CHAPTER 14 Bone Loss and Patterns of Bone Destruction
Although periodontitis is an infectious disease of the gingival tissue, changes that occur in bone are crucial because the destruction of bone is responsible for tooth loss.
The height and density of the alveolar bone are normally maintained by an equilibrium, regulated by local and systemic influences, between bone formation and bone resorption.12,14 When resorption exceeds formation, both bone height and bone density may be reduced.
The level of bone is the consequence of past pathologic experiences, whereas changes in the soft tissue of the pocket wall reflect the present inflammatory condition. Therefore the degree of bone loss is not necessarily correlated with the depth of periodontal pockets, severity of ulceration of the pocket wall, or presence or absence of pus.
Bone Destruction Caused by Extension of Gingival Inflammation
The most common cause of bone destruction in periodontal disease is the extension of inflammation from the marginal gingiva into the supporting periodontal tissues. The inflammatory invasion of the bone surface and the initial bone loss that follows mark the transition from gingivitis to periodontitis.
Periodontitis is always preceded by gingivitis, but not all gingivitis progress to periodontitis. Some cases of gingivitis apparently never become periodontitis, and other cases go through a brief gingivitis phase and rapidly develop into periodontitis. The factors that are responsible for the extension of inflammation to the supporting structures, thus initiating the conversion of gingivitis to periodontitis, are not clearly understood at this time.
The transition from gingivitis to periodontitis is associated with changes in the composition of bacterial plaque. In advanced stages of disease, the number of motile organisms and spirochetes increases, whereas the number of coccoid rods and straight rods decreases.34 The cellular composition of the infiltrated connective tissue also changes with increasing severity of the lesion (see Chapter 7). Fibroblasts and lymphocytes predominate in stage I gingivitis, whereas the number of plasma cells and blast cells increases gradually as the disease progresses. Seymour et al58,59 have postulated a stage of “contained” gingivitis in which T lymphocytes are preponderant; they believe that as the lesion becomes a B-lymphocyte lesion, it becomes progressively destructive.
Heijl and coworkers22 were able to convert, in experimental animals, a confined, naturally occurring chronic gingivitis into a progressive periodontitis by placing a silk ligature into the sulcus and tying it around the neck of the tooth. This induced ulceration of the sulcular epithelium, a shift in the inflammatory infiltrate from predominantly plasma cells to predominantly polymorphonuclear leukocytes, and osteoclastic resorption of the alveolar crest. The recurrence of episodes of acute destruction over time may be one mechanism leading to progressive bone loss in marginal periodontitis.
The extension of inflammation to the supporting structures of a tooth may be modified by the pathogenic potential of plaque and the resistance of the host. The latter includes immunologic activity and other tissue-related mechanisms, such as the degree of fibrosis of the gingiva, probably the width of the attached gingiva, and the reactive fibrogenesis and osteogenesis, that occur peripheral to the inflammatory lesion.52
Histopathology
Gingival inflammation extends along the collagen fiber bundles and follows the course of the blood vessels through the loosely arranged tissues around them into the alveolar bone67 (Figure 14-1). Although the inflammatory infiltrate is concentrated in the marginal periodontium, the reaction is a much more diffuse one, often reaching the bone and eliciting a response before evidence of crestal resorption or loss of attachment exists.41 In the upper molar region, inflammation can extend to the maxillary sinus, resulting in thickening of the sinus mucosa.40
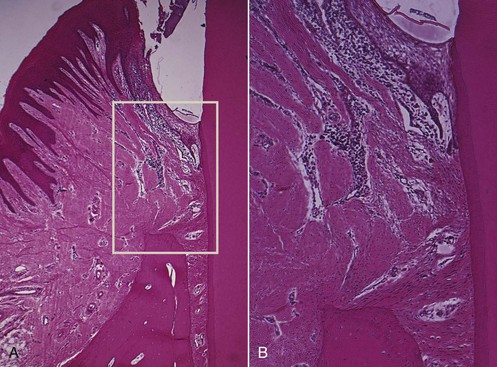
Figure 14-1 A, Area of inflammation extending from the gingiva into the suprabony area. B, Extension of inflammation along blood vessels and between collagen bundles.
Interproximally, inflammation spreads to the loose connective tissue around the blood vessels, through the fibers, and then into the bone through vessel channels that perforate the crest of the interdental septum at the center of the crest (Figure 14-2), toward the side of the crest (Figure 14-3), or at the angle of the septum. Also, inflammation may enter the bone through more than one channel. Less frequently, the inflammation spreads from the gingiva directly into the periodontal ligament and from there into the interdental septum1 (Figure 14-4).

Figure 14-2 Inflammation extending from the pocket area (top) between the collagen fibers, which are partially destroyed.
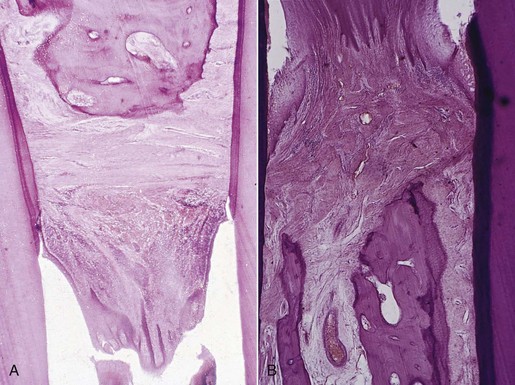
Figure 14-3 A, Extension of inflammation into the center of the interdental septum. Inflammation from the gingiva penetrates the transseptal fibers and enters the bone around blood vessel in the center of the septum. B, Cortical layer at the top of the septum has been destroyed, and the inflammation penetrates into the marrow spaces.
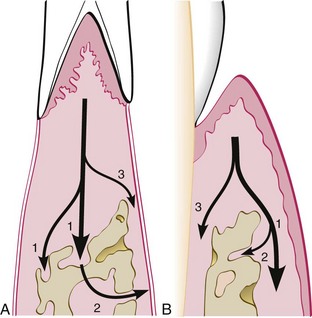
Figure 14-4 Pathways of inflammation from the gingiva into the supporting periodontal tissues in periodontitis. A, Interproximally, from the gingiva into the bone (1), from the bone into the periodontal ligament (2), and from the gingiva into the periodontal ligament (3). B, Facially and lingually, from the gingiva along the outer periosteum (1), from the periosteum into the bone (2), and from the gingiva into the periodontal ligament (3).
Facially and lingually, inflammation from the gingiva spreads along the outer periosteal surface of the bone (see Figure 14-4) and penetrates into the marrow spaces through vessel channels in the outer cortex.
Along its course from the gingiva to the bone, the inflammation destroys the gingival and transseptal fibers, reducing them to disorganized granular fragments interspersed among the inflammatory cells and edema.45 However, there is a continuous tendency to recreate transseptal fibers across the crest of the interdental septum farther along the root as the bone destruction progresses (Figure 14-5). As a result, transseptal fibers are present, even in cases of extreme periodontal bone loss.
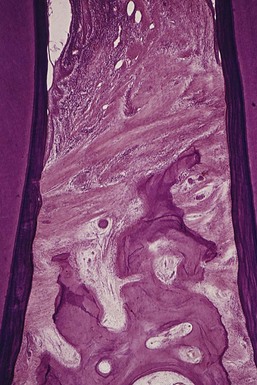
The dense transseptal fibers form a firm covering over the bone, which is encountered during periodontal flap surgery after the superficial granulation tissue is removed.50
After inflammation reaches the bone by extension from the gingiva (Figure 14-6), it spreads into the marrow spaces and replaces the marrow with a leukocytic and fluid exudate, new blood vessels, and proliferating fibroblasts (Figure 14-7). Multinuclear osteoclasts and mononuclear phagocytes increase in number, and the bone surfaces appear lined with Howship lacunae (Figure 14-8).
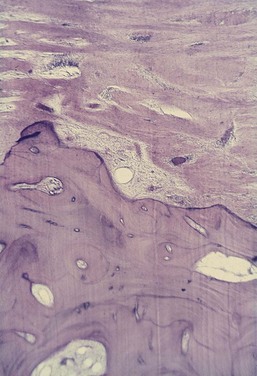

Figure 14-7 Interdental septum in human autopsy section. Extensive inflammatory infiltrate has invaded the marrow spaces, entering from both the mesial and the distal aspect. Fatty bone marrow has been replaced by inflammatory cells and fibrous marrow.
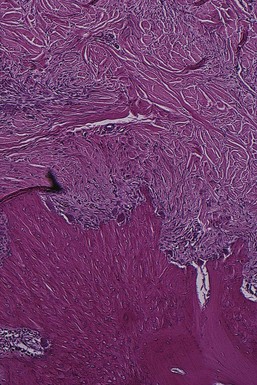
In the marrow spaces, resorption proceeds from within, causing a thinning of the surrounding bony trabeculae and enlargement of the marrow spaces, followed by destruction of the bone and a reduction in bone height. Normally, fatty bone marrow is partially or totally replaced by a fibrous type of marrow in the vicinity of the resorption.
Bone destruction in periodontal disease is not a process of bone necrosis.27 It involves the activity of living cells along viable bone. When tissue necrosis and pus are present in periodontal disease, they occur in the soft tissue walls of periodontal pockets, not along the resorbing margin of the underlying bone.
The amount of inflammatory infiltrate correlates with the degree of bone loss but not with the number of osteoclasts. However, the distance from the apical border of the inflammatory infiltrate to the alveolar bone crest correlates with both the number of osteoclasts on the alveolar crest and the total number of osteoclasts.51 Similar findings have been reported in experimentally induced periodontitis in animals.32
Radius of Action
Garant and Cho10 suggested that locally produced bone resorption factors may need to be present in the proximity of the bone surface to exert their action. Page and Schroeder,49 on the basis of Waerhaug’s measurements made on human autopsy specimens,64,65 postulated a range of effectiveness of about 1.5 to 2.5 mm in which bacterial plaque can induce loss of bone. Beyond 2.5 mm there is no effect; interproximal angular defects can appear only in spaces that are wider than 2.5 mm because narrower spaces would be destroyed entirely. Tal61 corroborated this with measurements in human patients.
Large defects greatly exceeding a distance of 2.5 mm from the tooth surface (as described in aggressive types of periodontitis) may be caused by the presence of bacteria in the tissues.6,10,54
Rate of Bone Loss
In a study of Sri Lankan tea laborers with no oral hygiene and no dental care, Löe et al36 found the rate of bone loss to average about 0.2 mm a year for facial surfaces and about 0.3 mm a year for proximal surfaces when periodontal disease was allowed to progress untreated. However, the rate of bone loss may vary, depending on the type of disease present. Löe et al35 identified the following three subgroups of patients with periodontal disease based on interproximal loss of attachment* and tooth mortality:
Periods of Destruction
Periodontal destruction occurs in an episodic, intermittent manner, with periods of inactivity or quiescence, and destructive periods that result in loss of collagen and alveolar bone and deepening of the periodontal pocket.
Periods of destructive activity are associated with subgingival ulceration and an acute inflammatory reaction, resulting in rapid loss of alveolar bone49,56; it was hypothesized that this coincides with the conversion of a predominantly T-lymphocyte lesion to one with a predominantly B-lymphocyte–plasma cell infiltrate.59 Microbiologically they are associated with an increase of the loose, unattached, motile, gram-negative, anaerobic pocket flora, while periods of remission coincide with the formation of a dense, unattached, nonmotile, gram-positive flora with a tendency to mineralize.43
It has also been suggested that the onset of periods of destruction coincide with tissue invasion by one or several bacterial species and is followed by an advanced local host defense that controls the attack.54
Mechanisms of Bone Destruction
The factors involved in bone destruction in periodontal disease are bacterial and host mediated. Bacterial plaque products induce the differentiation of bone progenitor cells into osteoclasts and stimulate gingival cells to release mediators that have the same effect.21,57 Plaque products and inflammatory mediators can also act directly on osteoblasts or their progenitors, inhibiting their action and reducing their numbers.
In addition, in rapidly progressing diseases such as aggressive periodontitis, bacterial microcolonies or single bacterial cells have been found between collagen fibers and over the bone surface, suggesting a direct effect.6,9,57
Several host factors released by inflammatory cells are capable of inducing bone resorption in vitro and play a role in periodontal disease. These include host-produced prostaglandins and their precursors, interleukin-1α (IL-1α) and IL-β, and tumor necrosis factor alpha (TNF-α).
When injected intradermally, prostaglandin E2 (PGE2) induces the vascular changes seen in inflammation; when injected over a bone surface, PGE2 induces bone resorption in the absence of inflammatory cells and with few multinucleated osteoclasts.18,26 In addition, nonsteroidal antiinflammatory drugs (NSAIDs), such as flurbiprofen and ibuprofen, inhibit PGE2 production, slowing bone loss in naturally occurring periodontal disease in beagle dogs and humans. This effect occurs without changes in gingival inflammation and rebounds 6 months after cessation of drug administration.25,68 (For more information on the host-mediated mechanisms of bone destruction, see Chapter 25.)
Bone Formation in Periodontal Disease
Areas of bone formation are also found immediately adjacent to sites of active bone resorption and along trabecular surfaces at a distance from the inflammation, in an apparent effort to reinforce the remaining bone (buttressing bone formation). This osteogenic response is clearly found in experimentally produced periodontal bone loss in animals.7 In humans, it is less obvious but has been confirmed by histometric4,5 and histologic studies.13
Autopsy specimens from individuals with untreated disease occasionally show areas in which bone resorption has ceased and new bone is being formed on previously eroded bone margins. This confirms the intermittent character of bone resorption in periodontal disease and is consistent with the varied rates of progression observed clinically in untreated periodontal disease.
The periods of remission and exacerbation (or inactivity and activity, respectively) appear to coincide with the quiescence or exacerbation of gingival inflammation, manifested by changes in the extent of bleeding, amount of exudate, and composition of bacterial plaque (see Chapter 23).
The presence of bone formation in response to inflammation, even in active periodontal disease, has an effect on the outcome of treatment. The basic aim of periodontal therapy is the elimination of inflammation to remove the stimulus for bone resorption and therefore allow the inherent constructive tendencies to predominate.
Bone Destruction Caused by Trauma from Occlusion
Another cause of bone destruction in periodontal disease is trauma from occlusion, which can occur in the absence or presence of inflammation (see Chapter 15).
In the absence of inflammation, the changes caused by trauma from occlusion vary from increased compression and tension of the periodontal ligament and increased osteoclasis of alveolar bone to necrosis of the periodontal ligament and bone and the resorption of bone and tooth structure. These changes are reversible in that they can be repaired if the offending forces are removed. However, persistent trauma from occlusion results in funnel-shaped widening of the crestal portion of the periodontal ligament, with resorption of the adjacent bone.33 These changes, which may cause the bony crest to have an angular shape, represent adaptation of the periodontal tissues aimed at “cushioning” increased occlusal forces, but the modified bone shape may weaken tooth support and cause tooth mobility.
When combined with inflammation, trauma from occlusion aggravates the bone destruction caused by the inflammation33 and results in bizarre bone patterns.
Bone Destruction Caused by Systemic Disorders
Local and systemic factors regulate the physiologic equilibrium of bone. When a generalized tendency toward bone resorption exists, bone loss initiated by local inflammatory processes may be magnified.
This systemic influence on the response of alveolar bone, as envisioned by Glickman12 in the early 1950s, considers a systemic regulatory influence in all cases of periodontal disease. In addition to the virulence of plaque bacteria, the nature of the systemic component, not its presence or absence, influences the severity of periodontal destruction. This concept of a role played by systemic defense mechanisms has been validated by the studies on immune deficiencies and host modulation in severely destructive types of periodontitis (see Chapters 25 and 37).
In recent years, interest has increased in the possible relationship between periodontal bone loss and osteoporosis.11 Osteoporosis is a physiologic condition of postmenopausal women, resulting in loss of bone mineral content and structural bone changes. Periodontitis and osteoporosis share a number of risk factors (e.g., aging, smoking, diseases, or medications that interfere with healing). Some studies show a relationship between skeletal density and oral bone density and between crestal height and residual ridge resorption, as well as a relationship between osteopenia and periodontitis, tooth mobility, and tooth loss19,23,24,55,63 (see Chapters 18 and 38).
Periodontal bone loss may also occur in generalized skeletal disturbances (e.g., hyperparathyroidism, leukemia, or histiocytosis X) by mechanisms that may be totally unrelated to the usual periodontal problem.
Factors Determining Bone Morphology in Periodontal Disease
Normal Variation in Alveolar Bone
Considerable normal variation exists in the morphologic features of alveolar bone (see Chapter 2), which affect the osseous contours produced by periodontal disease. The anatomic features that substantially affect the bone-destructive pattern in periodontal disease include the following:
For example, angular osseous defects cannot form in thin facial or lingual alveolar plates, which have little or no cancellous bone between the outer and inner cortical layers. In such cases the entire crest of the plate is destroyed, and the height of the bone is reduced (see Figure 14-9).
Exostoses
Exostoses are outgrowths of bone of varied size and shape. Palatal exostoses have been found in 40% of human skulls.46 They can occur as small nodules, large nodules, sharp ridges, spikelike projections, or any combination of these (Figure 14-10).42 Exostoses have been described in rare cases as developing after the placement of free gingival grafts.46
Trauma from Occlusion
Trauma from occlusion may be a factor in determining the dimension and shape of bone deformities. It may cause a thickening of the cervical margin of alveolar bone or a change in bone morphology (e.g., angular defects, buttressing bone) on which inflammatory changes will later be superimposed.
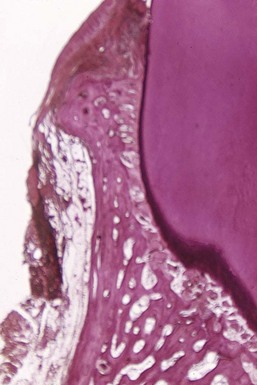
Buttressing Bone Formation
Bone formation sometimes occurs in an attempt to buttress bony trabeculae weakened by resorption. When it occurs within the jaw, it is termed central buttressing bone formation. When it occurs on the external surface, it is referred to as peripheral buttressing bone formation.13 The latter may cause bulging of the bone contour, which sometimes accompanies the production of osseous craters and angular defects (Figure 14-11).
Food Impaction
Interdental bone defects often occur where proximal contact is abnormal or absent. Pressure and irritation from food impaction contribute to the inverted bone architecture. In some cases, the poor proximal relationship may result from a shift in tooth position because of extensive bone destruction preceding food impaction. In such patients, food impaction is a complicating factor rather than the cause of the bone defect.
Aggressive Periodontitis
A vertical or angular pattern of alveolar bone destruction is found around the first molars in aggressive periodontitis. The cause of the localized bone destruction in this type of periodontal disease is unknown (see Chapter 18).
Bone Destruction Patterns in Periodontal Disease
Periodontal disease alters the morphologic features of the bone in addition to reducing bone height. An understanding of the nature and pathogenesis of these alterations is essential for effective diagnosis and treatment.48
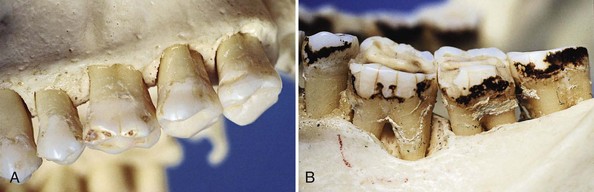
Horizontal Bone Loss
Horizontal bone loss is the most common pattern of bone loss in periodontal disease. The bone is reduced in height, but the bone margin remains approximately perpendicular to the tooth surface. The interdental septa and facial and lingual plates are affected, but not necessarily to an equal degree around the same tooth (Figure 14-12, A).
Bone Deformities (Osseous Defects)
Different types of bone deformities can result from periodontal disease. These usually occur in adults but have also been reported in human skulls with deciduous dentitions.28 Their presence may be suggested on radiographs, but careful probing and surgical exposure of the areas are required to determine their exact conformation and dimensions.
Vertical or Angular Defects
Vertical or angular defects are those that occur in an oblique direction, leaving a hollowed-out trough in the bone alongside the root; the base of the defect is located apical to the surrounding bone (Figures 14-12, B; 14-13; and 14-14). In most instances, angular defects have accompanying infrabony periodontal pockets; infrabony pockets, on the other hand always must have an underlying angular defect.
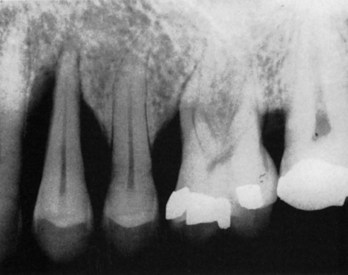
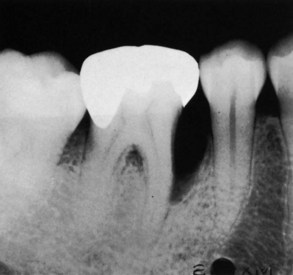
Figure 14-14 Angular defect on mesial surface of the first molar. Note also the furcation involvement.
Angular defects were classified by Goldman and Cohen on the basis of the number of osseous walls.16 Angular defects may have one, two, or three walls (Figures 14-15 to 14-18). The number of walls in the apical portion of the defect is often greater than that in its occlusal portion, in which case the term combined osseous defect is used (Figure 14-19).
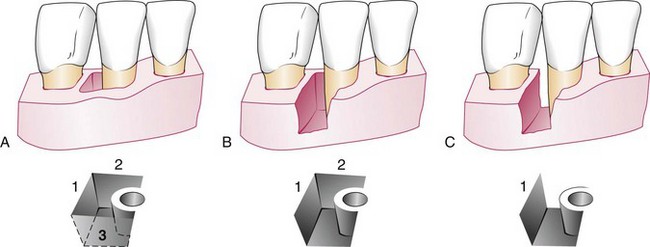
Figure 14-15 One-, two-, and three-walled vertical defects on right lateral incisor. A, Three bony walls: distal (1), lingual (2), and facial (3). B, Two-wall defect: distal (1) and lingual (2). C, One-wall defect: distal wall only (1).
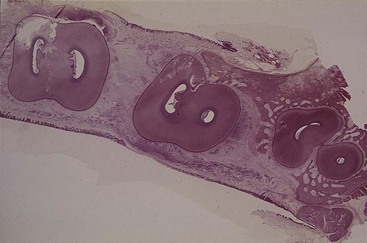
Figure 14-16 Horizontal section of lower molars at midroot level, showing a two-wall osseous defect distal to second molar.
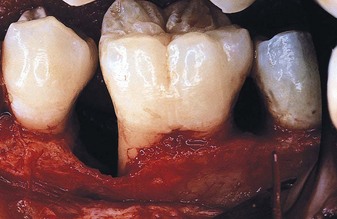
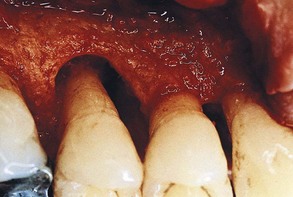
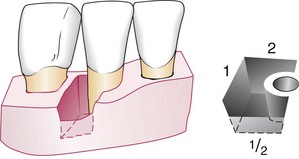
Figure 14-19 Combined type of osseous defect. Because the facial wall is half the height of the distal (1) and lingual (2) walls, this is an osseous defect with three walls in its apical half and two walls in the occlusal half.
Vertical defects occurring interdentally can generally be seen on the radiograph, although thick, bony plates sometimes may obscure them. Angular defects can also appear on facial and lingual or palatal surfaces, but these defects are not seen on radiographs. Surgical exposure is the only sure way to determine the presence and configuration of vertical osseous defects.
Vertical defects increase with age.44,47,69 Approximately 60% of persons with interdental angular defects have only a single defect.44 Vertical defects detected radiographically have been reported to appear most often on the distal44 and mesial surfaces.47 However, three-wall defects are more frequently found on the mesial surfaces of upper and lower molars.29
 Science Transfer
Science Transfer
Bone loss patterns associated with periodontal disease are varied, and the type of loss can be different in various areas in the same patient. Vertical bone loss is amenable to regenerative periodontal surgery using a variety of bone graft materials, bioactive molecules, and membranes. Often, more than one material may need to be used with larger wide defects to obtain good results. Horizontal bone loss and bone craters generally cannot be treated with regeneration, thus these lesions require flap surgery combined with osseous surgery.
Furcation bone loss is more difficult to treat than interproximal bone loss, and in advanced lesions of grade III furcation involvement, the prognosis may be so compromised that extraction and tooth replacement with dental implants should be done as soon as possible to maintain as much bone to support the implants.
Osseous Craters
Osseous craters are concavities in the crest of the interdental bone confined within the facial and lingual walls (Figure 14-20). Craters have been found to make up about one-third (35.2%) of all defects and about two-thirds (62%) of all mandibular defects and to occur twice as often in posterior segments as in anterior segments.37,38
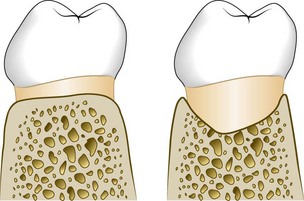
Figure 14-20 Diagrammatic representation of an osseous crater in a faciolingual section between two lower molars. Left, Normal bone contour. Right, Osseous crater.
The heights of the facial and lingual crests of a crater have been found to be identical in 85% of cases, with the remaining 15% being almost equally divided between higher facial crests and higher lingual crests.53
The following reasons for the high frequency of interdental craters have been suggested37,38,53:
Bulbous Bone Contours
Bulbous bone contours are bony enlargements caused by exostoses (see Figure 14-10), adaptation to function, or buttressing bone formation. They are found more frequently in the maxilla than in the mandible.
Reversed Architecture
Reversed architecture defects are produced by loss of interdental bone, including the facial plates and lingual plates, without concomitant loss of radicular bone, thereby reversing the normal architecture (Figure 14-21). Such defects are more common in the maxilla.44
Ledges
Ledges are plateaulike bone margins caused by resorption of thickened bony plates (Figure 14-22).
Furcation Involvement
The term furcation involvement refers to the invasion of the bifurcation and trifurcation of multirooted teeth by periodontal disease. The prevalence of furcation-involved molars is not clear.8,47 Although some reports indicate that the mandibular first molars are the most common sites and the maxillary premolars are the least common,30 other studies have found higher prevalence in upper molars.69 The number of furcation involvements increases with age.30,31
The denuded furcation may be visible clinically or covered by the wall of the pocket. The extent of involvement is determined by exploration with a blunt probe, along with a simultaneous blast of warm air to facilitate visualization (Figure 14-23).
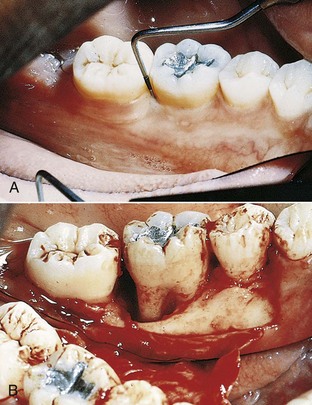
Figure 14-23 A, Molar with slightly inflamed gingiva clinically; however, it has a deep distal pocket. B, Flap elevation reveals extensive bone loss and furcation involvement.
(Courtesy Dr. Terry Fiori, Palo Alto, CA.)
Furcation involvements have been classified as grades I, II, III, and IV according to the amount of tissue destruction. Grade I is incipient bone loss, grade II is partial bone loss (cul-de-sac), and grade III is total bone loss with through-and-through opening of the furcation. Grade IV is similar to grade III but with gingival recession exposing the furcation to view.
Microscopically, furcation involvement presents no unique pathologic features. It is simply a phase in the rootward extension of the periodontal pocket. In its early stages, a widening of the periodontal space occurs, with cellular and fluid inflammatory exudation, followed by epithelial proliferation into the furcation area from an adjoining periodontal pocket. Extension of the inflammation into the bone leads to resorption and reduction in bone height. The bone-destructive pattern may produce horizontal loss, or angular osseous defects associated with intrabony pockets may exist (Figure 14-24). Plaque, calculus, and bacterial debris occupy the denuded furcation space.

The destructive pattern in a furcation involvement varies in different cases and with the degree of involvement. Bone loss around each individual root may be horizontal or angular, and frequently a crater develops in the interradicular area (Figure 14-25). Probing to determine the presence of these destructive patterns must be done horizontally and vertically around each involved root and in the crater area to establish the depth of the vertical component.
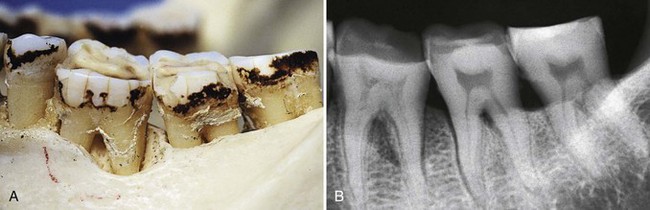
Figure 14-25 Photograph (A) and radiograph (B) of different degrees of bone loss in a skull. Furcation involvements in the first and second molar; deep angular bone loss in the distal root of the first molar; and interradicular and interdental craters in the second molar and between the second and third molars, respectively.
Furcation involvement is a stage of progressive periodontal disease and has its same etiology. The difficulty and sometimes the impossibility2,3 of controlling plaque in furcations are responsible for the presence of extensive lesions in this area.66
The role of trauma from occlusion in the etiology of furcation lesions is controversial. Some assign a key role to trauma, believing that furcation areas are most sensitive to injury from excessive occlusal forces.15 Others deny the initiating effect of trauma and consider that inflammation and edema caused by plaque in the furcation area tend to extrude the tooth, which then becomes traumatized and sensitive.60,66
Other factors that may play a role are the presence of enamel projections into the furcation,39 which occurs in about 13% of multirooted teeth, and the proximity of the furcation to the cementoenamel junction, which occurs in about 75% of cases of furcation involvement.31
The presence of accessory pulpal canals in the furcation area may extend pulpal inflammation to the furcation.20 This possibility should be carefully explored, particularly when mesial and distal bone retains its normal height. Accessory canals connecting the pulp chamber floor to the furcation have been found in 36% of maxillary first molars, 12% of maxillary second molars, 32% of mandibular first molars, and 24% of mandibular second molars.62
The diagnosis of furcation involvement is made by clinical examination and careful probing with a specially designed probe (see Chapter 30). Radiographic examination of the area is helpful, but lesions can be obscured by angulation of the beam and the radiopacity of neighboring structures (see Chapter 31).
For more detailed clinical considerations in the diagnosis and treatment of furcation involvement, see Chapter 63.
1 Akiyoshi M, Mori K. Marginal periodontitis: a histological study of the incipient stage. J Periodontol. 1967;38:45.
2 Bower RC. Furcation morphology relative to periodontal treatment: furcation entrance architecture. J Periodontol. 1979;50:23.
3 Bower RC. Furcation morphology relative to periodontal treatment: furcation root surface anatomy. J Periodontol. 1979;50:366.
4 Carranza FAJr. Histometric evaluation of periodontal pathology: a review of recent studies. J Periodontol. 1967;38:741.
5 Carranza FAJr, Cabrini RL. Histometric studies of periodontal tissues. Periodontics. 1967;5:308.
6 Carranza FAJr, Saglie R, Newman MG. Scanning and transmission electron microscopy study of tissue invading microorganisms in juvenile periodontitis. J Periodontol. 1983;54:598.
7 Carranza FAJr, Simes RJ, Mayo J, Cabrini RL. Histometric evaluation of periodontal bone loss in rats. J Periodontal Res. 1971;6:65.
8 Easley JR, Drennan GA. Morphological classification of the furca. J Can Dent Assoc. 1969;35:104.
9 Frank RM, Voegel JC. Bacterial bone resorption in advanced cases of human periodontitis. J Periodontal Res. 1978;13:251.
10 Garant PR, Cho MJ. Histopathogenesis of spontaneous periodontal disease in conventional rats. I. Histometric and histologic study. J Periodontal Res. 1979;14:297.
11 Geurs NC. Osteoporosis and periodontal disease. Periodontology 2000. 2007;44:29.
12 Glickman I. The experimental basis for the “bone factor” concept in periodontal disease. J Periodontol. 1951;20:7.
13 Glickman I, Smulow J. Buttressing bone formation in the periodontium. J Periodontol. 1965;36:365.
14 Glickman I, Wood H. Bone histology in periodontal disease. J Dent Res. 1942;21:35.
15 Glickman I, Stein RS, Smulow JB. The effects of increased functional forces upon the periodontium of splinted and nonsplinted teeth. J Periodontol. 1961;32:290.
16 Goldman HM, Cohen DW. The intrabony pocket: classification and treatment. J Periodontol. 1958;29:272.
17 Goodson JM, Haffajee AD, Socransky SS. The relationship between attachment level loss and alveolar bone loss. J Clin Periodontol. 1984;11:348.
18 Goodson JM, McClatchy K, Revell C. Prostaglandin-induced resorption of the adult calvarium. J Dent Res. 1974;53:670.
19 Groen JJ, Menczel J, Shapiro S. Chronic destructive periodontal disease in patients with presenile osteoporosis. J Periodontol. 1968;39:19.
20 Gutman JL. Prevalence, location and patency of accessory canals in the furcation region of permanent molars. J Periodontol. 1978;49:21.
21 Hausmann E. Potential pathways for bone resorption in human periodontal disease. J Periodontol. 1974;45:338.
22 Heijl L, Rifkin BR, Zander HA. Conversion of chronic gingivitis to periodontitis in squirrel monkeys. J Periodontol. 1976;47:710.
23 Jeffcoat MK. Osteoporosis: a possible modifying factor in oral bone loss. Ann Periodontol. 1998;3:312.
24 Jeffcoat MK, Lewis CE, Reddy MS, et al. Post-menopausal bone loss and its relationship to oral bone loss. Periodontol 2000. 2000;23:94.
25 Jeffcoat MK, Williams RC, Wachter WJ, et al. Flurbiprofen treatment of periodontal disease in beagles. J Periodontal Res. 1986;21:624.
26 Klein DC, Raisz LG. Prostaglandins: stimulation of bone resorption in tissue culture. Endocrinology. 1970;86:1436.
27 Kronfeld R. Condition of alveolar bone underlying periodontal pockets. J Periodontol. 1935;6:22.
28 Larato DC. Periodontal bone defects in the juvenile skull. J Periodontol. 1970;41:473.
29 Larato DC. Intrabony defects in the dry human skull. J Periodontol. 1970;41:496.
30 Larato DC. Furcation involvements: incidence of distribution. J Periodontol. 1970;41:499.
31 Larato DC. Some anatomical factors related to furcation involvements. J Periodontol. 1975;46:608.
32 Lindhe J, Ericsson I. Effect of ligature placement and dental plaque on periodontal tissue breakdown in the dog. J Periodontol. 1978;49:343.
33 Lindhe J, Svanberg G. Influence of trauma from occlusion on progression of experimental periodontitis in beagle dogs. J Clin Periodontol. 1974;1:3.
34 Lindhe J, Liljenberg B, Listgarten MA. Some microbiological and histopathological features of periodontal disease in man. J Periodontol. 1980;51:264.
35 Löe H, Anerud A, Boysen H, et al. Natural history of periodontal disease in man: rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. J Clin Periodontol. 1986;13:431.
36 Löe H, Anerud A, Boysen H, et al. The natural history of periodontal disease in man: the rate of periodontal destruction before forty years of age. J Periodontol. 1978;49:607.
37 Manson JD. Bone morphology and bone loss in periodontal disease. J Clin Periodontol. 1976;3:14.
38 Manson JD, Nicholson K. The distribution of bone defects in chronic periodontitis. J Periodontol. 1974;45:88.
39 Masters DH, Hoskins SW. Projection of cervical enamel into molar furcations. J Periodontol. 1963;35:49.
40 Moskow BS. A histomorphologic study of the effects of periodontal inflammation on the maxillary sinus mucosa. J Periodontol. 1992;63:674.
41 Moskow BS, Polson AM. Histologic studies on the extension of the inflammatory infiltrate in human periodontitis. J Clin Periodontol. 1991;18:534.
42 Nery EB, Corn H, Eisenstein IL. Palatal exostoses in the molar region. J Periodontol. 1977;48:663.
43 Newman MG. The role of Bacteroides melaninogenicus and other anaerobes in periodontal infections. Rev Infect Dis. 1979;1:313.
44 Nielsen JI, Glavind L, Karring T. Interproximal periodontal intrabony defects: prevalence, localization and etiological factors. J Clin Periodontol. 1980;7:187.
45 Ooya K, Yamamoto H. A scanning electron microscopic study of the destruction of human alveolar crest in periodontal disease. J Periodont Res. 1978;13:498.
46 Pack ARC, Gaudie WM, Jennings AM. Bony exostosis as a sequela to free gingival grafting: two case reports. J Periodontol. 1991;62:269.
47 Papapanou PN, Tonetti MS. Diagnosis and epidemiology of periodontal osseous lesions. Periodontol 2000. 2000;22:8.
48 Papapanou PN, Wennström JL, Grondahl K. Periodontal status in relation to age and tooth type: a cross-sectional radiographic study. J Clin Periodontol. 1988;15:469.
49 Page RC, Schroeder HE. Periodontitis in man and other animals: a comparative review. Basel: S Karger; 1982.
50 Prichard JF. Periodontal surgery: practical dental monographs. Chicago: Year Book Medical; 1961.
51 Rowe DJ, Bradley LS. Quantitative analyses of osteoclasts, bone loss and inflammation in human periodontal disease. J Periodontal Res. 1981;16:13.
52 Ruben M, Cooper SJ. Tissue factors modifying the spread of periodontal inflammation: a perspective. Contin Educ Dent. 1981;2:387.
53 Saari JT, Hurt WC, Briggs NL. Periodontal bony defects on the dry skull. J Periodontol. 1968;39:278.
54 Saglie RF, Rezende M, Pertuiset J, et al. Bacterial invasion during disease activity as determined by significant loss of attachment. J Periodontol. 1987;58:336. (abstract)
55 Salvi GE, Lawrence HP, Offenbacher S, et al. Sequence of risk factors on the pathogenesis of periodontitis. Periodontol 2000. 1997;14:173.
56 Schroeder HE, Lindhe J. Conditions and pathological features of rapidly destructive experimental periodontitis in dogs. J Periodontol. 1980;51:6.
57 Schwartz Z, Goultschin J, Dean DD, et al. Mechanisms of alveolar bone destruction in periodontitis. Periodontol 2000. 1997;14:158.
58 Seymour GJ, Dockrell HM, Greenspan JS. Enzyme differentiation of lymphocyte subpopulations in sections of human lymph nodes, tonsils, and periodontal disease. Clin Exp Immunol. 1978;32:169.
59 Seymour GJ, Powell RN, Davies WJR. Conversion of a stable T cell lesion to a progressive B cell lesion in the pathogenesis of chronic inflammatory periodontal disease: a hypothesis. J Clin Periodontol. 1979;6:267.
60 Svärdström G, Wennström JL. Prevalence of furcation involvements in patients referred for periodontal treatment. J Clin Periodontol. 1996;23:1093.
61 Tal H. Relationship between interproximal distance of roots and the prevalence of intrabony pockets. J Periodontol. 1984;55:604.
62 Vertucci FJ, Anthony RL. An SEM investigation of accessory foramina in the furcation and pulp chamber floor of molar teeth. Oral Surg. 1986;62:319.
63 Wactawski-Wende J, Grossi SG, Trevisan M, et al. The role of osteopenia in oral bone loss and periodontal disease. J Periodontol. 1996;67:1076.
64 Waerhaug J. The angular bone defect and its relationship to trauma from occlusion and downgrowth of subgingival plaque. J Clin Periodontol. 1979;6:61.
65 Waerhaug J. The infrabony pocket and its relationship to trauma from occlusion and subgingival plaque. J Periodontol. 1979;50:355.
66 Waerhaug J. The furcation problem: etiology, pathogenesis, diagnosis, therapy and prognosis. J Clin Periodontol. 1980;7:73.
67 Weinmann JP. Progress of gingival inflammation into the supporting structures of the teeth. J Periodontol. 1941;12:71.
68 Williams RC, Jeffcoat MK, Kaplan ML, et al. Flurbiprofen: a potent inhibitor of alveolar bone resorption in beagles. Science. 1985;227:640.
69 Wouters FR, Salonen LE, Helldén LB, et al. Prevalence of interproximal periodontal infrabony defects in an adult population in Sweden: a radiographic study. J Clin Periodontol. 1989;16:144.