells that are devoid of BMP4 surround the tip, where further branching proceeds. Mice heterozygous for a null mutation in BMP4 (+/−) manifest anomalies, including hypoplastic/dysplastic kidney, hydroureter, ectopic ureter, ureteral duplication, megaureter, ureterovesical junction obstruction, and reflux (Miyazaki et al, 2000, 2003).
Mammalian Kidney Organogenesis: New Advances
Gremlin 1 (GREM1) is an extracellular BMP antagonist that is expressed in the meso- and metanephric mesenchyme (Michos et al, 2004) (Fig. 117–2). GREM1 is upregulated in the mesenchyme around the origin of the UB prior to initiation of its outgrowth. BMP activity, at this time, is reduced locally. In the Grem1-deficient mouse embryo, metanephric development is disrupted at the stage of UB outgrowth initiation, resulting in bilateral renal agenesis. This inhibition of UB outgrowth causes progressive loss of Gdnf expression, resulting in apoptosis of the MM.
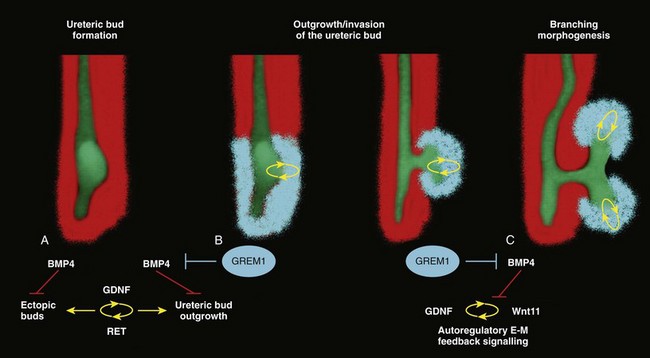
Figure 117–2 Reduction of BMP4 activity by gremlin 1 in the mesenchyme around the ureteric bud is essential to enable ureteric epithelial outgrowth, GDNF-RET– and WNT11-mediated epithelial-mesenchymal (e-m) feedback signaling, and branching morphogenesis. A, In mouse, the ureteric bud forms in the caudalmost part of the wolffian duct under the influence of GDNF-RET signaling. During this inductive period, Bmp4 is expressed by the mesenchyme enveloping the wolffian duct. High levels of mesenchymal BMP4 activity inhibit the formation of ectopic epithelial buds and epithelial branching at this stage (prior to E11.0). At this early stage, only low levels of Grem1 transcripts are detected (not shown). B, Expression of the BMP antagonist Grem1 is upregulated in the mesenchyme around the nascent ureteric bud, thereby locally reducing BMP4 signal transduction (around E11.75-11.0). This reduction of BMP4 activity by GREM1 enables initiation of ureteric bud outgrowth and its invasion into the metanephric mesenchyme. C, GREM1 is required to maintain and propagate expression of Wnt11 in the ureteric epithelial tip(s) and Gdnf in the mesenchyme by e-m feedback signaling.
(From Michos O, Goncalves A, Lopez-Rios J, et al. Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signaling during kidney branching morphogenesis. Development 2007;134:2397–405.)
To further examine the relationship of these various ligands and their effects on epithelial-mesenchymal interactions, Michos and colleagues (2007) cultured early Grem1-deficient mutant mouse kidney rudiments in medium supplemented with recombinant GREM1. The addition of GREM1 restored UB outgrowth and induced supernumerary epithelial buds that invaded the MM and initiated branching morphogenesis. At the molecular level, GREM1 replacement activated Wnt11 expression in the epithelial buds and upregulated Gdnf expression in the mesenchyme. Because genetic suppression of BMP4 activity in a Grem1-deficient mouse model completely restored kidney development, the local reduction of BMP4 activity by GREM1 was presumed to be critical to the initiation of UB outgrowth and kidney organogenesis.
Because BMP4 signaling by the mesenchyme surrounding the WD prevents formation of supernumerary epithelial buds, successful initiation of UB outgrowth most likely requires both antagonism of BMP4 by GREM1 in mesenchyme and signaling by GDNF from the MM to RET in the ureteric epithelium (Michos et al, 2007). In addition, autoregulatory feedback signaling between GDNF in the mesenchyme and WNT11 in the epithelial tips regulates branching morphogenesis. Grem1 is essential for both upregulation of Wnt11 in the ureteric epithelium and Gdnf expression in the mesenchyme and the establishment of epithelial-mesenchymal feedback signaling.
Gross Pathologic Description of Retroperitoneal Findings
In an extensive autopsy analysis by Ashley and Mostofi (1960), the kidneys were completely absent on gross inspection of the entire retroperitoneum. Occasionally, there was a small mass of poorly organized mesenchymal tissue containing primitive glomerular elements with only minute vascular branches from the aorta. Complete absence of the renal vessels was observed in about 25% of specimens with BRA in this series. Complete ureteral atresia was observed in 39 of the 42 cases of BRA, and partial ureteral absence was noted in 3. Only small projections with no demonstrable lumen alongside the bladder were noted. With complete absence of the ureter, a rudimentary kidney was discovered in only a few instances, supporting the concept of reciprocal induction.
The adrenal gland may appear flattened on ultrasonography but is rarely malpositioned or absent (Davidson and Ross, 1954; Hoffman et al, 1992). A normally located adrenal gland is expected, because the adrenal cortex develops from primitive mesoderm medial to the urogenital ridge and the medulla develops from ectodermal neural crest cells, while the metanephros is derived from the intermediate mesoderm.
Fused and/or horseshoe-shaped glands have been noted on prenatal ultrasound screening (Strouse et al, 2002). Potter (1965) noted that fused glands were often found in the presence of spinal anomalies. In a small number of autopsies, the gonads were absent, indicating the abnormality or insult occurred before the fifth week and affected the overall development of the urogenital ridge (Carpentier and Potter, 1959).
Key Points: Bilateral Renal Agenesis
In the Ashley and Mostofi series (1960), about 50% of cases of complete ureteral atresia showed complete absence of the bladder, and in the remainder of cases, a hypoplastic bladder was found consisting only of a muscular tube with a minute lumen. In Potter’s series (1965), the bladders were also hypoplastic and lacked ureteral orifices. A normally closed urachus was observed.
Abnormal development of the bladder is thought to be due to the lack of stimulation by fetal urine production, which starts at 10 to 12 weeks of gestation. Alternatively, it has been postulated that UB and WD structures migrating into the ventral cloacal region are needed to initiate normal bladder development. The absence of the UB, and not the lack of urine, may arrest bladder development (Levin, 1952; Katz and Chatten, 1974). This theory is supported by the fact that despite absence of bladder filling in bladder exstrophy, many of these bladders are functional following surgical closure alone, while the bladders associated with bilateral ureteral ectopia almost invariably require augmentation (Jayanthi et al, 1997, Gearhart and Matthews, 2007).
Phenotypic Features
Phenotypic features associated with BRA have been extensively described by Potter. These infants have low birth weights, ranging from 1000 to 2500 g, and intrauterine growth retardation due in part to low iron stores in the liver (Georgieff et al, 1996). At birth, oligohydramnios (absent or minimal amniotic fluid) is present. The characteristic facial appearance and deformity of the extremities distinguishes these children from normal newborns. The infants look prematurely senile and have “a prominent fold of skin that begins over each eye, swings down in a semi-circle over the inner canthus and extends onto the cheek” (Potter, 1946a, 1946b). This facial feature is a sine qua non of nonfunctioning renal parenchyma and suggests that its absence confirms the presence of at least one kidney (Fig. 117–3). The nose is blunted, and a prominent depression exists between the lower lip and chin. The ears appear to be low set, are drawn forward, and are often pressed against the side of the head, making the lobes seem unusually broad and exceedingly large. The ear canals are not dislocated, but the appearance of the ear lobes gives the impression that the ears are displaced downward. Periauricular pits and tags have been noted (Wang et al, 2001). The skin can be excessively dry and appears too loose for the body. This may be secondary to severe dehydration or lack of subcutaneous fat. The hands are relatively large and clawlike. The legs are often bowed and clubbed, with excessive flexion at the hip and knee joints (Saing et al, 1998; Carbillon et al, 2001; Das et al, 2002). Occasionally, the lower extremities are completely fused as seen with sirenomelia (Liatsikos et al, 1999). A lumbar meningocele with or without the Arnold-Chiari malformation and hydrocephalus is often observed (Davidson and Ross, 1954; Ashley and Mostofi, 1960). In Potter’s series (1965), anomalies of the gastrointestinal tract were found in 60% of fetuses.
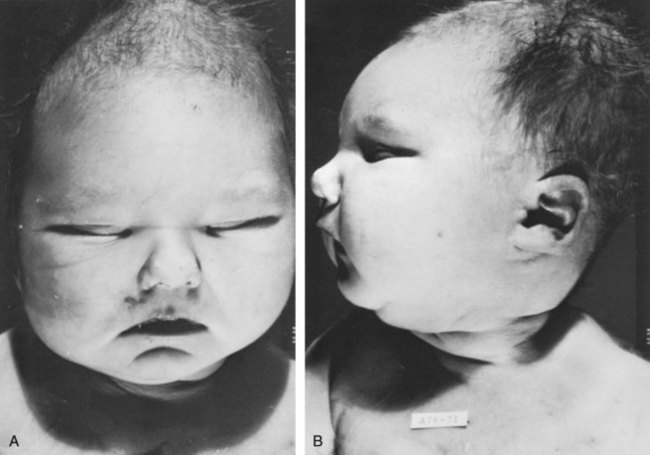
Figure 117–3 An anephric child who lived 2 days has the typical Potter facial appearance. A, Note the prominent fold and skin crease beneath each eye, blunted nose, and depression between lower lip and chin. B, The ears give an impression of being low set because lobes are broad and drawn forward, but actually the ear canals are located normally.
Anomalies of the external genitalia include absence of the scrotum and clitoral hypertrophy. Penile development is usually normal, but in a few cases, penile agenesis or a rudimentary penis and scrotum have been reported (O’Connor et al, 1993; Potter, 1965). Hypospadias is rare and does not appear to be related to the presence or absence of the testes. The testes are undescended in 43% of cases (Carpentier and Potter, 1959). Ashley and Mostofi (1960) found testicular agenesis in 10%. The vas deferens is normal in most cases, implying that the factor responsible for the renal agenesis influenced the UB only after it formed from a completely elongated WD or that the insult affected the induction of the MM.
There is a relatively high incidence of anomalies of the MD structures and ovaries (Carpentier and Potter, 1959). The ovaries are frequently hypoplastic or absent. The uterus is usually rudimentary or bicornuate but occasionally absent. The vagina is a short, blind-ending pouch or completely absent.
Role of Amniotic Fluid Production and Pulmonary Development
The characteristic facial abnormalities and limb features may result from deformations rather than malformations of structures due to the lack of “cushioning” from amniotic fluid (Fitch and Lachance, 1972; Thomas and Smith, 1974). This observation was confirmed by an experiment in nature in which one twin with BRA did not have the characteristic Potter facies because it shared the same amniotic sac with the second twin who had an adequate volume of amniotic fluid (Klinger et al, 1997). Fetal renal urine is the major source of amniotic fluid, accounting for more than 90% of its volume by the third trimester (Thomas and Smith, 1974; Chevalier and Roth, 2007), but the skin, gastrointestinal tract, and central nervous system also contribute small amounts, particularly before urine production begins at 10 to 12 weeks.
Pulmonary hypoplasia and a bell-shaped chest are common associated findings that were originally thought to be due to uterine wall compression of the thoracic cage as a result of oligohydramnios (Bain and Scott, 1960). Subsequently, it was postulated that the amniotic fluid alone was responsible for pulmonary development (Fitch and Lachance, 1972). However, this theory was rejected when they observed a significant reduction in the number of airway generations as well as a decrease in acini formation in these fetuses (Hislop et al, 1979). Pulmonary airway branching occurs between the 12th and 16th weeks of gestation (Reid, 1977). A reduction in the number of branches implies interference with this process before the 16th week of gestation. Hislop and colleagues (1979) suggested that the anephric fetus fails to produce proline, which is a prerequisite for collagen formation in the bronchiolar tree. The kidney is the primary source of proline (Clemmons, 1977). Thus pulmonary hypoplasia may result from the absence of renal parenchyma and not from diminished amniotic fluid. This hypothesis is supported by the finding of normal lungs in two infants with prolonged leakage of amniotic fluid beginning at a time when pulmonary hypoplasia would have been expected if the amniotic fluid alone were responsible for the defect (Perlman et al, 1976; Cilento et al, 1994).
Peters and colleagues (1991a) proposed a two-step process in pulmonary development, with a primary “renal growth factor” influencing early lung development and an amniotic fluid volume-dependent phase influencing later gestational lung growth. Smith and colleagues (2006) studied early lung development using a murine knockout model of renal agenesis/dysgenesis and anuria. They found that pulmonary development occurred early in embryogenesis, and fetal anuria and hypoplastic lung development preceded oligohydramnios. These observations support the two-step model proposed by Peters (1991a). Alternatively, oligohydramnios due to experimentally induced urinary obstruction is associated with pulmonary hypoplasia in fetal sheep, with initially normal renal function (Peters et al, 1991a, 1991b). Restoring amniotic fluid volume only partially restores lung growth. Therefore uropathy-associated pulmonary hypoplasia appears to be a result of oligohydramnios rather than renal dysfunction (Peters, 1991b).
Prenatal and Postnatal Diagnosis
BRA is being diagnosed by prenatal ultrasonography in the second and third trimesters, when severe oligohydramnios is noted and no renal parenchyma can be identified (Forys et al, 2003). Termination of the pregnancy has been considered when the diagnosis is certain (Rayburn and Laferla, 1986). Additional diagnostic findings include small lung volumes and chest diameter and abnormal adrenal gland appearance (Latini et al, 1998; Sepulveda et al, 1998; Heling et al, 2001; Strouse et al, 2002). The characteristic Potter facies and the presence of oligohydramnios are pathognomonic. Amnion nodosum—small, white, keratinized nodules on the surface of the amniotic sac—have been considered a placental hallmark of prolonged, severe, oligohydramnios. Recently, oligohydramnios was diagnosed in only 22% of cases of amnion nodosum, suggesting that it may not be a reliable sign of oligohydramnios. Nevertheless, the finding portends a very poor prognosis (Adeniran and Stanek, 2007).
Ninety percent of newborns void during the first day of life (Sherry and Kramer, 1955). In a study of 500 infants, every infant voided within the first 24 hours of life, regardless of the gestational age (Clarke, 1977). After the first 24 hours, anuria without distention of the bladder suggests BRA (Williams, 1974). However, most neonates with BRA who are born alive experience severe respiratory distress within the first 24 hours of life. When this becomes the focus of attention, the anuria may be initially unnoticed.
Postnatal Radiographic Evaluation
Renal ultrasonography is the most efficient way to identify the kidneys and bladder and confirm the presence or absence of urine production. The advent of power Doppler ultrasonography has been highly accurate in determining the status of the renal arteries, even in fetuses with oligohydramnios and suspected BRA (Sepulveda et al, 1998). The finding of a flattened adrenal gland in its normal location supports the diagnosis of an absent kidney (Hoffman et al, 1992). If abdominal ultrasonography is inconclusive, renal scintigraphy can be performed using 99mTc-dimercaptosuccinic acid (DMSA). The absence of uptake of the radionuclide in both renal fossae above background activity or in an ectopic location confirms the diagnosis of BRA. Historically, umbilical artery catheterization and an aortogram were performed when other modalities were not diagnostic.
Prognosis
About 40% of the affected neonates are stillborn. Of those neonates who are born alive, most do not survive beyond the first 24 to 48 hours due to respiratory distress associated with pulmonary hypoplasia. Survival subsequently depends on the rate at which renal failure develops. The longest-surviving child lived 39 days (Davidson and Ross, 1954).
Unilateral Renal Agenesis
Complete absence of one kidney occurs more frequently than BRA but is not easily detected from findings on physical examination. An isolated single umbilical artery has been associated with renal anomalies, including unilateral renal agenesis (URA) (Dursun et al, 2005). More recently, the largest study to date of neonates with an isolated single umbilical artery did not find an increased incidence of URA or other malformations and concluded that postnatal renal ultrasonography was not routinely warranted (Deshpande et al, 2009).
URA may remain undetected unless examination of the external genitalia and/or radiographic evaluation of the female or male pelvis for other reasons reveal an anomaly associated with renal agenesis. Over the past two decades, prenatal ultrasound examinations have been performed more routinely, and URA is being detected with increased frequency (Sipek et al, 1997). These imaging studies have also revealed that a substantial number of cases thought to be URA were a dysplastic or multicystic dysplastic kidney (MCDK) that had undergone involution prior to birth (Mesrobian et al, 1993; Hitchcock and Burge, 1994; Dell’Acqua et al, 2002; Hiraoka et al, 2002). A plain film of the abdomen supports this diagnosis if the splenic or hepatic flexure of the bowel is in its normal location and not in the ipsilateral renal fossa, suggesting that a dysplastic kidney or MCDK may have formed in the renal fossa before involuting. Curvilinear calcifications on a plain radiograph or computed tomography (CT) scan are another sign of a prior MCDK (Nakano et al, 1996). A flattened adrenal or the spleen (on the left) may be mistaken for a kidney in the 20-week structural ultrasound study, but at later gestational weeks, the diagnosis of URA becomes more apparent (Woolf and Hillman, 2006).
Incidence
Most autopsy series suggest that unilateral renal agenesis occurs once in 1100 births (Doroshow and Abeshouse, 1961). In an historical survey of excretory urograms, the incidence ranged between 1 in 1500 (Longo and Thompson, 1952) to 1 in 5000 (Wilson and Baird, 1985). Ultrasound screening of 280,000 school children in Taipei revealed the incidence of URA to be 1 in 1200 (Shieh et al, 1990). A similar incidence was found on prenatal screening in the Czech Republic (Sipek et al, 1997).
The high male predominance of BRA is not nearly as striking in the unilateral condition, with a male to female ratio of 1.8 : 1 (Doroshow and Abeshouse, 1961). Absence of a kidney occurs somewhat more frequently on the left side. A familial tendency has been noted (Arfeen et al, 1993; Selig et al, 1993; Cascio et al, 1999). Siblings within a single family and even monozygotic twins have been affected (Kohn and Borns, 1973; Uchida et al, 1990). In a study of several families, McPherson and colleagues (1987) concluded the inheritance of URA was autosomal dominant with a 50% to 90% penetrance. This inheritance pattern has been confirmed by others who evaluated families with more than one affected individual (Biedel et al, 1984; Roodhooft et al, 1984; Battin et al, 1993). For screening recommendations, see the section Incidence under Bilateral Renal Agenesis.
Genetic/Syndromic Associations
An absent kidney has been noted in a number of genetic disorders in which there is a deletion of several chromosomal loci: 8q13.3 (Pierides et al, 2002), 18q22.2 (Dowton et al, 1997), 22q11 (Anonymous, 1998; Stewart et al, 1999), as well as in X-linked and sporadic cases of Kallmann syndrome (Colquhoun-Kerr et al, 1999; Zenteno et al, 1999; Quinton et al, 2001). Several syndromes have been associated with URA, including Turner syndrome, Poland syndrome (Mace et al, 1972), Frazer syndrome (Fryns et al, 1997), BOR (brachio-oto-renal) syndrome (Pierides et al, 2002), DiGeorge anomaly (when associated with insulin-dependent diabetes mellitus in the mother) (Wilson et al, 1993; Novak and Robinson, 1994), dysmorphogenesis, and Kallmann syndrome. Abnormalities of the KAL1 locus at Xp22 in the X-linked autosomal-dominant disorder have a 40% incidence of URA (Say and Gerald, 1968; Colquhoun-Kerr et al, 1999; Zenteno et al, 1999; Quinton et al, 2001). Similarly, Townes-Brock syndrome with SALL1 deletions is associated with a high incidence of URA (Salerno et al, 2000; Nishinakamura et al, 2001; Sato et al, 2003, 2004). Twenty to 30 percent of children with the VACTERL association (Vertebral, imperforate Anus, Cardiac, Tracheo-Esophageal atresia, Renal, and Limb anomalies) have URA (Barry and Auldist, 1974; Kolon et al, 2000). Children with supernumerary nipples (Urbani and Betti, 1996) and disorders of the ears with hearing loss, especially if it is congenital (Huang et al, 2001), and preauricular pits (Pierides et al, 2002) have been thought to have an increased incidence of URA. Recently, studies have not shown a significant relationship between preauricular pits, minor ear tags, and URA (Arora and Pryce, 2004; Deshpande and Watson, 2006). Nonetheless, a screening renal ultrasonogram is recommended when these ear anomalies are found in the presence of other malformations. In addition, when more than one anomaly is present (e.g., ventricular septal defect [VSD] and an undescended testis) a screening renal ultrasonogram is prudent, but when specific complexes of anomalies associated with renal agenesis are present (for example, VACTERL-associated anomalies), a comprehensive radiographic review of all organ systems is mandatory.
Embryology
The embryologic basis for URA and BRA are thought to be similar. The etiology is most likely due to the UB, because increased RET mutations occur in humans with renal agenesis (Skinner et al, 2008). Complete absence of a bud or aborted ureteral development prevents reciprocal induction, which is critical for the development of the metanephric blastema into the definitive adult kidney. The metanephros is likely not to be responsible for the majority of cases, because the ipsilateral gonad (derived from adjacent mesenchymal tissue) is rarely absent, malpositioned, or nonfunctioning (Ashley and Mostofi, 1960). The high incidence of absent or malformed proximal WD structures in the male and anomalies of the MD structures in the female suggest that the embryologic insult affects the UB primarily in its early development and influences the development of WD derivatives. The abnormality most likely occurs no later than the fourth or fifth week of gestation, when the UB forms and the WD begins to develop into the ejaculatory duct, seminal vesicle, and vas deferens. The MD in the female begins its medial migration at this time, crossing over the WD (sixth week) during its differentiation into the fallopian tube, uterine horn and body, and proximal vagina (Woolf and Allen, 1953; Semmens, 1962; Yoder and Pfister, 1976).
Magee and colleagues (1979) proposed an embryologic classification to explain the association of URA and MD anomalies (Fig. 117–4). In type I URA, the insult occurs before the fourth week, and there is nondifferentiation of the urogenital ridge structures, including the MD and WD. If unilateral, a uterus consisting of a single MD (unicornate uterus) will form and will be associated with contralateral renal agenesis. In type II URA, the insult occurs early in the fourth week of gestation, affecting both the WD and the UB. Because it is critical that the MD maintains close contact with the WD for MD elongation and subsequent fusion, maldevelopment of the WD affects renal development, MD elongation, contact with the urogenital sinus (UGS), and subsequent fusion. Therefore a didelphys uterus will form with obstruction of the horn and vagina on the side of the URA. Finally, in type III URA, the insult occurs after the fourth week, and the WD and MD elongation and differentiation proceed normally. In this case, only the UB and metanephric blastema are affected, thereby resulting in isolated URA.
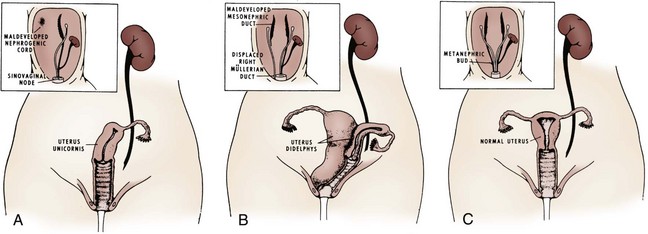
Figure 117–4 A to C, A proposed categorization of genital and renal anomalies in females. See text for details.
(From Magee MC, Lucey DT, Fried FA. A new embryologic classification for urogynecologic malformations: the syndromes of mesonephric duct induced müllerian deformities. J Urol 1979;121:265.)
Associated Genitourinary and Adrenal Anomalies
The ipsilateral ureter is completely absent in about 60% of the cases (Fortune, 1927; Collins, 1932; Ashley and Mostofi, 1960). In the Ashley and Mostofi series, 19 of 232 with URA had only a portion of the lower end of the ureter present. There were no normally developed ureters reaching the level of the normal kidney. In most cases of complete absence of the ureters, the bladder showed no evidence of a ureteric orifice with failure of ipsilateral trigone development (Ashley and Mostofi, 1960). Cell lineage studies using a murine model show that the trigone has a urogenital sinus origin and should form normally (Viana et al, 2007; Mendelsohn, 2009). The trigone may not be distinguishable from the surrounding detrusor when the intramural ureter is absent. Therefore the endoscopic appearance of the trigone in this setting has lead to the probable misnomer in the case of the “hemitrigone” (in association with complete ureteral agenesis) or “asymmetrical trigone” (in the presence of a partially developed ureter). Segmental ureteral atresia on one side has been associated with contralateral ureteral or renal ectopia (Limkakeng and Retik, 1972). Except for ectopia or malrotation, anomalies of the contralateral kidney are infrequent (Longo and Thompson, 1952; Chow et al, 2005) (Fig. 117–5). However, abnormalities of the contralateral ureter are not uncommon, including ureteropelvic and ureterovesical junction obstruction in 11% and 7%, respectively (Cascio et al, 1999), and reflux in 30% (Atiyeh et al, 1993; Cascio et al, 1999). Other urologic abnormalities are found in 65% with URA (Kaneyama et al, 2004).
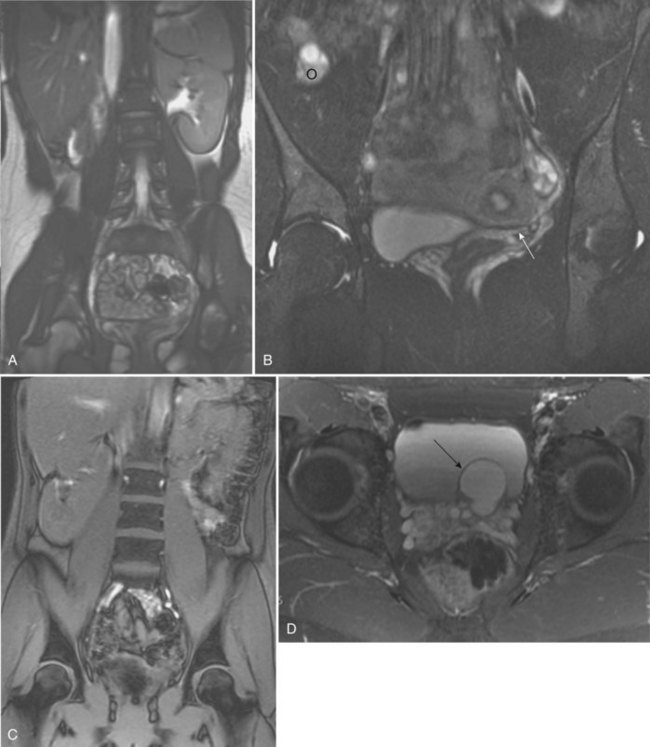
Figure 117–5 Magnetic resonance imaging (MRI) showing (A) coronal scout image of right renal agenesis with the bowel occupying the right renal fossa. B, Coronal T2 fat-saturated images show left unicornuate uterus (arrow), absent right cornua, and superior location of right ovary (o). C, Coronal scout image demonstrates left renal agenesis with bowel occupying the left renal fossa and right renal malrotation. D, Transverse T2 fat-saturated image of male pelvis shows left seminal vesicle cyst (arrow).
Although the ipsilateral adrenal gland may be flattened (Hoffman et al, 1992), adrenal agenesis occurs in fewer than 10% of autopsy reports (Fortune, 1927; Collins, 1932; Ashley and Mostofi, 1960) and in 17% of individuals with URA undergoing a CT scan (Kenney et al, 1985).
Genital anomalies are much more frequently observed. The incidence of a reproductive tract malformation for both sexes varies from 20% to 40% (Smith and Orkin, 1945; Doroshow and Abeshouse, 1961; Thompson and Lynn, 1966). Despite the predominance of males with URA, reproductive tract abnormalities in females occur in at least 25% to 50% compared with 10% to 15% in males. Regardless of sex, both gonads are usually normal. Therefore the different phenotypes that occur with URA may result from a primary urogenital ridge defect, which explains the finding of gonadal and adrenal agenesis in the minority of cases, or a primary defect in development of the UB and WD, which leads to the more common cases of URA and frequently observed abnormalities of the WD, MD, and their derivatives.
Anomalies in the Male
The testis and head of the epididymis, which contain the efferent ductules derived from the mesonephric tubules, are invariably present; all structures proximal to that point, which develop from the WD (the body and tail of the epididymis, vas deferens, seminal vesicle, ampulla, and ejaculatory duct), are absent in almost 50% (Radasch, 1908; Collins, 1932; Charny and Gillenwater, 1965; Ochsner et al, 1972). Donohue and Fauver (1989) reported 79% of adult males with an absence of the vas deferens have an absent ipsilateral kidney; left-sided lesions predominated with a ratio of 3.5 : 1. However, bilateral absence of the vas has been noted with URA (McCallum et al, 2001). Occasionally, the WD structures are rudimentary or ectopic rather than absent (Holt and Peterson, 1974). Ipsilateral cryptorchidism rarely occurs. In 1914, Zinner reported a seminal vesicle cyst in association with ipsilateral renal agenesis (Pereira et al, 2009). Seminal vesicle cysts secondary to obstruction of the ejaculatory duct are currently diagnosed with increasing frequency as pelvic ultrasound examinations are performed more often (Lopez-Garcia et al, 1998; Kaneyama et al, 2004). Six cases (5%) were noted among 119 boys who were found to have URA during ultrasound screening of schoolchildren (Shieh et al, 1990). A pelvic ultrasonogram or magnetic resonance imaging (MRI) in boys diagnosed with URA may demonstrate a seminal vesicle cyst (Van den Ouden et al, 1998, Seo et al, 2009) (see Fig. 117–5). In cases of seminal vesicle cysts and URA, the ureter may insert into the prostatic urethra or seminal vesicle. Cystic dysplasia of the rete testis, a rare benign condition, is often associated with ispsilateral renal anomalies, most commonly URA (Wojcik et al, 1997; Camassei et al, 2002).
In males, the diagnosis of URA should be suspected during a physical examination when the vas deferens or body and tail of the epididymis are impalpable. This may be more common in adults undergoing evaluation for infertility. In infants and young boys, URA should be considered when vasal and/or epididymal anomalies are incidentally found at the time of orchiopexy.
Anomalies in the Female
A variety of anomalies may result in the female from incomplete MD formation because of alterations in normal WD development. Approximately one fourth to one third of women with URA have an abnormality relating to WD development (Thompson and Lynn, 1966; Heinonen, 2004). Conversely, 43% of women with genital anomalies have URA (Semmens, 1962; Heinonen, 1997). The most common MD anomalies are a true unicornuate uterus with complete absence of the ipsilateral horn and fallopian tube or a bicornuate uterus with rudimentary development of the horn on the affected side (Candiani et al, 1997) (see Fig. 117–5). The fimbriated end of the fallopian tube, however, is usually fully formed and is analogous to the head of the epididymis in the male (Shumacker, 1938). Partial or complete midline fusion of the MD may result in a double (didelphys) or septate uterus with either a single or a duplicated cervix (Radasch, 1908; Fortune, 1927). Complete duplication or separation of the vagina, proximal vaginal atresia associated with a small introital dimple, and complete absence of the vagina have been reported (Woolf and Allen, 1953; D’Alberton et al, 1981). Obstruction of one side of a duplicated system is not uncommon, and unilateral hematocolpos or hydrocolpos associated with a pelvic mass and/or pain has been described in pubertal girls (Weiss and Dykhuizen, 1967; Vinstein and Franken, 1972; Gilliland and Dick, 1976; Wiersma et al, 1976; Yoder and Pfister, 1976). Smith and Laufer (2007) suggested the acronym OHVIRA to classify the syndrome of Obstructed Hemivagina and Ipsilateral Renal Anomaly. In rare instances, this anomalous condition has been mistaken for a large or infected Gartner duct cyst. Sometimes a true Gartner duct cyst has been found in a prepubertal girl in association with an ectopic ureter that is blind ending at its proximal end or one that is connected to a rudimentary kidney (Currarino, 1982). Six percent of girls with URA were found to have a Gartner cyst on mass screening of schoolchildren (Shieh et al, 1990). Infertility occurs in as many as 33% of affected women with renal agenesis and unicornuate uterus (Heinonen, 1997). When specific anomalies of the uterus, including congenital absence of the uterus, unicornuate uterus, and didelphic uterus, are found on ultrasonography or MRI, radiologic investigation of the urinary tract often demonstrates URA or other renal anomalies (Bryan et al, 1949; Phelan et al, 1953; Thompson and Lynn, 1966; Candiani et al, 1997; Heinonen, 1997, Govindarajan et al, 2008; Reichman and Laufer, 2010).
Another important anomaly often associated with URA is the Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH), which is a complex of malformations occurring in 1 in 5000 newborn females (Guerrier et al, 2006). This syndrome not only includes renal anomalies but also genital tract anomalies ranging from upper vaginal atresia to total müllerian agenesis in an otherwise phenotypically normal female with a normal 46, XX karyotype. There are two subtypes reported. Type I is the typical form characterized by the finding of only symmetrical muscular buds or müllerian remnants and normal fallopian tubes. Type II, which is the more common but considered the atypical form, is characterized by asymmetrical hypoplasia of one or two buds with or without dysplasia of the fallopian tubes. Most importantly, the atypical form is often associated with renal anomalies, primarily URA or ectopia of one or both kidneys and horseshoe kidney in about 40% to 60% (Guerrier et al, 2006). In addition, there can be cervico-thoracic anomalies, auditory defects, and digital anomalies. Duncan and colleagues (1979) reported on the most severe constellation of malformations and referred to this as the MURCS association or MÜllerian duct aphasia (96%), Renal aphasia or ectopic (86%), and Cardiothoracic Somite dysplasia (two to four anomalous vertebrae between C5-T1 (80%).
Anomalies of Other Organ Systems
Anomalies of other organ systems are found frequently in affected individuals. The more common sites involve the cardiovascular (30%), gastrointestinal (25%), and musculoskeletal (14%) systems (Emanuel et al, 1974) (Fig. 117–6). They include septal and valvular cardiac defects, imperforate anus and anal or esophageal strictures or atresia, and vertebral or phalangeal abnormalities (Jancu et al, 1976; Wheeler and Weaver, 2001; Rai et al, 2002). Dursun and colleagues (2005) found that 44% of individuals with a congenital solitary kidney, most of whom had URA, had various nonurologic anomalies, but they detected lower incidences of these problems (cardiovascular, 15%; gastrointestinal, 9%; neurologic, 3%; and hematologic, 6%) than previously reported by Emanuel. Chow and colleagues (2005) reported a similar incidence of 42%.
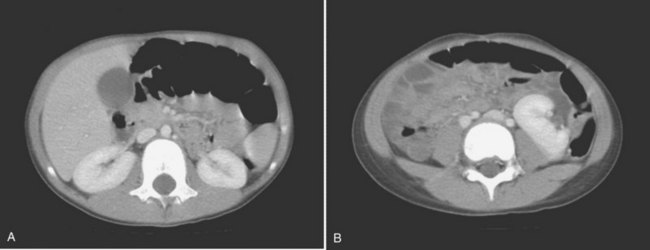
Figure 117–6 Contrast CT scan showing (A) right and left orthotopic kidneys. B, Right midabdominal malrotated supernumerary kidney.
Key Points: Unilateral Renal Agenesis
Diagnosis and Radiographic Evaluation
There are no specific symptoms that suggest an absent kidney. Previously, most reports were compiled from autopsy series, but now prenatal screening is detecting URA. The solitary kidney may begin to undergo compensatory hypertrophy in utero (Mandell et al, 1993; Hill et al, 2000). Recently, prenatal ultrasonography was used to determine compensatory hypertrophy in cases of fetal unilateral empty renal fossa (Cho et al, 2009). These investigators retrospectively measured the ratio of the anteroposterior (AP) and transverse (TR) diameters of the contralateral normal kidney in a set of patients: 12 with URA, 6 with MCDK, and 6 with pelvic kidneys, and these patients were compared with 20 normal controls. They found that when using 0.9 as the discriminating value of the ratio, there was 100% sensitivity, specificity, and accuracy.
Once URA is diagnosed, a retroperitoneal ultrasonogram with color Doppler will show absence of the kidney and ipsilateral renal vessels. A plain film of the abdomen showing the gas pattern of the splenic flexure in the left renal fossa suggests left renal agenesis, ectopia, or crossed ectopia (Mascatello and Lebowitz, 1976), whereas the gas pattern of the hepatic flexure positioned in the right renal fossa suggests congenital absence of the right kidney (Curtis et al, 1977). The diagnosis of URA usually can be confirmed with a DMSA scan showing absent uptake of the isotope on one side, with the contralateral kidney often showing compensatory hypertrophy (Hynes and Watkin, 1970; Cope and Trickey, 1982). A DMSA scan will also detect an ectopic (usually pelvic) or a crossed ectopic kidney in cases where the nonvisualized orthotopic kidney is thought to be absent (Volkan, 2003). In some cases, crossed fused ectopia may be difficult to distinguish from a congenital solitary kidney that has undergone compensatory hypertrophy or a solitary complete duplication. A small dysplastic kidney or MCDK may be misdiagnosed as URA when a kidney is not seen on ultrasonography (Mesrobian, 1993). In these cases, the colonic gas pattern will not be observed in the renal fossa. In addition, calcifications in the renal fossa may suggest an involuted MCDK (Nakano et al, 1996).
Ultrasonography, radionuclide scintigraphy, and MRI have replaced arteriography in diagnosing URA. When a fetus with other suspected organ anomalies undergoes MRI, an absent kidney can be confirmed (Dell’Acqua et al, 2002). URA has also been found incidentally during fluoroscopic monitoring of the renal fossa at the end of a cardiac catheterization or at the end of an echocardiogram. When URA is diagnosed, a voiding cystourethrogram should be performed, because there is a 28% incidence of contralateral reflux (Cascio et al, 1999; Kaneyama et al, 2004).
Special Considerations
The most common question many parents ask is “Will having only one kidney affect my child’s life, and will there be any restrictions on their activities?” Several North American studies examine the risks of injury to the pediatric kidney through sports. Psooy (2006, 2009) summarizes the results of these studies, which suggest that motor vehicle accidents as passenger and pedestrian result in more renal trauma than sports activities. In addition, bicycling, sledding, downhill skiing/snowboarding, and equestrian activities are the more common causes of high-grade renal trauma. These activities have a more than fivefold relative risk for head injury compared with renal injury. Psooy (2009) notes that children are not generally restricted from these activities on the basis of having “only one head.” Rice and the Council on Sports Medicine and Fitness of the American Academy of Pediatrics (2008) advise that each athlete needs individual assessment for their particular sport and that wearing protective padding may reduce the risk of injury to the solitary kidney, thus allowing participation in most sports.
Prognosis
In the past, there was no definitive evidence that a solitary kidney predisposes to increased susceptibility to other diseases (Shapiro et al, 2003). Reviews conducted in the preantibiotic era reported a high incidnce of glomerulonephritis, stones, and tuberculosis. The advent of antibiotics has reduced the incidence of morbidity and mortality for individuals with URA. In the Ashley and Mostofi series (1960), only 15% of the patients died because of renal disease. Renal trauma resulted in death for 5%; some patients in this group might have survived had there been two kidneys. However, because the source of the autopsy material included many military personnel, the potential risk of injury was accentuated. Therefore URA with a normal contralateral kidney is thought to be compatible with normal longevity and the contralateral kidney is not predisposed to greater than normal risks (Gutierrez, 1933; Dees, 1960).
In more contemporary studies, Rugui and colleagues (1986) found an increased occurrence of hypertension, hyperuricemia, and decreased renal function but no proteinuria in a small group of patients with congenital absence of one kidney. Only one patient had a renal biopsy showing focal glomerulosclerosis. This finding has been confirmed in other patients and is similar to observations in the remaining kidney in the hyperfiltration syndrome noted after unilateral nephrectomy (Kiprov et al, 1982; Nomura and Osawa, 1990; Brenner et al, 1996). Oldrizzi and colleagues (1991) reported on URA and progressive renal damage in 39 individuals with a mean age of 33 years. They found prevalence rates of 40% for hypertension, 46% for proteinuria, and 15% for impaired renal function. The limitation of this study is that it was composed of symptomatic patients, and there was no age-matched asymptomatic control population with URA.
Argueso and colleagues (1992) assessed 157 individuals with congenital URA with a mean age of 37 (age 2 to 84) at diagnosis and noted hypertension, proteinuria (>50 mg/day), and mild renal insufficiency in 47%, 19%, and 13%, respectively. Only 32 patients actually had their glomerular flow rate (GFR) measured. Of the 13 patients in their seventh to ninth decade, the mean GFR was 65 mL/minute per 1.73 m2, and the 4 who had renal insufficiency had a GFR between 32 to 53 mL/minute per 1.73 m2. Six of the 43 deceased patients in this study died from chronic renal failure. Only about 25% of patients had their proteinuria quantified, and less than a third had blood pressure measured. Recognizing these study deficiencies, the authors concluded that survival was not impaired in this group, because the overall patient survival curve was similar to the general population. In addition, others have reported that the ability of the kidney to excrete increased loads of protein was not impaired, even in patients with renal insufficiency or proteinuria (DeSanto et al, 1997).
In another study of 206 women with uterine abnormalities, 33 (16%) had URA and 19 had been pregnant and delivered (Heinonen, 2004; Heinonen, 1997). The control group was composed of 44 age-matched women with similar parity and similar uterine malformations but who had two normal kidneys. Eight (42%) of the 19 women with a uterine anomaly and URA (in at least one pregnancy) had gestational hypertension, pre-eclampsia, or gestational proteinuria, compared with only 8 (18%) of the women with two kidneys. The relative risk of gestational hypertension, pre-eclampsia, or gestational proteinuria was significantly higher in women with URA (RR = 2.3). Perinatal outcomes were similar in both groups, with no patient having proteinuria or chronic renal disease, but 2 of the 19 women (11%) with URA were started on long-term antihypertensive medication.
Current Concepts Regarding Prognosis in Adults with Unilateral Renal Agenesis
There is now evidence that subtle defects in UB branching can cause a reduced nephron number, which may lead to renal disease later in life (Brenner and Mackenzie, 1997; Costantini and Shakya, 2006; Chevalier, 2009). In humans, there is an eightfold range in normal variation in the number of glomeruli, that is, from 200,000 to 1.8 million (Hughson, 2003). Therefore it is more likely for an individual with nephron numbers at the lower end of the spectrum to be at greater risk for renal insufficiency at any age and from any etiology.
In the past, studies of the renal survival in children with Congenital Anomalies of the Kidney and Urinary Tract (CAKUT) have been difficult to perform because they involve decades of follow-up and the phenotypes of these disorders are not uniform (Pope et al, 1999; Kerecuk et al, 2008; Zaffanello et al, 2009). Recently, Sanna-Cherchi and colleagues (2009) evaluated the long-term functional outcomes in individuals with CAKUT, including those with URA from a single pediatric nephrology center. Three hundred and twelve patients with CAKUT who had a known defect of the number or size of at least one kidney were followed up to 20 years. Patients with isolated vesicoureteral reflux and duplications were excluded. Dialysis-free survival was evaluated, taking into consideration reflux, age at diagnosis, hypertension, proteinuria, and serum creatinine. Six subgroups were examined, including URA, unilateral and bilateral hypodysplasia, posterior urethral valves, multicystic dysplastic kidney, and horseshoe kidney. These investigators found that by 30 years of age, 19% of all patients were on dialysis, most of whom had posterior urethral valves, bilateral renal hypoplasia, or URA. Further analysis showed that patients with a solitary kidney had a 50% probability of requiring dialysis by 30 years of age. Notably, most of the patients with URA in this study were diagnosed during adolescence and had a normal creatinine level at the time of diagnosis. Interestingly, the patients diagnosed at birth had a slightly elevated creatinine level (0.68 mg/dL). These and other studies point out the fundamental differences in individuals with URA and adults who undergo unilateral nephrectomy. The number of nephrons in children with URA may be abnormally low, and those with fewer nephrons may be at greater risk to developing focal glomerulosclerosis.
Current suggestions for children with a normal-appearing single kidney include annual assessments of blood pressure and proteinuria, the hallmarks of a progressive decrease in glomerular filtration rate (Hegde and Coulthard, 2009). Laboratory evaluation, including a blood urea nitrogen and serum creatinine level, can be performed periodically unless abnormalities are denoted. If there is hypertension, proteinuria, and/or impaired renal function, a pediatric nephrologist should be consulted. They may suggest dietary changes, including limiting salt and avoiding excessive protein intake, depending on the age of the patient. Treatment with an angiotensin-converting enzyme inhibitor may also be indicated (Puddu et al, 2009).
Supernumerary Kidney
Parenchymal development was thought to be controlled by an unidentified substance that limited the amount of functioning renal tissue. Nature has created, albeit rarely, a condition in which three separate kidneys can form. In this condition, the two main kidneys are usually normal and equal in size, whereas the third is small. The supernumerary kidney is truly an accessory organ with its own collecting system, blood supply, and distinct encapsulated parenchymal mass. It may be either totally separate from the normal kidney on the same side or connected to it by loose areolar tissue (Geisinger, 1937). The ipsilateral ureters may be bifid or completely duplicated. The condition is not analogous to a single kidney with ureteral duplication in which the collecting systems drain portions of one parenchymatous mass surrounded by a single capsule.
Incidence
The true incidence of this anomaly cannot be determined, because it occurs very infrequently. About 100 cases have been reported since it was first described in 1656 (Sasidharan et al, 1976; MacPherson, 1987). It affects males and females equally but has a higher predilection for the left side (N’Guessan and Stephens, 1983). Four cases of bilateral supernumerary kidneys have been reported (Campbell, 1970; Oto et al, 2002).
Embryology and Molecular Mechanisms
A second UB or a branching from the initial UB appears as a necessary first step. Next, the nephrogenic anlage may divide into two metanephric tails, which separate entirely when induced to differentiate by the separate or bifid UBs (N’Guessan and Stephens, 1983). The two metanephroi develop only after being penetrated by the bifid or separate UBs. N’Guessan and Stephens do not accept that this condition is the result of widely divergent bifid or separate UBs. Geisinger (1937) proposed that the separate kidneys developed by fragmentation of a single metanephros or by linear infarction producing separate viable fragments that develop only when a second UB is present.
Kidney development is initiated when a single UB forms from the WD in response to GDNF secreted by the adjacent MM. Posterior restriction of Gdnf expression is critical for the development of a UB in the normal position, while another intercellular signaling system, including SLIT2 or its receptor ROBO2, is also important in ensuring that a single UB forms in the appropriate location. Grieshammer and colleagues (2004) showed that mutant mice lacking either SLIT2 or its receptor ROBO2 develop supernumerary UBs that are correlated with abnormal maintenance of Gdnf expression in anterior MM. The SLIT2/ROBO2 intercellular signaling system restricts, directly or indirectly, the extent of the Gdnf expression and plays a critical role in precisely positioning the site of kidney induction.
Description
The supernumerary kidney is a distinct mass of renal parenchyma that may be either completely separate or only loosely attached to the major kidney on the ipsilateral side. In about 60% of cases, it is located caudad to the dominant kidney, which is in its orthotopic position in the renal fossa. When a separate and distinct ureter is present, the supernumerary kidney is more likely to be cranial to the dominant kidney but caudal to the adrenal (Bernik et al, 2001). Occasionally, the supernumerary kidney lies either posterior or superior to the main kidney, or it may even be a midline structure anterior to the great vessels and loosely attached to each of the other two kidneys (see Fig. 117–6). The supernumerary kidney may become wedged between the lower poles of a right and left kidney, leading to the radiographic appearance of a “pseudohorseshoe” kidney (Macpherson, 1987). A pelvic supernumerary kidney has also been reported (Eberle et al, 2002).
Key Points: Supernumerary Kidney
The supernumerary kidney is reniform but generally smaller than the main ipsilateral kidney. In about one third of cases, the kidney or its collecting system is abnormal. In almost half of the reported cases, the collecting system is severely dilated with thin parenchyma suggesting obstruction.
The ureteral interrelationships on the side of the supernumerary kidney can be variable (Kretschmer, 1929). Convergence of the ipsilateral ureters distally to form a common stem and a single ureteral orifice occurs in 50% of the cases (Exley and Hotchkiss, 1944; N’Guessan and Stephens, 1983), which suggests “a bud off of a bud” situation. Two completely independent ureters, each with its own entrance into the bladder, are seen in the other 50% of cases. The Weigert-Meyer principle (see Chapter 111) usually is followed, but in 10%, the caudal kidney has a ureter that does not follow the rule and enters the trigone below the ipsilateral ureter (Tada et al, 1981) (Fig. 117–7). Rarely, the supernumerary kidney has a completely ectopic ureter opening into the vagina or introitus (Rubin, 1948; Carlson, 1950). Individual case reports have described calyceal communications between the supernumerary and the dominant kidney, or fusion of the dominant kidney’s ureter with the pelvis of the supernumerary kidney (Kretschmer, 1929) to create a single distal ureter that then enters the bladder (Fig. 117–8). The vascular supply to the supernumerary kidney is anomalous and depends on its position in relation to the major ipsilateral kidney. Most investigators believe that the blood supply to the individual parenchymal masses should be separate to consider this a true supernumerary kidney (Kaneoya et al, 1989).
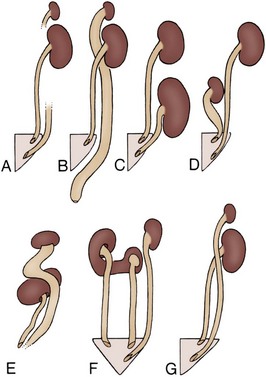
Figure 117–7 A to G, Various patterns of urinary drainage of supernumerary and ipsilateral kidneys when ureters are completely separated. All kidney positions are relative only and are depicted on the left side for ease of interpretation. Dashed lines indicate that detail was not defined.
(From N’Guessan G, Stephens FD. Supernumerary kidney. J Urol 1983;130:649.)
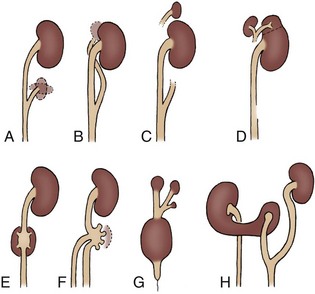
Figure 117–8 A to H, Various patterns of urinary drainage when ureters form a common stem. All kidney positions are relative only and are depicted on the left side for ease of interpretation. Dashed lines indicate that detail was not defined.
(From N’Guessan G, Stephens FD. Supernumerary kidney. J Urol 1983;130:649.)
Associated Anomalies
Usually, the ipsilateral and contralateral kidneys are normal. Except for an occasional ectopic orifice from the ureter draining the supernumerary kidney, no genitourinary abnormalities are present in any consistent pattern. A few of the case reports describe anomalies of other organ systems (Janda et al, 2009).
Symptoms
Although this anomaly is present at birth, it is rarely symptomatic but may become symptomatic in early adulthood. The average age at diagnosis was 36 years. Pain, fever, hypertension, and a palpable abdominal mass are the usual presenting complaints. Urinary infection or obstruction, or both, are the major conditions that lead to an evaluation. Ureteral ectopia from the supernumerary kidney may produce urinary incontinence, but this is extremely rare (Shane, 1942; Hoffman and McMillan, 1948).
A palpable abdominal mass secondary to development of carcinoma in the supernumerary kidney has been described in two patients. In 25% of all reported cases, the supernumerary kidney remains completely asymptomatic and is discovered only at autopsy (Carlson, 1950).
Diagnosis
If the supernumerary kidney is normal and asymptomatic, it is usually diagnosed when abdominal ultrasonography, CT, or MRI is performed for other reasons. The kidney may be inferior and distant enough from the ipsilateral kidney so that it does not alter the position of the normal kidney (Conrad and Loes, 1987). If it is in close proximity, it may displace the predominant kidney or its ureter very slightly.
A supernumerary kidney may become symptomatic from hydronephrosis due to obstruction or stone formation (Koureas et al, 2000). In this case, ultrasonography may demonstrate distortion of the normal ipsilateral kidney and ureter. If the collecting system is bifid, the dominant kidney on that side will usually be involved in the same disease process. If the ureters are separate, the ipsilateral kidney may show the effects of an abnormal supernumerary kidney. Voiding cystourethrography, ultrasonography, or MR urography, and even retrograde pyelography may be needed to help delineate the anomaly. Radionuclide imaging provides information about relative function in the supernumerary and the normal kidneys (Conrad and Loes, 1987). Cystoscopy reveals one or two ureteral orifices on the ipsilateral side, depending on whether the ureters are completely duplicated. Ureteral ectopia may exist in or outside of the bladder. Occasionally, a supernumerary kidney is not accurately diagnosed until the time of surgery or at autopsy, or it may mimic a duplication (Kaneoya et al, 1989).
Anomalies of Ascent
Simple Renal Ectopia
When the mature kidney fails to reach its normal location in the “renal” fossa, the condition is known as renal ectopia. The term is derived from the Greek words ek (“out”) and topos (“place”) and literally means “out of place.” It is to be differentiated from renal ptosis, in which the kidney initially is located in its proper place (and has normal vascularity) but moves downward in relation to body position. The ectopic kidney has never resided in the appropriate location. An ectopic kidney can be found in one of the following positions: pelvic, iliac, abdominal, thoracic, and contralateral or crossed (Fig. 117–9).
Figure 117–9 Incomplete ascent of kidney. The kidney may halt at any level of its ascent from the pelvis.
(From Gray SW, Skandalakis JE. The kidney and ureter. In: Gray SW, Skandalakis JE, editors. Embryology for surgeons. Philadelphia: WB Saunders; 1972.)
Incidence
Renal ectopia has been known to exist since it was described by 16th century anatomists, but it did not achieve clinical interest until the mid-19th century. Recently, with improved imaging, simple renal ectopia has been noted with increasing frequency.
The actual incidence among autopsy series varies from 1 in 500 (Campbell, 1930) to 1 in 1200 (Stevens, 1937; Thompson and Pace, 1937; Anson and Riba, 1939; Bell, 1946a), but the average occurrence is about 1 in 900 (Abeshouse and Bhisitkul, 1959). With increasing clinical detection, the incidence among hospitalized patients has approached the autopsy rate (Abeshouse and Bhisitkul, 1959). Autopsy studies reveal no significant difference in incidence between the sexes. A recent review of prenatal ultrasonograms detected an incidence of 0.003% in India, but this likely under-represents its true occurrence (Sanghvi et al, 1998). Clinically, renal ectopia is more readily recognized in females; they undergo uroradiologic evaluation more frequently than males because of their higher rate of UTI and/or associated genital anomalies (Thompson and Pace, 1937).
The left side is favored slightly over the right. Pelvic ectopia has been estimated to occur in 1 of 2100 to 3000 autopsies (Stevens, 1937). A solitary ectopic kidney occurs in 1 of 22,000 autopsies (Stevens, 1937; Hawes, 1950; Delson, 1975). By 1973, only 165 cases of a solitary pelvic kidney had been recorded (Downs et al, 1973). Bilateral ectopic kidneys are even more rarely observed and account for only 10% of all patients with renal ectopia (Malek et al, 1971) (Fig. 117–10).
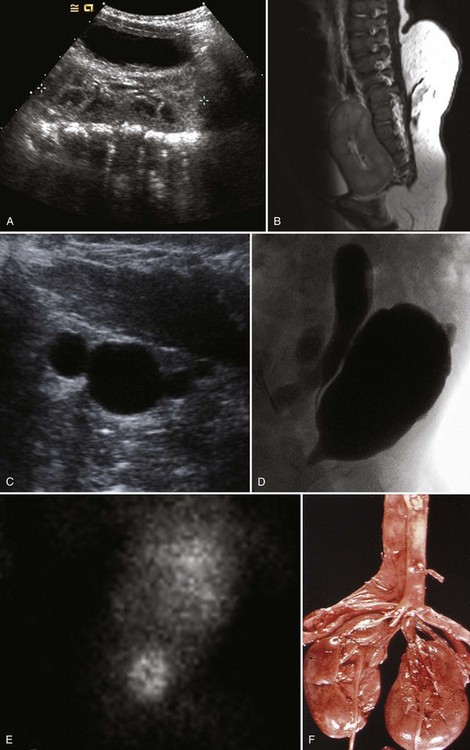
Figure 117–10 One-day-old boy with a right retrovesical pelvic kidney demonstrated on (A) transverse ultrasonogram of right pelvis. B, Sagittal MRI. Vertebral abnormalities and a portion of a lipomyelomeningocele are also observed. C, Longitudinal ultrasonogram of left multicystic dysplastic pelvic kidney. D, Voiding cystourethrogram shows reflux into dilated, tortuous right megaureter. E, Flow study of dimercaptosuccinic acid shows early activity only in the region of the right pelvic kidney and no uptake on the left. F, Postmortem specimen from a different case showing bilateral pelvic ectopia, anterior orientation of renal pelves, and anomalous blood supply from the aortic bifurcation.
(C, Courtesy of Dr. Sara Milla; F, from Weiss MA, Mills SE. Atlas of genitourinary tract disorders. Philadelphia: JB Lippincott; 1988.)
Embryology
The UB, arising from the WD at the end of the fourth week, grows craniad toward the urogenital ridge, acquiring a cap of metanephric blastema by the fifth week. The developing metanephric tissue and UB migrate cephalad, rotating medially on its long axis. The entire process is completed by the eighth week of gestation. Factors that may prevent the orderly ascent and rotation of kidneys include UB maldevelopment (Campbell, 1930), defective metanephric tissue that fails to induce ascent (Ward et al, 1965), genetic abnormalities, and maternal illnesses or teratogenic causes (Malek et al, 1971). A vascular barrier that prevents upward migration secondary to persistence of the fetal blood supply has also been postulated (Baggenstoss, 1951), but the existence of an “early” renal blood supply does not prevent the affected kidney’s movement to its ultimate position. Most probably, this is the end result, not the cause, of renal ectopia.
Description
The classification of ectopia is based on the position of the kidney within the retroperitoneum: The pelvic kidney opposite the sacrum and kidneys below the aortic bifurcation are the most common sites of ectopia; the lumbar kidney resides near the sacral promontory in the iliac fossa and anterior to the iliac vessels; and the abdominal kidney is above the iliac crest and adjacent to the second lumbar vertebra (see Fig. 117–10).
The ectopic kidney is usually smaller, and it may not conform to the usual reniform shape, due to the presence of fetal lobulations. The axis of the kidney is slightly medial or vertical, but it may be tilted as much as 90 degrees laterally so that it lies in a true horizontal plane. The renal pelvis is usually anterior (instead of medial) to the parenchyma, because the kidney has incompletely rotated. As a result, 56% of ectopic kidneys have a hydronephrotic collecting system. Half of these cases are due to obstruction of the ureteropelvic or the ureterovesical junction (70% and 30%, respectively), 25% from reflux grade III or greater, and 25% from the malrotation alone (Gleason et al, 1994). Overall, vesicoureteral reflux has been found in 30% of children with ectopic kidneys (Guarino et al, 2004).
The length of the ureter usually conforms to the position of the kidney, but occasionally, it is slightly tortuous. It is rarely redundant, in contrast to the ptotic kidney, in which the ureter has achieved its full length before the kidney drops. The ureter usually enters the bladder on the ipsilateral side with its orifice positioned normally, except for those unusual cases with ectopic ureters. The arterial and venous network is anomalous and its vascular pattern depends on the ultimate position of the kidney (Anson and Riba, 1939). There may be one or two main renal arteries arising from the distal aorta or from the aortic bifurcation, with one or more aberrant arteries emanating from the common or external iliac or even the inferior mesenteric artery. The kidney may be supplied entirely by multiple anomalous branches, none of which arises from the aorta. In no instance has the main renal artery arisen from the level of the aorta that would be its proper origin if the kidney were positioned normally.
Associated Anomalies
Although the contralateral kidney is usually normal, it is associated with a number of congenital defects. Malek and colleagues (1971) and Thompson and Pace (1937) reported the incidence of contralateral agenesis to be rather high (Chow et al, 2005). Bilateral ectopia occurs infrequently (10%) (see Fig. 117–10). Hydronephrosis secondary to obstruction or reflux may be seen in as many as 25% of nonectopic contralateral kidneys (Gleason et al, 1994).
Key Points: Simple Renal Ectopia
The most striking feature is the association of genital anomalies and ectopia. The incidence varies from 15% (Thompson and Pace, 1937) to 45% (Downs et al, 1973), depending on how carefully the patient is evaluated. Twenty to 66 percent of females have one or more of the following abnormalities of the reproductive organs: bicornuate or unicornuate uterus with atresia of one horn (McCrea, 1942), rudimentary or absent uterus and proximal and/or distal vagina (Tabisky and Bhisitkul, 1965; D’Alberton et al, 1981), and duplication of the vagina. Among male patients, 10% to 20% have a recognizable associated genital defect; undescended testes, duplication of the urethra, and hypospadias are the most common (Thompson and Pace, 1937). Fourteen percent of patients with a cloacal malformation have an ectopic kidney (Warne et al, 2002; Dursun et al, 2005).
Rarely is the adrenal gland absent or abnormally positioned. Twenty-one percent of individuals have anomalies of other organ systems (Downs et al, 1973); most involve the skeletal or cardiac systems.
Diagnosis
With the increasing use of various imaging modalities, the incidence of an asymptomatic ectopic kidney is increasing. Most ectopic kidneys are asymptomatic. Vague abdominal complaints or ureteral colic secondary to an obstructing stone are the most frequent symptoms leading to the diagnosis of an ectopic kidney. The abnormal position of the kidney results in a pattern of direct and referred pain that is atypical for colic and may be misdiagnosed as acute appendicitis or as pelvic inflammatory disease in female patients. Symptoms rarely occur due to adjacent organs to the ectopic kidney. Renal ectopia may also present with a UTI or a palpable abdominal mass. Seven cases of concomitant renal and ureteral ectopia presenting with urinary incontinence have been reported (Borer et al, 1993, 1998). The difficulty in diagnosing this condition is related to the poor function of these ectopic kidneys. The kidneys may be very small and/or dysplastic with essentially no function leading to the misdiagnosis of URA. DMSA scanning or MR urography may both be necessary to diagnose these unusual cases (Borer et al, 1998; Leitha, 1998; Pattaras et al, 1999) (see Fig. 117–10).
Malposition of the colon as observed with renal ageneisis will be observed in cases of ectopic lumbar or pelvic kidney. The diagnosis is made when the renal ultrasonogram fails to reveal a kidney in its orthotopic location. Many of these kidneys were not visualized in the past on an excretory urogram because they overlie the bony pelvis, which obscures the collecting system and leads to a misdiagnosis. With a carefully performed power color Doppler study, the main renal artery and intrarenal vasculature can be more easily delineated. Ultrasonography of the pelvic ectopic kidney demonstrated absence of renal sinus echoes, a normal finding associated with the extrarenal position of the pelvis and calyces (Barnewolt and Lebowitz, 1996).
Cystoscopy, if performed, will demonstrate ureteral orifices that are invariably normal unless the ureteral orifice is also ectopic. If surgery is indicated on an ectopic kidney, MR arteriography can be performed preoperatively to define the anatomy of the renal vasculature, which is especially important in cases of solitary ectopia.
Prognosis
The ectopic kidney is no more susceptible to disease than the normally positioned kidney, except for the development of hydronephrosis or urinary calculus formation (Gleason et al, 1994; Benchekroun et al, 2002). This may be due to the anteriorly placed pelvis and malrotation of the kidney, which may lead to impaired drainage of urine from a high insertion of the ureter to the pelvis or an anomalous vasculature that partially obstructs one of the major calyces or the upper ureter. In addition, there may be an increased risk of injury from blunt abdominal trauma, because the low-lying kidney is not protected by the rib cage.
Renovascular hypertension secondary to an anomalous blood supply has been reported, but a higher-than-normal incidence is yet to be proved. A recent study by van den Bosch and colleagues (2010) examined the urologic and nephrologic consequences of both simple and crossed renal ectopia. They found no adverse effects on blood pressure or kidney function during childhood. They did note that although global renal function of these kidneys was normal, the relative function of the ectopic kidney on dimercaptosuccinic acid scan (99mTc-DMSA) was 38% (interquartile range [IQR] 33% to 43%).
Anderson and Harrison (1965), in a review of pregnant women with renal ectopia, found no increased occurrence of difficult deliveries or maternal or fetal complications related to the ectopic kidney (Anderson and Harrison, 1965; Delson, 1975). Dystocia from a pelvic kidney is a very rare finding, but when it does occur, early recognition is mandatory and cesarean section is indicated. Although three cases of cancer in an ectopic kidney have been reported, there does not appear to be an increased risk for malignancy. No deaths have been directly attributable to the ectopic kidney, but in at least five instances, a solitary ectopic kidney has been mistakenly removed, because the kidney was thought to represent a pelvic malignancy (Downs et al, 1973). This should not happen with the current armamentarium of imaging techniques available to accurately diagnose this condition.
Cephalad Renal Ectopia
The kidney may be positioned more craniad than normal when there is an omphalocele (Pinckney et al, 1978). When the liver herniates into the omphalocele with the intestines, the kidneys continue to ascend until their ascent is arrested by the diaphragm. In all reported cases, both kidneys were affected and were positioned immediately beneath the diaphragm at the level of the 10th thoracic vertebra. The ureters are excessively long but otherwise normal. A color Doppler ultrasonogram or MR arteriography demonstrates that the origin of each renal artery is more cephalad than normal, and no other abnormality of the vascular network is present. Patients with this anomaly usually have no symptoms referable to the malposition, and urinary drainage is not impaired.
Thoracic Kidney
The rarest form of renal ectopia exists when the kidney is positioned considerably higher than normal. Intrathoracic ectopia denotes either a partial or a complete protrusion of the kidney above the level of the diaphragm into the posterior mediastinum (Fig. 117–11). Fewer than 5% of all patients with renal ectopia have an intrathoracic kidney, with an incidence of 1 : 13,000 at autopsy (Campbell, 1930). This condition must be differentiated from a congenital or traumatic diaphragmatic hernia in which other abdominal organs, as well as the kidney, have advanced into the chest cavity.
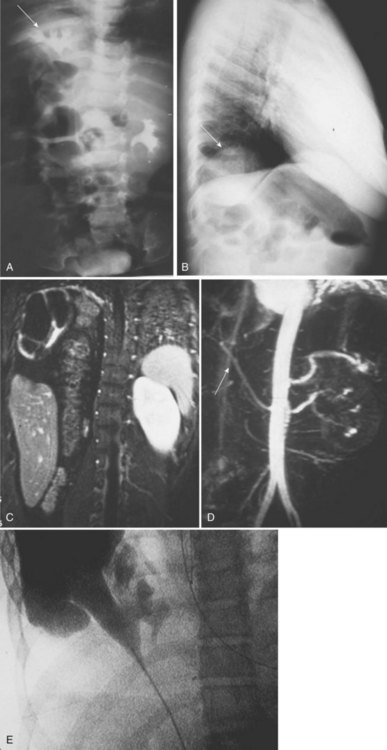
Figure 117–11 One-year-old girl with febrile urinary tract infection (UTI). A, Intravenous pyelogram shows a right thoracic kidney (arrow) and left orthotopic kidney. She was found to have bilateral vesicoureteral reflux and underwent bilateral ureteral reimplants. At age 16, she developed right back pain and chest tightness. B, Chest radiograph demonstrates right diaphragmatic eventration and a right intrathoracic kidney (arrow). C, MR urogram shows coronal T1 fat-saturation after contrast images of poorly functioning right hydronephrotic kidney located superior to the liver and inferior to the right lung with absence of the posterior right hemidiaphragm, permitting the colon to enter the right hemithorax. D, Angiographic sequence demonstrating right renal artery (arrow) arising from the aorta at the normal level of the left renal artery coursing superiorly to enter the right renal hilum. E, Intraoperative right retrograde pyelogram shows narrowing at the ureteropelvic junction.
(A, Courtesy of Dr. Terry Hensle.)
Incidence
Before 1940, this condition was only noted at autopsy (DeCastro and Shumacher, 1969). Since that time, at least 200 patients with a thoracic kidney have been reported in the literature (Donat and Donat, 1988; Lacasta Garcia et al, 1999), 4 of whom had bilateral thoracic kidneys (Berlin et al, 1957; Hertz and Shahin, 1969; Lundius, 1975; N’Guessan and Stephens, 1984; Liddell et al, 1989). There appears to be a slight left-sided predominance of 1.5 : 1, and the sex ratio favors males by 2 : 1 (Lozano and Rodriguez, 1975). This entity has been found on prenatal ultrasonography (Masturzo et al, 2001) and in all age groups, from a neonate (Shapira et al, 1965) to a 75-year-old man evaluated for prostatic hypertrophy (Burke et al, 1967), but it is most commonly detected in adults undergoing chest radiography for other reasons (Drop et al, 2003) (see Fig. 117–11).
Embryology
The kidney reaches its adult location by the end of the eighth week of gestation. At this time, the diaphragmatic leaflets are formed as the pleuroperitoneal membrane separates the pleural from the peritoneal cavity. Mesenchymal tissues associated with this membrane eventually form the muscular component of the diaphragm. It is uncertain whether delayed closure of the diaphragmatic anlage allows for protracted renal ascent above the level of the future diaphragm or whether the kidney overshoots its usual position because of accelerated ascent before normal diaphragmatic closure (Spillane and Prather, 1952; Burke et al, 1967; N’Guessan and Stephens, 1984). Delayed involution of mesonephric tissue has been proposed as a causative factor (Angulo et al, 1992), because intrathoracic kidneys occur in only 0.25% of patients with a diaphragmatic hernia (Donat and Donat, 1988). Renal angiography has demonstrated either a normal site (Lundius, 1975) or a more cranial origin (Franciskovic and Martincic, 1959) for the origin of the renal artery from the aorta supplying the thoracic kidney (see Fig. 117–11).
Description
The kidney is situated in the posterior mediastinum and usually has completed the normal rotation process. The renal contour and collecting system are normal. The kidney usually lies in the posterolateral aspect of the diaphragm in the foramen of Bochdalek. At this point, the diaphragm thins out and a flimsy membrane surrounds the protruding portion of kidney. Therefore the kidney is not within the pleural space (N’Guessan and Stephens, 1984). The lower lobe of the adjacent lung may be hypoplastic secondary to compression by the kidney mass. The renal vasculature and the ureter enter and exit from the pleural cavity through the foramen of Bochdalek.
Associated Anomalies
The ureter is elongated to accommodate the excessive distance to the bladder, but it never enters ectopically into the bladder or other pelvic sites. N’Guessan and Stephens (1984) analyzed 10 cases and determined that the adrenal gland typically occupies its normal location in most patients. In unilateral cases, the contralateral kidney is usually normal. No consistent anomalies have been described in other organ systems.
Symptoms
The vast majority of affected individuals are asymptomatic. Pulmonary and urinary symptoms are exceedingly rare. One case of a pyeloplasty in a thoracic kidney with ureteropelvic junction (UPJ) obstruction and presenting with flank pain has been reported (Hampton and Borden, 2002). A case depicted in Figure 117–11 shows severe hydronephrosis of a previously normal thoracic kidney, presumably due to intermittent obstruction by the muscular fibers of the diaphragm (Shapiro, personal communication).
Diagnosis
The diagnosis is most commonly made after a routine chest radiograph shows the affected hemidiaphragm slightly elevated. A smooth, rounded mass is seen extending into the chest near the midline on an anteroposterior film and along the posterior aspect of the diaphragmatic leaflet on a lateral view (see Fig. 117–11). A thoracic kidney may be found at the time of thoracotomy for a suspected mediastinal tumor (DeNoronha et al, 1974). Historically, excretory urography or renal scintigraphy (Williams et al, 1983) provided the diagnosis, while CT or MR urography is currently the imaging modality of choice. Rarely, cardiac or pulmonary arteriography reveals a thoracic kidney (Fusonie and Molnar, 1966).
Anomalies of Form and Fusion
Crossed Renal Ectopia with and without Fusion
When a kidney is located on the side opposite from that in which its ureter inserts into the bladder, the condition is known as crossed ectopia. Ninety percent of crossed ectopic kidneys are fused to their ipsilateral mate. Except for the horseshoe anomaly, they account for the majority of fusion defects. Fusion anomalies are usually diagnosed in children as part of a constellation of malformations, in young adults during evaluation for delayed menarche, and in the elderly as incidental findings (Glodny et al, 2008).
Fusion anomalies of the kidney were first logically categorized by Wilmer (1938), but McDonald and McClellan (1957) refined and expanded that classification to include crossed ectopia with fusion, crossed ectopia without fusion, solitary crossed ectopia, and bilaterally crossed ectopia (Fig. 117–12). The fusion anomalies have been designated as (1) unilateral fused kidney with inferior ectopia; (2) sigmoid, or S-shaped; (3) lump or cake; (4) L-shaped, or tandem; (5) disc, shield, or doughnut; and (6) unilateral fused kidneys with superior ectopia (Fig. 117–13). Although this classification has little clinical significance, it does provide insights into the embryology of renal ascent and rotation.
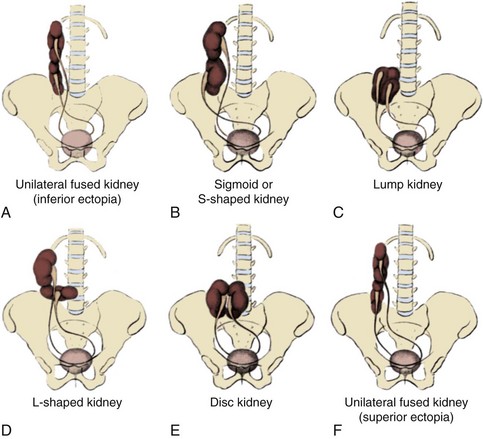
Incidence
The first reported case of crossed ectopia was described by Pamarolus in 1654. Abeshouse and Bhisitkul, in 1959, conducted the last significant review of the subject and reported 500 cases of crossed ectopia with and without fusion that presented primarily with clinical symptoms. Glodny and colleagues (2008) reported on 24 crossed fused ectopia found on a CT scan performed for nonurologic indications.
Sixty-two patients with crossed ectopia without fusion have been reported (Diaz, 1953; Winram and Ward-McQuaid, 1959). This represents approximately 10% of all crossed ectopic kidneys (Lee, 1949). The anomaly occurs more commonly in males with a ratio of 2 : 1, and left-to-right ectopia is seen three times more frequently than right-to-left ectopia (Lee, 1949).
Solitary crossed ectopia has been reported in 34 patients (Miles et al, 1985; Gu and Alton, 1991). Males predominate with a ratio of 2 : 1. The crossed ectopia involves migration of the left kidney to the right side with absence of the right kidney, rather than the reverse, with a ratio of almost 2 : 1 (Kakei et al, 1976). In most cases, the kidney fails to ascend and rotate completely. The left to right crossed kidney is rarely a multicystic dysplastic kidney. Bilateral crossed renal ectopia has been described in five patients (McDonald and McClellan, 1957; Abeshouse and Bhisitkul, 1959) and is considered the rarest form. Abeshouse and Bhisitkul (1959) compiled 443 reports of crossed ectopia with fusion and estimated its occurrence at 1 in 1000 live births. This figure varies with the type of fusion anomaly; the unilaterally fused kidney with inferior ectopia is the most common variety, whereas fusion with superior ectopia is the least common. The autopsy incidence has been calculated at 1 in 2000 (Baggenstoss, 1951). There is a slight male predominance (3 : 2), and a left-to-right crossover occurs somewhat more frequently than its counterpart.
Embryology
The UB enters the metanephric blastema adjacent to the anlage of the lumbosacral spine. During the next 4 weeks, the developing kidney comes to lie at the level of the L1-L3 vertebrae. Because the mechanisms responsible for normal complete ascent of the kidney during gestation are unknown, the cause of crossed ectopia is also unknown. Wilmer (1938) suggested that crossover occurs as a result of pressure from abnormally placed umbilical arteries that prevent cephalad migration of the renal unit, which then follows the path of least resistance to the opposite side.
Potter (1952) and Alexander and colleagues (1950) theorized that crossed ectopia is strictly a ureteral phenomenon, with the developing UB wandering to the opposite side and inducing differentiation of the contralateral nephrogenic anlage. Ashley and Mostofi (1960) deduced that strong but undetermined forces are responsible for renal ascent and that these forces attract one or both kidneys to their final place on the opposite side of the midline. Cook and Stephens (1977) postulated that crossover is the result of malalignment and abnormal rotation of the caudal end of the developing fetus, with the distal curled end of the vertebral column being displaced to one side or the other. As a result, either the cloaca and WD structures lie to one side of the vertebral column, allowing one ureter to cross the midline and enter the opposite nephrogenic blastema, or the kidney and ureter are transplanted to the opposite side of the midline during “normal” renal ascent (Hertz et al, 1977; Maizels and Stephens, 1979).
Kelalis and colleagues (1973) implicated teratogenic factors after they noted an increased incidence of associated genitourinary and other organ system anomalies. Finally, genetic influences may play a role, because familial inheritance of crossed renal ectopia has been reported (Greenberg and Nelsen, 1971; Hildreth and Cass, 1978; Rinat et al, 2001).
Fusion of the metanephric masses may occur when the renal anlagen are still in the true pelvis before or at the start of cephalad migration, or it may occur during the latter stages of ascent. The extent of fusion is determined by the proximity of the developing renal anlagen to one another. After fusion, advancement of the kidneys toward their normal location is impeded by midline retroperitoneal structures, the aortic bifurcation, the inferior mesenteric artery, and the base of the small bowel mesentery (Joly, 1940).
Description
Fusion of a crossed ectopic kidney is related to the time at which it comes in contact with its mate. The crossed kidney usually lies caudad to its normal counterpart on that side. It is likely that migration of each kidney begins simultaneously, but ascent of the ectopic renal unit lags behind because of crossover time. Therefore it is the superior pole of the ectopic kidney that usually joins with the inferior aspect of the normal kidney. Ascent continues until either the uncrossed kidney reaches its normal location or one of the retroperitoneal structures prevents further migration of the fused mass. The final shape of the fused kidneys depends on the time and extent of fusion and the degree of renal rotation that has occurred. No further rotation is likely once the two kidneys have joined. Therefore the position of each renal pelvis may provide a clue as to the chronology of the congenital defect. An anteriorly placed pelvis suggests early fusion, whereas a medially positioned renal pelvis indicates that fusion probably occurred after rotation was completed.
Ninety percent of crossed ectopic kidneys are fused with their mate. When they are not fused, the uncrossed kidney usually resides in its normal dorsolumbar location with proper orientation, while the ectopic kidney is inferior and in either a diagonal or a horizontal position with an anteriorly placed renal pelvis. The two kidneys are usually separated by a variable distance, and each is surrounded by its own capsule of Gerota fascia. In every case of crossed ectopia without fusion, the ureter from the normal kidney enters the bladder on the same side, and that of the ectopic kidney crosses the midline at the pelvic brim and enters the bladder on the contralateral side (Fig. 117–14).
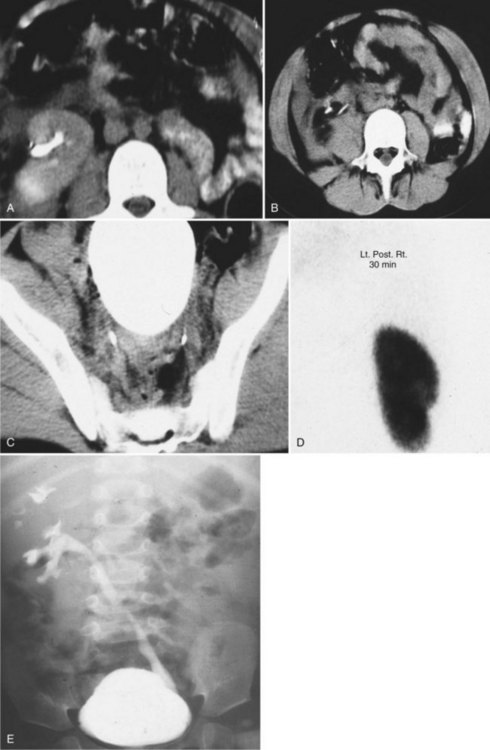
Figure 117–14 Twelve-year-old girl was struck by a motor vehicle. A, Contrast phase of CT scan shows absent left kidney with the bowel occupying the left renal fossa and a malrotated right kidney. B, Two contrast-filled ureters are seen in the midabdomen. C, The two contrast-filled ureters are noted in their orthotopic positions at the retrovesical level. D, Renal scan shows left to right crossed fused ectopia. E, Two-year-old girl with pyelonephritis. Voiding cystourethrogram shows bilateral vesicoureteral reflux and left to right crossed ectopia with colon occupying the left renal fossa.
In cases of solitary crossed ectopia, the kidney is usually located somewhat low but in the opposite renal fossa at the level of L1-L3 and is oriented anteriorly, having incompletely rotated on its vertical axis (Alexander et al, 1950; Purpon, 1963). When the kidney remains in the pelvis or ascends only to the lower lumbar region, it may assume a horizontal lie with an anteriorly placed pelvis because it has failed to rotate fully (Trabrisky and Bhisitkul, 1965). The ureter crosses the midline above the S2 vertebra and enters the bladder on the opposite side (Gu and Alton, 1991). The contralateral ureter, if present, is often rudimentary (Caine, 1956). The patient with bilateral crossed ectopia may have perfectly normal-appearing kidneys and renal pelves, but the ureters cross the midline at the level of the lower lumbar vertebrae (Abeshouse and Bhisitkul, 1959).
Key Points: Crossed Renal Ectopia with and without Fusion
Inferior Ectopic Kidney
Two thirds of all unilaterally fused kidneys involve inferior ectopia. The upper pole of the crossed kidney is attached to the inferior aspect of the normally positioned mate. Both renal pelves are anterior, so fusion probably occurs relatively early.
Sigmoid, or S-Shaped, Kidney
The sigmoid, or S-shaped, kidney is the second most common anomaly of fusion. The crossed kidney is inferior, with the two kidneys fused at their adjacent poles. Fusion of the two kidneys occurs relatively late, after complete rotation on the vertical axis has taken place. Therefore each renal pelvis is oriented correctly, and they face in opposite directions from one another. The lower convex border of one kidney is directly opposite the outer border of its counterpart, creating an S-shaped appearance to the entire renal outline. The ureter from the normal kidney courses downward anterior to the outer border of the inferior kidney, and the ectopic kidney’s ureter crosses the midline before entering the bladder.
Cake or Lump Kidney
The cake or lump kidney is a relatively rare form of fusion (Fig. 117–15). Extensive fusion has taken place over a wide margin of maturing renal anlage, resulting in one mass. The total kidney is irregular and lobulated. Usually, ascent progresses only as far as the sacral promontory, but in many instances, the kidney remains within the true pelvis. Both renal pelves are anterior, and they drain separate areas of parenchyma. The ureters do not cross. Only nine cases of pelvic cake kidney drained by a single ureter have been reported (Schwartz, 2010). Recently, a case of cake kidney drained by a single ureter associated with a unicornuate uterus, and another associated with bilateral absence of the vas deferens, have been reported (Rosenkrantz et al, 2010; Schwartz et al, 2010).
Figure 117–15 A, Lump or cake kidney showing the unusual anatomy, with the anterior blood supply coming from above and the ureters leaving from below. B, Ultrasonogram of cake kidney in sagittal view (space between calipers denotes renal length).
(A, Courtesy of Dr. H.S. Altman; B, courtesy of Dr. Shpetim Telegrafi.)
L-Shaped Kidney
The L-shaped, or tandem, kidney occurs when the crossed kidney assumes a transverse position at the time of its attachment to the inferior pole of the normal kidney. The crossed kidney lies in the midline or in the contralateral paramedian space anterior to the L4 vertebra. Rotation about the long axis of the kidney produces an inverted or a reversed pelvic position. The ureter from each kidney enters the bladder on its respective side.
Disc Kidney
Disc, shield, doughnut, or pancake kidneys are kidneys that have joined at the medial borders of each pole to produce a doughnut- or ring-shaped mass. More extensive fusion along the entire medial aspect of the kidneys creates a disc or shield shape. The lateral aspect of each kidney retains its normal contour. This type of fusion differs from the lump or cake kidney in that the reniform shape is better preserved owing to a less extensive degree of fusion. The pelves are anteriorly placed, and the ureters remain uncrossed. The collecting system drains each respective half of the kidney and does not communicate with the opposite side.
Superior Ectopic Kidney
The least common variety of renal fusion is the crossed ectopic kidney that lies superior to the normal kidney. The lower pole of the crossed kidney is fused to the upper pole of the normal kidney. The renal units retain a fetal orientation, with both pelves lying anteriorly, suggesting that fusion occurred very early.
Regardless of the type of fusion encountered, the vascular supply to the kidneys is variable and unpredictable. The crossed ectopic kidney is supplied by one or more branches from the aorta or common iliac artery (Rubinstein et al, 1976). The normal kidney frequently has an anomalous blood supply, with multiple renal arteries originating from various levels along the aorta. In one rare instance, Rubinstein and colleagues (1976) discovered that one renal artery crossed the midline to supply the tandem ectopic kidney. The solitary crossed ectopic kidney generally receives its blood supply from the aorta or iliac artery on the side which it is positioned (Tanenbaum et al, 1970).
Associated Anomalies
In all the types of fusion anomalies, the ureteral orifice associated with each kidney is usually orthotopic. Except for solitary crossed ectopia, most patients with crossed ectopia have a normal trigone (Magri, 1961; Tanenbaum et al, 1970; Yates-Bell and Packham, 1972). The incidence of an ectopic ureteral orifice from the crossed renal unit is about 3% (Abeshouse and Bhisitkul, 1959; Magri, 1961; Hendren et al, 1976). Occasionally, the ureter from the uncrossed renal segment of a fusion anomaly has an ectopic orifice (Hendren et al, 1976). Malek and Utz (1970) discovered a case of an ectopic ureterocele associated with the uncrossed kidney. Vesicoureteral reflux is noted in 20% of crossed ectopia and in 71% of bilateral crossed ectopia (Kelalis et al, 1973; Guarino et al, 2004) (see Fig. 117–14). Currarino and Weisbruch (1989) reported 10 cases of midline renal fusion in which a single ureter divided into two pelves that stretched across the midline to drain one respective half of the total parenchymatous mass. In 4 of the 10 cases, a second ureter was present that drained a separate duplex system on either the right or left side. Most of the affected individuals had an imperforate anus or abnormal vertebrae, or both.
Most orthotopic renal units are normal. If an abnormality exists, it usually involves the ectopic kidney and consists of cystic dysplasia, UPJ obstruction (29%), reflux (15%), or carcinoma (Abeshouse and Bhisitkul, 1959; Gerber et al, 1980; Caldamone and Rabinowitz, 1981; Macksood and James, 1983; Nussbaum et al, 1987; Gleason et al, 1994). Only four tumors have been reported, all of which were renal cell carcinomas (Stimac et al, 2004).
The highest incidence of associated anomalies occurs in children with solitary renal ectopia and involves both the skeletal system and genital organs (Miles et al, 1985; Gleason et al, 1994). This may be related more to renal agenesis than to the ectopic anomaly, per se. Fifty percent and 40% of solitary crossed renal ectopia have a skeletal or genital abnormality, respectively (Gu and Alton, 1991). The most common genital abnormality in the male is cryptorchidism or absence of the vas deferens; in the female, it is vaginal atresia or a unilateral uterine abnormality (Yates-Bell and Packham, 1972; Kakei et al, 1976). An imperforate anus has also been observed in 20% with solitary crossed ectopia.
In general, the occurrence of an associated anomaly in crossed renal ectopia, excluding solitary crossed ectopia, is low; the most frequent conditions are imperforate anus (4%), orthopedic anomalies (4%), skeletal abnormalities, and septal cardiovascular defects.
Symptoms
Most individuals with crossed ectopic anomalies have no symptoms. The defects are often discovered incidentally at autopsy, during routine perinatal ultrasound screening, or after bone scanning. When symptoms occur, they usually develop in the third or fourth decades of life and include vague lower abdominal pain, pyuria, hematuria, and urinary tract infection (UTI) (Gleason et al, 1994). It is believed that the abnormal kidney position and the anomalous blood supply may impede drainage from the collecting system, creating a predisposition to UTI and calculus formation (Collura et al, 2004). Romans and colleagues (1976) observed that when a stone in a crossed ureter causes colic, the pain is lateralized to the anephric side or the side of the embryonic origin of the ureter, while when there is pyelonephritis or obstruction at the UPJ in the crossed kidney, the lumbar pain is on the side of the kidneys. These observations suggest ureteral, not renal, migration as a causative factor in crossed ectopia.
An asymptomatic abdominal mass is the presenting sign in one third of cases (Abeshouse and Bhisitkul, 1959; Nussbaum et al, 1987). Hypertension occasionally leads to the finding of an ectopic fusion anomaly (Abeshouse and Bhisitkul, 1959), and in one case, hypertension was attributable to a vascular lesion in one of the anomalous vessels (Mininberg et al, 1971).
Diagnosis
In the past, the usual method of detection was by excretory urography, but currently, ultrasonography and radionuclide scintigraphy using 99mTc-DMSA have revealed more asymptomatic cases (Volkan et al, 2003).
Multidetector three-dimensional (3D) CT urography is excellent for delineating the renal parenchyma, collecting system, ureters, and vascular supply of the fused kidneys. The main limitation of this modality is the increased risk of significant radiation exposure, which is especially problematic for pediatric patients, pregnant women, and individuals requiring repeat examinations (Türkvatan et al, 2009). Magnetic resonance urography (MRU) and magnetic resonance arteriography (MRA) should be performed in children undergoing extensive surgery on an ectopic kidney. Cystoscopy and retrograde pyelography can be useful in mapping the collecting system and pattern of drainage.
Prognosis
Most individuals with crossed renal ectopia have normal longevity. However, those with an obstructive-appearing collecting system are at risk for development of UTI or renal calculi, or both (Kron and Meranze, 1949). Boatman and colleagues (1972a) noted that one third of their symptomatic patients required a pyelolithotomy for an obstructing stone. More recently, extracorporeal shock wave lithotripsy therapy and percutaneous nephrolithotomy have rendered most patients stone free (Semerci et al, 1997; Desai and Jasani, 2000). A rare case of renal cell carcinoma was recently reported in a solitary crossed ectopic kidney (Grotas and Phillips, 2009).
Horseshoe Kidney
The horseshoe kidney is the most common of all renal fusion anomalies. It should not be confused with asymmetrical or off-center fused kidneys, which may give the impression of being horseshoe shaped. The anomaly consists of two distinct renal masses lying vertically on either side of the midline and connected at their respective lower poles by a parenchymatous or fibrous isthmus that crosses the midplane of the body. It was first recognized during an autopsy by Da Carpi in 1522, but Botallo, in 1564, presented the first extensive description and illustration of a horseshoe kidney (Benjamin and Schullian, 1950).
Incidence
Horseshoe kidney occurs in 0.25% of the population, or about 1 in 400 persons (Dees, 1941; Nation, 1945; Bell, 1946b; Glenn, 1959; Campbell, 1970). A recent review of more than 15,000 radiologic imaging studies revealed an incidence of 1 in 666 individuals (Weizer et al, 2003). Horseshoe kidney is found more commonly in males with a ratio slightly greater than 2 : 1 (Basar et al, 1999; Weizer et al, 2003). The abnormality has been discovered in all age groups, ranging from fetal life to 80 years. In autopsy series, it is more prevalent in children (Segura et al, 1972). This early age prevalence is related to the high incidence of multiple congenital anomalies associated with the horseshoe kidney, some of which are incompatible with long-term survival (Scott, 2002).
Horseshoe kidneys have been reported in identical twins (Bridge, 1960) and among several siblings (David, 1974). It is doubtful that this anomaly represents a particular genetic predisposition, but it may be the result of a genetic expression with a low degree of penetrance (Leiter, 1972).
Embryology
The abnormality occurs between the fourth and sixth week of gestation, after the UB has entered the renal blastema. In view of the ultimate spatial configuration of the horseshoe kidney, the entrance of the UB had to have taken place before rotation and considerably before renal ascent (Fig. 117–16). Boyden (1931) described a 6-week-old embryo with a horseshoe kidney, the youngest fetus ever discovered with this anomaly. He postulated that at the 14-mm stage (4.5 weeks), the developing metanephric masses lie close to one another; any disturbance in this relationship might result in the masses joining at their inferior poles. A slight alteration in the position of the umbilical or common iliac artery could change the orientation of the migrating kidneys, leading to contact and fusion. It has been postulated that an abnormality in the formation of the tail of the embryo or another pelvic organ accounts for the fusion process (Cook and Stephens, 1977). Domenech-Mateu and Gonzales-Compta (1988), after studying a 16-mm human embryo, suggested that posterior nephrogenic cells migrate abnormally to form an isthmus, or connection, between the two developing kidneys to create the horseshoe shape.
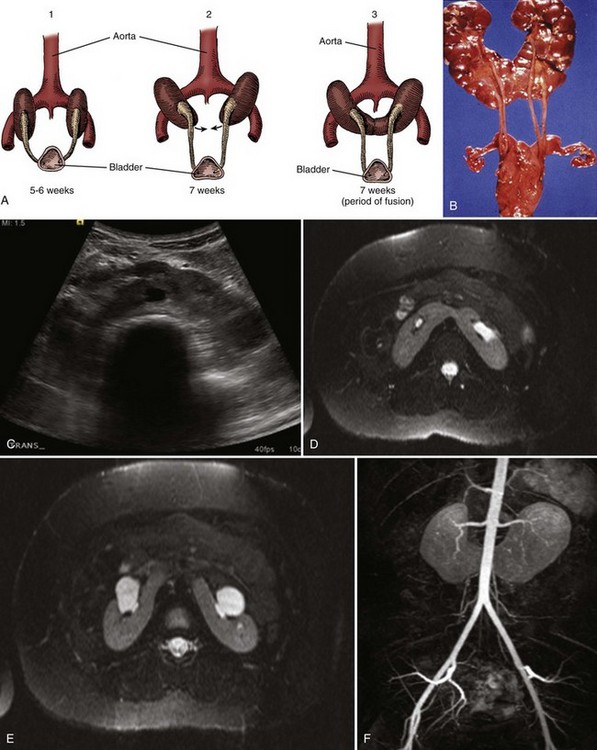
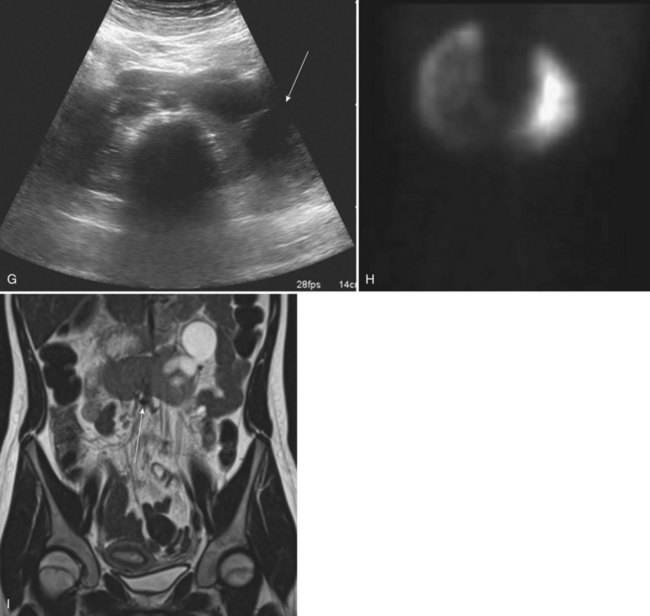
Figure 117–16 A, Embryogenesis of horseshoe kidney. The lower poles of the two kidneys touch and fuse as they cross the iliac arteries. Ascent is stopped when the fused kidneys reach the junction of the aorta and inferior mesenteric artery. B, Postmortem specimen showing horseshoe kidney with bilateral duplicated ureters. C, Ultrasonogram of horseshoe kidney at the level of the isthmus. D, MR urogram shows axial T2 fat-saturated image at the level of the isthmus. E, Axial T2 fat-saturated image demonstrates extrarenal pelves. F, Angiographic sequence shows variable blood supply to the kidney. G, Transverse ultrasonogram of 14-year-old girl with left flank pain found to have marked left hydronephrosis in a horseshoe kidney (arrow). H, MAG3 scan demonstrates left ureteropelvic junction obstruction. I, Coronal T2 images of MR urogram show the isthmus (arrow) and severe left hydronephrosis.
(A, From Benjamin JA, Schullian DM. Observation on fused kidneys with horseshoe configuration: the contribution of Leonardo Botallo [1564]. J Hist Med Allied Sci 1950;5:315, after Gutierrez, 1931; B, from Weiss, MA, Mills, SE. Atlas of genitourinary tract disorders. Philadelphia: JB Lippincott; 1988.)
Recently, Tripathi and colleagues (2010) investigated the role of axial structures, including the notochord and floor plate of the neural tube in metanephric kidney development. The notochord, which is replaced by the vertebral column in higher vertebrates, is essential for the formation of the floor plate. In addition, the sonic hedgehog (Shh) gene exists in these structures and is thought to affect renal development. Using a murine model, these investigators disrupted the notochord and neural plate, which resulted in kidney fusions. Then they only inactivated Shh in the notochord and floor plate, which also resulted in kidney fusion. These findings suggest that the notochord is not necessary for nephrogenesis but is required for correct positioning of the metanephric kidney, while the axial SHH signal is critical for kidney positioning along the mediolateral axis. These studies provide insights into the molecular basis for horseshoe kidney formation and provide an explanation for the increased incidence of horseshoe kidneys in children with vertebral and neural tube defects (see Associated Anomalies).
Description
There are several variations in the basic shape of the horseshoe kidney. In 95% of cases, the kidneys join at the lower pole, which occurs before the kidneys have rotated on their long axes. The pelves and ureters of the horseshoe kidney are usually anteriorly placed, crossing ventrally to the isthmus (see Fig. 117–16). Very rarely, the pelves are anteromedial, suggesting that fusion occurred after some rotation occurred. In a small subset, an isthmus connects both upper poles (Love and Wasserman, 1975). In addition, migration is usually incomplete, with the kidneys lying lower in the abdomen than normal. It is presumed that the inferior mesenteric artery prevents full ascent by obstructing the movement of the isthmus.
Generally, the isthmus is bulky and consists of parenchymatous tissue with its own blood supply (Glenn, 1959; Love and Wasserman, 1975). Occasionally, it is just a flimsy midline structure composed of fibrous tissue that tends to draw the renal masses close together. The isthmus is located adjacent to the L3 or L4 vertebra just below the origin of the inferior mesenteric artery from the aorta, resulting in the paired kidneys lying lower than normal in the retroperitoneum. The isthmus most often lies anterior to the aorta and vena cava, but it has been reported to pass between the inferior vena cava and the aorta or even behind both great vessels (Jarmin, 1938; Meek and Wadsworth, 1940; Dajani, 1966). In some instances, the anomalous kidneys are very low, anterior to the sacral promontory or even in the true pelvis behind the bladder (Campbell, 1970). A similar finding of fused pelvic kidneys is observed in the Foxd1 mouse mutant. These pelvic kidneys also show defects in UB branching and nephron formation (Levinson et al, 2005).
The calyces are normal in number and are atypical in orientation. Because the kidney fails to rotate, the calyces point posteriorly, and the axis of each pelvis remains in the vertical or obliquely lateral plane (on a line drawn from lower to upper poles). The lowermost calyces extend caudally or even medially to drain the isthmus and may overlie the vertebral column (Strauss et al, 2000).
The ureter may insert high on the renal pelvis and lie laterally, probably as the result of incomplete renal rotation. It courses downward and has a characteristic bend as it crosses over and anterior to the isthmus, a deviation that is proportionate to the thickness of the midline structure (Strauss et al, 2000). Despite upper ureteral angulation, the lower ureter usually enters the bladder normally and rarely is ectopic.
The blood supply to the horseshoe kidney can be quite variable. In 30% of cases, it consists of one renal artery to each kidney (Glenn, 1959) (see Fig. 117–16). Duplicate, or even triplicate, renal arteries may supply one or both kidneys. The blood supply to the isthmus and lower poles is also variable. The isthmus and adjacent parenchymal masses may receive a branch from each main renal artery, or they may have their own arterial supply from the aorta originating either above or below the level of the isthmus. Not infrequently, this area is supplied by branches from the inferior mesenteric, common or external iliac or sacral arteries (Boatman et al, 1971; Kolln et al, 1972). Three cases of retrocaval ureter and isthmus have been reported (Eidelman et al, 1978; Hefferman et al, 1978). A horseshoe kidney drained by a single circumcaval ureter has also been reported (Knutson and Hawas, 2004).
Associated Anomalies
The horseshoe kidney is frequently associated with other congenital anomalies. Boatman and colleagues (1972b) reported that almost one third of the 96 cases studied had at least one other abnormality. Many newborns and young infants with multiple congenital anomalies have a horseshoe kidney. The autopsy incidence of other anomalies is greater in children who die at birth or in early infancy than in those who reach adulthood (Zondek and Zondek, 1964; Scott, 2002). This implies that a horseshoe kidney occurs more often in association with other serious congenital anomalies. The organ systems most commonly affected include the skeletal, cardiovascular (primarily ventriculoseptal defects [Voisin et al, 1988]), and central nervous system. Horseshoe kidney is found in 3% of children with neural tube defects (Whitaker and Hunt, 1987). Anorectal malformations are frequently encountered in these patients. Horseshoe kidney is seen in approximately 20% of patients with trisomy 18, in 60% of females with Turner syndrome (Smith, 1970; Lippe et al, 1988), and with Townes-Brock syndrome in those who have a SALL1 transcription factor defect (Salerno et al, 2000).
Key Points: Horseshoe Kidney
Boatman and his colleagues (1972b) also discovered an increased occurrence of other genitourinary anomalies associated with a horseshoe kidney. Hypospadias and undescended testes occurred in 4% of males, and a bicornuate uterus or septate vagina or both were noted in 7% of the females.
Duplication of the ureter occurs in 10% of cases (Zondek and Zondek, 1964; Boatman et al, 1972b); in some cases this has been associated with an ectopic ureterocele (see Fig. 117–16). Vesicoureteral reflux has been noted in more than half of affected individuals (Segura et al, 1972; Pitts and Muecke, 1975; Cascio et al, 2002). UPJ dilation has been seen in over 20%, but on diuretic radionuclide scintigraphy, less than 20% have an obstructive pattern (Cascio et al, 2002; Kao et al, 2003). Cystic disease, including multicystic dysplasia in one half (the upper pole) of one side (Novak et al, 1977; Boullier et al, 1992) and the lower pole of one side (Shapiro, personal communication), and adult polycystic kidney disease, have been reported with horseshoe kidney (Gutierrez, 1934; Campbell, 1970; Pitts and Muecke, 1975; Correa and Paton, 1976). DMSA scanning in 22 patients revealed asymmetrical function in 63% (Kao et al, 2003). Stone formation has been commonly seen in horseshoe kidney. Metabolic stone evaluation in 11 of 37 of these stone formers revealed at least one abnormality in all cases, with an average of 2.68 abnormalities per 24-hour urine collection. Hypovolemia, hypercalciuria, and hypocitraturia were the most common metabolic defects (Raj et al, 2004). Because stone formation in the horseshoe kidney is likely due to an anatomic and metabolic etiology, metabolic evaluation should be performed to reduce the risk of recurrent calculus formation.
Symptoms
With increased ultrasound screening for renal anomalies, it is likely that at least 50% with horseshoe kidneys are asymptomatic rather than the 33% incidence previously reported (Glenn, 1959; Kolln et al, 1972). In most instances, the anomaly is an incidental finding at autopsy (Pitts and Muecke, 1975). Symptoms are typically related to hydronephrosis, infection, or calculus formation. The most common symptom is vague abdominal pain that may radiate to the lower lumbar region. Gastrointestinal complaints may also be present. The Rovsing sign is used to elicit right lower quadrant pain of appendicitis when pushing on the left lower quadrant by stretching the peritoneum. It is also used to describe patients with horseshoe kidney who have abdominal pain, nausea, and vomiting on hyperextension of the spine. UTIs occur in 30% of patients, and calculi have been noted in 20% to 80% (Glenn, 1959; Kolln et al, 1972; Pitts and Muecke, 1975; Evans and Resnick, 1981; Sharma and Bapna, 1986; Benchekroun et al, 1998). Five to 10 percent of horseshoe kidneys are confirmed after palpation of an abdominal mass (Glenn, 1959; Kolln et al, 1972).
Previously, up to one third of individuals with horseshoe kidney had hydronephrosis secondary to UPJ obstruction (Whitehouse, 1975; Das and Amar, 1984) (see Fig. 117–16). The high insertion of the ureter into the renal pelvis, its abnormal course anterior to the isthmus, and the anomalous blood supply to the kidney may individually or collectively contribute to this obstruction. In the modern era, horseshoe kidneys are frequently discovered incidentally, and their apparent hydronephrosis often shows a nonobstructed pattern on radionuclide scanning. Horseshoe kidneys have been detected after angiography for evaluation of an abdominal aortic aneurysm (Huber et al, 1990; deBrito et al, 1991).
Horseshoe kidneys are also incidentally observed on 99mTc bone scan as a result of renal uptake and excretion of the isotope (O’Brien et al, 2008).
Diagnosis and Radiographic Appearance
The horseshoe kidney does not by itself produce symptoms (Grandone et al, 1985). The clinical features from a diseased horseshoe kidney are often vague and nonspecific. In the absence of a palpable mass, the anomaly is not suspected until an imaging study is obtained. Prenatal ultrasonography is detecting most horseshoe kidneys (Sherer and Woods, 1992; Van Every, 1992). The classic radiologic features on a plain film of the abdomen include kidneys that are somewhat low lying and close to the vertebral column and have a vertical or outward axis with the lower poles being more medial than in the normal kidney (O’Brien et al, 2008). The kidneys may be observed by the delineation of the perinephric fat. Ultrasonography detects the isthmus joining the two lower poles of the kidneys in the midline. This may not always be observed, because the isthmus may be only a thin fibrous band. Ultrasound diagnosis is easily made by scanning horizontally along the midline in a craniocaudal direction, especially in children and thin patients. Radionuclide scanning demonstrates the abnormal axis of a horseshoe kidney. A continuous band across the midline is observed if the isthmus contains functioning parenchyma. CT and MRU can also be used to evaluate the horseshoe kidney. Both studies characterize the isthmus. MR angiography will accurately delineate the vascular anatomy for preoperative planning (O’Brien et al, 2008).
Prognosis
Although Smith and Orkin (1945) believed that horseshoe kidneys are almost always associated with disease, Glenn (1959) observed patients with horseshoe kidneys for an average of 10 years after discovery and found that almost 60% of these remained asymptomatic. Only 13% had persistent urinary infection or pain, and 17% developed recurrent calculi. When stone disease is present, extracorporeal shock wave lithotripsy and percutaneous nephrolithotomy yields stone-free rates of 68% and 87.5%, respectively (Kupeli et al, 1999; Raj et al, 2003). In Glenn’s series, no patients benefited from division of the isthmus for relief of pain; as a result, this idea has now been largely repudiated (Glenn, 1959; Pitts and Muecke, 1975).
Many disease processes have been described with a horseshoe kidney, but this likely reflects the relative frequency of the congenital defect. Kidney cancers have been reported in about 150 individuals with horseshoe kidney (Buntley, 1976; Hohenfellner et al, 1992; Schubert et al, 1998, Stimac et al, 2004). Renal cell carcinoma accounts for about half of these cases, although the incidence is no greater than that in the general population. Two cases of bilateral tumors have been reported (Romics et al, 2002). Renal pelvic tumors and Wilms tumor each accounted for about 25% of the total, with sarcoma and carcinoids occurring much less frequently. Overall, 41 of 8617 (0.48%) Wilms tumors in the National Wilms Tumor study occurred in horseshoe kidneys, mostly on the left side, rarely in the isthmus, and practically all with favorable histology (Neville et al, 2002). This incidence of Wilms tumor in horseshoe kidneys is 1.76 to 7.93 times higher than that expected in the general population (Mesrobian et al, 1985). Thirty-seven percent of these Wilms tumors were initially inoperable. However, with preoperative chemotherapy, most patients were salvaged, yielding a 75% preservation rate of renal parenchyma (Neville et al, 2002). Wilms tumor may originate in the isthmus (Beck and Hlivko, 1960), creating a very bizarre radiologic picture (Walker, 1977). Except for renal pelvic tumors, a surprisingly high number of renal cancers arise in the isthmus (Blackard and Mellinger, 1968). For this reason, it has been suggested that teratogenic factors are responsible for abnormal migration of nephrogenic cells to form an isthmus, which then leads to the horseshoe shape and the predisposition for the development of cancer in this portion of the kidney (Domenech-Mateu and Gonzales-Compta, 1988; Hohenfellner et al, 1992).
It has been suggested that the increased occurrence of chronic infection, obstruction, and stone formation explains the higher-than-expected incidence of renal pelvic tumors because the incidence exceeds that of the general population (Shoup et al, 1962; Castor and Green, 1975; Dische and Johnston, 1979). Survival from these tumors is related to the pathology and stage of the tumor at diagnosis and not to the renal anomaly (Murphy and Zincke, 1982). Surveillance for tumors in symptomatic horseshoe kidneys may be prudent.
Because a horseshoe kidney is located above the pelvic inlet, it should not adversely affect pregnancy or delivery (Bell, 1946b). Glomerulocystic disease has been reported in children younger than 1 year of age but does not appear to be related specifically to the horseshoe anomaly (Craver et al, 1993). The development of renal failure associated with adult polycystic kidney disease is not any greater in the presence of a horseshoe kidney (Correa and Paton, 1976). According to the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS, McDonald et al, 2000) and the Department of Health and Human Services 2000 Annual Report (U.S. Department of Health and Human Services, U.S. Scientific Registry of Transplant Recipients and the Organ Procurement and Transplantation Network, 2000) no individual with a horseshoe kidney received a renal transplant. A worldwide review from the Netherlands noted that 23 whole and 57 split horseshoe kidneys have been transplanted with initial failure rates of only 4.3% and 13.4% for en bloc and for split transplants, respectively. An overall 80% graft survival rate has been reported at 5 years (Stroosma et al, 2001).
Anomalies of Rotation
The kidney, as it assumes its final position in the “renal” fossa, orients itself so that the calyces point laterally and the pelvis faces medially. When this alignment is not exact, the condition is known as malrotation and is often described in conjunction with other renal anomalies, such as ectopia with or without fusion or horseshoe kidney. This discussion centers on malrotation as an isolated renal entity.
Incidence
The true incidence of this developmental anomaly cannot be accurately calculated, because minor degrees of malrotation are not reported. Campbell (1963) found renal malrotation in 1 of 939 autopsies, and Smith and Orkin (1945) noted 1 case per 390 hospital admissions. It is frequently observed in association with Turner syndrome (Gray and Skandalakis, 1972b). Males are affected twice as often as females, but there does not appear to be any predilection for one side or the other.
Embryology
It is thought that medial rotation of the collecting system occurs simultaneously with renal migration. The kidney starts to turn during the sixth week, just when it is leaving the true pelvis, and it completes this process by rotating 90 degrees toward the midline by the time ascent is complete at the end of the ninth week of gestation.
In 1912, Felix postulated that rotation is actually the result of unequal branching of successive orders of the budding ureteral tree, with two branches extending ventrally and one dorsally during each generation or division. Each ureteral branch then induces differentiation of the metanephrogenic tissue, surrounding it to encase it as a cap. More parenchyma develops ventrally than dorsally, and the pelvis seems to rotate medially. Weyrauch (1939) accepted this theory of renal rotation as the result of excessive ventral versus dorsal branching of the ureteral tree and concluded that the fault of malrotation lies entirely with the ureter. A late-appearing UB may insert into an atypical portion of the renal blastema, leading to a lessened propensity for the developing nephric tissue to shift. Late appearance of the UB is almost always associated with an aberrant origin from the WD; this translates into ureteral ectopia at the level of the lower urinary tract. Mackie and colleagues (1975), however, did not describe any malrotation anomalies in their study of renal ectopia. The renal blood supply does not appear to be the cause or a limiting factor in malrotation but rather follows the course of renal hyporotation, hyper-rotation, or reverse rotation.
Description
The kidney and renal pelvis normally rotate 90 degrees ventromedially during ascent. Weyrauch (1939), in a detailed study, categorized the various abnormal phases of medial and reverse rotation according to the position of the renal pelvis (Fig. 117–17).
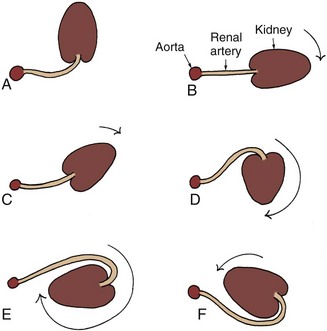
Figure 117–17 Rotation of the kidney during its ascent from the pelvis. The left kidney with its renal artery and the aorta are viewed in transverse section to show normal and abnormal rotation during its ascent to the adult site. A, Primitive embryonic position; hilus faces ventrad (anterior). B, Normal adult position; hilus faces mediad. C, Incomplete rotation. D, Hyper-rotation; hilus faces dorsad (posterior). E, Hyper-rotation; hilus faces laterad. F, Reverse rotation; hilus faces laterad.
(From Gray SW, Skandalakis JE. Embryology for surgeons. Philadelphia: WB Saunders; 1972.)
Ventral Position
The pelvis is ventral and in the same anteroposterior plane as the calyces, which point dorsally because they have undergone no rotation at all. This is the most common form of malrotation. Very rarely, this position may represent excessive medial rotation in which a complete 360-degree turn has occurred. In a case reported by Weyrauch (1939), the vasculature had rotated with the kidney and passed around dorsally and laterally to it before entering the anteriorly placed hilus.
Ventromedial Position
The pelvis faces ventromedially because of an incompletely rotated kidney. Excursion probably stops during the seventh week of gestation when the kidney and pelvis normally reach this position. The calyces thus point dorsolaterally.
Dorsal Position
Renal excursion of 180 degrees occurs to produce this rarest position. The pelvis is dorsal to the parenchyma, and the vessels pass behind the kidney to reach the hilum.
Lateral Position
When the kidney and pelvis rotate between 180 degrees to 360 degrees, or when reverse rotation of up to 180 degrees takes place, the pelvis faces laterally and the kidney parenchyma resides medially. The renal vascular supply provides the only clue to the actual direction of excursion. Vessels that course ventral to the kidney to enter a laterally or dorsolaterally placed hilum suggest reverse rotation, whereas a path dorsal to the kidney implies excessive ventral rotation. Both types of anomalous turning were cited in Weyrauch’s series (1939).
In cases of isolated malrotation, other characteristic features may be present. The kidney shape may be discoid, elongated, oval, or triangular, with flattened anterior and posterior surfaces. Fetal lobulations are invariably present and accentuated beyond normal limits. A dense amount of fibrous tissue encases the hilar area, possibly even distorting and fixing the pelvis. The UPJ may be distorted as well. The upper ureter initially courses laterally, and it, too, may be encased in this fibrous matting. The pelvis is elongated and narrow, and calyces, especially the superior calyx, may be stretched. The blood supply may vary widely, depending on the direction and degree of rotation. The vasculature may consist of a single vessel with or without multiple additional branches entering the parenchyma along the course of the renal artery. Also, there may be a polar vessel in conjunction with the main renal artery. The vascular orientation around the kidney provides the only clue to the type and extent of renal rotation.
Symptoms
Rotation anomalies, per se, do not produce any specific symptoms. The excessive amount of fibrous tissue encasing the pelvis, UPJ, and upper ureter, however, may lead to varying degrees of upper tract obstruction. Vascular compression from an accessory or main renal artery or distortion of the upper ureter or UPJ may contribute to impaired drainage. Symptoms of obstruction may occur during periods of increased urine production. Hematuria associated with hydronephrosis, infection, and calculus formation may also occur secondary to poor urinary drainage.
Diagnosis
The diagnosis should be considered when a renal calculus is detected in an abnormal location, but confirmation should be obtained from a renal ultrasonogram, CT, MRU, or retrograde pyelogram. These studies reveal the abnormal orientation of the renal pelvis and calyces, a flattened and elongated pelvis, a stretched superior calyx with blunting of the remaining calyces, and a laterally displaced upper third of the ureter. Bilateral malrotation is not uncommon and may lead to the diagnosis of a horseshoe kidney. However, careful inspection for an isthmus and observation of the lower pole renal outline should distinguish the two entities.
Anomalies of Renal Vasculature
Aberrant, Accessory, or Multiple Vessels
Knowledge of the anatomy of the renal blood supply is essential for good outcomes following renal surgery. The advent of aortography in the 1940s and 1950s spearheaded a systematic understanding of vascular anomalies (Graves, 1954, 1956; Anson and Kurth, 1955; Merklin and Michele, 1958; Anson and Daseler, 1961; Geyer and Poutasse, 1962).
The kidney is divided into various segments, each supplied by a single “end” arterial branch that usually courses from one main renal artery. Multiple renal arteries is the correct term to describe any kidney supplied by more than one vessel. The term anomalous vessels or aberrant vessels should be reserved for those arteries that originate from vessels other than the aorta or main renal artery. The term accessory vessels denotes two or more arterial branches supplying the same renal segment.
Incidence
Between 71% (Merklin and Michele, 1958) and 85% (Geyer and Poutasse, 1962) of kidneys have one artery that supplies the entire renal parenchyma. A slightly higher percentage of right-sided kidneys have a single renal artery compared with left-sided organs (Geyer and Poutasse, 1962). True aberrant vessels are rare, except in cases with renal ectopia, with or without fusion, and in individuals with a horseshoe kidney (Degani et al, 2010).
Embryology
The renal arterial tree is derived from three groups of primitive vasculature that coalesce to form the mature vascular pattern for all retroperitoneal structures. The cranial group consists of two pairs of arteries dorsal to the suprarenal gland that shift dorsally to form the phrenic artery. The middle group is made up of three pairs of vessels that pass through the suprarenal area and retain the same lateral position, becoming the adrenal artery. Finally, the caudal group of four paired arteries cross ventral to the suprarenal area and become the main renal artery. Sometimes they are joined by the most inferior pair from the middle group (Guggemos, 1962). It is believed that during renal migration this network of vessels selectively degenerates, and the remaining adjacent arteries assume a progressively more important function. By a process of elimination, one primitive renal arterial pair eventually becomes the dominant vessel, the completed process being dependent on the final position of the kidney (Graves, 1956). Polar arteries or multiple renal arteries to the normally positioned kidney represent a failure of complete degeneration of all primitive vascular channels. The multiple vessel pattern that has been described for renal ectopia should be considered as an arrested embryonic state for that particular renal position (Gray and Skandalakis, 1972a).
Description
On the basis of vascular supply, the renal parenchyma is divided into five segments: apical, upper, middle, lower, and posterior. The main renal artery divides initially into an anterior and posterior branch. The anterior branch almost always supplies the upper, middle, and lower segments of the kidney. The posterior branch invariably supplies the posterior and lower segments (Sampaio and Aragao, 1990a). The vessel to the apical segment has the greatest variation in origin; it arises from (1) the anterior division (43%), (2) the junction of the anterior and posterior divisions (23%), (3) the main stem renal artery or aorta (23%), or (4) the posterior division of the main renal artery (10%) (Graves, 1954). Rarely, the upper segment is supplied from a branch totally separate from the main renal artery (Merklin and Michele, 1958). The artrial and venous tree of the kidney and its relationship to the collecting system was beautifully depicted in endocasts by Sampaio and Aragao (1990a, 1990b). These investigations showed that the least likely areas to encounter vessels when entering the collecting system, either endourologically or with open surgery, is directly end-on through a fornix or inferiorly on the posterior aspect of the pelvis. Shoja and colleauges (2008) studied the perihilar (extraparenchymal) branching patterns and morphologies of the renal artery. A “fork” pattern with a common branching point (usually duplicated) was the most commonly observed extrarenal division and branching pattern of the main renal artery. They conclude that perihilar branching of the main renal artery was highly variable, with predictable patterns in the majority of kidneys. This information is very useful for the transplant surgeon when interpreting radiodiagnostics of the renal hilum.
The lower renal segment, however, is often supplied by an accessory vessel (Fig. 117–18). This vessel is usually the most proximal branch when it arises from the main renal artery or its anterior division (Graves, 1954). However, it may originate directly from the aorta near the main renal artery, or it may be aberrant, arising from the gonadal vessel. Merklin and Michele (1958) performed an extensive study of the variant renal and suprarenal blood supply. The venous drainage of the kidney has been studied by Sampaio and Aragao (1990b), who noted a close association between the inferior branch to the main renal vein and the anterior inferior aspect of the renal pelvis in 40% of kidneys. Although an endourologic incision for UPJ obstruction is now infrequently used, it should be performed laterally and posteriorly instead of anteriorly to avoid injury to this vessel.
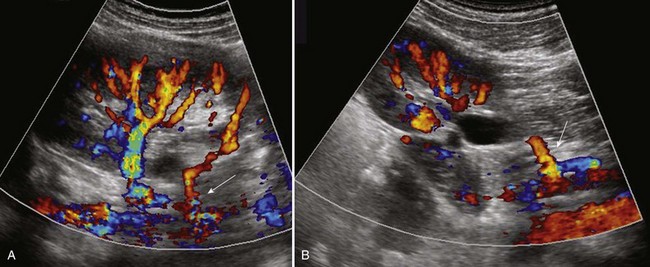
Figure 117–18 Nine-year-old girl with intermittent left flank pain, nausea, and vomiting. A, Color Doppler ultrasound study shows normal left renal artery and lower pole accessory vessel. B, Oblique sagittal view showing left lower pole accessory vessel supplying the lower pole of the kidney, causing intermittent left ureteropelvic junction obstruction.
Symptoms
Symptoms attributable to renal vascular anomalies are those that might result from inadequate urinary drainage. Multiple, aberrant, or accessory vessels may constrict an infundibulum, a major calyx, or the UPJ (Yen et al, 2004). Pain and hematuria secondary to hydronephrosis, UTI, or calculus may result.
Diagnosis
Historically, excretory urography would reveal multiple renal vessels or an aberrant artery (1) when a filling defect in the renal pelvis is consistent with an anomalous vascular pattern; (2) when hydronephrosis is noted along with a sharp cutoff in the superior infundibulum (Fraley, 1966, 1969); (3) when UPJ obstruction is seen in association with an angulated ureter near the renal pelvis and a kidney whose pole-to-pole axis is more vertical than normal; or (4) when differences are noted in the timing and concentration of one renal segment or in the entire kidney when compared with the opposite side (especially when hypertension is present). Renal angiography has been the gold standard for defining the vascular tree of the kidney, but less invasive imaging studies, including 3D power Doppler ultrasonography, CT, and MRI, provide precise anatomic resolution of vascular variants and associated disease states (Textor and Canzanello, 1996; Salcarga et al, 1999; Park et al, 2003; Degani et al, 2010).
Prognosis
None of these variations in the vascular tree, with the exception of a lower pole crossing vessel, increases the kidney’s susceptibility to disease. Hydronephrosis secondary to a vascular anomaly is a very rare finding, especially when one considers the relative frequency of all renal vascular variations. Hypertension is no more frequent in patients with multiple renal arteries than in those with a single vessel (Geyer and Poutasse, 1962).
Renal Artery Aneurysm
Aneurysmal dilation was the first disease process of the renal artery to be recognized (Poutasse, 1957); it was considered a rare occurrence until selective renal angiography became widely available. Since then, the overall incidence has been calculated to be between 0.1% and 0.3%. Abeshouse (1951) classified renal artery aneurysms as follows: saccular, fusiform, dissecting, and arteriovenous. The saccular aneurysm, a localized outpouching that communicates with the arterial lumen by a narrow or wide opening, is the most common type, accounting for 93% of all aneurysms (McKeil et al, 1966; Stanley et al, 1975; Hageman et al, 1978; Zinman and Libertino, 1982). Renal artery aneurysms have been associated with autosomal-dominant polycystic kidney disease (Schievink, 1998). When the aneurysm is located at the bifurcation of the main renal artery and its anterior and posterior divisions or at one of the more distal branchings, it is considered to be congenital in origin and is called the fusiform type (Poutasse, 1957). The presence of similar aneurysms at branching points in the vasculature of other organ systems attests to this possible origin (Lorentz et al, 1984). Acquired aneurysms may be located anywhere and may result from inflammatory, traumatic, or degenerative factors. A localized defect in the internal elastic tissue and the media allows the vessel to dilate at that point. It is a true aneurysm, because its walls are composed of most of the layers that make up the normal artery (Poutasse, 1957). The outpouchings may vary in size from 1 to 2 cm up to 10 cm (Garritano, 1957), but 90% are smaller than 2 cm. There is no absolute predilection for side, but the right appears to be favored, and bilateral aneurysms are seen in 15% (Pfeiffer et al, 2003).
Almost half of renal artery aneurysms are silent, especially in children (Sarker et al, 1991). Some produce symptoms at a later age, because the size of the aneurysm increases with time. Pain (15%), hematuria (microscopic and macroscopic) (30%), and hypertension (55%) secondary to compression of adjacent parenchyma or to altered blood flow within the vascular tree can occur (Glass and Uson, 1967; Bulbul and Farrow, 1992). The hypertension is renin-mediated and secondary to relative parenchymal ischemia (Lorentz et al, 1984). Thirteen cases of hydronephrosis secondary to an adjacent renal vessel aneurysm was noted (Miyagawa et al, 2001).
The diagnosis is suspected when a pulsatile mass is palpated in the region of the renal hilum or when a bruit is heard on abdominal auscultation. A wreathlike calcification in the area of the renal artery or its branches (30%) is highly suggestive (Silvis et al, 1956), but this finding is often missed on a plain abdominal radiograph (Bulbul and Farrow, 1992). Color Doppler imaging (Bunchman et al, 1991) will demonstrate decreased flow. Selective renal angiography (Cerny et al, 1968), digital subtraction angiography, color Doppler ultrasonography (Okamato et al, 1992), or MR angiography (Takebayashi et al, 1994) confirms the diagnosis.
Many asymptomatic renal artery aneurysms come to light during a workup of hypertension. Fifty percent are diagnosed when a renal arteriogram is performed for other reasons (Zinman and Libertino, 1982). CT also readily detects the lesion (Miyagawa et al, 2001; Nishimura et al, 2002). Generally, excision is recommended if (1) the hypertension cannot be easily controlled; (2) incomplete ringlike calcification is present; (3) the aneurysm is larger than 2.5 cm (Poutasse, 1975; Pfeiffer et al, 2003); (4) the patient is female and of child-bearing age, because rupture during pregnancy is a likely possibility (Cohen and Shamash, 1987); (5) the aneurysm increases in size on serial angiograms; or (6) an arteriovenous fistula is present. More than 30 cases of a ruptured aneurysm during pregnancy have been reported (Lacroix et al, 2001), so any aneurysm larger than 1.0 cm in younger women should be repaired (Henke and Stanley, 2003). The likelihood of spontaneous rupture (about 10%), with its dire consequences, dictates attentive treatment in the foregoing situations. Recent advances in endovascular techniques dictate early prophylactic treatment (Yamamoto et al, 1998).
Renal Arteriovenous Fistula
Although rare, renal arteriovenous fistulas (AVFs) have been diagnosed with increasing frequency since they were first described by Varela in 1928. Two types exist, congenital and acquired (Maldonado et al, 1964), with the latter (secondary to trauma, inflammation, renal surgery, or percutaneous needle biopsy) accounting for the recent increased incidence. Only the congenital variant is discussed here.
Fewer than 25% of all AVFs are congenital, with only 91 reported cases (Takaha et al, 1980). They are identifiable by their cirsoid configuration and multiple communications between the main or segmental renal arteries and venous channels (Crummy et al, 1965; Cho and Stanley, 1978). Although congenital, they rarely present clinically before the third or fourth decade. Women are affected three times as often as men, and the right kidney is involved slightly more often than the left (Cho and Stanley, 1978; Ishikawa et al, 2004). The lesion is usually located in the upper pole (45% of cases), but not infrequently, it may be found in the midportion (30%) or in the lower pole (25%) of the kidney (Yazaki et al, 1976).
The condition is thought to be either present at birth or results from a congenital aneurysm eroding into an adjacent vein (Thomason et al, 1972). The pathophysiology involved in the shunting of blood, which bypasses the renal parenchyma and rapidly joins the venous circulation and returns to the heart, results in a varied clinical picture. The symptoms are based on the age and size of the AVF (Messing et al, 1976).
The hemodynamic derangement often produces a loud bruit in 75% of cases. Diminished perfusion of renal parenchyma distal to the fistulous site leads to relative ischemia and renin-mediated hypertension in approximately 50% (McAlhany et al, 1971). The increased venous return and high cardiac output with concomitant diminution in peripheral resistance may result in left ventricular hypertrophy and subsequent high-output cardiac failure in 50% of cases (Maldonado et al, 1964). Macroscopic and microscopic hematuria occurs in more than 75% of affected individuals due to the proximity of the collecting system (Messing et al, 1976; Cho and Stanley, 1978; Montoya et al, 2004). Although flank or abdominal pain may be present, a mass is rarely palpable (10%).
In the past, excretory urography revealed diminished or absent function either in one segment or in the entire portion of the involved kidney (DeSai and DeSautels, 1973), an irregular filling defect in the renal pelvis or calyces (secondary to either clot or encroachment by the fistula), or calyceal distortion or obstruction distal to the site of the lesion (Gunterberg, 1968). Despite these specific radiographic features, an abnormality had been noted in only 50% of excretory urograms in the past. Three-dimensional Doppler ultrasonography and MR angiography are accurate and noninvasive tests (Mohaupt et al, 1999; Ishikawa et al, 2004), but selective renal arteriography or digital subtraction angiography is the most definitive method for diagnosing the lesion. A cirsoid appearance with multiple small, tortuous channels; prompt venous filling; and an enlarged renal, and possibly gonadal, vein are pathognomonic for arteriovenous malformation (AFM) (DeSai and DeSautels, 1973).
The symptomatic nature of this lesion, which causes progressive alterations in the cardiovascular system, often dictates surgical intervention. The congenital variant rarely behaves like its acquired counterpart, which may disappear spontaneously after several months. Nephrectomy, partial nephrectomy, vascular ligation (Boijsen and Kohler, 1962), selective embolization (Bookstein and Goldstein, 1973), and balloon catheter occlusion (Bentson and Crandalls, 1972) have been used to obliterate the fistula.