5 Molecular Diagnosis
Like the evidence left at the scene of a crime, the DNA (deoxyribonucleic acid), RNA (ribonucleic acid), or proteins of an infectious agent in a clinical sample can be used to help identify the agent. In many cases, the agent can be detected and identified in this way, even if it cannot be isolated or detected by immunologic means. New techniques and adaptations of older techniques are being developed for the analysis of infectious agents.
The advantages of molecular techniques are their sensitivity, specificity, and safety. From the standpoint of safety, these techniques do not require isolation of the infectious agent and can be performed on chemically fixed (inactivated) samples or extracts. Because of their sensitivity, very dilute samples of microbial DNA can be detected in a tissue, even if the agent is not replicating or producing other evidence of infection. These techniques can distinguish related strains on the basis of differences in their genotype (i.e., mutants). This is especially useful for distinguishing antiviral drug-resistant strains, which may differ by a single nucleotide.
Detection of Microbial Genetic Material
Electrophoretic Analysis of DNA and Restriction Fragment Length Polymorphism
The genome structure and genetic sequence are major distinguishing characteristics of the family, type, and strain of microorganism. Specific strains of microorganisms can be distinguished on the basis of their DNA or RNA or by the DNA fragments produced when the DNA is cleaved by specific restriction endonucleases (restriction enzymes). Restriction enzymes recognize specific DNA sequences that have a palindromic structure; an example follows:
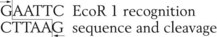
The DNA sites recognized by different restriction endonucleases differ in their sequence, length, and frequency of occurrence. As a result, different restriction endonucleases cleave the DNA of a sample in different places, yielding fragments of different lengths. The cleavage of different DNA samples with one restriction endonuclease can also yield fragments of many different lengths. The differences in the length of the DNA fragments among the different strains of a specific organism produced on cleavage with one or more restriction endonucleases is termed restriction fragment length polymorphism (RFLP).
DNA or RNA fragments of different sizes or structures can be distinguished by their electrophoretic mobility in an agarose or polyacrylamide gel. Different forms of the same DNA sequence and different lengths of DNA move through the mazelike structure of an agarose gel at different speeds, allowing their separation. The DNA can be visualized by staining with ethidium bromide. Smaller fragments (fewer than 20,000 base pairs), such as those from bacterial plasmids or from viruses, can be separated and distinguished by normal electrophoretic methods. Larger fragments, such as those from whole bacteria, can be separated only by using a special electrophoretic technique called pulsed-field gel electrophoresis.
RFLP is useful, for example, for distinguishing different strains of herpes simplex virus (HSV). Comparison of the restriction endonuclease cleavage patterns of DNA from different isolates can identify a pattern of virus transmission from one person to another or distinguish HSV-1 from HSV-2. RFLP has also been used to show the spread of necrotizing fasciitis produced by a strain of Streptococcus from one patient to other patients, an emergency medical technician, and the emergency department and attending physicians (Figure 5-1). Often, comparison of the 16S ribosomal RNA is used to identify different bacteria.
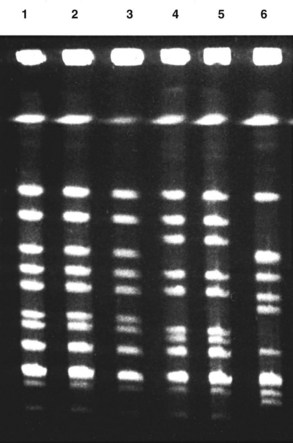
Figure 5-1 Restriction fragment length polymorphism distinction of DNA from bacterial strains separated by pulsed-field gel electrophoresis. Lanes 1 to 3 show Sma 1 restriction endonuclease-digested DNA from bacteria from two family members with necrotizing fasciitis and from their physician (pharyngitis). Lanes 4 to 6 are from unrelated Streptococcus pyogenes strains.
(Courtesy Dr. Joe DiPersio, Akron, Ohio.)
Nucleic Acid Detection, Amplification, and Sequencing
DNA probes can be used like antibodies as sensitive and specific tools to detect, locate, and quantitate specific nucleic acid sequences in clinical specimens (Figure 5-2). Because of the specificity and sensitivity of DNA probe techniques, individual species or strains of an infectious agent can be detected, even if they are not growing or replicating.
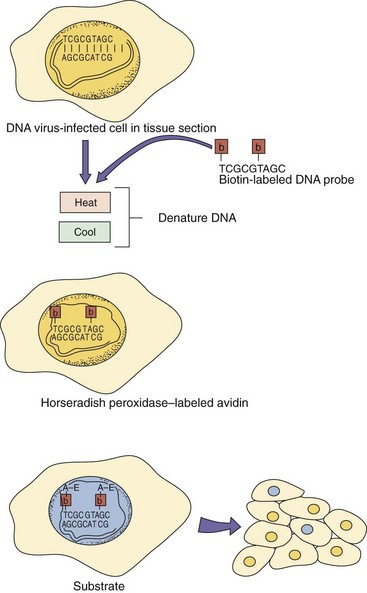
Figure 5-2 DNA probe analysis of virus-infected cells. Such cells can be localized in histologically prepared tissue sections using DNA probes consisting of as few as nine nucleotides or bacterial plasmids containing the viral genome. A tagged DNA probe is added to the sample. In this case, the DNA probe is labeled with biotin-modified thymidine, but radioactive agents can also be used. The sample is heated to denature the DNA and cooled to allow the probe to hybridize to the complementary sequence. Horseradish peroxidase-labeled avidin is added to bind to the biotin on the probe. The appropriate substrate is added to color the nuclei of virally infected cells. A, Adenine; b, biotin; C, cytosine; G, guanine; T, thymine.
DNA probes are chemically synthesized or obtained by cloning specific genomic fragments or an entire viral genome into bacterial vectors (plasmids, cosmids). DNA copies of RNA viruses are made with the retrovirus reverse transcriptase and then cloned into these vectors. After chemical or heat treatments melt (separate) the DNA strands in the sample, the DNA probe is added and allowed to hybridize (bind) with the identical or nearly identical sequence in the sample. The stringency (the requirement for an exact sequence match) of the interaction can be varied so that related sequences can be detected or different strains (mutants) can be distinguished. The DNA probes are labeled with radioactive or chemically modified nucleotides (e.g., biotinylated uridine) so that they can be detected and quantitated. The use of a biotin-labeled DNA probe allows the use of a fluorescent or enzyme-labeled avidin or streptavidin (a protein that binds tightly to biotin) molecule to detect viral nucleic acids in a cell in a way similar to how indirect immunofluorescence or an enzyme immunoassay localizes an antigen.
The DNA probes can detect specific genetic sequences in fixed, permeabilized tissue biopsy specimens by in situ hybridization. When fluorescent detection is used, it is called FISH: fluorescent in situ hybridization. The localization of cytomegalovirus (CMV)-infected (Figure 5-3) or papillomavirus-infected cells by in situ hybridization is preferable to an immunologic means of doing so and is the only commercially available means of localizing papillomavirus. There are now many commercially available microbial probes and kits for detecting viruses, bacteria, and other microbes.
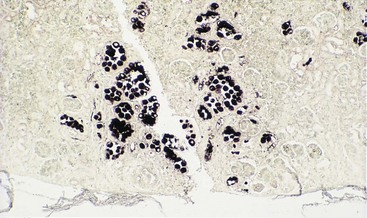
Figure 5-3 In situ localization of cytomegalovirus (CMV) infection using a genetic probe. CMV infection of the renal tubules of a kidney is localized with a biotin-labeled, CMV-specific DNA probe and is visualized by means of the horseradish peroxidase-conjugated avidin conversion of substrate, in a manner similar to enzyme immunoassay.
(Courtesy Donna Zabel, Akron, Ohio.)
Specific nucleic acid sequences in extracts from a clinical sample can be detected by applying a small volume of the extract to a nitrocellulose filter (dot blot) and then probing the filter with labeled, specific viral DNA. Alternatively, the electrophoretically separated restriction endonuclease cleavage pattern can be transferred onto a nitrocellulose filter (Southern blot—DNA : DNA probe hybridization), and then the specific sequence can be identified by hybridization with a specific genetic probe and by its characteristic electrophoretic mobility. Electrophoretically separated RNA (Northern blot—RNA : DNA probe hybridization) blotted onto a nitrocellulose filter can be detected in a similar manner.
The polymerase chain reaction (PCR) amplifies single copies of viral DNA millions of times over and is one of the newest techniques of genetic analysis (Figure 5-4). In this technique, a sample is incubated with two short DNA oligomers, termed primers, that are complementary to the ends of a known genetic sequence within the total DNA, a heat-stable DNA polymerase (Taq or other polymerase obtained from thermophilic bacteria), nucleotides, and buffers. The oligomers hybridize to the appropriate sequence of DNA and act as primers for the polymerase, which copies that segment of the DNA. The sample is then heated to denature the DNA (separating the strands of the double helix) and cooled to allow hybridization of the primers to the new DNA. Each copy of DNA becomes a new template. The process is repeated many (20 to 40) times to amplify the original DNA sequence in an exponential manner. A target sequence can be amplified 1,000,000-fold in a few hours using this method. This technique is especially useful for detecting latent and integrated virus sequences, such as in retroviruses, herpesviruses, papillomaviruses, and other DNA viruses.
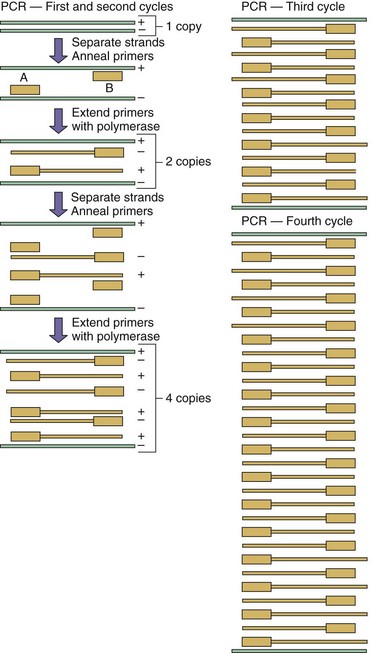
Figure 5-4 Polymerase chain reaction (PCR). This technique is a rapid means of amplifying a known sequence of DNA. A sample is mixed with a heat-stable DNA polymerase, excess deoxyribonucleotide triphosphates, and two DNA oligomers (primers), which complement the ends of the target sequence to be amplified. The mixture is heated to denature the DNA, then cooled to allow binding of the primers to the target DNA and extension of the primers by the polymerase. The cycle is repeated 20 to 40 times. After the first cycle, only the sequence bracketed by the primers is amplified. In the reverse transcriptase PCR technique, RNA can also be amplified after its conversion to DNA by reverse transcriptase. Labels A and B, DNA oligomers used as primers; + and −, DNA strands.
(Modified from Blair GE, Blair Zajdel ME: Biochem Educ 20:87–90, 1992.)
The RT-PCR (reverse transcriptase polymerase chain reaction) technique is a variation of PCR, and it involves the use of the reverse transcriptase of retroviruses to convert viral RNA or messenger RNA to DNA before PCR amplification. In 1993, hantavirus sequences were used as primers for RT-PCR to identify the agent causing an outbreak of hemorrhagic pulmonary disease in the Four Corners area of New Mexico. It showed the infectious agent to be a hantavirus.
Real-time PCR can be used to quantitate the amount of DNA or RNA in a sample after it is converted to DNA by reverse transcriptase. Simply put, the more DNA in the sample, the faster new DNA is made in a PCR reaction, and the reaction kinetics are proportional to the amount of DNA. The production of double-stranded DNA is measured by the increase in fluorescence of a molecule bound to the amplified double-strand DNA molecule or by other means. This procedure is useful for quantitating the number of human immunodeficiency virus (HIV) genomes in a patient’s blood to evaluate the course of the disease and antiviral drug efficacy.
The branched-chain DNA assay is a hybridization technique that is an alternative to PCR and RT-PCR for detecting small amounts of specific RNA or DNA sequences. This technique is especially useful for quantitating plasma levels of HIV RNA (plasma viral load). In this case, plasma is incubated in a special tube lined with a short complementary DNA (cDNA) sequence to capture the viral RNA. Another cDNA sequence is added to bind to the sample, but this DNA is attached to an artificially branched chain of DNA. On development, each branch is capable of initiating a detectable signal. This amplifies the signal from the original sample. The antibody capture solution hybridization assay detects and quantitates RNA : DNA hybrids using an antibody specific for the complex in a technique similar to an ELISA (enzyme-linked immunosorbent assay) (see Chapter 6).
Assay kits that use variations on the aforementioned techniques to detect, identify, and quantitate different microbes are commercially available.
DNA sequencing has become sufficiently fast and inexpensive to allow laboratory determination of microbial sequences for identification of microbes. Sequencing of the 16S ribosomal subunit can be used to identify specific bacteria. Sequencing of viruses can be used to identify the virus and distinguish different strains (e.g., specific influenza strains).
Detection of Proteins
In some cases, viruses and other infectious agents can be detected on the basis of finding certain characteristic enzymes or specific proteins. For example, the detection of reverse transcriptase enzyme activity in serum or cell culture indicates the presence of a retrovirus. The pattern of proteins from a virus or another agent after sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) can also be used to identify and distinguish different strains of viruses or bacteria. In the SDS-PAGE technique, SDS binds to the backbone of the protein to generate a uniform peptide structure and peptide length-to-charge ratio such that the mobility of the protein in the gel is inversely related to the logarithm of its molecular weight. For example, the patterns of electrophoretically separated HSV proteins can be used to distinguish different types and strains of HSV-1 and HSV-2. Antibody can be used to identify specific proteins separated by SDS-PAGE using a Western blot technique (see Chapter 47). The molecular techniques used to identify infectious agents are summarized in Table 5-1.
Table 5-1 Molecular Techniques
| Technique | Purpose | Clinical Examples |
|---|---|---|
| RFLP | Comparison of DNA | Molecular epidemiology, HSV-1 strains |
| DNA electrophoresis | Comparison of DNA | Viral strain differences (up to 20,000 bases) |
| Pulsed-field gel electrophoresis | Comparison of DNA (large pieces of DNA) | Streptococcal strain comparisons |
| In situ hybridization | Detection and localization of DNA sequences in tissue | Detection of nonreplicating DNA virus (e.g., cytomegalovirus, human papillomavirus) |
| Dot blot | Detection of DNA sequences in solution | Detection of viral DNA |
| Southern blot | Detection and characterization of DNA sequences by size | Identification of specific viral strains |
| Northern blot | Detection and characterization of RNA sequences by size | Identification of specific viral strains |
| PCR | Amplification of very dilute DNA samples | Detection of DNA viruses |
| RT-PCR | Amplification of very dilute RNA samples | Detection of RNA viruses |
| Real-time PCR | Quantification of very dilute DNA and RNA samples | Quantitation of HIV genome: virus load |
| Branched-chain DNA | Amplification of very dilute DNA or RNA samples | Quantitation of DNA and RNA viruses |
| Antibody capture solution hybridization DNA assay | Amplification of very dilute DNA or RNA samples | Quantitation of DNA and RNA viruses |
| SDS-PAGE | Separation of proteins by molecular weight | Molecular epidemiology of HSV |
DNA, Deoxyribonucleic acid; HIV, human immunodeficiency virus; HSV-1, herpes simplex virus-1; PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism; RNA, ribonucleic acid; RT-PCR, reverse transcriptase polymerase chain reaction; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Which procedure(s) can be used for the following analyses and why would that procedure be used?
1. Comparison of the major bacterial species present in the normal flora of a thin and an obese individual.
2. Comparison of the normal bacterial flora that is associated with chronic oral abscesses.
3. A 37-year-old man has flulike symptoms. A viral infection is suspected. The agent needs to be identified from a nasal wash sample.
4. The efficacy of antiretroviral therapy in an HIV-infected individual can be evaluated by quantitating the number of viral genomes in her blood.
5. A Pap smear is suspected to contain human papillomavirus (HPV) infection. How can HPV be detected in the sample?
6. A baby is born with microcephaly, and CMV is suspected. Urine contains cells with a characteristic CMV-infected morphology. How can CMV infection be verified?
7. Antiviral resistance and disease severity are analyzed for hepatitis C virus isolates from intravenous drug users.
1. The gene for 16S ribosomal RNA is amplified by PCR using universal primers that recognize large groups of bacteria, and then specific sequences within the gene are amplified and sequenced to determine individual bacteria and strains.
2. The gene for 16S ribosomal RNA is amplified by PCR using universal primers that recognize large groups of bacteria, and then specific sequences within the gene are amplified and sequenced to determine individual bacteria and strains.
3. RNA can be isolated from the samples, converted to DNA with reverse transcriptase and then amplified with a mixture of defined DNA primers by PCR (RT-PCR). The presence of specific viral sequences can then be detected by PCR using virus specific primers.
4. Quantitative RT-PCR can be used to determine the number of genome copies. If the individual is conscientious with their therapy, then the relevant viral genes can be sequenced to determine the nature of a resistant mutant.
5. In situ hybridization can be used to demonstrate the presence of HPV DNA sequences within the cells of the Pap smear.
6. In situ hybridization can be used to demonstrate the presence of CMV DNA sequences within the cells in the urine. PCR can also be used to detect viral sequences in the urine or the baby’s blood.
7. Viral genome sequences can be detected by RT-PCR analysis of RNA isolated from blood. Specific target genes can subsequently be amplified and then sequenced to determine the basis for the resistance.