CHAPTER 18 Aggressive Periodontitis
Aggressive periodontitis generally affects systemically healthy individuals less than 30 years of age, although patients may be older. Aggressive periodontitis may be universally distinguished from chronic periodontitis by the age of onset, the rapid rate of disease progression, the nature and composition of the associated subgingival microflora, alterations in the host’s immune response, and a familial aggregation of diseased individuals.17 In addition, a strong racial influence is observed in the United States (US); the disease is more prevalent among African Americans.26
Aggressive periodontitis describes three of the diseases formerly classified as “early-onset periodontitis.” They are localized aggressive periodontitis (LAP), which was formerly termed localized juvenile periodontitis (LJP), and generalized aggressive periodontitis (GAP), which encompasses the diseases previously classified as generalized juvenile periodontitis (GJP) and rapidly progressive periodontitis (RPP).
Localized Aggressive Periodontitis
Historical Background
In 1923, Gottlieb13 reported a patient with a fatal case of epidemic influenza and a disease that Gottlieb called “diffuse atrophy of the alveolar bone.” This disease was characterized by a loss of collagen fibers in the periodontal ligament and their replacement by loose connective tissue and extensive bone resorption, resulting in a widened periodontal space. The gingiva apparently was not involved. In 1928, Gottlieb14 attributed this condition to the inhibition of continuous cementum formation, which he considered essential for maintenance of the periodontal fibers. He then termed the disease “deep cementopathia” and hypothesized that this was a “disease of eruption” and that cementum initiated a foreign body response. As a result, it was postulated that the host attempted to exfoliate the tooth, resulting in the observed bone resorption and pocket formation.14
In 1938, Wannenmacher49 described incisor–first molar involvement and called the disease “parodontitis marginalis progressiva.” Several explanations evolved for the etiology and pathogenesis of this type of disease. Many authors considered this to be a degenerative, noninflammatory disease process12,34,45 and therefore gave it the name “periodontosis.” Other investigators denied the existence of a degenerative type of periodontal disease and attributed the changes observed to trauma from occlusion.6,33 Finally, in 1966, the World Workshop in Periodontics concluded that the concept of “periodontosis” as a degenerative entity was unsubstantiated and that the term should be eliminated from periodontal nomenclature.38 The committee did recognize that a clinical entity different from “adult periodontitis” might occur among adolescents and young adults.
 Science Transfer
Science Transfer
Localized aggressive periodontitis (LAP) affects primarily first molars and incisor teeth in adolescents with deep pockets and advanced bone loss. It occurs in less than 1% of adolescents. The exact etiology is unknown, but there may be a connection between the disease and the presence of the anaerobic gram-negative bacteria, Aggregatibacter actinomycetemcomitans, in the subgingival plaque biofilm. The rate of bone loss can be 3 to 4 times faster than that seen in chronic periodontitis, although in many cases the rate of bone loss is dramatically reduced when the patients reach 20 years of age or older.
Generalized aggressive periodontitis (GAP) usually occurs first in young adults and may be present in some populations in up to 8% of the adult population. Smoking may play a role because smokers with GAP have more teeth involved and more advanced pockets than nonsmokers.
The term “juvenile periodontitis” was introduced by Chaput and colleagues in 19678a and by Butler5 in 1969. In 1971, Baer2 defined it as “a disease of the periodontium occurring in an otherwise healthy adolescent which is characterized by a rapid loss of alveolar bone about more than one tooth of the permanent dentition. The amount of destruction manifested is not commensurate with the amount of local irritants.” In 1989 the World Workshop in Clinical Periodontics categorized this disease as LJP, a subset of the broad classification of early-onset periodontitis (EOP).8 Under this classification system, age of onset and distribution of lesions were of primary importance when making a diagnosis of LJP. Most recently, disease with the characteristics of LJP has been renamed localized aggressive periodontitis (LAP).
Clinical Characteristics
LAP usually has an age of onset at about puberty.22 Clinically, it is characterized as having “localized first molar/incisor presentation with interproximal attachment loss on at least two permanent teeth, one of which is a first molar, and involving no more than two teeth other than first molars and incisors”22 (Figure 18-1). The localized distribution of lesions in LAP is characteristic but as yet unexplained. The following possible reasons for the limitation of periodontal destruction to certain teeth have been suggested:
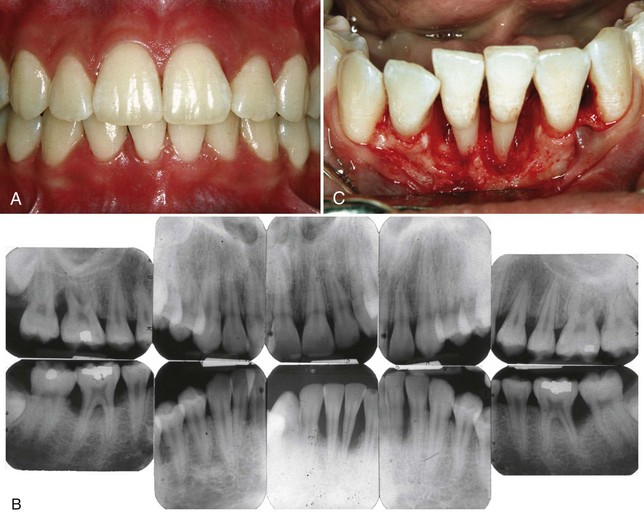
Figure 18-1 Localized aggressive periodontitis in 15-year-old black female patient who had a twin with a similar disease. A, Clinical view showing minimal plaque and inflammation except for localized inflammation on the distal side of the maxillary left central incisor and the mandibular right central incisor. B, Radiographs showing localized, vertical, angular bone loss associated with the maxillary and mandibular first molars and the mandibular central incisors. The maxillary incisors show no apparent involvement. C, Surgical appearance of the localized, vertical, angular bony defects affecting the mandibular incisors. Note the wide circumferential nature of the defects and the lack of calculus on the root surfaces.
A striking feature of LAP is the lack of clinical inflammation despite the presence of deep periodontal pockets and advanced bone loss (see Figure 18-1). Furthermore, in many cases the amount of plaque on the affected teeth is minimal, which seems inconsistent with the amount of periodontal destruction present.22 The plaque that is present forms a thin biofilm on the teeth and rarely mineralizes to form calculus.48 Although the quantity of plaque may be limited, it often contains elevated levels of A. actinomycetemcomitans, and in some patients, Porphyromonas gingivalis. The potential significance of the qualitative composition of the microbial flora in LAP is discussed later in the section on risk factors.
As the name suggests, LAP progresses rapidly. Evidence suggests that the rate of bone loss is about three to four times faster than in chronic periodontitis.2 Other clinical features of LAP may include (1) distolabial migration of the maxillary incisors with concomitant diastema formation, (2) increasing mobility of the maxillary and mandibular incisors and first molars, (3) sensitivity of denuded root surfaces to thermal and tactile stimuli, and (4) deep, dull, radiating pain during mastication, probably caused by irritation of the supporting structures by mobile teeth and impacted food. Periodontal abscesses may form at this stage, and regional lymph node enlargement may occur.30
Not all cases of LAP progress to the degree just described. In some patients the progression of attachment loss and bone loss may be self-arresting.22
Radiographic Findings
Vertical loss of alveolar bone around the first molars and incisors, beginning around puberty in otherwise healthy teenagers, is a classic diagnostic sign of LAP. Radiographic findings may include an “arc-shaped loss of alveolar bone extending from the distal surface of the second premolar to the mesial surface of the second molar”33 (see Figure 18-1, B). Bone defects are usually wider than usually seen with chronic periodontitis (see Figure 18-1, C).
Prevalence and Distribution by Age and Gender
The prevalence of LAP in geographically diverse adolescent populations is estimated at less than 1%. Most reports suggest a low prevalence, about 0.2%.26 Two independent radiographic studies of 16-year-old adolescents, one in Finland41 and the other in Switzerland,21 followed the strict diagnostic criteria delineated by Baer2 and reported a prevalence rate of 0.1%. A clinical and radiographic study of 7266 English adolescents 15 to 19 years old also showed a prevalence rate of 0.1%.40 In the US, a national survey of adolescents ages 14 to 17 reported that 0.53% had LAP.26 Blacks were at much higher risk for LAP, and black male teenagers were 2.9 times more likely to have the disease than black female adolescents. In contrast, white female teenagers were more likely to have LAP than white male adolescents. Several other studies have found the highest prevalence of LAP among black males,4,32,40 followed in descending order by black females, white females, and white males.32
LAP affects both males and females and is seen most frequently in the period between puberty and 20 years of age. Some studies have suggested a predilection for female patients, particularly in the youngest age groups,20 whereas others report no male-female differences in incidence when studies are designed to correct for ascertainment bias.18 (For additional epidemiologic data on LAP, see Chapter 5.)
Generalized Aggressive Periodontitis
Clinical Characteristics
GAP usually affects individuals under age 30, but older patients also may be affected.22 In contrast to LAP, evidence suggests that individuals affected with GAP produce a poor antibody response to the pathogens present. Clinically, GAP is characterized by “generalized interproximal attachment loss affecting at least three permanent teeth other than first molars and incisors.”22 The destruction appears to occur episodically, with periods of advanced destruction followed by stages of quiescence of variable length (weeks to months or years). Radiographs often show bone loss that has progressed since the radiographic examination.
As seen in LAP, patients with GAP often have small amounts of bacterial plaque associated with the affected teeth.22 Quantitatively, the amount of plaque seems inconsistent with the amount of periodontal destruction. Qualitatively, P. gingivalis, A. actinomycetemcomitans, and Tannerella forsythia (formerly Bacteroides forsythus) frequently are detected in the plaque that is present.46
Two gingival tissue responses can be found in cases of GAP. One is a severe, acutely inflamed tissue, often proliferating, ulcerated, and fiery red. Bleeding may occur spontaneously or with slight stimulation. Suppuration may be an important feature. This tissue response is believed to occur in the destructive stage, in which attachment and bone are actively lost.
In other cases the gingival tissues may appear pink, free of inflammation, and occasionally with some degree of stippling, although stippling may be absent (Figure 18-2, A). However, despite the apparently mild clinical appearance, deep pockets can be demonstrated by probing. Page and Schroeder36 believe that this tissue response coincides with periods of quiescence in which the bone level remains stationary.
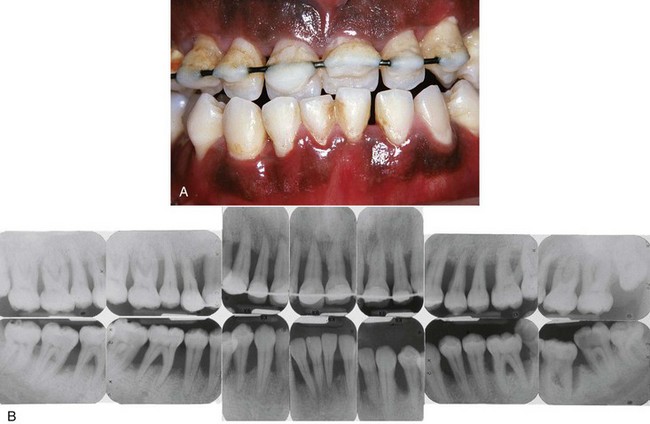
Figure 18-2 Severe generalized aggressive periodontitis in 22-year-old black male patient with a family history of early tooth loss through periodontal disease. A, Clinical view showing minimal plaque and inflammation. A provisional wire-and-resin splint had been placed by the general-practice dentist to stabilize the teeth. B, Radiographs showing the severe, generalized nature of the disease with all erupted teeth affected.
Some patients with GAP may have systemic manifestations, such as weight loss, mental depression, and general malaise.37 Patients with a presumptive diagnosis of GAP must have their medical histories updated and reviewed. These patients should receive medical evaluations to rule out possible systemic involvement. As seen with LAP, cases of GAP may be arrested spontaneously or after therapy, whereas others may continue to progress inexorably to tooth loss despite intervention with conventional treatment.
Radiographic Findings
The radiographic picture in GAP can range from severe bone loss associated with the minimal number of teeth, as described previously, to advanced bone loss affecting the majority of teeth in the dentition (see Figure 18-2, B). A comparison of radiographs taken at different times illustrates the aggressive nature of this disease. Page et al37 described sites in patients with GAP that demonstrated osseous destruction of 25% to 60% during a 9-week period. Despite this extreme loss, other sites in the same patients showed no bone loss.
Prevalence and Distribution by Age and Gender
In a study of untreated periodontal disease conducted in Sri Lanka by Löe et al,27 8% of the population had rapid progression of periodontal disease, characterized by a yearly loss of attachment of 0.1 to 1.0 mm. A US national survey of adolescents ages 14 to 17 reported that 0.13% had GAP.26 In addition, blacks were at much higher risk than whites for all forms of aggressive periodontitis, and male teenagers were more likely to have GAP than female adolescents (see Chapter 5).
Risk Factors for Aggressive Periodontitis
Microbiologic Factors
Although several specific microorganisms frequently are detected in patients with LAP (A. actinomycetemcomitans, Capnocytophaga spp., Eikenella corrodens, Prevotella intermedia, and Campylobacter rectus), A. actinomycetemcomitans has been implicated as the primary pathogen associated with LAP. As summarized by Tonetti and Mombelli,46 this link is based on the following evidence:
Not all reports support the association of A. actinomycetemcomitans and LAP. In some studies, A. actinomycetemcomitans either could not be detected in patients with this form of disease or could not be detected at the previously reported frequencies. Another study found elevated levels of P. gingivalis, Prevotella intermedia, Fusobacterium nucleatum, C. rectus, and Treponema denticola in patients with either localized or generalized aggressive disease, but no significant association was found between the presence of aggressive disease and A. actinomycetemcomitans.
In addition, A. actinomycetemcomitans often can be detected in periodontally healthy subjects, suggesting that this microorganism may be part of the normal flora in many individuals.
Electron microscopy studies of LAP have revealed bacterial invasion of connective tissue9,11 that reaches the bone surface.7 The invading flora has been described as morphologically mixed but composed mainly of gram-negative bacteria, including cocci, rods, filaments, and spirochetes.11 Using different methods, including immunocytochemistry, several tissue-invading microorganisms have been identified as A. actinomycetemcomitans, Capnocytophaga sputigena, Mycoplasma species, and spirochetes.39
Immunologic Factors
Some immune defects have been implicated in the pathogenesis of aggressive periodontitis. The human leukocyte antigens (HLAs), which regulate immune responses, have been evaluated as candidate markers for aggressive periodontitis. Although the findings with many HLAs have been inconsistent, HLA A9 and B15 antigens are consistently associated with aggressive periodontitis.
Several investigators10,23,24 have shown that patients with aggressive periodontitis display functional defects of polymorphonuclear leukocytes (PMNs), monocytes, or both. These defects can impair either the chemotactic attraction of PMNs to the site of infection or their ability to phagocytose and kill microorganisms. Current studies have also demonstrated a hyperresponsiveness of monocytes from LAP patients involving their production of prostaglandin E2 (PGE2) in response to lipopolysaccharide (LPS).43 This hyperresponsive phenotype could lead to increased connective tissue or bone loss caused by excessive production of these catabolic factors. Also, poorly functional inherited forms of monocyte FcγRII, the receptor for human immunoglobulin G2 (IgG2) antibodies, have been shown to be disproportionately present in patients with LAP.50 These PMN and monocyte defects may be induced by the bacterial infection or may be genetic in origin. Further studies are needed to characterize the origin of these cellular alterations.
Autoimmunity has a role in GAP, according to Anusaksathien and Dolby,1 who found host antibodies to collagen, deoxyribonucleic acid (DNA), and IgG. Possible immune mechanisms include an increase in the expression of major histocompatibility complex (MHC) class II molecules, HLA DR4,3 altered helper or suppressor T-cell function, polyclonal activation of B cells by microbial plaque, and genetic predisposition.
Genetic Factors
Results from several studies support the concept that all individuals are not equally susceptible to aggressive periodontitis.46 Specifically, several authors have described a familial pattern of alveolar bone loss and have implicated genetic factors in aggressive periodontitis.5,28,29,31 Segregational and linkage analyses of families with a genetic predisposition for LAP suggest that a major gene or set of genes play a role in LAP and are transmitted through an autosomal dominant mode of inheritance in US populations.31 It should be noted that most of the segregational studies were conducted in African-American populations, therefore other modes of inheritance may exist in different populations.
Evidence suggests that some immunologic defects associated with aggressive periodontitis may be inherited. For example, Van Dyke et al47 reported a familial clustering of the neutrophil abnormalities seen in LAP. This clustering suggests that the defect(s) may be inherited.46 Studies also have demonstrated that the antibody response to periodontal pathogens, particularly A. actinomycetemcomitans, is under genetic control and that the ability to mount high titers of specific, protective antibody (primarily IgG2) against A. actinomycetemcomitans may be race dependent.15
In summary, data support the concept that a gene or genes of major effect exists for aggressive periodontitis. Data also support a genetic basis for some of the immunologic defects seen in patients with aggressive periodontitis. However, it is unlikely that all patients affected with aggressive periodontitis have the same genetic defect. As summarized by Tonetti and Mombelli,46 “it seems that specific genes may be different in various populations and/or ethnic groups and therefore true heterogeneity in disease susceptibility may be present. The role of specific genes remains to be elucidated” (see Chapter 24).
Environmental Factors
The amount and duration of smoking are important variables that can influence the extent of destruction seen in young adults.46 Patients with GAP who smoke have more affected teeth and more loss of clinical attachment than nonsmoking patients with GAP.16 However, smoking may not have the same impact on attachment levels in younger patients with LAP.42
1 Anusaksathien O, Dolby AE. Autoimmunity in periodontal disease. J Oral Pathol Med. 1991;20:101.
2 Baer PN. The case for periodontosis as a clinical entity. J Periodontol. 1971;42:516.
3 Bonfil JJ, Dillier FL, Mercier P. A “case control” study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis: identification of types and subtypes using molecular biology. J Clin Periodontol. 1999;26:77.
4 Burmeister JA, Best AM, Palcanis KG, et al. Localized juvenile periodontitis and generalized severe periodontitis: clinical findings. J Clin Periodontol. 1984;11:181.
5 Butler JH. A familial pattern of juvenile periodontitis (periodontosis). J Periodontol. 1969;40:115.
6 Carranza FASr, Carranza FAJr. A suggested classification of common periodontal disease. J Periodontol. 1959;30:140.
7 Carranza FAJr, Saglie R, Newman MG. Scanning and transmission electron microscopy study of tissue invading microorganisms in localized juvenile periodontitis. J Periodontol. 1983;54:598.
8 Caton J. Consensus report: periodontal diagnosis and diagnostic aids. In: Proceedings of the World Workshop in Clinical Periodontics. Chicago: American Academy of Periodontology; 1989.
8a Chaput A, Held A-J, Palfer-Sollier M. Stomatologie. Paris: Flammarion; 1967.
9 Christersson LA, Albini B, Zambon J, et al. Demonstration of Actinobacillus actinomycetemcomitans in gingiva in localized juvenile periodontitis in humans. J Dent Res. 1983;62:255. (abstract)
10 Clark RA, Page RC, Wilde G. Defective neutrophil chemotaxis in juvenile periodontitis. Infect Immun. 1977;18:694.
11 Gillett R, Johnson NW. Bacterial invasion of the periodontium in a case of juvenile periodontitis. J Clin Periodontol. 1982;9:93.
12 Goldman HM. Similar condition to periodontosis in two spider monkeys. Am J Orthod. 1947;33:749.
13 Gottlieb B. Die diffuse Atrophy des Alveolarknochens. Z Stomatol. 1923;21:195.
14 Gottlieb B. The formation of the pocket: diffuse atrophy of alveolar bone. J Am Dent Assoc. 1928;15:462.
15 Gunsolley JC, Tew JG, Gooss CM, et al. Effects of race, smoking and immunoglobulin allotypes on IgG subclass concentrations. J Periodontal Res. 1997;32:381.
16 Haber J, Wattles J, Crowley M, et al. Evidence for cigarette smoking as a major risk factor for periodontitis. J Periodontol. 1993;64:16.
17 Hart TC. Genetic risk factors for early-onset periodontitis. J Periodontol. 1996;67:355.
18 Hart TC, Marazita ML, Schenkein HA, et al. No female preponderance in juvenile periodontitis after correction of ascertainment bias. J Periodontol. 1991;62:745.
19 Hillman JD, Socransky SS. Bacterial interference in the oral ecology of Actinobacillus actinomycetemcomitans and its relationship to human periodontosis. Arch Oral Biol. 1982;27:75.
20 Hormand J, Frandsen A. Juvenile periodontitis: localization of bone loss in relation to age, sex, and teeth. J Clin Periodontol. 1979;6:407.
21 Kronauer E, Borsa G, Lang NP. Prevalence of incipient juvenile periodontitis at age 16 years in Switzerland. J Clin Periodontol. 1986;13:103.
22 Lang N, Bartold PM, Cullinan M, et al. Consensus report: aggressive periodontitis. Ann Periodontol. 1999;4:53.
23 Lavine WS, Maderazo EG, Stolman J, et al. Impaired neutrophil chemotaxis in patients with juvenile and rapidly progressing periodontitis. J Periodontal Res. 1979;14:10.
24 Leino L, Hurttia H. A potential role of an intracellular signaling defect in neutrophil functional abnormalities and promotion of tissue damage in patients with localized juvenile periodontitis. Clin Chem Lab Med. 1999;37:215.
25 Lindskog S, Blomlof L. Cementum hypoplasia in teeth affected by juvenile periodontitis. J Clin Periodontol. 1983;10:443.
26 Löe H, Brown LJ. Early-onset periodontitis in the United States of America. J Periodontol. 1991;62:608.
27 Löe H, Anerud A, Boysen H, et al. Natural history of periodontal disease in man: rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. J Clin Periodontol. 1986;13:431.
28 Long JC, Nance WE, Waring P, et al. Early onset periodontitis: a comparison and evaluation of two modes of inheritance. Genet Epidemiol. 1987;4:13.
29 Lopez NJ. Clinical, laboratory and immunological studies of a family with a high prevalence of generalized prepubertal and juvenile periodontitis. J Periodontol. 1992;63:457.
30 Manson JD, Lehner T. Clinical features of juvenile periodontitis (periodontosis). J Periodontol. 1974;45:636.
31 Marazita ML, Burmeister JA, Gunsolley JC, et al. Evidence for autosomal dominant inheritance and race-specific heterogeneity in early-onset periodontitis. J Periodontol. 1994;65:623.
32 Melvin WL, Sandifer JB, Gray JL. The prevalence and sex ratio of juvenile periodontitis in a young racially mixed population. J Periodontol. 1991;62:330.
33 Miller SC. Precocious advanced alveolar atrophy. J Periodontol. 1948;19:146.
34 Orban B, Weinmann JP. Diffuse atrophy of alveolar bone. J Periodontol. 1942;13:31.
35 Page RC, Baab DA. A new look at the etiology and pathogenesis of early-onset periodontitis: cementopathia revisited. J Periodontol. 1985;56:748.
36 Page RC, Schroeder HE. Periodontitis in man and other animals: a comparative review. Basel: S Karger; 1982.
37 Page RC, Altman LC, Ebersole JL, et al. Rapidly progressive periodontitis: a distinct clinical condition. J Periodontol. 1983;54:197.
38 Ramfjord SP, Ash MM, Kerr DA, editors. World Workshop in Periodontics. Ann Arbor: University of Michigan, 1966.
39 Saglie FR, Carranza FAJr, Newman MG, et al. Identification of tissue invading bacteria in juvenile periodontitis. J Periodontal Res. 1982;17:452.
40 Saxby MS. Juvenile periodontitis: an epidemiologic study in the West Midlands of the United Kingdom. J Clin Periodontol. 1987;14:594.
41 Saxen L. Prevalence of juvenile periodontitis in Finland. J Clin Periodontol. 1980;7:177.
42 Schenkein JA, Gunsolley JC, Koertge TE, et al. Smoking and its effects on early-onset periodontitis. J Am Dent Assoc. 1995;126:1107.
43 Shapira L, Soskolone WA, Van Dyke TE, et al. Prostaglandin E2 secretion, cell maturation, and CD 14 expression by monocyte-derived macrophages from localized juvenile periodontitis patients. J Periodontol. 1996;67:224.
44 Slots J, Zambon JJ, Rosling BC, et al. Actinobacillus actinomycetemcomitans in human periodontal disease: association, serology, leukotoxicity, and treatment. J Periodontal Res. 1982;17:447.
45 Thoma KH, Goldman HM. Wandering and elongation of the teeth and pocket formation in paradontosis. J Am Dent Assoc. 1940;27:335.
46 Tonetti MS, Mombelli A. Early-onset periodontitis. Ann Periodontol. 1999;4:39.
47 Van Dyke TE, Schweinebraten M, Cinaciola LJ, et al. Neutrophil chemotaxis in families with localized juvenile periodontitis. J Periodontal Res. 1985;20:503.
48 Waerhaug J. Subgingival plaque and loss of attachment in periodontosis as well as observed in autopsy material. J Periodontol. 1976;47:636.
49 Wannenmacher E. Ursachen auf dem Gebiet der Paradentopathien. Zbl Gesant Zahn Mund Kieferheilk. 1938;3:81.
50 Wilson ME, Kalmar JR. FcγRIIa (CD32): a potential marker defining susceptibility to localized juvenile periodontitis. J Periodontol. 1996;67:323.
51 Zambon JJ, Christersson LA, Slots J. Actinobacillus actinomycetemcomitans in human periodontal disease: prevalence in patient groups and distribution of biotypes and serotypes within families. J Periodontol. 1983;54:707.