CHAPTER 7 Gingival Inflammation
Pathologic changes in gingivitis are associated with the presence of oral microorganisms attached to the tooth and perhaps in or near the gingival sulcus.
These organisms are capable of synthesizing products (e.g., collagenase, hyaluronidase, protease, chondroitin sulfatase, and endotoxin) that cause damage to epithelial and connective tissue cells, as well as to intercellular constituents such as collagen, ground substance, and glycocalyx (cell coat). The resultant widening of the spaces between the junctional epithelial cells during early gingivitis may permit injurious agents derived from bacteria, or bacteria themselves, to gain access to the connective tissue.10,48,52 Microbial products activate monocytes/macrophages to produce vasoactive substances such as prostaglandin E2 (PGE2), interferon (IFN), tumor necrosis factor (TNF), and interleukin-1 (IL-1).25,38 In addition, IL-1β alters properties of gingival fibroblasts by delaying their death via a mechanism-blocking apoptosis. This stabilizes the gingival fibroblast population in inflammation.58
Morphologic and functional changes in the gingiva during plaque accumulation have been thoroughly investigated, especially in beagle dogs and humans.39 A useful framework for organization and consideration of these data has been devised based on histopathologic, radiographic, and ultrastructural features and biochemical measurements.40,42 The sequence of events cumulating in clinically apparent gingivitis is categorized into initial, early, and established stages, with periodontitis designated as the advanced stage41 (Table 7-1). One stage evolves into the next, with no clear-cut dividing lines.
Despite extensive research, we still cannot distinguish definitively between normal gingival tissue and the initial stage of gingivitis.39 Most biopsies of clinically normal human gingiva contain inflammatory cells consisting predominantly of T cells, with very few B cells or plasma cells.39,53,54 These cells do not create tissue damage, but they appear to be important in the day-to-day host response to bacteria and other substances to which the gingiva is exposed.39 Therefore, under normal conditions, a constant stream of neutrophils is migrating from the vessels of the gingival plexus through the junctional epithelium, to the gingival margin, and into the gingival sulcus and oral cavity.46
 Science Transfer
Science Transfer
The initial lesion of gingivitis is focused on acute inflammation with vasodilation, edema, and accumulation of polymorphonuclear leukocytes. There is also ulceration of the epithelium lining the gingival sulcus so that bleeding on probing is present. In some patients, this is seen as early as 2 days after plaque accumulates and can be successfully treated by plaque and calculus removal. The plaque accumulation is then controlled by oral hygiene so that the gingiva can return to health within 7 to 10 days.
When gingivitis is established, the initial acute response is accompanied by chronic inflammation with emphasis on lymphocytic and plasma cell accumulation, capillary proliferation, and collagen destruction. This too can be reversed with initial therapy but will require a longer period for gingival tissues to heal and restore lost collagen fibers.
In some patients, gingivitis proceeds to periodontitis, which is much more difficult to treat. Therefore clinicians must be vigilant to detect the early stages of gingivitis and carry out effective treatment coupled with ongoing monitoring and therapy as part of a recall protocol.
Stage I Gingivitis: The Initial Lesion
The first manifestations of gingival inflammation are vascular changes consisting of dilated capillaries and increased blood flow. These initial inflammatory changes occur in response to microbial activation of resident leukocytes and the subsequent stimulation of endothelial cells. Clinically, this initial response of the gingiva to bacterial plaque (subclinical gingivitis28) is not apparent.
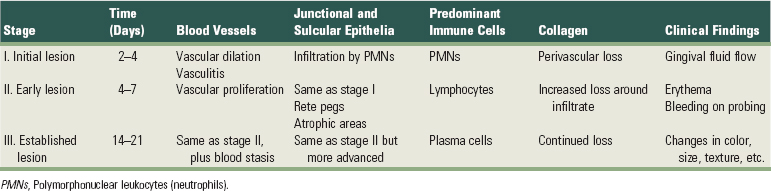
Microscopically, some classic features of acute inflammation can be seen in the connective tissue beneath the junctional epithelium. Changes in blood vessel morphologic features (e.g., widening of small capillaries or venules) and adherence of neutrophils to vessel walls (margination) occur within 1 week and sometimes as early as 2 days after plaque has been allowed to accumulate19,44 (Figure 7-1). Leukocytes, mainly polymorphonuclear neutrophils (PMNs), leave the capillaries by migrating through the walls (diapedesis, emigration)27,54,55 (Figure 7-2). They can be seen in increased quantities in the connective tissue, the junctional epithelium, and the gingival sulcus* (Figures 7-3 and 7-4). Exudation of fluid from the gingival sulcus19 and extravascular proteins are present.21,22
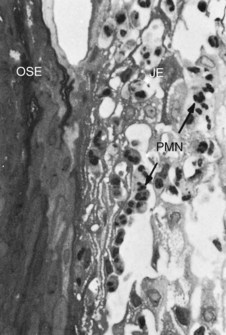
Figure 7-1 Human biopsy sample, experimental gingivitis. After 4 days of plaque accumulation, the blood vessels immediately adjacent to the junctional epithelium are distended and contain polymorphonuclear leukocytes (PMNs, neutrophils). Neutrophils have also migrated between the cells of the junctional epithelium (JE). OSE, Oral sulcular epithelium. (Magnification ×500.)
(From Payne WA, Page RC, Ogilvie AL, et al: J Periodontal Res 10:51, 1975.)
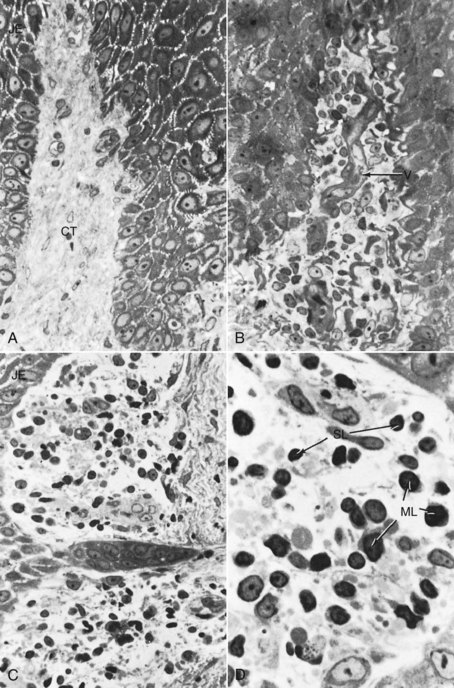
Figure 7-2 Human biopsy, experimental gingivitis. A, Control biopsy specimen from a patient with good oral hygiene and no detectable plaque accumulation. The junctional epithelium is at the left. The connective tissue (CT) shows few cells other than fibroblasts, blood vessels, and a dense background of collagen fibers. (×500.) B, Biopsy specimen taken after 8 days of plaque accumulation. The connective tissue is infiltrated with inflammatory cells, which displace the collagen fibers. A distended blood vessel (V) is seen in the center. (×500.) C, After 8 days of plaque accumulation, the connective tissue next to the junctional epithelium at the base of the sulcus shows a mononuclear cell infiltrate and evidence of collagen degeneration (clear spaces around cellular infiltrate). (×500.). D, The inflammatory cell infiltrate at higher magnification. After 8 days of plaque accumulation, numerous small (SL) and medium-sized (ML) lymphocytes are seen within the connective tissue. Most of the collagen fibers around these cells have disappeared, presumably as a result of enzymatic digestion. (×1250.)
(From Payne WA, Page RC, Ogilvie AL, et al: J Periodontal Res 10:51, 1975.)
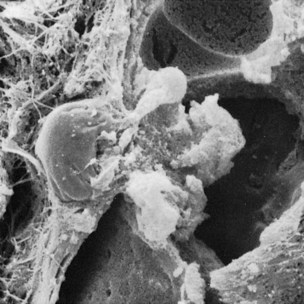
Figure 7-3 Scanning electron micrograph showing a leukocyte traversing the vessel wall to enter into the gingival connective tissue.
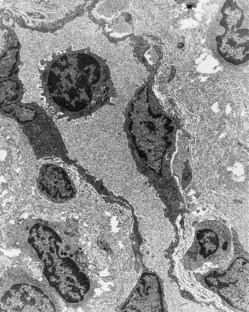
Figure 7-4 Early human gingivitis lesion. Area of lamina propria subjacent to the crevicular epithelium showing a capillary with several extravascular lymphocytes and one lymphocyte within the lumen. The specimen also exhibits considerable loss of perivascular collagen density. (×2500.)
(Courtesy Dr. Charles Cobb, Kansas City, MO.)
However, these findings are not accompanied by manifestations of tissue damage perceptible at the light microscopic or ultrastructural level; they do not form an infiltrate, and their presence is not considered to indicate pathologic change.39
Subtle changes can also be detected in the junctional epithelium and perivascular connective tissue at this early stage. For example, the perivascular connective tissue matrix becomes altered, and there is exudation and deposition of fibrin in the affected area.39 Also, lymphocytes soon begin to accumulate (see Figure 7-2, D). The increase in the migration of leukocytes and their accumulation within the gingival sulcus may be correlated with an increase in the flow of gingival fluid into the sulcus.4
The character and intensity of the host response determine whether this initial lesion resolves rapidly, with the restoration of the tissue to a normal state, or evolves into a chronic inflammatory lesion. If the latter occurs, an infiltrate of macrophages and lymphoid cells appears within a few days.
Stage II Gingivitis: The Early Lesion
The early lesion evolves from the initial lesion within about 1 week after the beginning of plaque accumulation.38,44 Clinically, the early lesion may appear as early gingivitis, and it overlaps with and evolves from the initial lesion with no clear-cut dividing line. As time goes on, clinical signs of erythema may appear, mainly because of the proliferation of capillaries and increased formation of capillary loops between rete pegs or ridges (Figure 7-5). Bleeding on probing may also be evident.1 Gingival fluid flow and the numbers of transmigrating leukocytes reach their maximum between 6 and 12 days after the onset of clinical gingivitis.28
Microscopic examination of the gingiva reveals a leukocyte infiltration in the connective tissue beneath the junctional epithelium, consisting mainly of lymphocytes (75%, with the majority T cells)44,51 but also composed of some migrating neutrophils, as well as macrophages, plasma cells, and mast cells. All the changes seen in the initial lesion continue to intensify with the early lesion.16,30,32,38,51 The junctional epithelium becomes densely infiltrated with neutrophils, as does the gingival sulcus, and the junctional epithelium may begin to show development of rete pegs or ridges.
The amount of collagen destruction increases12,30,51; 70% of the collagen is destroyed around the cellular infiltrate. The main fiber groups affected appear to be the circular and dentogingival fiber assemblies. Alterations in blood vessel morphologic features and vascular bed patterns have also been described.19,20
PMNs that have left the blood vessels in response to chemotactic stimuli from plaque components travel to the epithelium, cross the basement lamina, and are found in the epithelium, emerging in the pocket area (see Figure 7-3). PMNs are attracted to bacteria and engulf them in the process of phagocytosis (Figure 7-6). PMNs release their lysosomes in association with the ingestion of bacteria.24 Fibroblasts show cytotoxic alterations,43 with a decreased capacity for collagen.

Figure 7-6 Scanning electron micrograph of leukocyte emerging to pocket wall and covered with bacteria and extracellular lysosomes. EC, Epithelial cells; B, bacteria; L, lysosomes.
Meanwhile, on the opposite side of molecular events, collagen degradation is related to matrix metalloproteins (MMPs). Different MMPs are responsible for extracellular matrix remodeling within 7 days of inflammation, which is directly related to MMP-2 and MMP-9 production and activation.58
Stage III Gingivitis: The Established Lesion
Over time, the established lesion evolves, characterized by a predominance of plasma cells and B lymphocytes and probably in conjunction with the creation of a small gingival pocket lined with a pocket epithelium.50 The B cells found in the established lesion are predominantly of the immunoglobulin G1 (IgG1) and G3 (IgG3) subclasses.39
In chronic gingivitis, which occurs 2 to 3 weeks after the beginning of plaque accumulation, the blood vessels become engorged and congested, venous return is impaired, and the blood flow becomes sluggish (Figure 7-7). The result is localized gingival anoxemia, which superimposes a somewhat bluish hue on the reddened gingiva.18 Extravasation of erythrocytes into the connective tissue and breakdown of hemoglobin into its component pigments can also deepen the color of the chronically inflamed gingiva. The established lesion can be described as moderately to severely inflamed gingiva.
In histologic sections, an intense, chronic inflammatory reaction is observed. Several detailed cytologic studies have been performed on chronically inflamed gingiva.* A key feature that differentiates the established lesion is the increased number of plasma cells, which become the preponderant inflammatory cell type. Plasma cells invade the connective tissue not only immediately below the junctional epithelium but also deep into the connective tissue, around blood vessels, and between bundles of collagen fibers.6 The junctional epithelium reveals widened intercellular spaces filled with granular cellular debris, including lysosomes derived from disrupted neutrophils, lymphocytes, and monocytes (Figure 7-8). The lysosomes contain acid hydrolases that can destroy tissue components. The junctional epithelium develops rete pegs or ridges that protrude into the connective tissue, and the basal lamina is destroyed in some areas. In the connective tissue, collagen fibers are destroyed around the infiltrate of intact and disrupted plasma cells, neutrophils, lymphocytes, monocytes, and mast cells (Figure 7-9).
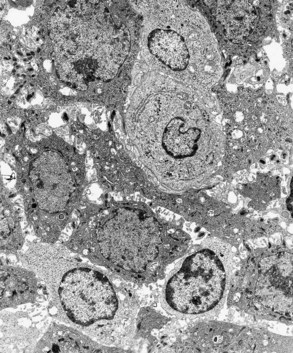
Figure 7-8 Established gingivitis in a human subject. Area of crevicular epithelium exhibiting enlarged intercellular spaces with numerous microvilli and desmosomal junctions. Several lymphocytes, both small and large, are seen migrating through the epithelial layer. (×3000.)
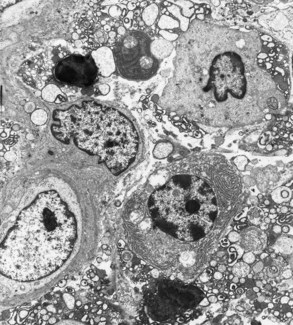
The predominance of plasma cells is thought to be a primary characteristic of the established lesion. However, several studies of human experimental gingivitis have failed to demonstrate plasma cell predominance in the affected connective tissues,7,8,53 including one study of 6 months’ duration.1 Increases in the proportions of plasma cells were evident with long-standing gingivitis, but the time for the development of the classic “established lesion” may exceed 6 months.
An inverse relationship appears to exist between the number of intact collagen bundles and the number of inflammatory cells.55 Collagenolytic activity is increased in inflamed gingival tissue17 by the enzyme collagenase. Collagenase is normally present in gingival tissues5 and is produced by some oral bacteria and by PMNs.
Enzyme histochemistry studies have shown that chronically inflamed gingivae have elevated levels of acid and alkaline phosphatase,60 β-glucuronidase, β-glucosidase, β-galactosidase, esterases,31 aminopeptidase,36,45 and cytochrome oxidase.9 Neutral mucopolysaccharide levels are decreased,57 presumably as a result of degradation of the ground substance.
Established lesions of two types appear to exist; some remain stable and do not progress for months or years,35,36,56 and others seem to become more active and to convert to progressively destructive lesions. Also, the established lesions appear to be reversible in that the sequence of events occurring in the tissues as a result of successful periodontal therapy seems to be essentially the reverse of the sequence of events observed as gingivitis develops. As the flora reverts from that characteristically associated with destructive lesions to that associated with periodontal health, the percentage of plasma cells decreases greatly, and the lymphocyte population increases proportionately.29,33
Stage IV Gingivitis: The Advanced Lesion
Extension of the lesion into alveolar bone characterizes a fourth stage known as the advanced lesion43 or phase of periodontal breakdown.28 This is described in detail in Chapters 13 and 14.
Microscopically, there is fibrosis of the gingiva and widespread manifestations of inflammatory and immunopathologic tissue damage.39 At the advanced stage, plasma cell presence dominates connective tissue and neutrophils continue dominating the junctional epithelium.
Patients with experimental gingivitis showed significantly more plaque accumulation, higher IL-1β, and lower IL-8 concentrations at 28 days.11
Gingivitis will progress to periodontitis only in individuals who are susceptible. Patients who had sites with consistent bleeding (gingival index [GI] = 2) had 70% more attachment loss than at sites that were not inflamed consistently (GI = 0). Teeth with noninflamed sites consistently had a 50-year survival rate of 99.5%, whereas teeth with consistently inflamed gingiva had a 63.4% survival rate over 50 years. Based on this longitudinal study on the natural history of periodontitis in a well-maintained male population, persistent gingivitis represents a risk factor for periodontal attachment loss and for tooth loss.23
However, whether periodontitis can occur without a precursor of gingivitis is not known at this time.
1 Amato R, Caton J, Polson A, et al. Interproximal gingival inflammation related to the conversion of a bleeding to a nonbleeding state. J Periodontol. 1986;57:63.
2 Attström R. Studies on neutrophil polymorphonuclear leukocytes at the dento-gingival junction in gingival health and disease. J Periodontal Res. 1971;8(suppl):1.
3 Attström R. The roles of gingival epithelium and phagocytosing leukocytes in gingival defense. J Clin Periodontol. 1975;2:25.
4 Attström R, Egelberg J. Emigration of blood neutrophils and monocytes into the gingival crevices. J Periodontal Res. 1970;5:48.
5 Beutner EH, Triftshauser C, Hazen SP. Collagenase activity of gingival tissue from patients with periodontal disease. Proc Soc Exp Biol Med. 1966;121:1082.
6 Brecx MC. Histophysiology and histopathology of the gingiva. J West Soc Periodontol. 1991;39:33.
7 Brecx MC, Frohlicher I, Gehr P, et al. Stereological observations on long-term experimental gingivitis in man. J Clin Periodontol. 1988;15:621.
8 Brecx MC, Lehmann B, Siegwart CM, et al. Observations on the initial stages of healing following human experimental gingivitis: a clinical and morphometric study. J Clin Periodontol. 1988;15:123.
9 Burstone MS. Histochemical study of cytochrome oxidase in normal and inflamed gingiva. Oral Surg Oral Med Oral Pathol. 1960;13:1501.
10 Caffesse RG, Nasjleti CE. Enzymatic penetration through intact sulcular epithelium. J Periodontol. 1976;47:391.
11 Deinzer R, Weik U, Kolb-Bachofen V, Herforth A. Comparison of experimental gingivitis with persistent gingivitis: differences in clinical parameters and cytokine concentrations. J Periodontal Res. 2007 Aug;4;2(4):318-324.
12 Flieder DE, Sun CN, Schneider BC. Chemistry of normal and inflamed human gingival tissues. Periodontics. 1966;4:302.
13 Freedman HL, Listgarten MA, Taichman NS. Electron microscopic features of chronically inflamed human gingiva. J Periodontal Res. 1968;3:313.
14 Fullmer HM, Gibson W. Collagenolytic activity in gingivae of man. Nature. 1966;209:728.
15 Garant PR, Mulvihill JE. The fine structure of gingivitis in the beagle. III. Plasma cell infiltration of the subepithelial connective tissue. J Periodontal Res. 1971;7:161.
16 Gavin JB. Ultrastructural features of chronic marginal gingivitis. J Periodontal Res. 1970;5:19.
17 Hancock EB, Cray RJ, O’Leary TJ. The relationship between gingival crevicular fluid and gingival inflammation: a clinical and histologic study. J Periodontol. 1979;50:13.
18 Hanioka T, Shizukuishi S, Tsunemitsu A. Changes in hemoglobin concentration and oxygen saturation in human gingiva with decreasing inflammation. J Periodontol. 1991;62:366.
19 Hock J, Nuki K. A vital microscopy study of the morphology of normal and inflamed gingiva. J Periodontal Res. 1971;6:81.
20 Kindlova M. Changes in the vascular bed of the marginal periodontium in periodontitis. J Dent Res. 1965;44:456.
21 Lamster IB, Hartley LJ, Vogel RI. Development of a biochemical profile for gingival crevicular fluid: methodological considerations and evaluation of collagen-degrading and ground substance–degrading enzyme activity during experimental gingivitis. J Periodontol. 1985;56:13.
22 Lamster IB, Vogel RI, Hartley LJ, et al. Lactate dehydrogenase, beta-glucuronidase and arylsulfatase activity in gingival crevicular fluid associated with experimental gingivitis in man. J Periodontol. 1985;56:139.
23 Lang NP, Schätzle MA, Löe H. Gingivitis as a risk factor in periodontal disease. J Clin Periodontol. 2009 Jul;36(Suppl 10):3-8.
24 Lange D, Schroeder HE. Cytochemistry and ultrastructure of gingival sulcus cells. Helv Odontol Acta. 1971;15:65.
25 Lindemann RA, Economou JS. Actinobacillus actinomycetemcomitans and Bacteroides gingivalis activate human peripheral monocytes to produce interleukin-1 and tumor necrosis factor. J Periodontol. 1988;59:728.
26 Levy BM, Taylor AC, Bernick S. Relationship between epithelium and connective tissue in gingival inflammation. J Dent Res. 1969;48:625.
27 Lindhe J, Socransky SS. Chemotaxis and vascular permeability produced by human periodontopathic bacteria. J Periodontal Res. 1979;14:138.
28 Lindhe J, Hamp SE, Löe H. Experimental periodontitis in the beagle dog. Int Dent J. 1973;23:432.
29 Lindhe J, Parodi R, Liljenberg B, et al. Clinical and structural alterations characterizing healing gingiva. J Periodontal Res. 1978;13:410.
30 Lindhe J, Schroeder HE, Page RC, et al. Clinical and stereologic analysis of the course of early gingivitis in dogs. J Periodontal Res. 1974;9:314.
31 Lisanti VF. Hydrolytic enzymes in periodontal tissues. Ann NY Acad Sci. 1960;85:461.
32 Listgarten MA, Ellegaard B. Experimental gingivitis in the monkeys: relationship of leukocyte counts in junctional epithelium, sulcus depth, and connective tissue inflammation scores. J Periodontal Res. 1973;8:199.
33 Listgarten MA, Hellden L. Relative distribution of bacteria at clinically healthy and periodontal diseased sites in humans. J Clin Periodontol. 1978;5:115.
34 Lorencini M, Silva JA, de la Hoz CLCarvalho HF, Stach-Machado DR. Changes in MMPs and inflammatory cells in experimental gingivitis. Histol Histopathol. 2009 Feb;24(2):157-166.
35 Lovdal A, Arno A, Waerhaug J. Incidence of clinical manifestations of periodontal disease in light of oral hygiene and calculus formation. J Am Dent Assoc. 1958;56:21.
36 Mori M, Kishiro A. Histochemical observation of aminopeptidase activity in the normal and inflamed oral epithelium. J Osaka Univ Dent School. 1961;1:39.
37 Oliver RC, Holm-Pederen P, Löe H. The correlation between clinical scoring, exudate measurements and microscopic evaluation of inflammation in the gingiva. J Periodontol. 1969;40:201.
38 Page RC. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodontal Res. 1991;26:230.
39 Page RC. Gingivitis. J Clin Periodontol. 1986;13:345.
40 Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease: a summary of current work. Lab Invest. 1976;34:235.
41 Page RC, Schroeder HE. Pathogenic mechanisms. In: Schluger S, Youdelis R, Page RC, editors. Periodontal disease: basic phenomena, clinical management and restorative interrelationships. Philadelphia: Lea & Febiger, 1977.
42 Page RC, Schroeder HE. Periodontitis in man and other animals: a comparative review. Basel: S Karger; 1982.
43 Page RC, Simpson DM, Ammons WF. Host tissue response in chronic inflammatory periodontal disease. IV. The periodontal and dental status of a group of aged great apes. J Periodontol. 1975;46:144.
44 Payne WA, Page RC, Ogilvie AL, et al. Histopathologic features of the initial and early stages of experimental gingivitis in man. J Periodontal Res. 1975;10:51.
45 Quintarelli G. Histochemistry of the gingiva. III. The distribution of amino-peptidase in normal and inflammatory conditions. Arch Oral Biol. 1960;2:271.
46 Ryder MI. Histological ultrastructural characteristics of the periodontal syndrome in the rice rat. I. General light microscopic observations and ultrastructural observations of initial inflammatory changes. J Periodontal Res. 1980;15:502.
47 Saglie R, Newman MG, Carranza FA Jr. Scanning electron microscopy study of leukocytes and their interaction with bacteria in human periodontitis. J Periodontol. 1982;53:752.
48 Saglie R, Newman MG, Carranza FAJr, et al. Bacterial invasion of gingiva in advanced periodontitis in humans. J Periodontol. 1982;53:217.
49 Schroeder HE. Transmigration and infiltration of leucocytes in human junctional epithelium. Helv Odontol Acta. 1973;17:6.
50 Schroeder HE, Graf-de Beer M, Attström R. Initial gingivitis in dogs. J Periodontal Res. 1975;10:128.
51 Schroeder HE, Munzel-Pedrazzoli S, Page RC. Correlated morphometric and biochemical analysis of gingival tissue in early chronic gingivitis in man. Arch Oral Biol. 1973;18:899.
52 Schwartz J, Stinson FL, Parker RB. The passage of tritiated bacterial endotoxin across intact gingival crevicular epithelium. J Periodontol. 1972;43:270.
53 Seymour GJ, Powell RN, Aitken JF. Experimental gingivitis in humans: a clinical and histological investigation. J Periodontol. 1983;54:522.
54 Seymour GJ, Powell RN, Cole KL, et al. Experimental gingivitis in humans: a histochemical and immunological characterization of the lymphoid cell subpopulations. J Periodontal Res. 1983;18:375.
55 Simpson DM, Avery BE. Histopathologic and ultrastructural features of inflamed gingiva in the baboon. J Periodontol. 1974;45:500.
56 Suomi JD, Smith LW, McClendon BJ. Marginal gingivitis during a sixteen-week period. J Periodontol. 1971;42:268.
57 Thilander H. Epithelial changes in gingivitis: an electron microscopic study. J Periodontal Res. 1968;3:303.
58 Vardar-Sengul S, Arora S, Baylas H, Mercola D. Expression profile of human gingival fibroblasts induced by interleukin-1beta reveals central role of nuclear factor-kappa B in stabilizing human gingival fibroblasts during inflammation. J Periodontol. 2009 May;80(5):833-849.
59 Wennstrom J, Heijl L, Lindhe J, et al. Migration of gingival leukocytes mediated by plaque bacteria. J Periodontal Res. 1980;15:363.
60 Winer RA, O’Donnell LJ, Chauncey HH, et al. Enzyme activity in periodontal disease. J Periodontol. 1970;41:449.